
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile conversion of ammonia to a nitride in a rhenium system that cleaves dinitrogen†
Gannon P.
Connor
 a,
Daniel
Delony
b,
Jeremy E.
Weber
a,
Brandon Q.
Mercado
a,
Julia B.
Curley
a,
Sven
Schneider
*b,
James M.
Mayer
*a and
Patrick L.
Holland
*a
a,
Daniel
Delony
b,
Jeremy E.
Weber
a,
Brandon Q.
Mercado
a,
Julia B.
Curley
a,
Sven
Schneider
*b,
James M.
Mayer
*a and
Patrick L.
Holland
*a
aDepartment of Chemistry, Yale University, New Haven, Connecticut, USA. E-mail: patrick.holland@yale.edu
bInstitute of Inorganic Chemistry, Georg-August-Universität Göttingen, Göttingen, Germany
First published on 4th March 2022
Abstract
Rhenium complexes with aliphatic PNP pincer ligands have been shown to be capable of reductive N2 splitting to nitride complexes. However, the conversion of the resulting nitride to ammonia has not been observed. Here, the thermodynamics and mechanism of the hypothetical N–H bond forming steps are evaluated through the reverse reaction, conversion of ammonia to the nitride complex. Depending on the conditions, treatment of a rhenium(III) precursor with ammonia gives either a bis(amine) complex [(PNP)Re(NH2)2Cl]+, or results in dehydrohalogenation to the rhenium(III) amido complex, (PNP)Re(NH2)Cl. The N–H hydrogen atoms in this amido complex can be abstracted by PCET reagents which implies that they are quite weak. Calorimetric measurements show that the average bond dissociation enthalpy of the two amido N–H bonds is 57 kcal mol−1, while DFT computations indicate a substantially weaker N–H bond of the putative rhenium(IV)-imide intermediate (BDE = 38 kcal mol−1). Our analysis demonstrates that addition of the first H atom to the nitride complex is a thermochemical bottleneck for NH3 generation.
Introduction
The interconversion of N2 and NH3 is important in fields that range from agriculture to sustainable energy.1,2 The heavy use of NH3 in fertilizer manufacturing has resulted in an extensive global infrastructure for its transportation and storage.3 Coupled with its high energy density, this makes NH3 an excellent candidate for a carbon-free chemical fuel, either through combustion or direct ammonia fuel cells (DAFCs).4–7 In order to realize this potential, it is beneficial to understand the individual steps of N–N and N–H bond formation and cleavage. One promising route to form the N–H bonds in NH3 from N2 is proton-coupled electron transfer (PCET).8a,9 Photo- or electrochemical energy may provide the necessary driving force for PCET-assisted N2 reduction using water as a source of protons and electrons, thus providing a sustainable strategy for converting N2 to NH3.10–16 A growing number of homogeneous systems catalytically achieve this difficult transformation utilizing PCET.17–31It is also important to understand the reverse reaction, NH3 oxidation to form N2. One application of this reaction is for releasing the chemical energy stored in N–H bonds for DAFC applications.6,7 In addition, the individual steps in NH3 oxidation to N2 are often the microscopic reverse of those used for PCET reduction of N2 and thus help to elucidate potential mechanisms for PCET-assisted reduction of N2 to NH3.32 In this context, it is relevant that many examples of chemical N–H bond oxidation from NH3-derived metal ammines yield metal nitride complexes.33–46 These systems utilize either chemical oxidants under basic conditions or H-atom abstracting (HAA) reagents for the ammine-to-nitride transformations.8b In some systems, electrochemical oxidation of ammine complexes yields metal–nitride products.41,42,44 Other systems can generate N2 as a product from the oxidation of NH3-derived ammine complexes, either through chemical36,40,47–49 or electrochemical49–52 methods. These include recently reported homogeneous systems that catalytically form N2 from NH3 through both chemical43–45 or electrochemical44,53–55 N–H bond oxidation. N–N bond formation can occur via bimetallic N–N coupling (e.g., between metal–NHx species or metal nitrides)40,44,45 or nucleophilic attack on a metal–NHx intermediate by NH3.43,53,54,56
Here, we study NH3 oxidation in a well-defined system that is also capable of reductive functionalization of N2via an N2-cleavage mechanism.57,58 Electrochemical reduction of (PNP)ReCl2 (1, PNP = N((CH2CH2)PtBu2)2) cleaves N2 to form the nitride complex (PNP)Re(N)Cl (2), which contains a nucleophilic nitride ligand (Scheme 1, black arrow).59–61 This nitride can be alkylated and reduced to give N–C containing products,62,63 but PCET reduction of the nitride in 2 to form NH3 (Scheme 1, grey arrows) was not observed because pincer protonation occurs rather than nitride protonation. Additional challenges are that the high energy of the lowest unoccupied molecular orbital (LUMO) of 2 prevents a reduction-first pathway, and that 2 is unreactive towards organic hydrogen-atom transfer (HAT) reagents or H2.59 In this manuscript, we evaluate the reverse reactions (Scheme 1, blue arrows) to elucidate the factors that prevent PCET nitride reduction in this system. This fundamental information may help to improve NH3 oxidation catalysis and to avoid bottlenecks in NH3 generation by future N2-cleaving systems, and importantly provides a thermochemical framework for nitrogen fixation products beyond ammonia.
 | ||
| Scheme 1 Cycles that represent reductive N2 splitting by (PNP)Re and PCET nitride reduction (gray cycle, not observed) or NH3 oxidation (blue cycle, studied here). | ||
Results
Binding and deprotonation of NH3
Introduction of 1 atm of NH3 gas to a solution of the dichloride complex 1 in benzene-d6 or tetrahydrofuran-d8 (THF-d8) results in an immediate color change from purple to brown. 1H NMR spectroscopy reveals the formation of a new Cs-symmetric product 3 in >95% yield (Scheme 2, top). The chemical shifts of 3 (Fig. S1†) are characteristic of a non-magnetic ground state non-, and the lack of noticeable temperature-independent paramagnetism, which is often observed in ReIII complexes,64,65 suggests that the two strongly π-donating amide ligands sufficiently destabilize the spin triplet state to give a well-isolated singlet ground state. A notable 1H resonance integrating to 2H is found at δ 12.7 ppm. A 1H–15N HSQC spectrum of a natural-abundance sample shows a 15N cross-peak from this resonance at δ –260 ppm (Fig. S2†), confirming that it corresponds to protons bound to N. This 15N chemical shift is significantly upfield from related nitride complexes (371–393 ppm) and closer to that for the protonated PNP backbone of [(HPNP)Re(N)Cl]+ (5) (−336 ppm).66 All spectroscopic signatures are consistent with the formulation of 3 as the amido complex (PNP)Re(NH2)Cl.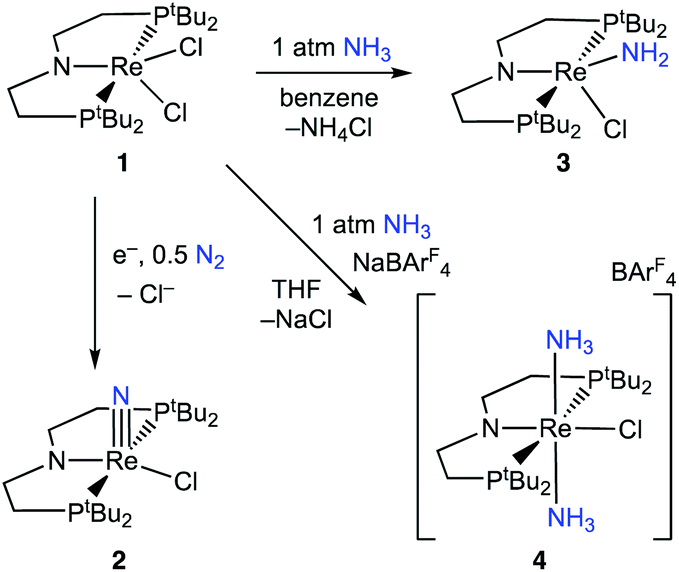 | ||
| Scheme 2 Reactivity of 1 with N2 and NH3. | ||
On a preparative scale, addition of 1 atm NH3 to a solution of 1 gives 3 as the major product, which is isolated from the reaction in 61% yield. The solid-state structure of 3 was elucidated via single crystal X-ray diffraction (XRD) and the N-bound hydrogen atoms were located in the Fourier map (Fig. 1). The Re–NH2 bond in 3 is 0.3 Å longer than the Re![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond in the nitride complex 2 (Table 1).66 In the supporting ligand, the (PNP)–Re bond is 0.1 Å shorter in 3 than in the Re–nitride complex 2, indicating increased π-bonding from the nitrogen of the pincer ligand in 3 (Table 1). The (PNP)–Re and Re–NH2 amide bond lengths in 3 are within 0.02 Å of each other with planar coordination of the nitrogen atoms in both cases (ΣPNP = 360°, ΣNH2 = 357°). The PNP and NH2 amides are oriented to π-donate into the same Re d orbital, which gives modest lengthening (0.04 Å) of the (PNP)–Re bond in 3 compared to the Re–dichloride complex 1.59 This also likely contributes to increased pyramidalization of the dialkylamide group (ΣPNP = 348°) in 2. These structural differences are accompanied by a change of the rhenium coordination geometry from square pyramidal (τ5 = 0.14) in complex 2, in which the coordination site trans to the nitride is open, toward trigonal bipyramidal (τ5 = 0.48) in complex 3.
N bond in the nitride complex 2 (Table 1).66 In the supporting ligand, the (PNP)–Re bond is 0.1 Å shorter in 3 than in the Re–nitride complex 2, indicating increased π-bonding from the nitrogen of the pincer ligand in 3 (Table 1). The (PNP)–Re and Re–NH2 amide bond lengths in 3 are within 0.02 Å of each other with planar coordination of the nitrogen atoms in both cases (ΣPNP = 360°, ΣNH2 = 357°). The PNP and NH2 amides are oriented to π-donate into the same Re d orbital, which gives modest lengthening (0.04 Å) of the (PNP)–Re bond in 3 compared to the Re–dichloride complex 1.59 This also likely contributes to increased pyramidalization of the dialkylamide group (ΣPNP = 348°) in 2. These structural differences are accompanied by a change of the rhenium coordination geometry from square pyramidal (τ5 = 0.14) in complex 2, in which the coordination site trans to the nitride is open, toward trigonal bipyramidal (τ5 = 0.48) in complex 3.
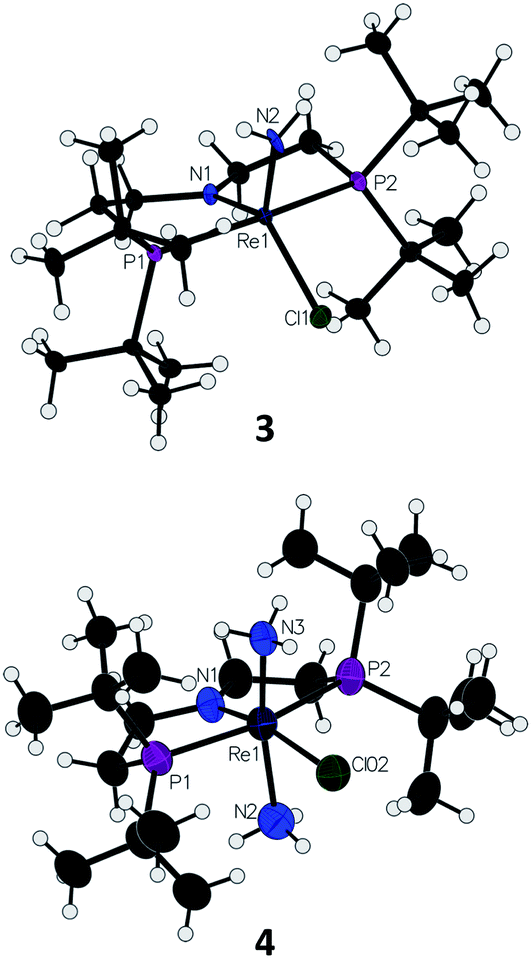 | ||
| Fig. 1 Solid-state structures of Re–amide complex 3 and Re(NH3)2 complex 4 (BArF4 ion omitted) with thermal ellipsoids at 50% probability. | ||
| Bond/angle | 2 | 3 | 4 |
|---|---|---|---|
| Re1–N1 | 2.033(6) | 1.936(3) | 1.894(5) |
| Re1–N2 | 1.643(6) | 1.959(3) | 2.150(5) |
| Re1–N3 | — | — | 2.193(6) |
| Re1–Cl1 | 2.441(2) | 2.384(1) | 2.495(2) |
| Re1–P1 | 2.443(2) | 2.397(1) | 2.424(2) |
| Re1–P2 | 2.435(2) | 2.382(1) | 2.425(2) |
| N1–Re1–N2 | 105.8(3) | 115.5(1) | 84.8(2) |
| N1–Re1–N3 | — | — | 165.6(2) |
| N2–Re1–N3 | 109.6(2) | ||
| N1–Re1–Cl1 | 106.5(2) | 108.8(1) | 83.4(1) |
| N2–Re1–Cl1 | 147.7(2) | 135.6(1) | 167.0(2) |
| N1–Re1–P1 | 100.4(2) | 95.1(1) | 91.0(1) |
| N1–Re1–P2 | 99.9(2) | 95.5(1) | 90.5(1) |
When 1 equiv. of NH3 gas was added to a solution of 1 in THF-d8 at −80 °C, 1H and 31P{1H} NMR spectra of the reaction showed a mixture of diamagnetic products (Fig. 2, middle). Addition of another 4 equiv. of NH3 gas (for a total of 5 equiv. NH3 per Re) resulted in full consumption of 1 and observation of 3 in 71% yield (Fig. 2, top). It is likely that dehydrohalogenation of the putative intermediate (PNP)Re(NH3)Cl2 by NH3 to form NH4Cl is required to drive the formation of 3. This implies that coordination to ReIII significantly increases the acidity of the N-bound protons.67
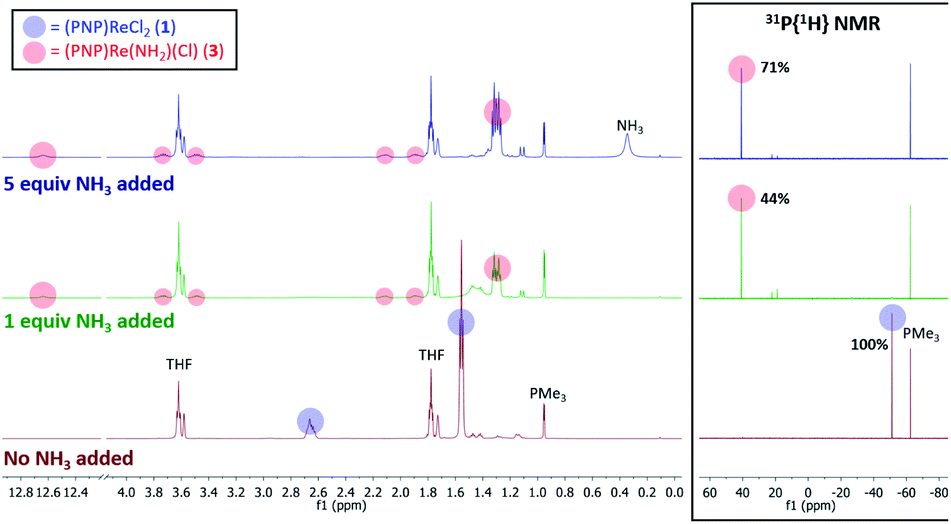 | ||
| Fig. 2 1H and 31P{1H} NMR spectra of 1 in THF-d8 without NH3 (bottom, maroon), with 1 equiv. NH3 added (middle, green), and with 5 equiv. NH3 added (top, blue). Spectroscopic yields reported vs. PMe3 in a capillary. | ||
Addition of 1 atm NH3 to a solution of 1 containing an equivalent of NaBArF4 (ArF = 3,5-bis(trifluoromethyl)phenyl) in THF-d8 at −80 °C resulted in a color change from purple to light green and formation of the new diamagnetic complex 4 by 1H and 31P{1H} NMR spectroscopy (see Scheme 2). In contrast to 3, complex 4 exhibits C2v symmetry, broadened resonances, and a new peak at δ = 5.47 ppm that integrates to 6H (Fig. S3†). Despite no identifiable cross-peaks in the 1H–15N HSQC spectrum of 4, N–H stretching bands were observed in the infrared (IR) spectrum at 3392, 3353, 3245, and 3174 cm−1. The molecular structure of 4 in the solid state shows the six-coordinate, cationic bis-ammine adduct [(PNP)Re(NH3)2Cl][BArF4] with a distorted octahedral geometry (Fig. 1). In comparison to 3, complex 4 shows lengthened Re–N bonds (2.172(5) Å vs. 1.959(3) Å) due to the lack of π-donation. With no strong π-donor ligands to compete with π-donation from the PNP amide, 4 contains a Re–PNP bond distance that is shorter than in 3 and 2 (Table 1). The flexibility of the PNP–Re interaction to accommodate the changes in ligand donor characteristics from ammine to nitride is also evident from the change in the PNP–Re bond lengths and PNP pyramidalization from 2–4.
Reactivity of [(PNP)Re(NH3)2Cl]+
To assess the plausibility of an ammine complex as an intermediate during formation of 3, a solution of 4 in THF-d8 was treated with 1 atm of NH3, which gave no reaction. However, addition of a slight excess of potassium hexamethyldisilazide (KHMDS) (Scheme 3) caused an immediate color change and formation of 3 as the major product in 61% yield, as judged by 1H and 31P{1H} spectroscopy (Fig. S4†). | ||
| Scheme 3 Reactivity of 4 with stoichiometric base or reductant. | ||
Cyclic voltammetry (CV) of 4 in THF under Ar shows irreversible redox processes, a reduction at Epc = −1.95 V vs. Cp2Fe+/0 and an oxidation at Epa = −0.58 V vs. Cp2Fe+/0 (Fig. S16†). The position of the reduction peak is similar to the reversible reduction of 1 under Ar at −2.00 V vs. Cp2Fe+/0.68 The first reduction of 1 under Ar was previously attributed to the formation of [(PNP)ReIICl2]−, which is followed by chloride dissociation to form (PNP)ReIICl which is subsequently reduced again.60 The difficulty of reducing 4 suggests that it is quite electron-rich despite its positive charge, but the lack of reversibility prevents further interpretation.
In an attempt to assess the species formed upon reduction, 4 was treated with a chemical reductant. Addition of 1.2 equiv. CoCp*2 to a solution of 4 in THF-d8 under N2 gave complete consumption of 4 but the Re amide 3 was formed (Fig. S5†). The spectroscopic yield of 3 was only 60%. The fate of the lost proton and electron in the formation of 3 remain unknown. Analysis of the headspace following the reaction showed no detectable amount of H2 (<1% yield).
N–H abstraction from Re–amide complex (PNP)Re(NH2)Cl
We hypothesized that abstraction of H atoms from 3 would lead to the nitride (Scheme 1, blue), by analogy with other reported systems.37–40,43,45,46 In the following, we assume that formal H˙ abstraction by the hydrogen atom abstraction (HAA) reagents is most likely concerted, based on the known difficulty of stepwise PCET pathways.8 Addition of 2 equiv. of either 2,4,6-tri-tert-butylphenoxyl radical (tBu3PhO˙) or 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO˙) as HAA reagents to a solution of 3 in THF-d8 or benzene-d6 at ambient temperature gives rapid and quantitative (>99%) formation of 2 (Scheme 4), as judged by 1H and 31P{1H} NMR spectroscopy (Fig. S6†). This is accompanied by the formation of 2 equiv. of tBu3PhOH or TEMPOH. These reagents have O–H bond dissociation free energies (BDFEO–H) of 74.4 and 65.5 kcal mol−1 in THF, respectively.8b,69 When 3 is mixed with only 1 equiv. of TEMPO˙, only half of 3 is consumed, showing that the second H-atom abstraction is more favorable than the first (Fig. S7†). The absence of reactivity of 2 with excess tBu3PhOH or TEMPOH supports this notion.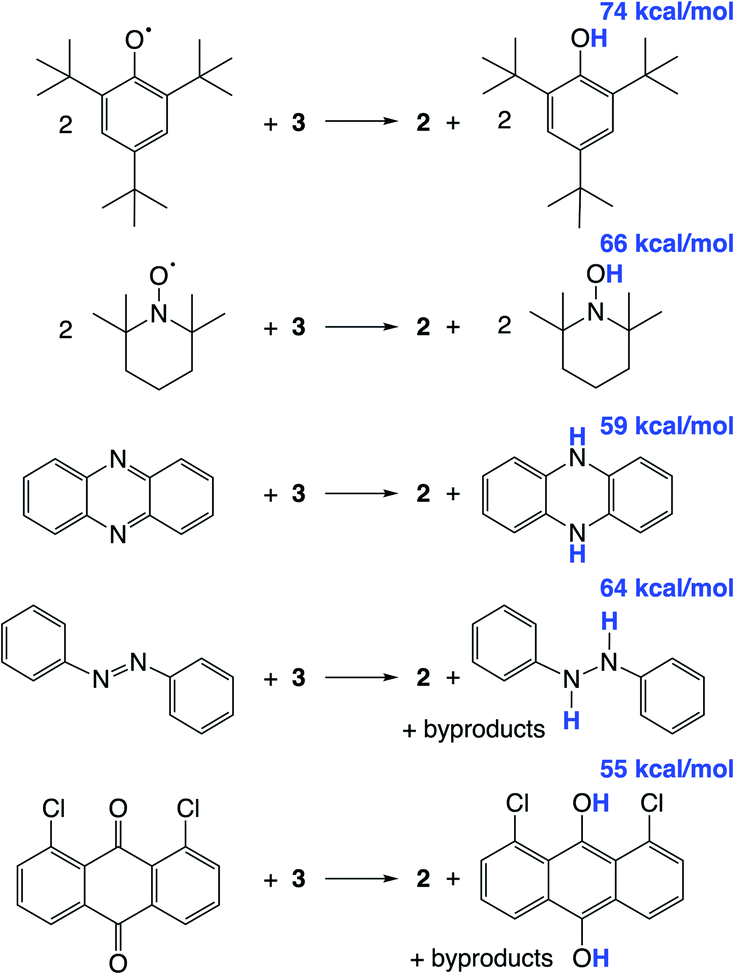 | ||
| Scheme 4 Reactivity of 3 with organic HAA reagents, with BDFEO–H or average BDFEX–H of the organic products in THF given in blue. | ||
Additional HAA reagents were used to further bracket the N–H bond strengths (Scheme 4). While 3 did not react with 5,10-phenazine (5 equiv) in THF-d8 at ambient temperature, heating to 80 °C gave quantitative (>98%) conversion to 2 and 5,10-dihydrophenazine (average BDFEN–H = 58.7 kcal mol−1 in MeCN69) after 21 h (Fig. S8†). Accordingly, no reaction was observed between 2 and 10 equiv. of 5,10-dihydrophenazine even after prolonged heating at 80 °C. Oxidation of 3 to form 2 was also observed when using 1.5 equiv. of azobenzene (54% yield of 2 after 72 h at 60 °C) and 1,8-dichloro-9,10-anthraquinone (67% yield of 2 after 4 d at ambient temperature). 1H NMR spectra of reaction mixtures showed the formation of 1,2-diphenylhydrazine and 1,8-dichloro-9,10-anthracenediol (average BDFEX–H = 60.9 [in MeCN] and 55.4 kcal mol−1 [THF]), respectively.69 However, these reactions form multiple products, so quantitative thermochemical information cannot be derived from the product formation in these cases. Quantification of the PCET thermochemistry was therefore carried out by titration calorimetry as detailed below.
Stepwise ET-PT from (PNP)Re(NH2)Cl
The reactivity of 3 with HAA reagents suggests that the N–H bonds in the amide ligand can be easily oxidized via concerted removal of an H-atom.8 We were also interested to determine whether the conversion to 2 is possible through stepwise PCET, with deprotonation and 1e− oxidation of 3.41,42,44 In order to test a PT-ET (proton transfer followed by electron transfer) pathway, a solution of 3 was mixed with up to 12 equiv. of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, pKa of conjugate acid = 16.9), phosphazene base P1-tBu-tris(tetramethylene) (pKa of conjugate acid = 20.2), or phosphazene base P4-tBu (pKa of conjugate acid = 33.9) in THF-d8 at ambient temperature.70 No reaction of 3 with any of these strong bases was observed by 1H and 31P{1H} NMR spectroscopy, indicating that the amide ligand in 3 is a poor Brønsted acid.In other tests, we explored whether a stepwise ET-PT pathway (electron transfer followed by proton transfer) is feasible. CV of 3 in THF shows an irreversible oxidation wave (Epa = −0.61 V vs. Cp2Fe+/0) at a scan rate of 100 mV s−1 (Fig. 3). However, increasing the scan rate to 1 V s−1 results in a distinct anodic shift of the oxidation event and increased reversibility, indicating chemical follow-up steps at a time-scale of the CV experiment. This potential is similar to those for the oxidation of both the dichloride complex 1 and the Re(NH3)2 complex 4.60 Further analysis of the CV has not been fruitful because of the lack of reversibility and formation of unknown byproducts (see below). However, electrolysis of a solution of 3 in the presence of 2,6-lutidine (pKa = 7.2 in THF70) at a potential of +0.6 V relative to the open circuit potential (OCP) resulted in steady passing of charge up to 2.2 equiv. e− (Fig. S15†) and a change in color from brown to orange. Rhenium(V) nitride complex 5, in which the backbone is protonated,66 was isolated from the post-electrolysis mixture in 69% isolated yield (Fig. S17†). The combination of removing an electron at −0.61 V and a proton with lutidine is thermodynamically equivalent to an “effective BDFE” of 56 kcal mol−1,69,71 so this e−/H+ removal is thermodynamically similar to the HAA reactions above.
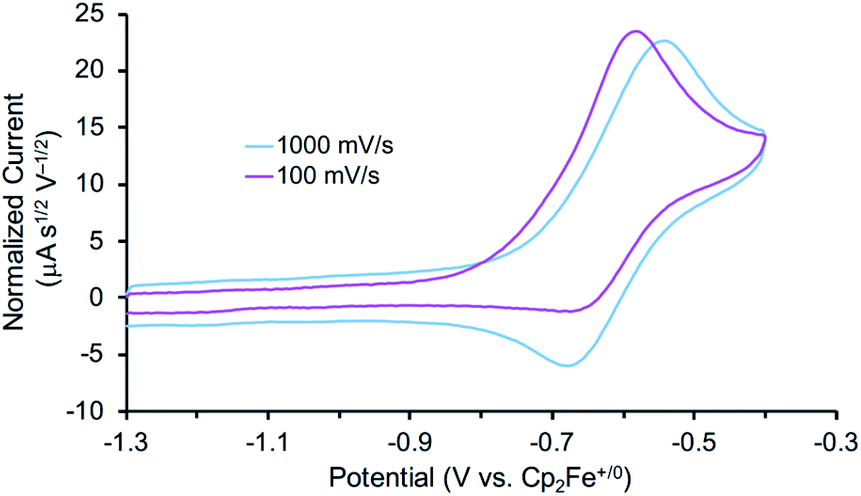 | ||
| Fig. 3 Cyclic voltammogram of the first oxidation of 3 (0.2 mM) in 0.2 M NBu4PF6 solution in THF under N2 using a glassy carbon working electrode, Pt wire auxiliary electrode, and Ag wire pseudoreference. Potentials referenced to Cp2Fe+/0 after the experiments. | ||
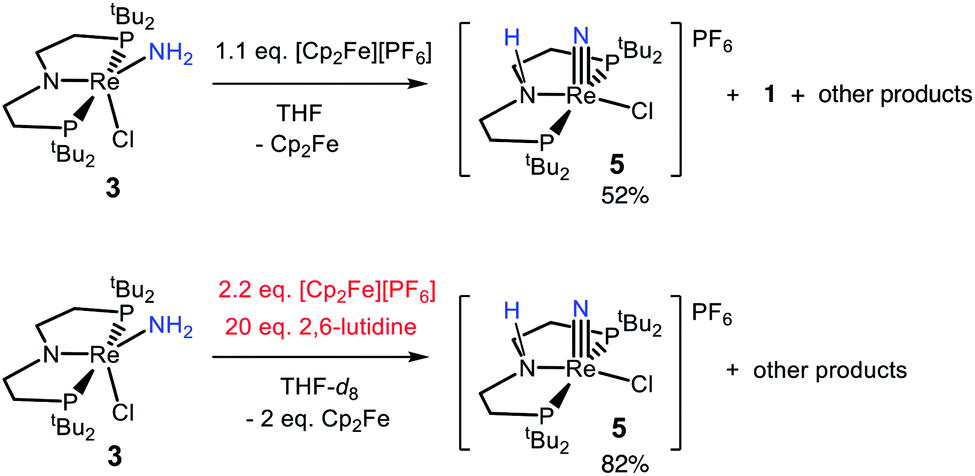
In an effort to identify oxidation products of 3, chemical oxidation was carried out with 1.1 equiv. of [Cp2Fe][PF6] in THF-d8 (Scheme 5). The major product identified from the resulting 1H and 31P{1H} NMR spectra was 5 in 50% yield (Fig. S9†).66 Furthermore, the ReIII-dichloride complex 1 was obtained in 25% yield, as well as a brown precipitate that could not be identified. The formation of both ReIII and ReV complexes from the 1e− oxidation of 3 implies disproportionation; however, these products are not formed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, implicating additional decomposition pathways. The product mixture that can be identified spectroscopically does not account for all of the Re, N, or H atoms present in the starting material. To test whether the missing H atoms could be released as H2 from weakened N–H bonds during the reaction, the reaction was repeated on a larger scale. Analysis of the THF-soluble products from the reaction showed formation of 1 and 5 in 17% and 52% yield, respectively, and no H2 was detected from analysis of the headspace (<1% yield).
:1 ratio, implicating additional decomposition pathways. The product mixture that can be identified spectroscopically does not account for all of the Re, N, or H atoms present in the starting material. To test whether the missing H atoms could be released as H2 from weakened N–H bonds during the reaction, the reaction was repeated on a larger scale. Analysis of the THF-soluble products from the reaction showed formation of 1 and 5 in 17% and 52% yield, respectively, and no H2 was detected from analysis of the headspace (<1% yield).
 | ||
| Scheme 5 Reactions of 3 with chemical oxidants. | ||
We also tested whether oxidation of 3 to a nitride could be facilitated by providing an exogenous base for the deprotonation steps and by using 2 equiv. of oxidant (Scheme 5). Accordingly, 20 equiv. 2,6-lutidine and 2.2 equiv. [Cp2Fe][PF6] were added to a solution of 3 in THF-d8, forming 5 in 82% spectroscopic yield, and no observable 1 (Fig. S10†). An unidentified brown solid was formed as a byproduct in both reactions, suggesting decomposition. The formation of unknown byproducts deterred us from further mechanistic analysis.
Calorimetric measurement and DFT calculations of N–H bond oxidation from (PNP)Re(NH2)Cl
Since the reaction of 3 with 2 equiv. tBu3PhO˙ to form 2 and 2 equiv. tBu3PhOH proceeds quantitatively, we chose this reaction for calorimetric determination of the reaction enthalpy.72,73 The titration of 3 with tBu3PhO˙ in THF using isothermal titration calorimetry (ITC) results in an exotherm of −50.9 kcal mol−1 until 2.0 equiv. of tBu3PhO˙ are added (Fig. S18†).33,73 Thus, on average, each abstraction of an H atom from 3 gives an enthalpy change of −25.4 kcal mol−1. Since the bond dissociation enthalpy of tBu3PhO–H in THF is 80.8 ± 1 kcal mol−1,‡ the average of the two BDEN–H values of 3 in THF is 55.4 ± 1 kcal mol−1.DFT calculations were used to obtain more insight into the thermodynamics of each PCET step from amide complex 3 to nitride complex 2. The B3LYP functional and def2-TZVP basis, together with standard solvent and dispersion corrections gave excellent agreement with the metrical parameters of the crystal structure of 3, and predicted the redox potential for 3+/0 (E = −0.65 V) close to the observed wave at −0.61 V in the experimental CV (see ESI†). Computation of the putative PCET intermediate confirmed that the S = 1/2 ReIV–imide (PNP)Re(NH)Cl (LRe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH) is the most stable tautomer; an isomeric amidorhenium(IV) complex, (PNP*)Re(NH2)Cl (PNP* = N(CHCH2PtBu2)(CH2CH2PtBu2)), with an unsaturated PNP backbone proved higher in free energy by 12 kcal mol−1. The optimized structure of LReNH shows a strongly bent parent imido ligand with a computed Re–N–H angle of 133°. Bending reduces the antibonding π-interaction of the multiply bonding imido ligand with the metal centred SOMO after reduction. In consequence, the Re–imide bond is considerably elongated (1.80 Å) with respect to the parent nitride (DFT: 1.66 Å).
NH) is the most stable tautomer; an isomeric amidorhenium(IV) complex, (PNP*)Re(NH2)Cl (PNP* = N(CHCH2PtBu2)(CH2CH2PtBu2)), with an unsaturated PNP backbone proved higher in free energy by 12 kcal mol−1. The optimized structure of LReNH shows a strongly bent parent imido ligand with a computed Re–N–H angle of 133°. Bending reduces the antibonding π-interaction of the multiply bonding imido ligand with the metal centred SOMO after reduction. In consequence, the Re–imide bond is considerably elongated (1.80 Å) with respect to the parent nitride (DFT: 1.66 Å).
Computations of the enthalpies associated with each sequential H-atom transfer from 3 to tBu3PhO˙ to give tBu3PhOH in THF gave an average calculated BDEN–H of 57 kcal mol−1 (Scheme 6).38,40,58 This is close to the calorimetrically determined average BDE of 55.4 ± 1 kcal mol−1. The free energy of conversion of 3 to 2via H-atom transfer to tBu3PhO˙ gave an average calculated BDFEN–H in 3 of 51 kcal mol−1. Further insight comes from the hypothetical 1e−/1H+ steps. The BDFEN–H for removing the first H from the amide ligand in 3 was calculated to be 69 kcal mol−1, which is substantially higher than the computed BDFEN–H of the second N–H bond (in LReNH), 33 kcal mol−1. These computations show that the N–H bond in LReNH is particularly weak, which is consistent with both the facile, irreversible oxidation of 3 with HAA reagents and the inability to observe this putative parent imido complex (see ESI†).
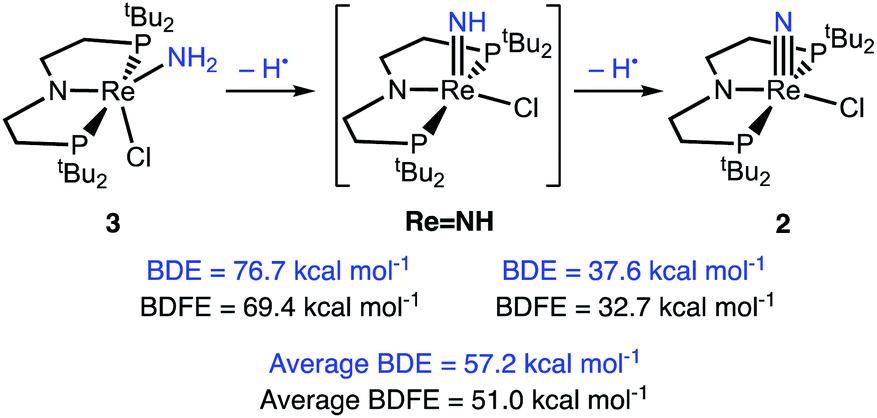 | ||
| Scheme 6 DFT (B3LYP/def2-TZVP) computed thermochemistry for the oxidation of 3 to 2via stepwise H-atom removal. | ||
Discussion
NH3 conversion to nitride with (PNP)Re
The conversion of NH3 to a nitride is quite facile in this system. Using excess NH3, complex 1 goes directly to the rhenium amide complex 3. The intermediate ReIII-ammine complex, potentially (PNP)Re(NH3)Cl2, has been neither spectroscopically observed nor isolated, which we attribute to rapid dehydrohalogenation by free NH3. This can be avoided through preparation of the cationic ReIII(NH3)2 complex 4, which can be deprotonated to form isolable complex 3. Further oxidation of 3 to Re–nitride complexes is also facile, forming 2 using either hydrogen atom abstracting (HAA) reagents or forming 5via 2e− chemical oxidation in the presence of a weak base. Thus, the complete conversion of NH3 to a nitride in this system is achievable in good yield using 2e− electrochemical oxidation in the presence of base (Scheme 7). From a functional standpoint, such facile formation of a nitride complex from NH3 is attractive considering the mild oxidation potential used (−0.6 V) and the possibility for excess NH3 to serve as an exogenous base.81 However, turnover to achieve catalytic NH3 oxidation to N2 would require a nitride coupling step.35,82,83 Although nitride coupling reactions between other late transition-metal nitrides bearing similar supporting pincer ligands have been reported,46,84–86 nitride coupling is not observed in this system due to the strong Re–nitride bond in complex 2.66 We attribute this to a thermodynamic difficulty because the reverse reaction, reductive N2 cleavage to form 2, is very exergonic. | ||
| Scheme 7 Full conversion of NH3 by 1 to a nitride in complex 5. | ||
The calorimetric titrations give an average bond enthalpy for the two N–H bonds in 3 of 55.4 ± 1 kcal mol−1 in THF. This is a rare example of an experimentally-derived bond energy in the context of NH3 oxidation.74 In contrast, almost all other literature values (Table 2; see also ref. 45 and 75) are estimated through bracketing experiments or computational models.32 The computationally derived average BDEN–H of 57 kcal mol−1 agrees with the experimental value, and the computations indicate that the analogous average BDFE (free energy) is 51 kcal mol−1. This value represents a substantial weakening from the BDFEN–H of free NH3 (100 kcal mol−1).8
| Complex | Solvent | BDFEN–H (kcal mol−1) | Reference | ||
|---|---|---|---|---|---|
| NH3 | NH2 | NH | |||
| a Bond dissociation enthalpies (BDE). | |||||
| (PNP)Re(NHx)Cl (3, computed) | THF | — | 69 | 33 | This work |
| (PNP)Re(NHx)Cl (3, experimental) | THF | 57(ave)a | This work | ||
| cis-(PONOP)Re(NHx)Cl2 | THF | — | 78 | 43 | 58 |
| (PNP)Ir(NHx) | Gas phase | — | 95a | 71a | 46 |
| trans-[(Ph-tpy)(PPh2Me)2Mo(NHx)]+ | THF | 46 | 64 | — | 74 and 75 |
| cis-[(Cp)(PPh2NtBu2)Mo(NHx)(CO)]+ | Et2O | 84 | 61 | — | 38 |
| [(PY5)Mo(NHx)]2+ | MeCN | 68 | 65 | 64 | 40 |
| [(Cp*)(PtBu2NPh2)Ru(NHx)]+ | THF | 83 | 89 | 72 | 43 |
| [(tpy)(NMe2bpy)Ru(NHx)]2+ | THF | 79 | 86 | — | 56 |
| (TMP)Ru(NHx)2 | C6H6 | 82 | 93 | 75 | 45 |
| [(tpy)(NMe2bpy)Fe(NHx)]2+ | THF | 82 | 90 | — | 56 |
| [(PhNCH2CH2)3N]Mo(NHx) | — | 52 | 64 | 42 | 76 |
| [(BP3)Fe(NHx)]+ | Et2O | — | 80 | 65 | 77 |
| (F)(H2PCH2CH2PH2)2Mo(NHx) | Benzene | 41 | 92 | 37 | 78 |
| (salen)Mn(NHx) | Gas phase | 85 | 84 | 60 | 79 |
| (η5-C5Me4SiMe3)2Ti(NHx) | Gas phase | 42 | 79 | — | 80 |
The oxidation of ammine complexes to form nitrides using HAA reagents is precedented in other systems using tBu3PhO˙ as the oxidant (forming tBu3PhOH).37–40,43,46 H-atom abstraction from the amide in 3 can be achieved with HAA reagents to form X–H bonds that are up to 15 kcal mol−1 weaker than the O–H bond in tBu3PhOH, though the phenazine reaction requires heating. The first HAA from 3 using organic reagents can be thermodynamically unfavorable by up to 10 kcal mol−1 because nitride formation is driven by the much more favorable second N–H oxidation.37,38,45
Though we have no experimental evidence for the intermediate imido complex LReNH, we considered its properties obtained from a DFT model. These computations indicate that the N–H bond in LReNH is especially weak, with a BDFE of 33 kcal mol−1. This bond is 36 kcal mol−1 weaker than the calculated first BDFEN–H of amido complex 3. Additionally, the N–H bond in LReNH is calculated to be 10 kcal mol−1 weaker than the 43 kcal mol−1 value computed for the closely related (PONOP)ReIVNH (PONOP = 2,6-bis-(diisopropylphosphinito)pyridine).58
As this last example shows, the thermochemistry of N–H bond forming and breaking is of particular interest for understanding how to achieve efficient N2 to NH3 interconversion. Table 2 compares our experimental values to literature values, typically derived from computational modelling. Complex 3 and its analogue (PONOP)Re(NH2)(Cl)2, both pincer-supported ReIII–NH2 complexes, have low BDFEN–H values. Interestingly, the ReIVNH compounds exhibit weaker imide N–H bonds than those of other metals. Some of the weakest N–H bonds were calculated for Mo–NH intermediates in the Chatt78 (37 kcal mol−1) and Schrock76 (42 kcal mol−1) systems, which can undergo complete N2 reduction to ammonia.79 Consistent with earlier systems (Table 2), amide intermediates consistently exhibit stronger N–H bonds than their corresponding imide intermediates. However, the difference between these two bond energies is particularly large in the Re systems, especially 3 where ΔBDFEN–H = 36 kcal mol−1. These values can be qualitatively rationalized by the very strong Re- and Mo-nitride bonds that arise when a d2 configuration is reached, and the less favorable M–N π bonding at higher d-electron counts.
Relevance to the PCET nitride reduction step of NRR
Nitride complex 2 is readily formed via electrochemical N2 cleavage,60 so the conversion of NH3 to the nitride ligand in 2 represents part of the reverse pathway from N2 to NH3 (Scheme 1). This would involve N2 cleavage to form 2 followed by 3e−/3H+ PCET reduction of the nitride. In a recent review, Chirik and coworkers highlighted the lack of data in the literature on the bond strengths of N–H bonds in NH2 and NH complexes in systems that perform N2 reduction,76 and the studies here are an important step toward understanding these species quantitatively. The thermochemical data from this study identify specific challenges associated with steps during the conversion of N2 to NH3.One clear challenge in the PCET reduction of 2 is formation of the first N–H bond, which would give a very weak bond in LReNH with a BDFEN–H of only 33 kcal mol−1.76,78,79 One approach that has been used to overcome this difficulty in literature systems is the use of potent acid/reductant pairs to form the weak N–H bonds,21,29,30,58,79,87 though this is complicated in the current system by the ease of protonation of the pincer amide group.66 In general, the instability of the imide species is identified as a key hindrance, because the imide intermediate must be accessed, even transiently, on the way to subsequent reduction that gives the amide species. The low driving force for PCET-assisted reduction of nitride 2 can be attributed to an overstabilization that results from strong ReN triple bonding.
Besides these thermochemical considerations, an encouraging result is that the amide complex 3 can be readily oxidized even though the first step (formation of ReNH) is uphill. The conversion of 3 to 2 by TEMPO, for instance, occurs within minutes even though the first step is thermodynamically uphill by 3 kcal mol−1. The ability of the first H˙ abstraction to proceed indicates that the barriers for the H-atom transfer reactions are low. Therefore, even less stable imide intermediates may be sufficient to allow rapid catalysis in the future.
Conclusions
Rhenium-amide and -ammine complexes have been isolated, and they are readily oxidized to the corresponding metal–nitride complex. The conversion of NH3 to a nitride by Re(PNP)Cl2 (1) to form Re(PNP)(N)Cl (2) represents the first example of an NH3-to-nitride transformation at Re. Since 2 can also be generated via electrochemical N2 cleavage, this system is relevant both to N2 reduction and ammonia oxidation. Facile, initial deprotonation of NH3 occurs upon coordination to 1 to form the amide complex Re(PNP)(NH2)Cl (3). The subsequent conversion to ReN can be accomplished with weak hydrogen atom acceptors that provide low driving force for the H˙ transfer, indicating that the reactions are kinetically facile.
Calorimetric measurements of the conversion of 3 to 2 with tBu3PhO˙ show that the average BDEN–H of 3 in THF is 55.4 ± 1 kcal mol−1. This is a rare experimental thermochemical measurement of N–H bond strengths relevant to N2/NH3 interconversions.76 Computations show that this average is derived from two disparate N–H bond strengths, as the second N–H BDE in 3 (77 kcal mol−1) is much stronger than the N–H BDE in the putative imide intermediate LReIVNH (38 kcal mol−1). The ability to form the weak N–H bond in this imide, even transiently, is identified as a crucial bottleneck for N2 to NH3 conversion in this system. The combination of bracketing,46 calorimetry and DFT is a powerful strategy that will continue to elucidate the steps of N2 reduction at metal complexes.
Data availability
Crystallographic data have been deposited at the CCDC with deposition numbers 2100974-2100975 and can be obtained from https://www.ccdc.cam.ac.uk/structures/. Other data supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization: G. P. C., J. M. M., P. L. H.; investigation and analysis: G. P. C., D. D., J. E. W., B. Q. M., J. B. C.; writing, interpretation: G. P. C., D. D., J. E. W., S. S., J. M. M., P. L. H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the U.S. National Science Foundation (P. L. H. and J. M. M., CHE-1665137, CHE-1954254 and DGE-1752134; J. E. W. and J. B. C., Graduate Research Fellowships) and the Deutsche Forschungsgemeinschaft (S. S., 389479699/RTG2455) for funding.Notes and references
- V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, MIT Press, Cambridge, MA, 2004 Search PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- J. N. Renner, L. F. Greenlee, K. E. Ayres and A. M. Herring, Electrochem. Soc. Interface, 2015, 24, 51–57 CrossRef CAS.
- R. F. Service, Science, 2018, 361, 120–123 CrossRef CAS PubMed.
- A. J. Martin, T. Shinagawa and J. Perez-Ramirez, Chem, 2019, 5, 263–283 CAS.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130–J3147 CrossRef CAS.
- O. Elishav, B. Mosevitzky Lis, E. M. Miller, D. J. Arent, A. Valera-Medina, A. Grinberg Dana, G. E. Shter and G. S. Grader, Chem. Rev., 2020, 120, 5352–5436 CrossRef CAS PubMed.
- (a) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed; (b) R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- G. Hochman, A. S. Goldman, F. A. Felder, J. M. Mayer, A. J. M. Miller, P. L. Holland, L. A. Goldman, P. Manocha, Z. Song and S. Aleti, ACS Sustainable Chem. Eng., 2020, 8, 8938–8948 CrossRef CAS.
- C. Rebreyend and B. de Bruin, Angew. Chem., Int. Ed., 2015, 54, 42–44 CrossRef CAS PubMed.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- H. Liu, L. Wei, F. Liu, Z. Pei, J. Shi, Z.-J. Wang, D. He and Y. Chen, ACS Catal., 2019, 9, 5245–5267 CrossRef CAS.
- Y. Wan, J. Xu and R. Lv, Mater. Today, 2019, 27, 69–90 CrossRef CAS.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- S. Kuriyama, K. Arashiba, K. Nakajima, H. Tanaka, N. Kamaru, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2014, 136, 9719–9731 CrossRef CAS PubMed.
- K. Arashiba, E. Kinoshita, S. Kuriyama, A. Eizawa, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 5666–5669 CrossRef CAS PubMed.
- K. Arashiba, A. Eizawa, H. Tanaka, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Bull. Chem. Soc. Jpn., 2017, 90, 1111–1118 CrossRef CAS.
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536–540 CrossRef CAS PubMed.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed.
- L. A. Wickramasinghe, T. Ogawa, R. R. Schrock and P. Muller, J. Am. Chem. Soc., 2017, 139, 9132–9135 CrossRef CAS PubMed.
- T. Itabashi, I. Mori, K. Arashiba, A. Eizawa, K. Nakajima and Y. Nishibayashi, Dalton Trans., 2019, 48, 3182–3186 RSC.
- A. Eizawa, K. Arashiba, A. Egi, H. Tanaka, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Chem.–Asian J., 2019, 14, 2091–2096 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2013, 136, 1105–1115 CrossRef PubMed.
- T. J. Del Castillo, N. B. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 5341–5350 CrossRef CAS PubMed.
- M. J. Chalkley, T. J. Del Castillo, B. D. Matson, J. P. Roddy and J. C. Peters, ACS Cent. Sci., 2017, 3, 217–223 CrossRef CAS PubMed.
- M. J. Chalkley, T. J. Del Castillo, B. D. Matson and J. C. Peters, J. Am. Chem. Soc., 2018, 140, 6122–6129 CrossRef CAS PubMed.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
- P. L. Dunn, B. J. Cook, S. I. Johnson, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 17845–17858 CrossRef CAS PubMed.
- K. Dehnicke and J. Strähle, Angew. Chem., Int. Ed. Engl., 1992, 31, 955–978 CrossRef.
- J. Du Bois, J. Hong, E. M. Carreira and M. W. Day, J. Am. Chem. Soc., 1996, 118, 915–916 CrossRef CAS.
- R. M. Clarke and T. Storr, J. Am. Chem. Soc., 2016, 138, 15299–15302 CrossRef CAS PubMed.
- M. Keener, M. Peterson, R. Hernandez Sanchez, V. F. Oswald, G. Wu and G. Menard, Chem. –Eur. J., 2017, 23, 11479–11484 CrossRef CAS PubMed.
- B. J. Cook, S. I. Johnson, G. M. Chambers, W. Kaminsky and R. M. Bullock, Chem. Commun., 2019, 55, 14058–14061 RSC.
- P. Bhattacharya, Z. M. Heiden, E. S. Wiedner, S. Raugei, N. A. Piro, W. S. Kassel, R. M. Bullock and M. T. Mock, J. Am. Chem. Soc., 2017, 139, 2916–2919 CrossRef CAS PubMed.
- G. W. Margulieux, M. J. Bezdek, Z. R. Turner and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 6110–6113 CrossRef CAS PubMed.
- S. I. Johnson, S. P. Heins, C. M. Klug, E. S. Wiedner, R. M. Bullock and S. Raugei, Chem. Commun., 2019, 55, 5083–5086 RSC.
- D. W. Pipes, M. Bakir, S. E. Vitols, D. J. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1990, 112, 5507–5514 CrossRef CAS.
- G. M. Coia, K. D. Demadis and T. J. Meyer, Inorg. Chem., 2000, 39, 2212–2223 CrossRef CAS PubMed.
- P. Bhattacharya, Z. M. Heiden, G. M. Chambers, S. I. Johnson, R. M. Bullock and M. T. Mock, Angew. Chem., Int. Ed., 2019, 58, 11618–11624 CrossRef CAS PubMed.
- K. Nakajima, H. Toda, K. Sakata and Y. Nishibayashi, Nat. Chem., 2019, 11, 702–709 CrossRef CAS PubMed.
- P. L. Dunn, S. I. Johnson, W. Kaminsky and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 3361–3365 CrossRef CAS PubMed.
- M. G. Scheibel, J. Abbenseth, M. Kinauer, F. W. Heinemann, C. Wuertele, B. de Bruin and S. Schneider, Inorg. Chem., 2015, 54, 9290–9302 CrossRef CAS PubMed.
- D. D. Thusius and H. Taube, J. Phys. Chem., 1967, 71, 3845–3857 CrossRef CAS.
- H. Taube and J. D. White, J. Phys. Chem., 1970, 74, 4142–4149 CrossRef CAS.
- J. D. Buhr and H. Taube, Inorg. Chem., 1979, 18, 2208–2212 CrossRef CAS.
- J. P. Collman, J. E. Hutchison, M. S. Ennis, M. A. Lopez and R. Guilard, J. Am. Chem. Soc., 1992, 114, 8074–8080 CrossRef CAS.
- O. Ishitani, P. S. White and T. J. Meyer, Inorg. Chem., 1996, 35, 2167–2168 CrossRef CAS PubMed.
- O. Ishitani, E. Ando and T. J. Meyer, Inorg. Chem., 2003, 42, 1707–1710 CrossRef CAS PubMed.
- M. D. Zott, P. Garrido-Barros and J. C. Peters, ACS Catal., 2019, 9, 10101–10108 CrossRef CAS.
- F. Habibzadeh, S. L. Miller, T. W. Hamann and M. R. Smith III, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2849–2853 CrossRef CAS PubMed.
- M. D. Zott and J. C. Peters, J. Am. Chem. Soc., 2021, 143, 7612–7616 CrossRef CAS PubMed.
- A. Najafian and T. R. Cundari, J. Phys. Chem. A, 2019, 123, 7973–7982 CrossRef CAS PubMed.
- S. J. K. Forrest, B. Schluschaß, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed.
- Q. J. Bruch, G. P. Connor, C.-H. Chen, P. L. Holland, J. M. Mayer, F. Hasanayn and A. J. M. Miller, J. Am. Chem. Soc., 2019, 141, 20198–20208 CrossRef CAS PubMed.
- I. Klopsch, M. Finger, C. Wuertele, B. Milde, D. B. Werz and S. Schneider, J. Am. Chem. Soc., 2014, 136, 6881–6883 CrossRef CAS PubMed.
- B. M. Lindley, R. S. van Alten, M. Finger, F. Schendzielorz, C. Würtele, A. J. M. Miller, I. Siewert and S. Schneider, J. Am. Chem. Soc., 2018, 140, 7922–7935 CrossRef CAS PubMed.
- R. S. van Alten, F. Wätjen, S. Demeshko, A. J. M. Miller, C. Würtele, I. Siewert and S. Schneider, Eur. J. Inorg. Chem., 2020, 2020, 1402–1410 CrossRef CAS PubMed.
- I. Klopsch, M. Kinauer, M. Finger, C. Würtele and S. Schneider, Angew. Chem., Int. Ed., 2016, 55, 4786–4789 CrossRef CAS PubMed.
- I. Klopsch, F. Schendzielorz, C. Volkmann, C. Würtele and S. Schneider, Z. Anorg. Allg. Chem., 2018, 644, 916–919 CrossRef CAS.
- A. Earnshaw, B. N. Figgis, J. Lewis and R. D. Peacock, J. Chem. Soc., 1961, 3132–3138, 10.1039/JR9610003132.
- J. Chatt, G. J. Leigh and D. M. P. Mingos, J. Chem. Soc. A, 1969, 1674–1680 RSC.
- G. P. Connor, B. Q. Mercado, H. M. C. Lant, J. M. Mayer and P. L. Holland, Inorg. Chem., 2019, 58, 10791–10801 CrossRef CAS PubMed.
- G. L. Hillhouse and J. E. Bercaw, J. Am. Chem. Soc., 1984, 106, 5472–5478 CrossRef CAS.
- B. M. Lindley, Q. J. Bruch, P. S. White, F. Hasanayn and A. J. M. Miller, J. Am. Chem. Soc., 2017, 139, 5305–5308 CrossRef CAS PubMed.
- C. F. Wise, R. G. Agarwal and J. M. Mayer, J. Am. Chem. Soc., 2020, 142, 10681–10691 CrossRef CAS PubMed.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 2019, 6735–6748 CrossRef CAS.
- C. R. Waidmann, A. J. M. Miller, C.-W. A. Ng, M. L. Scheuermann, T. R. Porter, T. A. Tronic and J. M. Mayer, Energy Env. Sci., 2012, 5, 7771–7780 RSC.
- D. Delony, M. Kinauer, M. Diefenbach, S. Demeshko, C. Würtele, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2019, 58, 10971–10974 CrossRef CAS PubMed.
- J. Abbenseth, D. Delony, M. C. Neben, C. Würtele, B. de Bruin and S. Schneider, Angew. Chem., Int. Ed., 2019, 58, 6338–6341 CrossRef CAS PubMed.
- M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730–733 CrossRef CAS PubMed.
- M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2018, 57, 2224–2228 CrossRef CAS PubMed.
- M. J. Bezdek, I. Pappas and P. J. Chirik, in Nitrogen Fixation, ed. Y. Nishibayashi, Springer International Publishing, Cham, 2017, pp. 1–21, DOI:10.1007/3418_2016_8.
- B. D. Matson and J. C. Peters, ACS Catal., 2018, 8, 1448–1455 CrossRef CAS PubMed.
- G. C. Stephan, C. Sivasankar, F. Studt and F. Tuczek, Chem. –Eur. J., 2008, 14, 644–652 CrossRef CAS PubMed.
- D. Wang, F. Loose, P. J. Chirik and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 4795–4799 CrossRef CAS PubMed.
- I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 13379–13389 CrossRef CAS PubMed.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130–J3147 CrossRef CAS.
- M. Keener, M. Peterson, R. Hernandez Sanchez, V. F. Oswald, G. Wu and G. Menard, Chem. –Eur. J., 2017, 23, 11479–11484 CrossRef CAS PubMed.
- W.-L. Man, T.-M. Tang, T.-W. Wong, T.-C. Lau, S.-M. Peng and W.-T. Wong, J. Am. Chem. Soc., 2004, 126, 478–479 CrossRef CAS PubMed.
- M. G. Scheibel, Y. Wu, A. C. Stückl, L. Krause, E. Carl, D. Stalke, B. de Bruin and S. Schneider, J. Am. Chem. Soc., 2013, 135, 17719–17722 CrossRef CAS PubMed.
- J. Abbenseth, M. Finger, C. Wuertele, M. Kasanmascheff and S. Schneider, Inorg. Chem. Front., 2016, 3, 469–477 RSC.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nature Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- M. J. Chalkley, P. H. Oyala and J. C. Peters, J. Am. Chem. Soc., 2019, 141, 4721–4729 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthesis, electrochemistry, calorimetry, computations, and crystallography; spectra. CCDC 2100974 and 2100975. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04503b |
| ‡ The BDFE of tBu3PhO–H in THF has recently been reported as 74.4 kcal mol−1,69 so the bond dissociation enthalpy (BDE) can be estimated as 80.7 kcal mol−1. This takes TS° for H˙ in THF to be 6.3 ± 0.2 kcal mol−1, the mean of the TS°(H˙) values for moderately polar aprotic solvents. |
| This journal is © The Royal Society of Chemistry 2022 |