
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of exceptional monomeric YPhos–PdCl2 complexes with high activities in coupling reactions†
Ilja
Rodstein
a,
Leif
Kelling
a,
Julian
Löffler
a,
Thorsten
Scherpf
a,
Abir
Sarbajna
 a,
Diego M.
Andrada
b and
Viktoria H.
Gessner
*a
a,
Diego M.
Andrada
b and
Viktoria H.
Gessner
*a
aFaculty of Chemistry and Biochemistry, Chair of Inorganic Chemistry II, Ruhr University Bochum, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: viktoria.gessner@rub.de
bGeneral and Inorganic Chemistry Department, University of Saarland, Campus C4.1, 66123 Saarbruecken, Germany
First published on 26th October 2022
Abstract
The use of well-defined palladium(II) complexes as precatalysts for C–X cross-coupling reactions has improved the use of palladium catalysts in organic synthesis including large-scale processes. Whereas sophisticated Pd(II) precursors have been developed in the past years to facilitate catalyst activation as well as the handling of systems with more advanced monophosphine ligands, we herein report that simple PdCl2 complexes function as efficient precatalysts for ylide-substituted phosphines (YPhos). These complexes are readily synthesized from PdCl2 sources and form unprecedented monomeric PdCl2 complexes without the need for any additional coligand. Instead, these structures are stabilized through a unique bonding motif, in which the YPhos ligands bind to the metal through the adjacent phosphine and ylidic carbon site. DFT calculations showed that these bonds are both dative interactions with the stronger interaction originating from the electron-rich phosphine donor. This bonding mode leads to a remarkable stability even towards air and moisture. Nonetheless, the complexes readily form monoligated LPd(0) complexes and thus the active palladium(0) species in coupling reactions. Accordingly, the YPhos–PdCl2 complexes serve as highly efficient precatalysts for a series of C–C and C–X coupling reactions. Despite their simplicity they can compete with the efficiency of more complex and less stable precatalysts.
Introduction
Palladium-catalyzed coupling reactions have become an indispensable tool in organic synthesis for the construction of C–C and C–X bonds under mild reaction conditions.1 Over the past two decades, the design of efficient catalysts and the establishment of reliable reaction protocols have turned them into robust methods also for large-scale industrial processes.2 Many advances in the field were based on the development of new ligands that allowed the tailoring of the catalyst properties to meet the requirements of different substrates and reaction conditions.3 Thus, intense research efforts have focused on the design of new ligands, above all phosphines and carbenes, to improve the reaction protocols with respect to substrate scope, temperature or catalyst loading.4 However, besides ligand design also the choice of palladium source has been recognized to have a profound impact on the efficiency of a catalyst system. Therefore, researchers have aimed at developing preformed palladium complexes as well-defined precatalysts.5In general, the optimal precatalyst should be air-stable, easy to handle and readily accessible from inexpensive and commercially available starting materials. Furthermore, catalyst activation should be facile and not lead to the formation of toxic or difficult to separate by-products which limit applications such as in the synthesis of pharmaceuticals or organic materials, where highly pure products are mandatory. Since monoligated Pd(0) complexes are usually the active species in coupling reactions,6 complexes with palladium in the zero oxidation state and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal:ligand ratio are the obvious choices. However, Pd(0) precursors such as Pd2dba3 often show varying qualities and limited stability particularly at ambient conditions,7 so that their phosphine complexes are usually only prepared in situ. In contrast, L2Pd complexes are usually more stable, but require higher reaction temperatures for the dissociation of the second ligand.8 Only a few other Pd(0) precatalysts have been developed to date.9 Palladium(I) complexes also readily form the active species, but suffer either from limited stability or are applicable to a limited number of phosphines or carbenes.10 Therefore, research endeavors have focused on Pd(II) complexes which are usually air-stable and thus easy to handle. Here, the design has focused on optimizing the generation of the LPd(0) species, leading to the development of a series of anionic coligands that facilitate the reduction process and minimize catalyst deactivation through formation of inactive metal species. Successfully applied complexes include η3-allyl systems such as allyl, crotyl or cinnamyl (A),11,12 indenyl (B)13 or methylnaphthyl (C)14 complexes as well as palladacycles15 above all based on the 2-aminobiphenyl structure (D, Fig. 1).16 Also oxidative addition complexes of type [LPd(Ar)Cl]x (x = 1 or 2) have been used as “on-cycle” precatalysts, as was first described by Shaughnessy in 2017.17 However, these complexes showed widely varying success as precatalysts and always lead to the formation of another coupling by-product in the amount of pre-catalyst used.18
:1 metal:ligand ratio are the obvious choices. However, Pd(0) precursors such as Pd2dba3 often show varying qualities and limited stability particularly at ambient conditions,7 so that their phosphine complexes are usually only prepared in situ. In contrast, L2Pd complexes are usually more stable, but require higher reaction temperatures for the dissociation of the second ligand.8 Only a few other Pd(0) precatalysts have been developed to date.9 Palladium(I) complexes also readily form the active species, but suffer either from limited stability or are applicable to a limited number of phosphines or carbenes.10 Therefore, research endeavors have focused on Pd(II) complexes which are usually air-stable and thus easy to handle. Here, the design has focused on optimizing the generation of the LPd(0) species, leading to the development of a series of anionic coligands that facilitate the reduction process and minimize catalyst deactivation through formation of inactive metal species. Successfully applied complexes include η3-allyl systems such as allyl, crotyl or cinnamyl (A),11,12 indenyl (B)13 or methylnaphthyl (C)14 complexes as well as palladacycles15 above all based on the 2-aminobiphenyl structure (D, Fig. 1).16 Also oxidative addition complexes of type [LPd(Ar)Cl]x (x = 1 or 2) have been used as “on-cycle” precatalysts, as was first described by Shaughnessy in 2017.17 However, these complexes showed widely varying success as precatalysts and always lead to the formation of another coupling by-product in the amount of pre-catalyst used.18
 | ||
| Fig. 1 Structures of state-of-the-art bench-stable palladium (II) precatalysts (L = phosphine or N-heterocyclic carbene). | ||
Surprisingly, the simplest Pd(II) precursor, PdCl2, has received less attention in the synthesis of defined precatalysts, although it usually provides stable complexes that do not liberate organic by-products upon activation. Although Kumada reported on the use of PdCl2(ddpf) in 1979,19 the later developed, more sophisticated monophosphines were rarely applied as their PdCl2 complexes.20 This may be due to the observation that the halide bridged [LPdX2]2 dimers often exhibited lower activities21 than mononuclear systems of type LPd(X2)L′ with an additional “throw-away” ligand as shown for example in case of the PEPPSI catalysts E.22
Recently, we reported on the synthesis and application of ylide-substituted phosphines (YPhos).23 These ligands feature enhanced donor properties due to the electron-releasing power of the ylide group which led to high activities in a variety of coupling reactions.24 In case of Buchwald–Hartwig aminations and Murahashi coupling reactions, an improved performance was observed with indenyl precatalysts of type B compared to in situ prepared catalysts from the phosphine and Pd2dba3, allowing for unprecedented activities.25 This improved performance led us to search for other simpler precatalysts, which are accessible from inexpensive precursors, more stable than previous precatalysts and do not produce an additional byproduct. Here, we report that simple PdCl2 complexes perfectly fulfil this purpose. The exceptional activity of these simple precatalysts is based on the unusual monomeric structure realized by the YPhos ligands through a unique kC,kP coordination mode.
Results and discussion
YPhos–PdCl2 synthesis, structures and properties
In search of efficient precatalysts for our YPhos ligands, we initially focused on using oxidative addition complexes which had repeatedly been shown to be suitable entry points into catalytic cycles.26 In case of p-tolyl chloride and bromide, the dimeric halide-bridged addition complexes were found to be easily accessible such as with the YPhos ligand, keYPhos (L1) and Pd2dba3 in the presence of an excess of aryl halide (Scheme 1).27 Both complexes 1 and 2 feature two characteristic doublets in the 31P{1H} NMR spectrum with large coupling constants of approx. 50 Hz. In contrast, switching to the sterically more demanding ortho-tolyl bromide, which we expected to form a monomeric oxidative addition complex with enhanced reactivity, led to the formation of palladium black along with a further new species featuring two signals with diminishing coupling constants between the two phosphorus centers in the keYPhos ligand. GC analysis of the reaction mixture showed the formation of the homo-coupled biaryl compound. The same observation was made using aryl iodides independent of their steric demand. XRD analysis finally provided unequivocal clarity about the composition of the newly formed compounds as simple Pd(II) dihalide complexes, L1·PdBr2 and L1·PdI2. To our surprise, these complexes formed monomeric structures even without an additional coligand such as pyridine (cf. the PEPPSI type catalysts). Instead, the palladium center is coordinated by the phosphine as well as the ylidic carbon center (Scheme 1). Despite many YPhos transition metal complexes have been isolated in the past years, such a coordination mode has never been observed before for any metal.24,28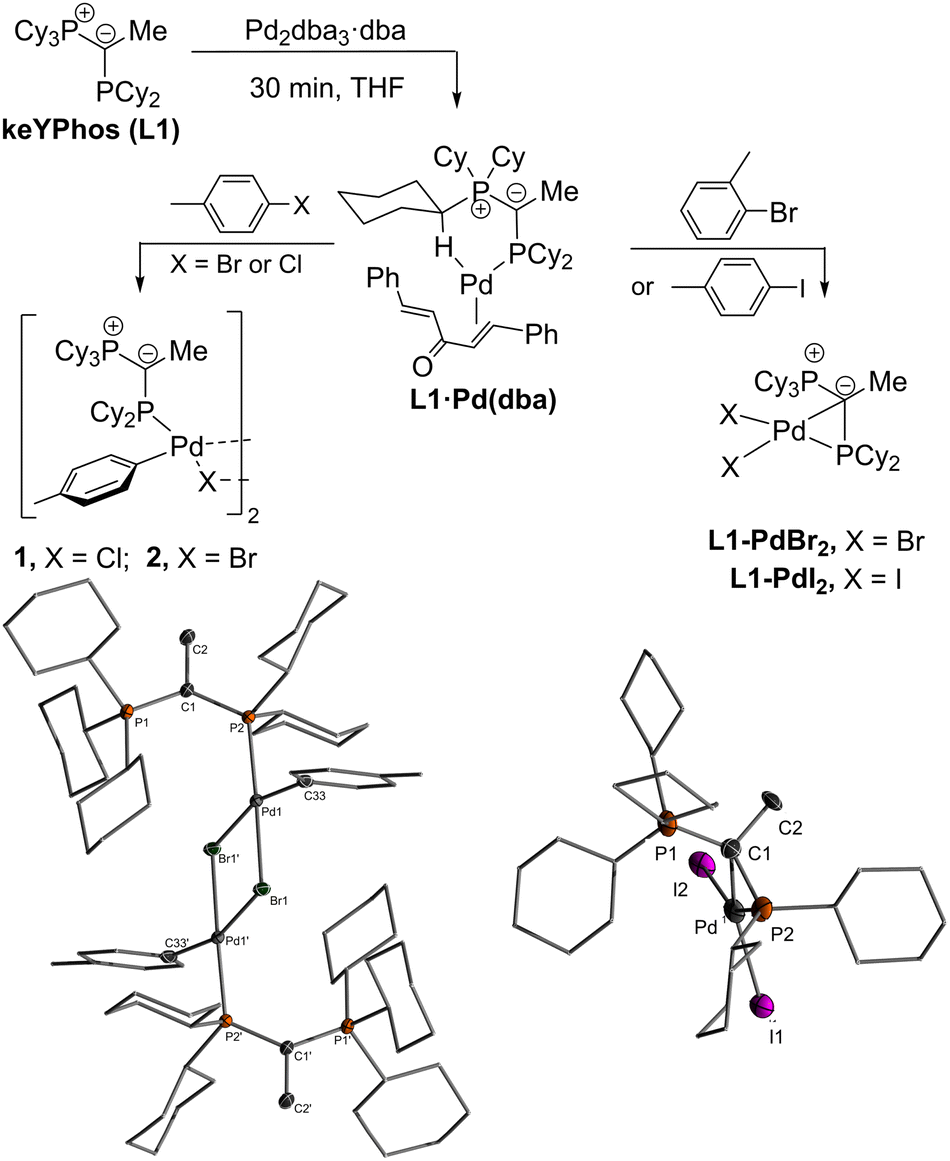 | ||
| Scheme 1 Reactivity of L1·Pd(dba) with different aryl halides. | ||
The unique structure of the YPhos Pd(II) dihalide complexes led us to develop a simpler preparation method starting directly from PdCl2. Simple mixing of the ligand with PdCl2 revealed to be problematic due to solubility issues. However, the more soluble precursors [(CH3CN)2PdCl2] or [(COD)PdCl2] gave L1·PdCl2 as a yellow solid by simple filtration from the reaction solution in yields of up to 96% (Scheme 2). Alternatively, L1·PdCl2 can also be prepared by in situ formation of [(CH3CN)2PdCl2] using a suspension of PdCl2 in a 1:1 mixture of THF and acetonitrile together with the ligand. The mixture of both solvents was necessary to account for the different solubilities of the ligand and the metal salt. Nonetheless, employment of the preformed acetonitrile complex gave slightly higher yields so that this method was employed to access the PdCl2 complexes with the other YPhos ligands joYPhos and pinkYPhos. For the bulkier trYPhos ligand, successful synthesis could only be achieved with [(COD)PdCl2]. Overall, all PdCl2 complexes could be isolated in excellent yields of greater than 80%.
 | ||
| Scheme 2 Synthesis of L·PdCl2 complexes with different YPhos ligands. | ||
To our delight, all L·PdCl2 complexes feature remarkably high stabilities even towards air and moisture and thus can be handled without any precautions. Storage of L·PdCl2 at ambient conditions led to no signs of decomposition even after several months. In solution however, only L3·PdCl2 and especially L4·PdCl2 with an aromatic backbone were stable toward oxygen and water for a longer time presumably due to the enhanced steric protection of the ylidic carbon center by the aryl substituents. This high stability of L·PdCl2 is beneficial for their handling and superior to previously reported YPhos allyl-type palladium precatalysts.25
Single crystals (Fig. 2) of L·PdCl2 with keYPhos, joYPhos and pinkYPhos could be obtained by diffusion of pentane into saturated DCM solutions. All complexes feature the same monomeric structure with the unique κC,κP bonding motif of the YPhos ligand as found for the PdBr2 and PdI2 complexes (Scheme 1). This motif contrasts the usual coordination mode of mono-phosphines towards PdX2, which usually leads to halide-bridged dimers or bisphosphine complexes. Monophosphine complexes are in general only formed in the presence of additional donor ligands (e.g.E). Yet, some other monophosphine complexes with a κC,κP bonding motif, in which the C-donor only weakly binds to the metal (e.g. no carbene), have been reported, amongst phosphines with an additional alkene moiety29 and bulky biaryl phosphines, which feature an additional arene metal interaction.30 A pallada cyclopropane structure however, has only once been reported by Bertrand and coworkers, yet for an (amino)(phosphino)carbene ligand.31 The molecular structures of L·PdCl2 feature a planarly coordinated palladium center, whose coordination environment strongly deviates from an ideal square-planar geometry due to the acute C–Pd–P angle of approx. 49° (Table 1). The P–Pd bond lengths (2.145(1) to 2.165(1) Å) are considerably shorter than those found in other YPhos palladium(II) complexes, such as the L·Pd(allyl)Cl systems or 1 and 2 (2.289(1) to 2.406(1) Å).25a The C–Pd bond length is slightly longer than common Pd–Ccarbene bonds (∼2.0 Å),22,31 but in the range of olefin complexes (e.g. 2.131(4) Å in L1·Pd(dba)),24b thus indicating a weak interaction. Despite the coordination of the ylide moiety to the palladium center, the ylidic center maintains its almost planar geometry. Thus, all L·PdCl2 complexes feature a sum of angles around the carbon atom greater than 354°. This corroborates with a weak Pd–C interaction, in line with the relatively long Pd–C distance.
 | ||
| Fig. 2 (a) Molecular structures of the PdCl2 complexes of keYPhos and joYPhos and (b) drawings of the C–H⋯Pd interaction observed in L3·PdCl2 and L4·PdCl2. Cy groups have been omitted for clarity. | ||
| L1·PdCl2 | L2·PdCl2 | L3·PdCl2 | L4·PdCl2 | |
|---|---|---|---|---|
| a n.d.: The signal could not be detected. | ||||
| δ P (PCy3) [ppm] | 37.1 | 38.7 | 35.3 | 38.2 |
| δ P (PR2) [ppm] | 48.6 | 83.2 | 53.7 | 57.4 |
| 2 J PP [Hz] | — | 4.7 | 3.9 | 5.7 |
| δ C (PCP) [ppm] | 14.2 | 20.0 | n.d.a | n.d.a |
| 1 J PC [Hz] | 41.1, 5.1 | 30.2, 12.7 | n.d.a | n.d.a |
| P2–Pd [Å] | 2.165(1) | — | 2.149(1) | 2.145(1) |
| C1–Pd [Å] | 2.122(5) | — | 2.187(3) | 2.186(3) |
| Pd–Cl1 [Å] | 2.412(1) | — | 2.436(1) | 2.421(1) |
| Pd–Cl2 [Å] | 2.394(1) | — | 2.338(1) | 2.351(1) |
| C1–P2 [Å] | 1.798(5) | — | 1.794(4) | 1.798(3) |
| P1–C1–P2 [°] | 124.8(3) | — | 124.3(2) | 129.4(2) |
| C1–Pd–P2 [°] | 49.6(1) | — | 48.9(1) | 49.1(1) |
| C1–P2–Pd [°] | 64.0(2) | — | 66.7(1) | 66.7(1) |
In solution, L·PdCl2 feature two singlets or a set of two doublets with very small coupling constants (2JPP < 6 Hz) in the 31P{1H} NMR spectrum (Table 1). The signals are substantially low-field shifted compared to those of the free ligands as a result of the coordination of the phosphine to the metal center. Likewise, the 13C NMR signals of the ylidic carbon atoms appear at lower field (14.2 ppm in L1·PdCl2 and −1.7 ppm in L1) with smaller coupling constants. Interestingly, in the 1H NMR spectrum of L3·PdCl2 and L4·PdCl2, the signal for one of the ortho hydrogen atoms of the aryl substituent in the ylide backbone appears at a remarkably lower field (approx. Δδ = 1.7 ppm, Fig. S3 and S4†). This can presumably be explained by a weak Pd–H interaction, which corroborates with short Pd–H distances found in the crystal structure. With 2.58(2) and 2.75(3) Å these distances are shorter or in the range of the sum of the van der Waals radii of palladium and hydrogen (2.73 Å) (Fig. 2b).
To gain further insights into the electronic structures of YPhos–PdCl2 complexes, we performed DFT calculations at the PBE0-D3(BJ)/def2-SVP32 level of theory (see the ESI† for details) using L1·PdCl2 as an outset. The equilibrium geometry is in very good agreement with the structure determined by SC-XRD (Fig. S50†), with the P–Pd bond (2.161 Å) being slightly shorter than the expected bond length for a P–Pd double bond length (2.19 Å), as observed with the experimental structure. In contrast, the C–Pd bond length (2.154 Å) deviates from the experimental measurement by ca. 0.03 Å and is clearly longer than expected for a single bond (1.95 Å). Furthermore, the structure exhibits only a small pyramidalization at the carbon atom (Σ∠C = 355°), supporting a weak bonding interaction.
We were particularly interested in the nature of the ylide-palladium chemical bond. Thus, we first analyzed the electronic structure by natural bond orbital (NBO) analysis. Table S25 in the ESI† gathers the most important calculated natural partial charges (Q) and Wiberg bond orders (P) of the bonding involved in the PdCl2 coordination. Natural population analysis (NPA) indicates an electron-rich C1 atom with −0.95e and a positively charged P2 atom with +1.22e. The complete moiety of PdCl2 bears a negative charge of −0.90e, mainly located at the chlorine atoms, which are due to the strong donation of the YPhos fragment. The Wiberg bond order values are consistent with a weak single bond character for both, the P–Pd (P = 0.54 a.u.) and the C–Pd (P = 0.34 a.u.) bond. The NBO analysis leads to a Lewis structure with a lone pair remaining on the C1 atom, supporting a dative interaction between the ylide and the palladium center and thus confirms that the ylide is functioning as a L-type rather than a X-type ligand.
This is further supported by the analysis of the electron density distribution with atoms in molecules (QTAIM).33Fig. 3 depicts the Laplacian distribution ∇2ρ(r) in the C1–Pd–P2 plane. The plot shows an electron accumulation on the phosphorus and carbon atoms localized on the σ-system with an electron density of the bond critical points (BCP) of P–Pd (ρBCP = 0.84 e Å−3) and C–Pd (ρBCP = 0.67 e Å−3). The corresponding delocalization index (DI) points toward weak single bonds, i.e. 0.93 and 0.78 for P–Pd and C–Pd, respectively. It is interesting to note that the electron density at the BCPs and the DI values both suggest that the stronger interaction between the ligand and palladium originates from the phosphine and not from the ylidic carbon center. This observation is well in line with the NBO analysis (see the ESI†) and can be explained by strong donor properties of the YPhos ligand. Notably, the ellipticity of the P–Pd bond fits to a single bond character (ε = 0.02), however, the C–Pd bond has a relatively high value, which is a consequence of a small hessian parameter on the three-membered ring plane.
 | ||
| Fig. 3 Laplacian distribution of the electron density of L1·PdCl2 (PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)/def2-SVP). Contour line diagrams of the Laplacian distribution ∇2ρ(r) in the Pd–C–P plane. Dashed red lines indicate areas of charge concentration (∇2ρ(r) < 0) while solid blue lines show areas of charge depletion (∇2ρ(r) > 0). The thick solid lines connecting the atomic nuclei are the bond paths and the small dots are the critical points. Bond critical points (in black), Ring critical points (in red). | ||
 | ||
| Fig. 4 Plot of deformation densities Δρ1–3 (iso value = 0.003) of the pairwise orbital interaction and shape of the most important occupied and vacant orbitals (iso value = 0.03) in YPhos–PdCl2 complex with the orbital interaction energies ΔEorb (in kcal mol−1) and their eigenvalues ν (in e). For the deformation densities, the direction of the charge flow is red → blue. The eigenvalues ν indicate the amount of donated (negative numbers) and accepted charge (positive numbers). The occupied orbitals are shown in yellow and blue for the different phases, while the unoccupied orbitals are in light yellow and light blue. | ||
More detailed information about the nature of the YPhos–PdCl2 coordination was obtained by the Energy Decomposition Analysis (EDA) method.34 EDA has proven to be a useful tool to assess the nature of the chemical bond in main group compounds and transition metal compounds.35,36 Nonetheless, a recent discussion has been placed about the path function nature of the energy components.36,37 Within the EDA scheme, the bond formation (De) between two (or more) fragments is dissected in the preparation energy (ΔEprep) and interaction energy (ΔEint). The interaction energy can be further divided into different physically meaningful energetic terms, namely, Pauli repulsion (ΔEPauli), dispersion (ΔEdisp), electrostatic (ΔEelstat) and orbital interaction (ΔEorb, for further details, see the ESI†). Table 2 shows the numerical results of the calculations where PdCl2 and the YPhos ligand are both in their neutral and singlet reference state as interacting fragments, although ionic interacting fragments is also possible. The EDA results reveal a strong interaction between the PdCl2(S) and YPhos(S) fragment (ΔEint = −142.1 kcal mol−1). The preparation energy is 39.9 kcal mol−1, which is mostly related to the geometrical deformation of the YPhos ligand (29.5 kcal mol−1). An examination of the ΔEint components suggests a large Coulomb interaction contribution (56.2%) and orbital interaction (40.5%), while the dispersion represents only 3.2% of the total stabilizing interactions. The specific deformation densities as well as the interacting orbitals are shown in Fig. 3 revealing a rather symmetrical bonding between the YPhos ligand and PdCl2, with equal contributions from the phosphine and the carbon site.
| L1 (S); PdCl2 (S) | |
|---|---|
| a Geometries optimized at the PBE0-D3(BJ)/def2-TZVP level of theory. b The value in parenthesis gives the percentage contribution to the total attractive interactions ΔEelstat + ΔEorb + ΔEdisp. c The values in parenthesis gives the percentage contribution to the total orbital interaction. | |
| ΔEint | −142.1 |
| ΔEPauli | 282.9 |
| ΔEdispb | −13.6 (3.2%) |
| ΔEelstatb | −239.0 (56.2%) |
| ΔEorb totalb | −172.3 (40.5%) |
| ΔEorb HF | 0.3 |
| ΔEorb | −172.6 |
| ΔEorb-σc | −103.1 (59.7%) |
| ΔEorb-πc | −15.2 (8.8%) |
| ΔEorb-σc | −13.9 (8.1%) |
| ΔEorb-polc | −11.7 (6.8%) |
| ΔEorb-restc | −40.4 (23.4%) |
| ΔEprep PdCl2 | 10.1 |
| ΔEprep L1 | 29.5 |
| ΔEprep total | 39.9 |
| D e | 102.2 |
| Fragment electron flow | 1.28e |
Synthesis and structure of YPhos-based palladate complexes
The unique monomeric structure of the PdCl2 complexes led us to explore whether they may also serve as precursors to mononuclear palladate complexes via cleavage of the Pd–C bond such as by HCl addition. Indeed, treatment of a solution of the L·PdCl2 complexes in DCM with an excess of aqueous HCl gave the zwitterionic products LH·PdCl3 as yellow solids in good yields between 62 and 80% (Route A, Scheme 3). Alternatively, the palladate complexes can be obtained in similar purities and yields (69 and 91%) directly from the α-phosphino phosphonium salts L·H and either [(CH3CN)2PdCl2] or [(COD)PdCl2] (Route B). However, in this case, the use of the chloride salt of L·H is strictly required to prevent halide scrambling and the formation of mixed bromo/chloro palladates. All LH·PdCl3 complexes are extremely robust solids, which do not react with oxygen and water. The stability is also maintained in DCM solutions, even upon addition of water or bubbling of oxygen through a solution of the palladate complexes. Similar to the PdCl2 complexes, the palladate complexes show a low solubility in ether or hydrocarbon solvents but are well soluble in chlorinated solvents.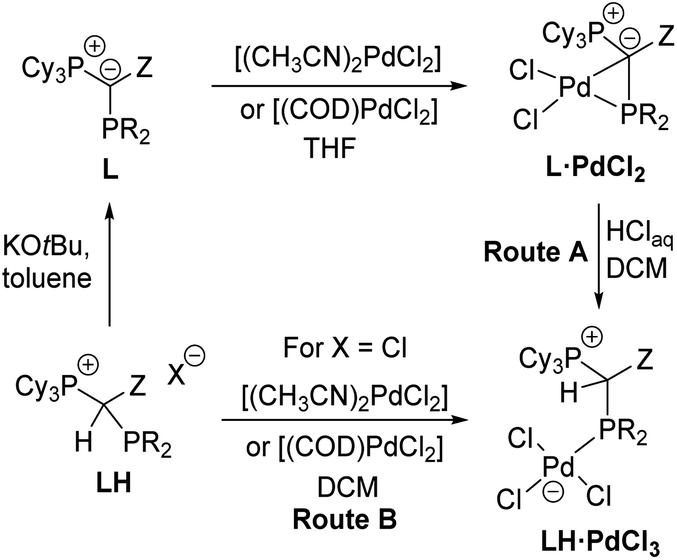 | ||
| Scheme 3 Preparation of the YPhos palladates LH·PdCl3. | ||
The palladate complexes were characterized by NMR, elemental and XRD analysis (Table 3). In solution, they are characterized by slightly larger 2JPP coupling constants (4.7–16.7 Hz) in the 31P{1H}NMR spectra in comparison to L·PdCl2. The phosphine signals are high-field shifted (e.g., from 53.7 ppm in L3·PdCl2 to 31.4 ppm in L3H·PdCl3), whereas the signals for the phosphonium group appeared at lower field than in L·PdCl2 (e.g., 35.3 ppm in L3·PdCl2 to 38.5 ppm in L3H·PdCl3) (see the ESI† for details). The 13C NMR signals of the central carbon center appear slightly more down-field shifted with smaller 1JPC coupling constants compared to the L·PdCl2 complexes due to the protonation of the former carbanionic center and the involved decrease in bond angles. The molecular structures (Fig. 5) confirmed the expected cleavage of the former Pd–C1 contact. This cleavage is accompanied by a significant increase of the C1–P2–Pd1 angle from approx. 65° in the PdCl2 complexes to 111.27(6)° to 116.5(1)° in LH·PdCl3. Also, the Pd–P2 bond elongates for example from approx. 2.165(1) Å in L1·PdCl2 to 2.268(1) Å in the corresponding palladate complex L1H·PdCl3. Furthermore, in contrast to L·PdCl2 the C1 carbon atom is now clearly pyramidalized with smaller P–C–P angles between 116.2(2)° and 121.2(1)°.
| L1H·PdCl3 | L2H·PdCl3 | L3H·PdCl3 | L4H·PdCl3 | |
|---|---|---|---|---|
| δ P (PCy3) [ppm] | 40.4 | 43.1 | 38.5 | 42.5 |
| δ P (PCy2) [ppm] | 30.5 | 66.9 | 31.4 | 37.0 |
| 2 J PP [Hz] | 4.7 | 10.5 | 14.5 | 16.7 |
| δ C (PCP) [ppm] | 20.7 | 27.9 | 34.7 | 33.0 |
| 1 J PC [Hz] | 36.8, — | 25.8, 7.5 | 36.4, 5.3 | 35.5, 7.9 |
| C1–P2 [Å] | 1.891(2) | 1.864(6) | 1.902(2) | 1.925(4) |
| P2–Pd [Å] | 2.2675(5) | 2.296(1) | 2.2737(6) | 2.286(1) |
| Pd–Cllongest [Å] | 2.3853(5) | 2.346(1) | 2.3750(6) | 2.357(1) |
| Pd–Clshortest [Å] | 2.3150(5) | 2.304(2) | 2.3128(7) | 2.311(1) |
| P1–C1–P2 [°] | 121.2(1) | 125.3(3) | 118.0(1) | 116.2(2) |
| C1–P2–Pd [°] | 111.27(6) | 111.9(2) | 115.67(7) | 116.5(1) |
 | ||
| Fig. 5 Molecular structures of palladates L1·PdCl3 and L3·PdCl3. | ||
Applications in catalysis
Motivated by the facile synthesis of L·PdCl2 and LH·PdCl3 and their high stability towards air and moisture, we next evaluated their potential as precatalysts. Their performance was compared with the in situ formed palladium(0) complex L·Pd(dba) and established precatalysts, i.e. the preformed palladium(II) precatalysts L·Pal, L·Pcin, and L·Pind (Fig. 6, see the ESI† for their synthesis).25b All complexes were tested in a series of palladium catalyzed C–N and C–C bond formation reactions (Fig. 5). | ||
| Fig. 6 YPhos precatalysts tested in coupling reactions (for YPhos definitions, see Scheme 2). L·Pd(dba) was formed in situ by stirring of ligand and Pd2dba3·dba for 1 h prior to catalysis. Due to its long-term instability L1·Pdcin was also formed in situ. The indenyl complex of L2 is not accessible due to steric reasons. | ||
Previous studies have shown that Buchwald–Hartwig aminations of aryl chlorides are efficiently catalyzed by YPhos complexes with high selectivities under mild conditions.25b To evaluate the potential of the L·PdCl2 and LH·PdCl3 complexes, two amination reactions with aryl chlorides of different steric profile were chosen, one with a secondary amine (4-chlorotoluene + piperidine) and one with a primary amine (2-chlorotoluene + n-butylamine). Earlier studies have revealed a higher activity of the η3-allyl-type YPhos palladium precatalysts compared to the in situ formed L·Pd(dba) complexes.25b To our delight, similar activities were reached with the simple PdCl2 complexes (Fig. 7a). Full conversion was reached in both reactions with 0.5 mol% using the PdCl2 complexes of keYPhos, trYPhos and joYPhos at room temperature. Also, at lower catalyst loadings higher yields than with the Pd(0) precursors L·Pd(dba) and similar yields than obtained with the allyl-systems were reached. Importantly, this performance was reached already at room temperature, thus showing that catalyst activation proceeds facile under the reaction conditions. The performance of L·PdCl2 is – despite the similarity of the type of precatalyst – clearly superior to that of the commercially available Pd-PEPPSI-IPent catalyst. Under the same reaction conditions as in Fig. 5A, this catalyst only achieves 36% conversion for the amination of 4-chlorotoluene with piperidine after 1 h (40% after 3 h) at room temperature (see the ESI† for details).
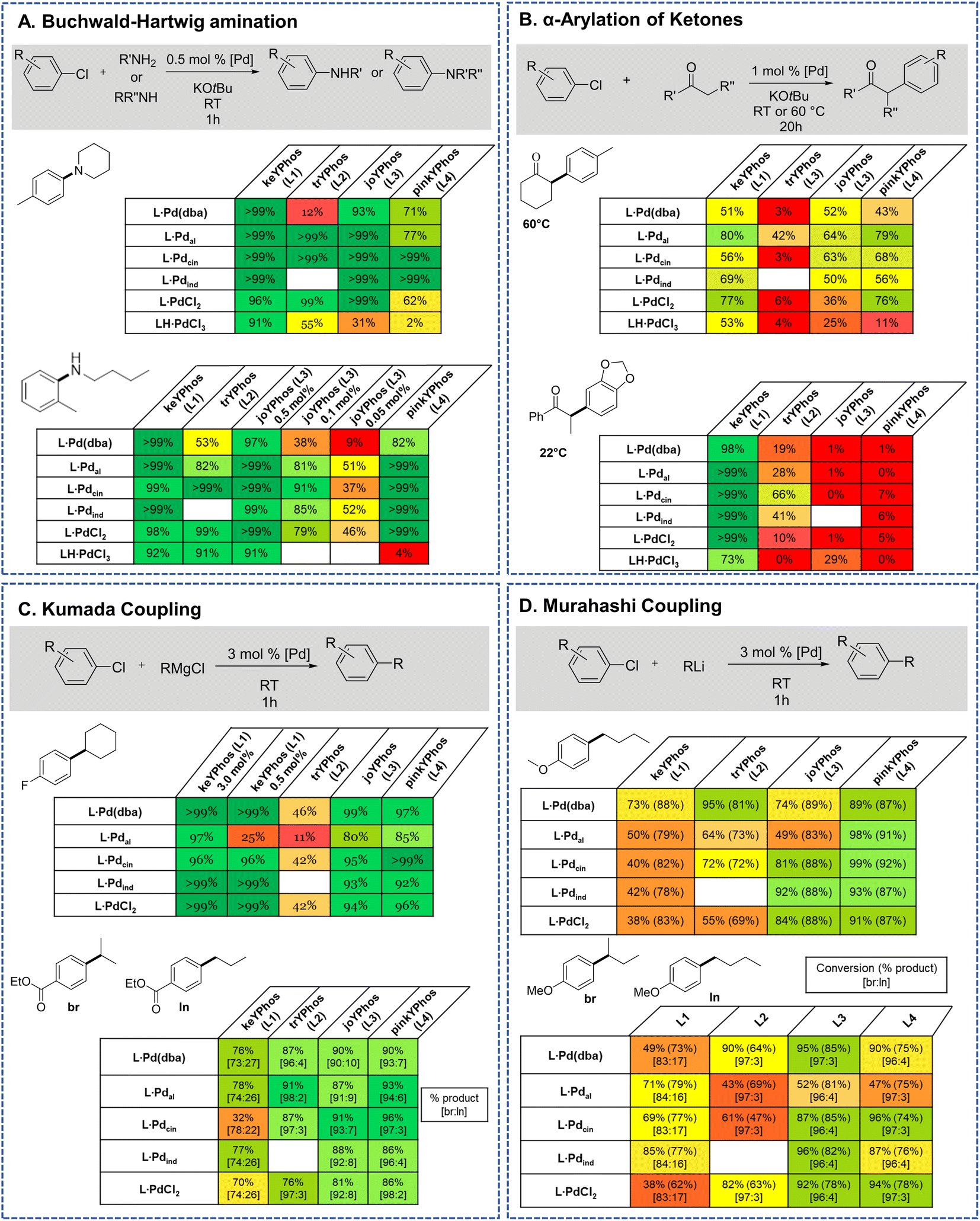 | ||
| Fig. 7 Comparison of the YPhos–PdCl2 and palladate complexes with other YPhos precatalysts (depicted in Fig. 4) in their application in (A) Buchwald–Hartwig aminations, (B) α-arylation of ketones, (C) Kumada and (D) Murahashi–Feringa coupling reactions. Detailed information about the reaction conditions are given in the ESI.† Yields are GC yields obtained after the given reaction time. | ||
In contrast to the PdCl2 complexes, the LH·PdCl3 complexes showed a lower activity. We assume that for the formation of the active species the palladate complexes first need to undergo dehydrohalogenation to the corresponding L·PdCl2 complex, since the cationic ligand would be too electron-poor to enable the oxidative addition of aryl chlorides at room temperature. However, deprotonation of the phosphonium backbone is presumably slow due to steric crowding, which results in the slow or incomplete catalyst activation. To improve the catalyst activation, different reaction conditions were tested, i.e. using different bases as well as chloride abstraction reagents. Yet, no reliable protocol could be established for all ligands. In general, best results were obtained with the smallest ligand, keYPhos, suggesting that steric shielding of the C1 carbon atom by the Z substituent indeed complicates deprotonation. This different shielding of the hydrogen atom at the C1 atom by the different ligand structures is illustrated by the van-der-Waals plots (Fig. S3†) of the molecular structures. Whereas H1 is still accessible from the side of the methyl group in L1, it is completely shielded by the ortho-tolyl group. Accordingly, L4·PdCl2 performed worst due to the more demanding ortho-tolyl group, which almost completely prevents catalyst activation with KOtBu as base within 1 h at room temperature. The slower activation of the palladate complexes and their moderate performance in comparison to the PdCl2 systems was also confirmed in kinetic studies. Whereas for the amination of p-tolylchloride with piperidine at room temperature less than 40% conversion were observed with the joYPhos complexL3H·PdCl3, the corresponding PdCl2 complex quickly formed the active species to reach full conversion already after 6 min reaction time (Fig. 8).
 | ||
| Fig. 8 Conversion time plots for the C–N coupling of p-toylchloride with piperidine with different precatalysts of joYPhos (L3). Conditions: 0.5 mol% precatalyst, chloride:amine = 1:1.1; 1.5 eq. KOtBu, THF, room temperature. | ||
This fast activation was also seen for the PdBr2 system indicating that there is no significant impact of the nature of the halide on the catalytic activity (see Fig. S6†). The activity of L3·PdCl2 compared well with the indenyl precatalyst L3·Pdind, which showed a slightly higher activity to reach full conversion already after 2 min. Nonetheless, L3·PdCl2 is clearly more active than the catalyst formed from the free ligand and Pd2(dba)3 which showed a longer induction period and only reached 97% conversion after 60 min. Overall, the PdCl2 complexes exhibit similarly high activities in C–N coupling reactions than the more complicated Pd(II) allyl-type complexes, thus representing a more stable and cost efficient alternative, which furthermore does not produce any further organic by-product.
The effectiveness of the L·PdCl2 complexes was also observed in the α-arylation of ketones, in which – according to previous studies38 – the keYPhos ligand performed best in comparison to the other YPhos ligands. Again, the corresponding palladate complex gave slightly lower yields. To our delight, also Kumada and Murahashi coupling reactions were easily catalyzed by the PdCl2 complexes, demonstrating that organolithium and Grignard reagents quickly produce the active species even at room temperature. Whereas for the Kumada coupling all ligands worked well, the Murahashi coupling was only effectively mediated by the aryl substituted YPhos ligands, joYPhos (L3) and pinkYPhos (L4). This observation also agrees well with the higher stability of these complexes observed in solution. We assume that this increased stability is the reason for the improved performance of these catalysts. In contrast, the more electron-rich PtBu2 substituted trYPhos ligand (L2) – which should facilitate the usually rate-limiting oxidative addition and thus exhibit higher activities – gave lower or inconsistent yields not only for the Murahashi but most other coupling reactions. This varying performance thus most likely originates from competing activity and stability trends.
To finally test the utility of the palladium chloride complexes in catalysis, we attempted the isolation of a series of different coupling products (Fig. 9). In case of the C–N couplings, perfect yields were obtained for a selection of aliphatic and aromatic amines with 0.5 mol% of the PdCl2 complex. Sole exception represents the 2-pyridyl benzylamine Aa, which could only be isolated in 48% yield under the reaction conditions (no optimization attempted). All other aryl amines could be isolated in high yields >90%. In case of the α-arylation of ketones the coupling worked well with electron-rich (Ba) and electron-poor (Bd) aryl chlorides. Lower yields were observed with sterically demanding chlorides, such as with the di-ortho-methyl substituted (Be) or naphthyl chloride (Bf). In case of the Kumada and Murahashi coupling moderate to good yields were reached without further optimizing the reaction conditions.39 Further substrates tested, but not isolated are given in the ESI (Fig. S1†).
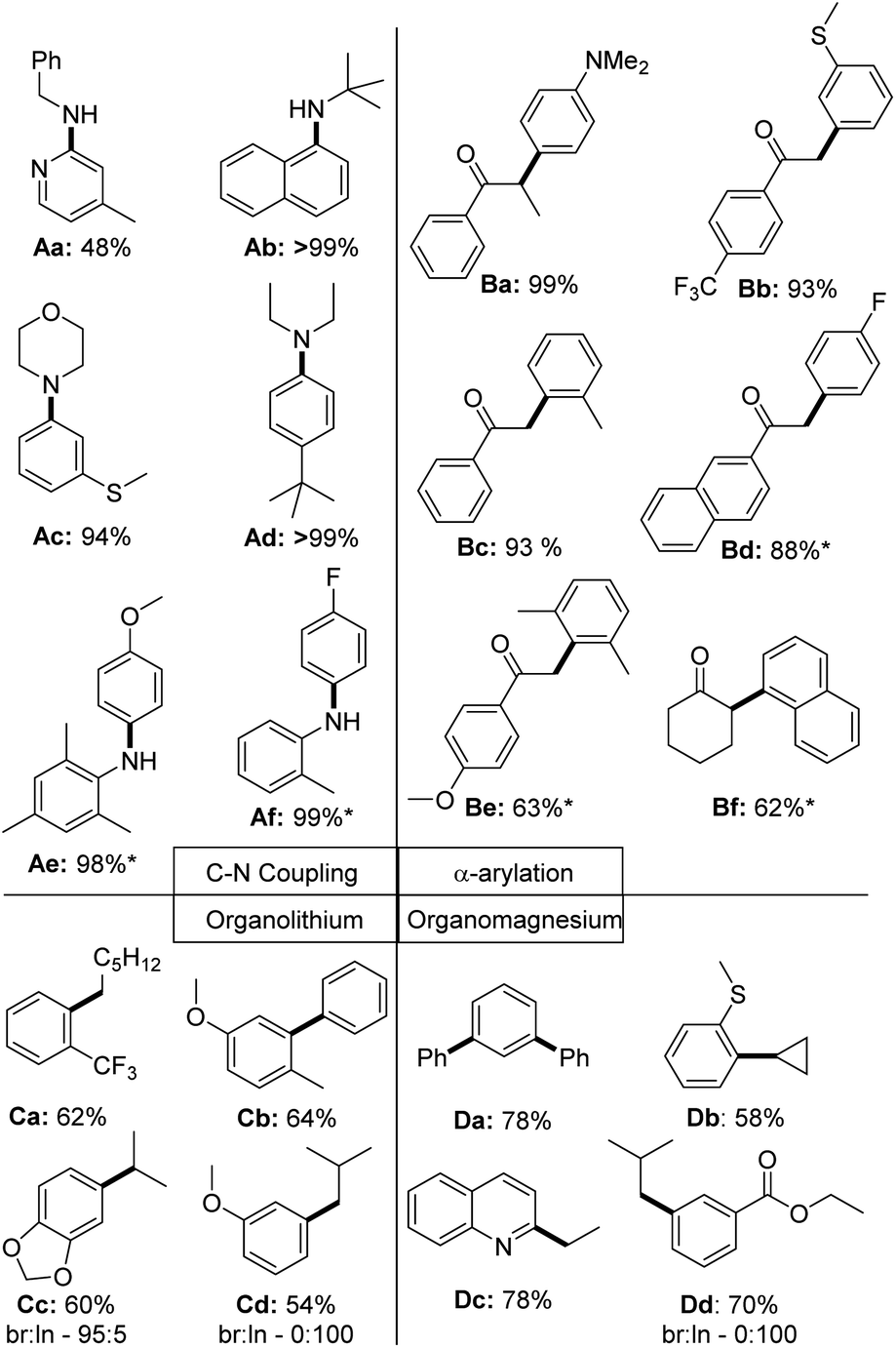 | ||
| Fig. 9 Scope of the application of PdCl2 complexes in C–N and C–C coupling reactions of aryl chlorides. * = Reaction was performed at 60 °C; br = branched product; ln = linear product. Used precatalyst: L1·PdCl2 for Ab, Ae, Af, Ba – Bf; L2·PdCl2 for Aa, Ad; L3·PdCl2 for Ac and L4·PdCl2 for Ca – Cd, Da – Dd. The same reactions conditions as used in the benchmarking reactions shown in Fig. 5 have been used. Further details are provided in the ESI.† | ||
Given the efficiency of the YPhos–PdCl2 complexes, we were interested in the mode of activation of these precatalysts. Preparation of the catalyst mixture already indicated a fast activation by addition of the base. To test whether treatment with a base already results in the reduction of Pd(II) to Pd(0), we performed stoichiometric experiments by treating the keYPhos (L1) PdCl2 complex in THF with two equiv. KOtBu. Addition of the base instantaneously resulted in the dissolution of the PdCl2 complex and the darkening of the reaction mixture. 31P NMR spectroscopy indicated the initial formation of a new species, which shows two doublets at 51.2 and 77.5 ppm with a coupling constant of 98 Hz (see the ESI† for details). This intermediate species reacts further to form palladium black along with the palladium(0) bisphosphine complex [(L1)2Pd],27 which is characterized by two signals at 18.9 and 30.3 ppm and which remains the main product after a longer reaction time. This observation clearly demonstrates that L·PdCl2 is readily reduced to Pd(0) species by the metal base. No further substrates, which might give rise to undesired byproducts, are necessary for the activation.
Conclusions
In conclusion, we reported on the formation of simple palladium dihalide complexes with different YPhos ligands and their application in Pd coupling chemistry. The PdCl2 complexes are easily generated from the free ligands and the metal salt precursors and form unusual monomeric structures through coordination of the metal by the phosphine and the ylidic carbon atom. The palladacyclopropane motif easily undergoes Pd–C bond cleavage such as by addition of HCl, thus giving rise to zwitterionic palladate complexes with a cationic phosphine ligand. The PdCl2 and palladate complexes of the YPhos ligands exhibit remarkable stabilities in the solid state and in solution even towards air and moisture, thus making them easy-to-handle and applicable precatalysts. Despite their simplicity and stability, the PdCl2 precatalysts exhibited remarkably activities in C–N and C–C coupling reactions. According to kinetic studies, catalyst activation proceeds fast under basic conditions at room temperature for the PdCl2 complexes, but slower for the palladate systems. Accordingly, the YPhos–PdCl2 showed a superior performance and could compete with more complex Pd(II) precatalysts reported earlier. Hence, these complexes represent easy to synthesize and more stable and cost efficient, but highly active alternatives to other specially designed precatalysts.Author contributions
I. R. has synthesized all complexes, performed most of the catalysis reactions and solved most of the crystal structures. L. K. has supported I. R. with the synthesis of the complexes. J. L. has performed the down-scaling reactions. T. S. and A. S. have solved some of the crystal structures. D. M. A. has performed the theoretical calculations. V. H. G. has designed the study and prepared the manuscript together with I. R.Conflicts of interest
A patent application has been filed on this work in collaboration with Umicore AG & Co KG.Acknowledgements
This work was Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV and by the Individual Research Grant DA1402/7-1. Furthermore, we thank Umicore AG & Co. KG for financial support and the donation of chemicals. LK thanks the Fonds der Chemischen Industrie for a PhD fellowship, AS the Alexander von Humboldt Foundation for a postdoctoral fellowship.Notes and references
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed.
- (a) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed; (b) P. Devendar, R.-Y. Qu, W.-M. Kang, B. He and G.-F. Yang, J. Agric. Food Chem., 2018, 66, 8914 CrossRef CAS PubMed.
- L.-C. Campeau and N. Hazari, Organometallics, 2019, 38, 3 CrossRef CAS PubMed.
- P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497 CrossRef CAS.
- (a) N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 1 CrossRef; (b) K. H. Shaughnessy, Isr. J. Chem., 2020, 60, 180 CrossRef CAS; (c) H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147 CrossRef CAS.
- (a) S. H. Newman-Stonebraker, S. R. Smith, J. E. Borowski, E. Peters, T. Gensch, H. C. Johnson, M. S. Sigman and A. G. Doyle, Science, 2021, 374, 301 CrossRef CAS PubMed; (b) F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8141 CrossRef CAS PubMed; (c) M. Ahlquist and P.-O. Norrby, Organometallics, 2007, 26, 550 CrossRef CAS.
- P. Weber, A. Biafora, A. Doppiu, H.-J. Bongard, H. Kelm and L. J. Gooßen, Org. Process Res. Dev., 2019, 23, 1462 CrossRef CAS.
- M. W. Hooper, M. Utsunomiya and J. F. Hartwig, J. Org. Chem., 2003, 68, 2861 CrossRef CAS PubMed.
- (a) M. G. Andreu, A. Zapf and M. Beller, Chem. Commun., 2000, 2475 RSC; (b) H. G. Lee, P. J. Milner and S. L. Buchwald, Org. Lett., 2013, 15, 5602 CrossRef CAS PubMed; (c) J. Huang, M. Isaac, R. Watt, J. Becica, E. Dennis, M. I. Saidaminov, W. A. Sabbers and D. C. Leitch, ACS Catal., 2021, 11, 5636 CrossRef CAS.
- (a) J. P. Stambuli, R. Kuwano and J. F. Hartwig, Angew. Chem., Int. Ed., 2002, 41, 4746 CrossRef CAS PubMed; (b) S. De, N. Sivendran, B. Maity, N. Pirkl, D. Koley and L. J. Gooßen, ACS Catal., 2020, 10(8), 4517 CrossRef CAS.
- (a) L. L. Hill, J. L. Crowell, S. L. Tutwiler, N. L. Massie, C. C. Hines, S. T. Griffin, R. D. Rogers, K. H. Shaughnessy, G. A. Grasa, C. C. C. Johansson Seechurn, H. Li, T. J. Colacot, J. Chou and C. J. Woltermann, J. Org. Chem., 2010, 75, 6477 CrossRef CAS PubMed; (b) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101 CrossRef CAS PubMed; (c) M. S. Viciu, R. F. Germaneau and S. P. Nolan, Org. Lett., 2002, 4, 4053 CrossRef CAS PubMed; (d) O. Navarro, H. Kaur, P. Mahjoor and S. P. Nolan, J. Org. Chem., 2004, 69, 3173 CrossRef CAS PubMed.
- A. J. DeAngelis, P. G. Gildner, R. Chow and T. J. Colacot, J. Org. Chem., 2015, 80, 6794 CrossRef CAS PubMed.
- P. R. Melvin, A. Nova, D. Balcells, W. Dai, N. Hazari, D. P. Hruszkewycz, H. P. Shah and M. T. Tudge, ACS Catal., 2015, 5, 3680 CrossRef CAS.
- N. Sivendran, N. Pirkl, Z. Hu, A. Doppiu and L. J. Gooßen, Angew. Chem., Int. Ed., 2021, 60, 25151 CrossRef CAS PubMed.
- (a) O. Navarro, R. A. Kelly III and S. P. Nolan, J. Am. Chem. Soc., 2003, 125, 16194 CrossRef CAS PubMed; (b) M. S. Viciu, R. A. Kelly III, E. D. Stevens, F. Naud, M. Studer and S. P. Nolan, Org. Lett., 2003, 5, 1479 CrossRef CAS PubMed; (c) G.-R. Peh, E. A. B. Kantchev, J.-C. Er and J. Y. Ying, Chem.–Eur. J., 2010, 16, 4010 CrossRef CAS PubMed; (d) L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392 CrossRef CAS PubMed.
- (a) M. R. Biscoe, B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 6886 CrossRef PubMed; (b) N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916 RSC; (c) N. C. Bruno, N. Niljianskul and S. L. Buchwald, J. Org. Chem., 2014, 79, 4161 CrossRef CAS PubMed.
- H. Hu, F. Qu, D. L. Gerlach and K. H. Shaughnessy, ACS Catal., 2017, 7, 2516 CrossRef CAS.
- Further examples of oxidative addition complexes as precatalysts: (a) B. T. Ingoglia and S. L. Buchwald, Org. Lett., 2017, 19, 2853 CrossRef CAS PubMed; (b) L. Chen, H. Francis and B. P. Carrow, ACS Catal., 2018, 8, 2989 CrossRef CAS.
- (a) T. Hayashi, M. Konishi and M. Kumada, Tetrahedron Lett., 1979, 1871 CrossRef CAS; (b) T. Hayashi, M. Konishi, Y. Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc., 1984, 106, 158 CrossRef CAS.
- (a) K. L. Barnett, J. R. Howard, C. J. Treager, A. T. Shipley, R. M. Stullich, F. Qu, D. L. Gerlach and K. H. Shaughnessy, Organometallics, 2018, 37, 1410 CrossRef CAS; (b) G. Xu, P. Gao and T. J. Colacot, ACS Catal., 2022, 12, 5123 CrossRef CAS.
- M.-T. Chen, D. A. Vicic, M. L. Turner and O. Navarro, Organometallics, 2011, 30, 5052 CrossRef CAS.
- (a) M. G. Organ, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien and C. A. Valente, Chem.–Eur. J., 2006, 12, 4749 CrossRef CAS PubMed; (b) M. G. Organ, G. A. Chass, D.-C. Fang, A. C. Hopkinson and C. Valente, Synthesis, 2008, 2008, 2776 CrossRef.
- (a) T. Scherpf, C. Schwarz, L. T. Scharf, J.-A. Zur, A. Helbig and V. H. Gessner, Angew. Chem., Int. Ed., 2018, 57, 12859 CrossRef CAS PubMed; (b) T. Scherpf, R. Wirth, S. Molitor, K.-S. Feichtner and V. H. Gessner, Angew. Chem., Int. Ed., 2015, 54, 8542 CrossRef CAS PubMed.
- (a) S. Lapointe, A. Sarbajna and V. H. Gessner, Acc. Chem. Res., 2022, 55(5), 770 CrossRef CAS PubMed; (b) P. Weber, T. Scherpf, I. Rodstein, D. Lichte, L. T. Scharf, L. J. Gooßen and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 3203 CrossRef CAS PubMed; (c) Z. Hu, X.-J. Wei, J. Handelmann, A.-K. Seitz, I. Rodstein, V. H. Gessner and L. J. Gooßen, Angew. Chem., Int. Ed., 2021, 60, 6778 CrossRef CAS PubMed.
- (a) T. Scherpf, H. Steinert, A. Großjohann, K. Dilchert, J. Tappen, I. Rodstein and V. H. Gessner, Angew. Chem., Int. Ed., 2020, 59, 20596 CrossRef CAS PubMed; (b) J. Tappen, I. Rodstein, K. McGuire, A. Großjohann, J. Löffler, T. Scherpf and V. H. Gessner, Chem.–Eur. J., 2020, 26, 4281 CrossRef CAS PubMed.
- B. T. Ingoglia and S. L. Buchwald, J. Org. Chem., 2017, 19, 2853 CAS.
- L. Scharf, I. Rodstein, M. Schmidt, T. Scherpf and V. H. Gessner, ACS Catal., 2020, 10, 999 CrossRef CAS PubMed.
- T. Scherpf, I. Rodstein, M. Paassen and V. H. Gessner, Inorg. Chem., 2019, 58, 8151 CrossRef CAS PubMed.
- M. N. Peñas-Defrutos, A. Vélez, E. Gioria and P. Espinet, Organometallics, 2019, 38, 4701 CrossRef.
- O. M. Demchuk, K. Kapłon, L. Mazur, D. Strzelecka and K. M. Pietrusiewicz, Tetrahedron, 2016, 72, 6668 CrossRef CAS.
- E. Teuma, C. Lyon-Saunier, H. Gornitzka, G. Mignani, A. Baceiredo and G. Bertrand, J. Organomet. Chem., 2005, 690, 5541 CrossRef CAS PubMed.
- (a) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS; (b) M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS; (c) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132 Search PubMed; (d) S. Grimme, S. Ehrlich and L. Goerig, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (e) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon, Oxford, 1990 Search PubMed.
- L. L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e13450 Search PubMed.
- W. Haider, D. M. Andrada, I.-A. Bischoff, V. Huch and A. Schäfer, Dalton Trans., 2019, 48, 14953–14957 RSC.
- (a) T. Yang, D. M. Andrada and G. Frenking, Mol. Phys., 2019, 117, 1306–1314 CrossRef CAS; (b) D. M. Andrada and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2020, 22, 22459–22464 RSC.
- J. Poater, D. M. Andrada, M. Solà and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2022, 24, 2344–2348 RSC.
- X.-Q. Hu, D. Lichte, I. Rodstein, P. Weber, A.-K. Seitz, T. Scherpf, V. H. Gessner and L. J. Gooßen, Org. Lett., 2019, 21, 7558 CrossRef CAS PubMed.
- Purification was found to be more difficult for these substrates due to their lower polarity, which hampered isolation by column chromatography using the same procedures as applied for the other compounds and similar boiling points of the products and starting materials..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all new compounds, NMR spectra of the isolated compounds, crystallographic and computational details. CCDC 2190326–2190335. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04523k |
| This journal is © The Royal Society of Chemistry 2022 |