
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterometallic bond activation enabled by unsymmetrical ligand scaffolds: bridging the opposites
R.
Govindarajan†
 ,
Shubham
Deolka†
and
Julia R.
Khusnutdinova
*
,
Shubham
Deolka†
and
Julia R.
Khusnutdinova
*
Coordination Chemistry and Catalysis Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, 904-0495, Okinawa, Japan. E-mail: juliak@oist.jp
First published on 28th October 2022
Abstract
Heterobi- and multimetallic complexes providing close proximity between several metal centers serve as active species in artificial and enzymatic catalysis, and in model systems, showing unique modes of metal–metal cooperative bond activation. Through the rational design of well-defined, unsymmetrical ligand scaffolds, we create a convenient approach to support the assembly of heterometallic species in a well-defined and site-specific manner, preventing them from scrambling and dissociation. In this perspective, we will outline general strategies for the design of unsymmetrical ligands to support heterobi- and multimetallic complexes that show reactivity in various types of heterometallic cooperative bond activation.
1. Introduction
Transition metal-catalyzed reactions played an essential role in the development of the modern chemical industry, allowing for the synthesis of commodity chemicals at large scales using energy-efficient protocols, mild conditions, and readily available starting materials.1 For example, the modern large scale production of ammonia used for fertilizers and as a commodity chemical relies on the heterogeneously catalyzed Haber–Bosch process.2 Ziegler–Natta polymerization by group 4 metal catalysts in combination with an organoaluminum co-catalyst is used to manufacture a large number of commercial plastics under mild conditions on a multimillion ton scale.3,4 Another Nobel prize-winning class of homogeneously catalyzed reactions are Pd-catalyzed C–C bond cross-coupling reactions that are widely used in organic synthesis and in the pharmaceutical industry.5 Both in heterogeneous and homogeneous catalytic reactions, the use of a combination of two or more metals acting as co-catalysts is not uncommon.6 For example, the most common protocol of the Sonogashira C–C bond cross-coupling of aryl or vinyl halides with terminal alkynes is based on the use of a palladium catalyst and a copper co-catalyst, the latter responsible for terminal acetylene activation and subsequent transmetalation of acetylide to Pd.7In nature, heteromultimetallic assemblies are often present in enzyme cofactors, facilitating challenging multielectron and multiproton transfer reactions in Fe/Mo and V/Fe nitrogenases, Ni/Fe hydrogenases, Fe/Ni and Cu/Mo-containing CO dehydrogenases, and many other metallo-proteins.6 In these enzymes, a suitable ligand environment enabling the assembly of such catalytically active, well-defined multimetallic active sites is optimized through evolution, and achieving the same level of control over multimetallic core assembly in artificial systems requires careful ligand design. At the same time, the simplicity, variability and robustness of the artificial synthetic systems is the key to their practical utilization in catalysis and in understanding the role of cooperativity between multiple metals.
For example, the active site of the oxygen-evolving complex (OEC) in photosystem II consists of an unsymmetrical Mn4CaOn cluster responsible for biological dioxygen generation (Scheme 1a). The synthesis of models for such a complicated heterometallic cluster has been a subject of many efforts over the decades. Two selected examples are shown in Scheme 1b and c. Polynucleating N,O-donor ligand 1.1 was utilized by Powell and co-workers to assemble a series of MMn4 (M = Ca, Na) clusters as structural models for the OEC.8 The trinucleating ligand framework 1.2, which was developed by Agapie and co-workers, contains three N,N,O-donor fragments tethered to a rigid 1,3,5-triarylbenzene spacer; it was employed in the stepwise synthesis of a [Mn3CaO4]6+ cluster with several types of bridging ligands used to interconnect the metal atoms.9 A complicated ligand architecture is not always a prerequisite for the formation of heterometallic clusters and a Mn4Ca cluster was also synthesized from very simple precursors, where the metal atoms are bridged only by oxo and pivalato-bridges.10
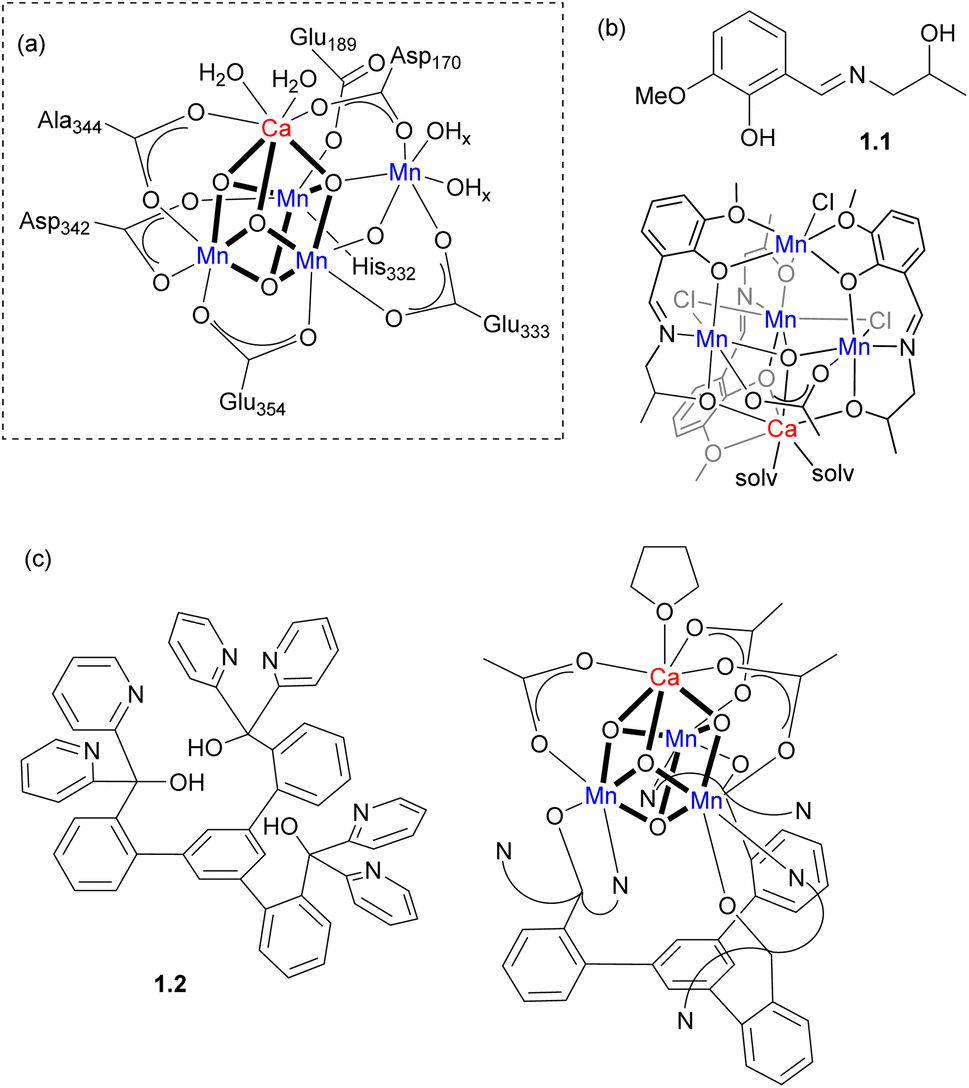 | ||
| Scheme 1 (a) The proposed structure of OEC.9 (b) Ligand 1.1 and Mn4Ca cluster supported by 1.1. (c) Ligand 1.2 and Mn3Ca cubane supported by 1.2. | ||
Thus, the rational design of ligands which support multimetallic assemblies often requires the presence of several bridging motifs that hold several metals together, preventing their dissociation into monometallic species, while also providing an unsymmetrical environment and different types of donor atoms to allow for site differentiation.
The main focus of this perspective is to highlight general principles of unsymmetrical ligand scaffold design for the support of heterobi- and multimetallic systems that can be employed in metal–metal cooperative bond activation or new materials design. Such unsymmetrical ligand scaffolds are designed to provide site selective positioning for the metal atoms to achieve the desired electronic and physical properties and the mode of bond activation. We will mostly focus on the heterometallic systems that show metal–metal bonding interactions (at least, prior to bond activation), although we will also discuss cases where two metals are present in close proximity and the presence or absence of metal–metal bonding hasn't been studied in detail. To limit the scope of the review, the majority of examples will describe heterobi- or multimetallic complexes containing at least one transition metal (TM) in a combination with another TM, post-transition metal, alkali or alkaline earth metal, or an f-block element. Only a few examples will be discussed that involve the combination of a transition metal with a metalloid, mostly for the sake of the comparison of reactivity or structural features with multimetallic complexes.
For the purpose of comparison and understanding the effect of bridging on a bimetallic activation mode, we will first discuss unbridged bimetallic complexes (Scheme 2a), where unsupported metal–metal bonds act as the main driving force holding the metals together. We will show that such unsupported systems may sometimes dissociate into monometallic or other types of multimetallic species in solution, which sometimes results in deactivation, but in some cases it may become the pathway to generate the active species in bimetallic catalysis.
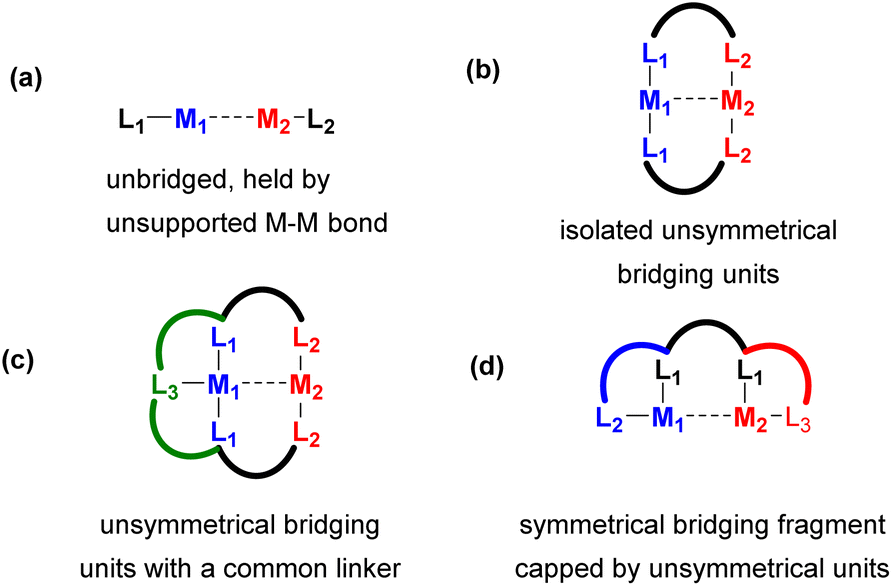 | ||
| Scheme 2 Commonly used motifs for unsymmetrical ligand scaffold design in heterobi- or multimetallic complexes: (a) unbridged complexes supported by mononucleating ligands and held by metal–metal interactions; (b) complexes containing one or more isolated unsymmetrical bridging units; (c) complexes in which several bridging units are linked with a common linker; (d) heterobimetallic complexes containing a bridging fragment capped by unsymmetrical coordinating units. | ||
Next, we will discuss various types of bi- or multiheterometallic complexes where two metals are bridged by a common ligand, typically based on a two- or three-atom neutral or anionic fragment that supports close metal–metal distances. Metal–metal interactions may or may not be present in such systems, depending on other factors such as the coordination environment at each metal center and the oxidation state, as will be discussed in Sections 2 and 3 that describe heterobimetallic complexes supported by bridging ligands.
One of the main approaches to site-selective assembly of heterometallic cores is the use of unsymmetrical bridging ligands (Scheme 2b). The presence of a suitable bridging fragment(s) provides stability against dissociation into monometallic species and allows for close proximity between two metals and heterometallic bonding interactions, which can be used as an important tool in cooperative catalysis and bond activation.
As the number of such bridging units often determine reactivity and vacant coordination site availability, another common approach involves linking two or three bridging units with a common linker to provide a well-defined coordination environment, such as in the ligand model shown in Scheme 2c.
Another approach to design an unsymmetrical ligand scaffold is to append a symmetrical bridging fragment with two unsymmetrical binding pockets at each (or only one) terminus (Scheme 2d). The last two approaches allow for a selective synthesis of heterobi- and multimetallic system in a site-selective manner.
In the final Section 4 of this perspective, we will show examples of polynucleating ligand designs which, via structural variations to the polynucleating ligand types shown in Scheme 2b–d, support polymetallic assemblies that contain more than two metal atoms.
To show the value of such systems for understanding metal–metal cooperativity in catalysis, we will discuss the examples of utilizing such heterometallic complexes in selective bond activation and catalysis, focusing on examples where both metals are required to observe reactivity, while an analogous monometallic species alone or even a combination of two different metal salts or precursors in the absence of polynucleating ligands, do not exhibit the same reactivity.11
There are multiple possible scenarios in which metal–metal cooperativity may manifest (Scheme 3), and it varies based on the nature of the metals, or the presence or absence of binucleating ligands.12 Two metals may participate in a concerted bond activation where the substrate's bond is broken between them. This was demonstrated for complexes containing polar metal–metal bonds, which are often reactive in selective polar covalent bond splitting. In contrast, Frustrated Lewis Pair (FLP)-type activation does not require the presence of a metal–metal bond prior to bond activation; in fact, metal–metal bonding in this case may even lead to lower reactivity. However, the combination of both a Lewis acidic and Lewis basic metals is required to achieve such cooperative bond activation processes. In another mode, only one metal participates directly in bond splitting, while it is influenced by the close proximity or bonding interactions with another metal acting as a “support”, which alters the active metal's substrate binding ability or Lewis acidity, or it can act as a redox reservoir. Many other scenarios may be envisioned, in which one of the metals may act as a docking site coordinating the substrate and bringing it closer to another metal, polarizing bonds via a π-coordination, or stabilizing the activation product by coordination, as will be discussed below in specific examples.
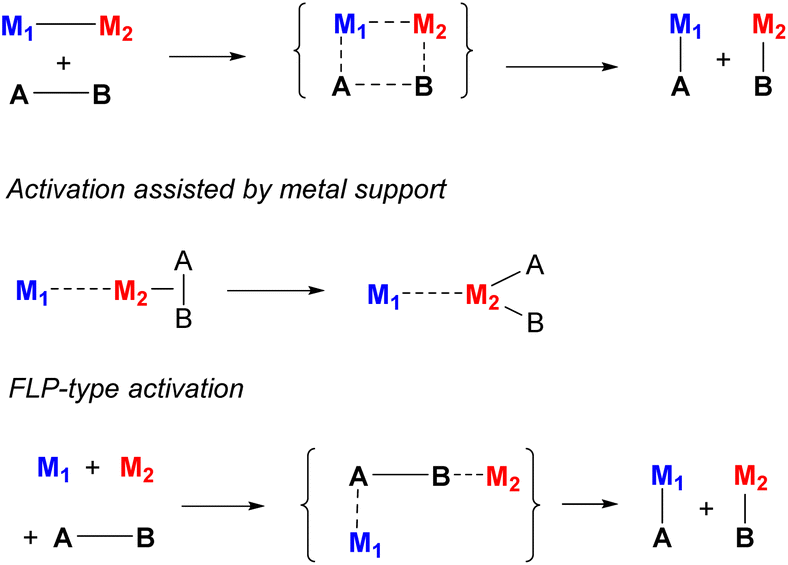 | ||
| Scheme 3 Selected examples of various types of heterometallic cooperative bond activation. | ||
2. Heterobimetallic complexes featuring unsupported metal–metal interactions
While the major focus of this perspective will be the design of polynucleating ligands, it is also necessary to mention an important class of heterobimetallic complexes that are held together solely or mainly by forming unsupported metal–metal interactions. Individual metal centers in these complexes may be supported by common mononucleating ligands such as carbonyls, N-heterocyclic carbenes (NHCs), β-diketiminates (nacnac), cyclopentadienyls (Cp), aryl or methyl groups, etc., although in some cases these ligands may also demonstrate interactions with both metals as will be discussed below. While more comprehensive reviews describing such unsupported metal–metal interactions in metal-only Lewis pairs have been published,13–15 these complexes will be discussed in this section in the context of various modes of the bimetallic bond activation and reactivity and compared with bridged bimetallic systems later.Bimetallic intermediates showing bonding interactions between two metals even in the absence of intentionally designed binucleating ligands may play an important role in catalysis where the presence of at least two different metal co-catalysts is required, especially at the transmetalation step where an aryl or alkyl group is transferred between two metals.16 The commonly accepted mechanism of Sonogashira C–C cross-coupling involves a Pd cycle and a Cu cycle, which merge at the transmetalation step, during which the acetylide group is transferred from Cu to Pd (Scheme 4a).7 Several research groups reported a number of Pd/Cu bimetallic organometallic complexes that serve as structural models of the transmetalation step and feature Pd and Cu atoms present in close proximity (Scheme 4b). For example, Chen and co-workers reported a cyclometalated aryl Pd complex 2.1, featuring a Pd⋯Cu distance of 2.55 Å, shorter than the sum of covalent radii (2.71 Å), suggesting bonding interactions between two metals.17 This complex was synthesized by mixing mononuclear precursors, cis-bis(1,10-benzo[h]quinolinato)palladium(II) and (IPr)CuIOTf (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene) followed by crystallization at low temperature, although the presence of bimetallic Pd/Cu species was also detected by electrospray mass spectrometry (ESI-MS). Interestingly, Cu also shows a relatively short distance to one of the carbon atoms of benzoquinolinate, showing that it essentially serves as the bridging aryl ligand between two metals, consistent with quantum theory of atoms in molecules (QTAIM) analysis. Another Pd/Cu complex 2.2 was reported by the Albéniz group, in which the acetylide ligand bridged two metal atoms by forming a σ-bond to Pd and π-bonding to Cu.18 In both cases, these complexes may be considered as structural, rather than functional models for the transmetalation step, as no C–C bond formation was ultimately observed. Interestingly, bridging Me group transfer was observed for a structurally related Pt dimethyl/Cu bimetallic complex using collision-induced dissociation (CID) mass-spectrometry.19
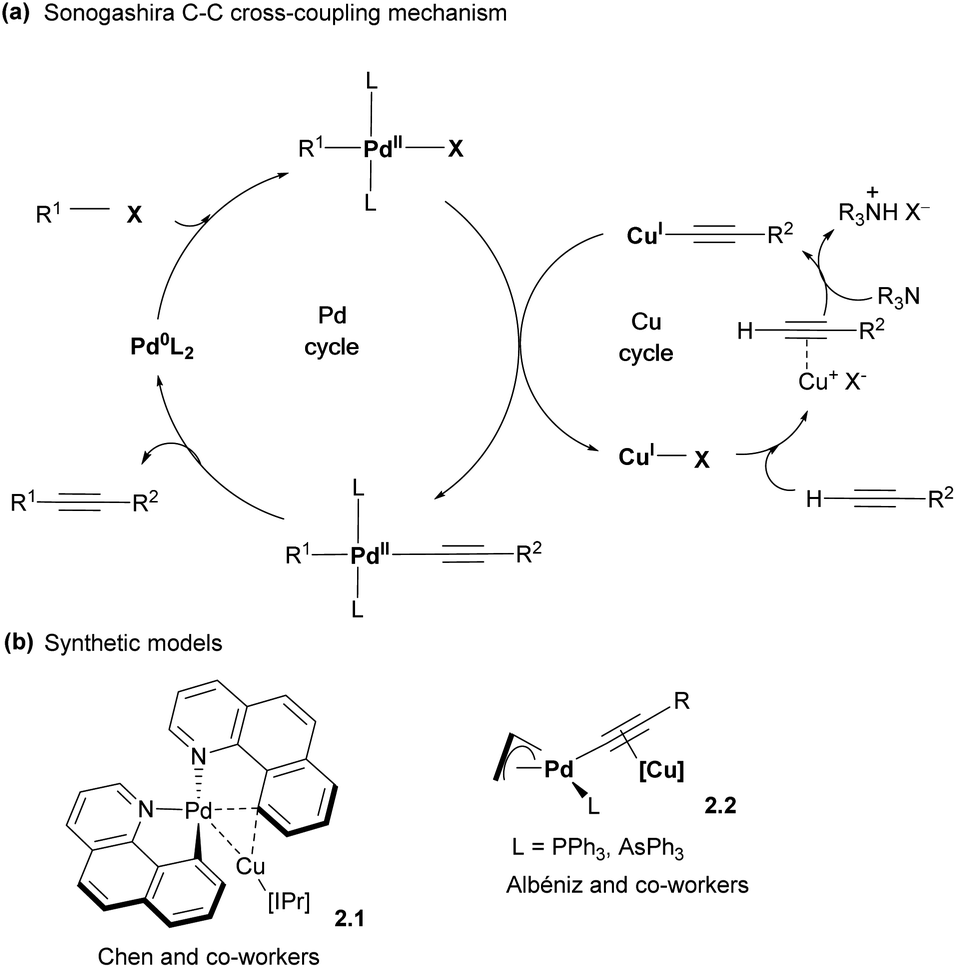 | ||
| Scheme 4 (a) General proposed mechanism for Sonogashira cross-coupling reactions. (b) Synthetic models reported in the literature. | ||
The complexes showing unsupported d8–d10 interactions between Pt (or Pd) and a d10 metal (CuI, AgI, ZnII) were also studied by the Chen and other groups for a wide range of organometallic compounds20–24 and the metal–metal bonding in these species was described as the donation from the filled d-orbital at the Pt (typically dz2) to the vacant orbital at the Lewis acidic metal center. The formation of such donor–acceptor bonding interactions between Lewis acidic and Lewis basic metals may significantly alter their reactivity as compared to the analogous monometallic systems. For example, the formation of unsupported Pt/Zn interactions in 2.5 and 2.6 is likely responsible for acceleration of the aryl–aryl reductive elimination reaction from Pt diaryl complexes 2.3 and 2.4 (Scheme 5). The transformation occurs after only 15 min at 60 °C in the presence of 10-fold access of ZnArF2, and does not occur in the absence of Zn. Interestingly, when the analogous diaryl Pt center is supported by the chelating diphosphine dmpe (1,2-bis(dimethylphosphino)ethane) ligand, a different geometry and reactivity pattern were observed leading to isolation of aryl-bridged 2.8 and aryl group exchange between Pt and Zn in complex upon heating. Based on density functional theory (DFT) studies, the bonding interactions in complex 2.5 mainly involve donation from the Pt-based dz2 orbital to the vacant Zn p-orbital along with back-donation from Zn to Pt–ligand antibonding orbital, while complex 2.8 shows the donation from both the Pt-based dz2 orbital and a π-system of aryl to Zn, as well as backdonation from the Zn d orbital to the Pt-bound aryls, further strengthening bridging aryl–Zn interactions. Upon aryl group transfer, the heterobimetallic core is disassembled, which is a common feature of many other heterobimetallic complexes showing unsupported metal–metal bonding, although in some cases such metal–metal interactions are proposed to re-form in the catalytic cycle.25
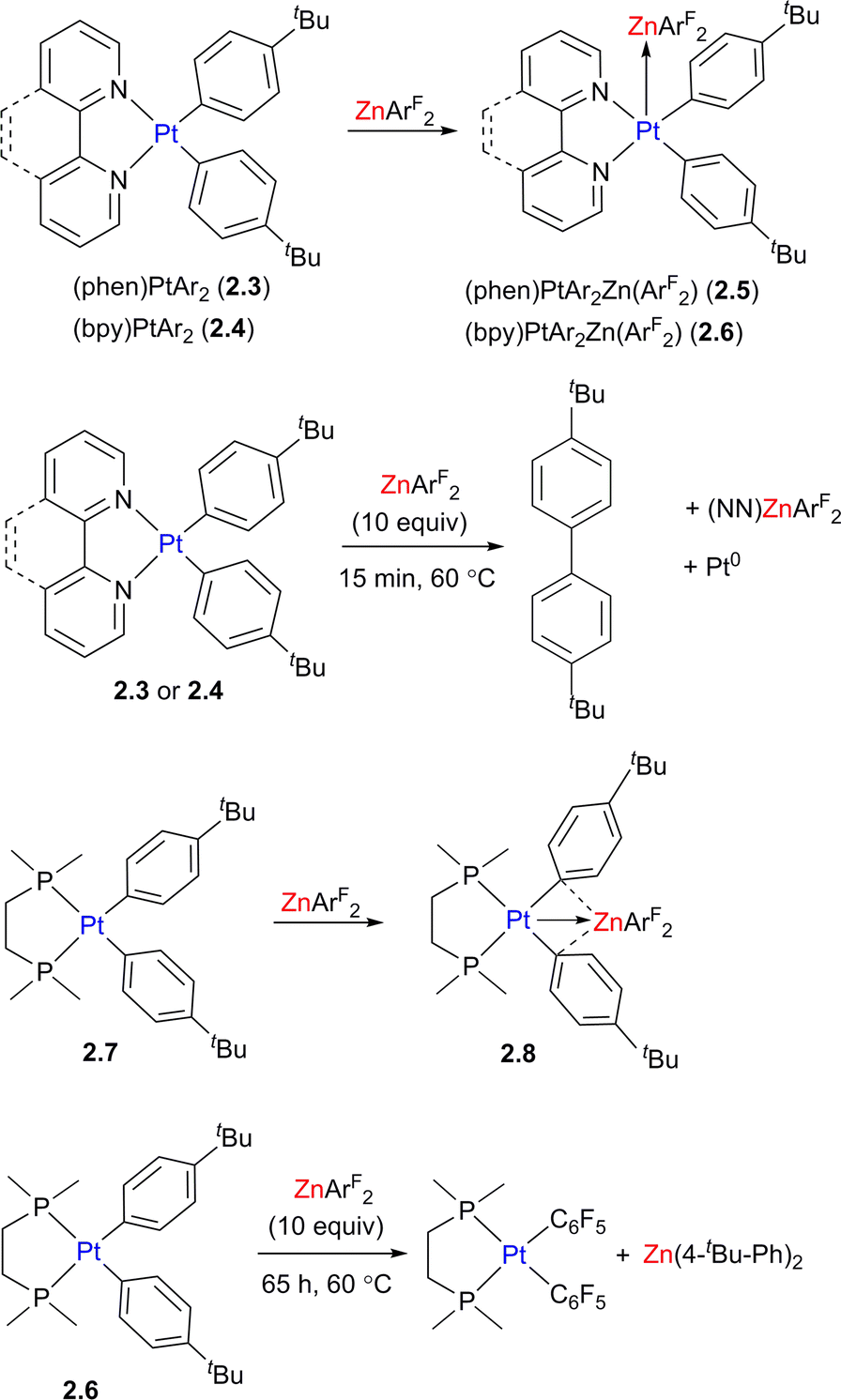 | ||
| Scheme 5 The structure and reactivity of Pt/Zn complexes as a function of the supporting ligand on Pt. | ||
For example, Mankad and co-workers reported catalysis using the family of bimetallic (NHC)Cu⋯metal carbonyl complexes.26 These complexes are typically obtained by reacting the (NHC)CuCl precursor with a suitable metal carbonyl precursor via salt metathesis (for example, Scheme 6a). The presence of unsupported Cu–[M] ([M] = M(Cp)n(CO)m, n = 0–1, m = 2–5; M = Cr, Mn, Co, Mo, Ru, W) bonding is evident from short Cu⋯M distances in the solid state and is supported by the computational studies that indicate the presence of a polarized M–M bond. Depending on the nature of the NHC ligand and nucleophilicity of the M counterpart, some (NHC)–M complexes were shown to be present in equilibrium concentrations along with isomeric {(NHC)2Cu}+{[M]2Cu}− species.26,27 Spectroscopic and structural analysis reveals that in (NHC)Cu⋯Fp (Fp = FeCp(CO)2) and several other complexes, the Cu atom shows secondary interactions with a “semi-bridging” carbonyl ligand, while such interactions are less prominent or absent in analogous complexes containing the Zn/Fe combination.27,28 The group's (IPr)Cu–FeCp(CO)2 complex 2.9 was used as a catalyst for photochemical arene C–H bond borylation (Scheme 6b).29 The proposed catalytic cycle involves metal–metal cooperative splitting of the B–H bond in pinacolborane to generate CpFe(CO)2(BPin) as the active borylating species and mononuclear (IPr)CuH. The bimetallic species is proposed to re-form following binuclear elimination of H2. Although monometallic Fe boryl complexes have been previously utilized for stoichiometric C–H bond borylation, catalytic turnover was not achieved due to the formation of an inactive [CpFe(CO)2]2 dimer,30,31 illustrating the importance of heterobimetallic cooperation in preventing undesired catalyst deactivation pathways. Later, the same Fe/Cu complex was utilized for regioselective 1,4-hydroboration of pyridines (Scheme 6c).32 The selectivity was explained by the synergistic activation via electrophilic FpBPin coordination to the N-atom of pyridine, which activates it towards nucleophilic attack by the mononuclear Cu hydride.
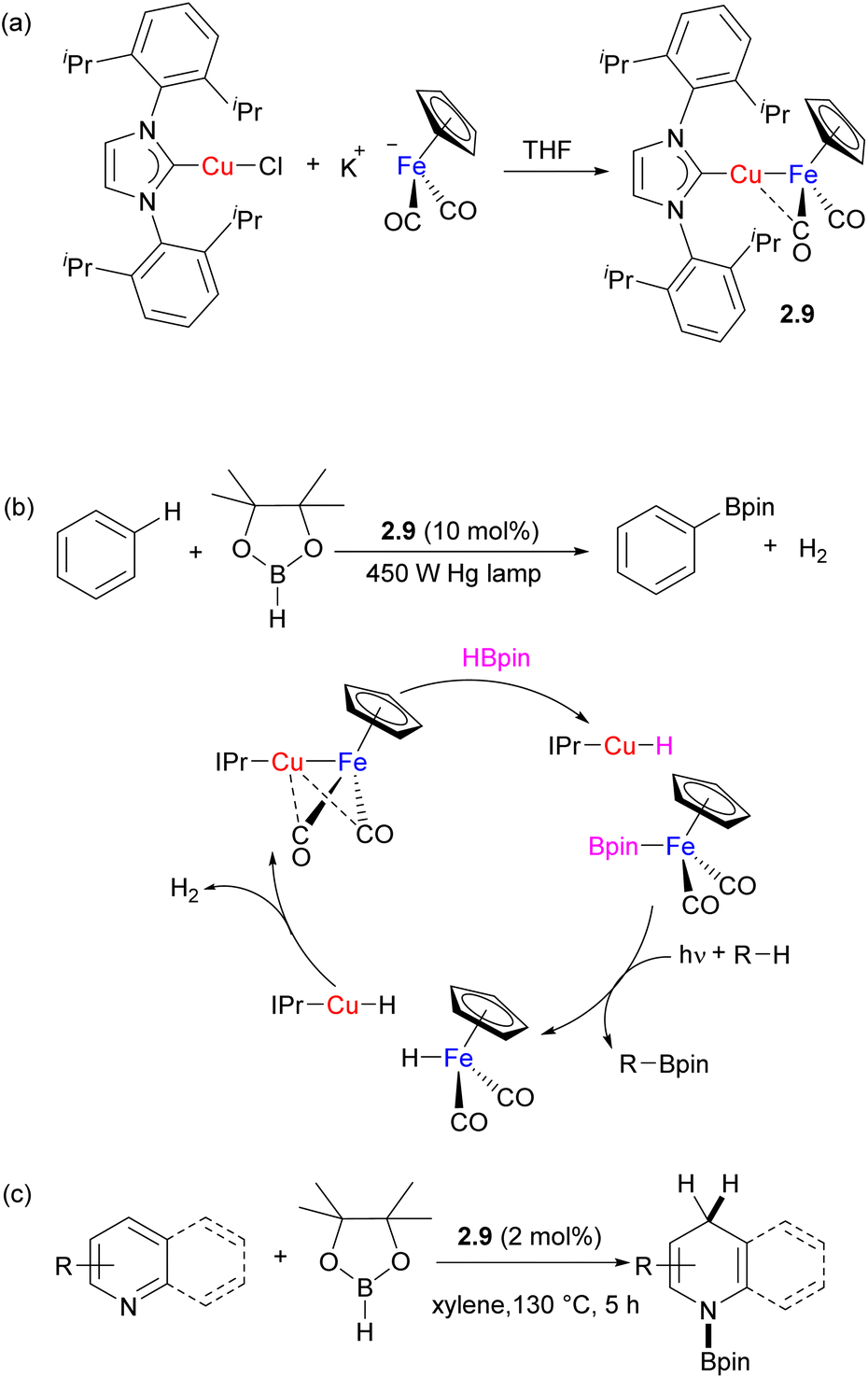 | ||
| Scheme 6 (a) Synthesis of Fe/Cu complex 2.9 complex; (b) catalytic arene C–H borylation and (c) catalytic activity in pyridine hydroboration. | ||
Using a similar synthetic approach, Zacchini and co-workers utilized a simple reaction between Na[Co(CO)4] and M(NHC)Cl (M = Cu, Ag, and Au) to prepare a series of heterobimetallic complexes featuring an unsupported metal–metal bond (Scheme 7).33 The outcome of the reaction was influenced by the nature of the metal, NHC ligand and the solvent. While Co/Cu and Co/Au complexes 2.10 and 2.11 were the only species formed, an analogous complex with silver is present in equilibrium with 2.13 and 2.14. The dissociation to form [Co(CO)4]− was also observed by IR spectroscopy in polar solvents such as MeCN or DMSO, reflecting the labile nature of unsupported metal–metal bonds in solution, especially in polar coordinating solvents. The X-ray structures reveal the presence of weak M⋯(CO) contacts with a carbonyl bound to Co, which may in part be imposed by steric requirements. However, an analysis of Wiberg Bond Indices (WBI) and “bridge asymmetry parameters” suggests semi-bridging interactions between M and CO.34,35 Natural Bond Orbital (NBO) analysis also reveals the presence of interactions between the Cu-based 3d and 4s orbitals and a π-antibonding CO orbital.
These complexes were examined in catalytic ammonia-borane (AB) dehydrogenation, where the best performance was shown by the bimetallic complex 2.10, while the trinuclear complex 2.14 also showed catalytic activity, and monometallic components alone, such as Na[Co(CO)4] or M(NHC)Cl, performed poorly. The authors proposed bimetallic activation of AB across the polar Co⋯Cu bond and interaction of the hydridic B–H with the positively charged Cu(NHC)+ and N–H with a negatively charged [Co(CO)4]− fragment.
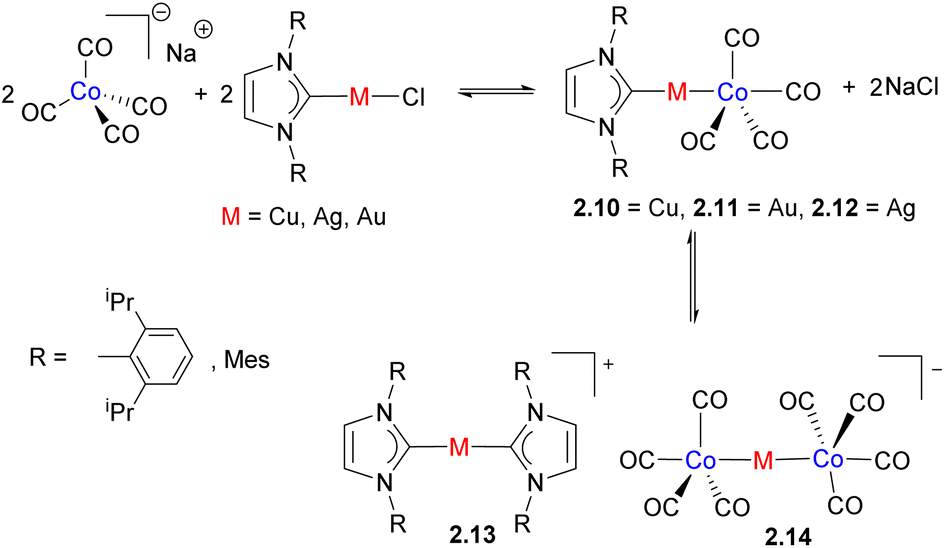 | ||
| Scheme 7 The formation of cobalt/coinage metals heterobimetallic complexes. | ||
Although substrate bond activation by heterobimetallic complexes with unsupported metal–metal bonds is accompanied by the complete cleavage of the bimetallic assembly and the formation of mononuclear complexes in many cases, if the substrate may also serve as a bridging ligand, the bimetallic assembly may be preserved, although metal–metal interactions are typically disrupted. This can be illustrated by several representative examples below describing CO2 activation across the polar metal–metal bond in Fe/Zr, Al/Au, Al/Zn, and Al/Fe complexes (Schemes 8–10).36–40 CO2 insertion into unsupported metal–metal bonds in Cp(CO)2M–Zr(Cl)Cp2 (M = Fe, Ru) was reported in 1994 by Cutler and co-workers.36 The Gade group extensively studied insertion of heteroallenes X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CY into unsupported Zr–M (M = Fe, Ru) bond where Zr is supported by a tripodal amido ligand.37 For example, the reaction of 2.15 with 1 equivalent of CO2, CS2 and other heteroallenes results in the formation of 2.16 (Scheme 8a). Based on the kinetic studies of the CS2 insertion, the authors proposed a mechanism of cooperative activation involving initial reversible Lewis acid–base interaction of CS2 with the Zr center, followed by the rate determining cleavage of Zr–Fe bond and insertion of CS2 (Scheme 8b).37 Interestingly, similar metallacarboxylate complexes could also be obtained by oxygen atom transfer to a carbonyl ligand from an organic substrate.41,42
CY into unsupported Zr–M (M = Fe, Ru) bond where Zr is supported by a tripodal amido ligand.37 For example, the reaction of 2.15 with 1 equivalent of CO2, CS2 and other heteroallenes results in the formation of 2.16 (Scheme 8a). Based on the kinetic studies of the CS2 insertion, the authors proposed a mechanism of cooperative activation involving initial reversible Lewis acid–base interaction of CS2 with the Zr center, followed by the rate determining cleavage of Zr–Fe bond and insertion of CS2 (Scheme 8b).37 Interestingly, similar metallacarboxylate complexes could also be obtained by oxygen atom transfer to a carbonyl ligand from an organic substrate.41,42
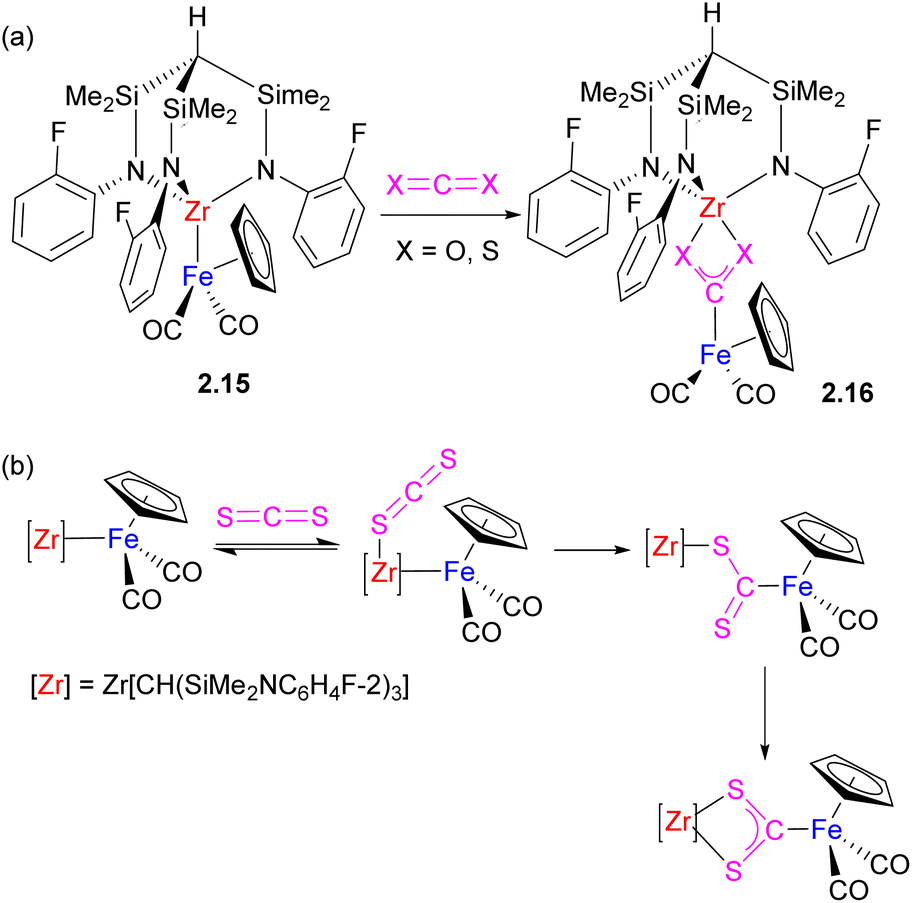 | ||
| Scheme 8 (a) Insertion of CO2 and CS2 into unsupported Fe–Zr bond. (b) Proposed mechanism of CS2 insertion. | ||
 | ||
| Scheme 9 Bimetallic CO2 activation of carbon dioxide by (a) Al/Au and (b) Al/Zn complexes. | ||
 | ||
| Scheme 10 CO2 and epoxide activation by Al/Fe heterobimetallic complex via metal–metal bond homolysis. | ||
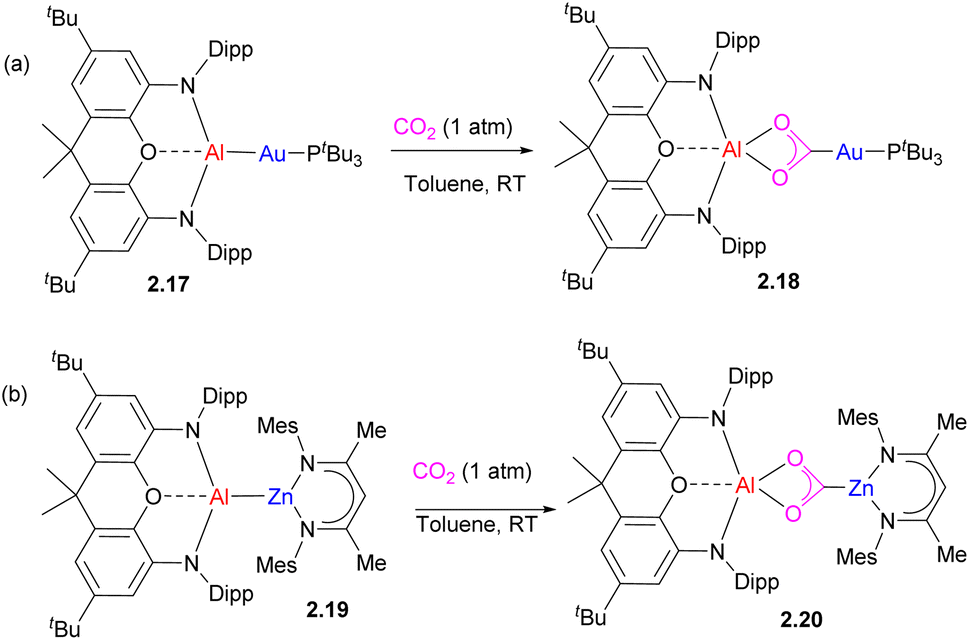
An interesting case of the analogous mode for CO2 insertion was reported for an Al/Au complex 2.17 leading to the formation of a metallacarboxylate 2.18 (Scheme 9a). Later on, the same group reported the analogous insertion of CO2 into unsupported Al–Zn to give 2.20 with Zn bound to a carbon atom.39 Based on the computational study of Al/Au complex 2.17, including QTAIM charge analysis, the authors proposed that Au binding to an extremely electron-donating aluminyl fragment induced nucleophilic character at the gold center leading to the observed reactivity with CO2.38 Interestingly, computational studies by Sorbelli et al. offer an alternative explanation, in which an Au–Al σ-bond is described as having an electron-sharing (rather than strongly polarized) nature, and a bimetallic cooperative CO2 activation is proposed via a diradical-like mechanism, assisted by electrophilic behaviour of Al.43 This alternative explanation suggests that a strongly polarized metal–metal bond may not be a prerequisite for CO2 activation.44
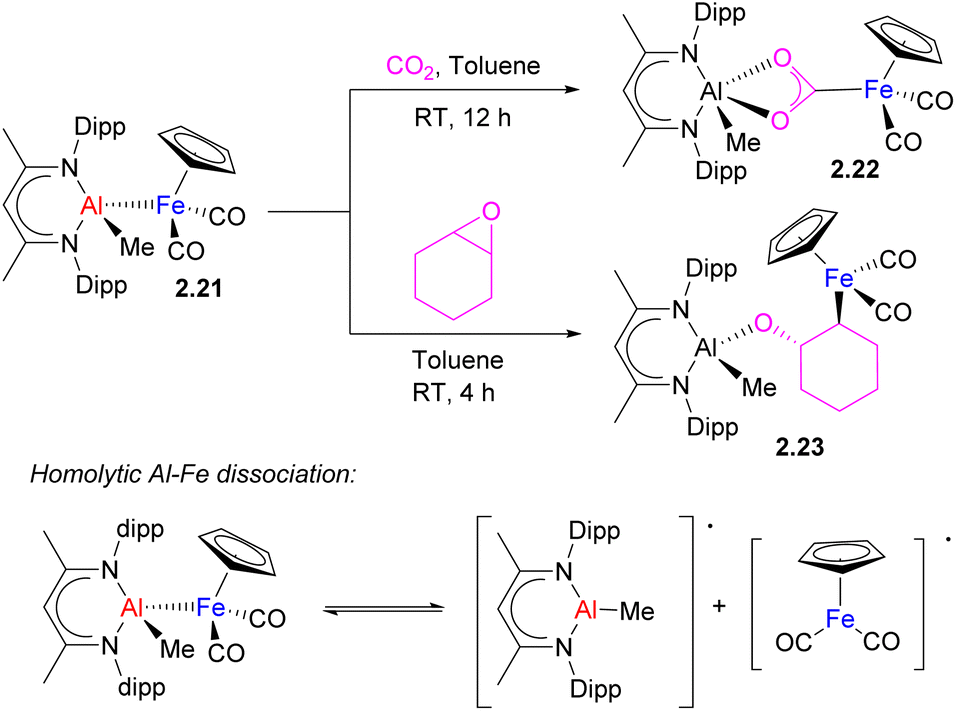
In this context, it is worth mentioning another example of a similar mode of CO2 insertion into an unsupported metal–metal bond leading to metallacarboxylate formation, which was proposed to occur through metallaradical formation. Recently, the Mankad group reported the insertion of CO2 into an Al–Fe bond in complex 2.21 to give a metallacarboxylate 2.22 with an Fe atom bound to the carbon of CO2 (Scheme 10).40 Based on mechanistic investigation, the authors proposed that homolytic Al–Fe bond dissociation precedes pairwise CO2 activation by Al- and Fe-containing a metallaradical pair. The C–O bond cleavage in epoxide by the same Al/Fe complex to give 2.23 was also proposed to occur via homolytic metal–metal bond cleavage which preceded substrate activation. The importance of sterics was also highlighted in promoting metal–metal bond homolysis through detailed computational analysis. Thus, the presence of a metal–metal interactions is not always a prerequisite for bimetallic bond activation and in this case, a M–M bonded complex may be considered as a dormant species that stabilizes highly reactive metallaradicals.
In an analogous manner, FLP type activation by Metal-Only Lewis Pairs (MOLP) does not require the presence of metal–metal interactions as a pre-requisite. For example, Campos reported facile H2 activation by a combination of Au(I) and Pt(0) complexes leading to the formation of hydride-bridged heterobimetallic (Scheme 11), homobimetallic, or Pt dihydride species, depending on ligand sterics and reaction conditions.45 Such facile H2 activation reactivity was not observed using each of the components alone. A detailed computational analysis by Campos and López-Serrano allowed them to rule out bimetallic activation via H2 addition across the Pt–Au bond and provided evidence for the metal-only FLP-type reactivity (Scheme 11).46 Similarly, H2 activation by Pt(0) complex in the presence of Zn(OTf)2 was proposed to occur through FLP-type reactivity.47
 | ||
| Scheme 11 H2 activation by Pt0/AuI system and possible modes of cooperative H2 activation. | ||
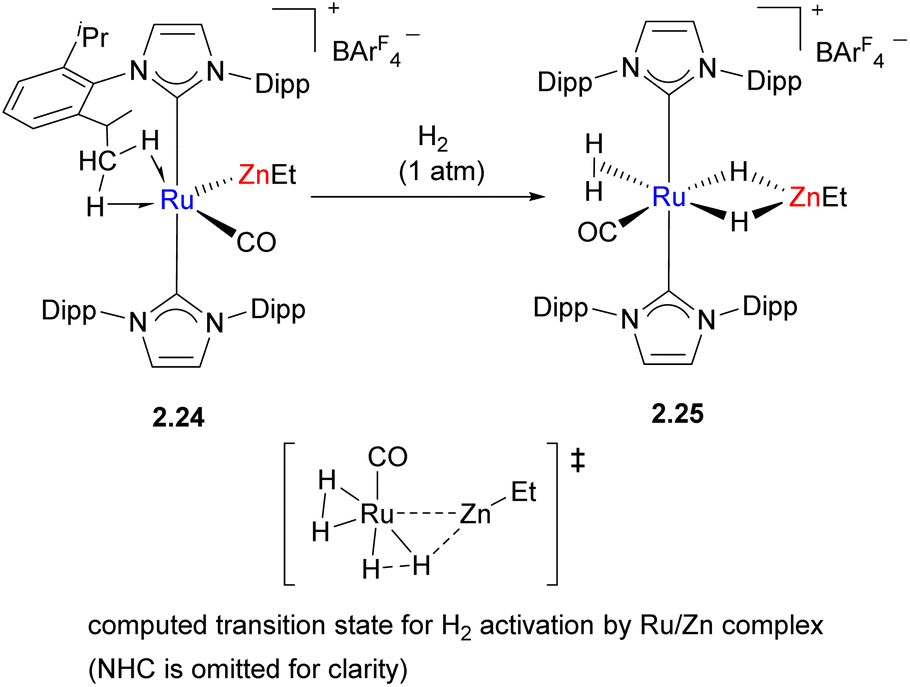
In another example of H2 activation, an unsupported Ru–Zn bond is present initially in the bimetallic complex 2.24, which reacts with H2 to give the hydride-bridged complex 2.25 (Scheme 12).48 However, detailed computational studies revealed that Ru is likely the only key player in this process which occurs via oxidative addition to Ru; the key H2 activation transition states suggests little or no interactions between H and Zn, which accepts a bridging hydride ligand after H–H bond cleavage has occurred. A structurally similar transition state was also found for the isomer in which Zn is unable to accept a hydride.
 | ||
| Scheme 12 The reactivity of Ru/Zn heterobimetallic complex 2.24 with hydrogen. | ||
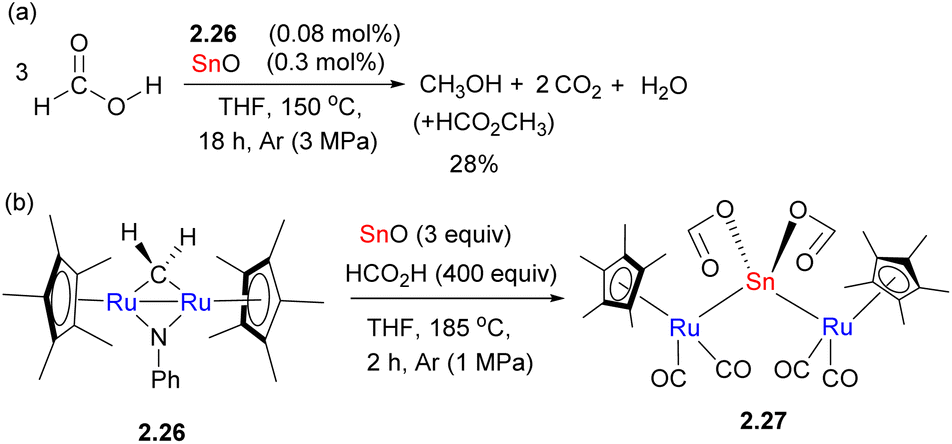
The exact nature of bimetallic participation is therefore not always easy to elucidate when unsupported metal–metal bonds are present in the active species. Fujita, Takemoto and Matsuzaka have recently shown that a combination of diruthenium catalyst 2.26 and Sn(II) oxide results in catalytic formic acid disproportionation to form methanol (and methyl formate) in up to 28% yield and a turnover number (TON) of 191 (Scheme 13a). The role of the trinuclear Ru–Sn–Ru complex 2.27 isolated from the reaction mixture that contained an unsupported Ru–Sn bond is not entirely clear (Scheme 13b), as the computational studies suggest that formic acid disproportionation may also be mediated by monomeric tin(II) formate alone. However, the experimental studies showed that catalytic conversion was not observed using tin(II) oxide only, suggesting that tin formate does not persist under catalytic conditions. Thus, the authors propose that the role of Ru in 2.27 may be stabilization of the monomeric tin formate via Ru–Sn bonds.49 Moreover, the control experiment in the absence of SnO results in a TON of 2, which indicates that Ru alone is not an active catalyst.
 | ||
| Scheme 13 Formic acid disproportionation to methanol catalyzed by Ru/Sn complex (a) and isolation of a trinuclear Ru–Sn–Ru complex (b). | ||
Overall, in this section, we discussed several representative examples of heterobimetallic complexes with unsupported metal–metal interactions. In some cases, metal–metal bonding is also supplemented by bridging or semi-bridging interactions with other ligands, such as CO, alkyl or aryl groups. Although the synthesis of these complexes may be quite straightforward from starting materials containing common cyclopentadienyl, nacnac, NHC or other mononucleating ligands, the solution reactivity of such complexes is complex to pin down due to metal–metal bond dissociation, disproportionation, and the formation of other mono- or other multimetallic species in solution. The types of bimetallic reactivity in such complexes differ depending on the system. In some cases, cooperative bimetallic bond activation across a polar metal–metal bond is proposed, while in other cases, prior dissociation of a metal–metal bond may be required to form the actual active monometallic or metallaradical species. Similarly, FLP type activation does not require (or may even be inhibited by) the formation of metal–metal bonding interactions. The presence of a metal–metal bond does not always imply the participation of the second metal at the crucial bond cleavage process, although it may greatly influence the resulting catalytic or stoichiometric reactivity.
3. Heterobimetallic complexes with bridging ligands
In this section we will discuss heterobimetallic compounds containing bridging ligands which may support bimetallic assembly and control site selectivity for coordination by careful choice of donor ligands and the overall ligand architecture.3.1. Heterobimetallic complexes with two-atom bridging ligand
Although complexes with monoatomic bridges are also known in bimetallic bond activation, such as in oxidative addition of H2 in an imido-bridged Zr/Ir complex reported by Bergman et al.50 or in bimetallic cooperation in sulfide-bridged [FeNi] hydrogenase51–53 that catalyzed reversible proton reduction/hydrogen oxidation, the use of unsymmetrical two- or three-atom bridges for heterobimetallic assembly formation presents a more convenient approach and allows for site selectivity control through the choice of donor atoms in the unsymmetrical bridge. The reactivity of the resulting bimetallic core is then often determined by the availability of vacant coordination sites for substrate activation. In an early example, Wolczanski et al. reported a bimetallic early/late transition metal complex formation bridged by alkoxyalkylphosphines to model a heterogeneous catalyst surface containing an electron-rich late TM/early TM oxide combination.54–56 The formation of a TM → TiIV (TM = Ni, Pd, Pt) dative bond was reported by Nagashima for complexes bridged with two phosphinoamide ligands, with the P-atom coordinating to the late TM and the N-atom bound to Ti.57 Such interactions and the influence of the second metal were reported to enhance electrophilicity of the η3-methallyl ligand at the late TM center, facilitating its reactivity towards nucleophilic attack. Simple phosphinoamide ligands were recently used to prepare a series of Pd/Ln (Ln = Sc, Y) complexes.58The utilization of phosphinoamides as bridging ligands to selectively construct heterobimetallic assemblies, especially those containing early TM and the first row late TM (Zr/Co and Ti/Co), was significantly expanded by Thomas and co-workers who demonstrated various applications of such complexes in bimetallic bond activation and catalysis.59–61 The common approach to the synthesis of such early/late TM phosphinoamide-bridged bimetallic complexes involves the treatment of the mononuclear Zr complex (“metalloligand”) with a late transition metal salt. In Zr precursor 3.1, the phosphinoamide ligand was shown to bind to Zr with both amide and phosphorus atoms in the solid state, while dynamic behaviour was observed in solution, involving reversible dissociation/coordination of three phosphorus atoms, making it a convenient precursor to assemble a bimetallic species by addition of a second metal.62 A representative example is shown in Scheme 14: the Zr phosphinoamide complex reacted with CoI2 to give a ZrIV/CoI bimetallic product in which a P-terminus selectively coordinates to the “soft” CoI center, and an amide terminus binds to a “hard” ZrIV.63 In this case, iodide likely served as a reductant; attempted reactions with metal salts having less accessible reduction potentials failed to give a bimetallic product.63 Metal–metal interactions facilitate further two-electron reduction at Co at potentials 0.6–0.9 V more positive than in the analogous monometallic systems.
 | ||
| Scheme 14 Synthesis of representative phosphinoamido Zr/Co complexes. | ||
Initial studies by Thomas et al. on C3-symmetric tris(phosphinoamide) ZrIV–Co−I complexes showed that they are highly reactive in a wide range of bond activation processes, exemplified below by reactivity with H2 (ref. 64) and a terminal alkyne.65 Notably, H2 oxidative addition across multiple metal–metal bond leads to significant lengthening of the Zr–Co bond distance in the product (2.4397(2) Å in 3.4 and 2.33(1) Å in 3.3), while one of the phosphinoamide ligands undergoes P–N bond cleavage (Scheme 15).64 Such ligand degradation or phosphinoamide ligand dissociation from Co resulting from bond activation eventually prompted the design of bis(phosphinoamido)-bridged Zr/Co complexes that showed greater reactivity due to the availability of additional coordination sites.66 Such bis(phosphinoamido)bridged complexes were reactive in H2 oxidative addition across the metal–metal bond to give 3.8a–b with a formally single bond between ZrIV and CoI, with both metals showing “closed” M–H–M interactions and significant metal–metal bonding. The complexes 3.7a–b were also catalytically active in alkene hydrogenation and alkyne semi-hydrogenation.
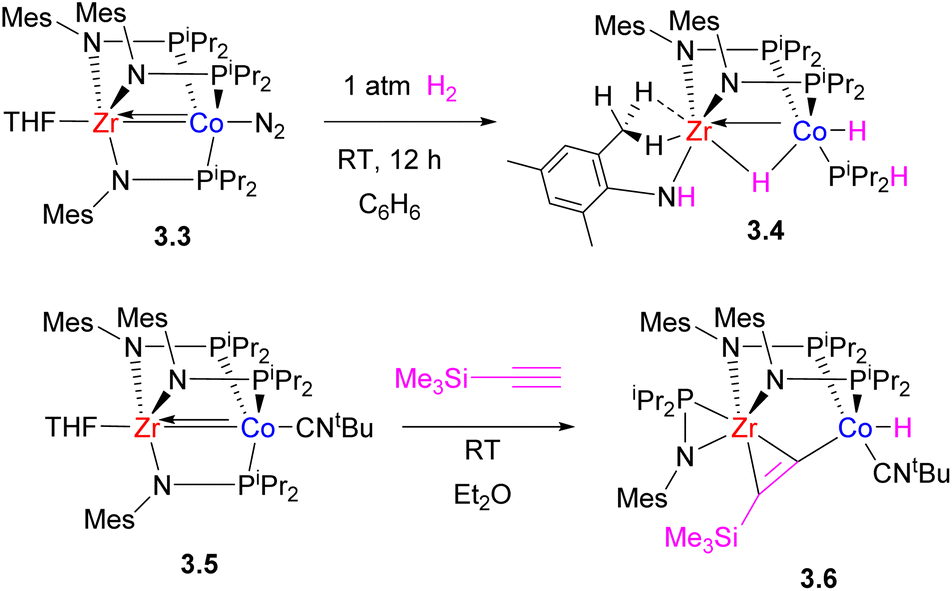 | ||
| Scheme 15 The reactivity of a tris(phosphinoamido) Zr/Co complex in H2 and terminal alkyne activation. | ||
Their high reactivity also allowed for the observation of bimetallic C–H bond activation with pyridine and terminal alkynes (Scheme 16).67
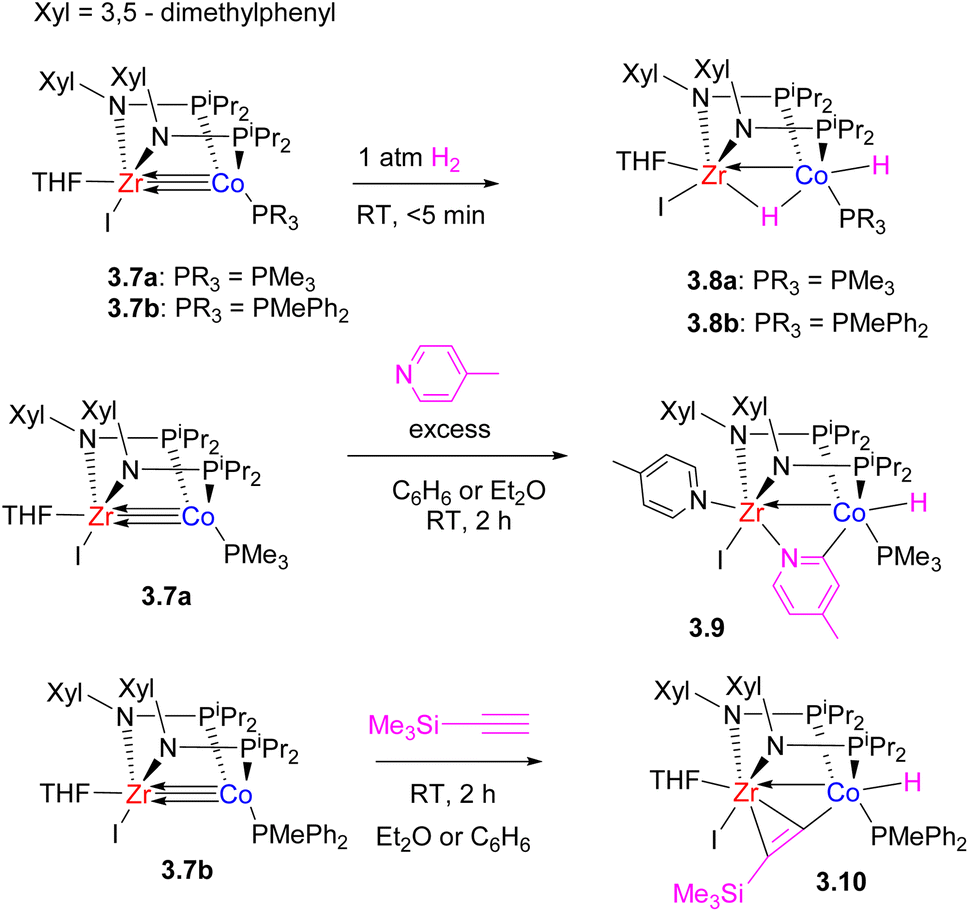 | ||
| Scheme 16 The reactivity of a bis(phosphinoamido) Zr/Co complex in H2 and C–H bond activation. | ||
Another type of two-atom bridged late TM/main group metal complex is the Pt/Al complex 3.11 obtained by treatment of the organoaluminum precursor with [Pt(PPh3)2(ethylene)]. It showed a significant degree of ring strain due to the short Pt⋯Al distance, 2.561(1) Å, which can be released in the CO2, H2 or amide reaction products (Scheme 17).68
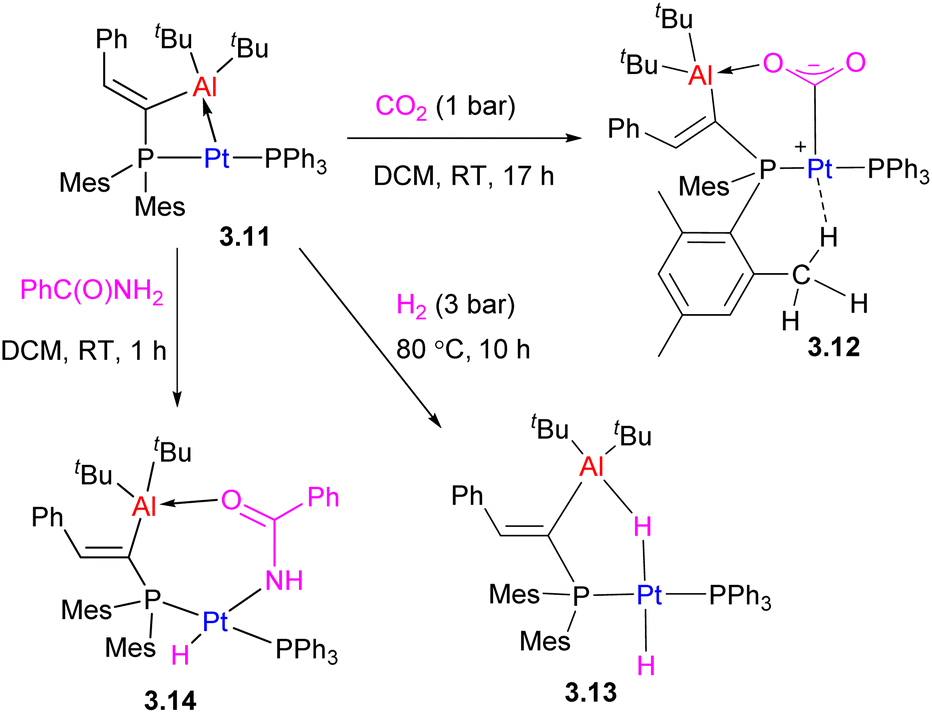 | ||
| Scheme 17 The bimetallic bond activation reactivity of a Pt/Al complex. | ||
3.2. Heterobimetallic complexes with three-atom unsymmetrical bridging ligands
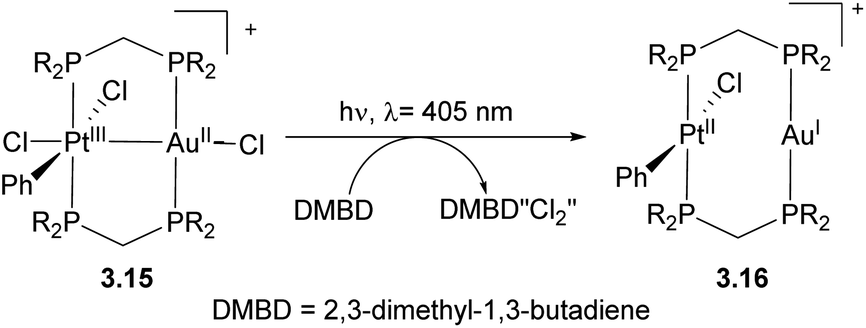
Bimetallic complexes in which metal–metal interactions are supported by a three-atom bridge are perhaps the most ubiquitous class, featuring a large number of homo- and heterobimetallic assemblies. The symmetrical anionic or neutral bridging ligands such as carboxylates, amidinates, naphthyridines, etc., are widely used for the construction of homobimetallic compounds, although in many cases such ligands may also act as mononucleating ligands binding in a κ2 or κ1 mode to a single metal center. However, the selective formation of heterobimetallic complexes supported by such symmetrical ligands is also reported. Shaw and co-workers utilized a general approach to prepare dppm-bridged late TM heterobimetallic complexes (dppm = diphenylphosphinomethane) by the treatment of a suitable monometallic phosphine complex with another metal source, with the equilibria between bi- and monodentate coordination of dppm to a single metal center dependant on steric requirements and the nature of alkyl ligands. The formation of heterobimetallic complexes likely involves the participation of species B in Scheme 18 with two dppm ligands binding in a monodentate fashion to a single Pt center.69–71 A similar approach was used by other groups for the preparation of heterobimetallics containing two late transition metals using symmetrical dppm or tfepma (bis(trifluoroethoxyl)phosphinomethylamine) as binucleating ligands.72,73 The bimetallic core in these complexes enables challenging multielectron redox transformations, in particular, photo-induced halogen reductive elimination that was extensively investigated by the Nocera group. The two-electron reductive elimination/oxidative addition in complexes with bimetallic d7⋯d9 or d8⋯d10 core is typically accompanied by changes in metal–metal interactions.74,75 For example, the oxidized form of a Pt/Au complex 3.15 features metal–metal interactions expected for a d7⋯d9 core with a Pt⋯Au bond distance of 2.6457(3) Å, which elongates upon its photoinduced reduction to a PtII/AuI complex 3.16 showing a Pt⋯Au distance of 2.9646(3) Å that suggests a lack of significant metal–metal bonding in 3.16, as expected for a d8⋯d10 core (Scheme 19).74 Although not directly related to the main focus of this perspective, it is worth mentioning that the formation of a redox active binuclear core combining a group 10 transition metal and a redox active metalloid has also been employed as a successful strategy to achieve halogen photoelimination from Pt/Sb, Pt/Te and Pd/Sb complexes, as reported by Gabbaï and co-workers.76–78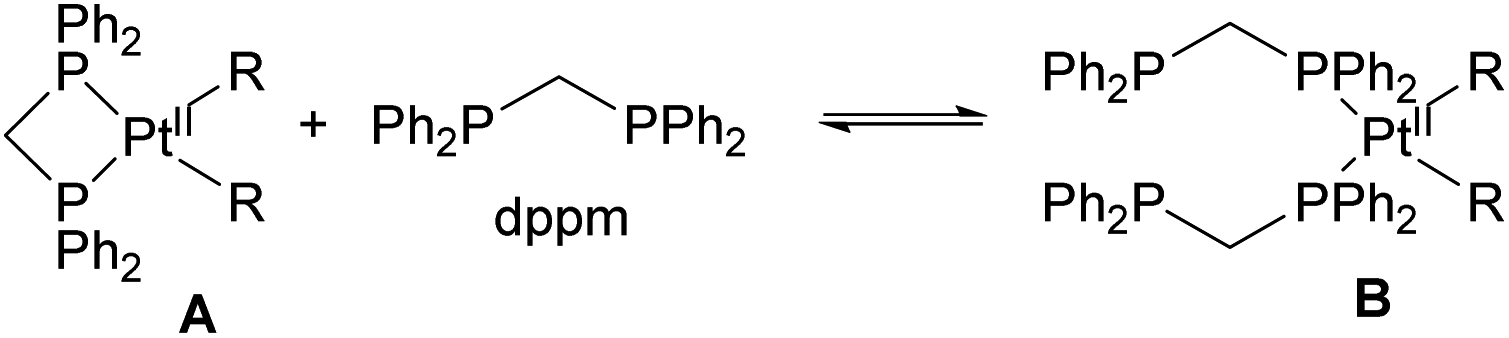 | ||
| Scheme 18 The equilibria in dppm Pt dialkyl or platinacycle complexes. | ||
 | ||
| Scheme 19 The reactivity of Pt/Au complexes in halogen photoelimination. | ||
Paddlewheel bimetallic dirhodium(II) complexes have found widespread applications in organic synthesis including development of enantioselective carbene reactions.79–82 While the predominant approach to controlling catalyst activity is based on bridging carboxylate ligand modification,83,84 recently the groups of Fürstner and Berry/Davies have shown that the formation of heterobimetallic Rh/Bi tetracarboxylate complexes is another way to tune reactivity in cyclopropanation and other reactions via modulation of electronic and steric properties at the rhodium center.85–87
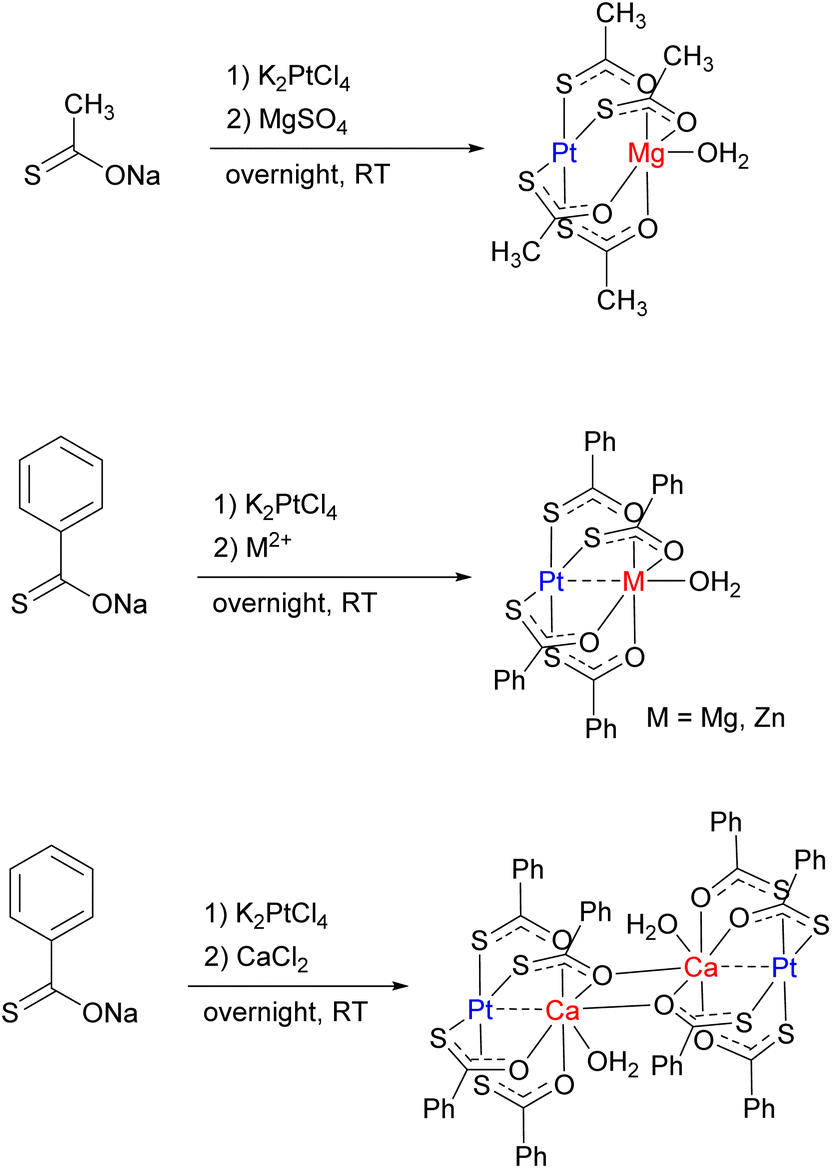
Simple unsymmetrical, isolated bridging ligands such as thiocarboxylate are also reported to support selective formation of heterobimetallic lantern-type complexes containing a “soft” TM (Pt) and a “hard” metal center M, typically an alkaline earth or 3rd row metal (M = Mg, Ca, Cr, Mn, Fe, Co, Ni, Zn) (Scheme 20). The selectivity is determined by binding of the “soft” metal center (Pt) to the “soft” S-donor site, while oxophilic, “hard” metal preferentially coordinates to the “hard” O-donor site.88–90 Spectroscopic and computational studies suggested that despite a short Pt⋯Mg distance enforced by the bridging ligands, no significant Pt⋯Mg interaction is present, because the Lewis acidity of the Mg center is satisfied by coordination to O-donors, while the Pt⋯Zn complexes do exhibit dative bonding, and Pt⋯Ca complexes represent an intermediate case, with an interaction but no significant metal–metal bonding.
 | ||
| Scheme 20 Lantern Pt/M complexes with thiocarboxylate ligands. | ||
Mixed phosphine/amide P,N-donor ligands have been widely used to selectively form heterobimetallic compounds with polarized metal–metal bonds, typically between late TM and an early TM or rare-earth (RE) element.91,92 For example, Lu and co-workers reported a series of bimetallic Ni0 complexes with GaIII or trivalent rare-earth (RE) metals93 (Scheme 21). Two synthetic approaches were used to build a bimetallic core: in the case of Ga complex 3.19d, the phosphine-bound Ni0 complex with lithiated ligand was obtained first, followed by treatment with GaCl3, while in case of Ni/RE pair, a rare-earth containing metalloligand 3.17 was first obtained, which was then metalated with a Ni0 precursor. The anodic peak potential of the resulting complexes varied in a wide range spanning 0.75 V, correlating with the Lewis acidity of a MIII ion and demonstrating the significant influence of metal–metal proximity on the redox properties of heterobimetallic species.
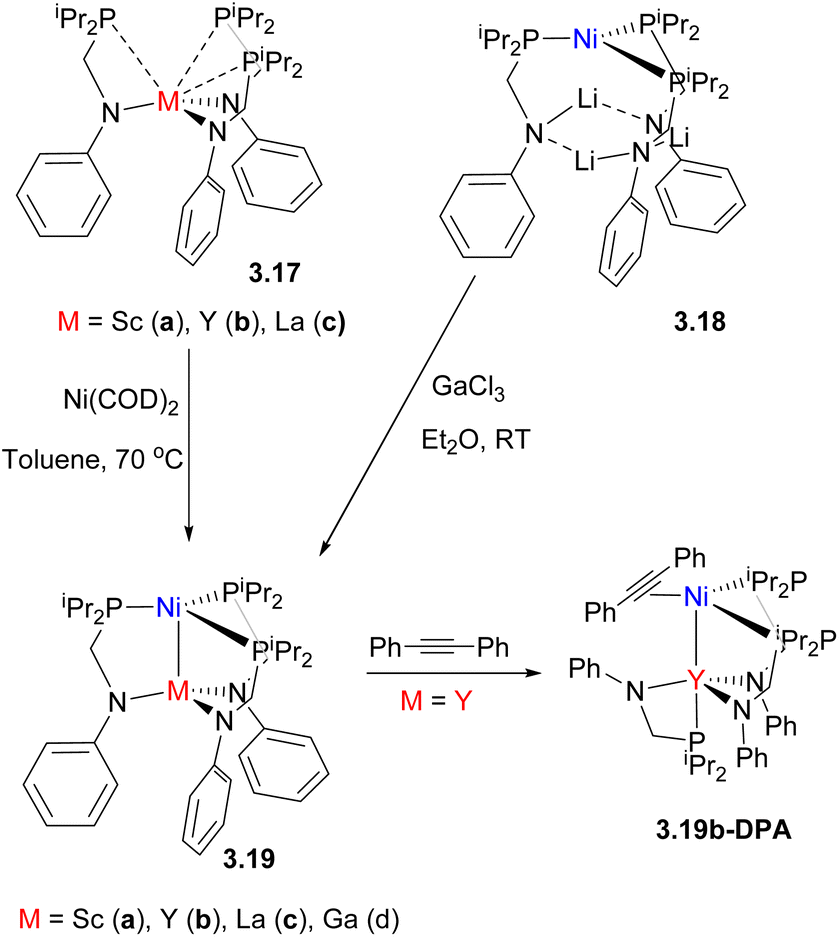 | ||
| Scheme 21 The formation of Ni–RE bimetallic complexes and the formation of a structurally characterized Ni/Y–alkyne adduct enabled by ligand hemilability. | ||
The resulting bimetallic complexes showed catalytic reactivity in selective semihydrogenation of diphenylacetylene to E-stilbene as the major product, outperforming Ni0/ligand or Ni/Li complex 3.18, which showed poor activity and low selectivity. Mechanistic studies suggest that the MIII “support” can tune the substrate-binding ability of Ni, resulting in different catalyst resting states: the strongly Lewis acidic GaIII ion promotes H2 binding to Ni to form experimentally observed, stable η2-H2 adduct, while the complexes with weakly Lewis acidic and large Y, Lu and La ions favor alkyne binding enabled by the ligand hemilability, such as in a structurally characterized Ni/Y-adduct with 1,2-diphenylacetylene, 3.19b-DPA (Scheme 21).
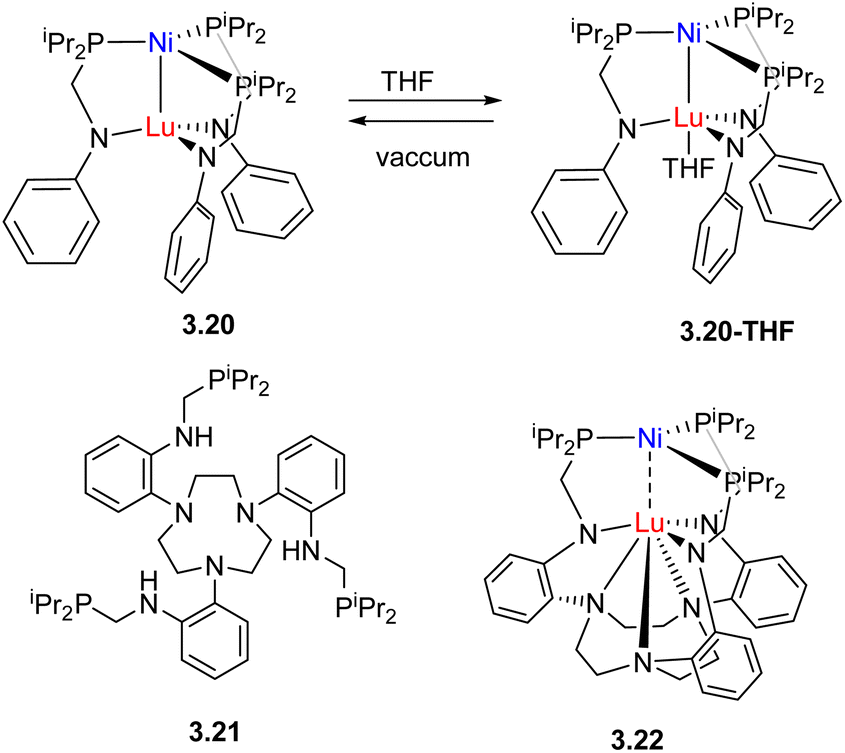
In an alternative approach, Lu and co-workers used the combination of the same pair of metals, Ni and Lu, however, the strength of metal–metal interactions was controlled by tuning the coordination environment around Lu in complexes 3.20, 3.20-THF and 3.22 with more coordinatively saturated Lu showing diminished Ni⋯Lu interactions (Scheme 22).94 This in turn affected the ability of Lu to serve as a Lewis acid, causing changes in the Ni0/NiI oxidation potential, H2 binding (η2-H2 adducts were observed for 3.20 and 3.20-THF, but barely detected for 3.22) and catalytic activity in styrene hydrogenation, with complex 3.20 outperforming complex 3.22 by a factor of 4.
 | ||
| Scheme 22 A series of Ni/Lu complexes with Ni/Lu interaction strength controlled by coordination environment around Lu. | ||
Interestingly, using only two equivalents of phosphine/amide ligand provided a pincer-type (PAlP)Ni complex 3.23, highly reactive in oxidative addition of aryl halide and H2 across the metal–metal bond and ortho-directed C–H bond activation in pyridine N-oxide.95 Depending on the nature of added donors, reversible intramolecular aryl group transfer between Ni and Al was also observed (Scheme 23).
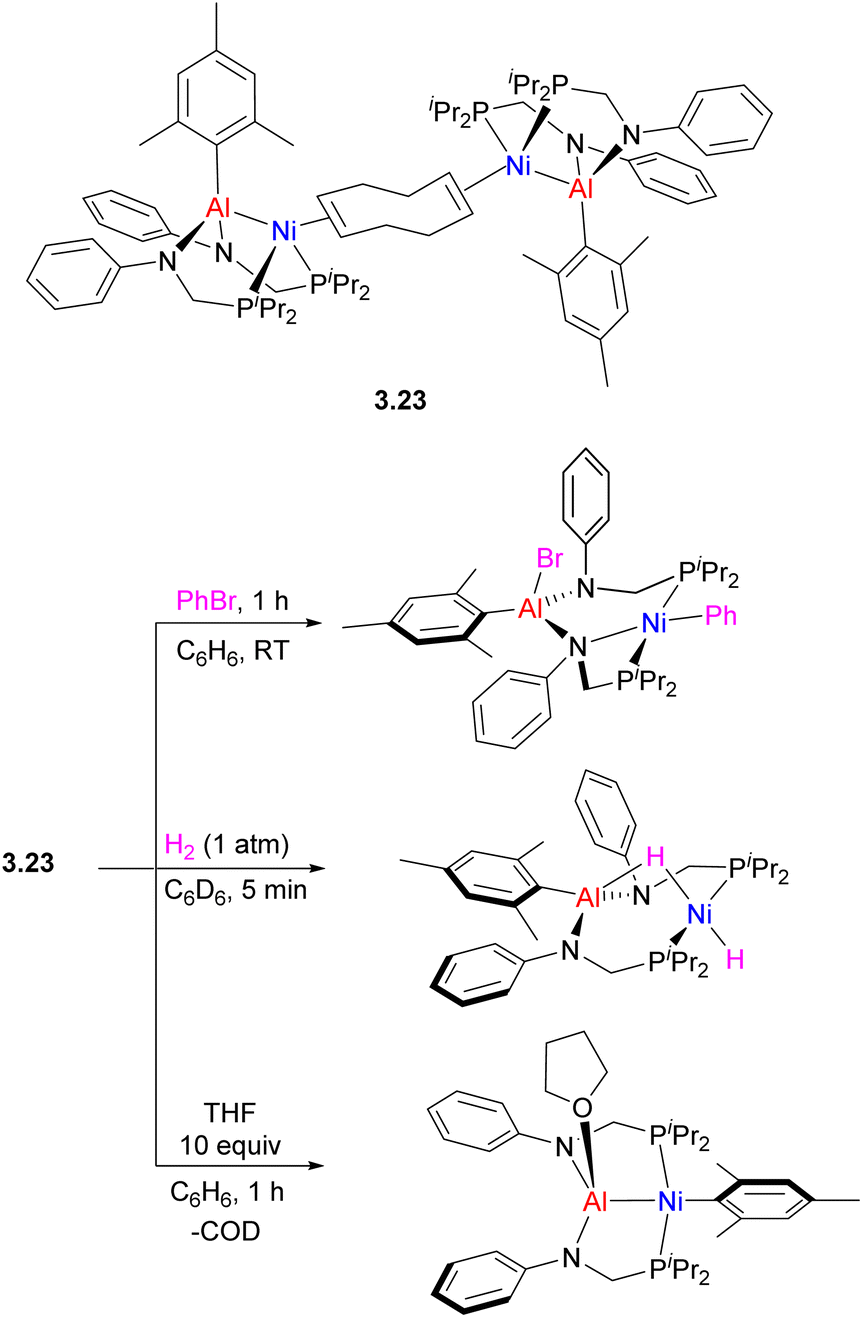 | ||
| Scheme 23 Reactivity of (PAlP)Ni complex in oxidative addition and mesityl group transfer from Al to Ni. | ||
The strength of metal–metal interactions may also be tuned by controlling the coordination environment at the late TM component, or by redox-changes at the early TM center (Scheme 24). For example, in Ti/Ni complex 3.24 reported by Tonks and co-workers, metal–metal bonding is disrupted upon coordination of CO due to Ni → CO backbonding effectively competing with Ni → Ti dative bonding. One-electron reduction to TiIII also leads to a weakening of Ni → Ti bonding.96
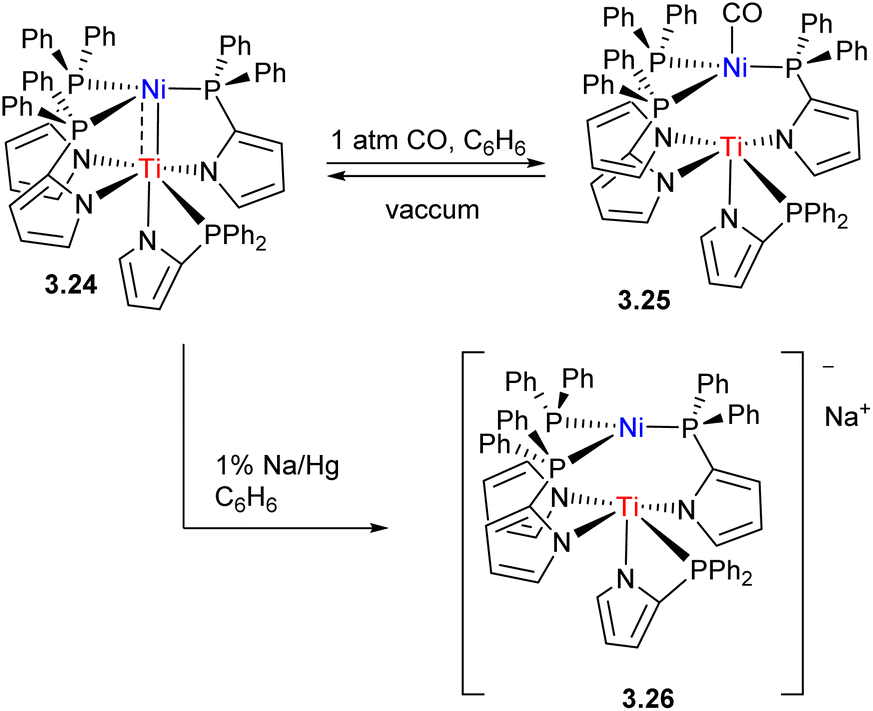 | ||
| Scheme 24 Metal–metal bond disruption in Ni/Ti complexes upon CO coordination to Ni and reduction of Ti. | ||
The examples discussed above demonstrate that simple binucleating ligands such as phosphinoamides, phosphinomethylamides, etc., show a great variety of coordination modes. They may act as mononucleating (often dynamic) ligands binding to one metal center, or as binucleating ligands connecting two metals together. Moreover, as can be seen from several examples discussed above, the number of such binucleating fragments greatly influences the reactivity of a bimetallic core as determined by vacant site availability for substrate activation. Thus, it's not surprising that tethering several classical binucleating motifs to a common linker has been used as an effective strategy to create polynucleating ligands with a well-defined coordination environment to host two metal centers in a site-selective manner connected by a pre-defined number (typically two or three) of bridging units. Some examples of such ligands (3.21 in Scheme 22) have already been discussed above, in the context of comparison with bimetallics supported by isolated binucleating ligands.
Another example of tethering common amide/phosphine-donor ligands to an amine linker is shown in Scheme 25, which provides a well-defined “soft” phosphine pocket to host “soft”, late transition metals, and a “hard” triamide pocket suitable for “hard” Lewis acidic early TM, rare earth or main group metals. Such differentiation between the two binding sites allows for selective stepwise metalation to obtain a series of structurally similar bimetallic complexes 3.27–3.28, allowing a detailed study into the influence of the “supporting metal” residing in the amide pocket on the reactivity of the late TM. One such well-defined polynucleating ligand developed by Lu allowed for the tuning of first row late TM reactivity, particularly in H2 and N2 activation.97–99 More recently, Rh/In complexes 3.29–3.30 supported by this ligand have been shown to be catalytically active in hydrogenolysis of aryl C–F bonds.100,101 Based on mechanistic studies, a catalytic cycle was proposed based on a Rh−I/RhI redox cycle, in which the Rh → In interaction is proposed to stabilize the reactive Rh−I species is responsible for Ar–F bond cleavage.
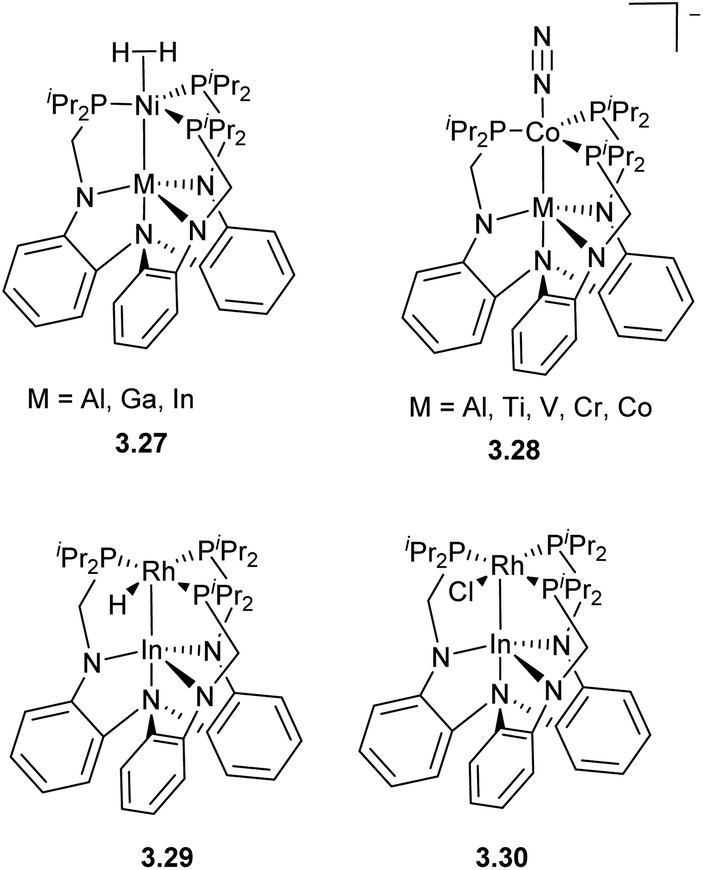 | ||
| Scheme 25 Stabilization of series of heterobimetallic complexes in tethered ligand. | ||
Tethering two three-atom P,N-donor binucleating scaffolds was used to develop several types of pincer-type metalloligands to support late TM bonds with group 13 elements (Al, Ga, In).102,103 Iwasawa and Takaya reported 6,6′′-bis(phosphino)terpyridine as a convenient ligand scaffold to stabilize Pd–E (E = Al, Ga, In) bonds.104 The derived Pd complex with “PAlP” pincer-type aluminyl metalloligand showed high catalytic reactivity for hydrosilylation of CO2 (Scheme 26). This is reminiscent of another type of Pd complexes with diphosphino metalloligands containing Zn, Li or Cu reported by Tauchert and co-workers, which show excellent catalytic activity in CO2 hydrosilylation.105 In these complexes the ligand framework is formed through the attachment of phosphine donors to the tris(picolyl)amine fragment containing Li+, Cu+ or Zn2+ ions.105,106
 | ||
| Scheme 26 A series of Pd complexes with “PEP” “metalloligands and CO2 hydrosilylation catalyzed by a Pd(PAlP) complex. | ||
A different type of tethered ligand with a tertiary amine linker connecting two P,N-units was developed by the Nakao group (Scheme 27), supporting the formation of Rh/Al complexes.102,103 These complexes were then extensively studied for bimetallic bond activation and catalysis. The in situ reduction of Rh/Al precursor complex 3.32 with KC8 in fluorobenzene results in C–F bond activation across the Rh–Al bond, which proceeds even at −30 °C.107 The DFT-calculated mechanisms suggests a cooperative C–F bond activation at the Al–Rh sigma-bond. This was further developed into a catalytic magnesiation of aryl fluorides catalyzed by 3.32, and the resulting arylmagnesium reagents were further reacted with CO2 to give carboxylic acids after acidic workup, or it could be trapped with other electrophiles. The reaction did not proceed without a Rh/Al catalyst or in the presence of a mononuclear [Rh(μ-Cl)(2,5-norbornadiene)]2/Et2AlCl combination.
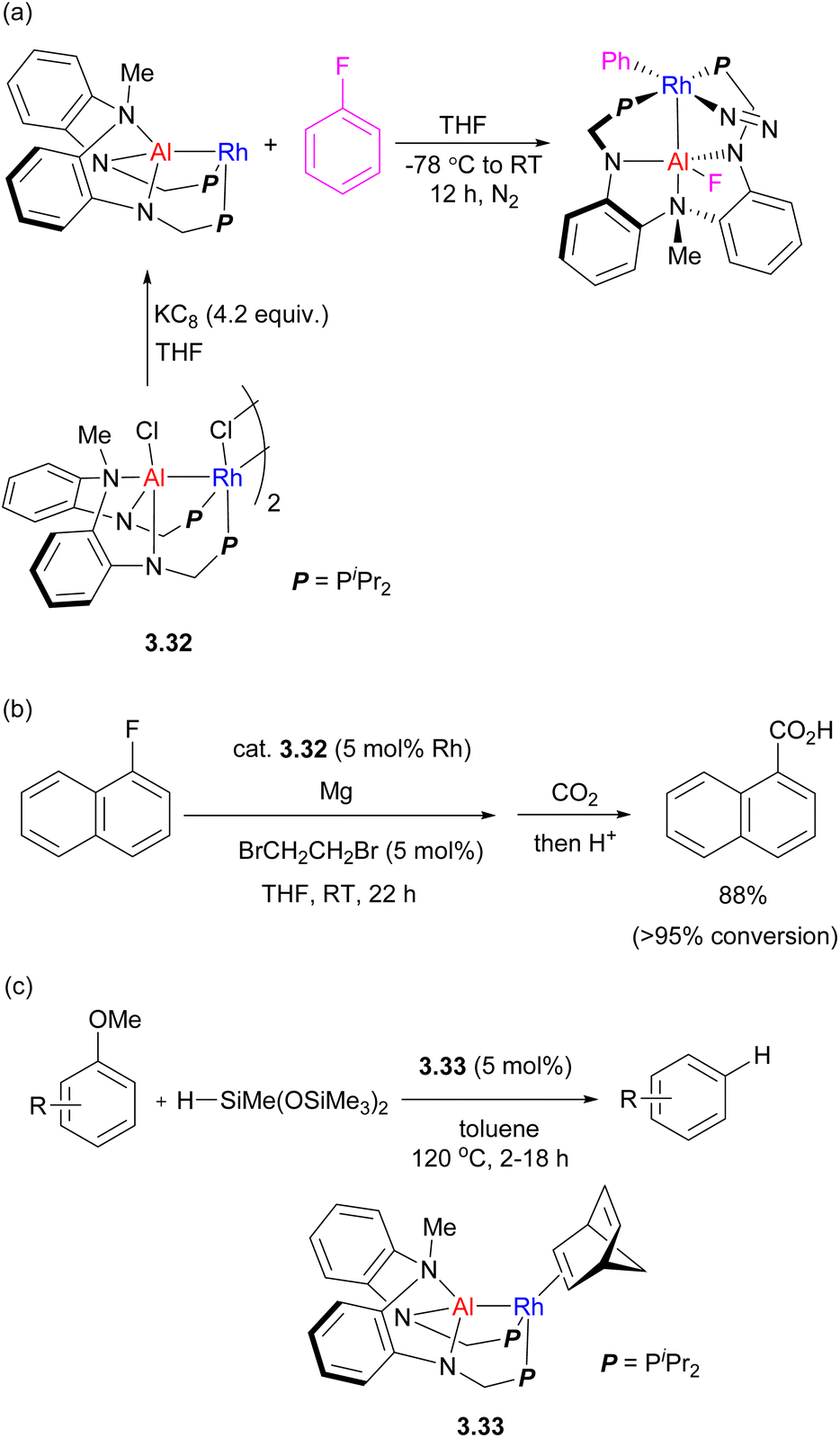 | ||
| Scheme 27 (a) Fluorobenzene activation by an Al/Rh heterobimetallic complex. (b) Rh/Al catalyzed aryl fluoride magnesiation. (c) Catalytic C–O bond reduction catalyzed by Rh/Al complex. | ||
Selective C–O bond reduction of anisole derivatives catalyzed by an analogous Rh–Al complex was also developed, in which bimetallic C–O bond activation was proposed.108
3.3. Heterobimetallic complexes with unsymmetrical capping units for bridging ligands
An alternative approach to designing unsymmetrical ligand scaffolds is through the use of a symmetrical bridging fragment, supplemented by two inequivalent capping units providing two distinctive binding pockets with different geometrical and electronic preferences (Scheme 2d). This allows for the use of common symmetrical binucleating motifs, while the presence of the unsymmetrical “capping” units may provide another useful synthetic tool to vary coordination environments around each metal.Another potential advantage is that the presence of well-defined binding pockets on each side allows to obtain heterobimetallic complexes with only one bridging fragment present, in contrast to the majority of the examples discussed above featuring two, three or even four unsymmetrical tethered or untethered bridges between the two metals. Thus, this approach may lead to heterobimetallic complexes with potentially many vacant coordination sites, that can then activate substrates or incorporate a variety of other organometallic ligands.
1,8-Naphthyridines have been employed as common binucleating ligands for many decades, however, mainly for the construction of homobi- or multimetallic complexes, due to the intrinsic difficulty of differentiating between two identical N-donor sites.109–114 Although naphthyridine ligands appended by two chelating capping units have been reported, in many cases two identical functional groups are introduced in 2,7-positions for the construction of well-defined homobimetallic complexes,115–120 with a few representative examples shown in Scheme 28.121–127
 | ||
| Scheme 28 Representative symmetrical ligand scaffolds bearing a binucleating 1,8-naphthyridine core. | ||
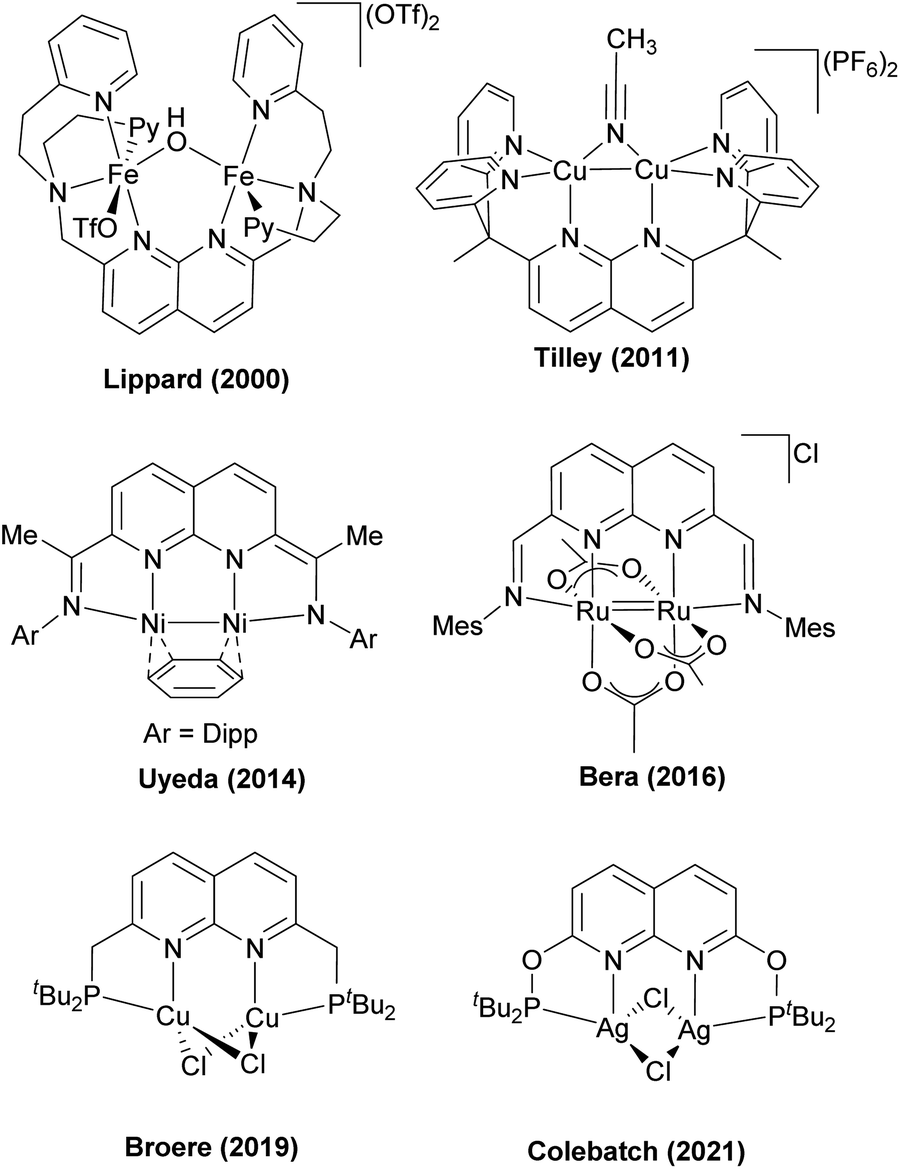
Recently, in 2018 the Tilley group designed an unsymmetrical version of the naphthyridine-based ligand, 3.34, in which two binding pockets were present, one containing a single phosphine donor, and another supplemented by two pyridines, which was then used to prepare a series of heterobimetallic complexes (Scheme 29). These complexes were synthesized by the addition of a Cu precursor to the solution containing 3.34 and one equivalent of a divalent metal chloride salt. A “hard” Lewis acid, divalent metal cation selectively occupied a dipyridyl pocket, while a “soft” CuI ion binds to the phosphine terminus, giving a chloride-bridged bimetallic core. However, no significant metal–metal interactions or bimetallic bond activation were reported for these systems, presumably due to the coordination of two bridging chloro-ligands.128
 | ||
| Scheme 29 Synthesis of 3d transition metal-based heterobimetallic complexes stabilized by unsymmetrically capped naphthyridine ligand. | ||
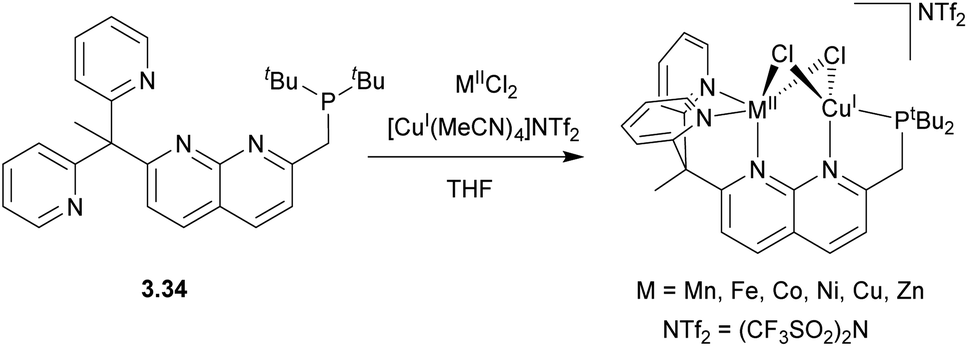
In 2017, our group reported a naphthyridinone-based ligand capped with a picolylamine chelating group, which will be discussed in more detail in Section 4.129 Simple modification of this ligand using ClPtBu2 and a base led to the formation of the unsymmetrical 3.35 with two well-defined binding pockets, one terminated with a phosphinite arm and another containing a picolylamine arm (Scheme 30).130 The reaction of this ligand with a PtII dimethyl precursor results in the selective PtII coordination to the “soft” P-pocket. At the same time, the reaction with PtIV trimethyl precursor results in its selective binding to a “harder” picolylamine fragment, which also provides a more favourable coordination environment by stabilizing an octahedral PtIV center via fac-coordination of the three N-donors. Interestingly, upon oxidation of 3.36 with MeI, the Pt atom selectively migrates from the P-pocket to the N-pocket of the ligand.
 | ||
| Scheme 30 Synthesis of the unsymmetrical naphthyridine-based ligand and selective formation of monometallic PtII, PtIV, and Pt/Cu heterobimetallic complexes. | ||
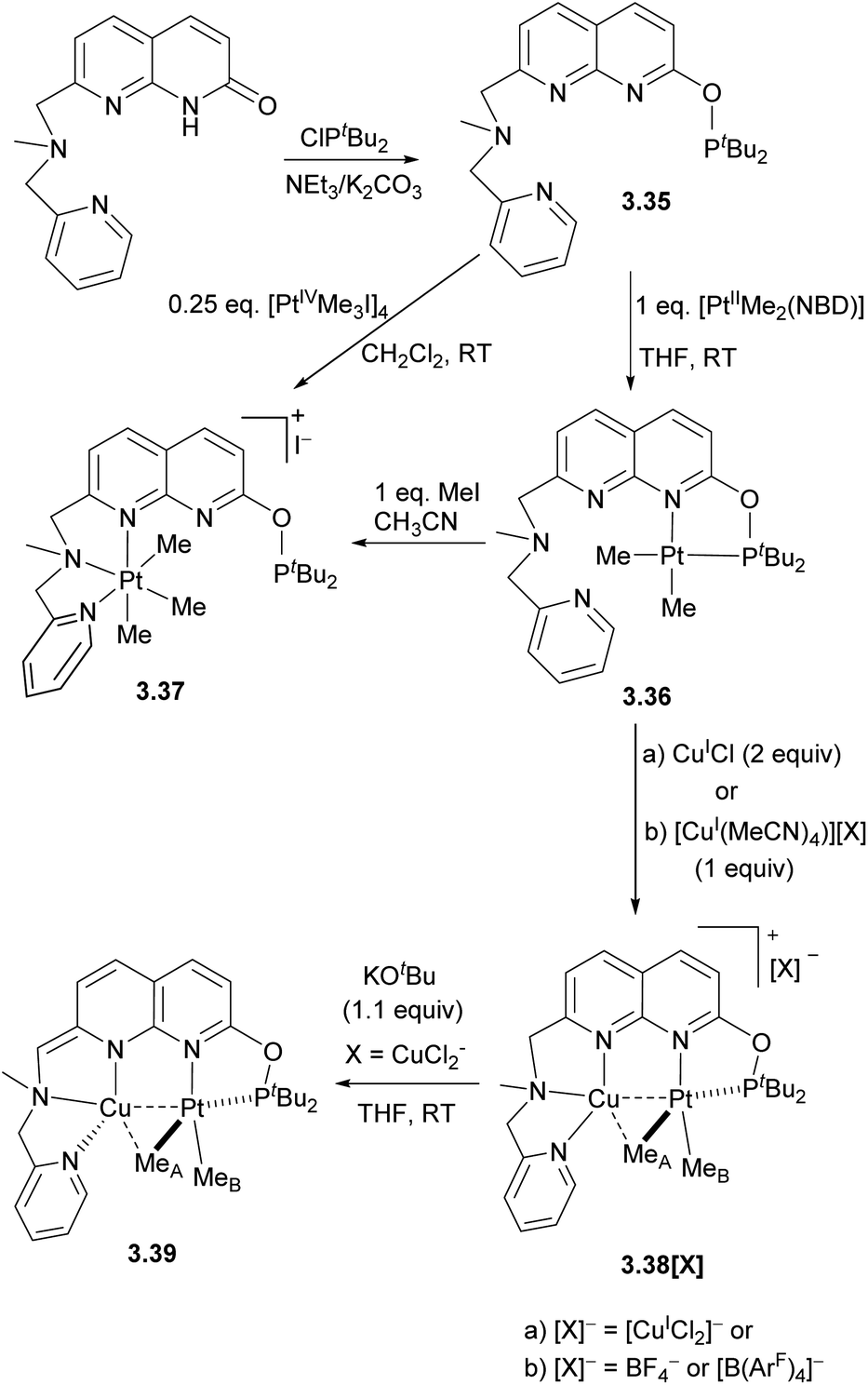
To obtain heterobimetallic complexes, the pre-formed 3.36 PtII complex was combined with a copper(I) precursor to give a series of Pt/Cu complexes 3.38[X] with various counter anions. Deprotonation of the amine-CH2 arm in complex 3.38[CuCl2] generated a neutral complex 3.39, in which the Cu center coordinates to a formally anionic amide donor of the dearomatized naphthyridine backbone. The X-ray structures of 3.38[X] show Cu⋯Pt distances of 2.6119(3)–2.6486(3) Å, which are shorter than the sum of covalent radii (2.68 Å).131,132 The CuI ion also has a relatively short Cu–C distances of 2.160(3)–2.362(9) Å, suggesting a CuI interaction with a proximal MeA group (Scheme 30). Notably, in neutral complex 3.39, longer Pt⋯Cu and Cu⋯MeA distances are present, indicative of weaker metal–metal and Cu–Me interactions.
QTAIM analysis of the cationic 3.38 and neutral 3.39 revealed the presence of bond critical points (bcp) between Pt and Cu in all complexes, however, the bcp between Cu and the bridging MeA group is only present in a cationic 3.38. NBO analysis reveals that metal–metal interactions involve donation from the Pt-based d-orbital to a vacant Cu(s) orbital, as well as weak back-donation from the Cu-based d-orbital to the antibonding σ(Pt–MeA) orbital. The Me group in 3.38 acts as an unsymmetrically bridging ligand between Pt and Cu, showing significant donation from the Pt–MeA σ-bonding orbital to a vacant Cu(s) orbital. Overall, this is consistent with the presence of a three-center two-electron binding mode in 3.38 that can be described as a donor–acceptor interactions between the Pt–MeA bridging group and the Lewis acidic Cu center, further supported by the Pt⋯Cu bonding interaction.
The close proximity of Cu significantly affects the reactivity of the Pt center in complexes 3.38[X], stabilizing the bimetallic core against undesired “rollover” cyclometallation, which occurs in the Pt-only complex 3.36 but not in 3.38[BF4] under identical conditions (Scheme 31a). Transmetalation of an electron-deficient aryl group from the [B(ArF)4]− anion ([B(ArF)4]− = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) (Scheme 30b) and terminal alkyne activation (Scheme 31c) were also observed in Pt/Cu complexes, but not in the monometallic 3.36. The DFT-calculated mechanism of terminal alkyne activation suggests that the Cu center acts as a docking site for substrate coordination, bringing it into proximity to the Pt center by forming an intermediate π-complex with an alkyne, I1, which also leads to polarization of the C–H bond and facile oxidative addition to form PtIV/Cu intermediate I2 (Scheme 31d).130
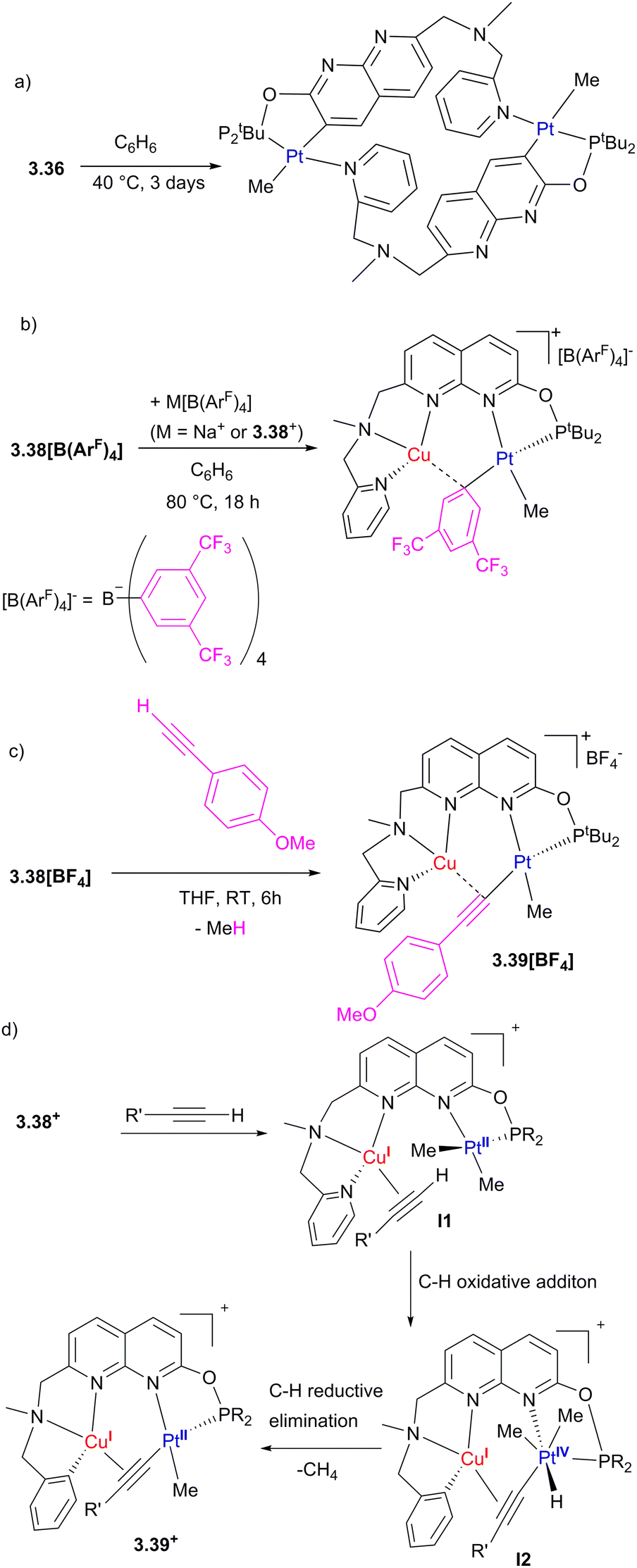 | ||
| Scheme 31 Reactivity of Pt/Cu complexes: (a) rollover cyclometalation of a monometallic 3.36. (b) Aryl group transfer from the [B(ArF)4]− (B(ArF)4 = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) to 3.38+. (c) Reactivity of 3.38+ with a terminal alkyne. (d) Proposed mechanism of alkyne activation by 3.38+. | ||
Recently, we also reported the formation of Pt/Zn and Pt/Ca complexes following an analogous stepwise reaction of the pre-formed Pt complex 3.36 with Zn or Ca salts (Scheme 32).133 In case of Zn complexes, different coordination numbers at Zn could be obtained depending on the nature of the anion: when more coordinating triflate was used, Zn was bound to three N-donors and oxygen of triflate, while in the presence of less coordinating NTf2−, Zn binds to three N-donors. The coordinatively unsaturated Zn atom in 3.41 exhibits a significantly shorter Zn–C distance to the bridging MeA group (2.157(7) Å), indicative of a stronger interaction, as compared to complex 3.40 (2.388(4) Å). Therefore, these two complexes may be seen as snapshots of the hypothetical transmetalation process between Zn and Pt, showing how the degree of bridging Me group interaction with Zn varies depending on zinc's coordinative saturation. At the same time, the Ca atom shows essentially no interaction with either Pt or the bridging Me group as further confirmed through QTAIM and NBO computational analysis.
 | ||
| Scheme 32 Stepwise synthesis of heterobimetallic Pt/Zn and Pt/Ca complexes. | ||
Pt/Zn complexes 3.40 and 3.41 react with H2, boranes and phenylsilane to form a hydride-bridged Pt–H–Zn complex 3.43, with liberation of methane (Scheme 33). Such reactivity was not observed under analogous conditions using the Pt-only complex alone, or using a combination of the model Pt complex 3.44 with a mononucleating P,N-donor and Zn triflate additive, indicating that H2, activation does not involve FLP-type reactivity47 and the presence of these two metals in close proximity is required. QTAIM and NBO analysis showed that a bcp is present between Pt and Zn atoms in complex 3.40, while it is absent in complexes 3.41 and 3.43 where Zn forms stronger interaction with a bridging Me group or hydride, and essentially no Pt⋯Ca or Ca⋯Me interactions are present in complex 3.42.
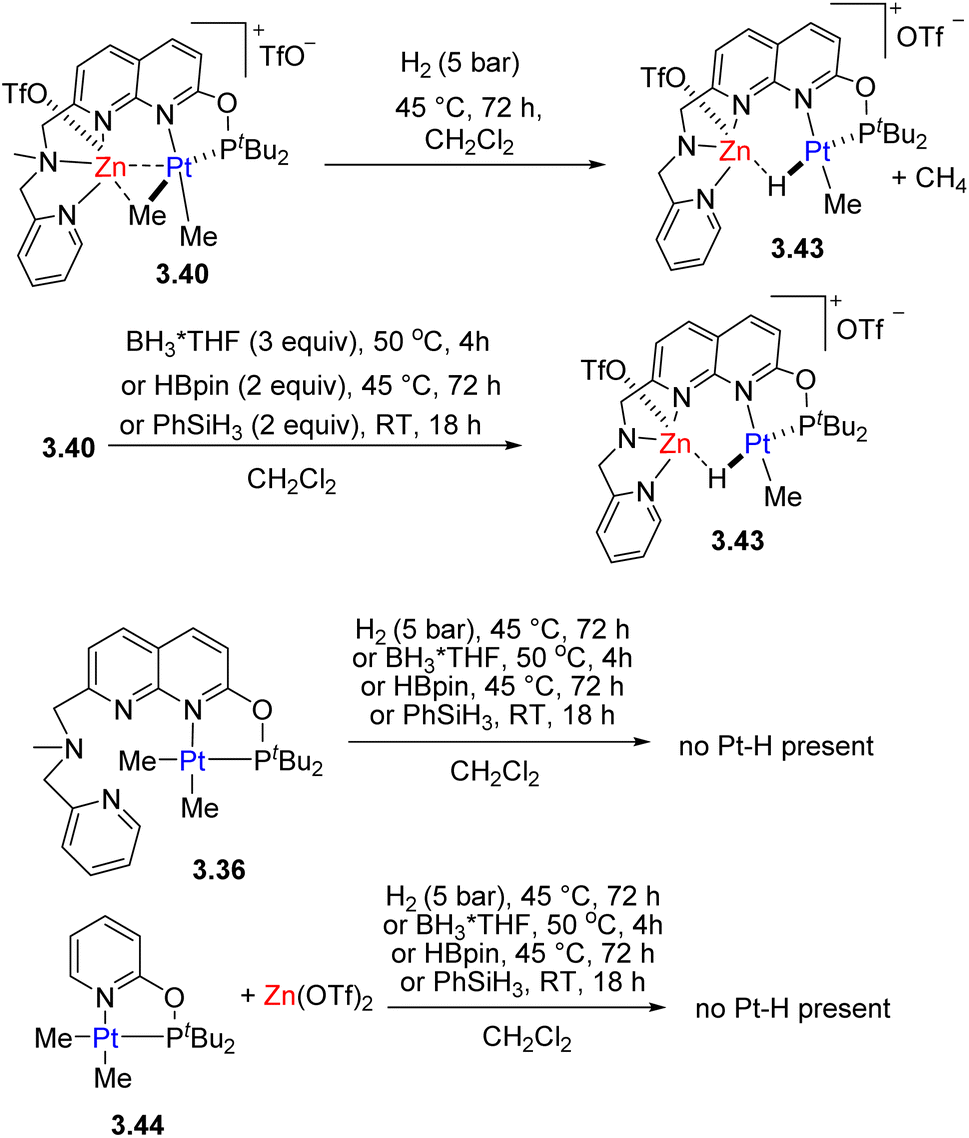 | ||
| Scheme 33 Reactivity of Pt/Zn complexes and their monometallic analogues in H2, Si–H and B–H bond activation. | ||
Similar to the Pt/Cu complexes discussed above, 3.40 also reacted with a terminal alkyne to give an acetylide-bridged complex. The presence of a Lewis acidic Zn center also leads to facile and selective protonolysis of only one of the Pt–Me groups, even in the presence of a few equivalents of water or alcohols.133
Overall, in this section, we have considered several approaches to the design of unsymmetrical ligand scaffolds to construct heterobimetallic complexes in a selective manner. In the simplest approach, two- or three-atom unsymmetrical binucleating ligands act as isolated bridging ligands between two metal centers. The nature of the donor atoms (typically a combination of one soft and one hard donor) is a useful tool to control the site selectivity of metal incorporation, especially when a late TM is used in a combination with a Lewis acidic main group metal, rare-earth element, or early TM. Stepwise synthesis using a monometallic precursor, usually a “hard” metal forming dynamic “metalloligand” species, followed by the addition of the soft metal precursor, is a frequently used approach, although the opposite order of addition or in situ formation from a mixture of two metal precursors have also been reported. The reactivity of the resulting complexes strongly depends on the number of binucleating ligands, typically showing higher reactivity if a more open bimetallic core is present with easily accessible coordination sites for substrate binding and activation. To control the number of binucleating ligands, the bridging fragments may be linked together with a (coordinating) capping unit, which led to the development of a class of pincer-type “metalloligands”. Alternatively, even a symmetrical bridging scaffold such as naphthyridine may be appended with one or two unsymmetrical “caps”, creating soft and hard pockets with different geometrical and electronic preferences, thus enabling site selectivity for two different metals. The presence of two well-defined binding pockets enables the synthesis of bimetallic systems with accessible coordination sites for bimetallic activation, avoiding the presence of multiple bridging fragments such as in “lantern” type complexes which contain several bridging ligands.
The examples discussed above demonstrate that strength of metal–metal, as well as metal–bridging ligand interactions in heterobimetallic complexes is determined by the coordination environment at the metal centers, showing that strongly coordinating ligands may disrupt metal–metal bonding and/or diminish Lewis acidity of one of the metal centers. The strength and bond order for metal–metal interactions are also controlled by redox activity, in extreme cases leading to disruption of the metal–metal bond.
Bimetallic cooperation in these systems may occur via cooperative bond splitting across a polar metal–metal bond, where both metals participate in bond cleavage/bond formation. Alternatively, especially when the second metal is protected by the polynucleating ligand's capping unit, one of the metals acts as a “support” without directly participating in bond activation, but can strongly affect redox properties and substrate binding ability at the reactive metal center.
4. Higher nuclearity heteromultimetallic systems
Although it is not possible to cover all possible types of ligand scaffolds and metal combinations used to support multinuclear metal complexes, in this section we will focus on several common strategies used to obtain, in a selective manner, multinuclear complexes (where the number of metal atoms exceeds 2) containing at least two different kinds of metals. For a more comprehensive overview of multinuclear chains and metal clusters and their reactivity in catalysis, the readers is referred to other books or reviews that categorize polynuclear metal complexes by bridging ligand type, catalytic applications, or metal combinations.6,134–136A common approach involves linking several (two or three) classical bridging fragments together using a common coordinating capping unit; for example, as illustrated by ligands 4.1–4.5 (Scheme 34). This systematic approach has been successfully used by Zhu, Maron and co-workers to obtain a series of bi- and multinuclear heterometallic complexes featuring late TM-uranium single or multiple bonds.137,138
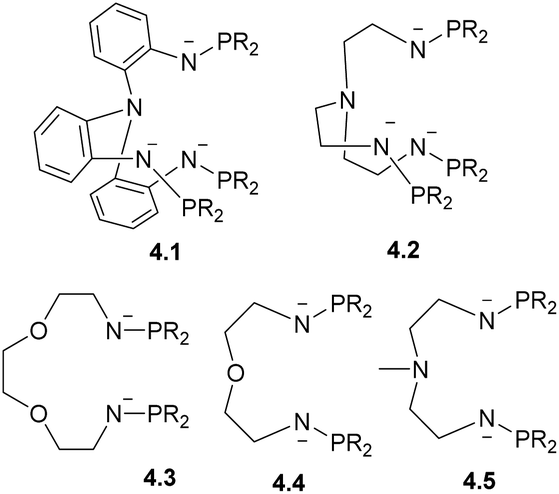 | ||
| Scheme 34 Trianionic and dianionic ligands to support uranium-late TM complexes developed by Zhu, Maron and co-workers. | ||
Trianionic and dianionic ligands of this type were used for stepwise construction of the U/TM complexes (TM = Ni, Pd, Pd) featuring several U–TM bonds via treatment of the pre-formed mononuclear uranium complex with a late TM precursor (Ni(COD)2, Pd(PPh3)4, Pt(COD)2 or [RhCl(COD)]2), with the TM ligating selectively to the P-donor of P,N-bridging fragment.138–140
The Rh/U cluster 4.6 was obtained by treatment of a U-containing metalloligand with [RhCl(COD)]2 (Scheme 35). The complex 4.6 or the product of its reduction, 4.7, was shown to induce challenging N2 bond cleavage to form 4.8, which was not observed in the absence of Rh.141 Based on frontier orbital analysis, the authors proposed that electronic communication between Rh and U plays an important role in the N2 stepwise reduction process, although no direct bonding of N2 to Rh is observed.
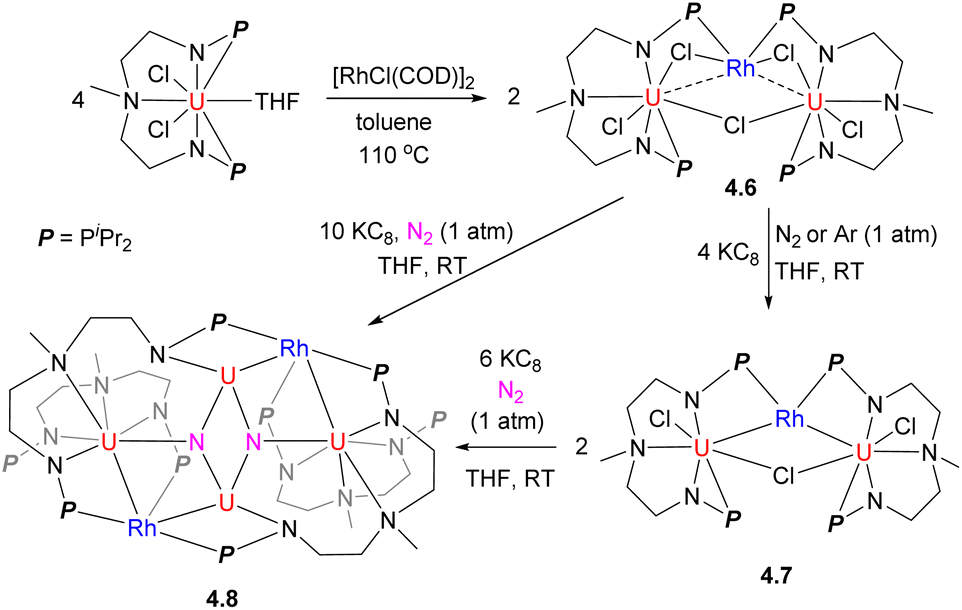 | ||
| Scheme 35 The synthesis of U/Rh complex and its reactivity in N2 splitting. | ||
The tris(2-aminoethyl)amine(tren)-containing polynucleating ligand was recently reported to support bi- and multinuclear complexes. A series of bi- and tetranuclear U/Fe and U/Co complexes 4.10–4.13 were obtained, starting from the U-containing metalloligand 4.9 reacted with Co or Fe salts, and their subsequent treatment with KC8 (Scheme 36).142 The U/Co complex 4.12 was catalytically active in selective hydroboration of terminal alkynes to give α-vinylboronates with good yields and regioselectivities.
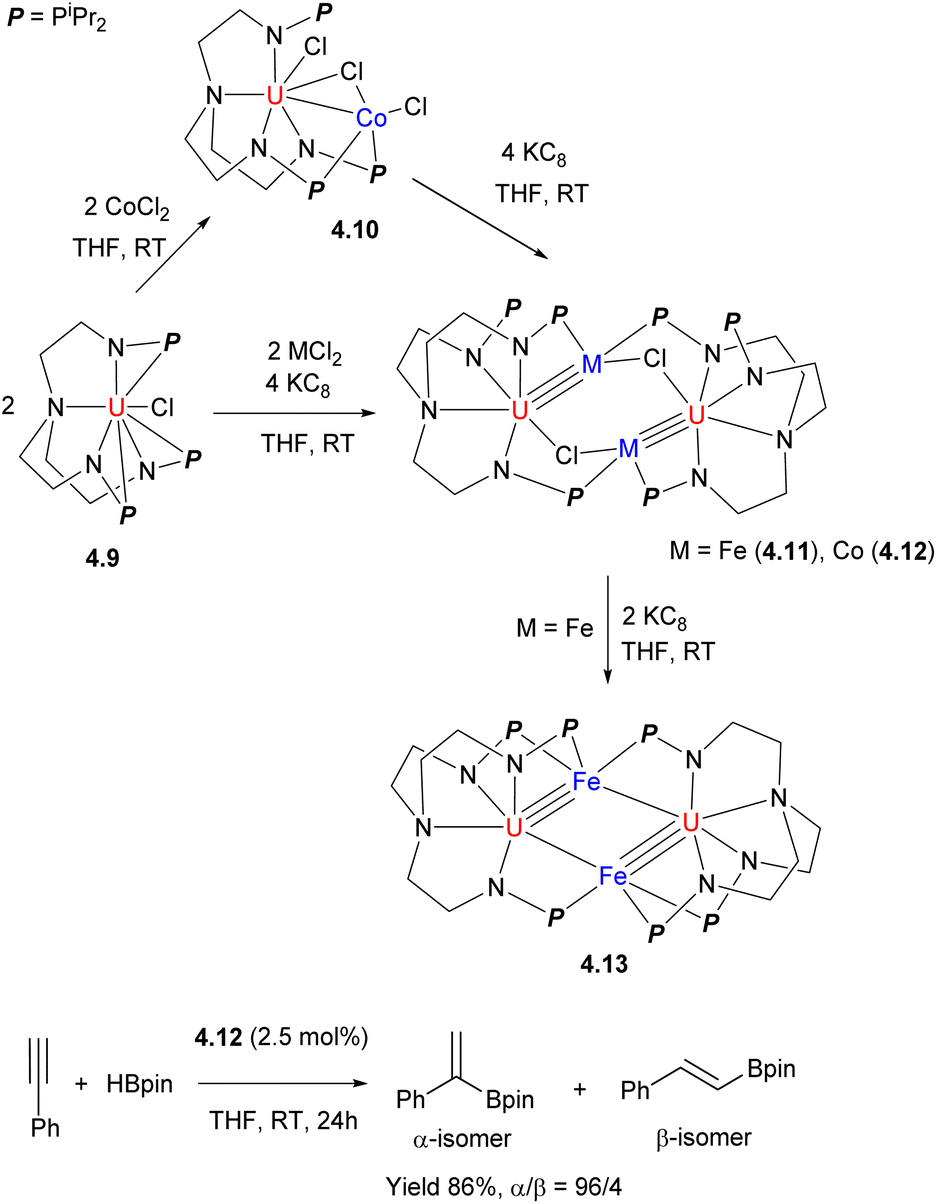 | ||
| Scheme 36 Formation of U/M (M = Co, Fe) complexes and their reactivity in catalytic hydroboration of a terminal alkyne. | ||
Simple modification of the common nacnac-type ligand with a long phosphine arm to give an unsymmetrical NNP ligand, allowed for the selective synthesis of triangular Zn2M (M = Ni, Pd, Pt, Cu+, Ag+) clusters 4.17–4.19 reported by Xu and co-workers (Scheme 37).143–145 Two alternative synthetic strategies were developed: in the presence of a Pd0 complex, dehydrocoupling of ZnII hydrides 4.14 gives a triangular Zn2Pd complex 4.17.145 Dehydrocoupling of ZnII hydrides was also achieved using Ni(CO)2(PPh3)2 catalyst, and the treatment of ZnI–ZnI dimer 4.15 with zero-valent group 10 precursors also produced analogous triangular clusters with the retention of a Zn–Zn bond to give 4.17–4.19. Computational studies suggest that dizinc acts as an η2-ligand for Pd, resembling an η2-H2 adduct formation. According to NBO analysis, the bonding interactions within the Zn2Pd cluster 4.17 include the donation from Pd-based 4d orbital to a σ*(Zn–Zn) orbital as well as donation from the σ(Zn–Zn) to an sp-hybrid orbital on Pd (86% 5s and 12% 5p).
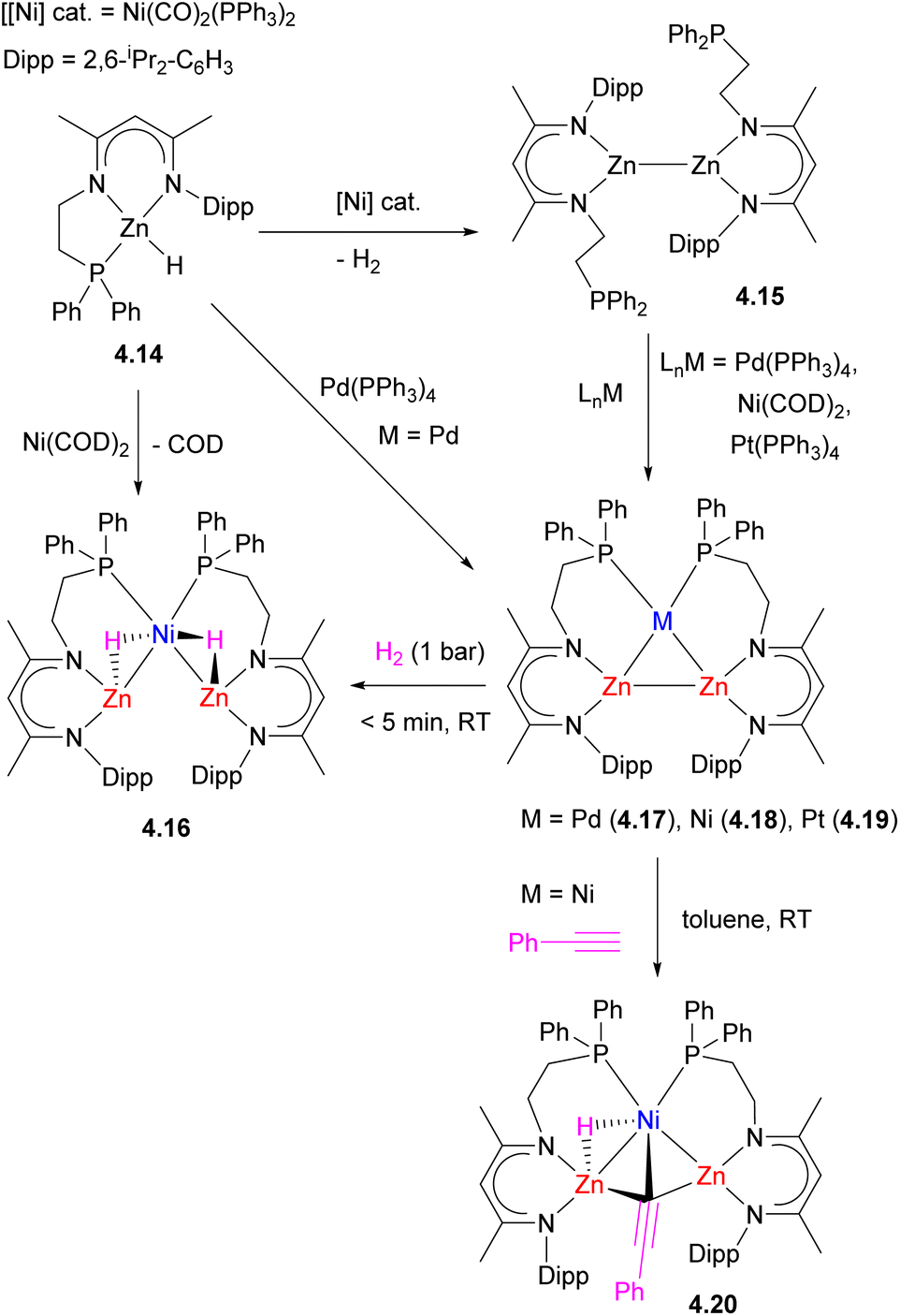 | ||
| Scheme 37 Synthesis and reactivity of Zn2M triangular clusters. | ||
The triangular Zn2Ni cluster 4.18 was reactive towards H2 and phenylacetylene C–H bond splitting, resulting in Zn–Zn bond cleavage and the formation of trinuclear hydride and acetylide bridged complexes 4.16 and 4.20.
While the formation of heteromultimetallic complexes combining two first row TM may be complicated due to solution lability and the potential formation of stoichiometric mixtures, such lability may be utilized as a tool to build more complex heterometallic clusters by metal substitution. For example, treatment of Fe cluster 4.21 supported by a polynucleating amide-donor ligand with 2 or 5 equiv. of CoCl2 afforded mixed Fe/Co clusters 4.22–4.24 resulting from the substitution of one or two irons, respectively, within a trinuclear core (Scheme 38).146 As unambiguous assignment of Fe and Co in these complexes is not possible by X-ray single crystal diffraction, the metal substitution in these complexes was confirmed by a number of other methods, including paramagnetic 1H NMR, 57Fe Mössbauer, X-ray fluorescence, and magnetometry analysis.
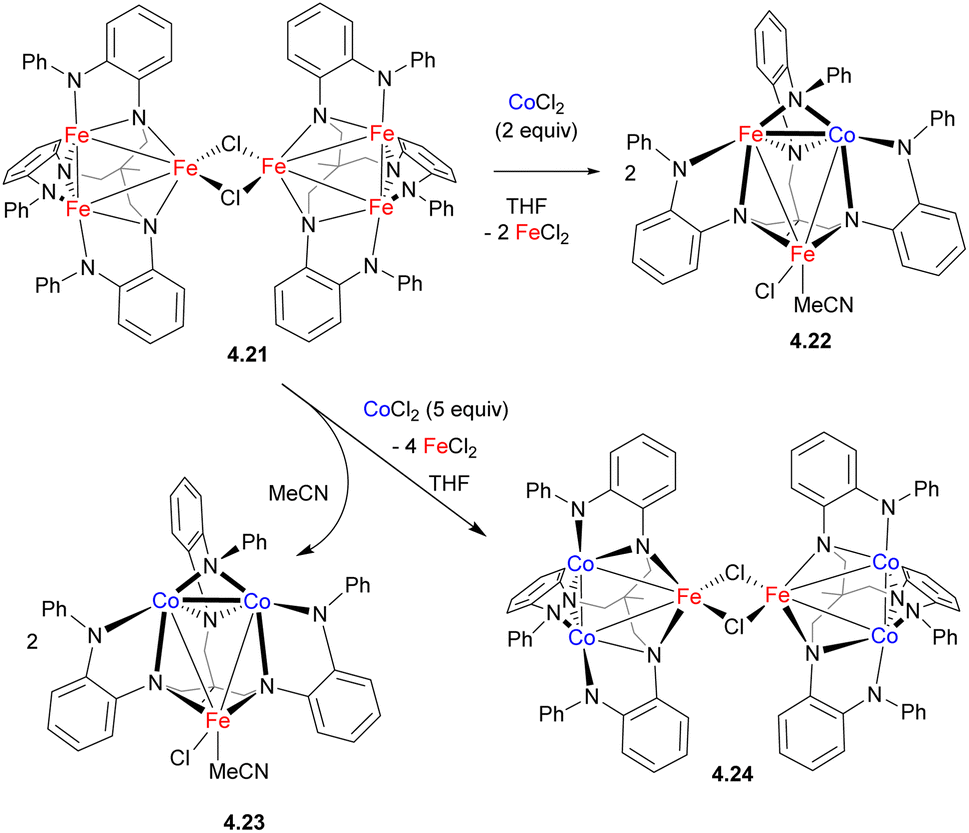 | ||
| Scheme 38 Formation of Fe/Co polynuclear complexes via metal substitution. | ||
While the examples discussed above feature a non-linear arrangement of metals in the cluster, the formation of linear metal chains is of particular interest for the study of electronic communication between the metals in the chain. Extended metal atom chains (EMACs) have been widely studied due to their potential applications as “molecular wires” in single molecule conductivity studies, with many types of polynucleating ligands (polyamidopyridyls, polyenes, polyphosphines, etc.) developed over many decades to support well-defined homomultimetallic chains.147–152
Accordingly, selective incorporation of two (or more) different metals may potentially be used as a tool to tune catalytic reactivity and physical properties, although controlling site-selectivity becomes particularly challenging.
One of the strategies to obtain infinite 1D heterometallic metal chains is based on HOMO–LUMO interactions between several unbridged metal-containing units. For example, the paddlewheel compound [RhII2(O2CCH3)4] (d7–d7) features a vacant σ*orbital that can accept an electron from the filled dz2 orbital in pivalamidate (piam) PtII (d8) in complex 4.25, leading to the formation of a tetranuclear Pt–Rh2–Pt chain in complex 4.26 (Scheme 39). This strategy was further expanded to build infinite 1D chains containing two or even more metals,153–155 such as 4.28, formed by a combination of the trinuclear Pt–Cu–Pt complex 4.27 and a [RhII2(O2CCH3)4] paddlewheel unit.156
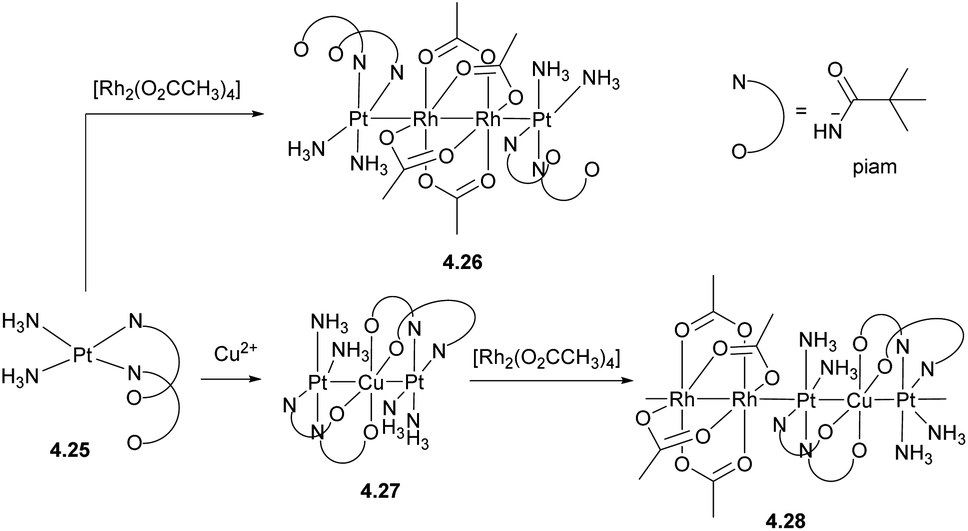 | ||
| Scheme 39 Formation of polymetallic Pt/Rh and Pt/Rh/Cu linear chain complexes. | ||
To obtain finite, solution-stable metal chains with a defined number and order of metal atoms, well-defined ligand scaffolds have been developed, with a few representative examples shown in Scheme 40. One strategies to build heteromultimetallic chains is based on the hard/soft Lewis acid/base approach similar to that used for the formation of binuclear complexes, exemplified here by complexes 4.29–4.30.157,158 Ligands containing donor sites with appropriate Lewis base properties have also been used to selectively obtain multimetalic cores via stepwise metal incorporation. A series of trimetallic complexes containing a metal–metal multiply bonded group in combination with another transition metal were obtained by the Berry group using this method (for example, complex 4.31).159–162 An interesting approach was used by Tanase et al. to obtain a series of Pd/Pt tetranuclear complexes 4.32 supported by a tetraphosphine ligand via stepwise replacement of the Pd with Pt in a pre-formed metal chain.163
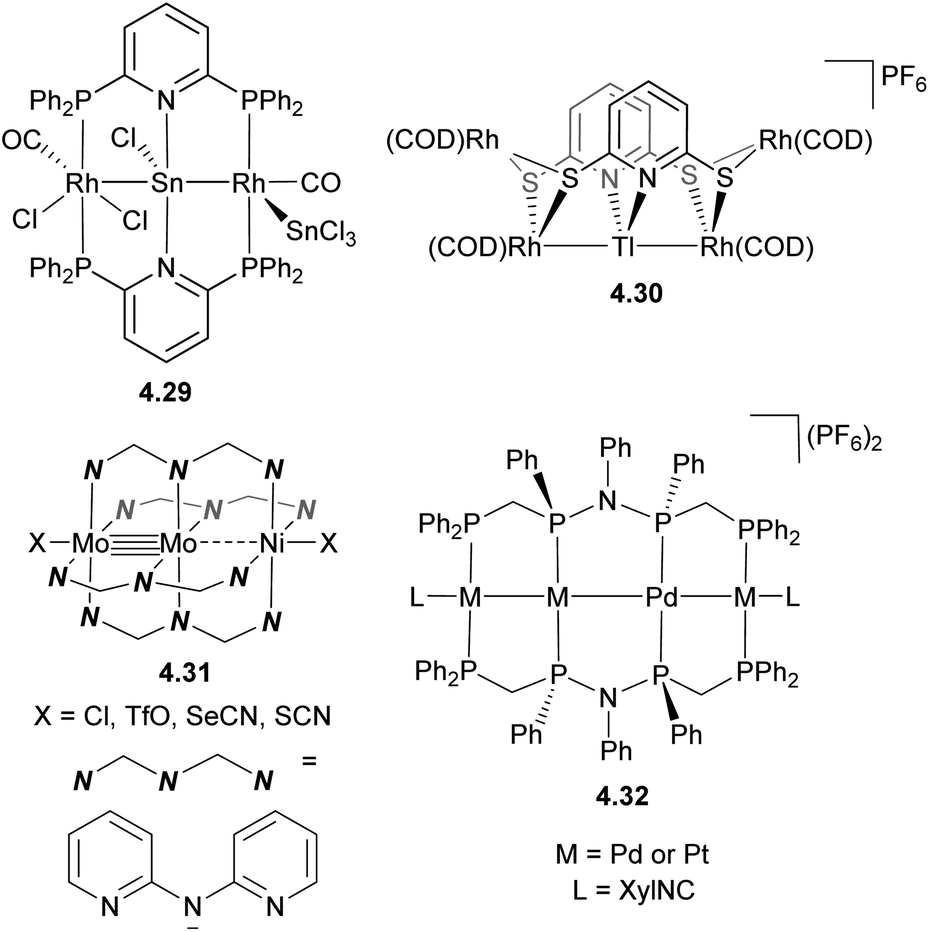 | ||
| Scheme 40 Representative examples of heteromultimetallic complexes containing linear arrangement of metal atoms. | ||
In a contrast to the symmetrical ligands typically used for such multinuclear complexes, which can typically bind to a pre-defined, fixed number of metal atoms (Scheme 40), our group has developed unsymmetrical dynamic ligand scaffolds 4.33a and 4.33b, containing a naphthyridinone backbone that can act as a versatile bridging fragment of variable denticity capped with a chelating mono- or dipicolylamine units (Scheme 41).129
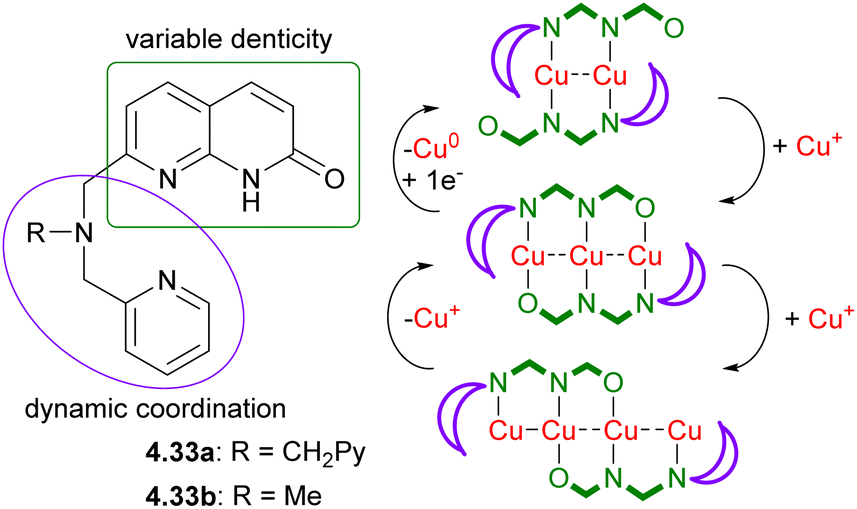 | ||
| Scheme 41 Dynamic polynucleating ligands based on naphthyridone backbone and schematic representation of the stepwise growth of multicopper(I) chains by “sliding” naphthyridonate fragments along the growing chain. | ||
These ligands allowed for the stepwise growth of multicopper(I) chains (from two to three and four metal atoms), enabled by “sliding” of naphthyridonate fragments along the metal chain as schematically represented in Scheme 41. Moreover, stepwise metal chain deconstruction by reducing copper string length from four to three and two was also achieved by removing Cu atoms when changing solvent polarity or by chemical reduction.129
We then expanded the application of these ligands to the stepwise synthesis of heteromultimetallic Pd/Cu complexes with variable chain lengths and a precise positioning of the metal atoms, where Pd formed an inner core and Cu was preferentially present in the terminal positions, where the appropriate tetrahedral coordination environment was provided by the picolylamine capping units.164 The stepwise synthesis is shown in Scheme 42, starting from a bimetallic dipalladium(II) core formed by treatment with a cyclometalated benzo[h]quinolyl Pd dimer 4.34, and followed by a reaction with one equivalent of [Cu(MeCN)4]+ to give a trinuclear PdPdCu core in 4.36a[X] and 4.36b[X], which can be further expanded to give a tetranuclear CuPdPdCu core in 4.37a[X]2 and 4.37b[X]2 by treatment with another equivalent of [Cu(MeCN)4]+ (or by simultaneous treatment of a Pd2 complex with two equivalents of [Cu(MeCN)4]+). Similar to multicopper chains reported previously, such dynamic metal chain growth is enabled by “sliding” of naphthyridonate along the metal chain.
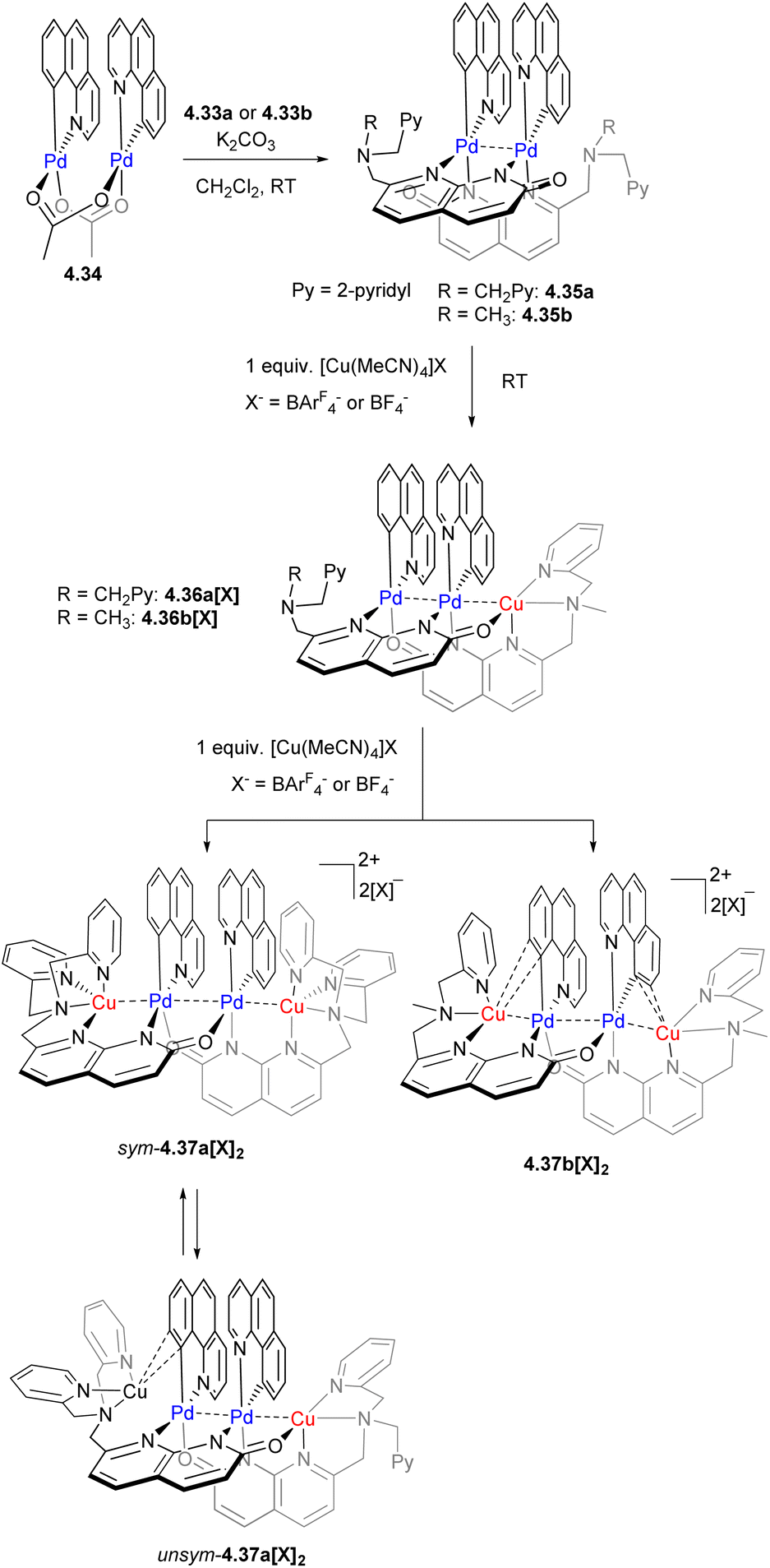 | ||
| Scheme 42 Synthesis of Pd/Cu heteromultimetallic complexes. | ||
A dynamic coordination environment around the Cu center results in the formation of two isomers, unsym-4.37a[X]2 and sym-4.37a[X]2, with short Cu⋯Pd distances present only in the unsymmetrical isomer unsym-4.37a[X]2. The reactivity comparison along the series of several tri- and tetranuclear Pd/Cu complexes showed that only complexes featuring the relatively short Pd⋯Cu distances of 2.65–2.67 Å react with a terminal acetylene (Scheme 43). The product of alkyne activation by 4.37a[X]2 was isolated as a binuclear Pd/Cu complex 4.38[X], with acetylide forming a σ-bond to Pd and a π-bond to Cu, while the N-atom of the naphthyridonate acts as an internal base deprotonating the terminal acetylene C–H bond. Moreover, the resulting complex 4.38[X] undergoes a C(sp)–C(sp2) elimination in the presence of phosphines. The mechanistic studies through DFT, combined with ab initio molecular dynamics calculations and metadynamics simulations, suggest a multistep mechanism via initial alkyne coordination to CuI, followed by C–H deprotonation and the formation of a σ-bonded CuI–acetylide complex, followed by transmetalation to Pd to give an intermediate where an acetylide ligand forms a σ-bond to PdII and a π-bond to CuI. The ability of the ligand to support the dynamic behaviour of this multimetallic assembly during this multistep process is crucial for the transformation.164 Thus, these Pd/Cu complexes serve as a functional model of Pd/Cu intermediates in Sonogashira coupling, mimicking both alkyne activation and C–C bond reductive elimination steps, although the latter step eventually required disassembly of the Pd/Cu core into monometallic species.
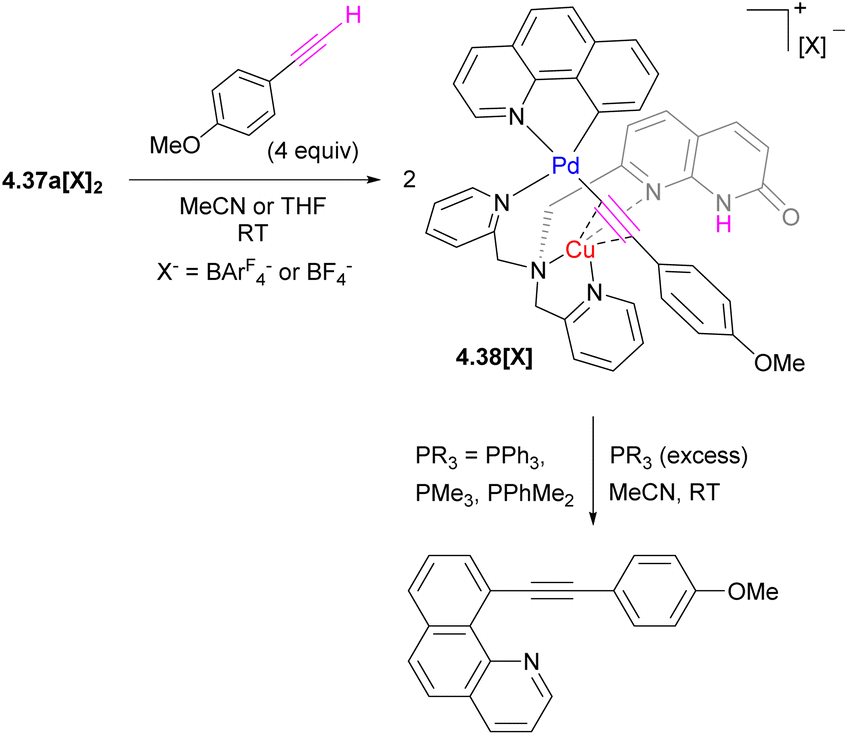 | ||
| Scheme 43 Alkyne activation and C–C elimination reactivity of 4.37a[X]2. | ||
We were also able to achieve a precise “reverse order” Pd/Cu chain by modifying the capping unit, using the phosphine-containing ligand 4.39 (Scheme 44).165 By contrast to 4.33a and 4.33b, the treatment of 4.39 with the same Pd precursor leads to selective coordination in the soft P,N-binding pocket, while further treatment by [Cu(MeCN)4]+ results in the formation of a PdCuCuPd chain where the internal core contains two Cu atoms. Interestingly, the number of metal atoms could also be controlled by the presence of coordinating anions: when CuCl was used, a binuclear PdCuCl core was obtained. QTAIM analysis reveals the presence of bcp between Pd and Cu in both 4.41 and 4.42, with a larger value for the electron density at the bcp in complex 4.41 as compared to 4.42, suggesting a stronger metal–metal interaction, consistent with the shorter Pd⋯Cu bond distances in 4.41. NBO analysis showed that moderate donation is present from a Pd-based d-orbital to a Cu-based s-orbital, consistent with metallophilic d8–d10 metal interactions.
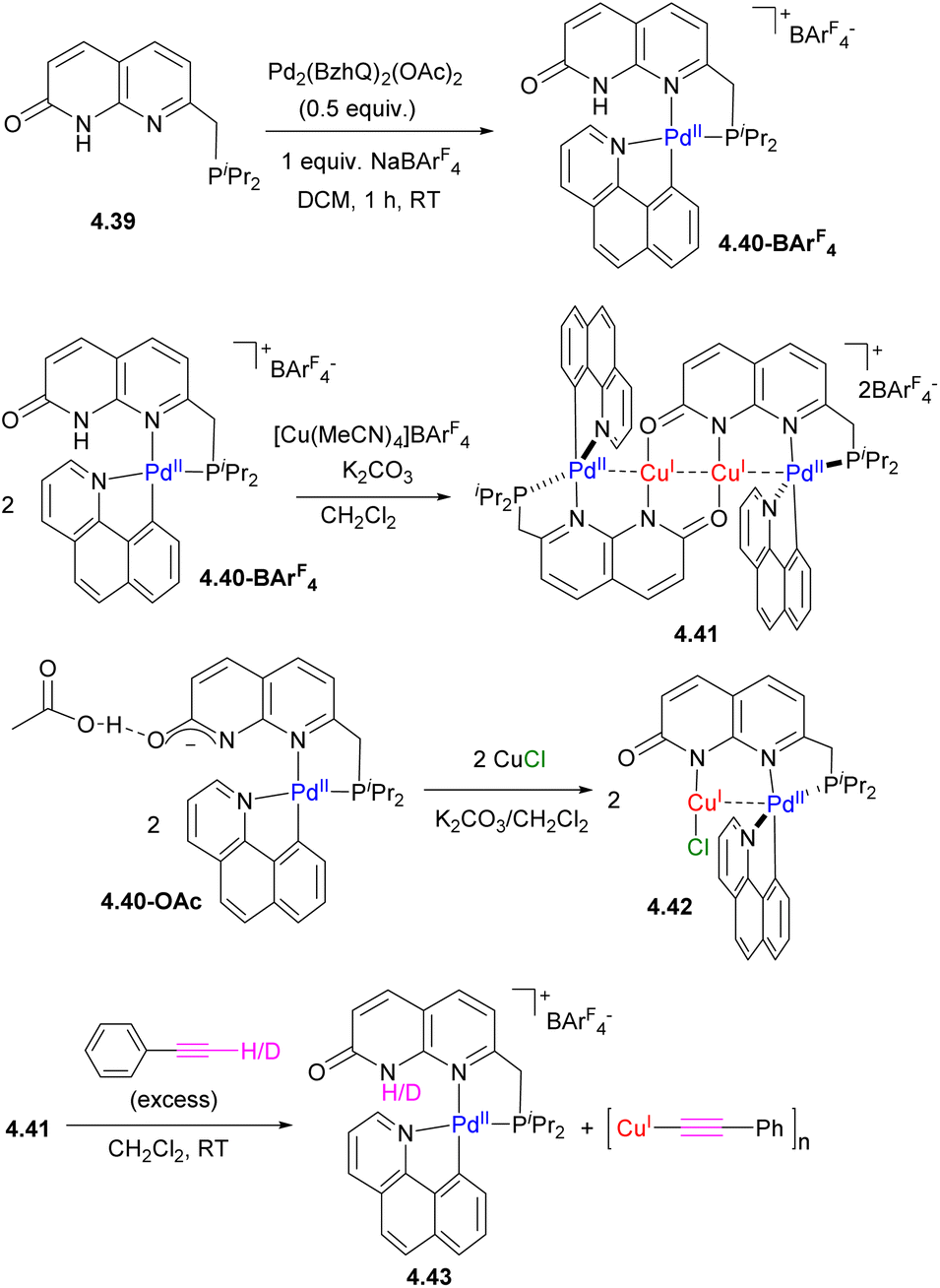 | ||
| Scheme 44 The formation of “inverse order” Pd/Cu chain and its reactivity with phenylacetylene. | ||
By contrast to complex 4.37a, terminal alkyne activation generates an acetylide complex σ-bound to Cu, accompanied by the disassembly of the multimetallic core. This striking difference in reactivity in a “reverse” order PdCuCuPd complex 4.41, as compared to CuPdPdCu complex 4.37a, is likely due to greater lability of the inner Cu2 core and the greater chelating ability of the capping P,N unit.165
5. Conclusions and outlook
In conclusion, we have outlined a variety of approaches to the selective formation of heteromultimetallic complexes. In the simplest approach, an unsupported metal–metal bond is used to assemble a bimetallic (or multimetallic) core in the absence of bridging ligands. Here bond activation often results in the disassembly of the bimetallic core and formation of monometallic species. Lability of the metal–metal bond may also result in disproportionation reactions and the formation of a mixture of mono- and multinuclear species.Therefore, the use of unsymmetrical polynucleating ligand scaffolds bridging two or more metal centers together is a commonly used approach to build systems with a well-defined heterometallic core. In addition to stabilization against dissociation into monomeric species (or other types of homo- or heteromultimetallic assemblies), a well-defined, differentiated coordination environment provided by the unsymmetrical polynucleating ligand scaffolds allows for control over site selectivity during metal incorporation. This is most commonly accomplished via the use of unsymmetrical bridges with a “soft”/“hard” donor atom. Tethering two or three unsymmetrical bridges together with a common linker provides another pathway to build a well-defined heteromultimetallic core.
An alternative method to build an unsymmetrical ligand scaffold is based on using an unsymmetrically substituted polynucleating fragment that provides two well-defined binding pockets with different steric and electronic properties. Finally, using polynucleating dynamic ligand scaffolds capped by a chelating unit only from one side allowed for the dynamic formation of homo- and heteromultimetallic chains in a stepwise manner.
The availability of coordination sites is an important consideration and factor in the utilization of well-defined heteromultimetallic complexes in catalytic or stoichiometric bond activation via metal–metal cooperation. We have discussed a variety of possible scenarios in which heteromultimetallic complexes can be used for selective bimetallic bond activation enabled by the presence of two metals in close proximity, where the same reactivity was not exhibited by mono/multi-metallic systems alone or in the absence of a supporting polynucleating ligand.
Ultimately, such variety of approaches to ligand design allows to synthetically access multimetallic assemblies with well-defined geometries and site-selective positioning of metal atoms, with structural complexity and functional diversity resembling multimetallic sites in metalloenzymes that catalyze a variety of highly selective catalytic processes. While the development of homogeneous transition metal-based catalysts has historically relied on structural variations in the organic ligand attached to the metal, the ability to use metal–metal interactions in addition to metal–ligand interactions provides an indispensable tool to modulate reactivity in a greater variety of ways. Multimetallic systems often show properties that are difficult to achieve using organic ligand variation alone due to a significantly expanded scope of elements that can be used to modulate the desired properties. The second metal can vary electrophilicity via incorporation of positively charged Lewis acidic center. Multielectron redox transformations may also be mediated via incorporation of several redox active metals. Finally, the approach may enable new metal–metal cooperative modes of substate activation. Just as the organic ligand modification became a routine tool in the optimization of homogeneous catalysts, rational approaches to varying metal–metal interactions should eventually become a common tool in organometallic chemistry and catalysis to modulate reactivity in a greater variety of ways. The design of well-defined, unsymmetrical polynucleating ligands helps to achieve this goal by bridging metals from any part of the periodic table.
Author contributions
R. G. and S. D. assembled the examples, prepared the figures and wrote the initial draft. J. K. edited and revised the manuscript and the figures. All coauthors participated in the discussion of the outline and editing process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Eugene Khaskin and Dr R. R. Fayzullin for checking the manuscript and helpful discussions. We also thank all previous and current group members engaged in working on multimetallic complexes and ligand design, especially Dr Orestes Rivada-Wheelaghan.Notes and references
- P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Springer, Netherlands, 2004 Search PubMed.
- J. Humphreys, R. Lan and S. Tao, Adv. Sustainable Syst., 2021, 2, 2000043 Search PubMed.
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef PubMed.
- R. Mülhaupt, Macromol. Chem. Phys., 2003, 204, 289–327 CrossRef.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- I. J. Hewitt, J.-K. Tang, N. T. Madhu, R. Clérac, G. Buth, C. E. Anson and A. K. Powell, Chem. Commun., 2006, 2650–2652 RSC.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS.
- B. Chatterjee, W.-C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329–4346 CrossRef CAS PubMed.
- G. Jansen, M. Schubart, B. Findeis, L. H. Gade, I. J. Scowen and M. McPartlin, J. Am. Chem. Soc., 1998, 120, 7239–7251 CrossRef CAS.
- L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658–2678 CrossRef CAS PubMed.
- M. H. Pérez-Temprano, J. A. Casares and P. Espinet, Chem.–Eur. J., 2012, 18, 1864–1884 CrossRef PubMed.
- R. J. Oeschger and P. Chen, J. Am. Chem. Soc., 2017, 139, 1069–1072 CrossRef CAS PubMed.
- I. Meana, P. Espinet and A. C. Albéniz, Organometallics, 2014, 33, 1–7 CrossRef CAS.
- M.-E. Moret, D. Serra, A. Bach and P. Chen, Angew. Chem., Int. Ed., 2010, 49, 2873–2877 CrossRef CAS PubMed.
- G. J. Arsenault, C. M. Anderson and R. J. Puddephatt, Organometallics, 1988, 7, 2094–2097 CrossRef CAS.
- T. Yamaguchi, F. Yamazaki and T. Ito, J. Am. Chem. Soc., 2001, 123, 743–744 CrossRef CAS PubMed.
- M. Baya, Ú. Belío, D. Campillo, I. Fernández, S. Fuertes and A. Martín, Chem.–Eur. J., 2018, 24, 13879–13889 CrossRef CAS PubMed.
- E. Paenurk, R. Gershoni-Poranne and P. Chen, Organometallics, 2017, 36, 4854–4863 CrossRef CAS.
- R. J. Oeschger and P. Chen, Organometallics, 2017, 36, 1465–1468 CrossRef CAS.
- A. L. Liberman-Martin, D. S. Levine, M. S. Ziegler, R. G. Bergman and T. D. Tilley, Chem. Commun., 2016, 52, 7039–7042 RSC.
- D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC.
- S. Banerjee, M. K. Karunananda, S. Bagherzadeh, U. Jayarathne, S. R. Parmelee, G. W. Waldhart and N. P. Mankad, Inorg. Chem., 2014, 53, 11307–11315 CrossRef CAS PubMed.
- M. K. Karunananda, F. X. Vázquez, E. E. Alp, W. Bi, S. Chattopadhyay, T. Shibata and N. P. Mankad, Dalton Trans., 2014, 43, 13661–13671 RSC.
- T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258–17261 CrossRef CAS PubMed.
- K. M. Waltz, C. N. Muhoro and J. F. Hartwig, Organometallics, 1999, 18, 3383–3393 CrossRef CAS.
- K. M. Waltz, X. He, C. Muhoro and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 11357–11358 CrossRef CAS.
- H.-C. Yu, S. M. Islam and N. P. Mankad, ACS Catal., 2020, 10, 3670–3675 CrossRef CAS.
- C. Cesari, B. Berti, F. Calcagno, C. Lucarelli, M. Garavelli, R. Mazzoni, I. Rivalta and S. Zacchini, Organometallics, 2021, 40, 2724–2735 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- S. R. Parmelee and N. P. Mankad, Dalton Trans., 2015, 44, 17007–17014 RSC.
- J. R. Pinkes, B. D. Steffey, J. C. Vites and A. R. Cutler, Organometallics, 1994, 13, 21–23 CrossRef CAS.
- H. Memmler, U. Kauper, L. H. Gade, I. J. Scowen and M. McPartlin, Chem. Commun., 1996, 1751–1752 RSC.
- J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed.
- M. M. D. Roy, J. Hicks, P. Vasko, A. Heilmann, A.-M. Baston, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 22301–22306 CrossRef CAS PubMed.
- S. Sinhababu, M. R. Radzhabov, J. Telser and N. P. Mankad, J. Am. Chem. Soc., 2022, 144, 3210–3221 CrossRef CAS PubMed.
- M. Lutz, M. Haukka, T. A. Pakkanen and L. H. Gade, Organometallics, 2001, 20, 2631–2634 CrossRef CAS.
- L. H. Gade, M. Schubart, B. Findeis, S. Fabre, I. Bezougli, M. Lutz, I. J. Scowen and M. McPartlin, Inorg. Chem., 1999, 38, 5282–5294 CrossRef CAS.
- D. Sorbelli, L. Belpassi and P. Belanzoni, J. Am. Chem. Soc., 2021, 143, 14433–14437 CrossRef CAS PubMed.
- J. Li, M. Hermann, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 8611–8614 CrossRef CAS PubMed.
- J. Campos, J. Am. Chem. Soc., 2017, 139, 2944–2947 CrossRef CAS PubMed.
- N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Chem.–Eur. J., 2020, 26, 5982–5993 CrossRef CAS PubMed.
- N. Hidalgo, C. Romero-Pérez, C. Maya, I. Fernández and J. Campos, Organometallics, 2021, 40, 1113–1119 CrossRef CAS PubMed.
- I. M. Riddlestone, N. A. Rajabi, J. P. Lowe, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, J. Am. Chem. Soc., 2016, 138, 11081–11084 CrossRef CAS PubMed.
- H. Fujita, S. Takemoto and H. Matsuzaka, ACS Catal., 2021, 11, 7460–7466 CrossRef CAS.
- A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 3822–3835 CrossRef CAS.
- A. Volbeda, E. Garcin, C. Piras, A. L. de Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989–12996 CrossRef CAS.
- M. Kampa, M.-E. Pandelia, W. Lubitz, M. van Gastel and F. Neese, J. Am. Chem. Soc., 2013, 135, 3915–3925 CrossRef CAS PubMed.
- E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557–566 CrossRef CAS PubMed.
- G. S. Ferguson, P. T. Wolczanski, L. Parkanyi and M. C. Zonnevylle, Organometallics, 1988, 7, 1967–1979 CrossRef CAS.
- G. S. Ferguson and P. T. Wolczanski, Organometallics, 1985, 4, 1601–1605 CrossRef CAS.
- L. M. Slaughter and P. T. Wolczanski, Chem. Commun., 1997, 2109–2110 RSC.
- H. Tsutsumi, Y. Sunada, Y. Shiota, K. Yoshizawa and H. Nagashima, Organometallics, 2009, 28, 1988–1991 CrossRef CAS.
- J. Du, Y. Zhang, Z. Huang, S. Zhou, H. Fang and P. Cui, Dalton Trans., 2020, 49, 12311–12318 RSC.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev., 2012, 54, 1–40 CrossRef CAS.
- C. M. Thomas, Comments Inorg. Chem., 2011, 32, 14–38 CrossRef CAS.
- J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115–5127 RSC.
- T. Sue, Y. Sunada and H. Nagashima, Eur. J. Inorg. Chem., 2007, 2007, 2897–2908 CrossRef.
- B. P. Greenwood, S. I. Forman, G. T. Rowe, C.-H. Chen, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2009, 48, 6251–6260 CrossRef CAS PubMed.
- C. M. Thomas, J. W. Napoline, G. T. Rowe and B. M. Foxman, Chem. Commun., 2010, 46, 5790–5792 RSC.
- J. W. Beattie, C. Wang, H. Zhang, J. P. Krogman, B. M. Foxman and C. M. Thomas, Dalton Trans., 2020, 49, 2407–2411 RSC.
- K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, ACS Catal., 2019, 9, 3153–3164 CrossRef CAS.
- N. H. Hunter, E. M. Lane, K. M. Gramigna, C. E. Moore and C. M. Thomas, Organometallics, 2021, 40, 3689–3696 CrossRef CAS.
- M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CrossRef CAS PubMed.
- P. G. Pringle and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1982, 81–82 RSC.
- D. M. McEwan, P. G. Pringle and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1982, 859–861 RSC.
- P. G. Pringle and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1984, 849–853 RSC.
- C. Xu, G. K. Anderson, L. Brammer, J. Braddock-Wilking and N. P. Rath, Organometallics, 1996, 15, 3972–3979 CrossRef CAS.
- A. J. Esswein, J. L. Dempsey and D. G. Nocera, Inorg. Chem., 2007, 46, 2362–2364 CrossRef CAS PubMed.
- T. R. Cook, A. J. Esswein and D. G. Nocera, J. Am. Chem. Soc., 2007, 129, 10094–10095 CrossRef CAS PubMed.
- T. R. Cook, B. D. McCarthy, D. A. Lutterman and D. G. Nocera, Inorg. Chem., 2012, 51, 5152–5163 CrossRef CAS PubMed.
- H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 10866–10869 CrossRef CAS PubMed.
- T.-P. Lin and F. P. Gabbaï, J. Am. Chem. Soc., 2012, 134, 12230–12238 CrossRef CAS PubMed.
- S. Sahu and F. P. Gabbaï, J. Am. Chem. Soc., 2017, 139, 5035–5038 CrossRef CAS PubMed.
- M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–936 CrossRef CAS PubMed.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed.
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed.
- A. Padwa, D. J. Austin, A. T. Price, M. A. Semones, M. P. Doyle, M. N. Protopopova, W. R. Winchester and A. Tran, J. Am. Chem. Soc., 1993, 115, 8669–8680 CrossRef CAS.
- L. R. Collins, M. van Gastel, F. Neese and A. Fürstner, J. Am. Chem. Soc., 2018, 140, 13042–13055 CrossRef CAS PubMed.
- L. R. Collins, S. Auris, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2019, 58, 3557–3561 CrossRef CAS PubMed.
- Z. Ren, T. L. Sunderland, C. Tortoreto, T. Yang, J. F. Berry, D. G. Musaev and H. M. L. Davies, ACS Catal., 2018, 8, 10676–10682 CrossRef CAS.
- F. G. Baddour, A. S. Hyre, J. L. Guillet, D. Pascual, J. M. Lopez-de-Luzuriaga, T. M. Alam, J. W. Bacon and L. H. Doerrer, Inorg. Chem., 2017, 56, 452–469 CrossRef CAS.
- F. G. Baddour, S. R. Fiedler, M. P. Shores, J. A. Golen, A. L. Rheingold and L. H. Doerrer, Inorg. Chem., 2013, 52, 4926–4933 CrossRef CAS PubMed.
- S. A. Beach and L. H. Doerrer, Acc. Chem. Res., 2018, 51, 1063–1072 CrossRef CAS PubMed.
- J. Du, X. He, D. Hong, S. Zhou, H. Fang and P. Cui, Dalton Trans., 2022, 51, 8777–8785 RSC.
- P. Cui, C. Wu, J. Du, G. Luo, Z. Huang and S. Zhou, Inorg. Chem., 2021, 60, 9688–9699 CrossRef CAS PubMed.
- B. L. Ramirez and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 5396–5407 CrossRef CAS PubMed.
- B. L. Ramirez, P. Sharma, R. J. Eisenhart, L. Gagliardi and C. C. Lu, Chem. Sci., 2019, 10, 3375–3384 RSC.
- B. J. Graziano, M. V. Vollmer and C. C. Lu, Angew. Chem., Int. Ed., 2021, 60, 15087–15094 CrossRef CAS PubMed.
- P. L. Dunn, R. K. Carlson, L. Gagliardi and I. A. Tonks, Dalton Trans., 2016, 45, 9892–9901 RSC.
- R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100–111 CrossRef CAS.
- J. T. Moore, S. Chatterjee, M. Tarrago, L. J. Clouston, S. Sproules, E. Bill, V. Bernales, L. Gagliardi, S. Ye, K. M. Lancaster and C. C. Lu, Inorg. Chem., 2019, 58, 6199–6214 CrossRef CAS PubMed.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS PubMed.
- J. T. Moore and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 11641–11646 CrossRef CAS PubMed.
- R. C. Cammarota, M. V. Vollmer, J. Xie, J. Ye, J. C. Linehan, S. A. Burgess, A. M. Appel, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 14244–14250 CrossRef CAS PubMed.
- N. Hara, K. Semba and Y. Nakao, ACS Catal., 2022, 12, 1626–1638 CrossRef CAS.
- N. Hara, T. Saito, K. Semba, N. Kuriakose, H. Zheng, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2018, 140, 7070–7073 CrossRef CAS PubMed.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 6074–6077 CrossRef CAS PubMed.
- P. Steinhoff, M. Paul, J. P. Schroers and M. E. Tauchert, Dalton Trans., 2019, 48, 1017–1022 RSC.
- P. Steinhoff, R. Steinbock, A. Friedrich, B. G. Schieweck, C. Cremer, K.-N. Truong and M. E. Tauchert, Dalton Trans., 2018, 47, 10439–10442 RSC.
- I. Fujii, K. Semba, Q.-Z. Li, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2020, 142, 11647–11652 CrossRef CAS PubMed.
- R. Seki, N. Hara, T. Saito and Y. Nakao, J. Am. Chem. Soc., 2021, 143, 6388–6394 CrossRef CAS PubMed.
- W.-C. Chang, C.-W. Chang, M. Sigrist, S.-A. Hua, T.-J. Liu, G.-H. Lee, B.-Y. Jin, C.-h. Chen and S.-M. Peng, Chem. Commun., 2017, 53, 8886–8889 RSC.
- R. H. Ismayilov, F. F. Valiyev, Y. Song, W.-Z. Wang, G.-H. Lee, S.-M. Peng and B. A. Suleimanov, Polyhedron, 2018, 144, 75–81 CrossRef CAS.
- C.-H. Yu, M.-S. Kuo, C.-Y. Chuang, G.-H. Lee, S.-A. Hua, B.-Y. Jin and S.-M. Peng, Chem.–Asian J., 2014, 9, 3111–3115 CrossRef CAS PubMed.
- J.-H. Kuo, T.-B. Tsao, G.-H. Lee, H.-W. Lee, C.-Y. Yeh and S.-M. Peng, Eur. J. Inorg. Chem., 2011, 2011, 2025–2028 CrossRef.
- C. Krämer, S. Leingang, O. Hübner, E. Kaifer, H. Wadepohl and H.-J. Himmel, Dalton Trans., 2016, 45, 16966–16983 RSC.
- A. N. Desnoyer, A. Nicolay, P. Rios, M. S. Ziegler and T. D. Tilley, Acc. Chem. Res., 2020, 53, 1944–1956 CrossRef CAS PubMed.
- I. Dutta, S. De, S. Yadav, R. Mondol and J. K. Bera, J. Organomet. Chem., 2017, 849–850, 117–124 CrossRef CAS.
- J. K. Bera, N. Sadhukhan and M. Majumdar, Eur. J. Inorg. Chem., 2009, 2009, 4023–4038 CrossRef.
- C. He and S. J. Lippard, Tetrahedron, 2000, 56, 8245–8252 CrossRef CAS.
- C. He and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 184–185 CrossRef CAS.
- J. P. Collin, A. Jouaiti, J. P. Sauvage, W. C. Kaska, M. A. McLoughlin, N. L. Keder, W. T. A. Harrison and G. D. Stucky, Inorg. Chem., 1990, 29, 2238–2241 CrossRef CAS.
- W. R. Tikkanen, C. Krueger, K. D. Bomben, W. L. Jolly, W. Kaska and P. C. Ford, Inorg. Chem., 1984, 23, 3633–3638 CrossRef CAS.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- I. Dutta, A. Sarbajna, P. Pandey, S. M. W. Rahaman, K. Singh and J. K. Bera, Organometallics, 2016, 35, 1505–1513 CrossRef CAS.
- C. He, A. M. Barrios, D. Lee, J. Kuzelka, R. M. Davydov and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 12683–12690 CrossRef CAS.
- T. C. Davenport and T. D. Tilley, Angew. Chem., Int. Ed., 2011, 50, 12205–12208 CrossRef CAS PubMed.
- Y.-Y. Zhou, D. R. Hartline, T. J. Steiman, P. E. Fanwick and C. Uyeda, Inorg. Chem., 2014, 53, 11770–11777 CrossRef CAS PubMed.
- E. Kounalis, M. Lutz and D. L. J. Broere, Chem.–Eur. J., 2019, 25, 13280–13284 CrossRef CAS PubMed.
- A. R. Delaney, L.-J. Yu, M. L. Coote and A. L. Colebatch, Dalton Trans., 2021, 50, 11909–11917 RSC.
- A. Nicolay and T. D. Tilley, Chem.–Eur. J., 2018, 24, 10329–10333 CrossRef CAS PubMed.
- O. Rivada-Wheelaghan, S. L. Aristizábal, J. López-Serrano, R. R. Fayzullin and J. R. Khusnutdinova, Angew. Chem., Int. Ed., 2017, 56, 16267–16271 CrossRef CAS PubMed.
- S. Deolka, O. Rivada-Wheelaghan, S. L. Aristizábal, R. R. Fayzullin, S. Pal, K. Nozaki, E. Khaskin and J. R. Khusnutdinova, Chem. Sci., 2020, 11, 5494–5502 RSC.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- R. Govindarajan, S. Deolka, E. Khaskin, R. R. Fayzullin, S. Pal, S. Vasylevskyi and J. R. Khusnutdinova, Chem.–Eur. J., e202201639, DOI:10.1002/chem.202201639.
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds between Metal Atoms, Springer, New York, 2006 Search PubMed.
- S. T. Liddle, Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties, Wiley, 2015 Search PubMed.
- P. Braunstein and A. A. Danopoulos, Chem. Rev., 2021, 121, 7346–7397 CrossRef CAS PubMed.
- Q. Zhu, W. Fang, L. Maron and C. Zhu, Acc. Chem. Res., 2022, 55, 1718–1730 CrossRef CAS PubMed.
- G. Feng, M. Zhang, D. Shao, X. Wang, S. Wang, L. Maron and C. Zhu, Nat. Chem., 2019, 11, 248–253 CrossRef CAS PubMed.
- G. Feng, K. N. McCabe, S. Wang, L. Maron and C. Zhu, Chem. Sci., 2020, 11, 7585–7592 RSC.
- K. Shi, I. Douair, G. Feng, P. Wang, Y. Zhao, L. Maron and C. Zhu, J. Am. Chem. Soc., 2021, 143, 5998–6005 CrossRef CAS PubMed.
- X. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Zhu, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS PubMed.
- P. Wang, I. Douair, Y. Zhao, R. Ge, J. Wang, S. Wang, L. Maron and C. Zhu, Chem, 2022, 8, 1361–1375 CAS.
- M. Chen, S. Jiang, L. Maron and X. Xu, Dalton Trans., 2019, 48, 1931–1935 RSC.
- Y. Cai, S. Jiang, L. Dong and X. Xu, Dalton Trans., 2022, 51, 3817–3827 RSC.
- S. Jiang, Y. Cai, A. Carpentier, I. del Rosal, L. Maron and X. Xu, Inorg. Chem., 2022, 61, 8083–8089 CrossRef CAS PubMed.
- E. V. Eames, R. Hernández Sánchez and T. A. Betley, Inorg. Chem., 2013, 52, 5006–5012 CrossRef CAS PubMed.
- P.-J. Chen, M. Sigrist, E.-C. Horng, G.-M. Lin, G.-H. Lee, C.-h. Chen and S.-M. Peng, Chem. Commun., 2017, 53, 4673–4676 RSC.
- A. Nicolini, R. Galavotti, A.-L. Barra, M. Borsari, M. Caleffi, G. Luo, G. Novitchi, K. Park, A. Ranieri, L. Rigamonti, F. Roncaglia, C. Train and A. Cornia, Inorg. Chem., 2018, 57, 5438–5448 CrossRef CAS PubMed.
- R. H. Ismayilov, W.-Z. Wang, G.-H. Lee, C.-Y. Yeh, S.-A. Hua, Y. Song, M.-M. Rohmer, M. Bénard and S.-M. Peng, Angew. Chem., Int. Ed., 2011, 50, 2045–2048 CrossRef CAS PubMed.
- M. Yamashita, S. Horiuchi, K. Yamamoto and T. Murahashi, Dalton Trans., 2019, 48, 13149–13153 RSC.
- T. Tanase, K. Nakamae, H. Miyano, Y. Ura, Y. Kitagawa, S. Yada, T. Yoshimura and T. Nakajima, Chem.–Eur. J., 2021, 27, 12010 CrossRef.
- N. V. S. Harisomayajula, B.-H. Wu, D.-Y. Lu, T.-S. Kuo, I.-C. Chen and Y.-C. Tsai, Angew. Chem., Int. Ed., 2018, 57, 9925–9929 CrossRef CAS PubMed.
- K. Uemura, Dalton Trans., 2017, 46, 5474–5492 RSC.
- K. Uemura and R. Miyake, Inorg. Chem., 2020, 59, 1692–1701 CrossRef CAS PubMed.
- K. Uemura, Y. Aoki and A. Takamori, Dalton Trans., 2022, 51, 946–957 RSC.
- K. Uemura and M. Ebihara, Inorg. Chem., 2013, 52, 5535–5550 CrossRef CAS PubMed.
- A. L. Balch, H. Hope and F. E. Wood, J. Am. Chem. Soc., 1985, 107, 6936–6941 CrossRef CAS.
- M. A. Casado, J. J. Pérez-Torrente, J. A. López, M. A. Ciriano, F. J. Lahoz and L. A. Oro, Inorg. Chem., 1999, 38, 2482–2488 CrossRef CAS.
- J. A. Chipman and J. F. Berry, Chem. Rev., 2020, 120, 2409–2447 CrossRef CAS PubMed.
- M. Nippe and J. F. Berry, J. Am. Chem. Soc., 2007, 129, 12684–12685 CrossRef CAS PubMed.
- J. A. Chipman and J. F. Berry, Inorg. Chem., 2018, 57, 9354–9363 CrossRef CAS PubMed.
- D. W. Brogden and J. F. Berry, Inorg. Chem., 2015, 54, 7660–7665 CrossRef CAS PubMed.
- T. Tanase, M. Tanaka, M. Hamada, Y. Morita, K. Nakamae, Y. Ura and T. Nakajima, Chem.–Eur. J., 2019, 25, 8219–8224 CrossRef CAS PubMed.
- O. Rivada-Wheelaghan, A. Comas-Vives, R. R. Fayzullin, A. Lledós and J. R. Khusnutdinova, Chem.–Eur. J., 2020, 26, 12168–12179 CrossRef CAS PubMed.
- O. Rivada-Wheelaghan, S. Deolka, R. Govindarajan, E. Khaskin, R. R. Fayzullin, S. Pal and J. R. Khusnutdinova, Chem. Commun., 2021, 57, 10206–10209 RSC.
Footnote |
| † These authors made equal contributions. |
| This journal is © The Royal Society of Chemistry 2022 |