
Constructing anisotropic conical graphene aerogels with concentric annular structures for highly thermally conductive phase change composites towards efficient solar–thermal–electric energy conversion†
Hao-Yu
Zhao
ab,
Chao
Shu
a,
Peng
Min
b,
Changjun
Li
a,
Wenchao
Deng
b,
Jing
Yang
c,
Xiaofeng
Li
 *a and
Zhong-Zhen
Yu
*b
*a and
Zhong-Zhen
Yu
*b
aBeijing Key Laboratory of Advanced Functional Polymer Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: xfli@mail.buct.edu.cn
bState Key Laboratory of Organic-Inorganic Composites, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: yuzz@mail.buct.edu.cn
cCollege of Chemistry and Chemical Engineering, Hunan Normal University, Changsha 410081, China
First published on 5th October 2022
Abstract
Although organic phase change materials can reversibly store and release latent heat during their phase change processes, their weak solar–thermal conversion ability, low thermal conduction, and poor structural stability seriously hinder their applications for solar–thermal energy conversion as well as thermal energy utilization. Herein, anisotropic high-quality conical graphene aerogels (HCGAs) with concentric annular structures are constructed to enhance thermal conduction, solar–thermal energy conversion, and shape stability of phase change materials for solar–thermal–electric energy conversion applications. By regulating the orientation of graphene oxide liquid crystals, the resultant graphene sheets are aligned from the apex to the bottom of the cone, providing efficient heat transfer along the vertical direction of the phase change composites. An optimal HCGA/tetradecanol phase change composite with 7.05 wt% of graphene achieves a high through-plane thermal conductivity of 4.54 W m−1 K−1 with a high latent heat of 206.1 J g−1. Benefiting from the larger solar light-absorption surface than conventional square/cylindrical phase change composites and the rapid heat transfer of the anisotropic high-quality graphene conduction network, the conical phase change composite achieves a high solar–thermal energy conversion and storage efficiency of 84.0%. Furthermore, a solar–thermal–electric generator is assembled with the conical phase change composite array, exhibiting maximum output voltages of 261 and 1214 mV under solar light intensities of 100 and 500 mW cm−2, respectively. Even after the removal of the solar light, the voltage output can still be continued by releasing the stored thermal energy.
Introduction
The continual consumption of fossil resources results in increasingly serious issues of resource shortage and environmental pollution, which can be alleviated by utilizing renewable and clean solar energy.1 On the basis of solar–thermal energy conversion, the solar energy can be converted to thermal energy and then stored in organic phase change materials, including tetradecanol,2 octadecanol,3 paraffin wax,4–7 and polyethylene glycols.8–11 Although phase change materials can store and release large amounts of latent heat by melting and solidifying,12–15 conventional phase change materials have several drawbacks: (i) weak solar light absorption, and no solar–thermal energy conversion ability; (ii) low inherent thermal conductivity, which adversely affects the energy storage and release rates; and (iii) poor structural stability, which cannot guarantee the long-term reliability during repeated cycles.16–19 These drawbacks seriously compromise the applications of phase change materials for solar energy conversion and storage.20The drawbacks of the organic phase change materials can be solved by compounding with three-dimensional (3D) graphene aerogels as highly thermally conductive skeletons.21–24 The continuous thermal conduction pathways of the graphene aerogels can significantly enhance the thermal conductivity of phase change materials,25 thereby improving the thermal energy storage and release rates. Moreover, the black graphene sheets exhibit high solar light absorption and satisfactory solar–thermal energy conversion efficiency.26,27 In addition, the hydrophobic and porous graphene skeleton can adequately accommodate phase change materials and suppress their leakage, improving shape stability of the phase change materials.28–30
The two-dimensional graphene sheets with high thermal and electrical conductivities31–34 can be aligned anisotropically to form anisotropic graphene aerogels for enhancing heat transfer along one specific direction of the phase change composites.35–39 As an example, Min et al. fabricated an anisotropic aerogel of high-quality graphene by unidirectionally freezing an aqueous suspension of polyamic acid salt and graphene oxide (GO) sheets to form vertically aligned monolith, followed by freeze-drying, imidization of polyamic acid salt at 300 °C, and graphitization at 2800 °C. The fabricated graphene aerogel/paraffin wax phase change composite exhibits a highly anisotropic thermal conductivity of 8.87 W m−1 K−1 with an exceptional latent heat retention of ∼98.7%.14 An et al. reported a vertically aligned graphene hybrid foam with a concentric circular structure by hydrothermal reduction of GO in the presence of high-quality graphene nanoplatelets (GNPs) followed by air-drying and graphitization, and its epoxy composite possesses an ultrahigh through-plane thermal conductivity of 35.5 W m−1 K−1.40 Apparently, anisotropic graphene aerogels are well suitable for increasing thermal conductivities of phase change composites along the vertical direction, allowing phase change composites to have higher energy storage and release rates for solar–thermal energy conversion and storage, and enabling the phase change composites to be more homogeneous in temperature and phase state during the solar–thermal energy conversion and storage.
Solar–thermal conversion phase change composites can be assembled with temperature-difference power-generation-sheets to form solar–thermal–electric (STE) generators for realizing solar–thermal–electric energy conversion and enhancing the solar energy utilization.41–47 Yang et al. assembled a GNP/boron nitride/polyethylene glycol phase change composite with a power-generation-sheet to obtain a STE generator with a maximum output voltage of ∼123 mV under a simulated solar light irradiation of 400 mW cm−2.48 Cao et al. used GO-enhanced phase change composites for solar–thermal energy conversion, and the maximum output voltage of the STE generator reached 144 mV under a solar light intensity of 200 mW cm−2.49 Liu et al. applied the convex lens to a STE generator, and the maximum power density could be up to 40.28 W m−2 under real sunlight irradiation.15 Currently, considerable efforts are devoted to improving the output voltage and power density of STE generators.
When graphene aerogel-based phase change composites are used for solar–thermal–electric conversion, their cylindrical/square shapes have obvious drawbacks. The lower temperature of the bottom of a phase change composite than that of its top surface due to the top-down input of the thermal energy, the heat dissipation by the large side area of the phase change composite, and the heat loss during the heat transfer50 would unfavorably decrease the temperature difference between the hot and cold source sides of the power-generation-sheets, causing low output voltages.51
To avoid the drawbacks of conventional cylindrical/square-shaped phase change composites, we design high-quality conical graphene aerogels (HCGAs) with concentric annular structures by controlling the orientation of GO liquid crystals during the hydrothermal self-assembly of aqueous GO/GNP suspensions followed by freeze-drying and graphitizing at 2800 °C. Subsequently, the thermally conductive HCGA/tetradecanol phase change composites are fabricated by compounding HCGAs with molten tetradecanol using a vacuum-assisted impregnation approach. At an initial GO/GNP mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, the HCGA/tetradecanol phase change composite exhibits a high thermal conductivity of 4.54 W m−1 K−1 at the graphene content of 7.05 wt%. Benefiting from the low graphene loading, the phase change composite achieves a high phase change enthalpy of 206.1 J g−1 and a high latent heat retention rate of 94.5%, indicating its excellent energy storage performance. Because the entire conical surface can be used for solar light absorption and solar–thermal energy conversion, the conical phase change composite has an excellent solar–thermal conversion performance with a solar–thermal conversion and storage efficiency of 84.0%. By assembling the 3 × 3 array of the conical phase change composites with a thermoelectric device, the resultant STE generator under the circumstance of dissipating heat in the air exhibits maximum output voltages of ∼261 and ∼1214 mV under solar light intensities of 100 and 500 mW cm−2, respectively. Even after the removal of the solar light, the voltage output can be continued by releasing the stored thermal energy. Different heat dissipation environments are adopted to enhance the performances of the STE generator, and the potential applications of the solar–thermal–electric energy conversion device are explored.
:2, the HCGA/tetradecanol phase change composite exhibits a high thermal conductivity of 4.54 W m−1 K−1 at the graphene content of 7.05 wt%. Benefiting from the low graphene loading, the phase change composite achieves a high phase change enthalpy of 206.1 J g−1 and a high latent heat retention rate of 94.5%, indicating its excellent energy storage performance. Because the entire conical surface can be used for solar light absorption and solar–thermal energy conversion, the conical phase change composite has an excellent solar–thermal conversion performance with a solar–thermal conversion and storage efficiency of 84.0%. By assembling the 3 × 3 array of the conical phase change composites with a thermoelectric device, the resultant STE generator under the circumstance of dissipating heat in the air exhibits maximum output voltages of ∼261 and ∼1214 mV under solar light intensities of 100 and 500 mW cm−2, respectively. Even after the removal of the solar light, the voltage output can be continued by releasing the stored thermal energy. Different heat dissipation environments are adopted to enhance the performances of the STE generator, and the potential applications of the solar–thermal–electric energy conversion device are explored.
Experimental
Materials
Pristine natural graphite (100 mesh) and GNPs (99.5%) were supplied by Huatai Lubricant & Sealing (China) and Shanghai Chaoqian New Materials Technology, respectively. Sulfuric acid (98%), potassium permanganate (99.5%), hydrochloric acid (37%), and hydrogen peroxide (30%) were bought from Beijing Chemical Reagents (China). Sodium nitrate, polyvinyl pyrrolidone (PVP, K16-18), potassium hydroxide (KOH, 99.5%), and 1-tetradecanol were purchased from Aladdin, Aladdin Industrial, Tianjin Fuchen Chemical Reagents, and Shanghai Macklin Biochemicals, respectively.Preparation of conical graphene hydrogels and conical high-quality graphene aerogels
Graphite oxide was synthesized from graphite flakes with a modified Hummers' method.52 12.5 mL of an aqueous suspension of GO (10 mg mL−1) was obtained by ultrasonication of an aqueous suspension of graphite oxide for 10 min. In addition, 0.02 g of PVP was dissolved in 10 mL of deionized water, and the resultant PVP solution was mixed with GNPs by ultrasonication for 30 min. After the two aqueous suspensions of GO and GNPs were mixed, 2.5 mL of the 1.32 M KOH solution was added. The resultant mixture was homogenized for 30 min, and poured into an autoclave with a conical polytetrafluoroethylene negative mold for hydrothermal treatment at 160 °C for 3 h, generating conical reduced graphene oxide (RGO)/GNP hydrogels. The resulting hydrogels were dialyzed in deionized water for 3 days to remove residual KOH, and freeze-dried under vacuum at −60 °C for 3 days to obtain conical graphene aerogels. The conical graphene aerogel with an initial GO/GNP mass ratio of 1:2 was designated as CGA2. To improve the quality, the conical graphene aerogels were thermally annealed at 2800 °C under an argon atmosphere for 2 h to form HCGAs. The HCGAs with initial GO/GNP mass ratios of 1:1 and 1:2 were designated as HCGA1 and HCGA2, respectively. For comparison, a cylindrical high-quality graphene aerogel with an initial GO/GNP mass ratio of 1:2 was prepared with the similar methodology without the conical polytetrafluoroethylene negative mold inside the autoclave, and designated as HGA2. The isotropic HCGA with an initial GO/GNP mass ratio of 1:2 was prepared using the similar methodology without the use of the KOH in the aqueous GO/GNP suspension and designated as iHCGA2.
Fabrication of thermally conductive phase change composites
Tetradecanol phase change composites were fabricated by a vacuum-assisted infiltration process. Graphene-based aerogels were impregnated in molten tetradecanol under vacuum at 80 °C for 12 h, cooled to ambient temperature under a natural environment, and trimmed off the excess tetradecanol, obtaining HCGA1/tetradecanol, HCGA2/tetradecanol, CGA2/tetradecanol, HGA2/tetradecanol, and iHCGA2/tetradecanol composites.Solar–thermal energy conversion measurements
CEL-HXUV300 Xe lamp equipped with a standard AM1.5 G optical filter was applied as a simulated solar light source, whose light intensity was calibrated by a CEL-NP2000 optical power meter. The as-prepared phase change composites were exposed to simulated sunlight with a solar light intensity of 100 mW cm−2 (1 sun). To characterize the temperature changes of the phase change composites with time during solar–thermal conversion and cooling processes, two thermocouples (Node 1 and Node 2) were inserted into the top and bottom of phase change composites, respectively. The temperatures were recorded every 10 s by an Onset HOBO data logging system. Infrared images were taken with a FLIR E40 infrared camera to visually reflect the temperature changes during the solar–thermal conversion process.Solar–thermal–electric energy conversion measurements
The STE generator was assembled by using HCGA2/tetradecanol phase change composites, a Xinquan Electronic TGM-263-1.4–1.8 temperature-difference power-generation-sheet, and heat dissipation aluminum fins. The bottom of the 3 × 3 array of phase change composites was bonded to the hot source side of the power-generation-sheet with a QM850 thermally conductive silver silicone grease. The cold source side of the power-generation-sheet was bonded to the heat dissipation aluminum fins with the silver silicone grease. The aluminum fins were placed in the air. The water with an initial temperature of 20 °C, and the ice/water mixture were also used to create different heat dissipation environments. To characterize the temperature changes of the phase change composites during the solar–thermal–electric conversion and the cooling process, a thermocouple was inserted into the sample from the top to record the temperatures every 10 s by a data logging system. The output voltages and output currents of the STE generator were measured and recorded by a Fluke 15B+ multimeter and a Keithley DMM7510 7½ digit graphical sampling multimeter. To visualize the output power, the STE generator was connected to an external circuit assembled with light-emitting diodes (LEDs), breadboards, and wires.Characterization
The morphologies of aerogels were characterized with a Hitachi S4700 scanning electron microscope (SEM). X-ray diffraction (XRD) patterns were obtained with a Rigaku SmartLab SE X-ray diffractometer at a generator voltage of 40 kV. Raman mappings were recorded using a Renishaw inVia Raman microscope at an excitation wavelength of 633 nm. An area of 40 × 40 μm2 was automatically scanned with an XY stage using a step size of 1 μm, and the Raman spectra were recorded at every point. The lateral size and thickness of GO and GNPs were characterized using a DMFASTSCAN2-SYS atomic force microscope. Thermal diffusivities of tetradecanol and its phase change composites were measured on a Netzsch LFA467 light flash apparatus at 25 °C. Densities of tetradecanol and its phase change composites were measured using a Mettler Toledo electronic balance with a density determination kit 33360. Specific heat capacities and latent heats of tetradecanol and its phase change composites were measured with a TA Q20 differential scanning calorimeter (DSC) at a scanning rate of 10 °C min−1 in a nitrogen atmosphere. Thermal conductivities of tetradecanol and its phase change composites were calculated with the equation of K = α × ρ × Cp, where α is the thermal diffusivity, ρ is the density, and Cp is the specific heat capacity. Thermogravimetric analysis (TGA) curves were obtained with a TA Q50 thermogravimetric analyzer at a heating rate of 10 °C min−1 in a nitrogen atmosphere. The diffuse reflection and transmission of tetradecanol and its phase change composites were measured by a Shimadzu UV-3600i Plus spectrophotometer.
Results and discussion
Construction and microstructures of graphene-based conical aerogels and their thermally conductive phase change composites
Fig. 1 illustrates the preparation process of the GO-derived HCGA skeleton and its thermally conductive tetradecanol phase change composite. The GO sheets with a large lateral size of ∼70 μm and thickness of ∼2.5 nm (Fig. S1†) are synthesized by oxidizing graphite flakes with a modified Hummers' method.52 Commercially available GNPs are ∼20 μm in lateral size and ∼3.5 nm in thickness (Fig. S2†). After adding KOH, the mixture of the GO and GNP suspensions is homogenized and poured into an autoclave equipped with a mold for hydrothermal reduction to generate a conical RGO/GNP hydrogel. The HCGA is generated by freeze-drying the RGO/GNP hydrogel followed by graphitization at 2800 °C under an inert atmosphere. Thermally conductive HCGA/tetradecanol phase change composites are obtained by vacuum-assisted infiltration of the HCGA skeletons with tetradecanol melt. | ||
| Fig. 1 Schematic illustrating the fabrication process of conical HCGA skeleton and its thermally conductive tetradecanol phase change composite. | ||
To prepare a conical hydrogel with anisotropically aligned RGO sheets, the large GO sheets used in the aqueous GO/GNP/KOH suspension with a GO concentration of 5 mg mL−1 can form GO liquid crystals, resulting in the anisotropic arrangement of GO sheets.53 The presence of KOH could bring about electrostatic repulsion between GO sheets, and the mutual exclusion between the GO sheets allows them to align along the inner surface of the mold,54 forming the conical structure with concentric annular orientation in the horizontal direction, and the GO sheets are arranged from the apex to the bottom of the cone along the vertical direction (Fig. 1). During the hydrothermal process, the GO sheets are partially reduced to RGO sheets, and the restored π–π conjugation interactions of RGO sheets enable them to assemble into a 3D network,55,56 and the dispersed GNP sheets have to be distributed inside the RGO skeleton.40
Fig. 2a and b show digital photographs of HCGA2 skeleton and HCGA2/tetradecanol phase change composite. Both HCGA2 and its tetradecanol phase change composite are in black color, beneficial for effective solar light absorption. SEM images (Fig. 2c–f) confirm the concentric annular conical aerogel microstructure of HCGA2. The graphene walls in the HCGA2 tend to orientate concentrically and annularly in the horizontal direction with a layer spacing of ∼10 μm, and the GNPs are uniformly distributed in the skeleton (Fig. 2c and d), while the HCGA2 exhibits the alignment of the graphene sheets from the apex to the bottom of the cone in the vertical direction (Fig. 2e and f). In addition, the low magnification SEM images clearly show the concentric circular arrangement of the graphene sheets in the horizontal direction (Fig. S3†). It is also seen that the graphene walls are not separate but bridged by graphene sheets, demonstrating the formation of 3D interconnecting graphene networks, which facilitates heat transfer in both vertical and horizontal directions. Digital photographs of the cross-section of HCGA2 also demonstrate the concentric circular orientation trend of the graphene sheets in the horizontal direction, and its side-section photograph confirms the orientation in the vertical direction (Fig. S4a–c†). In addition, the bottom photograph of the HCGA2/tetradecanol shows the concentric circular orientation of the graphene sheets inside the phase change composite (Fig. S4d†).
 | ||
| Fig. 2 Digital photographs of (a) HCGA2 and (b) HCGA2/tetradecanol phase change composite. SEM images of (c, d) cross-section and (e, f) side-section of the conical HCGA2. | ||
To monitor the evolution of graphene quality during the fabrication process, Fig. 3 shows Raman mapping images of GO, RGO, GNP, CGA2 and HCGA2. As shown in Fig. 3a–e, the color of the area close to blue indicates a low intensity ratio of D and G bands (ID/IG) and a low defect density of sheets, while the color close to red implies a high ID/IG value and a high defect density. The colors of the Raman mappings of GO and GNP are uniform (Fig. 3a and c), demonstrating the stable quality of the raw materials. In particular, the Raman mapping of GNP is dark blue, reflecting its excellent quality.
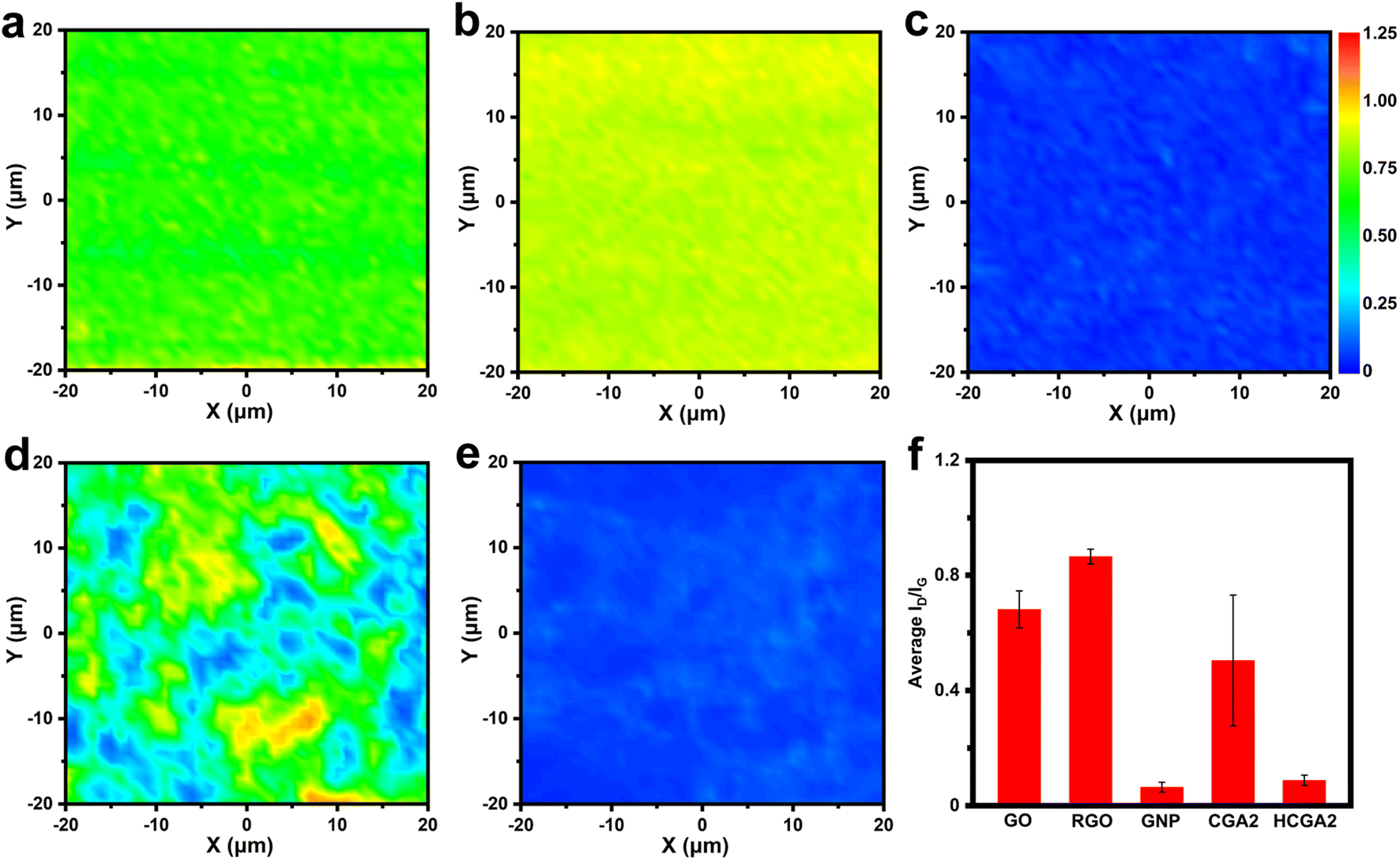 | ||
| Fig. 3 Raman mappings of (a) GO, (b) RGO, (c) GNP, (d) CGA2, and (e) HCGA2. The blue, green, and red colors correspond to their ID/IG values of 0, ∼0.6, and ∼1.25, respectively. (f) Average ID/IG histograms of GO, RGO, GNP, CGA2 and HCGA2. | ||
The comparison of Fig. 3b with Fig. 3a reveals the increase in the defect density after hydrothermal treatment of GO due to the removal of oxygen-containing functional groups on GO.57 The uneven color of the Raman mapping of CGA2 (Fig. 3d) derives from the presence of RGO and GNP with a significant difference in defect density. After graphitizing at 2800 °C in an inert atmosphere, in contrast, the Raman mapping of HCGA2 exhibits uniform dark blue (Fig. 3e), verifying that the high-temperature thermal annealing can heal the defects of the RGO and hence improve the quality of graphene in the aerogel.58Fig. 3f enumerates the average ID/IG values of the Raman mapping regions of GO, RGO, GNP, CGA2 and HCGA2. The ID/IG values of GO, RGO and CGA2 are in a high range, indicating that more defects exist on the graphene sheets without the high-temperature annealing treatment, which would cause serious phonon scattering at the defect regions and result in heat loss.59 On the contrast, HCGA2 exhibits an extremely low ID/IG value, which is the reflection of its high quality of graphene sheets and implies the greatly enhanced heat conduction of the HCGA2 skeleton.
Fig. 4a manifests XRD patterns of GO, GNP, CGA2, and HCGA2. The GO exhibits a typical diffraction peak at ∼12°, reflecting its layer spacing of ∼0.74 nm,60 much larger than that of pristine graphite of 0.34 nm, because of the oxygen-containing functional groups on the lamellae of GO; whereas, the GNP shows a sharp peak at 26.4°, corresponding to a layer spacing of 0.34 nm. The disappearance of the typical diffraction peak of GO and the appearance of a sharp peak at 26.4° in the XRD patterns of CGA2 and HCGA2 demonstrate that the hydrothermal treatment and the high-temperature graphitization can sufficiently remove the oxygen-containing groups of GO sheets.61
 | ||
| Fig. 4 (a) XRD patterns of GO, GNP, CGA2, and HCGA2. (b) Thermal conductivities of tetradecanol and graphene aerogel/tetradecanol composites. (c) TGA curves of HCGA1/tetradecanol, and HCGA2/tetradecanol composites. (d) Heating and (e) cooling DSC curves of tetradecanol, HCGA1/tetradecanol, and HCGA2/tetradecanol composites. (f) Comparison of thermal conductivity and latent heat retention rate of the phase change composite with those reported in the literature. | ||
Thermally conductive and thermophysical properties of phase change composites
Fig. 4b shows the thermal conductivities of tetradecanol and graphene aerogel/tetradecanol composites. The through-plane thermal conductivities of the HCGA/tetradecanol composites increase with the GNP contents. The HCGA2/tetradecanol phase change composite exhibits a high thermal conductivity of 4.54 W m−1 K−1, which is enhanced by 1193% as compared to that of tetradecanol (0.38 W m−1 K−1). The excellent through-plane thermal conductivity of the HCGA/tetradecanol composite can be attributed to four aspects: (i) the interconnecting 3D graphene network reduces the interfaces between the graphene sheets and the tetradecanol for minimizing heat loss at the interfaces, and facilitates the heat transfer to the entire phase change composite;62 (ii) the introduction of GNPs densifies the thermally conductive network and provides more thermal conduction pathways for rapid heat transfer inside the phase change composite;63,64 (iii) the arrangement of the graphene sheets in the vertical direction provides efficient thermal conduction pathways for heat transfer from the top to the bottom of the HCGA/tetradecanol composite;65 and (iv) the greatly enhanced graphene quality by the graphitization prevents phonon scattering and promotes the heat transfer on the graphene sheets.66To confirm the influence of the anisotropic structure of the aerogels on the through-plane thermal conductivity, the iHCGA2/tetradecanol phase change composite with a random arrangement of graphene sheets is fabricated for comparison, and its thermal conductivity is 3.76 W m−1 K−1, lower than that of the HCGA2/tetradecanol counterpart, indicating that the orientation of the graphene sheets in the vertical direction is positive for the enhancement in through-plane thermal conductivity of the phase change composite. In addition, the positive contribution of the graphitization is well reflected by the fact that the thermal conductivity of the HCGA2/tetradecanol is 3.65-fold of the CGA2/tetradecanol in the absence of the graphitization. The TGA analysis reveals that the total graphene/GNP content in the graphene aerogel/tetradecanol composites increases with the GNP content. The loading of graphene/GNP in the HCGA2/tetradecanol is ∼7.05 wt% (Fig. 4c), which is still at a low loading level.41,48,67,68
Fig. 4d and e show DSC curves of tetradecanol, HCGA1/tetradecanol, and HCGA2/tetradecanol. The comparison of these curves indicates that the peak shapes and peak positions of the graphene aerogel/tetradecanol composites are almost identical to those of the tetradecanol during the heating and cooling processes, demonstrating that no chemical reaction occurs between the graphene aerogel and tetradecanol during the compounding process.69 Notably, there are two obvious exothermic peaks during the cooling of tetradecanol and graphene aerogel/tetradecanol phase change composites, representing the liquid–solid transition and the solid–solid transition.70 Table S1† lists the melting temperature (Tm), melting enthalpy (ΔHm), crystallization temperature (Tc), and crystallization enthalpy (ΔHc) of tetradecanol and its phase change composites. HCGA2/tetradecanol has a Tm of 35.8 °C, which is very close to that of tetradecanol (35.6 °C), indicating that the introduction of HCGA2 hardly affects the phase transition behaviour of tetradecanol.71 On the basis of the ΔHm of tetradecanol (218.0 J g−1), the ΔHm of HCGA2/tetradecanol is calculated to be 206.1 J g−1 with a high latent heat retention rate of 94.5%, which is ascribed to the low graphene/GNP loading in the HCGA2/tetradecanol. Moreover, the DSC curves of the HCGA2/tetradecanol after 50 and 100 heating–cooling cycles almost coincide with the first cycle (Fig. S5†), indicating that the phase change composite possesses excellent cyclic stability in terms of thermophysical properties and high reliability in long-cycle operation.
As the introduction of graphene could cause the decrease in phase change enthalpy of the phase change materials, the high graphene loading would compromise the energy storage capacity of the phase change composite.13 By promoting the quality of graphene and controlling the graphene loading at a low level, the HCGA2/tetradecanol has an excellent thermal conductivity while retaining a high phase change enthalpy of the tetradecanol matrix. Fig. 4f shows the thermal conductivities and latent heat retention rates of phase change composites reported in recent years.8,15,27,30,41,67,71–74 Apparently, the HCGA2/tetradecanol possesses a relatively high thermal conductivity and an outstanding latent heat retention rate, which is well suitable for solar–thermal energy conversion and thermal energy storage.
Solar–thermal energy conversion behavior of the phase change composites
To evaluate the solar–thermal energy conversion behavior of phase change composites, the temperatures are measured when the phase change composites are irradiated with simulated sunlight. Fig. 5a and d illustrate the measurement of solar–thermal energy conversion performances of the phase change composites, which are exposed to the simulated sunlight with an intensity of 100 mW cm−2. Two thermocouples (Node 1, and Node 2) are inserted into the top and bottom of the samples to measure the top temperature (Tt) and the bottom temperature (Tb) of the samples. Fig. 5b shows the temperature–time curves of the HCGA2/tetradecanol. At the beginning of the solar light exposure (Stage a), the temperature of the HCGA2/tetradecanol rises rapidly, demonstrating its excellent light responsiveness, and the Tb is slightly higher than the Tt at this stage. In the melting process (Stage b), the temperature of the HCGA2/tetradecanol tends to be stable. The tetradecanol is converted from solid to liquid, and the heat generated by the solar–thermal energy conversion is mainly stored as the latent heat of the tetradecanol instead of the sensible heat reflected in the temperature increase. Simultaneously, the gap between Tb and Tt increases as the phase change proceeds, and the temperature difference reaches ∼0.6 °C. After the phase change is complete, the temperature of the HCGA2/tetradecanol continues to rise rapidly (Stage c) until the equilibrium of heat exchange with the environment (Stage d), at which Tb is still higher than Tt with a temperature difference of ∼0.9 °C, and the temperature of the HCGA2/tetradecanol reaches ∼60 °C. At Stage e, as the light is removed, the temperature drops dramatically due to the absence of the light energy input. A solidification platform (Stage f) occurs when the temperature drops to the liquid–solid phase transition temperature. After a continued temperature decrease in Stage g, the second phase change platform (Stage h) appears. The two phase-transition platforms in the cooling stages correspond to the two exothermic peaks of the HCGA2/tetradecanol (Fig. 4e). Finally, the temperature drops to the ambient temperature (Stage i). Almost during the whole process of the solar–thermal energy conversion, the Tb of the HCGA2/tetradecanol is higher than its Tt, which is because the conical shape enables the external surface of the lower part of the sample to have a large solar light-absorbing area (Fig. S6†). Meanwhile, the temperature difference is maintained at a low absolute value, which indicates that the thermal conductivity of the HCGA2/tetradecanol can fully meet the requirement for its solar–thermal conversion application. The heating process of the HCGA2/tetradecanol under the solar light irradiation intensity of 100 mW cm−2 can be observed visually by the infrared photographs (Fig. 5c). The solar–thermal conversion performances demonstrate that the HCGA2/tetradecanol can overcome the problem that the Tb of a conventional phase change composite is much lower than the Tt because a conventional phase change composite usually has a low thermal conductivity and only its upper surface can be exposed to the solar light, which also cause inhomogeneous phase state of the phase change composite and the inability of the bottom of the material to complete the phase change, and thus hindering the ability of the solar–thermal conversion phase change composite to store and utilize heat.75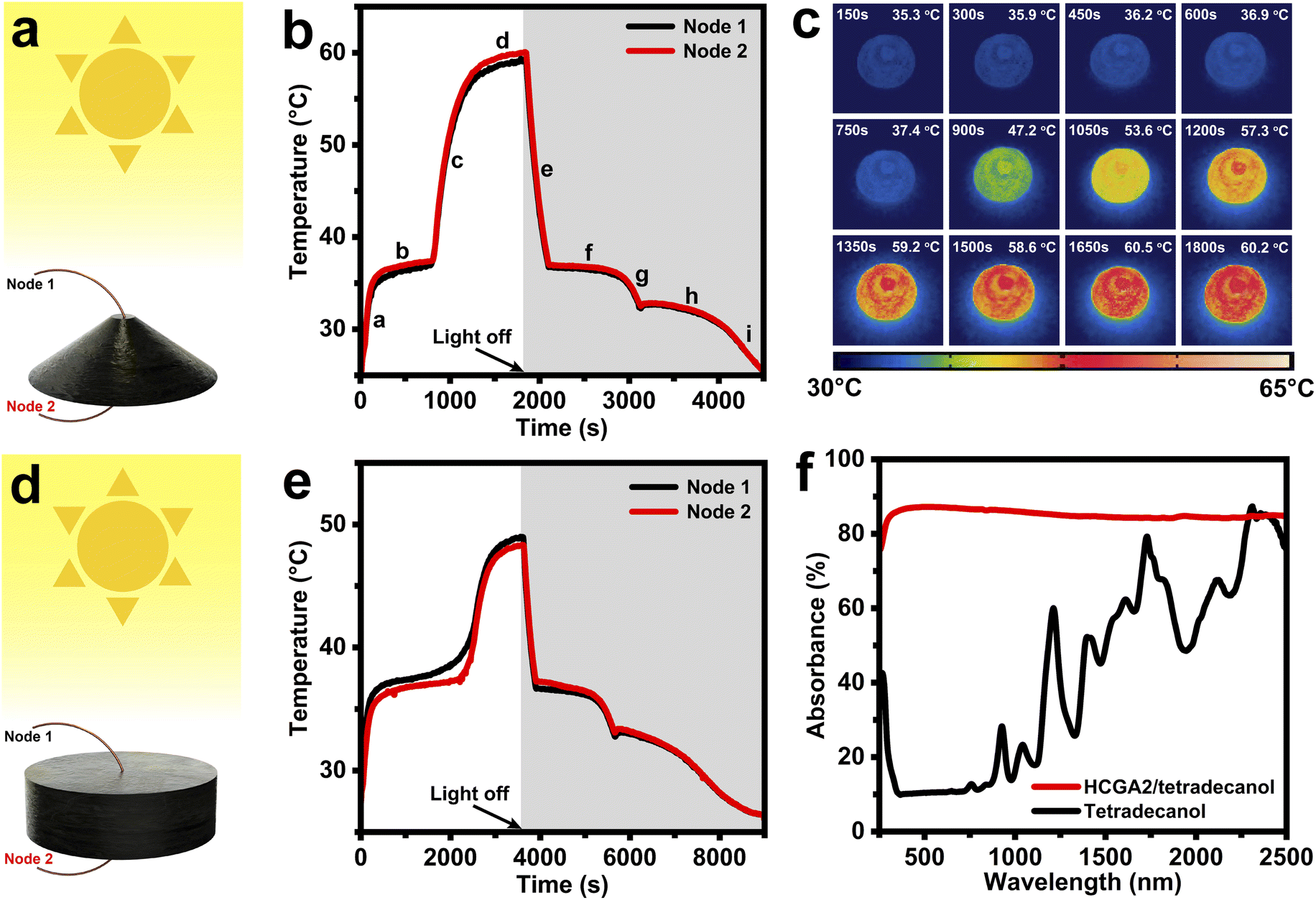 | ||
| Fig. 5 Schematics of the solar–thermal conversion characterization of (a) HCGA2/tetradecanol and (d) HGA2/tetradecanol. Temperature–time curves of the bottom and top of (b) HCGA2/tetradecanol and (e) HGA2/tetradecanol under a solarlight intensity of 100 mW cm−2. (c) Infrared photographs of HCGA2/tetradecanol during the solar–thermal conversion process under a light intensity of 100 mW cm−2. (f) Absorbance spectra of HCGA2/tetradecanol and tetradecanol. | ||
Besides, the higher temperature of the bottom of the HCGA2/tetradecanol that contacts the hot side of the power-generation-sheet is favorable for improving the output voltage of the STE generator.76 The solar–thermal conversion and thermal energy storage efficiency (λ) of a phase change composite is calculated by the equation: λ = (m × ΔH + m × Cp × ΔT)/(P × A × t), where m, ΔH, and Cp are the mass, melting enthalpy, and specific heat capacity of the phase change composite, respectively. ΔT is the temperature increase during the melting process, P is the solar light intensity, A is the projected area of the light-receiving surface, and t is the duration of the melting process. The beginning and ending positions of the melting process are determined by a linear fitting method (Fig. S7†). As calculated, the λ of the HCGA2/tetradecanol is up to 84.0% under a light intensity of 100 mW cm−2.
For comparison, the solar–thermal conversion performances of the cylindrical HGA2/tetradecanol under the same light intensity (100 mW cm−2) are characterized, and its temperature–time curves are shown in Fig. 5e. In the irradiation process, as the lower part of the cylindrical HGA2/tetradecanol cannot receive the solar light, the heat can only be generated by the upper surface and transferred to the lower part of the sample, during which the heat loss occurs inevitably. Different from the HCGA2/tetradecanol, the HGA2/tetradecanol cannot keep the lower part at a high temperature. As the smaller effective surface area of the HGA2/tetradecanol than that of the HCGA2/tetradecanol, and the inevitable heat dissipation from the side surface of the HGA2/tetradecanol to the environment, the λ of HGA2/tetradecanol is only 75.2%. The beginning and ending positions of the melting process are determined by a linear fitting method (Fig. S8†), and the maximum temperature is ∼49 °C. Clearly, the conical shape endows the HCGA2/tetradecanol with a better solar–thermal conversion performances. As a comparison experiment, the temperature of the conical tetradecanol is measured under the light intensity of 100 mW cm−2 (Fig. S9†). The maximum temperature of the tetradecanol is only ∼34 °C, and its phase transition cannot occur, leading to no heat storage. These results confirm that the solar–thermal conversion of tetradecanol is improved remarkably by the HCGA2. Fig. 5f compares the light absorbances of the HCGA2/tetradecanol and tetradecanol in the whole wavelength range of 250–2500 nm. The absorbance of HCGA2/tetradecanol remains almost above 85%, much higher than that of tetradecanol, indicating the excellent light absorption ability of the HCGA2/tetradecanol.
Solar–thermal–electric energy conversion performances of STE generators
Fig. 6a illustrates the assembly of a STE generator, in which the 3 × 3 HCGA2/tetradecanol composite array and the heat dissipation aluminum fins are connected to the hot and cold source sides of the power-generation-sheet, respectively. The schematic of the components and their connections in the solar–thermal–electric energy conversion measurements is shown in Fig. S10.† When exposed to the solar light, the temperature of the HCGA2/tetradecanol rises because of the solar–thermal energy conversion. In contrast, the temperature of the aluminum fins is lower, resulting in a temperature difference between the two sides of the power-generation-sheet and thus leading to the output voltage of the STE generator. The output voltage is proportional to the temperature difference.77 To create different heat dissipation environments, the aluminum fins can be placed in the air, in the water with an initial temperature of 20 °C, and in an ice/water mixture.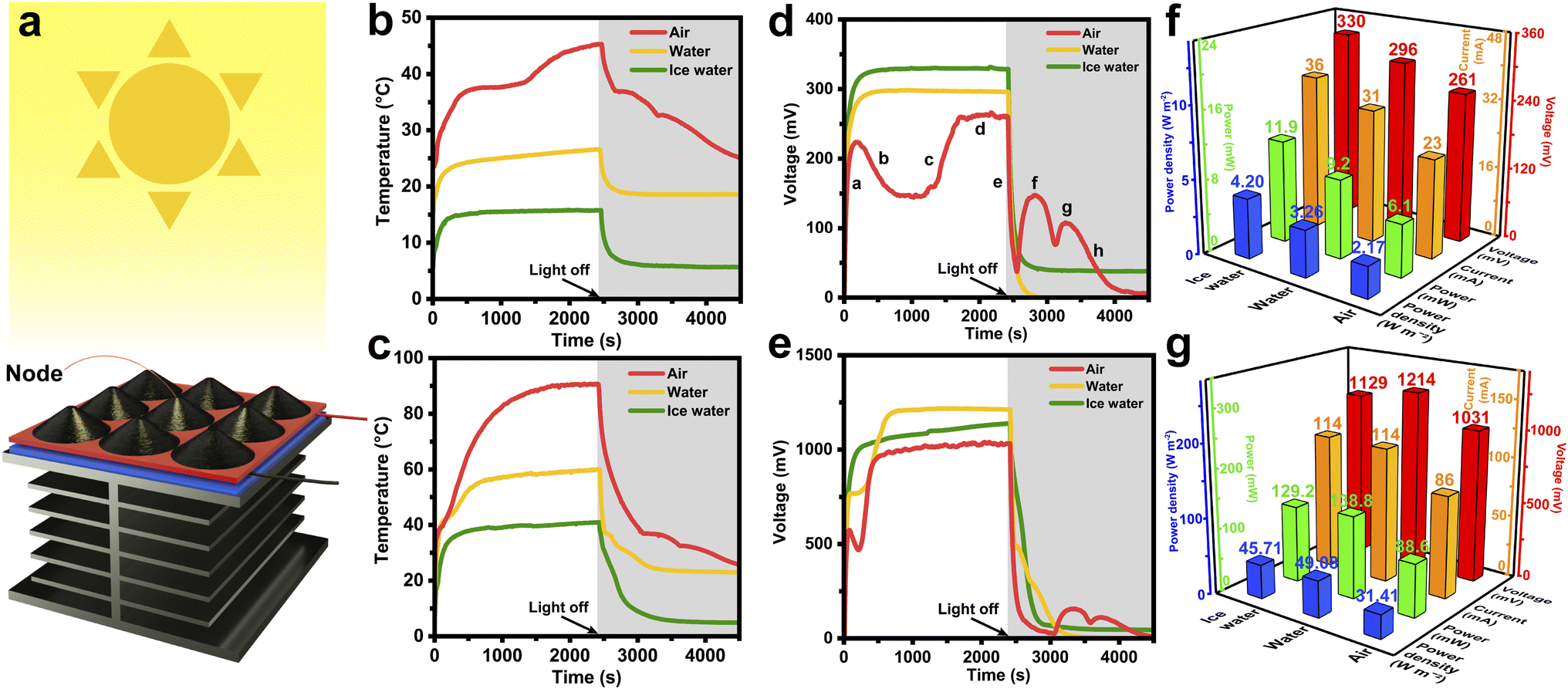 | ||
| Fig. 6 (a) Schematic of the STE generator and solar–thermal–electric energy conversion characterization. Temperature–time curves of HCGA2/tetradecanol employed in the STE generator under light intensities of (b) 100 mW cm−2 and (c) 500 mW cm−2 in three heat dissipation environments. Voltage-time curves of the STE generator under light intensities of (d) 100 mW cm−2 and (e) 500 mW cm−2 in three heat dissipation environments. The maximum output voltage, output current, output power and power density histograms of the STE generator under solar light intensities of (f) 100 mW cm−2 and (g) 500 mW cm−2 in three heat dissipation environments. | ||
Fig. 6b shows the temperature–time curves of the phase change composites under a light intensity of 100 mW cm−2. When the aluminum fins are placed in the air, under the solar light irradiation, the temperature of the phase change composites rises to the phase change temperature, and the phase transition occurs to achieve the heat storage. The maximum temperature of the phase change composite is ∼45 °C because a part of the heat converts to electrical energy, and a part of the heat transfers to the aluminum fins via the power-generation-sheet and dissipates to the environment. The red curve in Fig. 6d is the output voltage–time curve of the STE generator for dissipating heat in the air under a light intensity of 100 mW cm−2. In the initial irradiation stage (Stage a), the temperature of the phase change composites rises rapidly, resulting in an increase in the temperature difference between the hot and cold source sides of the power-generation-sheet and a sharp rise in the output voltage. The temperature of the phase change composites is almost constant during the phase transition, but the heat still transfers to the cold side of the power-generation-sheet, causing the decrease in temperature difference between the hot and cold sides, and hence the output voltage tends to drop in Stage b. After completing the phase transition of the phase change composites, the temperature rises rapidly again, resulting in a sharp increase in output voltage (Stage c). Subsequently, the device reaches a stable state of the temperature and the output voltage (Stage d).
After stopping the solar light irradiation, the output voltage of the STE generator drops steeply due to the absence of the energy input (Stage e). As the temperature of the phase change composites remains constant after dropping to the solidification platform while the aluminum fins are still dissipating heat, the temperature difference between the hot and cold sides of the power-generation-sheet still increases, leading to the increase in output voltage in the absence of solar light (Stage f). This result demonstrates that the released thermal energy from the phase change composite can be converted to electrical energy. Similarly, the second output voltage rise (Stage g) is achieved by releasing the energy at the second solidification platform. Finally, the phase change composite is gradually cooled to room temperature, and the STE generator stops outputting voltage (Stage h). The voltage–time curves demonstrate that the STE generator can still output voltage for 2100 s in the absence of the solar light after the solar light irradiation for 2400 s under a light intensity of 100 mW cm−2 when the aluminum fins are placed in the air. The storage and release energy capability of the phase change composites is crucial for endowing the STE generator to output voltage continuously at the daytime and night. The maximum output voltage can be up to ∼261 mV. In the other two heat dissipation environments, as a large amount of heat is absorbed by the environments, the temperature of the phase change composite cannot reach the phase change temperature, and the latent heat of the phase change composite cannot be utilized for heat storing and releasing, which is reflected in the rapid voltage drop after stopping the solar light irradiation.
Furthermore, when the STE generator is exposed to a high light intensity of 500 mW cm−2, the temperatures of the phase change composite and the output voltages of the STE generator are measured (Fig. 6c and e). Thanks to the large improvement in the input energy, the temperature of the phase change composite can reach the phase change temperature in all three heat dissipation environments, and the STE generators can still output the voltage for some time after stopping the solar light irradiation. Fig. S11 and S12† manifest the output voltage and output current curves for all working environments, demonstrating that the voltage and current have the same trend. Fig. 6f and g compare the maximum output voltage, output current, output power, and power density of STE generators in the three heat dissipation environments under light intensities of 100 and 500 mW cm−2. The highest output voltage of ∼330 mV is achieved when the heat dissipation environment is the ice water at the light intensity of 100 mW cm−2, which is attributed to the largest temperature difference between the hot and cold sides of the STE generator when the aluminum fins are placed in the ice water. The output power and power density of the STE generator under this condition can reach 11.9 mW and 4.20 W m−2, respectively. When the light intensity increases to 500 mW cm−2, the phase change composite can receive more solar energy and convert it to heat energy, resulting in a particularly high temperature; whereas, the aluminum fins still maintain a low temperature by dissipating the heat in the water. The resultant large temperature difference between the hot and cold sides of the power generator sheet leads to a maximum output voltage of the STE generator (∼1214 mV). The output power of the STE generator under this working condition can reach 138.8 mW with a power density of 49.08 W m−2. Table S2† manifests that the maximum output voltage and current of the STE generators are much higher than those reported at the same light intensities.47 Even at low light intensities, the maximum output voltage and current of the STE generators are superior to the reported performances at high light intensities.41,42,44,47–49
To demonstrate a practical application, the STE generator is exposed to light with an intensity of 1200 mW cm−2, and served as a power supply for an external circuit, which enables LED bulbs to emit light (Video S1†), and the measured voltage can be ∼2.02 V (Fig. S13†). Actually, the enhanced light intensities can be readily achieved in practical application scenarios by concentrating solar beams with convex lenses and concave mirrors.78,79 In addition, the heat dissipation condition of the ice/water mixture simulates the use of the STE generator in cold environments (e.g., high-latitude regions, and high-altitude glacier areas), confirming the feasibility of the STE generator for solar–thermal–electric energy conversion in such cold regions.
Conclusions
Anisotropic and conical-shaped high-quality graphene aerogels with concentric annular structures are constructed by hydrothermal treatment of the GO/GNP suspensions followed by freeze-drying and graphitizing at 2800 °C for enhancing thermal conduction, solar–thermal energy conversion, and shape stability of the tetradecanol phase change material. By controlling the orientation of GO liquid crystals during their self-assembly, the resultant conical aerogels exhibit anisotropic structures and achieve efficient heat transfer along their vertical direction. The graphitization at 2800 °C is efficient in removing the residual oxygen-containing groups from the RGO sheets and healing the defects of the RGO sheets, leading to high-quality thermally conductive graphene networks. The HCGA2/tetradecanol phase change composite exhibits a high thermal conductivity of 4.54 W m−1 K−1 with a high latent heat retention of 94.5%. Under a solar light irradiation of 100 mW cm−2, the phase change composite achieves a high solar–thermal energy conversion and storage efficiency of 84.0%. Furthermore, the STE generator assembled with the 3 × 3 phase change composite array and a thermoelectric device exhibits a maximum output voltage of ∼261 mV when dissipating heat in the air under a solar light intensity of 100 mW cm−2, and can still have a voltage output after stopping the solar light irradiation. Under a solar light intensity of 500 mW cm−2, the maximum output voltage of the STE generator can be up to 1214 mV, demonstrating the great potential of the conical phase change composite for solar–thermal–electric energy conversion applications.Author contributions
Hao-Yu Zhao: investigation, data curation, formal analysis, writing-original draft. Chao Shu: investigation. Peng Min: methodology. Changjun Li: data curation. Wenchao Deng: investigation. Jing Yang: methodology. Xiaofeng Li: methodology, supervision. Zhong-Zhen Yu: writing-review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (U1905217, 52090034, 51773008), and the Fundamental Research Funds for the Central Universities (XK1802) is gratefully acknowledged.References
- Y. Wang, B. Tang and S. Zhang, Adv. Funct. Mater., 2013, 23, 4354–4360 CrossRef.
- J.-L. Zeng, J. Gan, F.-R. Zhu, S.-B. Yu, Z.-L. Xiao, W.-P. Yan, L. Zhu, Z.-Q. Liu, L.-X. Sun and Z. Cao, Sol. Energy Mater. Sol. Cells, 2014, 127, 122–128 CrossRef.
- X. Wang, X. Cheng, Y. Li, G. Li and J. Xu, Sol. Energy, 2019, 179, 128–134 CrossRef.
- G. Cheng, X. Wang and Y. He, Appl. Therm. Eng., 2020, 178, 115560 CrossRef CAS.
- J. Huang, B. Zhang, M. He, X. Huang, G. Wu, G. Yin and Y. Cui, J. Mater. Sci., 2020, 55, 7337–7350 CrossRef CAS.
- M. Weng, S. Liu, J. Su, W. Xu, J. Huang, W. Tan, Y. Liu and Y. Min, Eng. Sci., 2022, 19, 301–311 Search PubMed.
- X. Hu, H. Wu, X. Lu, S. Liu and J. Qu, Adv. Compos. Hybrid Mater., 2021, 4, 478–491 CrossRef CAS.
- F. Xue, X. Z. Jin, X. Xie, X. D. Qi, J. H. Yang and Y. Wang, Nanoscale, 2019, 11, 18691–18701 RSC.
- G. Yang, L. Zhao, C. Shen, Z. Mao, H. Xu, X. Feng, B. Wang and X. Sui, Sol. Energy Mater. Sol. Cells, 2020, 209, 110441 CrossRef CAS.
- H. Cheng, L. Xing, Y. Zuo, Y. Pan, M. Huang, A. Alhadhrami, M. M. Ibrahim, Z. M. El-Bahy, C. Liu, C. Shen and X. Liu, Adv. Compos. Hybrid Mater., 2022, 5, 755–765 CrossRef CAS.
- Y. Cao, M. Weng, M. H. H. Mahmoud, A. Y. Elnaggar, L. Zhang, I. H. El Azab, Y. Chen, M. Huang, J. Huang and X. Sheng, Adv. Compos. Hybrid Mater., 2022, 5, 1253–1267 CrossRef CAS.
- J. Yang, Y. C. Zhou, L. Y. Yang, C. P. Feng, L. Bai, M. B. Yang and W. Yang, Adv. Funct. Mater., 2022, 32, 2200792 CrossRef CAS.
- P. Liu, F. An, X. Lu, X. Li, P. Min, C. Shu, W. Li and Z.-Z. Yu, Compos. Sci. Technol., 2021, 201, 108492 CrossRef CAS.
- P. Min, J. Liu, X. Li, F. An, P. Liu, Y. Shen, N. Koratkar and Z.-Z. Yu, Adv. Funct. Mater., 2018, 28, 1805365 CrossRef.
- D. Liu, C. Lei, K. Wu and Q. Fu, ACS Nano, 2020, 14, 15738–15747 CrossRef.
- J. Shi, W. Aftab, Z. Liang, K. Yuan, M. Maqbool, H. Jiang, F. Xiong, M. Qin, S. Gao and R. Zou, J. Mater. Chem. A, 2020, 8, 20133–20140 RSC.
- G. Li, X. Zhang, J. Wang and J. Fang, J. Mater. Chem. A, 2016, 4, 17042–17049 RSC.
- J. Yang, X. Li, S. Han, Y. Zhang, P. Min, N. Koratkar and Z.-Z. Yu, J. Mater. Chem. A, 2016, 4, 18067–18074 RSC.
- J. Huang, Y. Luo, M. Weng, J. Yu, L. Sun, H. Zeng, Y. Liu, W. Zeng, Y. Min and Z. Guo, ES Mater. Manuf., 2021, 13, 23–39 CAS.
- H.-Y. Zhao, M.-Y. Yu, J. Liu, X. Li, P. Min and Z.-Z. Yu, Nano-Micro Lett., 2022, 14, 129 CrossRef CAS.
- S. Ye, Q. Zhang, D. Hu and J. Feng, J. Mater. Chem. A, 2015, 3, 4018–4025 RSC.
- M. Su, G. Han, J. Gao, Y. Feng, C. He, J. Ma, C. Liu and C. Shen, Chem. Eng. J., 2021, 427, 131665 CrossRef.
- F. Xue, Y. Lu, X.-D. Qi, J.-H. Yang and Y. Wang, Chem. Eng. J., 2019, 365, 20–29 CrossRef CAS.
- X. Hu, H. Wu, S. Liu, S. Gong, Y. Du, X. Li, X. Lu and J. Qu, Eng. Sci., 2022, 17, 1–27 CAS.
- J. Yang, G.-Q. Qi, R.-Y. Bao, K. Yi, M. Li, L. Peng, Z. Cai, M.-B. Yang, D. Wei and W. Yang, Energy Storage Mater., 2018, 13, 88–95 CrossRef.
- D. Jaque, L. M. Maestro, B. D. Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. M. Rodríguez and J. G. Solé, Nanoscale, 2014, 6, 9494–9530 RSC.
- H. Wu, S. Deng, Y. Shao, J. Yang, X. Qi and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 46851–46863 CrossRef CAS.
- J. Shen, P. Zhang, L. X. Song, J. P. Li, B. Q. Ji, J. J. Li and L. Chen, Composites, Part B, 2019, 179, 107545 CrossRef.
- B. Mu and M. Li, Sci. Rep., 2018, 8, 8878 CrossRef PubMed.
- H. Y. Wu, S. T. Li, Y. W. Shao, X. Z. Jin, X. D. Qi, J. H. Yang, Z. W. Zhou and Y. Wang, Chem. Eng. J., 2020, 379, 122373 CrossRef.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef PubMed.
- D. Yan, H.-B. Zhang, Y. Jia, J. Hu, X.-Y. Qi, Z. Zhang and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2012, 4, 4740–4745 CrossRef PubMed.
- F.-Y. Yuan, H.-B. Zhang, X. Li, H.-L. Ma, X.-Z. Li and Z.-Z. Yu, Carbon, 2014, 68, 653–661 CrossRef.
- J. Liu, Y. Liu, H.-B. Zhang, Y. Dai, Z. Liu and Z.-Z. Yu, Carbon, 2018, 132, 95–103 CrossRef.
- P. Liu, X. Li, P. Min, X. Chang, C. Shu, Y. Ding and Z.-Z. Yu, Nano-Micro Lett., 2021, 13, 22 CrossRef PubMed.
- Z. Bo, H. R. Zhu, C. Y. Ying, H. C. Yang, S. H. Wu, J. Kong, S. L. Yang, X. Wei, J. H. Yan and K. F. Cen, Nanoscale, 2019, 11, 21249–21258 RSC.
- W. Zhao, J. Kong, H. Liu, Q. Zhuang, J. Gu and Z. Guo, Nanoscale, 2016, 8, 19984–19993 RSC.
- J. D. Renteria, S. Ramirez, H. Malekpour, B. Alonso, A. Centeno, A. Zurutuza, A. I. Cocemasov, D. L. Nika and A. A. Balandin, Adv. Funct. Mater., 2015, 25, 4664–4672 CrossRef CAS.
- Y. Li, W. Wei, Y. Wang, N. Kadhim, Y. Mei and Z. Zhou, J. Mater. Chem. C, 2019, 7, 11783–11789 RSC.
- F. An, X. Li, P. Min, P. Liu, Z.-G. Jiang and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 17383–17392 CrossRef CAS.
- J. Yang, L.-S. Tang, R.-Y. Bao, L. Bai, Z.-Y. Liu, W. Yang, B.-H. Xie and M.-B. Yang, J. Mater. Chem. A, 2016, 4, 18841–18851 RSC.
- J. Yang, P. Yu, L. S. Tang, R. Y. Bao, Z. Y. Liu, M. B. Yang and W. Yang, Nanoscale, 2017, 9, 17704–17709 RSC.
- C. B. Yu, S. H. Yang, S. Y. Pak, J. R. Youn and Y. S. Song, Energy Convers. Manage., 2018, 169, 88–96 CrossRef CAS.
- J. Yang, L. S. Tang, R. Y. Bao, L. Bai, Z. Y. Liu, B. H. Xie, M. B. Yang and W. Yang, Sol. Energy Mater. Sol. Cells, 2018, 174, 56–64 CrossRef.
- X. Zhang, G. Yang, L. Zong, M. Jiang, Z. Song, C. Ma, T. Zhang, Y. Duan and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 1378–1386 CrossRef.
- Y. Zhang, K. Wu and Q. Fu, Adv. Funct. Mater., 2021, 32, 2109255 CrossRef.
- L. Zhang, R. Li, B. Tang and P. Wang, Nanoscale, 2016, 8, 14600–14607 RSC.
- J. Yang, L. S. Tang, R. Y. Bao, L. Bai, Z. Y. Liu, W. Yang, B. H. Xie and M. B. Yang, Chem. Eng. J., 2017, 315, 481–490 CrossRef CAS.
- R. Cao, D. Sun, L. Wang, Z. Yan, W. Liu, X. Wang and X. Zhang, J. Mater. Chem. A, 2020, 8, 13207–13217 RSC.
- G. J. Chen, Y. P. Su, D. Y. Jiang, L. J. Pan and S. Li, Appl. Energy, 2020, 264, 114786 CrossRef CAS.
- H. Mamur, M. R. A. Bhuiyan, F. Korkmaz and M. Nil, Renewable Sustainable Energy Rev., 2017, 82, 4159–4169 CrossRef.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- S. Gambhir, R. Jalili, D. L. Officer and G. G. Wallace, NPG Asia Mater., 2015, 7, e186 CrossRef CAS.
- B. W. Yao, J. Chen, L. Huang, Q. Q. Zhou and G. Q. Shi, Adv. Mater., 2016, 28, 1623–1629 CrossRef CAS PubMed.
- R. Yu, Y. Shi, D. Yang, Y. Liu, J. Qu and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 21809–21819 CrossRef CAS PubMed.
- H. Wei, A. Li, D. Kong, Z. Li, D. Cui, T. Li, B. Dong and Z. Guo, Adv. Compos. Hybrid Mater., 2021, 4, 86–95 CrossRef CAS.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- X.-H. Li, P. Liu, X. Li, F. An, P. Min, K.-N. Liao and Z.-Z. Yu, Carbon, 2018, 140, 624–633 CrossRef CAS.
- B. S. Lee, J. Phys.: Condens. Matter, 2018, 30, 295302 CrossRef PubMed.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- W. Xie, F. Yao, H. Gu, A. Du, Q. Lei, N. Naik and Z. Guo, Adv. Compos. Hybrid Mater., 2022, 5, 1003–1016 CrossRef CAS.
- J. Yang, X. Li, S. Han, R. Yang, P. Min and Z.-Z. Yu, J. Mater. Chem. A, 2018, 6, 5880–5886 RSC.
- G.-Q. Qi, J. Yang, R.-Y. Bao, Z.-Y. Liu, W. Yang, B.-H. Xie and M.-B. Yang, Carbon, 2015, 88, 196–205 CrossRef.
- T. Yang, Z. Jiang, H. Han, X. Cai, Y. Liu, X. Zhang, X. Zhang, Y. Ren and J. Hu, Composites, Part B, 2021, 205, 108509 CrossRef.
- F. An, X. Li, P. Min, H. Li, Z. Dai and Z.-Z. Yu, Carbon, 2018, 126, 119–127 CrossRef.
- W. Y. Zhang, Q. Q. Kong, Z. C. Tao, J. C. Wei, L. J. Xie, X. Y. Cui and C. M. Chen, Adv. Mater. Interfaces, 2019, 6, 8 Search PubMed.
- J. Yang, L.-S. Tang, L. Bai, R.-Y. Bao, Z. Liu, B.-H. Xie, M.-B. Yang and Y. Wei, ACS Sustainable Chem. Eng., 2018, 6, 6761–6770 CrossRef.
- S. Wu, T. Li, Z. Tong, J. Chao, T. Zhai, J. Xu, T. Yan, M. Wu, Z. Xu, H. Bao, T. Deng and R. Wang, Adv. Mater., 2019, 31, 1905099 CrossRef.
- Z. Zhang and X. Fang, Energy Convers. Manage., 2006, 47, 303–310 CrossRef.
- L. Carreto, A. R. Almeida, A. C. Fernandes and W. L. C. Vaz, Biophys. J., 2002, 82, 530–540 CrossRef.
- J. Ding, X. Wu, X. Shen, S. Cui and X. Chen, Appl. Surf. Sci., 2020, 534, 147612 CrossRef.
- X. Wei, X.-Z. Jin, N. Zhang, X.-D. Qi, J.-H. Yang, Z.-W. Zhou and Y. Wang, Carbohydr. Polym., 2021, 253, 117290 CrossRef PubMed.
- J. Yang, E. Zhang, X. Li, Y. Zhang, J. Qu and Z.-Z. Yu, Carbon, 2016, 98, 50–57 CrossRef.
- H. Liao, W. Chen, Y. Liu and Q. Wang, Compos. Sci. Technol., 2020, 189, 108010 CrossRef.
- Z. Wang, Z. Tong, Q. Ye, H. Hu, X. Nie, C. Yan, W. Shang, C. Song, J. Wu, J. Wang, H. Bao, P. Tao and T. Deng, Nat. Commun., 2017, 8, 1478 CrossRef PubMed.
- R. Ahiska, H. Mamur and M. Ulis, J. Fac. Eng. Archit. Gazi Univ., 2011, 26, 889–896 Search PubMed.
- D. Kraemer, B. Poudel, H.-P. Feng, J. C. Caylor, B. Yu, X. Yan, Y. Ma, X. Wang, D. Wang, A. Muto, K. McEnaney, M. Chiesa, Z. Ren and G. Chen, Nat. Mater., 2011, 10, 532–538 CrossRef PubMed.
- Y. Gao, M. Zhang, Y. Cui, D. Bao, F. Xu, X. Shen, Y. Zhu and H. Wang, J. Mater. Chem. A, 2022, 10, 10452–10465 RSC.
- Y. F. Nassar, S. A. Yousif and A. A. Salem, Desalination, 2007, 209, 177–181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: AFM images and height profile of GO and GNPs; SEM images and digital photographs of the samples; Tm, ΔHm, Tc and ΔHc of the samples; DSC curves of HCGA2/tetradecanol after heating–cooling cycles; temperature difference between the bottom and top of HCGA2/tetradecanol during the solar–thermal energy conversion; determination of the beginning positions and ending positions of the melting processes of HCGA2/tetradecanol and HGA2/tetradecanol; temperature–time curves of the bottom of HCGA2/tetradecanol and tetradecanol; schematic of the components in the solar–thermal–electric energy conversion measurements; voltage–time curves and current–time curves of STE generators; digital photographs of LED bulbs lit by the STE generator and the output voltage; comparison of maximum output voltages and output currents of the STE generators in this work with those reported in the literature. See DOI: https://doi.org/10.1039/d2ta06457j |
| This journal is © The Royal Society of Chemistry 2022 |