
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLive-cell RNA imaging using the CRISPR-dCas13 system with modified sgRNAs appended with fluorescent RNA aptamers†
Heng
Tang
a,
Junran
Peng
a,
Shuang
Peng
a,
Qi
Wang
a,
Xin
Jiang
a,
Xiaocheng
Xue
a,
Yanxin
Tao
b,
Limin
Xiang
a,
Quanjiang
Ji
 c,
Song-Mei
Liu
b,
Xiaocheng
Weng
*a and
Xiang
Zhou
*a
c,
Song-Mei
Liu
b,
Xiaocheng
Weng
*a and
Xiang
Zhou
*a
aCollege of Chemistry and Molecular Sciences, Key Laboratory of Biomedical Polymers of Ministry of Education, The Institute for Advanced Studies, Hubei Province Key Laboratory of Allergy and Immunology, Wuhan University, Wuhan, 430072, Hubei, P. R. China. E-mail: xzhou@whu.edu.cn; xcweng@whu.edu.cn
bDepartment of Clinical Laboratory, Center for Gene Diagnosis, and Program of Clinical Laboratory Medicine, Zhongnan Hospital of Wuhan University, Wuhan, 430072, Hubei, P. R. China
cSchool of Physical Science and Technology, ShanghaiTech University, Shanghai, 201210, P. R. China
First published on 8th November 2022
Abstract
The development of RNA imaging strategies in live cells is essential to improve our understanding of their role in various cellular functions. We report an efficient RNA imaging method based on the CRISPR-dPspCas13b system with fluorescent RNA aptamers in sgRNA (CasFAS) in live cells. Using modified sgRNA attached to fluorescent RNA aptamers that showed reduced background fluorescence, this approach provides a simple, sensitive way to image and track endogenous RNA with high accuracy and efficiency. In addition, color switching can be easily achieved by changing the fluorogenic dye analogues in living cells through user-friendly washing and restaining operations. CasFAS is compatible with orthogonal fluorescent aptamers, such as Broccoli and Pepper, enabling multiple colors RNA labeling or intracellular RNA–RNA interaction imaging. Finally, the visualization of severe fever with thrombocytopenia syndrome virus (SFTSV) was achieved by CasFAS, which may facilitate further studies on this virus.
Introduction
Spatiotemporal localization of RNAs is associated with their cellular functions; thus, RNA imaging methods are needed to reveal their location and dynamics. Although fluorescence in situ hybridization (FISH) is effective in detecting and quantifying RNA molecules in fixed cells, developing other technologies to visualize RNAs in living cells is still needed.1 The MS2-MCP system and stem-loop labeling by molecular beacons are most frequently used to visualize RNAs inside cells.2 However, laborious genetic manipulations are needed to fuse the multiplex aptamer sequences to target transcripts, which may interfere with the structure and function of tagged RNA, impeding its broad application to track RNA.Recently, various technology-based clustered regularly interspaced short palindromic repeats (CRISPRs) have been expanded for RNA imaging in living cells. The excellent performance of CRISPRs in gene editing and regulation indicates its precise nucleic acid targeting ability,3 enabling efficient RNA imaging by deactivated Cas (dCas) proteins. For instance, in addition to recognizing and visualizing double-stranded DNA,4–6 the complex of dCas9 and single-guide RNA (sgRNA) can also be stabilized by the artificial protospacer-adjacent motif (PAM) oligonucleotide to target RNA, which has been explored to track the bulk movement of highly abundant mRNAs and observe their accumulation in stress granules.7 Several other CRISPR-dCas9 imaging systems have also been developed to label and track RNA,8–10 but the reliance on the PAM sequence is still a significant limitation of the CRISPR-dCas9 system for RNA study.
A family of single-stranded RNAs (ssRNAs) targeting ribonucleases, CRISPR-Cas, has provided a more straightforward and effective way to study endogenous RNA transcripts without PAM generation.11 Cas13 can be programmed to degrade unique RNA sequences within the transcriptome,12 and catalytically deactivated Cas13 (dCas13) can be fused to a range of effector domains to perform RNA sequence-specific operations, such as A-to-I editing, modulating translation, or splicing.13,14 Previous studies established that the CRISPR-Cas13 system can be fused with fluorescent proteins for robust real-time RNA imaging and tracking, providing a promising new suite of tools for RNA imaging in live cells.12,15–17 However, it is suggested to bind and track RNAs with a sufficient number of sequence repeats or aggregate multiplex copies.
Fluorescent RNA aptamers, which have fluorogenic dyes that fluoresce upon binding, are a powerful tool for live-cell RNA imaging. The fluorescence turn-on response of the cell-permeable chemical reagents enables RNA detection with a high signal-to-noise ratio (SNR) to highlight the location of RNA aptamers. Although many fluorescent RNA aptamers with a broad range of emissions from cyan to red have been developed,18–22 the approach requires exogenous insertion of dozens of hairpins into the RNA of interest, similar to the MS2-MCP system, raising the concern of using this system in studying the behaviors of endogenous RNAs. It is reported that Cas9 and sgRNA modified with fluorescent RNA aptamer could label genome DNA,5 but such modification has not been proven to be practicable in dCas13 system.
Here, we suspected that if the CRISPR-dCas13 sgRNA is tagged with fluorescent RNA aptamers, the advantages of precise RNA recognition by the dCas13 system and the low background fluorescent aptamer can be integrated to achieve more efficient RNA visualization. Compared with the strategy of direct fusion of dCas13 with fluorescent proteins, this approach may provide several advantages. First, a previous study reported that sgRNAs are highly unstable and quickly degraded in the cellular environment without binding with target nucleic acids, which may eliminate the background fluorescence induced by the unbound sgRNA-fluorescent aptamer conjugate.4,5,16 Second, for multiplex color assays or simultaneous RNA labeling, previous dCas13-based methods needed different orthogonal dCas13 orthologues, which may be unfriendly for cellular transfection due to the large size of Cas proteins. In addition, to date, only dPspCas13b and dPguCas13b have exhibited credible RNA imaging efficiency among current dCas13 homologs,15,16 providing minimal options for multiplex detection strategies. In our design, different kinds of fluorescent RNA aptamers, such as Spinach, Broccoli or Pepper (with a series of fluorophores spanning from cyan to red), can be attached to sgRNA, allowing multicolor RNA imaging simultaneously using only one type of dCas13 protein. Furthermore, compared with the traditional fluorescent RNA aptamer assay, no genetic manipulation of the target RNA is required. The stability of the dCas13-aptamer-modified sgRNA-target RNA complex can provide long-lasting RNA labeling and imaging efficacy.
Hence, in this work, we introduce an efficient RNA imaging method in live cells based on the dPspCas13b system by modifying sgRNAs appended with fluorescent RNA aptamers, named CasFAS (CRISPR-dCas13 system with fluorescent RNA aptamers in sgRNA) (Scheme 1A). This method is easily programmable, and less invasive, conferring high-sensitivity, and precise targeting efficiency. Moreover, the easily adjustable sgRNA scaffold with multicolor fluorescent RNA aptamers offers additional advantages for simultaneous visualization of endogenous untagged RNA transcripts in live cells, allowing extended applications such as the detection of RNA–RNA interactions.
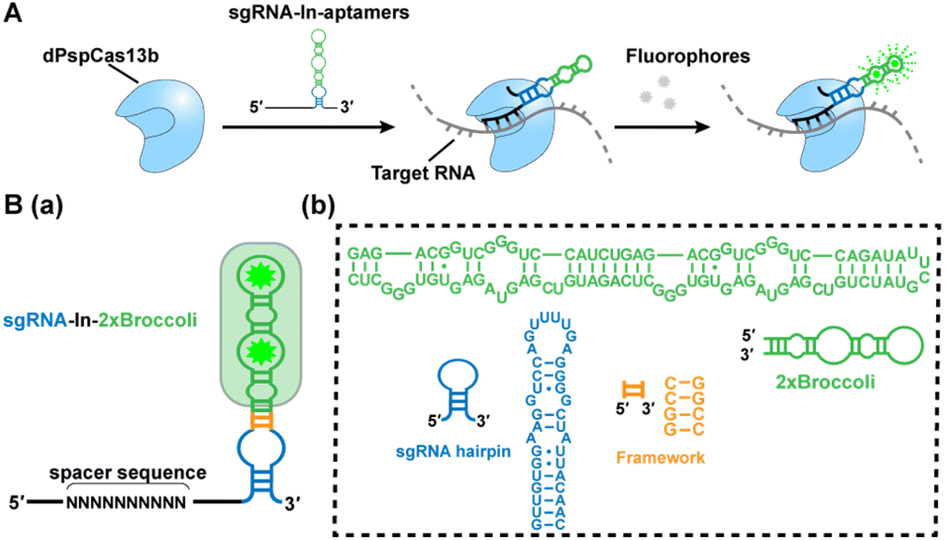 | ||
| Scheme 1 Development of the CRISPR-dPspCas13b system with fluorescent RNA aptamers in sgRNA (CasFAS). (A) Schematic drawing of the CasFAS: CRISPR-dCas13 system binds with sgRNA containing fluorescent RNA aptamers and target RNA. (B) (a) Overview of sgRNA-In-2 × Broccoli designs. The structure of dCas13b sgRNA's DR loop (blue), framework (F, orange) and 2 × Broccoli (green) with DFHBI-1T (green star). (b), Modules and their sequence illustrations of sgRNA-In-2 × Broccoli. | ||
Results and discussion
Design and in vivo optimization of modified sgRNA
We chose the well-known fluorescent RNA aptamer Broccoli in a preliminary study. The Broccoli aptamer can bind and activate nonfluorescent (Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-2-methyl-1-(2,2,2-trifluoroethyl)-1H-imidazole-5(4H)-one (DFHBI-1T) to emit green fluorescence. First, the sgRNA scaffold was fused with two tandem Broccoli aptamers (2 × Broccoli) at the 3′ end of sgRNA, named sgRNA-3′-2 × Broccoli (Fig. S1†). In the absence of dPspCas13b, the fluorescence of 2 × Broccoli-modified sgRNA was too low to be detected inside the cell (Fig. S2†, top row), indicating that the sgRNA alone is extremely unstable without the formation of the dPspCas13b/sgRNA complex. Instead, in the presence of successfully expressed dPspCas13b protein (Fig. S3†), obvious fluorescent signals were observed (Fig. S2†, middle row). This might be caused by the improved stability after binding with the dCas13 protein to enhance the abundance of sgRNAs, which was further verified by RT-qPCR analysis and consistent with previous reports (Fig. S4†).4,5,16Next, we investigated the attached modes between the sgRNA scaffold and RNA aptamer in two other ways. One is called sgRNA-3′-F-2 × Broccoli, containing a linker sequence between the sgRNA and 2× Broccoli; the other is to insert the 2× Broccoli sequence into the loop region of the sgRNA, named sgRNA-In-2 × Broccoli (Scheme 1B and Fig. S1†). By comparing the colocalization between the green fluorescence of sgRNA modified with aptamer and the red fluorescence of dPspCas13b fused with red fluorescence protein (RFP), we found that the fluorescent signals and colocalization were not satisfactory for sgRNA-3′-2 × Broccoli without linker sequences (Fig. S2†, middle row). Instead, bright fluorescence intensity, better colocalization and higher abundance of sgRNA were observed for sgRNA-3′-F-2 × Broccoli and sgRNA-In-2 × Broccoli, indicating the rapid formation of the dCas13b-sgRNA-aptamer complex (Fig. S2 bottom row, S5 and S6† left), which suggests that the linker sequence may be essential to maintain the activities of aptamer-modified sgRNA. Interestingly, based on RT-qPCR assay, the abundance of sgRNA-In-2 × Broccoli in the presence of dPspCas13b increased almost ∼30-fold compared with no dPspCas13b binding. In contrast, the level of sgRNA-3′-F-2 × Broccoli increased only ∼10-fold, indicating that aptamer insertion in the loop of the sgRNA may lead to better stability (Fig. S6†, right). This result was consistent with a previous study using the CRISPR-Cas9 system in which the efficiency of aptamer insertion in the loop was better than that at the 3′ end of the sgRNA scaffold.4,5 Structural simulation of the dPspCas13b-sgRNA complex showed that the sgRNA loop was located on the outside of the complex, suggesting that engineering of the loop region may not the affect function of dCas13b (Fig. S7†). Taken together, the sgRNA-In-2× Broccoli was chosen for the subsequent studies due to its superior performance.
Labeling GCN4 repeats and MUC4 mRNAs by sgRNA-In-2 × Broccoli
After examining the fluorescence induced by our CasFAS system, we evaluated the ability of CasFAS to image RNA inside cells. We initially introduced constructs containing 24 × GCN4 elements as the target, whose abundance of repeats was suitable for preliminary experiments to investigate the feasibility and sensitivity of this system in living cells.23 To verify the specificity of the target, we designed sgRNA targeting the repeat region of GCN4 mRNA (called GCN4-sgRNA-In-2 × Broccoli, Fig. 1A) and no target spacer gRNA (called gNC, Fig. S1†). As shown in Fig. 1B (top and middle row), gRNA signals were degraded within 24 hours after transfection. Not surprisingly, GCN4-sgRNA-In-2× Broccoli at the 24 × GCN4 transfected cell exhibited brighter fluorescence intensity than cells without 24 × GCN4 (Fig. 1B, top and bottom row and S8†). The real-time qPCR analysis further revealed a higher level of gRNA in cells transfected with 24 × GCN4 and GCN4-sgRNA-In-2 × Broccoli (Fig. 1C). This might be caused by the improved stability after binding with the dCas13 protein and the target RNA to enhance the abundance of sgRNAs. The colocalization with the results of single-molecule FISH (smFISH) further confirmed the accuracy of the CasFAS system in GCN4 labeling (Fig. S9†).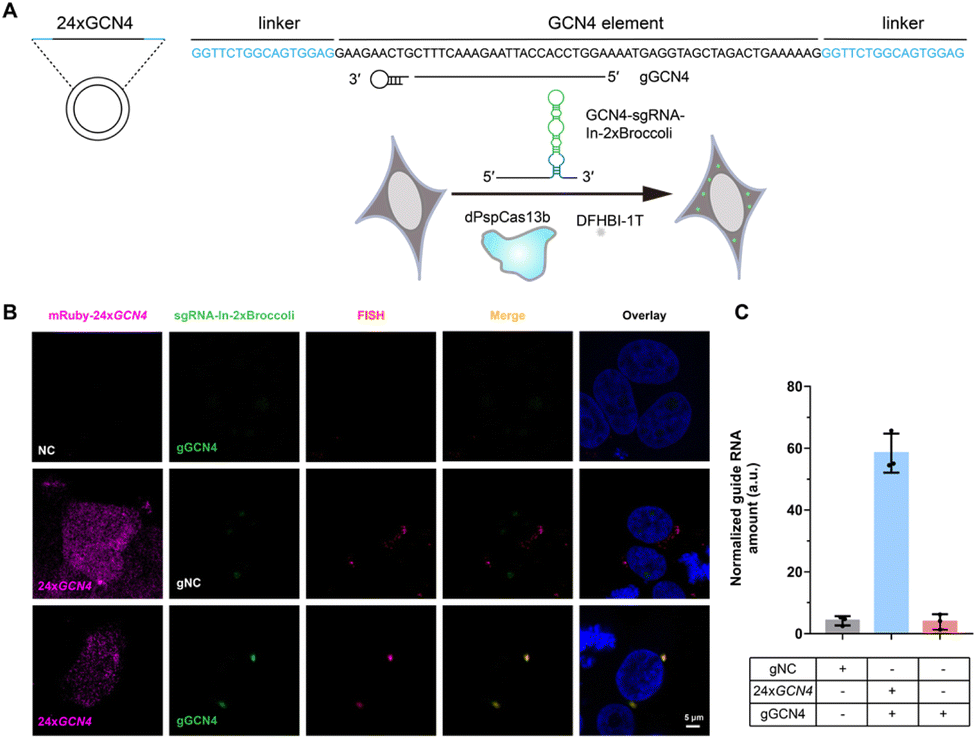 | ||
| Fig. 1 Labeling GCN4 Repeats by sgRNA-In-2 × Broccoli. (A) Schematic drawing of the GCN4 repeats labeling by CasFAS system. (B) Representative images of sgRNA-In-2 × Broccoli labeling exogenous 24 × GCN4 colocalized with smFISH in fixed HEK 293T cells. Upper row, negative control of exogenous 24 × GCN4, the cells were transfected with pGCN4-sgRNA-In-2 × Broccoli (gGCN4: with spacer sequence targeting GCN4) and pdPspCas13b-2 × NLS; middle row, the cells were transfected with pmRuby-24 × GCN4, psgRNA-In-2 × Broccoli (gNC: as a negative control of spacer sequence) and pdPspCas13b-2 × NLS; bottom row, the cells were cotransfected with pmRuby-24 × GCN4, pGCN4-sgRNA-In-2 × Broccoli (gGCN4) and pdPspCas13b-2 × NLS. The cell nucleus was dyed by Hoechst. (C) RT-qPCR analysis of GCN4-sgRNA-In-2 × Broccoli RNA levels in (B). All data are presented as the mean ± s.d.; n = 3 independent experiments. | ||
Encouraged by successful GCN4 imaging, we further asked about the feasibility of endogenous mRNAs imaging using this method. MUC4 is membrane mucin that is abundantly expressed in many epithelia, where it is proposed to play a protective role. It has become a candidate clinical marker for its aberrant expression in many epithelial tumors.24 Thus, transcript analysis is vital for understanding MUC4 expression and functions. We used sgRNA targeting the repeat region in exon 2 of MUC4 mRNA (Fig. 2A).25,26 Cell transfected with MUC4-sgRNA-In-2 × Broccoli exhibited brighter fluorescence intensity than those transfected with gNC (Fig. 2B, top and middle row). Next, we depleted the MUC4 transcript using multiple small interfering RNA (siRNAs), which is proved by RT-qPCR experimental results showing a drastic downregulation of MUC4 expression (Fig. S10†). Unsurprisingly, we observed a loss of fluorescence intensity of the cells transfected with siRNA, also verifying the specificity of targeting (Fig. 2B, bottom row). Moreover, MUC4 knock down by siRNA treatment result in a low level of sgRNA in cells, which is similar to the cells only transfected with gNC (Fig. 2C). At the same time, the statistical analysis of colocalization data between smFISH and sgRNA-In-2 × Broccoli confirmed that CasFAS could recognize and label MUC4 mRNA (Fig. S11†).
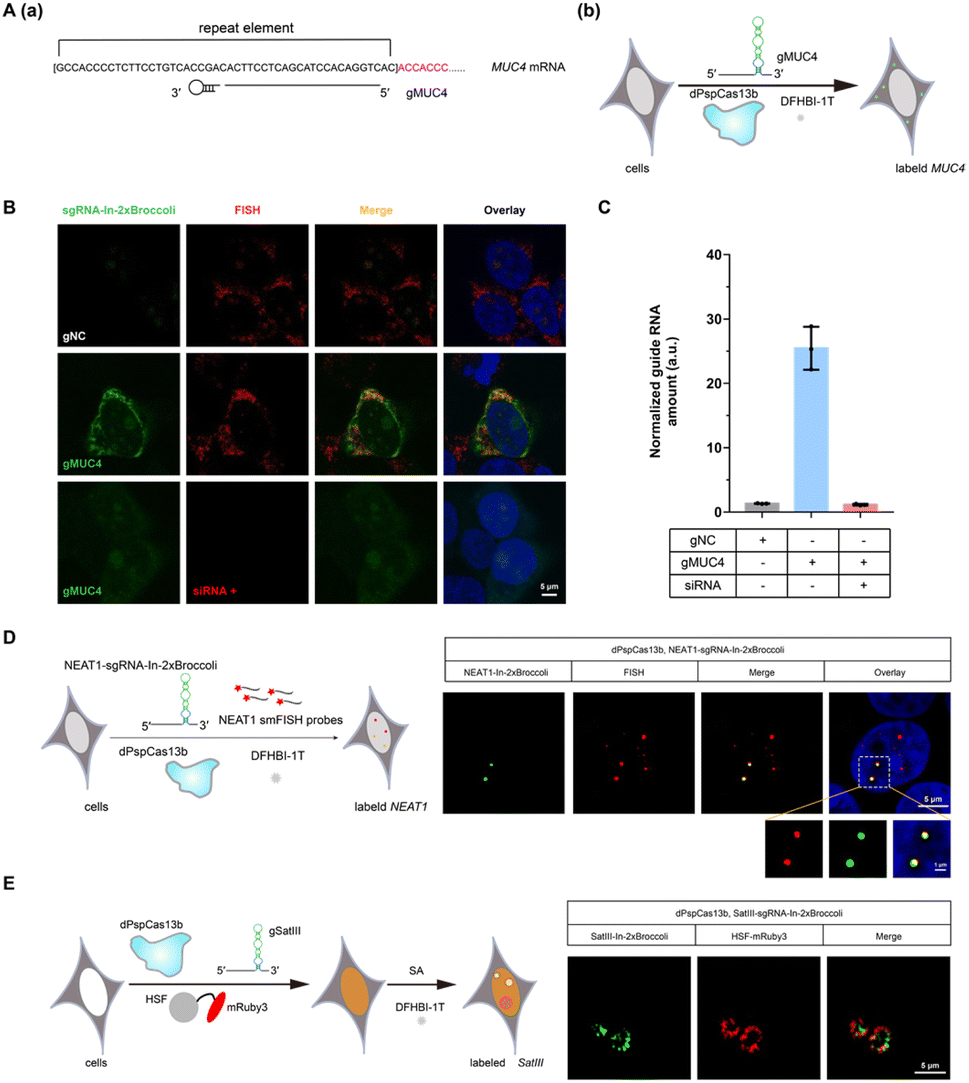 | ||
| Fig. 2 CRISPR-dPspCas13b system with fluorescent RNA aptamers in sgRNA (CasFAS) system for MUC4, lncRNA NEAT1 and SATIII RNA labeling. (A) (a) Illustration of gRNAs used in labeling MUC4 by CasFAS; (b) overview of MUC4-sgRNA-In-2 × Broccoli mediated MUC4 labeling. (B) Colocalization of MUC4-sgRNA-In-2 × Broccoli and smFISH for endogenous MUC4 mRNA in fixed HEK 293T cells. Upper row, negative control, the cells were transfected with psgRNA-In-2 × Broccoli (gNC: as a negative control of spacer sequence) and pdP-spCas13b-2 × NLS; (Middle row) pMUC4-sgRNA-In-2 × Broccoli (gMUC4: with spacer sequence targeting MUC4) and pdPspCas13b-2 × NLS was transfected; (Bottom row) MUC4 mRNA was depleted by siRNA-induced gene silencing, and the cells were transfected with pMUC4-sgRNA-In-2 × Broccoli (gMUC4) and pdPspCas13b-2 × NLS. The nucleus was dyed by Hoechst. (C) MUC4-sgRNA-In-2 × Broccoli RNA levels in the presence or absence of siRNA which silence MUC4 were measured by RT-qPCR. All data are presented as the mean ± s.d.; n = 3 dependent experiments. (D) Left, schematic of lncRNA NEAT1 labeling by NEAT1-sgRNA-In-2 × Broccoli. Right, representative images of NEAT1-sgRNA-In-2 × Broccoli labeling NEAT1 (green) colocalized with smFISH in HEK 293T cells. (E) Left, schematic of SatIII labeling by SatIII-sgRNA-In-2 × Broccoli under SA treatment. Right, representative images of SatIII (green) and HSF1-mRuby3 (red) upon SA (100 μM, 6 h) treatment. | ||
Visualization of NEAT1 in live cells and dynamic tracking SATIII
In recent years, long noncoding RNAs (lncRNAs) have gained widespread attention for their crucial roles in gene regulation during development and disease.27 For instance, nuclear paraspeckle assembly transcript 1 (NEAT1), a lncRNA that interacts with various RNAs, such as MALAT1,28 is frequently overexpressed in human tumors and recognized as a potential diagnostic biomarker and therapeutic target.29–32 Therefore, tracking NEAT1 can help us to visualize the dynamic interaction with RNA or protein to understand the biological mechanisms in live cells. We next asked whether our method can be used to image this kind of lncRNA with a complex structure and complicated interactome (Fig. 2D, left). By comparing the signal overlap among NEAT1-sgRNA-In-2 × Broccoli and smFISH (Fig. 2D, right), we determined that CasFAS was capable of labeling lncRNA NEAT1.Many RNA-protein assemblies will be formed in eukaryotic cells after exposure to stressful conditions. Long noncoding RNA Satellite III (SatIII), whose expression is induced upon thermal stress, is gathered after heat shock transcription factor 1 (HSF1)-dependent transcription to form nuclear stress bodies (nSBs), resulting in heat shock-induced HSF1 aggregation.33 Moreover, it has been reported that SatIII RNA also accumulates in nSBs after chemical stresses, such as sodium arsenite (SA) treatments.33 Imaging approaches are helpful in revealing the components and dynamics of ribonucleoprotein (RNP) granules, and are expected to shed light on the interpretation of their functions. To determine whether our system is suitable for observing RNA in nuclear stress bodies, we designed sgRNA based on previous findings to target SatIII lncRNA (Fig. 2E, left).34,35 With 1 min-interval time-lapse imaging of the SatIII-sgRNA-In-2 × Broccoli under SA treatments, we observed apparent aggregated signals compared with the untreated condition (Video S1†). This specific aggregation of SatIII was similar to the red fluorescence of HSF1-mRuby3 (Fig. 2E, right), indicating that SatIII and HSF1 form nuclear stress bodies. These results show that CasFAS is also competent in investigating lncRNAs and tracking the formation of RNP granules.
CasFAS systems was compatible with RNA aptamer Pepper
One of the advantages of the CasFAS system is that the fluorescent RNA aptamer can be easily changed by other kinds of RNA aptamer species through the convenient replacement of the sequences encoding RNA aptamer motifs. Next, we changed the fluorescent RNA aptamer part of CasFAS to another kind of RNA aptamer, Pepper, which has shown the advantages as more diminutive size, brighter and more stable fluorescence with a broad range of emission maxima spanning from cyan to red (Fig. 3A and S12†).22 Similar to sgRNA-Broccoli, no detectable fluorescence signal was observed in the absence of dPspCas13b (Fig. 3B, first row). With the GFP fluorophore-like synthetic dye HBC530, green fluorescence appeared after binding with dPspCas13b (Fig. 3B, second row). Benefitting from the small size of the aptamers and chemical fluorophores, it is possible to integrate multiplex RNA aptamers to obtain a higher fluorescence brightness and SNR. Thus, we designed a series of modified sgRNAs with 2 × Pepper, 4 × Pepper and 8 × Pepper aptamers and investigated their performances by an in vitro fluorescence assay. As the number of aptamers increased, the fluorescence signal gradually increased (Fig. S12–S15†). Due to the most vigorous fluorescence intensity, 8 × Pepper was chosen for use in the subsequent study for imaging in live cells. Compared with sgRNA-In-Pepper (Fig. 3B, second row), the sgRNA-In-8 × Pepper with more aptamers generated a noticeably stronger fluorescent signal, which is consistent with the results of the in vitro fluorescence assay, indicating its potential to investigate low-abundance RNA in living cells (Fig. 3B, third row). It is worth mentioning that both sgRNA-In-Pepper and sgRNA-In-8 × Pepper exhibited colocalization with dPspCas13b-RFP. Similar to the previous results of Broccoli, the abundance of sgRNA-In-8 × Pepper transcripts inside the cell was much higher than the levels of sgRNA-3′-F-8 × Pepper and sgRNA-3′-8 × Pepper in the presence of dPspCas13b, once again confirming the stability of sgRNA with an insertion aptamer in the loop region (Fig. S16†). The microscopy and FACS results of sgRNA-In-8 × Pepper illustrate that in addition to Broccoli, the CasFAS system was also compatible with other kinds of fluorescent RNA aptamers, such as Pepper (Fig. 3B and S17†).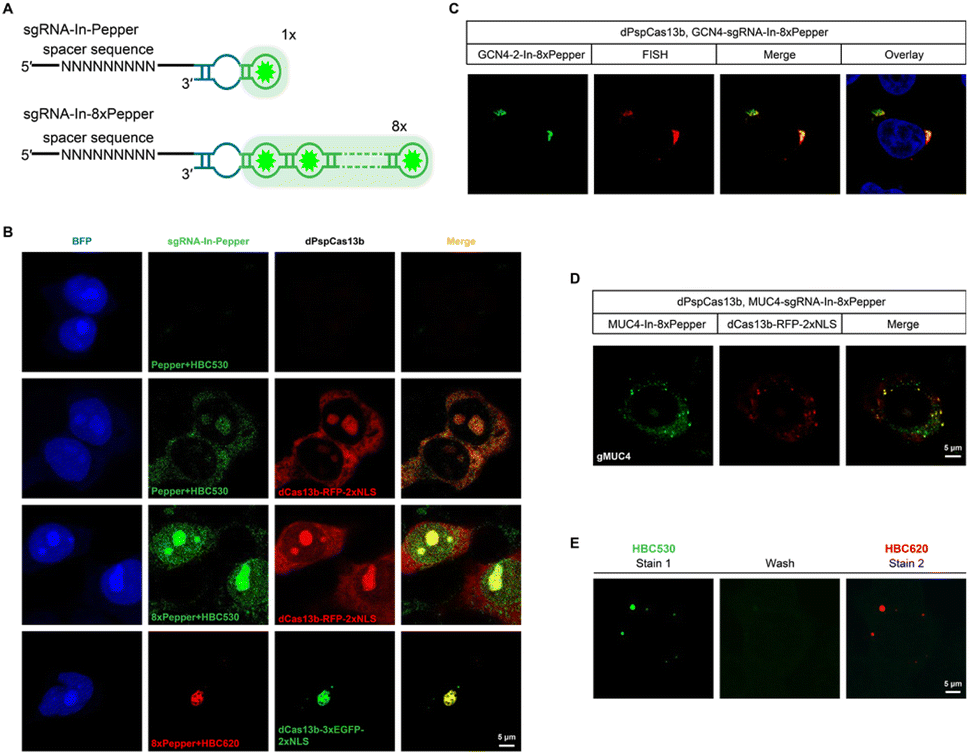 | ||
| Fig. 3 CasFAS system with the Pepper RNA aptamer. (A) Diagram of the strategy for multiplexed Pepper RNA aptamers tagged to sgRNA. (B) Visualization of sgRNA-In-Pepper expressed in live HEK 293T cells, which is reported by BFP (blue, first column). The first row, sgRNA-In-Pepper (HBC530, green) in the absence of dPspCas13b-RFP (red) transfection was used as an internal control. The second and third rows, sgRNA-In-Pepper and sgRNA-In-8 × Pepper in cells expressing dPspCas13b-RFP were characterized for direct comparison. The fourth row, localization of sgRNA-In-8 × Pepper (HBC620, red) and dPspCas13b-3 × EGFP (green). (C) GCN4 repeats visualized by GCN4-sgRNA-In-8 × Pepper with sgRNAs and smFISH-labeled signals in live HEK 293T cells. The nucleus was dyed by Hoechst. (D) Representative images are illustrating MUC4 mRNAs labeled by MUC4-sgRNA-In-8 × Pepper with sgRNAs, colocalized with RFP signal tagging dPspCas13b, in HEK 293T cells. (E) Color switching of β-actin-sgRNA-In-8 × Pepper using different fluorophores in live HEK 293T cells. Cell expressing β-actin-sgRNA-In-8 × Pepper and dPspCas13b was initially labeled with 1 μM HBC530 and imaged. The cells were then washed twice with a fresh medium to remove HBC530 fluorophores followed by staining with 1 μM HBC620 and the same cell was imaged. Two independent experiments were carried out with similar results. | ||
The color of CasFAS systems can be switched by dye replacement
Next, we aimed to determine the RNA-targeting ability of the CasFAS system with the Pepper aptamer in living cells. Both exogenous GCN4 and endogenous MUC4 mRNA were successfully labeled and imaged by the sgRNA-Pepper scaffold and were accompanied by distinct fluorescent signals. Their imaging accuracy was separately confirmed by colocalization with smFISH probes, suggesting competent RNA imaging capabilities (Fig. 3C and D). The most attractive merit of Pepper is that a broad spectral range from cyan to red of HBC analogues was built to generate a palette of fluorescence using only one kind of aptamer. Then, we wanted to ask whether the fluorescent color of CasFAS can be switched by changing only the fluorogenic dyes. For example, the green fluorescence reduced by HBC530 can be converted to red when replaced with chromophore HBC620. As expected, the red fluorescence of sgRNA-Pepper can be observed accordingly, which could also colocalize with the green fluorescent signal by dPspCas13b-GFP (Fig. 3B, fourth row). Benefitting from sharing the identical aptamer sequences of HBC530 and HBC620, we next asked whether a simple fluorescence switch can be achieved by replacing only the fluorescent reagent through washing and restaining processes in a living cell. To our satisfaction, the CasFAS system with sgRNA-In-8 × Pepper permits easy conversion of the fluorescence emission from a green signal to red by only changing HBC530 to HBC620 in the culture medium (Fig. S18†), which further realized β-actin imaging with the color changing (Fig. 3E). Generally, the CasFAS platform enables easier color switching in living cells, which cannot be realized by other CRISPR-based methods. Compared to fluorescent protein-based CRISPR imaging, the flexibility of CasFAS in RNA labeling and fluorescent conversion may possess more excellent potential applications for creating stable cell lines or transgenic animals.Multicolor RNA labeling using CasFAS system
The CRISPRainbow system, which attaches the RNA hairpin (MS2, PP7 and boxB) at the 3′-end of sgRNA to recruit hairpin binding domain fused fluorescent proteins, can realize multicolor labeling of chromosomal loci simultaneously.6 We next asked whether the CasFAS system can produce a “rainbow” to achieve multicolor RNA imaging by combining different kinds of fluorescent RNA aptamers. We designed two ways to perform multicolor endogenous RNA imaging. One is to assemble two RNA aptamers into one sgRNA, such as sgRNA-In-2 × Broccoli-2 × Pepper (Fig. 4A and S19†). The other is to use two kinds of modified sgRNAs simultaneously, such as the employment of sgRNA-In-2 × Broccoli and sgRNA-In-8 × Pepper together (Fig. 4B). The results showed that these approaches could visualize the MUC4 mRNA in different colors, resulting in the expected dual-color RNA imaging platform (Fig. 4A and B). Compared with the previous CRISPRainbow system using a pair of RNA hairpin and hairpin-binding domain-fused fluorescent proteins, the smaller size and flexibility of fluorescent aptamers endow the CasFAS system with superiority in achieving multicolor imaging in an easier way.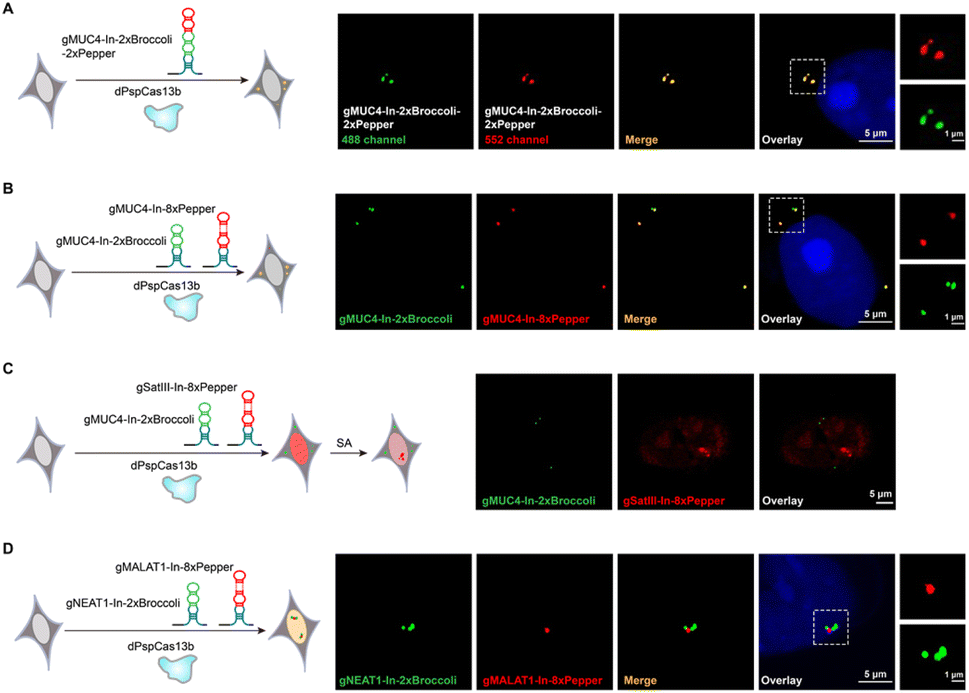 | ||
| Fig. 4 Localization and live tracking of multiple RNAs simultaneously. (A) Left, overview of dual-color labeling of MUC4. Right panels, representative images showing unique sites in MUC4 labeled simultaneously using MUC4-sgRNA-In-2 × Broccoli-2 × Pepper (green/red). (B) Overview and representative images of dual-color labeling of MUC4 by MUC4-sgRNA-In-2 × Broccoli (green) and MUC4-sgRNA-In-8 × Pepper (red) in single cells. (C) Left, an overview of dual-color labeling of MUC4 and SatIII. Right panel, pSatIII-sgRNA-In-8 × Pepper and pMUC4-sgRNA-In-2 × Broccoli were cotransfected into HEK 293T cells. Each color was dedicated to one RNA locus: green for MUC4 and red for SatIII. (D) Tracking the RNA interaction by cotransfecting pNEAT1-In-2 × Broccoli (green) and pMALAT1-In-8 × Pepper (red) in HEK 293T cells. All experiments were incubated with 10 μM DFHBI-1T and 1 μM HBC 620. Two in-dependent experiments were carried out with similar results. | ||
To simultaneously image different RNAs in the same cell, the conventional method deploys different dCas protein organoids, each fusing or recruiting with specific fluorescent proteins.15,16,36 However, two main concerns limit its application. One concern is the lack of enough dCas orthologues with sufficient efficiency for nucleic acid labeling and imaging. A panel of eight dCas13 homologues was recently screened to image and track RNA, but only two were competent.19 Second, each dCas protein should pair with its specific sgRNA, resulting in too many plasmids to burden the transfection process. However, in the CasFAS system, only one kind of dCas protein is needed. For different targets, multiplex sgRNAs with varying guide sequences paired with various fluorescent RNA aptamers can achieve simultaneous additional RNA imaging without the above two concerns. Thus, we performed multicolor RNA labeling by pooling NEAT1-sgRNA-In-2 × Broccoli-2 × Pepper, β-actin-sgRNA-In-8 × Pepper and MUC4-sgRNA-In-2 × Broccoli together in HEK 293T cells. As shown in Fig S20†, we observed three colors in an individual cell (NEAT1, orange; MUC4, green; β-actin, red), indicating that these three systems are sufficiently orthogonal to discriminate between different RNAs. Moreover, the CasFAS system can also orthogonally track the dynamics of RNA in live cells. For this purpose, we performed MUC4 labeling with MUC4-sgRNA-In-2 × Broccoli and SatIII labeling with SatIII-sgRNA-In-8 × Pepper. In the untreated control cells, both fluorescent signals of MUC4 and SatIII were observed with different colors and locations, while no aggregation was observed for SatIII. After treatment with SA, we succeeded in tracking the formation of nuclear bodies by aggregating SatIII with no effect on the fluorescent signal of MUC4 (Fig. 4C). In general, sgRNAs with fluorescent RNA aptamers for different RNA targets exhibited sufficient specificity to recognize their own target, showing the capability to simultaneously discriminate different RNAs in the same cell. It is worth emphasizing that, unlike previous studies that need multiple dCas proteins and their unique sgRNAs, CasFAS is more straightforward, requiring only one kind of dCas protein, and can achieve multicolor imaging and dynamic tracking with excellent orthogonality.
Intracellular RNA–RNA interaction imaging using CasFAS system
Interactions between RNA molecules play crucial roles in many fundamental cellular activities.27 However, technologies for visualizing intracellular RNA–RNA interactions (RRIs) are lacking.Imaging RNA–RNA interactions in the cellular space is attractive for helping us to understand and verify these interactions more directly. Previous findings have discovered an interaction between the lncRNA NEAT1 and MALAT1, which was further confirmed by smFISH.36 Therefore, we wondered whether our CasFAS system can also observe this RNA–RNA interaction in live cells. For this purpose, we next constructed an RNA–RNA interaction tracking strategy simultaneously using MALAT1-sgRNA-In-8 × Pepper and NEAT1-sgRNA-In-2 × Broccoli. As shown in the microscopic image, NEAT1-sgRNA-In-2 × Broccoli, which was designed to target the 5′ regions of NEAT1, presented apparent green fluorescence that distinctly colocalized with the red signals of MALAT1-sgRNA-In-8 × Pepper (Fig. 4D). The result is consistent with previous reports that MALAT1 can bind the 5′ region of NEAT1, indicating the ability of CasFAS to monitor intracellular RNA–RNA interactions.36
Visualization of severe fever with thrombocytopenia syndrome virus (SFTSV) in 293T cells with CasFAS
Severe fever with thrombocytopenia syndrome virus (SFTSV), an emerging tick-borne virus, can cause acute febrile illness in humans with thrombocytopenia and hemorrhagic complications.37,38 Due to a high mortality rate and no effective therapy or vaccine is yet available for clinical use, it is necessary to expand the toolbox for understanding the virus.39,40 Previously, we performed transcriptomic and epigenetic profile studies on SFTSV-infected clinic samples.41 It is also significant to visualize the mRNA of SFTSV distribution, which may facilitate the understanding pathogenic mechanism of this virus.42 To generate a system that efficiently and easily targets SFTSV, we developed a single vector that expresses multiple sgRNA-In-2 × Broccoli and designed the strategy based on Golden Gate assembly (Fig. 5A and Table. S1†). As a proof of principle, we cloned three crRNAs targeting SFTSV into the vector. To test our system, 293T cells were infected with SFTSV 48 h post-infection (p.i.) with an MOI of 1, as well as uninfected control cells. To confirm the SFTSV-infection, we performed real-time PCR analysis of the SFTSV-NP mRNA (Fig. 5B). This revealed a high level of SFTSV-NP mRNA in SFTSV-infected 293T cells (SFTSV) compared with the control cells (MOCK), indicating the successful infection. As shown in Fig. 5C (top row and middle), in uninfected and gNC samples, cells displayed only weak and diffuse fluorescent signal. By contrast, in infected cells with SFTSV-sgRNA-In-2 × Broccoli, a large proportion of cells showed very strong and localized signal. These data show that CasFAS can simultaneously visualize SFTSV RNA in cells. We provide a new technique for research on SFTSV biology, which may potentially be used in studying virus–host interactions in live cells for diagnostics and drug screening.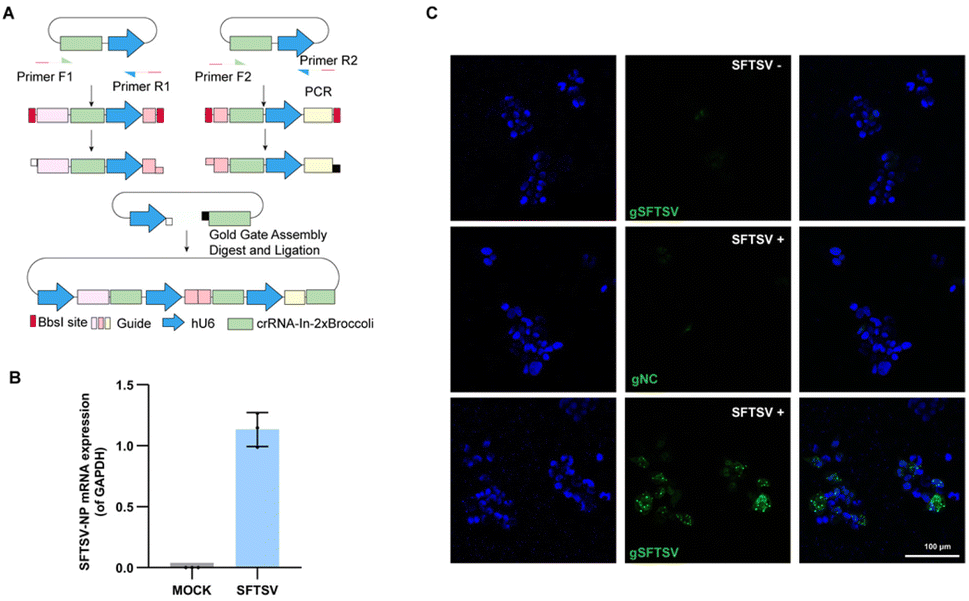 | ||
| Fig. 5 Visualizing thrombocytopenia syndrome virus (SFTSV) RNA with CasFAS. (A) Schematic design of one-step method to generate a multiple targeting SFTSV system. Dual human U6 promoter and crRNA-In-2 × Broccoli constructs were synthesized and then cloned into PC0043. PCR amplified template of crRNA-In-2 × Broccoli and U6 promoter was indicated in the box. (Primer sequences are shown in Table. S1†). (B) RT-PCR analysis for SFTSV-NP mRNA expression level of SFTSV-infected 293T cells and uninfected 293T cells (MOCK). (The SFTSV-NP mRNA expression level was normalized by GAPDH). (n = 3 independent experiments, data are represented as mean values ± SD, black dots represent individual data points). (C) Images of uninfected (top row) and infected 293T cells (middle and bottom row). Nuclei are labeled with Hoechst (blue). Top row, uninfected control cells with gSFTSV-In-2 × Broccoli (gSFTSV: with spacer sequences targeting SFTSV-NP mRNA). Middle row, psgRNA-In-2 × Broccoli (gNC: as negative control of spacer sequences) was transfected into infected cells. Bottom row, pgSFTSV-In-2 × Broccoli (gSFTSV) was transfected in infected cells. 293T cells infected with SFTSV (MOI 1.0, 48 h p.i.). | ||
Conclusions
Over the years, technical efforts in RNA-seq have revolutionized our understanding of RNA, which accompanies a remarkable variety of biological functions. However, tools for RNA visualization in living cells lag behind the methods for RNA profiling. Recently, CRISPR systems have become widely popular for their use in intracellular RNA imaging and applications in the genome or transcriptome editing. The nuclease-dead versions of Cas9 and Cas13 can target RNA and efficiently light up the RNAs of interest while fusing with the fluorescent proteins, although PAM sequences are needed for dCas9. The strategy of CRISPR-fluorescent protein conjugates has demonstrated their power for RNA imaging and dynamic tracking in live cells but still has sufficient space for improvement. The significant fluorescence background induced by unbound fluorescent proteins requires multiplex fusions to achieve an optimal SNR. In addition, multiplex RNA imaging needs many more orthogonal CRISPR-Cas orthologues, which likely await further discovery and screening.In addition to the Cas protein, the sgRNA components have also been shown to be the ideal object for modifications by various functional groups, endowing other talents besides their targeting capability, such as for imaging purposes by the attachment of fluorescent RNA aptamers. Previous studies established that sgRNA is extremely unstable in the absence of dCas9. It is reasonable that the unbound fluorescent aptamer-tagged sgRNA will be degraded rapidly to reduce the fluorescence background. The progress of fluorescent RNA aptamers with increased brightness and photostability encouraged us to couple the RNA-targeting capability of the CRISPR-dCas13b system, generating a CasFAS system to image RNA in living cells. We demonstrate that CasFAS is capable of labeling and tracking endogenous untagged RNA transcripts with high efficiency and accuracy. In addition, the success visualization for SFTSV mRNA indicated CasFAS can further be used to provide more information about this virus, and even help us better understand, diagnose and fight this virus.
Due to the small size of fluorescent dyes, CasFAS has the advantage of assembling multiple fluorescent units together. For instance, to achieve better RNA imaging results, the traditional method is to fuse or recruit multiple fluorescent components such as fluorescent proteins to enhance the fluorescence intensity. Some concerns have been raised that large fluorescent proteins with hundreds of amino acids may have unpredictable effects.34 It is easier to integrate multiple RNA aptamers without the need for linker sequences between two aptamer units. Theoretically, the whole size of multiple assembled RNA aptamers may be further reduced by optimizing the length of the stem in the junction. Our results showed that increasing the number of aptamers can improve the fluorescent signals without affecting their colocalization ability (Fig. 3B).
Furthermore, the continuous increase in the number of fluorescent light-up aptamers endows the CasFAS system with greater flexibility for multicolor or multiple RNA imaging simultaneously. It is worth emphasizing that just one kind of dCas13b protein can pair with various modified sgRNAs with different guide sequences and attached fluorescent aptamers (Fig. 4A–C). This advantage reduces the impact of the lack of enough CRISPR-Cas homologues. In addition, chemical fluorescent dyes can be modified with various groups with different electronic properties, which will change their fluorescence emission wavelength. Thus, one kind of fluorescent aptamer, such as Pepper, can bind a broad spectral range of fluorescent dyes to emit fluorescence from cyan to red.20 Benefitting from the feature that the same aptamer can couple with different substrates, we realized a simple color-switch operation in live cells through washing and restaining procedures (Fig. 3E and S18†), indicating the versatility of the CasFAS system that may further improve the accuracy of fluorescent signals.
The CasFAS system is feasible for many RNA imaging applications, such as RNA aggregation in stress granules and visualization of RNA–RNA interactions and virus RNA (Fig. 4D, 5C and Video S1†). Even during dynamic processes, such as stress granule formation, the total orthogonality between Broccoli and Pepper enables them to specifically target their RNA target with high efficiency (Fig. 4C). CasFAS has been suggested as an ideal RNA labeling and imaging tool in live cells based on its simplicity, dynamic range, sensitivity, and flexibility. We anticipate that with the development of fluorescent RNA aptamers, the utility and applicability of RNA visualization by CasFAS can be further enhanced to understand the cellular RNA dynamics and function.
Author contributions
X. Z., X. W. and H. T. conceived this study. X. Z., X. W. and H. T. designed the experiments. H. T. designed the modified sgRNAs scaffold. H. T. and J. P. performed all experiments and data analysis. X. C. helped with preparing the plasmids. Q. W. and S. P. helped with living-cell image analysis. Y. T. and Song-Mei Liu provided materials and helped for viral (SFTSV) RNA imaging. Quanjiang Ji helped with the three-dimensional simulation assay of the dPspCas13b and sgRNA complex. X. Z., X. W. and H. T. wrote the manuscript with help from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21822704, 91940304, 21778040 and 22177087 to X. W.; 91753201, 21721005, and 91940000 to X. Z.).Notes and references
- A. M. Femino, F. S. Fay, K. Fogarty and R. H. Singer, Science, 1998, 280, 585–590 CrossRef CAS PubMed.
- D. R. Larson, D. Zenklusen, B. Wu, J. A. Chao and R. H. Singer, Science, 2011, 332, 475–478 CrossRef CAS PubMed.
- B. Wiedenheft, S. H. Sternberg and J. A. Doudna, Nature, 2012, 482, 331–338 CrossRef CAS PubMed.
- H. Ma, L. C. Tu, A. Naseri, M. Huisman, S. Zhang, D. Grunwald and T. Pederson, J. Cell Biol., 2016, 214, 529–537 CrossRef CAS PubMed.
- H. Ma, L. C. Tu, A. Naseri, Y. C. Chung, D. Grunwald, S. Zhang and T. Pederson, Nat. Methods, 2018, 15, 928–931 CrossRef CAS PubMed.
- H. Ma, L. C. Tu, A. Naseri, M. Huisman, S. Zhang, D. Grunwald and T. Pederson, Nat. Biotechnol., 2016, 34, 528–530 CrossRef CAS PubMed.
- D. A. Nelles, M. Y. Fang, M. R. O'Connell, J. L. Xu, S. J. Markmiller, J. A. Doudna and G. W. Yeo, Cell, 2016, 165, 488–496 CrossRef CAS PubMed.
- B. Chen, S. Deng, T. Ge, M. Ye, J. Yu, S. Lin, W. Ma and W. Z. Song, Protein Cell, 2020, 11, 641–660 CrossRef CAS PubMed.
- Y. Liu, S. Li, L. Zhang, Q. Zhao, N. Li and Y. Wu, RSC Adv., 2020, 10, 28037–28040 RSC.
- M. Wang, K. Chen, Q. Wu, R. Peng, R. Zhang and J. R. Li, Anal. Chem., 2020, 92, 2468–2475 CrossRef CAS PubMed.
- S. Chen, R. Wang, S. Peng, S. Xie, C. Lei, Y. Huang and Z. Nie, Chem. Sci., 2022, 13, 2011–2020 RSC.
- O. O. Abudayyeh, J. S. Gootenberg, P. Essletzbichler, S. Han, J. Joung, J. J. Belanto, V. Verdine, D. B. T. Cox, M. J. Kellner, A. Regev, E. S. Lander, D. F. Voytas, A. Y. Ting and F. Zhang, Nature, 2017, 550, 280–284 CrossRef PubMed.
- D. B. T. Cox, J. S. Gootenberg, O. O. Abudayyeh, B. Franklin, M. J. Kellner, J. Joung and F. Zhang, Science, 2017, 358, 1019–1027 CrossRef CAS PubMed.
- S. Konermann, P. Lotfy, N. J. Brideau, J. Oki, M. N. Shokhirev and P. D. Hsu, Cell, 2018, 173, 665–676 CrossRef CAS PubMed.
- L. Yang, Y. Wang, S. Q. Li, R. W. Yao, P. F. Luan, H. Wu, G. G. Carmichael and L. L. Chen, Mol. Cell, 2019, 76, 981–997 CrossRef CAS PubMed.
- H. F. Wang, M. Nakamura, T. R. Abbott, D. H. Zhao, K. W. Luo, C. Yu, C. M. Nguyen, A. Lo, T. P. Daley, M. La Russa, Y. X. Liu and L. S. Qi, Science, 2019, 365, 1301–1305 CrossRef CAS PubMed.
- L. Z. Yang, B. Q. Gao, Y. Huang, Y. Wang, L. Yang and L. L. Chen, Cell Insight, 2022, 1, 100044 CrossRef.
- E. Braselmann, A. J. Wierzba, J. T. Polaski, M. Chrominski, Z. E. Holmes, S. T. Hung, D. Batan, J. R. Wheeler, R. Parker, R. Jimenez, D. Gryko, R. T. Batey and A. E. Palmer, Nat. Chem. Biol., 2018, 14, 964–971 CrossRef CAS PubMed.
- J. S. Paige, K. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed.
- W. Song, R. L. Strack, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 1198–1201 CrossRef CAS PubMed.
- X. Chen, D. Zhang, N. Su, B. Bao, X. Xie, F. Zuo, L. Yang, H. Wang, L. Jiang, Q. Lin, M. Fang, N. Li, X. Hua, Z. Chen, C. Bao, J. Xu, W. Du, L. Zhang, Y. Zhao, L. Zhu, J. Loscalzo and Y. Yang, Nat. Biotechnol., 2019, 37, 1287–1293 CrossRef CAS PubMed.
- M. E. Tanenbaum, L. A. Gilbert, L. S. Qi, J. S. Weissman and R. D. Vale, Cell, 2014, 159, 635–646 CrossRef CAS PubMed.
- M. A. Hollingsworth and B. J. Swanson, Nat. Rev. Cancer, 2004, 4, 45–60 CrossRef CAS PubMed.
- B. Chen, L. A. Gilbert, B. A. Cimini, J. Schnitzbauer, W. Zhang, G. W. Li, J. Park, E. H. Blackburn, J. S. Weissman, L. S. Qi and B. Huang, Cell, 2013, 155, 1479–1491 CrossRef CAS PubMed.
- S. Nollet, N. Moniaux, J. Maury, D. Petitprez, P. Degand, A. LAine, N. Porchet and J. P. Aubert, Biochem. J., 1998, 332, 739–748 CrossRef CAS PubMed.
- C. Adriaens, L. Standaert, J. Barra, M. Latil, A. Verfaillie, P. Kalev, B. Boeckx, P. W. Wijnhoven, E. Radaelli, W. Vermi, E. Leucci, G. Lapouge, B. Beck, S. Nakagawa, T. Hirose, A. A. Sablina, D. Lambrechts, S. Aerts, C. Blanpain and J. C. Marine, Nat. Med., 2016, 22, 861–868 CrossRef CAS PubMed.
- J. A. West, C. P. Davis, H. Sunwoo, M. D. Simon, R. I. Sadreyev, P. I. Wang, M. Y. Tolstorukov and R. E. Kingston, Mol. Cell, 2014, 55, 791–802 CrossRef CAS PubMed.
- L. Chen and G. G. Carmichael, Mol. Cell, 2009, 35, 467–478 CrossRef CAS PubMed.
- T. Naganuma, S. Nakagawa, A. Tanigawa, Y. F. Sasaki, N. Goshima and T. Hirose, EMBO J., 2012, 31, 4020–4034 CrossRef CAS PubMed.
- J. A. West, M. Mito, S. Kurosaka, T. Takumi, C. Tanegashima, T. Chujo, K. Yanaka, R. E. Kingston, T. Hirose and C. Bond, J. Cell Biol., 2016, 214, 817–830 CrossRef CAS PubMed.
- C. M. Clemson, J. N. Hutchinson, S. A. Sara, A. W. Ensminger, A. H. Fox, A. Chess and J. B. Lawrence, Mol. Cell, 2009, 33, 717–726 CrossRef CAS PubMed.
- R. Valgardsdottir, I. Chiodi, M. Giordano, A. Rossi, S. Bazzini, C. Ghigna, S. Riva and G. Biamonti, Nucleic Acids Res., 2008, 36, 423–434 CrossRef CAS PubMed.
- C. Jolly, L. Konecny, D. L. Grady, Y. A. Kutskova, J. J. Cotto, R. I. Morimoto and C. Vourc'h, J. Cell Biol., 2002, 156, 775–781 CrossRef CAS PubMed.
- D. Mateju, B. Eichenberger, F. Voigt, J. Eglinger, G. Roth and J. A. Chao, Cell, 2020, 183, 1801–1812 CrossRef CAS PubMed.
- Z. Cai, C. C. Cao, L. Ji, R. Ye, D. Wang, C. Xia, S. Wang, Z. C. Du, N. J. Hu, X. H. Yu, J. Chen, L. Wang, X. G. Yang, S. M. He and Y. C. Xue, Nature, 2020, 582, 432–437 CrossRef CAS PubMed.
- B. Xu, L. Liu, X. Huang, H. Ma, Y. Zhang, Y. Du, P. Wang, X. Tang, H. Wang, K. Kang, S. Zhang, G. Zhao, W. Wu, Y. Yang, H. Chen, F. Mu and W. Chen, PLoS Pathog., 2011, 7, e1002369 CrossRef CAS PubMed.
- S. J. Park, Y. I. Kim, A. Park, H. I. Kwon, E. H. Kim, Y. J. Si, M. S. Song, C. H. Lee, K. Jung, W. J. Shin, J. Zeng, Y. Choi, J. U. Jung and Y. K. Choi, Nat. Microbiol., 2018, 4, 438–446 CrossRef PubMed.
- M. Honda, D. Ninomiya, T. Sakai, T. Senba and S. Kaneyuki, J. Infect. Dis., 2014, 209, 816–827 CrossRef PubMed.
- L. K. McMullan, S. M. Folk, A. J. Kelly, A. MacNeil, C. S. Goldsmith, M. G. Metcalfe, B. C. Batten, C. G. Albariño, S. R. Zaki, P. E. Rollin, W. L. Nicholson and S. T. Nichol, N. Engl. J. Med., 2012, 367, 834–841 CrossRef CAS PubMed.
- Y. Wang, S. Han, R. Ran, A. Li, H. Liu, M. Liu, Y. Duan, X. Zhang, Z. Zhao, S. Song, X. Weng, S. M. Liu and X. Zhou, Nat. commun., 2021, 12, 5629 CrossRef CAS PubMed.
- J. Zhang, H. Li, B. Lin, X. Luo, P. Yin, T. Yi, B. Xue, X. L. Zhang, H. Zhu and Z. Nie, J. Am. Chem. Soc., 2021, 143, 19317–19329 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04656c |
| This journal is © The Royal Society of Chemistry 2022 |