
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular encapsulation of redox-active monomers to enable free-radical polymerisation†
Stefan
Mommer
 ,
Kamil
Sokołowski
,
Magdalena
Olesińska
,
Zehuan
Huang
and
Oren A.
Scherman
*
,
Kamil
Sokołowski
,
Magdalena
Olesińska
,
Zehuan
Huang
and
Oren A.
Scherman
*
Melville Laboratory for Polymer Synthesis, Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: oas23@cam.ac.uk
First published on 7th June 2022
Abstract
Extended polymeric structures based on redox-active species are of great interest in emerging technologies related to energy conversion and storage. However, redox-active monomers tend to inhibit radical polymerisation processes and hence, increase polydispersity and reduce the average molecular weight of the resultant polymers. Here, we demonstrate that styrenic viologens, which do not undergo radical polymerisation effectively on their own, can be readily copolymerised in the presence of cucurbit[n]uril (CB[n]) macrocycles. The presented strategy relies on pre-encapsulation of the viologen monomers within the molecular cavities of the CB[n] macrocycle. Upon polymerisation, the molecular weight of the resultant polymer was found to be an order of magnitude higher and the polydispersity reduced 5-fold. The mechanism responsible for this enhancement was unveiled through comprehensive spectroscopic and electrochemical studies. A combination of solubilisation/stabilisation of reduced viologen species as well as protection of the parent viologens against reduction gives rise to the higher molar masses and reduced polydispersities. The presented study highlights the potential of CB[n]-based host–guest chemistry to control both the redox behavior of monomers as well as the kinetics of their radical polymerisation, which will open up new opportunities across myriad fields.
Polyviologens are redox-active polymers based on N-substituted bipyridinium derivatives which have emerged as promising materials for energy conversion and storage.1–5 Their physicochemical properties can be adjusted through copolymerisation of the redox-active viologen monomers.6–8 The resultant materials are stable, water soluble and exhibit fast electron transfer kinetics. Polyviologens have been commonly fabricated through step-growth polymerisation in linear and dendritic architectures,9–13 as supramolecular polymers,14–16 networks,6,17,18 and covalent organic frameworks.19,20 Alternatively, anionic/cationic or metathesis-based polymerisations are used to avoid interference of radical-stabilising monomers with the radical initiators, however, these techniques are highly water- and/or oxygen-sensitive.21,22 When free-radical polymerisation (FRP) is conducted in the presence of viologen species, its reduction can cause a depletion of active radicals and thus disruption of the polymerisation process. Despite varying solvents, comonomers and initiator loadings, the direct FRP of viologen-containing monomers remains therefore limited to molar masses of 30 kDa.23–25 Accessing higher molar masses has been possible via post-polymerisation modification,26–28 which has impacted the electrochemical properties of the resultant materials.29,30 Alternative strategies to access higher molar masses of redox-active polymers and control their polymerisation are highly desirable.
Incorporation of cucurbit[n]uril (CB[n]) macrocycles have lead to a variety of functional materials through host–guest chemistry.31–34 Moreover, the redox chemistry of viologens can be modulated through complexation with CB[n].35–38 Specifically, CB[n] (n = 7, 8) can tune the redox potential of pristine viologens and efficiently sequester monoreduced viologen radical cations, avoiding precipitation in aqueous environments. Further to this, we recently demonstrated that the viologen radical cation is stabilised by −20 kcal mol−1 when encapsulated in CB[7].39
Consequently, we envisioned that incorporating CB[n]s as additives prior to polymerisation could (i) overcome current limits in accessible molar masses, (ii) increase control over FRP of viologen-based monomers through encapsulation and (iii) enable separation of radical species avoiding aggregation.
Here, we demonstrate a new approach to control FRP of redox-active monomers leading to high molar masses and decreased dispersity of the resultant polymers. In absence of CB[n], co-polymerisation of the N-styryl-N′-phenyl viologen monomer 12+ and N,N-dimethylacrylamide (DMAAm) only occurs at high initiator loadings (>0.5 mol%, Fig. 1a), leading to low molecular weights and high polydispersity. Using our synthetic approach, 12+ is efficiently copolymerised with DMAAm in the presence of CB[n] (n = 7, 8) macrocycles resulting in control of the polymer molar mass across a broad range, 4–500 kDa (Fig. 1b). Finally, CB[n] are successfully removed from the polymer via competitive host–guest binding and dialysis. Spectroscopic and electrochemical studies revealed that solubilisation/stabilisation of the reduced species and/or shielding of the redox-active monomers from electron transfer processes was responsible for this enhancement.
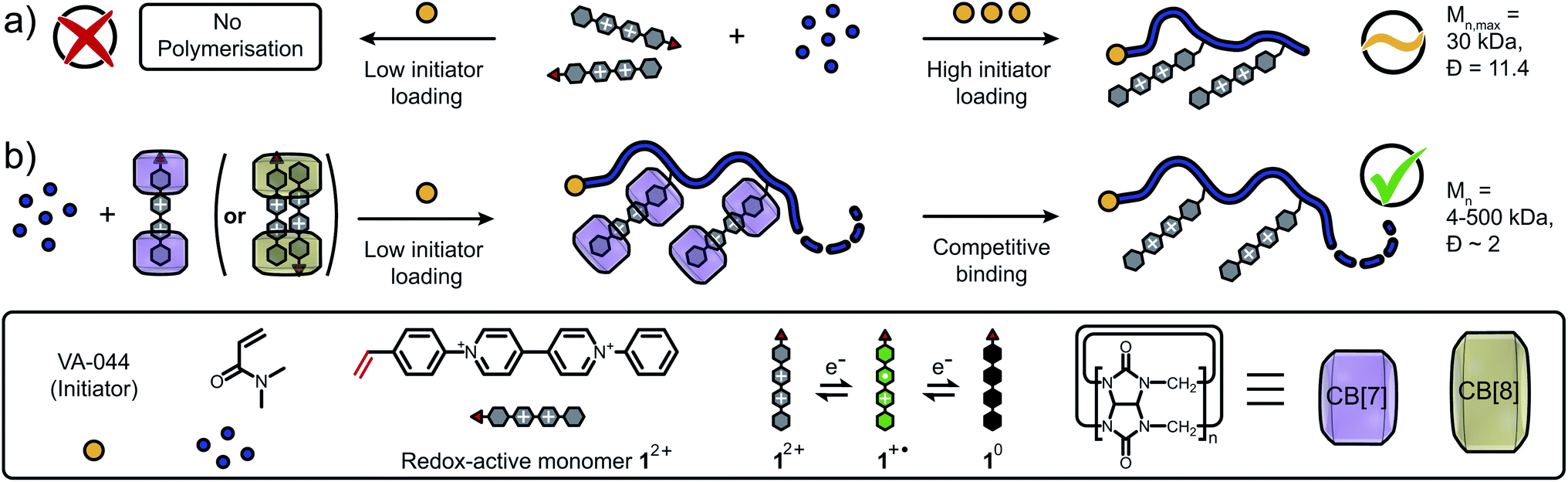 | ||
| Fig. 1 Schematic representation of the investigated polymerisation. (a) Conventional free radical polymerisation either completely fails to copolymerise redox-active monomers (low initiator loading) or delivers copolymers with limited molar masses and high dispersities (high initiator loading). (b) CB[n]-mediated protection suppresses interference of viologen monomers with radicals formed through the initiation process facilitating copolymerisation. The molar mass of the resulting copolymers is readily tunable via the amount of present CB[n] macrocycles and the CB[n] is post-synthetically removed via competitive binding to yield the final copolymer with desired molar mass. Cl− counter-ions are omitted for clarity. | ||
Recent studies on symmetric aryl viologens demonstrated 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 binding modes with CB[8] and high binding constants (up to Ka ∼ 1011 M−2).40,41 Incorporation of polymerisable vinyl moieties, in combination with the relatively static structure of their CB[n] host–guest complexes, was postulated to allow polymerisation without unfavorable side reactions. The asymmetric N-styryl-N′-phenyl viologen monomer 12+ prepared for this study (Fig. S1a and S2–S13†) displays a linear geometry and was predicted to bind CB[n] (n = 7, 8) in a 2:1 and 2:2 binding fashion (Fig. S1b†).40,42 Binding modes between CB[n] (n = 7, 8) and 12+ were investigated through titration experiments (1H NMR and ITC) which confirmed the formation of 1·(CB[7])2 and (1)2·(CB[8])2 (see Fig. S25 and S26†). 1H NMR titration of CB[7] with 12+ demonstrates encapsulation of both aryl moieties (including the vinyl group) through upfield chemical shifts of the respective signals (Fig. 2a). Similar upfield shifts were observed for CB[8] (Fig. 2c). Different para-aryl substituents (vinyl vs. hydrogen) resulted in either head-to-tail or head-to-head (1)2·(CB[8])2 dimers (Fig. S1b and S26†), a previously reported phenomenon.43 Nonetheless, the reversible nature of the complex renders the vinyl group temporarily available for copolymerisation. In the presence of CB[8], 12+ yields polymer molar masses of up to 500 kDa as its complexation is more robust. ITC data confirmed binding stoichiometry, with binding constants of Ka = 2.64 × 106 M−1 for 1·(CB[7])2 and Ka = 9.02 × 1010 M−2 for (1)2·(CB[8])2 (Table S2, Fig. S29a and b†).
:2 binding modes with CB[8] and high binding constants (up to Ka ∼ 1011 M−2).40,41 Incorporation of polymerisable vinyl moieties, in combination with the relatively static structure of their CB[n] host–guest complexes, was postulated to allow polymerisation without unfavorable side reactions. The asymmetric N-styryl-N′-phenyl viologen monomer 12+ prepared for this study (Fig. S1a and S2–S13†) displays a linear geometry and was predicted to bind CB[n] (n = 7, 8) in a 2:1 and 2:2 binding fashion (Fig. S1b†).40,42 Binding modes between CB[n] (n = 7, 8) and 12+ were investigated through titration experiments (1H NMR and ITC) which confirmed the formation of 1·(CB[7])2 and (1)2·(CB[8])2 (see Fig. S25 and S26†). 1H NMR titration of CB[7] with 12+ demonstrates encapsulation of both aryl moieties (including the vinyl group) through upfield chemical shifts of the respective signals (Fig. 2a). Similar upfield shifts were observed for CB[8] (Fig. 2c). Different para-aryl substituents (vinyl vs. hydrogen) resulted in either head-to-tail or head-to-head (1)2·(CB[8])2 dimers (Fig. S1b and S26†), a previously reported phenomenon.43 Nonetheless, the reversible nature of the complex renders the vinyl group temporarily available for copolymerisation. In the presence of CB[8], 12+ yields polymer molar masses of up to 500 kDa as its complexation is more robust. ITC data confirmed binding stoichiometry, with binding constants of Ka = 2.64 × 106 M−1 for 1·(CB[7])2 and Ka = 9.02 × 1010 M−2 for (1)2·(CB[8])2 (Table S2, Fig. S29a and b†).
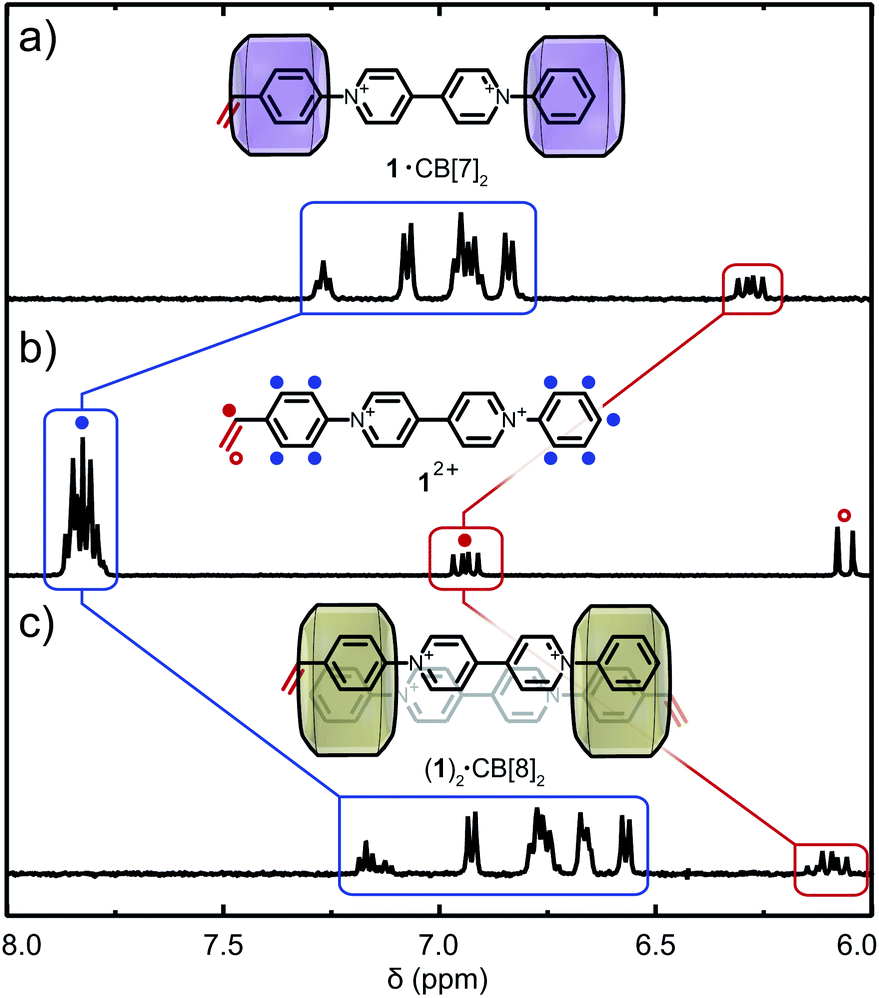 | ||
| Fig. 2 Supramolecular complexation of 12+ and CB[n]. 1H NMR spectra of 12+ at (a) χCB[7] = 2, (b) χCB[n] = 0 and (c) χCB[8] = 1 in D2O. Cl− counter-ions are omitted for clarity. | ||
The free radical copolymerisation of 12+ and DMAAm ([M] = 2 M), in the absence of CB[n], was based on optimised DMAAm homopolymerisations (Fig. S14 and S15†) and full conversion was confirmed by 1H NMR spectroscopy (Table S1 and Fig. S16†). 12+ was maintained at 1 mol% relative to DMAAm and by varying the radical initiator concentration molar masses of up to 30 kDa with broad dispersities (Đ = 11.4) were obtained (Fig. S17†). Lower initiator concentrations (<0.25 mol%) limited polymerisation (Mn = 3.7 kDa) and size exclusion chromatography elution peaks exhibited extensive tailing, suggesting that 12+ engages in radical transfer processes.
To verify our hypothesis that CB[n] macrocycles can modulate the redox behavior of 12+, FRP of 12+ and DMAAm was conducted with varying amounts of CB[n] (n = 7, 8) (Fig. 3, S18 and S20†). Full conversion of all monomers including their successful incorporation into the polymer was verified via1H NMR spectroscopy and SEC (Fig. S18 and S21–S23†). Using CB[7], the molar mass of the copolymers was tunable between Mn = 3.7–160 kDa (Fig. 3b and S21a†). Importantly, in the presence of CB[8], a broad range of molar masses Mn = 3.7–500 kDa were accessible for 0 < χCB[8] < 1.2 (Fig. S20 and S21b†). Increasing the CB[n] (n = 7, 8) concentration caused dispersity values to converge to Đ = 2.2 (χCB[8] = 1.2, χ is the ratio of CB[n] to the redox-active monomer, Fig. S20†). The copolymers were purified by addition of adamantylamine (competitive binder) prior to dialysis to deliver CB[n]-free redox-active copolymers (Fig. S23†).
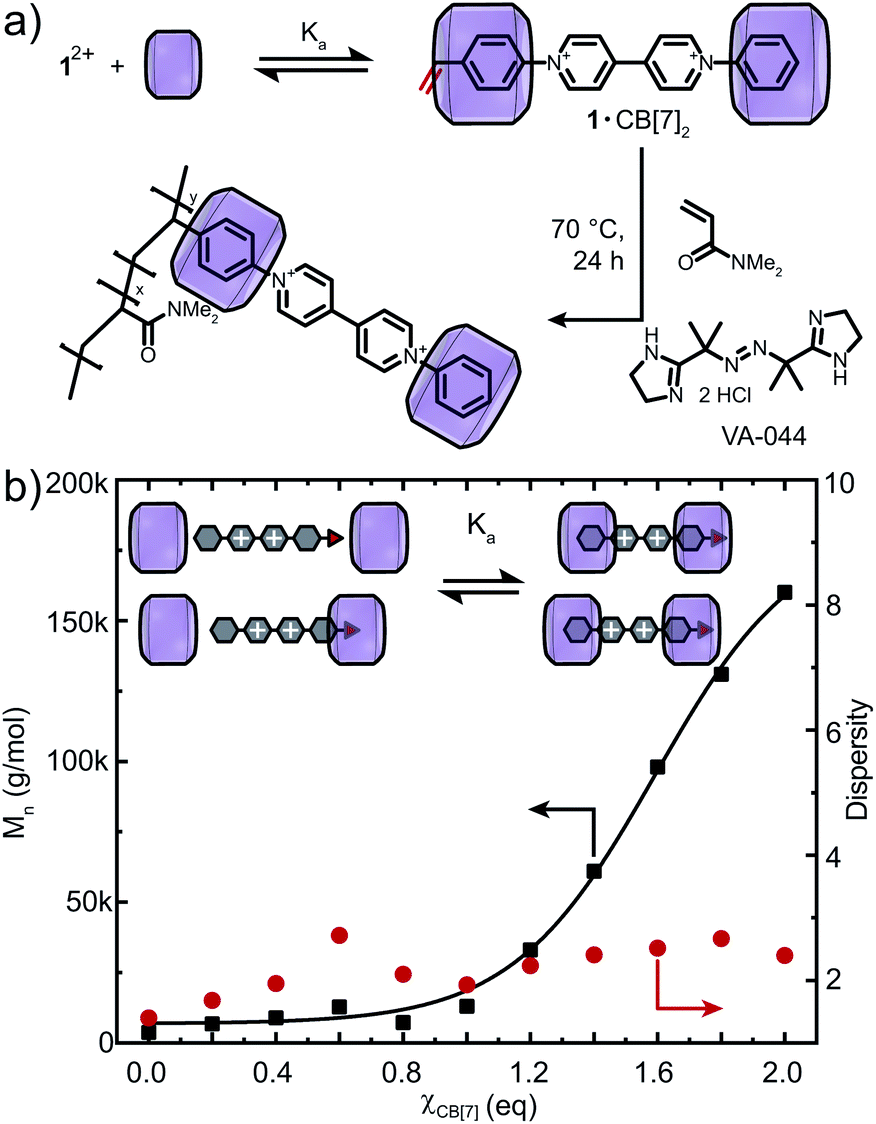 | ||
| Fig. 3 (a) In situ copolymerisation of DMAAm with 12+ and CB[7]. (b) Molar mass and dispersity vs. amount of CB[7] in the system. Fitted curve is drawn to guide the eye. Cl− counter-ions are omitted for clarity. | ||
The range of molar masses obtainable through addition of CB[n] (n = 7, 8) correlated with the measured Ka (Fig. 3b and S20†). Binding of 12+ to CB[8] was stronger and therefore lower concentrations of CB[8] were required to shift the binding equilibrium and mitigate disruption of the polymerisation. Dispersity values reached a maximum at χCB[7] = 0.6 or χCB[8] = 0.3, suggesting 1+˙ is only partially encapsulated. Consequently, higher CB[n] concentrations can enable FRP with lower initiator concentrations (0.10 mol%, Fig. S19†), which demonstrates the major role of complexation to modulate electron accepting properties of 12+.
The redox-active monomer 12+ can engage with propagating primary radicals (Pm˙) to either be incorporated into the growing polymer chain (Pm–12+˙) or to abstract an electron deactivating it (Pm). This deactivation likely occurs through oxidative termination producing 1+˙ (energetic sink), inactive oligo- and/or polymer chains (Pm) and a proton H+, causing retardation of the overall polymerisation. Oxidative terminations have been previously observed in aqueous polymerisations of methyl methacrylate, styrenes and acrylonitriles that make use of redox initiator systems.44–47 Another example by Das et al. investigated the use of methylene blue as a retarder, with the primary radical being transferred to a methylene blue electron acceptor via oxidative termination, altogether supporting the outlined mechanism of our system (extended discussion see ESI, Section 1.4†).48
The process of retardation can, however, be successfully suppressed, when monomer 12+ is encapsulated within CB[n] macrocycles. Herein the formation of 1·(CB[7])2 or (1)2·(CB[8])2 results in shielding of the redox-active component of 12+ from other radicals within the system, hampering other electron transfer reactions. This inhibits termination and results in extended polymerisation processes leading to higher molar mass polymers through mitigation of radical transfer reactions. Moreover, suppressing the formation of 1+˙ through supramolecular encapsulation minimises both π and σ dimerisation of the emerging viologen radical species,39 preventing any further reactions that could impact the molar mass or polydispersity of the resulting polymers.
Cyclic voltammetry (CV) and UV-Vis titration experiments were conducted to provide insight into the impact of CB[n] on the redox behavior and control over FRP of 12+. Excess of CB[n] (n = 7, 8) towards 12+ resulted in a complete suppression of electron transfer processes (Fig. S31 and S32†). Initially, 12+ shows a quasi-reversible reduction wave at −0.44 V forming 1+˙ (Fig. 4a). Increasing χCB[7], this reduction peak decreases and shifts towards more negative potentials (−0.51 V, χCB[7] = 1) accompanied by the formation of 12+·(CB[7])1. A second cathodic peak emerges at −0.75 V due to the increased formation of 12+·(CB[7])2. At χCB[7] = 2, this peak shifts to −0.80 V, where it reaches maximum intensity, once 12+·(CB[7])2 is the dominating species in solution. When 2 < χCB[7] < 4, the intensity of the reduction peak decreases and the complexation equilibrium is shifted towards the bound state, complete suppression of the reduction peak occurs at χCB[7] = 4. Similarly, the oxidation wave intensity is reduced by 95% at χCB[7] = 4 causing suppression of potential oxidative radical transfer processes (Fig. 4c).
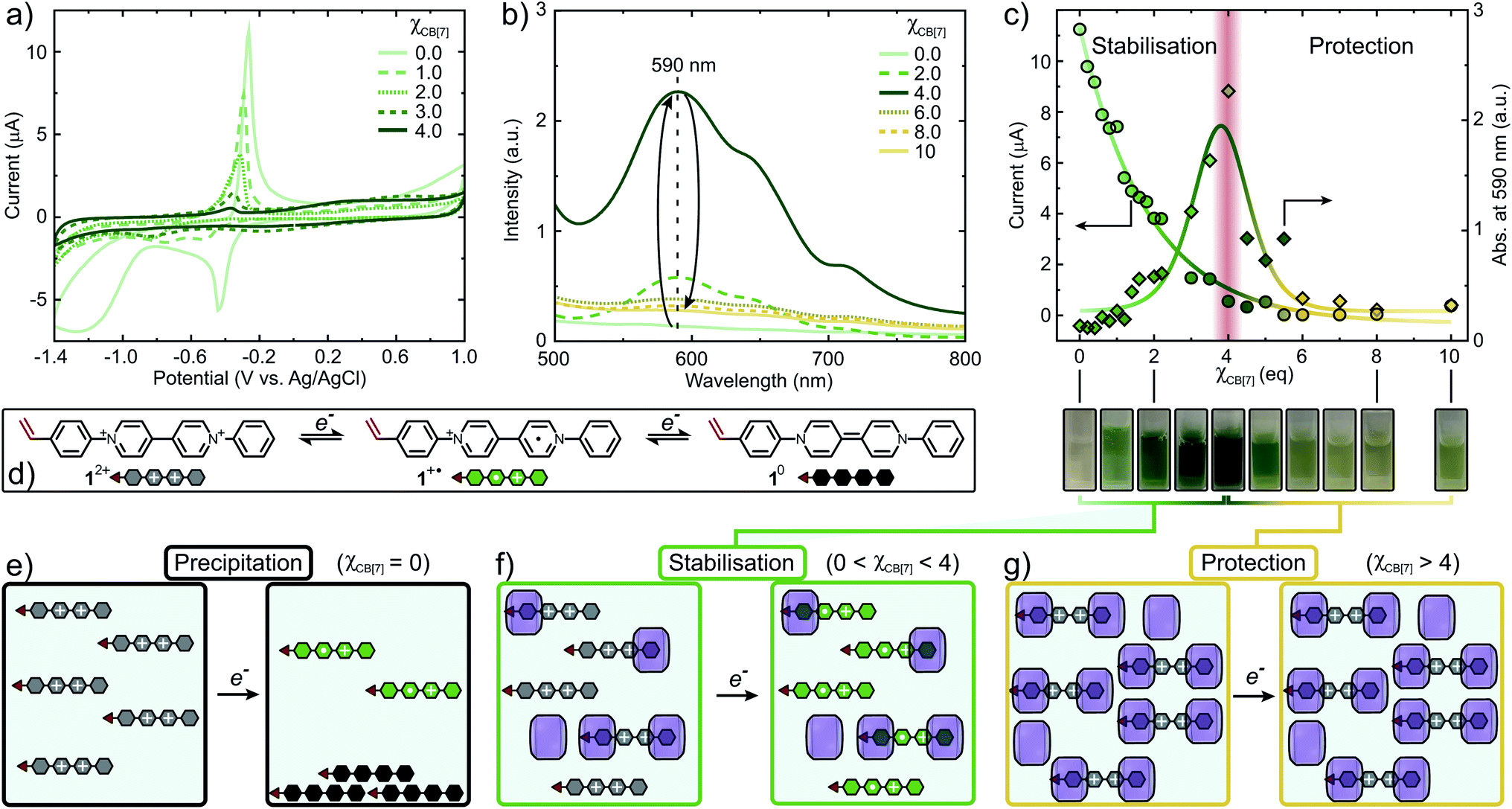 | ||
| Fig. 4 Mechanism of the CB[n]-mediated (n = 7, 8) strategy for the controlled copolymerisation of redox-active monomer 12+. (a) Cyclic voltammogram with varying amounts of CB[7]. (b) UV-Vis titration of 12+ with varying amounts of CB[7]. (c) Intensity decay of the oxidation peak at −0.27 V and change in absorption maximum of 1+˙ at 590 nm vs. χCB[7]. (d) Electron transfer processes of 12+ to generate 1+˙ and 10. (e) Reduction of 12+ resulting in precipitation of 10. (f) Stabilisation of 1+˙ through encapsulation with CB[7]. (g) Protection of 12+ from redox processes through CB[7]-mediated encapsulation. | ||
The concentration of 1+˙ can be monitored using UV-Vis (Fig. 4b and S34†).49 Absorbance at 590 nm (λmax) vs. χCB[7] was plotted and the concentration of 1+˙ increases, reaching a maximum at χCB[7] = 4 (Fig. 4c). When χCB[7] > 4, a decrease in concentration of 1+˙ was observed. We postulate the following mechanism: at χCB[7] = 0, 12+ is reduced to produce high concentrations of 1+˙ that partially disproportionates to form 10, which precipitates (Fig. 4e and S34†). When 0 < χCB[7] < 4, increasing amounts of green 1+˙ are stabilised through encapsulation within CB[7] suppressing disproportionation (Fig. 4c (cuvette pictures), Fig. 4f). For χCB[7] > 4, 12+ is protected from reduction through encapsulation (Fig. 4g).
To further demonstrate applicability of this strategy, we chose another viologen-based monomer 22+ for copolymerisation (Fig. 5a). As opposed to 12+, CB binds predominantly to the styryl moiety of 22+ (Fig. S27 and S28†).50 ITC data showed that 22+ binds CB[7] in a 1:1 fashion with a binding affinity of Ka = 2.32 × 106 M−1 (Fig. S30 and Table S2†). Monomer 22+ was also analysed via CV and showed three reversible reduction waves at −0.91 V, −0.61 V (viologen) and 0.40 V (styrene). Similar to 12+, excess CB[7] selectively protects the molecule from redox processes, while the vinyl moiety remains accessible (Fig. 5a, S33c and d†). For CB[8], only partial suppression of electron transfer processes was observed (Fig. S33e and f†). We therefore chose CB[7] as an additive to increase control over FRP of 22+ (Fig. 5b). Copolymerisation of 22+ (1 mol%) and DMAAm ([M] = 2 M) at χCB[7] = 0 resulted in Mn = 28 kDa. When χCB[7] = 0.1, 0.2 or 0.3, Mn increased gradually from 124 to 230 and 313 kDa, respectively, demonstrating the potential of this strategy for FRP of redox-active monomers. Higher percentages of CB[7] led to copolymers with presumably higher molar masses causing a drastic decrease in solubility that prevented further analysis. Investigations on a broader spectrum of such copolymers, including those with higher contents of 22+ are currently ongoing.
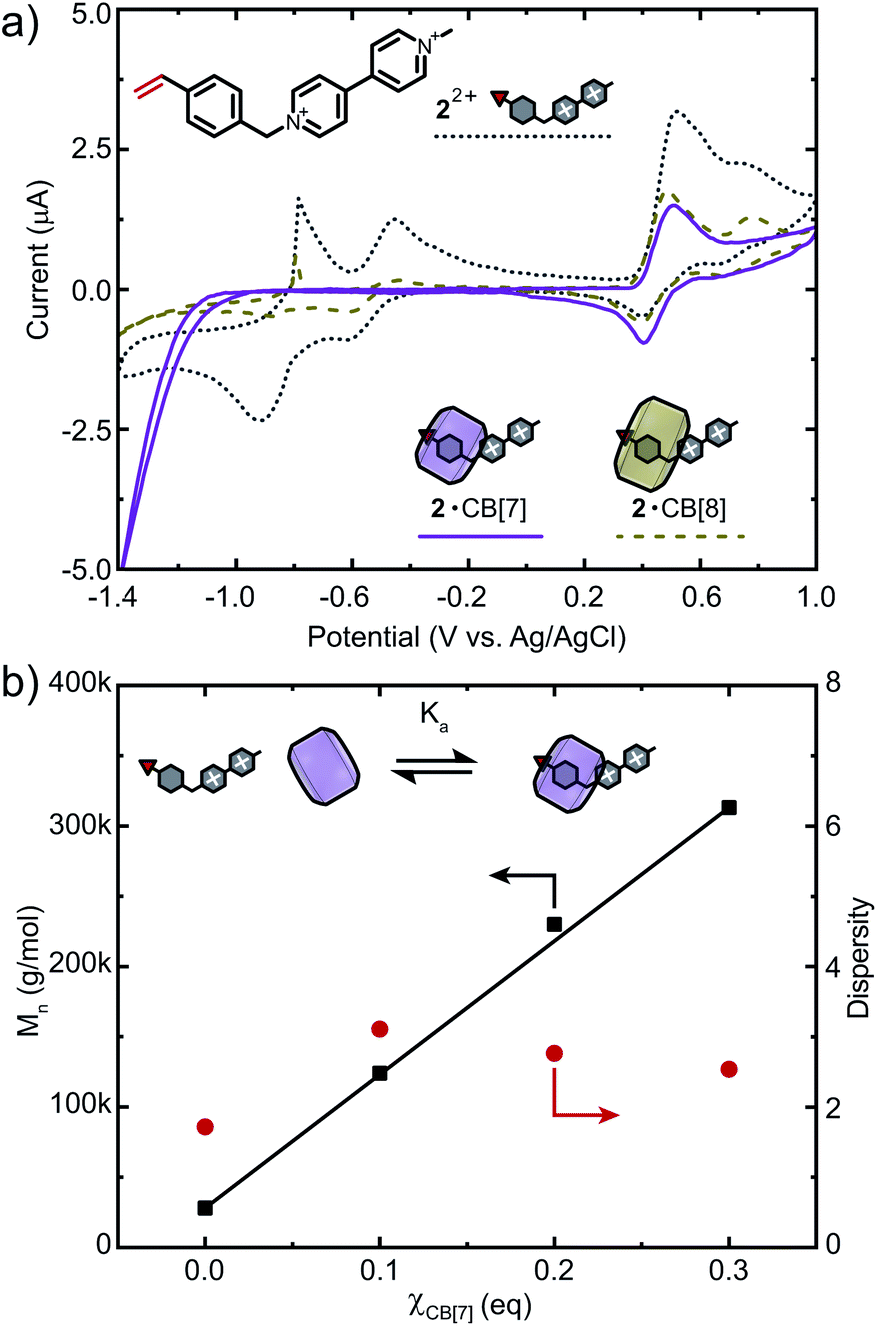 | ||
| Fig. 5 (a) Cyclic voltammogram of viologen-containing monomer 22+ and its complexation with CB[n] (n = 7, 8) at a concentration of 1 mM using a scan rate of 10 mV s−1 in 0.1 mM NaCl solution. (b) Molar mass and dispersity of 22+-containing copolymers vs. equivalents of CB[7]. Cl− counter-ions are omitted for clarity. | ||
In conclusion, we report a supramolecular strategy to induce control over the free radical polymerisation of redox-active building blocks, unlocking high molar masses and reducing polydispersity of the resulting polymers. Through the use of CB[n] macrocycles (n = 7, 8) for the copolymerisation of styrenic viologen 12+, a broad range of molar masses between 3.7–500 kDa becomes accessible. Our mechanistic investigations elucidated that the redox behavior of monomer 12+ is dominated by either CB[n]-mediated stabilisation of monoradical cationic species or protection of the encapsulated pyridinium species from reduction. In the stabilisation regime (χCB[7] < 4), 12+ is reduced to form the radical cation 1+˙, which is subsequently stabilised through CB[7] encapsulation. Upon reaching a critical concentration of CB[7] (χCB[7] > 4), the system enters a protection-dominated regime, where reduction of 12+ is suppressed and the concentration of 1+˙ diminishes. The resulting copolymers can be purified by use of a competitive binder to remove CB[n] macrocycles from the product. This strategy was successfully translated to a structurally different redox-active monomer that suffered similar limitations. We believe that the reported strategy of copolymerisation of redox-active monomers will open new avenues in the synthesis of functional materials for energy conversion and storage as well as for applications in electrochromic devices and (nano)electronics.
Data availability
Data for this paper, including NMR, UV-Vis, CV and ITC are available at https://doi.org/10.17863/CAM.85780.Author contributions
Conceptualization: SM, KS, MO, OAS data curation: SM, KS, ZH, OAS formal analysis: SM, KS, ZH, OAS funding acquisition: OAS supervision: OAS writing – original draft: SM, KS, ZH writing – review & editing: SM, KS, OAS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. thanks the Newton International Fellowship. K. S., Z. H. & O. A. S. thank EPSRC Programme Grant NOtCH (EP/L027151/1) for financial support. Z. H. and O. A. S. acknowledge ERC CoG grant CAM RIG (726470).References
- K. Koshika, N. Chikushi, N. Sano, K. Oyaizu and H. Nishide, Green Chem., 2010, 12, 1573–1575 RSC.
- M. Burgess, J. S. Moore and J. Rodríguez-López, Acc. Chem. Res., 2016, 49, 2649–2657 CrossRef CAS PubMed.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS.
- K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K.-C. Ho, J. Mater. Chem. C, 2019, 7, 4622–4637 RSC.
- Y. Y. Lai, X. Li and Y. Zhu, ACS Appl. Polym. Mater., 2020, 2, 113–128 CrossRef CAS.
- N. Sano, W. Tomita, S. Hara, C.-M. Min, J.-S. Lee, K. Oyaizu and H. Nishide, ACS Appl. Mater. Interfaces, 2013, 5, 1355–1361 CrossRef CAS PubMed.
- S. Sen, J. Saraidaridis, S. Y. Kim and G. T. R. Palmore, ACS Appl. Mater. Interfaces, 2013, 5, 7825–7830 CrossRef CAS PubMed.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- L. A. Godínez, R. Castro and A. E. Kaifer, Langmuir, 1996, 12, 5087–5092 CrossRef.
- J. B. Stepp and J. Schlenoff, J. Electrochem. Soc., 1997, 144, L155–L158 CrossRef CAS.
- C. Ronconi, J. F. Stoddart, V. Balzani, M. Baroncini, P. Ceroni, C. Giansante and M. Venturi, Chem.–Eur. J., 2008, 14, 8365–8373 CrossRef CAS PubMed.
- Y. Takahashi, N. Hayashi, K. Oyaizu, K. Honda and H. Nishide, Polym. J., 2008, 40, 763–767 CrossRef CAS.
- Y. Wang, M. Frasconi, W.-G. Liu, Z. Liu, A. A. Sarjeant, M. S. Nassar, Y. Y. Botros, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 876–885 CrossRef CAS PubMed.
- T. Ogoshi, Y. Nishida, T.-a. Yamagishi and Y. Nakamoto, Macromolecules, 2010, 43, 7068–7072 CrossRef CAS.
- X. Ji, S. Dong, P. Wei, D. Xia and F. Huang, Adv. Mater., 2013, 25, 5725–5729 CrossRef CAS PubMed.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed.
- F. Amir, K. P. Liles, A. O. Delawder, N. D. Colley, M. S. Palmquist, H. R. Linder, S. A. Sell and J. C. Barnes, ACS Appl. Mater. Interfaces, 2019, 11, 24627–24638 CrossRef CAS PubMed.
- F. Amir, X. Li, M. C. Gruschka, N. D. Colley, L. Li, R. Li, H. R. Linder, S. A. Sell and J. C. Barnes, Chem. Sci., 2020, 11, 10910–10920 RSC.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS PubMed.
- J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- H. Nishide, S. Iwasa, Y.-J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- K. Zhang, M. J. Monteiro and Z. Jia, Polym. Chem., 2016, 7, 5589–5614 RSC.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- T. Janoschka, S. Morgenstern, H. Hiller, C. Friebe, K. Wolkersdörfer, B. Häupler, M. D. Hager and U. S. Schubert, Polym. Chem., 2015, 6, 7801–7811 RSC.
- Z. Wang and N. V. Tsarevsky, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1173–1182 CrossRef CAS.
- N. Oyama, T. Ohsaka, H. Yamamoto and M. Kaneko, J. Phys. Chem., 1986, 90, 3850–3856 CrossRef CAS.
- M. Burgess, E. Chénard, K. Hernández-Burgos, G. Nagarjuna, R. S. Assary, J. Hui, J. S. Moore and J. Rodríguez-López, Chem. Mater., 2016, 28, 7362–7374 CrossRef CAS.
- T. Škorjanc, D. Shetty, M. A. Olson and A. Trabolsi, ACS Appl. Mater. Interfaces, 2019, 11, 6705–6716 CrossRef PubMed.
- G. Nagarjuna, J. Hui, K. J. Cheng, T. Lichtenstein, M. Shen, J. S. Moore and J. Rodríguez-López, J. Am. Chem. Soc., 2014, 136, 16309–16316 CrossRef CAS PubMed.
- W. Xue, H. Mutlu and P. Theato, Eur. Polym. J., 2020, 130, 109660 CrossRef CAS.
- H. Zou, J. Liu, Y. Li, X. Li and X. Wang, Small, 2018, 14, 1802234 CrossRef PubMed.
- Q. Li, J. Sun, J. Zhou, B. Hua, L. Shao and F. Huang, Org. Chem. Front., 2018, 5, 1940–1944 RSC.
- Cucurbituril-based Functional Materials, ed. D. Tuncel, The Royal Society of Chemistry, 2020, pp. P001–P289 Search PubMed.
- Q. Li, K. Jie and F. Huang, Angew. Chem., Int. Ed., 2020, 59, 5355–5358 CrossRef CAS PubMed.
- H.-J. Kim, W. S. Jeon, Y. H. Ko and K. Kim, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5007–5011 CrossRef CAS PubMed.
- A. Trabolsi, M. Hmadeh, N. M. Khashab, D. C. Friedman, M. E. Belowich, N. Humbert, M. Elhabiri, H. A. Khatib, A.-M. Albrecht-Gary and J. F. Stoddart, New J. Chem., 2009, 33, 254–263 RSC.
- M. C. Lipke, T. Cheng, Y. Wu, H. Arslan, H. Xiao, M. R. Wasielewski, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 3986–3998 CrossRef CAS PubMed.
- M. M. MacInnes, B. R. Cousineau, S. M. Youngs, K. Sinniah, J. J. Reczek and S. Maldonado, J. Electrochem. Soc., 2019, 166, H825–H834 CrossRef CAS.
- K. Sokołowski, J. Huang, T. Földes, J. A. McCune, D. Xu, B. de Nijs, R. Chikkaraddy, S. Collins, E. Rosta, J. J. Baumberg and O. A. Scherman, Nat. Nanotechnol., 2021, 16, 1121–1129 CrossRef PubMed.
- G. Wu, M. Olesińska, Y. Wu, D. Matak-Vinkovic and O. A. Scherman, J. Am. Chem. Soc., 2017, 139, 3202–3208 CrossRef CAS PubMed.
- G. Wu, I. Szabó, E. Rosta and O. A. Scherman, Chem. Commun., 2019, 55, 13227–13230 RSC.
- Y. Song, X. Huang, H. Hua and Q. Wang, Dyes Pigm., 2017, 137, 229–235 CrossRef CAS.
- G. Wu, Y. J. Bae, M. Olesińska, D. Antón-García, I. Szabó, E. Rosta, M. R. Wasielewski and O. A. Scherman, Chem. Sci., 2020, 11, 812–825 RSC.
- M. D. Fernandez and G. M. Guzmán, Eur. Polym. J., 1990, 26, 301–307 CrossRef CAS.
- C. Billaud, M. Sarakha and M. Bolte, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3997–4005 CrossRef CAS.
- M. D. Fernández and G. M. Guzmán, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 3703–3720 CrossRef.
- G. V. Reddy, M. V. Umamaheswari, V. Vidya and V. Krishnaswamy, Polym. Int., 1994, 34, 279–287 CrossRef CAS.
- N. K. Das and B. M. Mandal, Polymer, 1982, 23, 1653–1658 CrossRef CAS.
- M. R. Geraskina, A. S. Dutton, M. J. Juetten, S. A. Wood and A. H. Winter, Angew. Chem., Int. Ed., 2017, 56, 9435–9439 CrossRef CAS PubMed.
- H. Ji, F. Liu and S. Sun, Sci. World J., 2013, 452056 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02072f |
| This journal is © The Royal Society of Chemistry 2022 |