
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation–π and hydrophobic interaction controlled PET recognition in double mutated cutinase – identification of a novel binding subsite for better catalytic activity†
Anjima Jamesa and
Susmita De *b
*b
aDepartment of Applied Chemistry, Cochin University of Science and Technology, Thrikkakara, Kochi 682 022, Kerala, India
bDepartment of Chemistry, Center for Computational Chemistry & Drug Discovery, University of Calicut, Calicut University P.O., Malappuram 673 635, Kerala, India. E-mail: dr_susmita_de@uoc.ac.in
First published on 15th July 2022
Abstract
Accelerated hydrolysis of polyethylene terephthalate (PET) by enzymatic surface modification of various hydrolases, which would not degrade the building blocks of PET in order to retain the quality of recycled PET, is a promising research area. Many studies have been reported to identify mutations of different hydrolases that can improve PET degradation. Recently, the mutation of glycine and phenyl alanine with alanine in cutinase was found to improve the activity of PET degradation 6-fold. Yet, a deep insight into the overall structural basis as well as the explicit role played by the amino acid residues for PET degradation is still elusive, which is nevertheless important for comparative analyses, structure–function relations and rational optimization of the degradation process. Our molecular dynamics simulations coupled with quantum mechanical study demonstrate that mutations of anchor residue phenyl alanine to alanine at the PET binding cleft of cutinase unveiled a distal yet novel binding subsite, which alters the nature of dispersive interaction for PET recognition and binding. The phenyl alanine engages in π–π interaction with the phenyl ring of PET (−8.5 kcal mol−1), which on one side helps in PET recognition, but on the other side restricts PET to attain fully extended conformations over the entire binding cleft. The loss of π–π interaction due to mutation of phenyl alanine to alanine is not only compensated by the favourable cation–π and hydrophobic interactions from the arginine residues (−17.1 kcal mol−1) found in the newly discovered subsite, but also favours the fully extended PET conformation. This subsequently impacts the overall increased catalytic activity of mutated cutinase.
Introduction
Polymeric esters of terephthalic acid or PET, which is a polyester comprising terephthalic acid (TPA) and ethylene glycol (EG) linked via ester bonds, constitute a good portion of the plastics industry due to their massive production at low cost.1–4 The chemical structure of these plastics makes them durable and in turn also resistant to natural processes of degradation,5 meaning harsh chemical conditions as well as high temperature are required to degrade them.6,7 Hence, they are a potential threat to human health.8 However, it is nearly impossible to replace plastics with other materials throughout the various industries. Therefore, it is essential to adopt the concept of reduce, recycle and reuse for a sustainable future, but not much recycling and reuse happens.9 PET has hydrolysable ester bonds, which can in-principle be cleaved by esterase enzymes, and this makes PET an achievable target for biological recycling. PET can be bio-degraded to monomeric products such as bis(hydroxyethyl)terephthalic acid (BHET), mono(hydroxyethyl)terephthalic acid (MHET), terephthalic acid (TPA) and ethylene glycol (EG), which can be reused for the reproduction of recycled PET of high quality.10 Thus, enzymatic degradation of PET offers a green and promising alternative with regeneration of monomers, thereby adding to the economy.The century long exposure of PET to the ecosystems has provided a long time for the bacteria in the nature to evolve and utilize PET as a novel carbon source.10 In 2016 for the first time, Yoshida et al. reported an enzyme, IsPETase from Ideonella sakaiensis, which can cleave PET at 30 °C.10 IsPETase, belonging to hydrolase superfamily, shows high substrate specificity to PET, though it suffers from low reactivity and low thermal stability.11,12 Since its discovery, a lot of research has been focusing on improving the efficiency and thermal stability by introducing mutations13–17 and other strategies.18,19 There are approximately 69 IsPETase like enzymes from phylogenetically different organisms, which can in principle degrade the ester linkage present in PET.14,20–22 Studies to improve the performance of these enzymes is indeed a valuable research area.23–25 In the year 2022, a novel enzyme PHL7 was discovered by Zimmermann and co-workers, which shows promising activity for large scale degradation of PET.26,27 In the same year, application of machine learning techniques are also used to design FAST-IsPETase,28 which shows higher activity.28
Thus, unsurprisingly, the hydrolase family of enzymes from evolutionary diverse sources is greatly explored for the reactivity towards PET degradation. Among them, the bacterial thermophilic cutinase family have emerged as one of the suitable candidate for further research owing to its structural similarity and stability at high temperature between 60–84.7 °C,29,30 which is much higher than the melting temperature (Tm) of IsPETase (46.8 °C)14 and above the glass transition temperature (Tg = 65–70 °C) of PET.31 The earlier reports pointed out, that the most reactive bacterial cutinases towards PET are leaf compost32 (LC-cutinase or LCC) and TfCut2 from Thermobifida fusca.33 The reactivity calculated by weight loss of low crystalline PET at 65–70 °C with LCC and G62A mutated TfCut2 (ref. 15) after 24 h are 24% and 20% respectively.11 Structural similarity of TfCut2 (51%) with IsPETase is slightly more than that of LCC (49%), which makes TfCut2 a valid target for our study.23 Moreover, the amino acid residues involved in the cleavage of ester bond in TfCut2 are the same as that of IsPETase.12
Muller et al. in 2005 first reported the PET degradation by TfCut2 with a degradation rate of 2.5 nmol−1 min−1 cm−2.33 Later in 2016, Barth et al. reported TfCut2 could be inhibited by degradation products MHET and BHET, with MHET being the major inhibitor.15 Though, degradation action of IsPETase is higher than that of WT TfCut2 at their optimum temperatures on PET film,10 a notable increase in activity was seen with the G62A mutation on TfCut2.15 The mutant G62A decreased the binding constant by ∼5.5 fold leading to relieved product inhibition by MHET than in WT TfCut2. In 2019, Furukawa and co-workers added another mutation F209A to the single mutant resulting in G62A/F209A double mutant (DM) TfCut2 with a PET degradation rate of 15.0 nmol−1 min−1 cm−2.11 Interestingly, in 2018 Austin et al. reported a double mutant W130H/S209F IsPETase, which mimics the active site of TfCut2 cutinase showing a higher PET degradation rate.13 However, the enzymatic PET degradation process is yet to be used in a large scale.34,35 Therefore, to improve the bio-degradability of PET, it is very important to identify the important amino acid residues present in the enzyme responsible for the interaction with PET and at the same time classify the nature of interaction these amino acid residues exhibits. This process would help engineering of PET degrading enzymes with higher reactivity.
In general, it is observed that mutations can increase the activity of all these enzymes. However, a systematic approach in predicting appropriate mutations to achieve optimum PET degradation is elusive. Thus, our study aims to correlate the increased PET degradation activity of DM TfCut2 with the structure-based role of the mutation, which not only is instrumental for understanding the catalytic mechanism but it will also offer avenues for the applications of genetic engineering strategies to improve the efficiency of enzymes. In doing this, the major challenge is the complexity in characterizing all plausible approaching pathways that can lead to different PET binding conformations on the target protein. In order to address this, the detailed structural information as well as different types of interactions on different PET binding states at the molecular level was analysed based on Molecular Dynamics Simulations and Quantum Mechanical calculations to gain a deep understanding and rational control of PET binding. We have chosen the TfCut2 (WT) and its double mutated (DM) variant G62A/F209A and a tetrameric PET, 2-hydroxyethyl-(monohydroxyethyl terephthalic acid)4 (2-HE(MHET)4) for our theoretical study.
Computational details
Structure preparation of proteins and PET
The PDB structure 4CG1 (resolution 1.40 Å) was used as starting structure for WT TfCut2.36 The missing residue, Asp246 was added using SWISS-PROT.37 Hydrogens were added using AMBER2038,39 leap program corresponding to the protonation states predicted by H++40 at pH 7. The protein structure thus obtained was minimized for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps using the steepest descend-algorithm41 using the CUDA version of pmemd program in AMBER20.38 G62A/F209A TfCut2 (ref. 11) structure was prepared from the above WT TfCut2 structure by introducing mutations via PyMOL.42 2-Hydroxyethyl-(monohydroxyethyl terephthalic acid)4 or 2-HE(MHET)4 was taken as a representative of PET polymer.14 Note that, earlier reports indicate that the tetrameric PET is long enough to establish contact with the entire binding cleft of TfCut2.14 The tetrameric PET structure is minimized using the B3LYP43 functional and 6-31+g(d)44,45 basis set using Gaussian 09 program package.46 The molecular electrostatic surface potential (MESP) of the protein was calculated using PDB2PQR47,48 online server followed by continuum electrostatics calculations employed in the Poisson–Boltzmann solver (ABPS).49
000 steps using the steepest descend-algorithm41 using the CUDA version of pmemd program in AMBER20.38 G62A/F209A TfCut2 (ref. 11) structure was prepared from the above WT TfCut2 structure by introducing mutations via PyMOL.42 2-Hydroxyethyl-(monohydroxyethyl terephthalic acid)4 or 2-HE(MHET)4 was taken as a representative of PET polymer.14 Note that, earlier reports indicate that the tetrameric PET is long enough to establish contact with the entire binding cleft of TfCut2.14 The tetrameric PET structure is minimized using the B3LYP43 functional and 6-31+g(d)44,45 basis set using Gaussian 09 program package.46 The molecular electrostatic surface potential (MESP) of the protein was calculated using PDB2PQR47,48 online server followed by continuum electrostatics calculations employed in the Poisson–Boltzmann solver (ABPS).49
Molecular docking
The minimized tetrameric PET and protein structure was used for performing 11 sets of docking on each protein viz. WT TfCut2 and G62A/F209A TfCut2 with PET tetramer using Autodock Vina.50 The 99 PET conformers thus generated were classified using a distance cutoff of 5 Å between any ester carbonyl carbon-atom of PET to the hydroxy–oxygen atom of the catalytic serine. These structures were further grouped using a PET RMSD cut-off of 2 Å. This procedure resulted in 19 and 16 different poses of PET with WT and DM TfCut2 respectively, which were subsequently used for Molecular Dynamics Simulations to investigate the conformational landscape.Parametrization of PET and molecular dynamics simulations
PET was parametrized using the antechamber51 module of AMBER2038 using AM1-BCC52 charge model and the Generalized AMBER Force Field (GAFF).53 The protein part was treated with the ff14SB54 forcefield. Proteins with docked tetrameric PET was placed in 0.15 M NaCl with TIP3P55,56 water in a truncated octahedron box. Non bonded interactions were restricted to 12 Å cut-off and long range electrostatic interactions were treated using the particle mesh Ewald method.57 Solvated protein-PET system was relaxed by minimization of 10000 steps using the steepest descend-algorithm.58 The system was gradually heated in 100 K increments from 0 K to 300 K with positional restraints on the protein and tetrameric PET system for 120 ps. Positional restraint was then slowly reduced from 12 kcal mol−1 Å−2 to zero for the protein, but by retaining a 0.5 kcal mol−1 Å−2 restraint on PET, which was then released followed by a 200 ns production run on each of the structures obtained from docking study. The 4 trajectories where PET moved out of the binding site were not considered further, while the rest was run for 1 μs simulation. This resulted in 15 and 12 simulations each of one μs duration for WT and DM TfCut2, respectively. Also, a total of 1 μs MD simulation on WT and DM proteins without PET were performed.
All MD simulations were performed at a constant pressure of 1 bar in the NPT59 ensemble. Hydrogen-mass repartition60 along with SHAKE61 algorithm has been applied on bonds involving hydrogen atoms. VMD,62 CPPTRAJ63 and the Qt version of xmgrace64 were used for trajectory analysis. The simulations were clustered into different groups by the average-linkage hierarchical clustering algorithm65 in AMBER20. Clustering using this algorithm is reported to be a reliable approach for analysing the MD trajectories.65 The trajectories obtained after clustering were subjected to binding energy calculations using the Molecular Mechanics-Poisson Boltzmann Surface Area66 (MM-PBSA) method. Though this binding energy calculation does not include the entropic contribution to ligand-binding affinity, it is still reported to give a good correlation with the experimental values.67 Normal-mode analysis was carried out on extended and folded conformers of PET with WT and DM protein systems using the nabnmode module of AMBER to estimate the conformational entropy contributions to the binding free energy using Kongsted method (Fig. S11†).68 This follows the same trend as the MM-PBSA energies, indicating an steady entropy contribution for all the conformers. Further to highlight the most important residues involved in the ligand recognition, MM-PBSA per residue energy decomposition analysis was also carried out.
Quantitative estimation of PET binding energy
The lower order symmetry adapted-perturbation theory calculations, SAPT069,70 which comes with a good error cancellation with jun-cc-pvDZ71 basis was employed with density fitting techniques and core electron freezing70 using the quantum chemistry package PSI4.72 The total interaction energy in SAPT is a sum of energy terms arising from electrostatic, exchange, induction and dispersion effects.70 Further, PET binding energy was calculated including dispersion73 corrections at the B3LYP-D3/6-31+g(d) level of theory by correcting basis set super position error using Gaussian 09 program package.46Results and discussion
The amino acid residues involved directly in the ester bond cleavage of PET are conserved in wild type (WT) and double mutated (DM) cutinase, yet the catalytic activity differs significantly. Hence, the catalytic activity of these enzyme is hugely dependent upon the molecular recognition of the substrate to the PET binding site through multiple weak non-covalent interactions. Concomitantly, the extent of these weak interactions depends upon the orientation of the amino-acid side chain residues and the binding poses of the substrate. A deeper understanding of the complementary molecular reactivity and molecular recognition of PET by cutinase can be obtained by the detailed analysis of the plausible PET binding poses on the protein surface.PET binding poses on WT and DM TfCut2
In order to gain a preliminary understanding, we have extensively explored 99 possible binding poses of tetrameric PET the with the PET binding cleft of each of WT and DM TfCut2 by molecular docking using AutoDock Vina.42 The DM TfCut2 has an average binding affinity of −6.5 kcal mol−1 (range: −5.0 to −7.5 kcal mol−1) which is a little higher than the average binding affinity of WT TfCut2, −5.5 kcal mol−1 (range: −4.2 to −7.2 kcal mol−1). 46 conformations show a docking score higher than the average binding affinity in WT TfCut2, while 72 conformations of tetrameric PET in DM TfCut2 have a docking score higher than the average. However, all the binding poses with higher docking score are not identical, which warrants more thorough analysis of the different binding poses to enable structural basis for enhanced degradation due to mutation.The observed PET binding poses from docking can be broadly classified into three categories, (i) extended, where the entire tetrameric PET maintains contact with the entire binding cleft (ii) semi-extended where the tetrameric PET does not bind throughout the entire region of binding cleft and (iii) folded, where the tetrameric PET forms a loop like structure, binding only to a very small region (Fig. 1). Austin et al.13 and Joo et al.14 in two separate works had reported that tetrameric PET adopting an extended conformation in IsPETase shows higher docking score as well as higher reactivity. Likewise, we have identified the extended PET conformers among the docked structures for both WT and DM TfCut2. Only 15 docked poses exhibit extended conformation in WT TfCut2 (Fig. 2a), while as much as 51 docked poses exhibit extended conformation in DM TfCut2 (Fig. 2b). Thus, the binding site cleft of DM TfCut2 seems more suitable to extended PET conformations, whereas folded PET conformations are more common for WT TfCut2. A comprehensive analysis of the docked conformations of PET indicates that PET binds to three-fourths of the active site (up to F209) in a similar fashion in both WT and DM TfCut2. The visual inspection revealed that DM TfCut2 uses an unprecedented terminal region of the protein for binding PET in the extended conformation as compared to WT TfCut2 (Fig. 2). Earlier binding studies did not indicate PET binding to such residues.13,14,36,74–77 At this point we might reason that, the mutation of the aromatic Phe209 (F209) in WT to hydrophobic Ala209 (A209) in DM cutinase might have affected the conformation of PET binding on the surface of cutinase. However, further analysis is required to elucidate this striking difference in the mode of PET binding upon mutation.
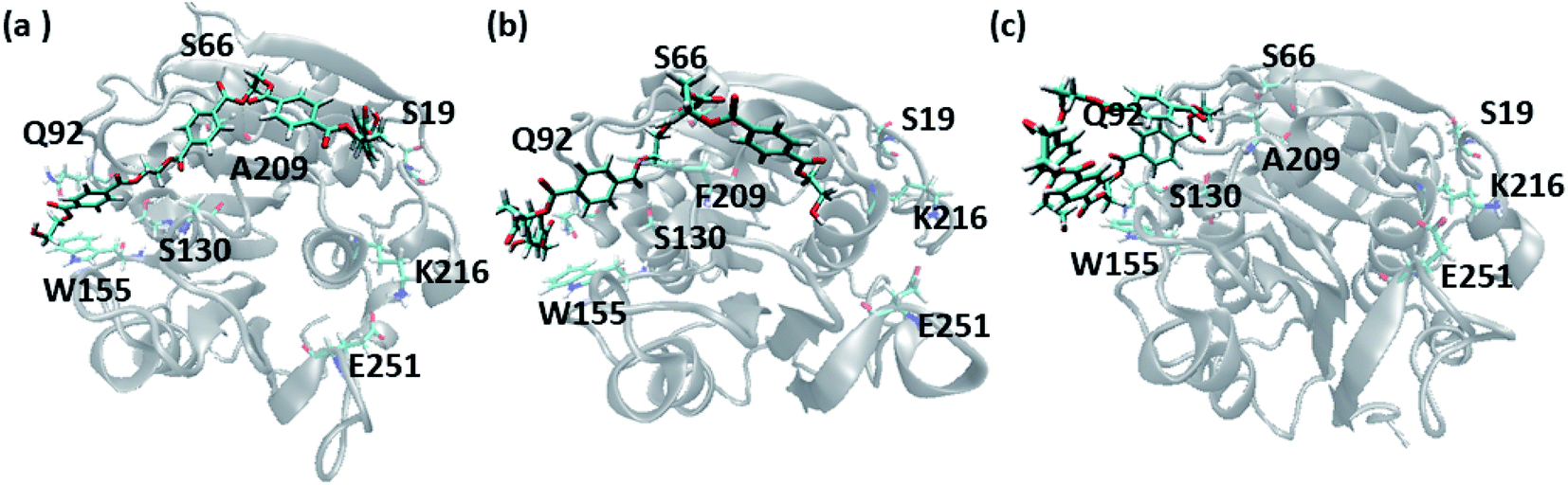 | ||
| Fig. 1 Representative PET binding poses in cutinase identified as (a) extended (b) semi-extended and (c) folded. The important amino acid residues are marked. | ||
 | ||
| Fig. 2 Extended docked conformers of tetrameric PET in (a) 15 in WT TfCut2 (b) 51 in DM TfCut2. The region in the dotted circle indicates the terminal portion of the binding site. | ||
From theoretical mechanistic studies on the enzymatic PET hydrolysis,78–82 it was proposed that the hydroxyl group of Ser130 of the catalytic triad initiates the attack to the carbonyl carbon of the ester bond, followed by the stabilization of the negative charge by the two back bone nitrogen atoms of Tyr60 and Met131 of the oxyanion hole.83 Thus, in order to identify the best binding poses of PET with WT and DM TfCut2 in terms of catalytic activity, it is very important to analyse the proximity of the carbonyl carbon-atom of PET to Ser130 that lie in the subsite S1. Thus, a distance cut off of 5 Å is used to identify 62 and 65 PET poses respectively for WT and DM TfCut2. These structures were further grouped together by a PET RMSD cut-off of 2 Å, and one representative pose with the highest docking score was selected from each group. This resulted in shortlisting of 19 and 16 PET complex of WT TfCut2 and DM TfCut2 respectively (Fig. S2 and S3†), which were further subjected to detailed analysis to identify what might be responsible for PET binding to novel sub-site in DM cutinase.
Analysis of structure and electrostatic potential of WT & DM TfCut2
A closer look at the entire PET binding cleft of cutinase reveals that it constitutes of 36 amino acid residues (Fig. 3). These amino acid residues can be divided into two categories, the ones that are directly involved in the catalytic cleavage of the ester bonds or catalytic residues, the others that are involved mainly in the recognition and binding of PET on the protein surface or anchor residues. One can classify the entire cleft into four major subsites viz. S1, S2, S3 and S4, where each subsite can roughly accommodate a monomeric PET unit. A similar description of the entire binding cleft was also provided for PET degrading enzyme IsPETase.14,77 However, our docking studies have identified an unprecedented binding subsite, named as S4′ that seems an alternative to S4. Note that, the binding of PET into S4′ was not previously reported. Also, the presence of Arg18 at the extreme end of S4′ have close resemblance with the binding sub-site S4 of IsPETase, where Arg251 marks the end of the binding cleft.14 For easy identification purpose, the amino acid residues in each subsite are colour coded as green (S1), yellow (S2), red (S3), blue (S4) and cyan (S4′) respectively for both WT and DM TfCut2 (Fig. 3a and b). Fig. 3c and d indicates that, the conventional binding cleft constituted by subsites S1, S2, S3 and S4 has a narrow and inverted V-shape (Q92, W155, F209/A209, E251, K216, S66), while the binding cleft constituted by subsites S1, S2, S3 and S4′ appears to be wider and linear (Q92, W155, F209/A209, K216, S19, S66). Also, the distance between the C-alpha atoms of the terminal residues shows that the mutation does not alter the overall size and shape of the cavity. | ||
| Fig. 3 Cartoon representation of (a) WT TfCut2 and (b) DM TfCut2 with amino acid residues in subsites S1, S2, S3, S4 and S4′ shown in green, yellow, red, blue and cyan licorice representations respectively. Plot of molecular electrostatic potential (MESP) for (c) WT TfCut2 and (d) DM TfCut2 with potential over the range −8.0 to +8.0kT/e. | ||
The plot of the molecular electrostatic potential (MESP) on the van der Waals surface clearly indicates that the S1, S2 and S3 subsites are mostly neutral with slight negative region in S1 and S3 due to the presence of aromatic Trp155 (W155), Tyr60 (Y60) and Phe209 (F209) residues while the S4 subsite is mainly polar with both electrophilic and nucleophilic regions near Lys216 (K216) and Glu251 (E251) respectively (Fig. 3c). Similar to S4, the alternate subsite S4′ also contains both nucleophilic (Trp69 and Glu72) and electrophilic (Arg73 and Arg18) residues, indicating similarity in the molecular electrostatic potential. The mutations of Gly62 by Ala62 in S2 and Phe209 by Ala209 in S3 did not change the overall MESP. However, local decrease in the electron density near the Phe209 in WT TfCut2 is observed after the mutation (Fig. 3d).
Molecular-dynamics simulations on WT and DM (G62A/F209A) TfCut2 with and without PET
1 μs Molecular Dynamics simulations was carried out on WT and DM TfCut2 with and without PET to identify the amino acid residues and their corresponding nature of interactions responsible for PET recognition. The average RMSD value of the WT and DM TfCut2 without the tetrameric PET is 2.60 Å and 1.93 Å respectively (Fig. 5a), depicting overall stability of the proteins. The RMSD of the average structures generated after 1 μs Molecular Dynamics simulations on WT and DM TfCut2 without PET, showed an average RMSD of 1.33 Å of the DM TfCut2 with respect to the WT variant. The RMSF analyses indicates higher fluctuation of the N-terminal and C-terminal residues (Fig. 4b and S1†). The low RMSD of the average structures (0.83 Å) by excluding the N-terminal and C-terminal residues highlights the importance of the local conformational changes for PET recognition. | ||
| Fig. 4 Root mean square deviations (RMSD) and root mean square fluctuation (RMSF) in Å of the 1 μs Molecular Dynamics Simulation of WT TfCut2 (black) and DM G62A/F209A TfCut2 (red). The average RMSD is indicated by a line parallel to the x-axis in corresponding axis. | ||
To explore the binding interaction of PET with the side-chain in WT and DM TfCut2, 1 μs MD simulation was run on each of the selected 15 and 12 PET conformers for the WT and DM TfCut2 respectively. The analysis of the RMSD (Fig. S4 and S5†) shows that though the RMSD of the protein–PET complexes is significantly higher, the RMSD of protein alone in the protein–PET complex is similar to that of the proteins. Thus, the significant change in the RMSD of the protein–PET complex is mainly contributed from the different conformations adopted by PET in the PET binding cleft. Looking at the trajectories, conversion between the folded and extended conformers of PET is widely observed, validating the possible binding of both the conformers in the binding cleft. In addition, the RMSF plot of the cavity residues with and without PET indicates that the subsites S1 and S3 undergo greater extent of conformational changes in WT TfCut2, while subsite S1, S4/S4′ undergo major changes in DM TfCut2 for PET binding (Fig. S6†).
We have further used hierarchical average-linkage clustering implemented in AMBER20 to identify the major conformational isomers of PET from the MD simulation trajectories of the PET bound WT and DM TfCut2. Clustering of all the trajectories for WT and DM TfCut2 produced 78 and 67 clusters respectively (Fig. 5 and 6). The clustering analysis of WT TfCut2 clearly indicates that, majority of the trajectories yield a single preferred conformation, which is corroborated well with the minimal fluctuation observed in the RMSD and RMSF (Fig. S4–S6†). The WT TfCut2 shows a clear preference for PET conformers that are interacting with the Phe209 of the S3 subsite leading to either a semi-extended or a folded conformation. There are only a few instances where PET attain a fully extended conformation by binding throughout all the four subsites S1–S4/S1–S4′. The binding affinity of all the PET conformers in each cluster was calculated using MM-PBSA analysis, which ranges from −15.2 kcal mol−1 to −38.1 kcal mol−1 (Fig. 5). Among the prominent clusters, the higher binding energy is mostly observed for the semi-extended PET conformers, which indicates the prominent role of Phe209.
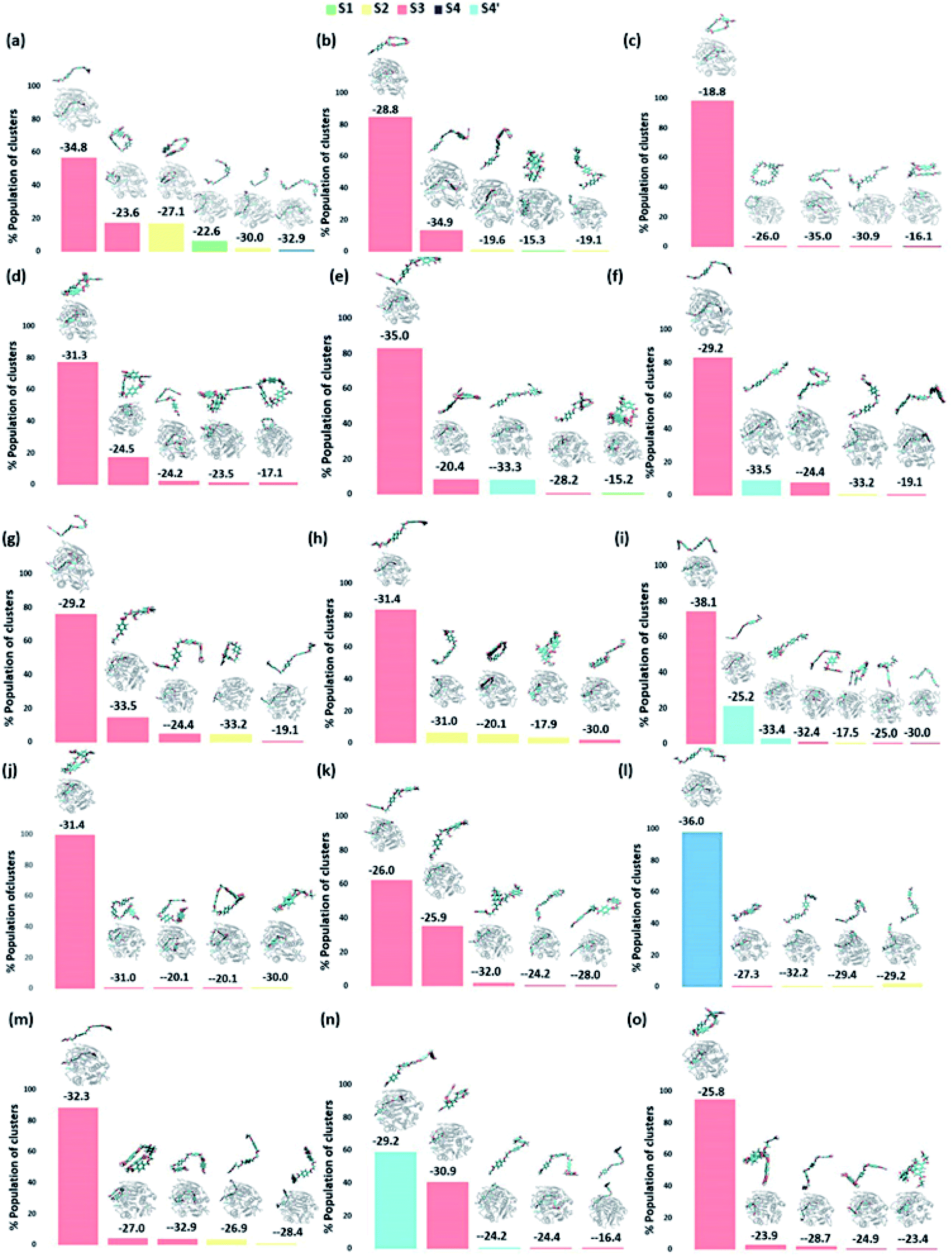 | ||
| Fig. 5 The output of clustering on 15 different WT TfCut2-PET systems with the population percent of each cluster shown in Y axis. The average structure of each cluster along with its binding energy (kcal mol−1) obtained via MM-PBSA calculations is shown above the bar corresponding to each cluster. A zoomed image of PET is shown on top of each average structure. | ||
 | ||
| Fig. 6 The output of clustering on 12 different DM TfCut2-PET systems with the population percent of each cluster shown in Y axis. The average structure of each cluster along with its binding energy (kcal mol−1) obtained via MM-PBSA calculations is shown above the bar corresponding to each cluster. A zoomed image of PET is shown on top of each average structure. | ||
On the contrary, the clustering analysis on the trajectories of DM TfCut2 reveals conformational flexibility of PET in the binding cavity leading to more than one prominent clusters. This corroborates with the higher RMSD and RMSF as compared to WT cutinase (Fig. 6). Note that, majority of them indicates binding to the hitherto unidentified subsite S4′ leading to fully-extended conformations more often than WT TfCut2. Also, the binding energy of PET in DM TfCut2 ranges from −10.2 to −48.8 kcal mol−1 and the binding energies for the fully extended PET conformers binding to S1–S4′ subsites are noticeably higher. Therefore, the higher PET affinity of DM TfCut2 than WT variant can possibly result from binding to the novel subsite S4′, which also leads to extended PET conformations. Note that, the extended PET conformations in the binding cleft has been linked with higher activity of IsPETase.13,14 Thus, extended PET conformations with relatively higher binding energy might lead to higher activity in TfCut2 as well.
In order to identify the residues that might play important role in energetically favouring extended PET conformations over the folded ones, we have selected the trajectories corresponding to extended and folded conformations having the five highest and five lowest binding energies in both WT and DM TfCut2. The average structures of these 20 conformers are given in Fig. S7 and S8.† Note that, fully-extended PET conformations are only observed for DM TfCut2, while the WT TfCut2 exhibits semi-extended conformations. The Fig. S9, S10 and S12† show the contact plot analysis, MM-PBSA per-residue binding energy and comparison of average per residue binding energy with the average close contact respectively calculated using MM-PBSA method for each subsite on the extended and folded PET for WT and DM TfCut2. The pair-wise binding energy for PET with all the residues of the entire PET binding cleft of WT and DM TfCut2 (S1, S2, S3, S4 and S4′) was plotted in Fig. 7. We can clearly see different interaction energy pattern for the WT and DM TfCut2 as well as for the extended and folded PET. In general, the residues with highest number of close contacts have higher interaction energy with PET. We have also performed zero order Symmetry Adapted Perturbation Theory (SAPT0) analysis to get a quantitative picture of different energy components contributing to the total energy.
 | ||
| Fig. 7 Pairwise energy contribution of residues with PET for five extended and folded clusters of (a) WT TfCut2 extended PET conformer (b) WT TfCut2 folded PET conformer (c) DM TfCut2 extended PET conformer and (d) DM TfCut2 folded PET conformer calculated using MM-PBSA method. | ||
The S1 residues Ser130 from the catalytic triad and Tyr60, Met131 from oxyanion hole that are essential for the ester bond cleavage of PET as well as the anchor residues such as hydrophobic Ile178 and aromatic Trp155 that are essential for PET binding exhibit more close contact and higher energy contribution irrespective of the conformational change and mutation (Fig. S9, S10 and Movie S1†). Thus, the residues in S1 can be considered as the most important for the PET recognition and cleavage by cutinases. Hence, any mutation at S1 might compromise the recognition and degradation of PET. Indeed studies on IsPETase, which also contain the same residues, shows decline in activity upon mutation of S1 residues.14,84
Tyr60 of the oxyanion hole is a non-polar aromatic residue with a PhOH functional group. Three main interactions viz., displaced π-stacking (PETPh⋯PhY60), T-stacking (PETC–H⋯PhY60 and Y60C–H⋯PhPET) are mainly observed between Tyr60 and PET along with hydrogen bonding between amide hydrogen of Tyr60 and carbonyl oxygen of PET (PETO⋯H–NY60) (−10.7, −7.8 and −7.0 kcal mol−1 in Fig. 8a–c and Movie S1†). Note that, the hydroxyl group of Tyr60 does not interact with non-polar functional groups in PET. The other two residues, Met131 and Ser130, responsible for the ester bond cleavage show lesser number of interactions and less energy contribution as compared to Tyr60. The non-polar Met131 is involved in weak M131C–H⋯πPET, hydrophobic M131C–H⋯H–CPET and PETO⋯H–NM131 hydrogen bonding (−4.3 kcal mol−1, Fig. 8d) and the polar Ser130 show occasional hydrogen bonding interaction S130O–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPET with the carbonyl group of the PET (−6.2 kcal mol−1, Fig. 8e) in the neutral state, which resembles the state prior to proton abstraction by His208 in the catalytic cycle.78,80 The aromatic residue Trp155, which is responsible for anchoring the phenyl ring of PET to S1, shows PETC–H⋯πw155 (−8.1 kcal mol−1, Fig. 8f) interaction and weak T-stacking interaction with PET (−5.0 kcal mol−1, Fig. 8g). The hydrophobic residue Ile178 shows mainly weak I178C–H⋯πPET interaction with one or both phenyl rings of PET (−6.5 kcal mol−1, Fig. 8h). The interaction energy of Tyr60, Ile178 and Trp155 are found to be highest in the folded PET conformer with double mutated TfCut2, explaining the overall higher interaction energy for the folded PET conformer in DM TfCut2 as compared to WT TfCut2.
CPET with the carbonyl group of the PET (−6.2 kcal mol−1, Fig. 8e) in the neutral state, which resembles the state prior to proton abstraction by His208 in the catalytic cycle.78,80 The aromatic residue Trp155, which is responsible for anchoring the phenyl ring of PET to S1, shows PETC–H⋯πw155 (−8.1 kcal mol−1, Fig. 8f) interaction and weak T-stacking interaction with PET (−5.0 kcal mol−1, Fig. 8g). The hydrophobic residue Ile178 shows mainly weak I178C–H⋯πPET interaction with one or both phenyl rings of PET (−6.5 kcal mol−1, Fig. 8h). The interaction energy of Tyr60, Ile178 and Trp155 are found to be highest in the folded PET conformer with double mutated TfCut2, explaining the overall higher interaction energy for the folded PET conformer in DM TfCut2 as compared to WT TfCut2.
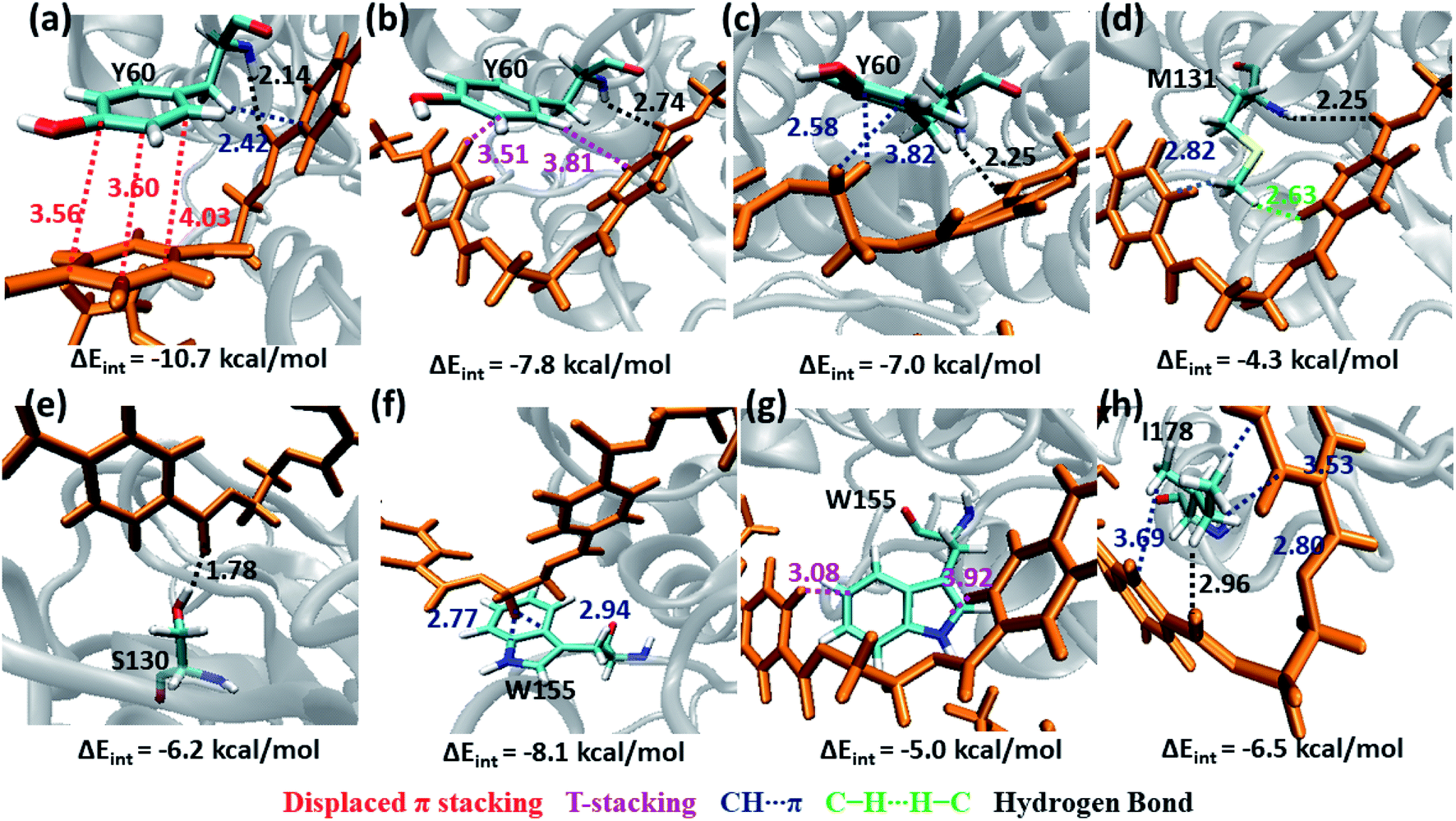 | ||
| Fig. 8 Snapshots showing major interactions in subsite S1. The cartoon structure of the protein is shown in silver with the target amino acid residues and PET shown in cyan and orange licorice representation respectively. The various interactions C–H⋯π, displaced-π stacking, T-stacking, C–H⋯H–C and hydrogen bonds are shown in blue, red, purple, green and black colors respectively. This color code for interactions and residues will be followed in the following sections. The single letter code indicating the amino acids are Tyr (Y), Met (M), Ser (S), Trp (W), and Ile178 (I). | ||
Other than Ser130, the major contribution to the total interaction energy with PET comes from dispersion component indicating the importance of hydrophobic interaction (Fig. S13 and S14†). However, the polar Ser130 have electrostatic component as major contributor to the total interaction energy (Fig. S13 and S14†). Note that, even though the energy corresponding to the individual interaction energy is very less, a significant contribution arises from the cooperative stabilizing effect of multiple non-covalent interactions leading to preferential PET recognition.
Weak hydrophobic C–H⋯H–C interactions of PET are mainly observed with Gly59 and Gly62 (−4.9 and −1.1 kcal mol−1, Fig. 9a and b) from S2. The mutation of Gly62 with Ala62 (−1.5 kcal mol−1, Fig. 9c) does not affect the interaction energy, hence might not play important role in PET binding and cleavage. Indeed, the major role of G62A mutation was reported to assist in release of product.74 Polar hydroxyl group of Thr61 shows hydrogen bonding PETCO⋯H–OT61, PETC–O⋯H–OT61 interactions and the methyl group is involved in PETC–H⋯H–CT61 and T61C–H⋯πPET interactions (−4.3 kcal mol−1, Fig. 9d). Occasional close contact with the S2 residue His129 is observed in WT and DM TfCut2. Unlike the aromatic residue Phe209 in S3, His129 does not participate in stronger π–π interaction due to the unfavourable conformation perpendicular to the phenyl ring of PET, instead shows weak H129C–H⋯H–CPET and H129CO⋯H–CPET interactions (−4.2 kcal mol−1, Fig. 9e). Note that, mutation of Trp159 in WT IsPETase with His159 increases the activity significantly.13 Thus, S2 residues in general show less contact and hence lower interaction energy with PET as compared to S1, irrespective of the conformational change of PET as well as G62A mutation of TfCut2. Also, the presence of non-polar residues in S2 is corroborated with the dispersion interaction being the major energy component (Fig. S13 and S14†).
 | ||
| Fig. 9 Snapshots showing major interactions in subsite S2. The color code is same as the S1 interactions. The single letter code indicating the amino acids are Gly (G), Ala (A), Thr (T) and His (H). | ||
The next subsite that shows significant close contact with PET is S3, where the aromatic residue Phe209 in WT TfCut2 exhibit greater contact with both the semi-extended and folded PET conformers through displaced π and T-stacking interactions with the phenyl ring of PET (−8.5 kcal mol−1, Fig. 10a and Movie S2†). When F209 is mutated to aliphatic Ala209, the π–π interactions are no longer present rather weak hydrophobic C–H⋯H–C and hydrogen bonding PETCO⋯H–CA209 interactions are observed, which is also reflected in the significant lowering of the interaction energy upon mutation (−1.9 kcal mol−1, Fig. 10b and Movie S3†). In addition, semi-extended PET in WT TfCut2 exhibit major interaction with polar Asn212 and hydrophobic Ile213 from S3, which are not found in folded PET conformers (Movie S4†). Polar Asn212 shows hydrogen bonding N212CO⋯H–CPET, PETCO⋯H–NN212 as well as N212C–H⋯πPET and PETC–H⋯H–NN212 interactions (−6.8 kcal mol−1, Fig. 10c), while nonpolar Ile213 is involved in I213C–H⋯πPET and I213C–H⋯H–CPET interactions (−6.8 kcal mol−1, Fig. 10d). However, mutation of aromatic residue Phe209 to hydrophobic Ala209 had reduced the close contact of both extended and folded PET with S3 of DM TfCut2, while maintaining the close contact with Asn212 and Ile213 in the extended form.
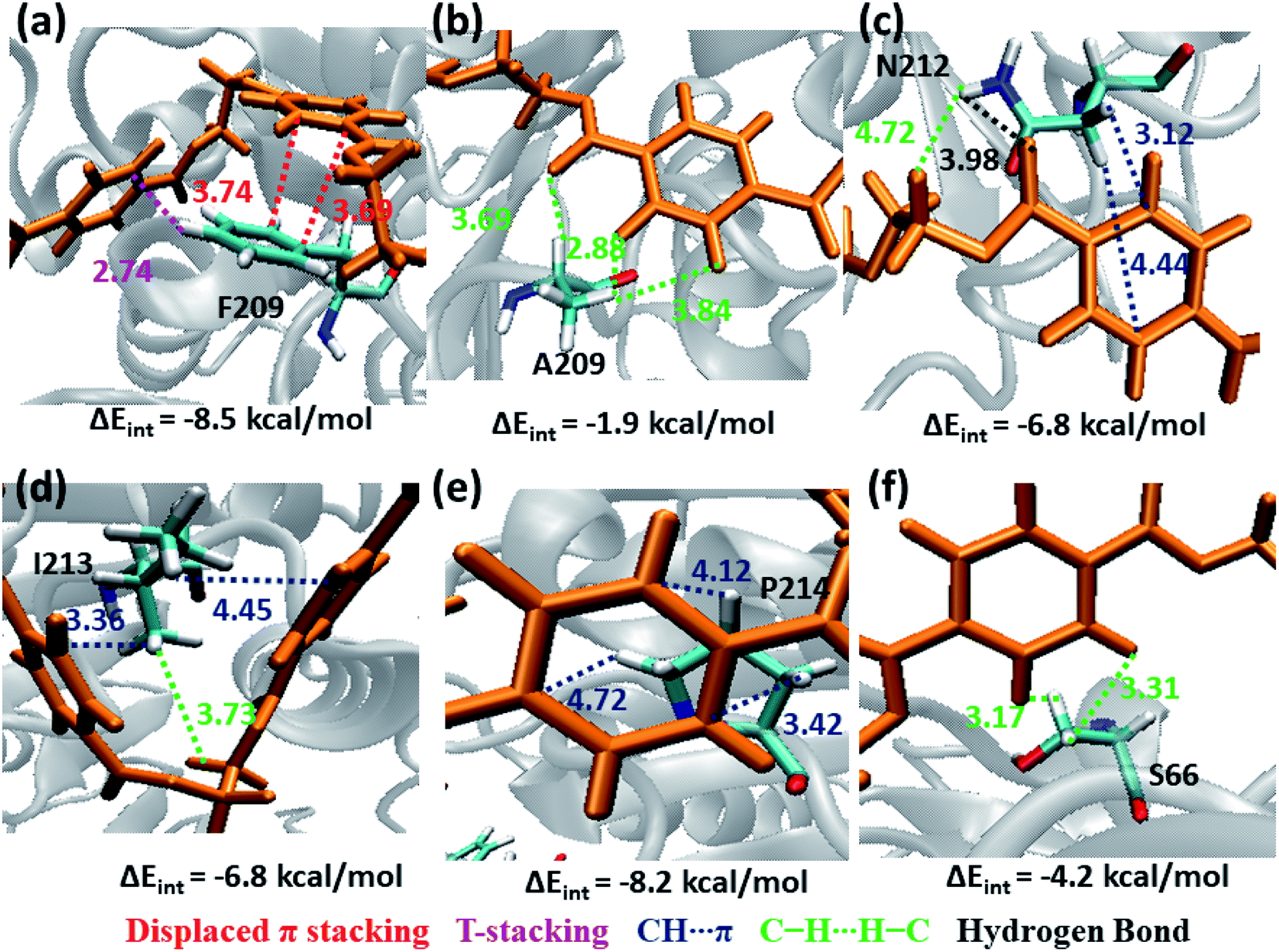 | ||
| Fig. 10 Snapshots showing major interactions in subsite S3 with color code same as previous cases. The single letter code indicating the amino acids are Phe (F), Ala (A), Asn (N), Ile (I) and Pro (P). | ||
In addition, Pro214, which is situated at the end of S3 shows significantly high close contact and interaction energy contribution due to P214C–H⋯πPET interaction, indicating the preference of the extended PET in DM TfCut2 (−8.2 kcal mol−1, Fig. 10e) as compared to semi-extended PET in WT TfCut2. Ser66 on the other hand, though a polar residue, mainly shows weak hydrophobic interactions S66C–H⋯H–CPET in both WT and DM cutinase (−4.2 kcal mol−1, Fig. 10f). Likewise, the contribution from the dispersion energy is major in all residues other than polar Asn212, where electrostatic component is higher (Fig. S13 and S14†).
Note that, no residues from S4 in both WT and DM TfCut2 show any close contact with either semi-extended, extended or folded PET conformers. Accordingly, the energy contribution of S4 residues is minimum (Fig. S12†). The striking difference in terms of close contact with the extended PET in DM TfCut2 as compared to semi-extended PET in WT Tfcut2 is observed for the novel S4′ subsite residues. The MESP analysis (Fig. 3c and d) revealed that the subsite S4′ contains both polar and non-polar residues. The energy contribution from nonpolar aliphatic residues Ile67, Ala68, Leu70 and Gly71, as well as polar Ser19 is not significant. However, the aromatic residue Trp69 shows T-stacking and W69C–H⋯πPET interactions with the phenyl ring of PET (−5.6 kcal mol−1, Fig. 11a). The carboxylic group of Glu72 interacts with PET via E72CO⋯H–CPET and hydrophobic C–H⋯H–C interactions (−2.0 kcal mol−1, Fig. 11b). Arg73 shows strong hydrogen bonding interaction with the hydrogen of guanidino group and carboxyl oxygen of PET (PETCO⋯H–NR73) and PETC–H⋯H–NR73 interactions −17.1 kcal mol−1, Fig. 11c). The terminal Arg18 having major contribution shows cation–π interaction with the phenyl ring of PET in addition to the PETCO⋯H–C hydrogen bonding (−9.9 kcal mol−1, Fig. 11d and Movie S5†). The SAPT0 breakdown of the total interaction energy indicates that the major contribution comes from the dispersion components except for polar Arg residues, which have high electrostatic component. The significantly higher close contact and interaction energy with polar S4′ residues like Arg18, Trp69, Arg73 and Glu72 indicates that, the S4′ plays a vital role in attaining favourable fully extended PET conformation in DM TfCut2, which might be prevented by the π–π interaction of the aromatic Phe209 in S3 of WT TfCut2.
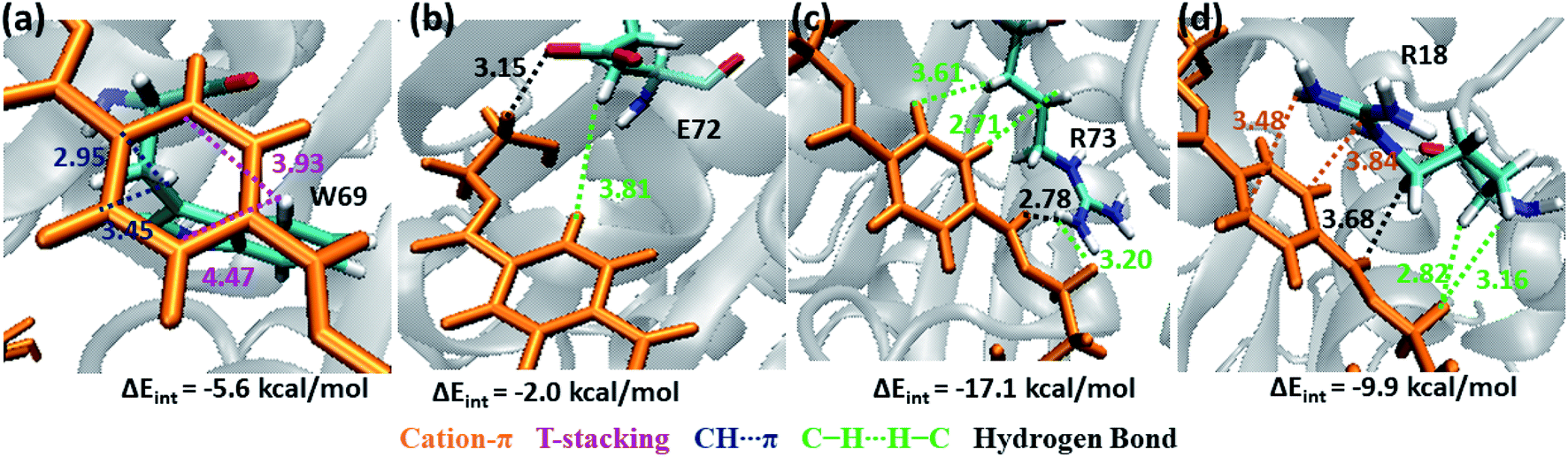 | ||
| Fig. 11 Snapshots showing major interactions in subsite S4′ (color code same as above). Cation–π interactions which are unique in S4′are shown in orange color. The single letter code indicating the amino acids are Trp (W), Glu (E) and Arg (R). | ||
From these results, one may infer that S1 is important for both binding and cleavage of PET and mutation in S1 might cause reduction in the PET degradation activity either due to poor binding or poor cleavage. In accordance with that, S1 shows very high binding energy irrespective of the conformational change as well as mutation (Fig. S14†). The next important subsite is S3, which plays very important role in anchoring the aromatic phenyl ring of PET with Phe209 by π–π interaction in WT TfCut2. However, note that, this interaction also restricts PET to attain a favourable extended conformation as well as might delay the sequential approach of the ester bonds close to the catalytic residues situated in S1 (Scheme 1). The loss of π–π interaction in DM cutinase due to F209A mutation is compensated by cation–π, hydrogen bonding and hydrophobic interactions from the Arg18, Trp69, Arg73 and Glu72 residues in S4′, which not only gives higher binding energy but also assist the fully extended PET conformation. A combination of these factors may in turn facilitate higher degree of PET degradation by DM TfCut2. Therefore, an optimum balance between the two opposing effect viz. the intrinsic PET affinity to the enzyme surface and PET binding to the active site seems to control the extent of degradation. While our manuscript was under review, Kari and Westh have showed that Sabatier principle might be helpful in rationalizing the optimum enzymatic PET degradation processes,25 which supports our analysis.
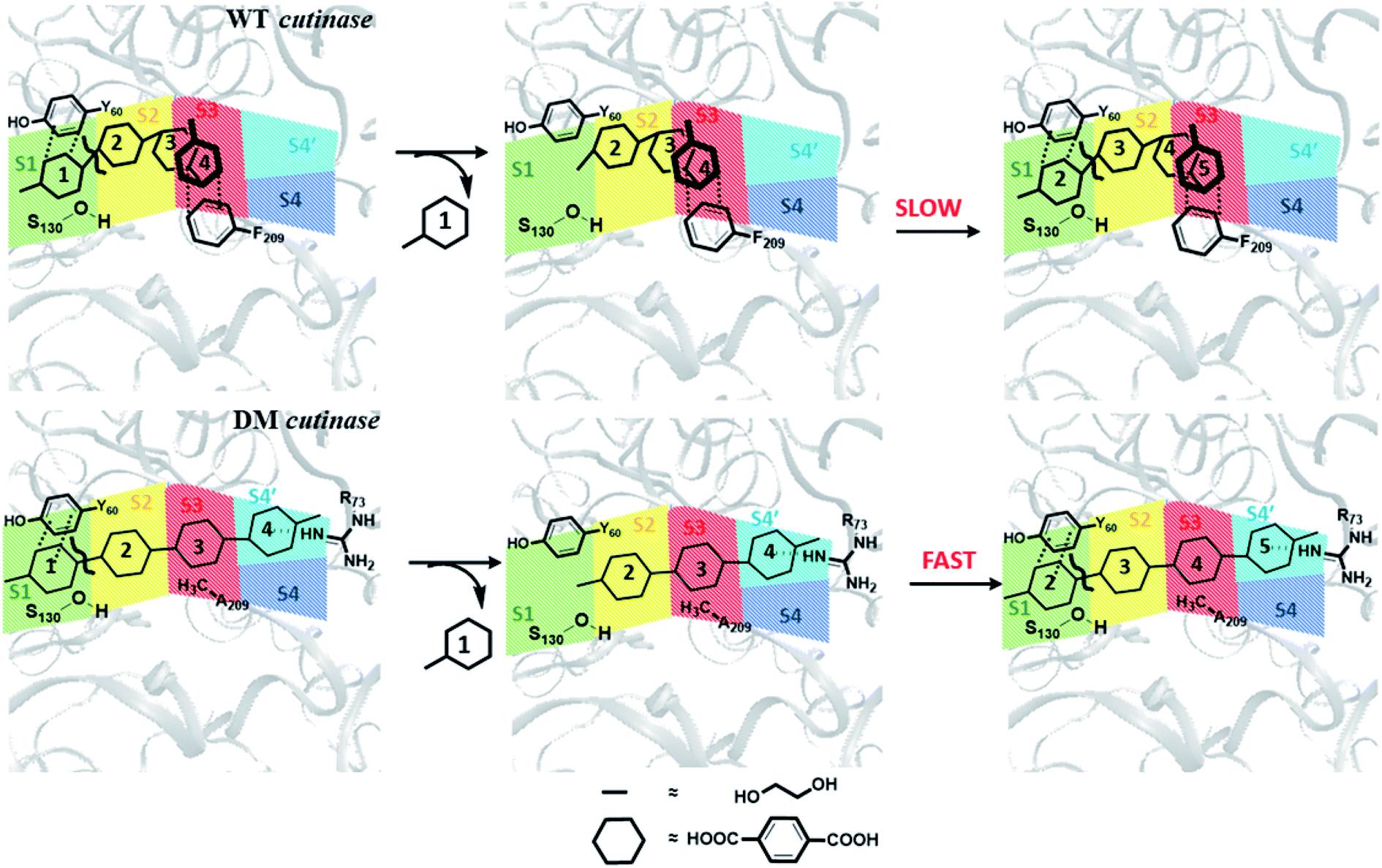 | ||
| Scheme 1 Schematic representation of plausible PET degradation pathway in WT and DM cutinase. The subsites are color coded as green, yellow, red, blue and cyan respectively for S1, S2, S3, S4 and S4′. The TPA and EG moieties of the PET polymer are presented with hexagons and lines, respectively. | ||
Conclusions
In this study, we have explored the different conformational states and binding affinity of the tetrameric PET with the amino acid residues from the PET binding cleft of wild type and double mutated cutinase using molecular dynamics simulations and quantum mechanical approaches. We demonstrate that mutations of anchor residue phenyl alanine to alanine at the PET binding cleft of cutinase alters the conformation of PET on the binding surface of the mutated cutinase demonstrating structural adaptability of ligand on the protein surface. This unveiled a distal yet novel binding subsite, which alters the nature of dispersive interaction for PET recognition and binding. The phenyl alanine engages in π–π interaction with the phenyl ring of PET (−8.5 kcal mol−1), which at one side helps in PET recognition, but at the other side restricts PET to attain fully extended conformations over the entire binding cleft. The loss of π–π interaction due to mutation of phenyl alanine to alanine is not only compensated by the favourable cation–π and hydrophobic interactions from the arginine residues (−17.1 kcal mol−1) found in the newly discovered subsite, but also favours the fully extended PET conformation. Thus, a perfect balance between the PET recognition and binding by the anchor residues and attainment of suitable conformation for the cleavage would likely be the key for a higher PET degrading activity. Clearly, further investigation is needed to identify the best possible mutations to be engineered for more efficient enzymatic PET degradation.Author contributions
SD designed the study. AJ performed the simulations and calculations. SD and AJ did the analysis of the data, discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
SD thanks partial financial support from DST (IFA12-CH-76, CRG/2019/002891), seed grant from CUSAT for new research initiative and UGC-SAP, DST-PURSE grant at CUSAT. The computer centre at CUSAT is also acknowledged for providing the computational facility set up using the DST-PURSE fund. AJ thanks CUSAT for research fellowship.References
- A. E. Brown and K. A. Reinhart, Science, 1971, 173, 287–293 CrossRef CAS PubMed.
- U. T. Bornscheuer, Science, 2016, 351, 1154–1155 CrossRef CAS PubMed.
- L. Ji, Appl. Mech. Mater., 2013, 312, 406–410 Search PubMed.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS.
- H. K. Webb, J. Arnott, R. J. Crawford and E. P. Ivanova, Polymers, 2013, 5, 1–18 CrossRef.
- I. Marshall and A. Todd, Trans. Faraday Soc., 1953, 49, 67–78 RSC.
- D. Paszun and T. Spychaj, Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- C. M. Rochman, M. A. Browne, B. S. Halpern, B. T. Hentschel, E. Hoh, H. K. Karapanagioti, L. M. Rios-Mendoza, H. Takada, S. Teh and R. C. Thompson, Nature, 2013, 494, 169–171 CrossRef CAS PubMed.
- R. Wei and W. Zimmermann, Microb. Biotechnol., 2017, 10, 1308–1322 CrossRef CAS PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara and K. Miyamoto, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- M. Furukawa, N. Kawakami, A. Tomizawa and K. Miyamoto, Sci. Rep., 2019, 9, 16038 CrossRef PubMed.
- F. Kawai, ChemSusChem, 2021, 14, 4115–4122 CrossRef CAS PubMed.
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Proc. Natl. Acad. Sci., 2018, 115, E4350–E4357 CrossRef CAS PubMed.
- S. Joo, I. J. Cho, H. Seo, H. F. Son, H.-Y. Y. Sagong, T. J. Shin, S. Y. Choi, S. Y. Lee and K.-J. J. Kim, Nat. Commun., 2018, 9, 382 CrossRef PubMed.
- R. Wei, T. Oeser, J. Schmidt, R. Meier, M. Barth, J. Then and W. Zimmermann, Biotechnol. Bioeng., 2016, 113, 1658–1665 CrossRef CAS PubMed.
- H. F. Son, I. J. Cho, S. Joo, H. Seo, H.-Y. Sagong, S. Y. Choi, S. Y. Lee and K.-J. Kim, ACS Catal., 2019, 9, 3519–3526 CrossRef CAS.
- H. F. Son, S. Joo, H. Seo, H.-Y. Sagong, S. H. Lee, H. Hong and K.-J. Kim, Enzyme Microb. Technol., 2020, 141, 109656 CrossRef CAS PubMed.
- Y. Cui, Y. Chen, X. Liu, S. Dong, Y. Tian, Y. Qiao, R. Mitra, J. Han, C. Li and X. Han, et al., ACS Catal., 2021, 11, 1340–1350 CrossRef CAS.
- M. Furukawa, N. Kawakami, K. Oda and K. Miyamoto, ChemSusChem, 2018, 11, 4018–4025 CrossRef CAS PubMed.
- Y. Liu, C. Liu, H. Liu, Q. Zeng, X. Tian, L. Long and J. Yang, Front. Bioeng. Biotechnol., 2022, 10, 865787 CrossRef PubMed.
- C. M. Carr, B. F. R. de Oliveira, S. A. Jackson, M. S. Laport, D. J. Clarke and A. D. W. Dobson, Front. Microbiol., 2022, 13, 888343 CrossRef PubMed.
- E. Nikolaivits, G. Taxeidis, C. Gkountela, S. Vouyiouka, V. Maslak, J. Nikodinovic-Runic and E. Topakas, J. Hazard. Mater., 2022, 434, 128900 CrossRef CAS PubMed.
- K. E. Kosiorowska, P. Biniarz, A. Dobrowolski, K. Leluk and A. M. Mirończuk, Sci. Total Environ., 2022, 831, 154841 CrossRef CAS PubMed.
- L. Aer, Q. Jiang, I. Gul, Z. Qi, J. Feng and L. Tang, Environ. Res., 2022, 212, 113472 CrossRef CAS PubMed.
- J. Arnling Bååth, K. Jensen, K. Borch, P. Westh and J. Kari, JACS Au, 2022, 2, 1223–1231 CrossRef PubMed.
- C. Sonnendecker, J. Oeser, P. K. Richter, P. Hille, Z. Zhao, C. Fischer, H. Lippold, P. Blázquez-Sánchez, F. Engelberger, C. A. Ramírez-Sarmiento, T. Oeser, Y. Lihanova, R. Frank, H. G. Jahnke, S. Billig, B. Abel, N. Sträter, J. Matysik and W. Zimmermann, ChemSusChem, 2022, 15, e202101062 CAS.
- R. Frank, D. Krinke, C. Sonnendecker, W. Zimmermann and H.-G. Jahnke, ACS Catal., 2022, 12, 25–35 CrossRef CAS.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662–667 CrossRef CAS PubMed.
- J. Then, R. Wei, T. Oeser, A. Gerdts, J. Schmidt, M. Barth and W. Zimmermann, FEBS Open Bio, 2016, 6, 425–432 CrossRef CAS PubMed.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M.-L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- A. N. Shirke, C. White, J. A. Englaender, A. Zwarycz, G. L. Butterfoss, R. J. Linhardt and R. A. Gross, Biochemistry, 2018, 57, 1190–1200 CrossRef CAS PubMed.
- S. Sulaiman, S. Yamato, E. Kanaya, J.-J. Kim, Y. Koga, K. Takano and S. Kanaya, Appl. Environ. Microbiol., 2012, 78, 1556–1562 CrossRef CAS PubMed.
- R.-J. Müller, H. Schrader, J. Profe, K. Dresler and W.-D. Deckwer, Macromol. Rapid Commun., 2005, 26, 1400–1405 CrossRef.
- R. Wei, D. Breite, C. Song, D. Gräsing, T. Ploss, P. Hille, R. Schwerdtfeger, J. Matysik, A. Schulze and W. Zimmermann, Adv. Sci., 2019, 6, 1900491 CrossRef PubMed.
- F. Kawai, T. Kawabata and M. Oda, Appl. Microbiol. Biotechnol., 2019, 103, 4253–4268 CrossRef CAS PubMed.
- C. Roth, R. Wei, T. Oeser, J. Then, C. Föllner, W. Zimmermann and N. Sträter, Appl. Microbiol. Biotechnol., 2014, 98, 7815–7823 CrossRef CAS PubMed.
- A. Bairoch and B. Boeckmann, Nucleic Acids Res., 1991, 19, 2247–2249 CrossRef CAS PubMed.
- T.-S. Lee, D. S. Cerutti, D. Mermelstein, C. Lin, S. LeGrand, T. J. Giese, A. Roitberg, D. A. Case, R. C. Walker and D. M. York, J. Chem. Inf. Model., 2018, 58, 2043–2050 CrossRef CAS PubMed.
- D. Case, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. Cheatham III, V. W. D. Cruzeiro, T. Darden, R. Duke, G. Giambasu, M. Gilson, H. Gohlke, A. Götz, R. Harris, S. Izadi, S. A. Iz mailov, K. Kasavajhala, A. Kovalenko, P. A. Kollman, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue and D. M. York, AMBER 2020, 2020 Search PubMed.
- J. C. Gordon, J. B. Myers, T. Folta, V. Shoja, L. S. Heath and A. Onufriev, Nucleic Acids Res., 2005, 33, W368–W371 CrossRef CAS PubMed.
- B. Widrow and J. McCool, IEEE Trans. Antennas Propag., 1976, 24, 615–637 CrossRef.
- Schrödinger, LLC, The {PyMOL} Molecular Graphics System, version ∼ 1.8, 2015 Search PubMed.
- A. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- A. Frisch, et al., Gaussian 09, Revision B. 01, Gaussian, Inc., Wallingford, USA, 2009, p. 25 Search PubMed.
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, Nucleic Acids Res., 2004, 32, W665–W667 CrossRef CAS PubMed.
- T. J. Dolinsky, P. Czodrowski, H. Li, J. E. Nielsen, J. H. Jensen, G. Klebe and N. A. Baker, Nucleic Acids Res., 2007, 35, W522–W525 CrossRef PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci., 2001, 98, 10037–10041 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Am. Chem. Soc., 2001, 222, 651–656 Search PubMed.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- W. Jorgensen, J. Chandrasekhar, J. Madura, R. Impey and M. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- P. Florová, P. Sklenovský, P. Banáš and M. Otyepka, J. Chem. Theory Comput., 2010, 6, 3569–3579 CrossRef PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- A. Cauchy, et al., Comp. Rend. Sci., 1847, 25, 536–538 Search PubMed.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS.
- C. W. Hopkins, S. Le Grand, R. C. Walker and A. E. Roitberg, J. Chem. Theory Comput., 2015, 11, 1864–1874 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- P. J. Turner, XMGRACE, Version 5.1. 19, Cent. Coast. Land-Margin Res. Oregon Grad. Inst. Sci. Technol., Beaverton, OR, 2005 Search PubMed.
- J. Shao, S. Tanner, N. Thompson and T. Cheatham, J. Chem. Theory Comput., 2007, 3, 2312–2334 CrossRef CAS PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- H. Gohlke and D. A. Case, J. Comput. Chem., 2004, 25, 238–250 CrossRef CAS PubMed.
- J. Kongsted and U. Ryde, J. Comput.-Aided Mol. Des., 2008, 23, 63 CrossRef PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno and C. D. Sherrill, J. Chem. Phys., 2014, 140, 94106 CrossRef PubMed.
- A. K. Wilson, T. van Mourik and T. H. Dunning, J. Mol. Struct.: THEOCHEM, 1996, 388, 339–349 CrossRef CAS.
- R. M. Parrish, L. A. Burns, D. G. A. Smith, A. C. Simmonett, A. E. DePrince, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. Di Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. Schaefer, K. Patkowski, R. A. King, E. F. Valeev, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS PubMed.
- L. Goerigk, in Non-Covalent Interactions in Quantum Chemistry and Physics: Theory and Applications, ed. A. Otero de la Roza and G. A. DiLabio, Elsevier, 2017, pp. 195–219 Search PubMed.
- T. Fecker, P. Galaz-Davison, F. Engelberger, Y. Narui, M. Sotomayor, L. P. Parra and C. A. Ramírez-Sarmiento, Biophys. J., 2018, 114, 1302–1312 CrossRef CAS PubMed.
- Q. Dong, S. Yuan, L. Wu, L. Su, Q. Zhao, J. Wu, W. Huang and J. Zhou, Bioresour. Bioprocess., 2020, 7, 37 CrossRef.
- B. Guo, S. R. Vanga, X. Lopez-Lorenzo, P. Saenz-Mendez, S. R. Ericsson, Y. Fang, X. Ye, K. Schriever, E. Bäckström, A. Biundo, R. A. Zubarev, I. Furó, M. Hakkarainen and P.-O. Syrén, ACS Catal., 2022, 12, 3397–3409 CrossRef CAS.
- C. H. S. da Costa, A. M. dos Santos, C. N. Alves, S. Martí, V. Moliner, K. Santana and J. Lameira, Proteins: Struct., Funct., Bioinf., 2021, 89, 1340–1352 CrossRef CAS PubMed.
- S. Boneta, K. Arafet and V. Moliner, J. Chem. Inf. Model., 2021, 61, 3041–3051 CrossRef CAS PubMed.
- B. K. Ge, G. M. Hu and C. M. Chen, Chin. J. Phys., 2021, 73, 331–339 CrossRef CAS.
- C. Jerves, R. P. P. Neves, M. J. Ramos, S. da Silva and P. A. Fernandes, ACS Catal., 2021, 11, 11626–11638 CrossRef CAS.
- S. Feng, Y. Yue, M. Zheng, Y. Li, Q. Zhang and W. Wang, ACS Sustainable Chem. Eng., 2021, 9, 9823–9832 CrossRef CAS.
- M. Zheng, Y. Li, W. Dong, W. Zhang, S. Feng, Q. Zhang and W. Wang, ACS Sustainable Chem. Eng., 2022, 10, 7341–7348 CrossRef CAS.
- S. Longhi and C. Cambillau, Biochim. Biophys. Acta, 1999, 1441, 185–196 CrossRef CAS.
- X. Han, W. Liu, J. Huang, J. Ma, Y. Zheng, T. Ko, L. Xu, Y. Cheng, C. Chen and R. Guo, Nat. Commun., 2017, 8, 2106 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03394a |
| This journal is © The Royal Society of Chemistry 2022 |