
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of pre-analytical variables on Raman and FTIR spectral content of lymphocytes†
Jade F.
Monaghan
 ab,
Daniel
Cullen
ab,
Claire
Wynne
c,
Fiona M.
Lyng‡
*ab and
Aidan D.
Meade‡
*ab
ab,
Daniel
Cullen
ab,
Claire
Wynne
c,
Fiona M.
Lyng‡
*ab and
Aidan D.
Meade‡
*ab
aSchool of Physics, Clinical and Optometric Sciences, Technological University Dublin, Central Quad, City Campus, Grangegorman, D07 XT95, Ireland. E-mail: aidan.meade@tudublin.ie; fiona.lyng@tudublin.ie
bRadiation and Environmental Science Centre, Focas Research Institute, Technological University Dublin, Aungier Street, D02 HW71, Ireland
cSchool of Biological, Health and Sports Sciences, Technological University Dublin, Central Quad, City Campus, Grangegorman, D07 XT95, Ireland
First published on 26th September 2023
Abstract
The use of Fourier transform infrared (FTIR) and Raman spectroscopy (RS) for the analysis of lymphocytes in clinical applications is increasing in the field of biomedicine. The pre-analytical phase, which is the most vulnerable stage of the testing process, is where most errors and sample variance occur; however, it is unclear how pre-analytical variables affect the FTIR and Raman spectra of lymphocytes. In this study, we evaluated how pre-analytical procedures undertaken before spectroscopic analysis influence the spectral integrity of lymphocytes purified from the peripheral blood of male volunteers (n = 3). Pre-analytical variables investigated were associated with (i) sample preparation, (blood collection systems, anticoagulant, needle gauges), (ii) sample storage (fresh or frozen), and (iii) sample processing (inter-operator variability, time to lymphocyte isolation). Although many of these procedural pre-analytical variables did not alter the spectral signature of the lymphocytes, evidence of spectral effects due to the freeze–thaw cycle, in vitro culture inter-operator variability and the time to lymphocyte isolation was observed. Although FTIR and RS possess clinical potential, their translation into a clinical environment is impeded by a lack of standardisation and harmonisation of protocols related to the preparation, storage, and processing of samples, which hinders uniform, accurate, and reproducible analysis. Therefore, further development of protocols is required to successfully integrate these techniques into current clinical workflows.
1. Introduction
In recent years, RS and FTIR spectroscopies have demonstrated their potential in translational and clinical applications such as screening, diagnosis, monitoring, and prognosis.1–8 This is mainly due to these complementary techniques sharing the same advantages, i.e., being non-destructive, label free, cost-effective modalities for analysis for biological samples which require minimal sample preparation. However, to exploit the clinical potential of these bio-analytical techniques, protocols first need to be developed to facilitate uniform and reliable spectroscopic analysis, to ensure discovery of robust spectral profiles with a high degree of sensitivity and specificity.Blood is already a commonly collected biosample in a clinical setting and is an invaluable source of information for spectral profiling of patients.9 Blood collection is minimally invasive, low-cost, and provides a temporal snap-shot of the biomolecular status of the patient.10 Blood contains erythrocytes, leukocytes (neutrophils, eosinophils, basophils, monocytes and lymphocytes; B, T, NK cells) and platelets which are suspended in plasma.11 Using density gradient centrifugation, it is possible to isolate peripheral blood mononuclear cells (PBMCs) from whole blood. This heterogeneous fraction is made up of immune cell subsets of dendritic cells (1–2%), monocytes (10–20%), NK cells (5–20%), and T (75–85%) and B lymphocytes (5–10%).12
Due to recent technological and computational advancements, FTIR and RS can identify and differentiate subpopulations of lymphocytes and monocytes.13,14 These techniques have also been demonstrated to be capable of differentiating between resting and activated lymphocytes, in addition to monocytes and macrophages.15–18 These proof-of-concept studies demonstrate that both RS and FTIR are invaluable tools for PBMC analysis. Other studies have shown that RS and FTIR analysis of PBMCs and pure lymphocytes holds great promise for clinical real-world applications in the analysis of malignancies, infectious diseases, immune response, occupational contaminants, drug response, radiotherapeutic response, and for retrospective radiobiological dosimetry.19–31
The potential of both RS and FTIR spectroscopy as a robust clinical tool is clear; however, both types of analysis are far from being translated into a clinic environment as currently there are no standardised protocols relating to sample preparation, storage, and processing. The biomolecular quality and composition of biological samples can be altered by environmental or technical pre-analytical variability, analytical variability and post-analytical variability.32 The nature of sample processing in the pre-analytical phase offers a window of susceptibility for the introduction of sample variance and has been deemed the most vulnerable phase of testing with up to 61.9% of laboratory errors originating at this point.33,34 The pre-analytical phase in this context can be defined as all procedures prior to the commencement of spectroscopic analysis.35
RS and FTIR datasets contain highly detailed information relating to the biomolecular composition of a sample, variability from sample preparation approach induced at the pre-analytical phase could possibly mask clinically important spectral features and/or be mistaken for a clinically significant finding. To the best of our knowledge, this study is the first of its kind, as no other study in the literature explores the biochemical variability in RS and FTIR of lymphocytes from multiple processing parameters at various stages of lymphocyte sample preparation.
2. Materials and methods
2.1. Ethical approval
Informed consent was obtained from healthy volunteers and peripheral blood was collected by venous blood sampling from three male human donors between 28 and 35 years of age. Therefore, for each pre-analytical variable investigated, three peripheral blood samples were obtained and analysed. All sample collections were carried out in accordance with the 1964 Helsinki Declaration36 and approved by the Technological University Dublin Research Ethics Committee (REC number 15-32). Volunteer information was anonymised prior to analysis.In summary, variables investigated in this study that have the potential to impact lymphocyte spectral information included:
Sample preparation: (1) the Vacuette® and S-Monovette® blood collection systems, (2) 21 and 23 gauge (G) needles, (3) EDTA (ethylenediaminetetra-acetic acid), lithium heparin (LH) and sodium citrate (SC) Vacuette® anticoagulant blood collection tubes. Sample storage: (1) freezing samples (frozen versus non-frozen lymphocytes). Sample processing: (1) different operators processing samples (inter-operator variability), and (2) lymphocytes isolated within 1 hour or after 24 hours post-sample collection (Fig. 1).
 | ||
| Fig. 1 Schematic depiction of the experimental design of each pre-analytical consideration. | ||
Unless otherwise indicated in the methodological summary (Table 1), peripheral blood samples were collected using the Vacuette® blood collection system (Greiner Bio-One, Stonehouse, UK) with Vacuette® LH anticoagulant (Greiner Bio-One), 21 gauge needle (Greiner Bio-One) and immediately processed by the same operator. All volunteer lymphocyte samples for each pre-analytical variable were analysed by FTIR and RS, except for FTIR analysis of lymphocytes collected with different needle gauges. This exception was due to the extended elapsed time between sample fixation and recording caused by the COVID-19 campus closure. As a result, there is a possibility that protein degradation occurred at an accelerated rate, which could have been misinterpreted as a spectroscopic effect related to the investigated needle gauges.37 For each pre-analytical variable, 10 mL of peripheral blood was collected from each volunteer. However, due to the availability of volunteers, samples were not obtained from the same three volunteers for each pre-analytical variable.
| Pre-analytical variable | Collection system | Anticoagulant | Needle gauge | Operator (n) | Time to lymphocyte isolation post-sample collection | Freezing samples |
|---|---|---|---|---|---|---|
| Vac = Vacuette®, SM = S-Monovette®. A detailed summary of the experimental design of each pre-analytical variable can be found in the ESI.† | ||||||
| Blood collection systems | Vac and SM | LH | 21 G | 1 | 1 h | Non-frozen |
| Needle gauges | Vac | LH | 21 G and 23 G | 1 | 1 h | Non-frozen |
| Anticoagulant | Vac | LH, SC, and EDTA | 21 G | 1 | 1 h | Non-frozen |
| Freezing samples | Vac | LH | 21 G | 1 | 1 h | Frozen and non-frozen |
| Inter-operator variability | Vac | LH | 21 G | 2 | 1 h | Non-frozen |
| Sample processing time | Vac | LH | 21 G | 1 | 1 h and post-24 h | Non-frozen |
2.2. Preparation of PBMC culture
PBMC culture preparation was carried out as previously described by Maguire et al.29 In a 50 mL polypropylene tube (Thermo Fisher Scientific Inc., MA, USA), 6 mL of room temperature Dulbecco's phosphate buffered saline (DPBS; Sigma Aldrich LLC, MO, USA) and volunteer blood were mixed. In a new 50 mL polypropylene tube, 15 mL of Histopaque-1077 (Sigma Aldrich LLC) was added and the mixture of DPBS and peripheral blood was pipetted on top, allowing the mixture to gently run down the side of the tube. Sample tubes were centrifuged at 400g for 30 minutes at 18 °C with deceleration set to zero to allow the whole blood to be separated into different fractions, i.e., density centrifugation. The PBMC layer was gently aspirated using a 5 mL pipette (SARSTEDT, Germany) and washed in 50 mL polypropylene tubes by adding DPBS to a total volume to 30 mL and mixed by inversion. The sample was then centrifuged at 400g for 4 minutes at 18 °C with the acceleration set to nine. The washing step was repeated twice more with 5 mL of DPBS added to the cell pellet at each wash, bringing the total number of washes to three. After the last wash, DPBS was removed, and the cell pellet was resuspended in 5 mL of full medium (RPMI + 12.5% vol/vol FBS + 2 mM L-glutamine; Sigma Aldrich LLC). The resuspended cell pellet was transferred to a 25 cm2 tissue culture flask (SARSTEDT) and supplemented with 2.5% vol/vol (50 μL) of phytohaemagglutinin (PAA Laboratories, West Yorkshire, UK). PBMCs were incubated with a Thermo Scientific Forma 320 CO2 incubator (Thermo Fisher Scientific Inc.) for 68–72 hours at 37 °C with 5% CO2. The flasks were placed in a horizontal orientation to facilitate the separation of lymphocytes and monocytes by plastic adherence.2.3. Calcium fluoride (CaF2) slide preparation
Lymphocytes were brought into suspension by gently swirling the tissue culture flasks. The supernatant containing the lymphocytes was aspirated into a new 30 mL polypropylene tube. The tissue culture flasks were then washed with 5 mL of DPBS. The DPBS was decanted into 30 mL polypropylene tubes (Thermo Fisher Scientific Inc.) and the washing step was repeated twice more. The sample tubes were centrifuged at 400g for 5 minutes at 18 °C with the deceleration and acceleration set to 9. The supernatant was decanted, and the cell pellet was resuspended in 200 μL of 2% vol/vol paraformaldehyde (PFA; AppliChem GmbH, Darmstadt, Germany). The cells were fixed for 10 minutes at room temperature. From each sample suspension, 20 μL was aliquoted onto a labelled CaF2 slide (Crystran Ltd, Dorset, UK), and allowed to settle and attach to the slide for 5 minutes, after which the liquid supernatant was removed. Cells were then washed in deionised water three times. Finally, excess deionised water was removed, and the sample was allowed to air dry.2.4. Acquisition of Raman spectra
Raman spectral acquisition was carried out using a protocol described previously.31 Before sample analysis, room temperature was set at 18 °C for 20 minutes. RS was performed using a Horiba Jobin Yvon LabRAM HR800 UV micro-Raman spectrometer (Horiba UK Ltd, Middlesex, UK), equipped with a cooled charge-coupled device (CCD) array detector with 1024 × 256 pixels and Peltier cooled to −70 °C. The Raman spectrometer was also equipped with a 660 nm solid-state diode laser that directed 100 mW power at the sample. An initial calibration of the detector used 520.8 cm−1 of crystalline silicon, as here pixels are assigned wavenumber values in the CCD camera.38 A 100× objective (numerical aperture = 0.9) and a 600 line per nm diffraction grating (centred at 1450 cm−1) was used. The confocal hole and filter transmission were set to 100 μm and 100%, respectively. Spectra (n = 3) of a known standard, 1,4-bis(2-methylstyryl)benzene (Sigma Aldrich LLC) were recorded with a 0.3 seconds integration time and averaged over 3 integrations per spectrum and used for wavenumber calibration in post-processing.39 Single cell spectra were recorded using a 4 × 4 μm raster scan of the centre of each cell with a 20 seconds integration time and averaged over 3 integrations, along with a spectral resolution of 1 cm−1. The Raman acquisition of the samples was performed over the spectral range of 1800–400 cm−1. Cells were visually inspected before recording to ensure mostly lymphocytes were included in subsequent Raman analysis. Between 25 and 35 spectra were recorded from each sample. The variation in the number of spectra recorded per sample is due to variability in sample quality. The CaF2 slides, with fewer recorded lymphocyte spectra, experienced a higher degree of cell loss during the final washing steps with deionized water, as outlined in Section 2.3. Spectral acquisition was carried out using LabSpec v.6 (Horiba, UK).2.5. Raman spectra pre-processing
All pre-processing of Raman spectra was performed in Matlab version R2017b (MathWorks Inc., MA, USA) and Toolbox v.8.0 (Eigenvector Research Inc., MA, USA) using in-house algorithms. All Raman spectra were wavenumber calibrated corrected with recorded spectra of 1,4-bis(2-methylstyryl)benzene. Following this, the Raman spectra were truncated to include the region of 1795–408 cm−1. Baseline removal was performed using a rubberband method40,41 and spectra were smoothed using a Savitzky–Golay filter (5th order polynomial, 15 point window, or 14 wavenumbers). Finally, all Raman spectra were standardised using vector normalisation. An example of the spectral pre-processing steps carried out on lymphocyte Raman spectra is provided in ESI Fig. 1.†2.6. Acquisition of FTIR spectra
The FTIR spectra were collected using a PerkinElmer Spotlight 400 mid-infrared imaging system (PerkinElmer, Waltham, MA) using a liquid-nitrogen cooled mercury cadmium (MCT) 16 × 1 linear array detector. This system is equipped with an AutoImage microscope system operating with a 40× Cassegrain objective. The FTIR acquisition of the samples was performed in transmission mode, over the spectral range of 4000–800 cm−1 and with an interferometer speed of 1.0 cm s−1. An aperture size of 100 μm × 100 μm was selected as this aperture size produced the spectra with the best signal to noise ratio. A total of 128 scans per pixel were recorded and averaged at a spectral resolution of 4 cm−1 to maximise the signal-to-noise ratio.42 Before each sample measurement, 256 background scans per pixel were recorded in an empty space on the CaF2 side to remove contributions from carbon dioxide and water vapour.43 For each sample, 25–35 spectra were recorded with each spectrum recorded from around 30–50 lymphocytes. As mentioned above, cells were visually inspected prior to recording to ensure that mainly lymphocytes were included in subsequent FTIR analysis. Spectral acquisition was performed using Spectrum-IMAGE-Spotlight-400 software v.10.5.4 (PerkinElmer, MA).2.7. FTIR spectra pre-processing
All pre-processing of FTIR spectra was carried out in OCTAVVS (Open Chemometrics Toolkit for Analysis and Visualisation of Vibrational Spectroscopy Data) version 0.1.17, in Python version 3.9.13.44First, atmospheric correction was performed to remove H2O and CO2 spectral contributions. Next, the distorted baseline due to Mie scattering was corrected using the resonant Mie scattering correction (RMieS) algorithm developed by Bassan et al.45 The settings selected within the RMieS algorithm include 20 iterations, 8 orthogonal principal components, and a matrigel reference spectrum. Following this, the FTIR spectra were truncated to the fingerprint region from 1800–900 cm−1. The spectral baseline was removed using a rubberband fitting method.40,41 Finally, prior to spectroscopic analysis, spectra were standardised using vector normalisation. The second derivative spectra were calculated using the Savitzky–Golay algorithm (2nd order polynomial, 9-point window, or 32 wavenumbers). The resulting spectra were used for multivariate analysis. An example of the spectral pre-processing steps carried out on lymphocyte FTIR spectra is provided in ESI Fig. 2.†
2.8. PCA
Principal Components Analysis (PCA) is an unsupervised dimensionality reduction technique and is used extensively in the identification of clusters, patterns, and outliers in spectroscopic datasets. Here, principal components analysis (PCA) was used for exploratory data visualisation.As PCA is sensitive to outliers, these must first be identified and removed before analysis is carried out. In this study, outlier removal was approached using the popular Hotelling T2 test as it provides an easily interpreted visual of outliers present within the spectroscopic datasets.46 Normalised RS and FTIR spectral datasets corresponding to a processing technique in each pre-analytical consideration were analysed for outliers via Hotelling T2 test. The Hotelling T2 test is the multivariate alternative to Student's T-test to assess the difference in the means of the data.47 In this study, 10 PCs were included in the Hotelling's T2 model and a 95% confidence limit was selected. Samples with a T2 range that fell outside of this range were removed from further analysis. Hotelling's T2 range represents the sum of normalised squares, which can also be explained as the distance of the sample from the centre to the projection of the sample onto the number of principal components included in the PCA model.48 A T2 score of 0 is obtained when the sample is projected onto the multivariate centre of the model.
For Raman spectra, PCA was performed on spectra acquired over the fingerprint region from 1795–408 cm−1, while for FTIR spectra, PCA was performed on the second derivative spectra over the fingerprint region from 1800–900 cm−1. For both Raman and FTIR spectra, PCA was performed with 10 principal components. There is no consensus on the number of PCs to include in a PCA model of spectroscopic datasets. However, lower PCs represent the most common information in the dataset while the opposite is said for higher PCs. Inclusion of the latter in a PCA model would involve the measurement of noise and hence, have been excluded from our analysis.49 The inclusion of 10 PCs produced a model describing X% of the variance in the data, whilst reducing its dimensionality substantially. PCA outputs were represented by 3-D score plots and 2-D score plots. For the latter, the covariance ellipse (95% confidence) was used to highlight the extent of the variance in each dataset.
3. Results and discussion
3.1. Sample preparation
Lymphocyte FTIR spectra of PBMCs drawn using the Vacuette® system were highly uniform as evidenced by the narrow standard deviation around each mean spectrum (Fig. 2A), and the similarly condensed dispersion in the PC scores (Fig. 2C). In contrast, the FTIR spectra of lymphocytes collected through the S-Monovette® blood collection system showed substantial variability over the wavenumber range of 994–1124 cm−1 in their mean spectra. This region of the spectrum is tentatively attributed to vibrations associated with glycogen, collagen, C–O–C stretching (in nucleic acids and phospholipids), DNA (due to PO2− vibrations), symmetric stretching of phosphate groups in phospholipids, and C–O stretching of phosphodiester and ribose. Similarly, the PC scores for this group depicted broad dispersion, reinforcing the variability in the spectra in this sample, while the loadings to PC1 (Fig. 2E) are associated with spectral regions dominated by contributions from nucleic acids and proteins (from 964–1092 cm−1 and 1518–1580 cm−1).
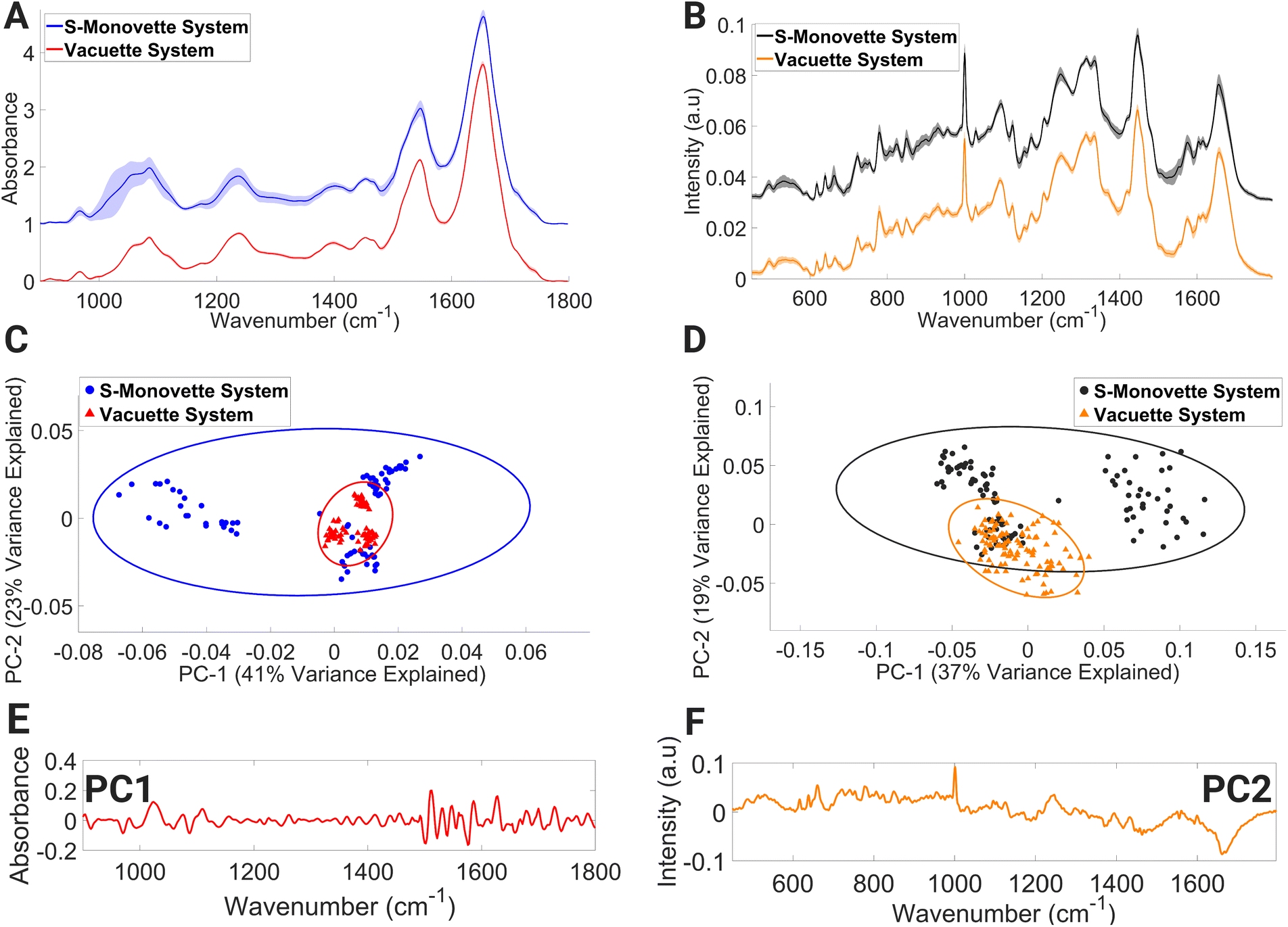 | ||
| Fig. 2 Mean FTIR (A) and Raman (B) spectra of lymphocytes collected with S-Monovette® or Vacuette® blood collection systems, PCA score plot of FTIR (C) and Raman (D) spectra, PC1 loading of FTIR spectra (E) and PC2 loading of Raman spectra (F). Covariance ellipses (95% confidence limit) in (C) and (D) are shown for each data class. | ||
A similar trend was also observed in the mean lymphocyte Raman spectra (Fig. 2B) for lymphocytes collected with the S-Monovette® system. Here, Raman bands tentatively assigned to ring breathing of tryptophan, guanine and thymine (648–674 cm−1), single-bond stretching vibrations of amino acids and polysaccharides (845–857 cm−1), amide III and lipids (1233–1269 cm−1), amide II, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) of carotenoids and tryptophan (1498–1587 cm−1), and ν(CO) of amide I and CC stretching of lipids displayed considerable variance compared to similar bands for the Raman spectra of lymphocytes collected using the Vacuette® system. The spectral variance seen in the PC score plot (Fig. 2D) is reflective of the observations in the FTIR spectra, with spectral regions of the loadings (Fig. 2F) dominated by Raman signatures from amide II β-sheet, amide I α-helix confirmation and lipids (1626–1700 cm−1). Compared to the aspiration method, i.e., manual gentle gradual drawing of the plunger, it has previously been shown that the effect of shear forces inflicted on the blood flow through the needle during the blood draw process via a conventional vacuum system can result in an increased rate of haemolysis in serum samples.50,51 The spectral profiles indicative of individual biochemical characteristics were detected in our FTIR lymphocyte spectral datasets and the S-Monovette® lymphocyte Raman spectra. This is particularly evident in the PCA plots (Fig. 2C and D) of the S-Monovette® lymphocyte spectral datasets, where a clear separation into three distinct subclusters is observed, with each subcluster representing a healthy volunteer.
C) of carotenoids and tryptophan (1498–1587 cm−1), and ν(CO) of amide I and CC stretching of lipids displayed considerable variance compared to similar bands for the Raman spectra of lymphocytes collected using the Vacuette® system. The spectral variance seen in the PC score plot (Fig. 2D) is reflective of the observations in the FTIR spectra, with spectral regions of the loadings (Fig. 2F) dominated by Raman signatures from amide II β-sheet, amide I α-helix confirmation and lipids (1626–1700 cm−1). Compared to the aspiration method, i.e., manual gentle gradual drawing of the plunger, it has previously been shown that the effect of shear forces inflicted on the blood flow through the needle during the blood draw process via a conventional vacuum system can result in an increased rate of haemolysis in serum samples.50,51 The spectral profiles indicative of individual biochemical characteristics were detected in our FTIR lymphocyte spectral datasets and the S-Monovette® lymphocyte Raman spectra. This is particularly evident in the PCA plots (Fig. 2C and D) of the S-Monovette® lymphocyte spectral datasets, where a clear separation into three distinct subclusters is observed, with each subcluster representing a healthy volunteer.
Furthermore, while Vacuette® lymphocyte Raman spectra are suggestive of a similar effect from shear forces in PBMCs, we did not observe an associated increase in haemolysis.
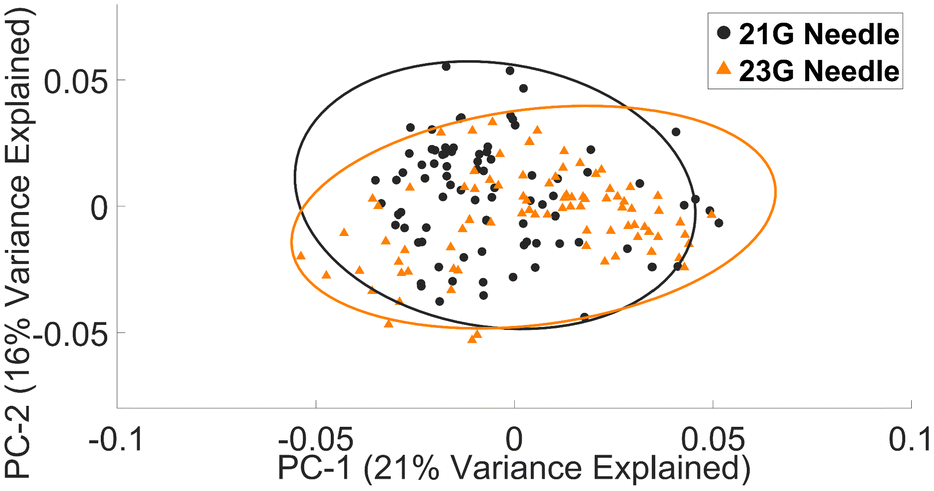 | ||
| Fig. 3 PCA plot of Raman lymphocyte spectra collected with a 21 G or 23 G needle. | ||
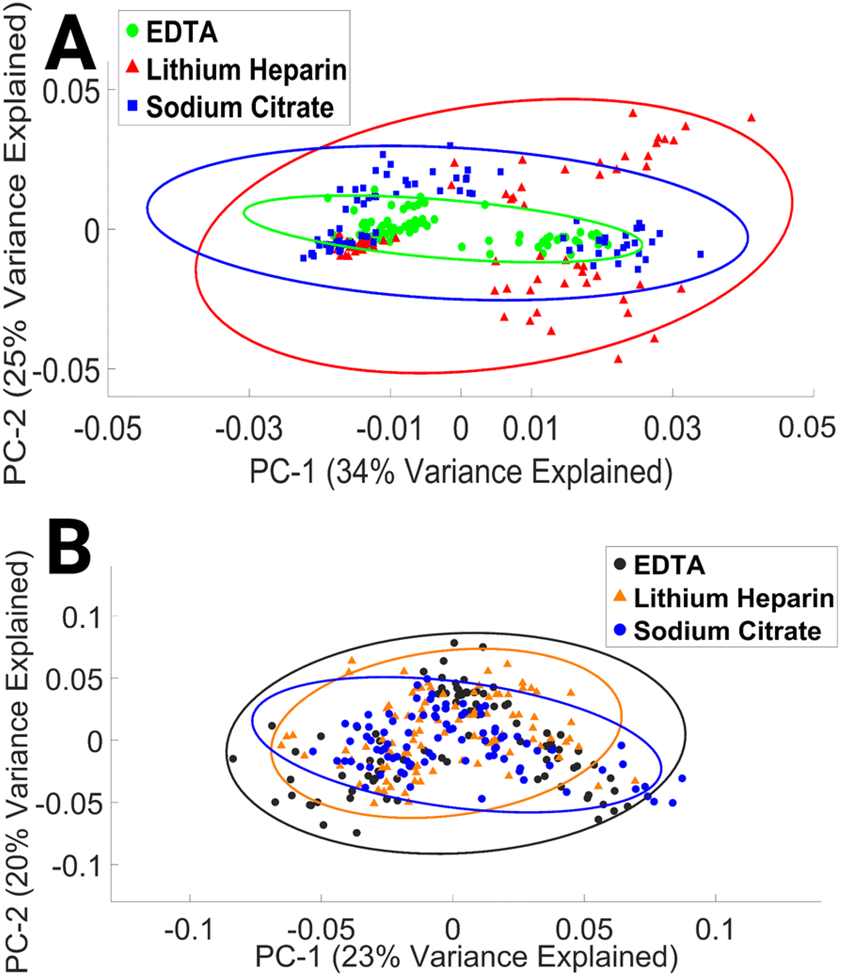 | ||
| Fig. 4 PCA score plot of FTIR (A) and Raman (B) spectra of lymphocytes collected with EDTA, LH, and SC anticoagulant. Covariance ellipses (95% confidence limit) in (A) and (B) are shown for each data class. | ||
To our knowledge, this study is the first to assess the impact of EDTA, LH, and SC anticoagulant on the spectral integrity of lymphocytes. The results presented here indicate that the choice of anticoagulant has a minimal effect on the spectral properties of lymphocytes. This finding may be explained by time in culture and subsequent numerous washing steps.
3.2. Sample storage
Freezing cells at −80 °C can have a significant impact on the functionality and structure of biomolecules such as lipids, proteins, and nucleic acids due to changes in hydrophobic and hydrophilic interactions.57 These changes may affect the content of FTIR and Raman spectra, which provide information on the molecular composition and structure of cells. However, it is not yet clear how a single freeze–thaw cycle influences the biochemical information of lymphocytes obtained from these spectroscopic techniques.
The mean FTIR and Raman spectra of non-frozen lymphocytes, i.e. processed immediately after the drawing of blood and frozen lymphocytes, i.e. a single freeze–thaw cycle can be seen in ESI Fig. 5.†
The PCA data of the FTIR spectra (Fig. 5A) cluster by donor and by preservation method, with clustering occurring along the PC1 axis. PC1 explains 62% of the variance between the data sets. The spectral spread across the negative PC1 axis (Fig. 5C) is due to the spectral regions consisting of contributions from C–O ribose (984–1000 cm−1), glycogen, collagen, and phosphodiester groups of nucleic acids (1034–1046 cm−1), β-sheet structure of amide I and CC of thymine, adenine (1630–1646 cm−1), amide I and νCC cis of lipids, fatty acids (1660–1670 cm−1), amide I and CO guanine deformation N–H in plane (1680–1692 cm−1). Variations in amide I vibrations that originate in the protein backbone indicate changes in the structure of secondary proteins and suggest instability of biomolecules after freezing at −80 °C.58
 | ||
| Fig. 5 PCA score plot of FTIR (A) and Raman (B) spectra of non-frozen (processed immediately) and frozen lymphocytes (single freeze–thaw cycle), PC1 loading of FTIR lymphocyte spectra (C) and PC1 loading of Raman lymphocyte spectra (D). Covariance ellipses (95% confidence limit) in (A) and (B) are shown for each data class. | ||
The PCA score plot of Raman spectra (Fig. 5B) demonstrates discrimination occurring across the PC1 axis, which accounts for 18% of the variance. Negative PC1 loadings (Fig. 5D) correspond to spectral regions dominated by contributions from O–P–O stretching of DNA (769–812 cm−1), phospholipids (1077–1107 cm−1), tryptophan and lipids (1361–1388 cm−1), DNA (1476–1495 cm−1), and phenylalanine, tyrosine, and cytosine (1563–1602 cm−1). In the literature, a decrease in protein, lipids, and DNA after freezing has previously been reported in plasma, calf thymus DNA, domestic cat oocytes and embryos, and prostate tumour cells.54,57,59–61 To increase the rigour of this research, it would be necessary to replicate these findings using a larger data set. Furthermore, this study did not account for several factors, such as the impact of multiple freeze–thaw cycles, long-term storage, and the temperature and time required for effective lymphocyte thawing, on the spectral content of lymphocytes. Investigating these aspects of lymphocyte storage would be of great interest for future research, especially since it is common practice in clinical settings to resample patient biological material due to the low volume of biological material usually obtained.
3.3. Sample processing
Within the mean FTIR spectral data sets of both operators (Fig. 6A), large variations in spectral regions associated with contributions from vibrational modes of C–C and C–O of deoxyribose (956–976 cm−1), νC–O of carbohydrates, DNA, glycogen, phosphodiester, and phospholipids (998–1144 cm−1) were observed. For operator 1, large intensity variation was observed in the range (1492–1540 cm−1); this spectral intensity trend was not found in the operator 2 dataset. Within the operator 2 dataset, large intensity-related variation was also observed in the spectral region dominated by molecular signatures from amide II, C–N stretching of cytosine and guanine (1256–1368 cm−1), α-sheet of amide II (1542–1552 cm−1), amide II (1566–1600 cm−1), CO of thymine and guanine (1700–1722 cm−1), and CO stretching of lipids and fatty acid ester bond (1728–1746 cm−1).
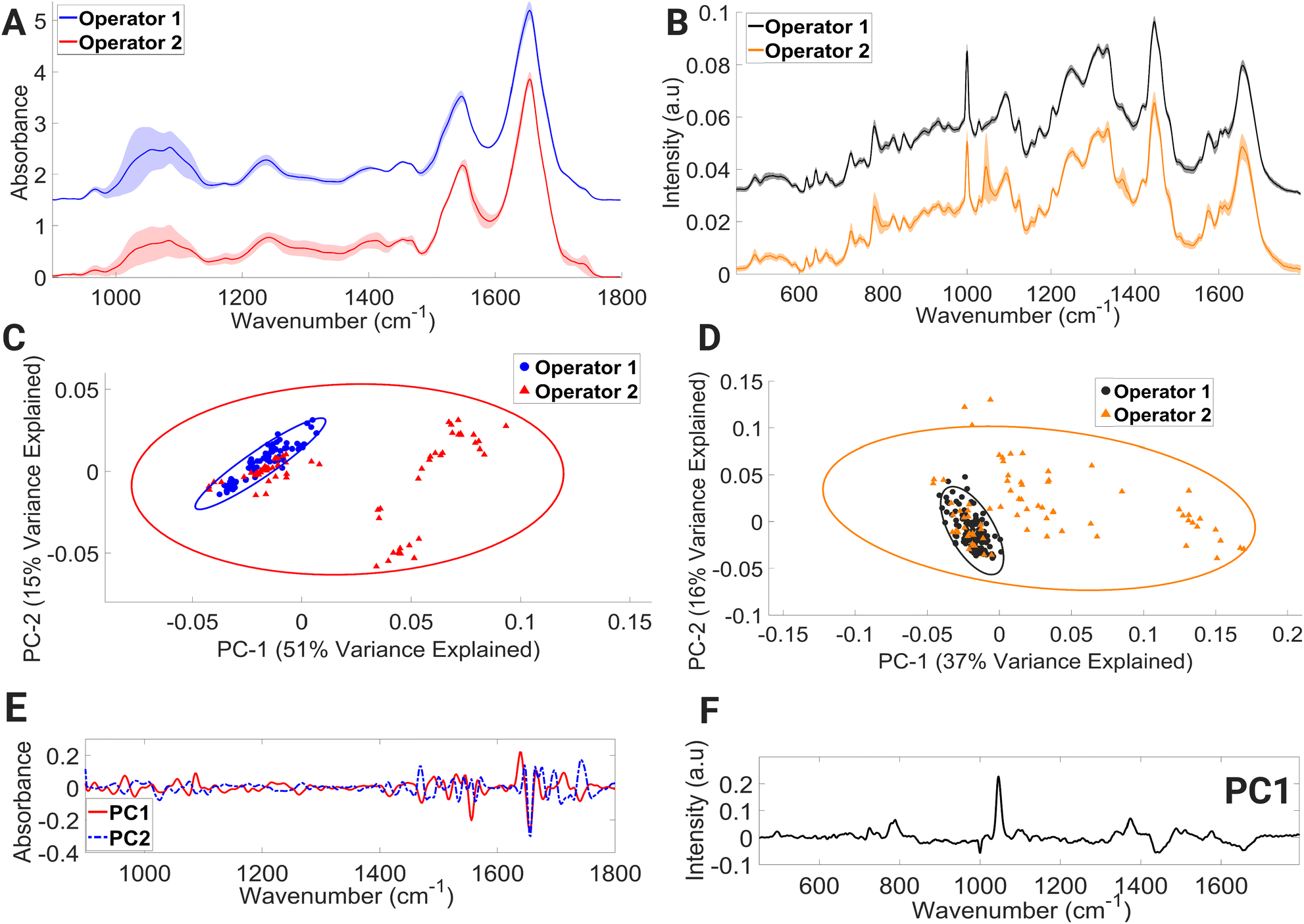 | ||
| Fig. 6 Mean FTIR (A) and Raman (B) spectra of lymphocytes processed by operator 1 and 2, PCA score plot of FTIR (C) and Raman (D) spectra, PC1 and PC2 loading of FTIR spectra (E), PC1 loading of Raman spectra (F). Covariance ellipses (95% confidence limit) in (C) are shown for each data class. | ||
PCA (Fig. 6C) showed overlapping covariance ellipses with operator 1 forming a tight spectral datapoint cluster indicating spectral homogeneity. However, the spectral spread within the operator 2 data set can be seen across the positive PC1 and negative PC2 axis, accounting for 51% and 15% of the variance, respectively. Positive PC1 loadings (Fig. 6E) correspond to the spectral regions consisting of contributions from β-sheet structure of amide I, while negative PC2 loadings (Fig. 6E) correspond to spectral regions associated with α-helix of amide I. This change in protein confirmation and/or protein concentration is most likely due to the handling technique of operator 2. The process of isolating peripheral PBMCs from whole blood using density gradient separation is a multistep procedure that demands a high level of technical proficiency and attention to detail from the operator. Any deviation from protocols during the various washing and centrifugation steps can negatively impact PBMCs, ultimately leading to alterations in the spectral integrity of lymphocytes.
The mean Raman spectra of lymphocytes processed by operators 1 and 2 are shown in Fig. 6B. Visually, operator 2 contains major spectral-intensity related variation in the regions consisting of contributions from ring breathing modes of uracil, thymine, and cytosine (775–796 cm−1), ring breathing modes of thymine, guanine, and adenine (1360–1380 cm−1), amide I and CC stretch of lipids (1649–1669 cm−1), and triglycerides, lipids, and CH2 bending mode of proteins and lipids (1441–1452 cm−1). The most prevalent spectral intensity-related variation can be observed over the wavenumber range of 1037–1057 cm−1. A PCA model (Fig. 6D) was further constructed to aid in interpretation of these spectral alternations. The PCA plot shows that the spectral data points of operator 1 hug the central axis, while operator 2 shows spread across the positive PC1 axis, with PC1 accounting for 37% of variance. The positive PC1 loadings (Fig. 6F) display the largest variance in the region of 1037–1057 cm−1. However, it is shown in these results that with proper training and due diligence, an analyst can consistently generate accurate results while minimising any impact on the spectral integrity of the lymphocytes, as evidenced by the results produced by operator 1.
N of adenine and cytosine (1492–1554 cm−1), and vibrational modes of amide I and nucleic acids (1640–1658 cm−1).
 | ||
| Fig. 7 Mean FTIR (A) and Raman (B) spectra of lymphocytes processed immediately and 24 hours after collection, PCA score plot of FTIR (C) and Raman (D) spectra, PC2 loading of FTIR spectra (E), PC1 loading of Raman spectra (F). Covariance ellipses (95% confidence limit) in (C) are shown for each data class. | ||
The PCA score plot of FTIR spectra (Fig. 7C) displays two distinct spectral data class clusters and shows discrimination across the PC2 axis, which accounts for 10% of variance between the data classes. Negative PC2 loadings (Fig. 7E) are dominated by spectral regions associated with collagen & phosphodiester groups of nucleic acids (1026–1052 cm−1), amide I (CO stretching) and CO, CN, N–H of adenine, thymine, guanine, and cytosine (1642–1652 cm−1), amide I and νCC cis of lipids and fatty acids (1660–1668 cm−1), and amide I (1688–1700 cm−1). While positive PC2 loadings are dominated with spectral regions with contributions from carbohydrates: glucose and glycogen (1010–1026 cm−1), C–O stretching of deoxyribose (1052–1074 cm−1), C–O stretching vibration of C–OH group of ribose (1110–1136 cm−1), amide II (1530–1542 cm−1), and amide I (1652–1660 cm−1). The highest variance exists for amide I, nucleic acids, and carbohydrates.
The processing of lymphocytes after 24 hours of a venous blood draw results in the introduction of variance in spectral regions dominated by contributions from numerous Raman molecular fingerprints (Fig. 7B) such as glycogen and carotenoids (1133–1167 cm−1), CH2/CH3 twisting of lipids and collagen, (1301–1321 cm−1), CH3 band (1387–1407 cm−1), cytosine, adenine, amide II and tryptophan (1499–1562 cm−1), and amide I and CC stretch of lipids (1647–1671 cm−1). Processed within 1 hour, spectral datapoints are spread into three distinct clusters across the PC2 and PC3 axes, while processed after 24 hours, spectral datapoints are spread across the PC2 axis while also demonstrating spread across the negative PC1 axis. The first three PCs account for 41%, 23% and 13%, respectively (Fig. 7D). Negative PC1 loadings (Fig. 7F) consist of spectral regions tentatively assigned to Raman signatures representing C–N stretching of protein vibrations, glycogen, and carotenoids (1111–1167 cm−1), CH3 band (1380–1411 cm−1), and ring breathing mode of DNA bases and amide II (1498–1560 cm−1). Based on the Raman spectra presented here, it is possible to detect Raman signatures that highlight protein-related structural changes and cellular stress in lymphocytes processed after 24 hours.
Whole blood collection often occurs at a remote location from the laboratory where sample processing takes place, resulting in a delay between collection and laboratory processing.62 According to the European Directorate for the Quality of Medicines & Healthcare,63 it is standard practice in Europe to store blood for up to 24 hours at ambient temperature. However, a recent study by Hope et al.64 has shown that storage of blood samples for 24 hours at ambient temperature does not negatively impact PBMC viability and total viable cell numbers when compared to samples stored at 4 °C. Despite these previous findings, 24 hours ambient storage of whole blood can introduce variance in FTIR spectra, particularly in the 1002–1072 cm−1 region, which can have a detrimental effect on downstream spectroscopic analysis and classification. Therefore, spectroscopists working with whole blood samples stored at 24 hours at ambient temperature must exercise caution and consider this factor when developing and interpreting machine learning models. The findings of the 24 hours processed lymphocytes analysed by RS revealed variability in their biochemical response, despite being processed under identical experimental conditions. This suggests that lymphocytes processed in this manner may exhibit varying levels of cell injury, which could be attributed to suboptimal storage conditions. These findings underscore the need for further investigation using a larger cohort in order to fully assess the impact of processing lymphocytes after 24 hours on intra-sample spectral variation.
It is acknowledged that lymphocyte isolation from PBMCs by density gradient separation does not ensure a pure lymphocyte population will be obtained following cell culture. This issue can be minimised by selecting cells on visual characteristics with the coupled microscope before recording with the Raman and FTIR instrumentation.
4. Conclusion
To successfully implement FTIR and RS in clinical settings, it is essential to identify sources of pre-analytical variability, establish standardised protocols, and ensure the accuracy, quality, and reproducibility of spectral data. Our study investigated pre-analytical variables related to sample preparation, storage, and processing, and the results confirmed the following:• The S-Monovette® blood collection system and anticoagulants had minimal impact on FTIR and Raman spectral outcomes.
• Vacuette®-collected lymphocytes had little impact on lymphocyte FTIR spectral quality, but Raman spectra of lymphocytes possessed signals of cellular injury.
• Common needles (21 G or 23 G) had little impact on the spectral profile of lymphocytes.
• Although DMSO was used as a cryoprotectant to prevent cellular membrane damage, FTIR and RS can detect biochemical differences present in cells after a single freeze–thaw cycle at −80 °C.
• Adequate training is a critical element for spectral reproducibility, and although the issue is not fully resolved in this current study, a foundation is provided to improve reproducibility in spectral measurements.
• Processing blood samples for lymphocyte isolation 24 hours after collection had a significant negative impact on the integrity of lymphocyte spectra, particularly in the case of FTIR analysis.
Developing a standardised protocol for lymphocyte spectroscopic analysis will ensure fitness of use of lymphocyte samples, eliminate the influence of external factors, reduce costs associated with addressing pre-analytical errors, and enhance the credibility of results in future studies. This study is limited by its small sample size, and further research is needed to establish fully standardised protocols.
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
A. D. M., F. M. L., J. F. M. and D. C. conceived the study, with A. D. M. and F. M. L. supervising the study. C. W. assisted D. C. with the collection of volunteer blood. J. F. M. and D. C. performed FTIR and Raman analyses, respectively. J. F. M. performed multivariate analysis, manuscript writing, and design of the illustrations. A. D. M. developed in-house software for the purpose of spectral calibration and analysis. All authors contributed equally to reviewing and editing the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
J. F. M. is funded by Fiosraigh Enterprise Award co-funded by TU Dublin and St Luke's Institute for Cancer Research. D. C. was funded by a TU Dublin Fiosraigh scholarship.References
- D. Traynor, S. Duraipandian, R. Bhatia, K. Cuschieri, C. M. Martin and J. J. O’Leary, et al., The potential of biobanked liquid based cytology samples for cervical cancer screening using Raman spectroscopy, J. Biophotonics, 2019, 12(7), e201800377 CrossRef PubMed.
- F. M. Lyng, D. Traynor, I. R. M. Ramos and F. Bonnier, Raman spectroscopy for screening and diagnosis of cervical cancer, Anal. Bioanal. Chem., 2015, 407(27), 8279–8289 CrossRef CAS PubMed.
- M. S. Nogueira, L. B. Leal, W. D. Marcarini, R. L. Pimentel, M. Muller and P. F. Vassallo, et al., Rapid diagnosis of COVID-19 using FT-IR ATR spectroscopy and machine learning, Sci. Rep., 2021, 11(1), 15409 CrossRef CAS PubMed.
- A. G. Carota, B. Campanella, R. Del Carratore, P. Bongioanni, R. Giannelli and S. Legnaioli, Raman spectroscopy and multivariate analysis as potential tool to follow Alzheimer's disease progression, Anal. Bioanal. Chem., 2022, 414(16), 4667–4675 CrossRef CAS PubMed.
- A. Salman, G. Sebbag, S. Argov, S. Mordechai and R. K. Sahu, Early detection of colorectal cancer relapse by infrared spectroscopy in “normal” anastomosis tissue, J. Biomed. Opt., 2015, 20(7), 075007 CrossRef PubMed.
- A. Shrivastava, L. M. Aggarwal, C. Murali Krishna, S. Pradhan, S. P. Mishra and S. Choudhary, et al., Diagnostic and prognostic application of Raman spectroscopy in carcinoma cervix: A biomolecular approach, Spectrochim. Acta, Part A, 2021, 250, 119356 CrossRef CAS PubMed.
- M. S. Vidyasagar, K. Maheedhar, B. M. Vadhiraja, D. J. Fernendes, V. B. Kartha and C. M. Krishna, Prediction of Radiotherapy Response in Cervix Cancer by Raman spectroscopy: a pilot study, Biopolymers, 2008, 89(6), 530–537 CrossRef CAS PubMed.
- L. Lovergne, D. Ghosh, R. Schuck, A. A. Polyzos, A. D. Chen and M. C. Martin, et al., An infrared spectral biomarker accurately predicts neurodegenerative disease class in the absence of overt symptoms, Sci. Rep., 2021, 11(1), 15598 CrossRef CAS PubMed.
- C. C. Compton, J. A. Robb, M. W. Anderson, A. B. Berry, G. G. Birdsong and K. J. Bloom, et al., Preanalytics and precision pathology: Pathology practices to ensure molecular integrity of cancer patient biospecimens for precision medicine, Arch. Pathol. Lab. Med., 2019, 143(11), 1346–1363 CrossRef CAS PubMed.
- A. L. Mitchell, K. B. Gajjar, G. Theophilou, F. L. Martin and P. L. Martin-Hirsch, Vibrational spectroscopy of biofluids for disease screening or diagnosis: Translation from the laboratory to a clinical setting, J. Biophotonics, 2014, 7(3–4), 153–165 CrossRef CAS PubMed.
- C. M. Carter, Alterations in Blood Components, Compr. Toxicol., 2018, 249–293 CAS.
- C. R. Kleiveland, Peripheral Blood Mononuclear Cells, in The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models, ed. K. Verhoeckx, P. Cotter, I. López-Expósito, C. Kleiveland, T. Lea and A. Mackie, et al., Springer, Cham, 2015 Search PubMed.
- N. Chaudhary, T. N. Que Nguyen, A. Maguire, C. Wynne and A. D. Meade, Comparison of sample preparation methodologies towards optimisation of Raman spectroscopy for peripheral blood mononuclear cells, Anal. Methods, 2021, 13(8), 1019–1032 RSC.
- M. Verdonck, S. Garaud, H. Duvillier, K. Willard-Gallo and E. Goormaghtigh, Label-free phenotyping of peripheral blood lymphocytes by infrared imaging, Analyst, 2015, 140(7), 2247–2256 RSC.
- B. R. Wood, B. Tait and D. McNaughton, Fourier-transform infrared spectroscopy as a tool for detecting early lymphocyte activation: a new approach to histocompatibility matching, Hum. Immunol., 2000, 61(12), 1307–1314 CrossRef CAS PubMed.
- A. I. Mazur, J. L. Monahan, M. Miljković, N. Laver, M. Diem and B. Bird, Vibrational spectroscopic changes of B-lymphocytes upon activation, J. Biophotonics, 2013, 6(1), 101–109 CrossRef CAS PubMed.
- N. Chaudhary, T. N. Q. Nguyen, D. Cullen, A. D. Meade and C. Wynne, Discrimination of immune cell activation using Raman micro-spectroscopy in an in-vitro & ex-vivo model, Spectrochim. Acta, Part A, 2021, 248, 119118 CrossRef CAS PubMed.
- N. Pavillon, A. J. Hobro, S. Akira and N. I. Smith, Noninvasive detection of macrophage activation with single-cell resolution through machine learning, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(12), E2676–E2685 CrossRef PubMed.
- A. H. Agbaria, G. Beck Rosen, I. Lapidot, D. H. Rich, M. Huleihel and S. Mordechai, et al., Differential Diagnosis of the Etiologies of Bacterial and Viral Infections Using Infrared Microscopy of Peripheral Human Blood Samples and Multivariate Analysis, Anal. Chem., 2018, 90(13), 7888–7895 CrossRef CAS PubMed.
- A. H. Agbaria, G. Beck, I. Lapidot, D. H. Rich, J. Kapelushnik and S. Mordechai, et al., Diagnosis of inaccessible infections using infrared microscopy of white blood cells and machine learning algorithms, Analyst, 2020, 145(21), 6955–6967 RSC.
- A. H. Agbaria, G. B. Rosen, I. Lapidot, D. H. Rich, S. Mordechai and J. Kapelushnik, et al., Rapid diagnosis of infection etiology in febrile pediatric oncology patients using infrared spectroscopy of leukocytes, J. Biophotonics, 2020, 13(2), e201900215 CrossRef PubMed.
- A. Pistiki, A. Ramoji, O. Ryabchykov, D. Thomas-Rüddel, A. T. Press and O. Makarewicz, et al., Biochemical Analysis of Leukocytes after In Vitro and In Vivo Activation with Bacterial and Fungal Pathogens Using Raman Spectroscopy, Int. J. Mol. Sci., 2021, 22(19), 10481 CrossRef CAS PubMed.
- A. Ramoji, D. Thomas-Rüddel, O. Ryabchykov, M. Bauer, N. Arend and E. J. Giamarellos-Bourboulis, et al., Leukocyte Activation Profile Assessed by Raman Spectroscopy Helps Diagnosing Infection and Sepsis, Crit. Care Explor., 2021, 3(5), e0394 CrossRef PubMed.
- P. Leszczenko, A. Borek-Dorosz, A. M. Nowakowska, A. Adamczyk, S. Kashyrskaya and J. Jakubowska, et al., Towards Raman-Based Screening of Acute Lymphoblastic Leukemia-Type B (B-ALL) Subtypes, Cancers, 2021, 13(21), 5483 CrossRef CAS PubMed.
- E. Gavgiotaki, G. Filippidis, I. Zerva, G. Kenanakis, E. Archontakis and S. Agelaki, et al., Detection of the T cell activation state using nonlinear optical microscopy, J. Biophotonics, 2019, 12(3), e201800277 CrossRef PubMed.
- D. K. D. do Nascimento Santos, B. R. da Silva Barros, L. M. Aguiar de Souza, I. J. da Cruz Filho, V. M. B. de Lorena and C. M. L. de Melo, et al., Immunostimulatory and antioxidant activities of a lignin isolated from Conocarpus erectus leaves, Int. J. Biol. Macromol., 2020, 150, 169–177 CrossRef PubMed.
- L. Xiao, S. Shelake, M. Ozerova, K. M. Balss, K. Amin and A. Tsai, Spectral Markers for T Cell Death and Apoptosis—A Pilot Study on Cell Therapy Drug Product Characterization Using Raman Spectroscopy, J. Pharm. Sci., 2021, 110(12), 3786–3793 CrossRef CAS PubMed.
- P. Wen, X. Wei, G. Liang, Y. Wang, Y. Yang and L. Qin, et al., Long-term exposure to low level of fluoride induces apoptosis via p53 pathway in lymphocytes of aluminum smelter workers, Environ. Sci. Pollut. Res., 2019, 26(3), 2671–2680 CrossRef CAS PubMed.
- A. Maguire, I. Vegacarrascal, L. White, B. McClean, O. Howe and F. M. Lyng, et al., Analyses of Ionizing Radiation Effects In Vitro in Peripheral Blood Lymphocytes with Raman Spectroscopy, Radiat. Res., 2015, 183(4), 407–416 CrossRef CAS PubMed.
- A. D. Meade, A. Maguire, J. Bryant, D. Cullen, D. Medipally and L. White, et al., Prediction of DNA damage and G2 chromosomal radio-sensitivity ex vivo in peripheral blood mononuclear cells with label-free Raman micro-spectroscopy, Int. J. Radiat. Biol., 2019, 95(1), 44–53 CrossRef CAS PubMed.
- D. Cullen, J. Bryant, A. Maguire, D. Medipally, B. McClean and L. Shields, et al., Raman spectroscopy of lymphocytes for the identification of prostate cancer patients with late radiation toxicity following radiotherapy, Transl. Biophotonics, 2020, 2(4), e201900035 CrossRef.
- C. Ellervik and J. Vaught, Preanalytical variables affecting the integrity of human biospecimens in biobanking, Clin. Chem., 2015, 61(7), 914–934 CrossRef CAS PubMed.
- G. Lima-Oliveira, W. Volanski, G. Lippi, G. Picheth and G. C. Guidi, Pre-analytical phase management: a review of the procedures from patient preparation to laboratory analysis, Scand. J. Clin. Lab. Invest., 2017, 77(3), 153–163 CrossRef PubMed.
- P. Carraro and M. Plebani, Errors in a Stat Laboratory: Types and Frequencies 10 Years Later, Clin. Chem., 2007, 53(7), 1338–1342 CrossRef CAS PubMed.
- P. Bonini, M. Plebani, F. Ceriotti and F. Rubboli, Errors in Laboratory Medicine, Clin. Chem., 2002, 48(5), 691–698 CrossRef CAS.
- The World Medical Association, The Declaration of Helsinki, Ethical Principles for Medical Research Involving Human Subjects, 1964.
- A. I. Mazur, et al., Evaluating different fixation protocols for spectral cytopathology, Part I, Anal. Chem., 2012, 84(3), 1259–1266 CrossRef CAS PubMed.
- L. T. Kerr, K. Domijan, I. Cullen and B. M. Hennelly, Applications of Raman spectroscopy to the urinary bladder for cancer diagnostics, Photonics Lasers Med., 2014, 3(3), 193–224 Search PubMed.
- ASTM International, ASTM E1840-96 Standard Guide for Raman Shift Standards for Spectrometer Calibration, 2014.
- S.-J. Baek, et al., Baseline correction using asymmetrically reweighted penalised least squares smoothing, Analyst, 2015, 140(1), 250–257 RSC.
- S. Wartewig, IR and Raman Spectroscopy: Fundamental Processing, Wiley, 1st edn, 2003 Search PubMed.
- K. Z. Liu, M. Xu and D. A. Scott, Biomolecular characterisation of leucocytes by infrared spectroscopy, Br. J. Haematol., 2007, 136(5), 713–722 CrossRef CAS PubMed.
- A. G. Theakstone, C. Rinaldi, H. J. Butler, J. M. Cameron, L. R. Confield and S. H. Rutherford, et al., Fourier-transform infrared spectroscopy of biofluids: A practical approach, Transl. Biophotonics, 2021, 3(2), e202000025 CrossRef.
- C. Troein, S. Siregar, M. Op De Beeck, C. Peterson, A. Tunlid and P. Persson, OCTAVVS: A Graphical Toolbox for High-Throughput Preprocessing and Analysis of Vibrational Spectroscopy Imaging Data, Methods Protoc., 2020, 3(2), 34 CrossRef CAS PubMed.
- P. Bassan, A. Kohler, H. Martens, J. Lee, H. J. Byrne and P. Dumas, et al., Resonant Mie Scattering (RMieS) correction of infrared spectra from highly scattering biological samples, Analyst, 2010, 135(2), 268–277 RSC.
- C. Wang, Y. Long, W. Li, W. Dai, S. Xie and Y. Liu, et al., Exploratory study on classification of lung cancer subtypes through a combined K-nearest neighbour classifier in breathomics, Sci. Rep., 2020, 10(1), 5880 CrossRef CAS PubMed.
- M. Aslam and O. H. Arif, Multivariate Analysis under Indeterminacy: An Application to Chemical Content Data, J. Anal. Methods Chem., 2020, 2020, e1406028 Search PubMed.
- J. Kuligowski, G. Quintás, C. Herwig and B. Lendl, A rapid method for the differentiation of yeast cells grown under carbon and nitrogen-limited conditions by means of partial least squares discriminant analysis employing infrared micro-spectroscopic data of entire yeast cells, Talanta, 2012, 99, 566–573 CrossRef CAS PubMed.
- J. R. Beattie and F. W. L. Esmonde-White, Exploration of principal component analysis: Deriving principal component analysis using spectra, Appl. Spectrosc., 2021, 75(4), 361–375 CrossRef CAS PubMed.
- G. Lippi, P. Avanzini, R. Musa, F. Sandei, R. Aloe and G. Cervellin, Evaluation of sample haemolysis in blood collected by S-Monovette using vacuum or aspiration mode, Biochem. Med., 2013, 23(1), 64–69 CrossRef PubMed.
- G. Lippi, P. Bonelli, L. Bonfanti and G. Cervellin, The use of S-Monovette is effective to reduce the burden of haemolysis in a large urban emergency department, Biochem. Med., 2015, 25(1), 69–72 CrossRef PubMed.
- A. Wollowitz, P. E. Bijur, D. Esses and E. John Gallagher, Use of Butterfly Needles to Draw Blood Is Independently Associated With Marked Reduction in Haemolysis Compared to Intravenous Catheter, Acad. Emerg. Med., 2013, 20(11), 1151–1155 CrossRef PubMed.
- G. Lippi, G. L. Salvagno, M. Montagnana, G. Brocco and G. C. Guidi, Influence of the needle bore size used for collecting venous blood samples on routine clinical chemistry testing, Clin. Chem. Lab. Med., 2006, 44(8), 1009–1014 CAS.
- L. Lovergne, P. Bouzy, V. Untereiner, R. Garnotel, M. J. Baker and G. Thiéfin, et al., Biofluid infrared spectro-diagnostics: Pre-analytical considerations for clinical applications, Faraday Discuss., 2016, 187, 521–537 RSC.
- M. Martin, D. Perez-Guaita, D. Andrew, J. Richards, B. Wood and P. Heraud, The effect of common anticoagulants in detection and quantification of malaria parasitemia in human red blood cells by ATR-FTIR spectroscopy, Analyst, 2017, 142(8), 1192–1199 RSC.
- J. E. Lovelock and M. W. Bishop, Prevention of freezing damage to living cells by dimethyl sulphoxide, Nature, 1959, 183(4672), 1394–1395 CrossRef CAS PubMed.
- W. F. Wolkers, S. K. Balasubramanian, E. L. Ongstad, H. C. Zec and J. C. Bischof, Effects of freezing on membranes and proteins in LNCaP prostate tumor cells, Biochim. Biophys. Acta, 2007, 1768(3), 728–736 CrossRef CAS PubMed.
- R. Arunkumar, C. J. Drummond and T. L. Greaves, FTIR Spectroscopic Study of the Secondary Structure of Globular Proteins in Aqueous Protic Ionic Liquids, Front. Chem., 2019, 7(74) CAS.
- E. Galyuk, R. Wartell, Y. Dosin and D. Lando, DNA Denaturation Under Freezing in Alkaline Medium, J. Biomol. Struct. Dyn., 2009, 26, 517–523 CrossRef CAS PubMed.
- K. A. Okotrub, V. I. Mokrousova, S. Y. Amstislavsky and N. V. Surovtsev, Lipid Droplet Phase Transition in Freezing Cat Embryos and Oocytes Probed by Raman Spectroscopy, Biophys. J., 2018, 115(3), 577–587 CrossRef CAS PubMed.
- J. C. Bischof, W. F. Wolkers, N. M. Tsvetkova, A. E. Oliver and J. H. Crowe, Lipid and protein changes due to freezing in Dunning AT-1 cells, Cryobiology, 2002, 45(1), 22–32 CrossRef CAS PubMed.
- P. F. van der Meer and D. de Korte, The Effect of Holding Times of Whole Blood and Its Components During Processing on In Vitro and In Vivo Quality, Transfus. Med. Rev., 2015, 29(1), 24–34 CrossRef PubMed.
- European Directorate for the Quality of Medicines & Healthcare, Guide to the preparation, use and quality assurance of blood components, European Pharmacopoeia, Council of Europe, Strasbourg, 19th edn, 2017 Search PubMed.
- C. M. Hope, D. Huynh, Y. Y. Wong, H. Oakey, G. B. Perkins and T. Nguyen, et al., Optimization of Blood Handling and Peripheral Blood Mononuclear Cell Cryopreservation of Low Cell Number Samples, Int. J. Mol. Sci., 2021, 22(17), 9129 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00686g |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |