
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArsenic in Lake Geneva (Switzerland, France): long term monitoring, and redox and methylation speciation in an As unpolluted, oligo-mesotrophic lake†
Montserrat
Filella
 *a,
Sebastian
Wey
a,
Tomáš
Matoušek
b,
Mathieu
Coster
c,
Juan-Carlos
Rodríguez-Murillo
d and
Jean-Luc
Loizeau
a
*a,
Sebastian
Wey
a,
Tomáš
Matoušek
b,
Mathieu
Coster
c,
Juan-Carlos
Rodríguez-Murillo
d and
Jean-Luc
Loizeau
a
aDepartment F.-A. Forel for Environmental and Aquatic Sciences, University of Geneva, Boulevard Carl-Vogt 66, CH-1205 Geneva, Switzerland. E-mail: montserrat.filella@unige.ch; Sebastian.Wey@etu.unige.ch; jean-luc.loizeau@unige.ch
bInstitute of Analytical Chemistry of the Czech Academy of Sciences, Veveří 97, 602 00 Brno, Czech Republic. E-mail: matousek@biomed.cas.cz
cService de l’écologie de l’Eau, Geneva, Switzerland. E-mail: mathieu.coster@etat.ge.ch
dMuseo Nacional de Ciencias Naturales, CSIC, Serrano 115 dpdo, E-28006 Madrid, Spain. E-mail: jcmurillo@mncn.csic.es
First published on 4th February 2023
Abstract
Arsenic speciation was followed monthly along the spring productivity period (January–June 2021) in the Petit Lac (76 m deep) and in April and June 2021 in the Grand Lac (309.7 m deep) of Lake Geneva (Switzerland/France). Lake Geneva is presently an oligo-mesotrophic lake, and As-unpolluted. The water column never becomes anoxic but the oxygen saturation at the bottom of the Grand Lac is now below 30% owing to lack of water column mixing since 2012. Thus, this lake offers excellent conditions to study As behaviour in an unpolluted, oxic freshwater body. The following ‘dissolved’ As species: iAs(III), iAs(III + V), MA(III), MA(III + V), DMA(III + V), and TMAO were analysed by HG-CT-ICP-MS/MS. Water column measurements were complemented with occasional sampling in the main rivers feeding the lake and in the interstitial waters of a sediment core. The presence of MA(III) and TMAO and the predominance of iAs(V) in lake and river samples has been confirmed as well as the key role of algae in the formation of organic species. While the total ‘dissolved’ As concentrations showed nearly vertical profiles in the Petit Lac, As concentrations steadily increase at deeper depths in the Grand Lac due to the lack of mixing and build up in bottom waters. The evaluation of 25 years of monthly data of ‘dissolved’ As concentrations showed no significant temporal trends between 1997 and 2021. The observed seasonal character of the ‘dissolved’ As along this period coincides with a lack of seasonality in As mass inventories, pointing to a seasonal internal cycling of As species in the water column with exchanges between the ‘dissolved’ and ‘particulate’ (i.e., algae) fractions.
Environmental significanceThe presence and fate of arsenic in aquatic environments have been widely studied, but its chemical speciation and cycling in lakes have received relatively little attention so far. Moreover, most of the lakes studied were shallow, eutrophic, and heavily polluted with arsenic. The present study investigates the behaviour of arsenic in a natural environment at background concentrations, thus providing a basis to better assess the fate of this element in polluted environments. To this end, we carefully applied state-of-the-art analytical techniques to the waters of a deep, oligo-mesotrophic, arsenic unpolluted lake that never becomes anoxic. Collected data on the speciation of arsenic each month during the spring productivity period show that the production and presence of reduced and methylated As species are mainly biota related. In addition, the availability of 25 years of monthly data on arsenic concentrations in this lake allows us to contextualize our findings and provides additional insight into the behaviour of arsenic. |
Introduction
Arsenic (As) is among the trace elements receiving the most public and scientific attention mainly because of its occurrence in contaminated groundwater and association with human health problems. Even though the toxicity of As has been known for a long time,1 the massive pollution of drinking water by As in Bangladesh, where as many as 35–50 million people were exposed to toxic levels,2,3 put As pollution on the environmental agenda. Moreover, the problem is not restricted to Bangladesh but is widespread across the planet.4Natural sources of environmental As include volcanism, hydrothermal activity, and weathering of rocks that contain As-bearing minerals. Anthropogenic activities such as ore smelting, fossil fuel combustion, and the use of agricultural chemicals are also sources of As in the environment. Geogenic As contamination of groundwater rather than pollution linked to the intentional use of the element or to the exploitation of other chemical elements is the largest public health issue for As.
Arsenic can exist in four oxidation states: As(−III), As(0), As(III), and As(V). Elemental As is rare and traces of arsines have been detected only in gases released from anoxic systems.5 In aqueous solutions, As is predominantly found in oxidation states III and V. Trivalent inorganic As, iAs(III), is in the form of As(OH)3 over the whole environmental pH range, and iAs(V) as H2AsO4− and HAsO42−, depending on the water pH (pK 7.03, http://jess.murdoch.edu.au/jess_home.htm). The thermodynamically stable form of As in oxygenated water is iAs(V) and, thus, negligible concentrations of the reduced form should be present in oxic waters. In practice, however, although iAs(V) is the dominant species, iAs(III) is always present. The redox speciation of As has also been found to be far from thermodynamic equilibrium in anoxic and sulfidic environments.6 Arsenic speciation determines the solubility and the mobility of inorganic As. In the aquatic environment, iAs(V) and iAs(III) show different behaviours: iAs(V) has a higher tendency than iAs(III) to be adsorbed onto the surface of minerals and, therefore, the mobility of iAs(V) in natural water is constrained, leaving iAs(III) as the more mobile As oxyanion.7 It is widely accepted that As speciation in surface waters is linked to photosynthetic activity6 and that consecutive steps of As(V) reduction to As(III) and oxidative methylation by cells is a detoxification mechanism widespread in bacteria,8 algae,9,10 mammals,11,12etc., with As(V) probably entering cells by following phosphate pathways due to structural similarities between the compounds.
Arsenic is also extensively present as an organometallic species in natural waters. Commonly detected methylated species in aqueous environments include methylarsonate (MA(V)), dimethylarsinate (DMA(V)), and trimethylarsine oxide (TMAO). Methylated As(III) species include methylarsonite (MA(III)) and dimethylarsinite (DMA(III)). The terms we use for the various As compounds can be found in Table SI1†13 and in the abbreviation list. The presence of these compounds is linked to the toxicity of inorganic As that makes algae undergo different processes to reduce As toxicity, including As(V) reduction, methylation, transformation into arsenosugars or arsenolipids, chelation of As(III) with glutathione and phytochelatins, as well as excretion from cells.14
Even if the presence and fate of As in the environment have been extensively studied, its behaviour in lakes has comparatively received scant attention. Published studies are summarized in Table 1. Previously studied lakes were generally shallow, eutrophic and polluted by As, and the number of sampling campaigns was generally low and sampling frequency limited. In this study, with the general objectives of (i) improving our understanding of As cycling in freshwaters by expanding the type of freshwater bodies studied, and (ii) evaluating the impact of aspects such as sampling frequency and the meaning and quality of analytical data used on the results obtained, we have mainly focused on two aspects: (i) tracking As speciation along the spring productivity period in an oligo-mesotrophic, As-unpolluted lake, and (ii) analyzing the temporal trends of As concentrations in the lake thanks to the existence of 25 years of monthly data of ‘dissolved’ As concentrations – rather uncommon information.
| Lake | Lake depth/m | Characteristics | Water sampled | # Sampling campaigns | Filtration | Technique | Inorganic species | Organic species | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a The majority of the samples were only analysed for iAs(III) and iAs(V). b Species called UV-As(III + V), UV-MA, UV-DMA were also measured. | |||||||||
| Pavin, France | 90 | Permanently stratified | Profile | 1: Dec | 0.4 μm | HG-AAS | iAs(III) and iAs(V) | — | 15 |
| 9 lakes, US | 20 | Seasonally anoxic | Surface | 1 | 0.4 μm | HG-AAS | iAs(III) and iAs(V) | MA and DMA | 16 |
| Davis Creek Reservoir, US | Profile | 5: Jul, Sep, Oct, Dec, and Feb | |||||||
| Greifen, Switzerland | Max: 17.7 | Seasonally anoxic | Profile | Monthly: Aug 89 to Jan 91 | 0.45 μm | HG-AAS | iAs(III) and iAs(V) | MA(III + V) and DMA(III + V)a | 17 |
| Aberjona watershed | Strongly As polluted | Surface | 0.45 μm | HG-AAS | iAs(III) and iAs(V) | MA(III + V) and DMA(III + V) | 18 | ||
| Hall's Brook | Average: 3 | 1 (5 locations) | |||||||
| Upper Mystic | ∼20 | Eutrophic | Profile | 11: Fall-spring | |||||
| Lower Mystic | ∼20 | Eutrophic | Profile | 6: Fall-spring | |||||
| Upper Mystic, US | 25 | Historically polluted | Profile | Several (unclear when) | 0.45 μm | HG-AAS | iAs(III) and iAs(V) | DMA(V) mentioned | 19 |
| Meg, Keg and Peg, Canada | Max: < 2 | Strongly As polluted | Surface | 1 | Not mentioned | HG-AAS | iAs(III) and iAs(V) | MA, DMA, TMAO, and MnAsIII(SR)3−n (n = 1, 2, 3) | 20 |
| Biwa, Japan; dredged area South basin | ca. 12 | Profile | Feb–Oct 1993 and apr–Dec 1994 | 0.45 μm | HG-AAS | iAs(III) and iAs(V) | MA(III), MA(V), DMA(III), and DMA(V) | 21 | |
| Biwa, Japan | Average: 44 | Mesotrophic, eutrophic | Profile | Monthly: 2 years | 0.45 μm | HG-AAS | iAs(III) and iAs(V) | MA(III + V), MA(III), DMA(III + V), and DMA(III) | 22 |
| 18 lakes, Japan | Mostly shallow, median: 4.1 | Eutrophic | Surface | 2: Summer & winter | 0.45 μm | HG-AAS | iAs(III) and iAs(V) | MA and DMAb | 23 |
| Mohawk, US | 1–7 | As polluted | Profile | 2: Sep & Oct | 0.45 μm | Not mentioned | iAs(III) and iAs(V) | MA and DMA | 24 |
| Taihu, China | Average: 2 | From hypereutrophic to mesotrophic | Surface | 4: Autumn, summer, winter, and spring | 0.45 μm | HPLC-ICP-MS | iAs(III) and iAs(V) | MA(V) and DMA(V) | 25 |
| 4 small subarctic lakes, Canada | 0.8–6.9 | As polluted, ice-covered | Surface | Several, covering 2 open water and 2 ice covered seasons | 0.45 μm | HG-AAS | iAs(III) and iAs(V) | — | 26 |
| Yamdrok, Tibet | Median: 30, max: 60 | As polluted, cold lake | Surface | 2: dry & wet seasons | 0.45 μm | HPLC-HG-AFS | iAs(III) and iAs(V) | DMA (only in one sample) | 27 |
| Aha reservoir, China | Average: 13 | Surface | 4 seasons | 0.45 μm | HPLC-ICP-MS | iAs(III) and iAs(V) | MA and DMA | 28 | |
Materials and methods
Study site
Lake Geneva is a freshwater lake of glacial origin on the border of Switzerland and France. With a surface area of 580.1 km2 and a volume of 89 km3,29 it is among the biggest freshwater lakes in Central Europe. The lake is often considered to consist of two parts (Fig. 1): the Petit Lac, that represents a relatively shallow part of smaller surface area to the west (max. depth: 76 m and 14% of the surface area), and the Grand Lac, that constitutes the major part in terms of water surface and volume (max. depth: 309.7 m and 96% of the volume). The lake water residence time is 11.3 years.29 Lake Geneva's catchment area includes some of the highest summits in the Alps and covers almost 8000 km2. The main characteristics of the lake and its watershed are summarised in Table SI2†.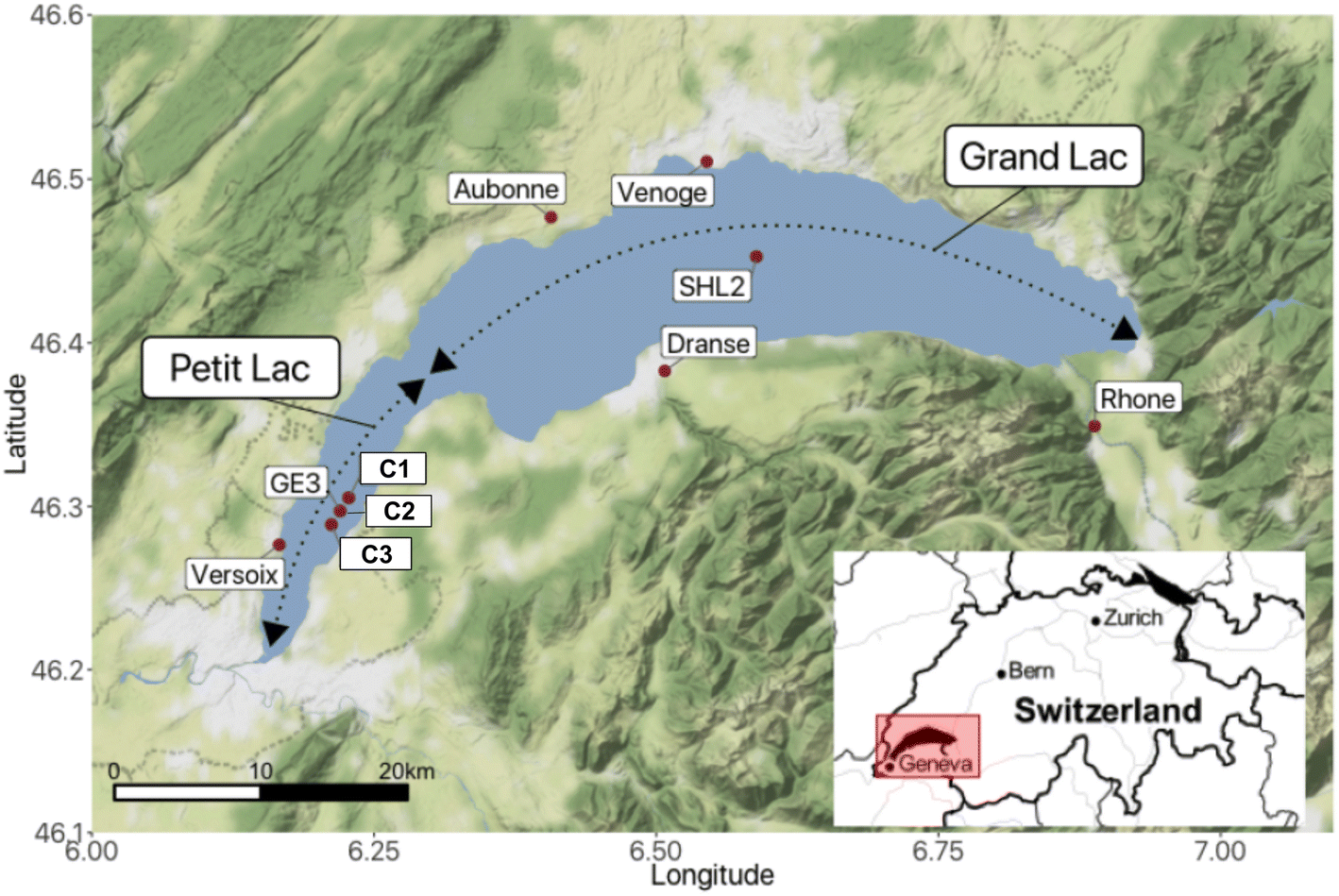 | ||
| Fig. 1 Lake Geneva (Switzerland/France) with sampling locations for rivers, lake and sediments. | ||
Lake Geneva has a monomictic regime with infrequent complete overturns in recent decades. After the last entire mixing of the water column in 2012, mixing in the Grand Lac has been limited to the upper layer (∼130–150 m depth), preventing oxygen from reaching the lake bottom through mixing, i.e., oxygen concentrations at the bottom never exceeded 4 mg L−1 during the entire year after 2016 and sediments remained poorly oxygenated.29 The Petit Lac mixes entirely once a year, and the build up of anoxia has never been observed.
Lake Geneva undergoes regular physicochemical, biological and micropollutant monitoring. Samples across the water column are taken once a month in the Petit Lac at sampling site GE3 (6.2197° E/46.2994° N) by the Service de l'Écologie de l'Eau of the Canton of Geneva (SECOE), Switzerland. Similar analyses are conducted at least once a month at the sampling location SHL2 (6.5887° E/46.4527° N), which corresponds to the deepest point of the lake.29
The phytoplankton community in Lake Geneva shows a seasonal pattern.30 In early spring, a first phytoplankton bloom is followed by an increasing zooplankton community that exerts heavy grazing pressure on the phytoplankton. This pattern leads to a clear water phase which usually occurs in June. However, global warming seems to make this phenomenon appear earlier and, in recent years, the clear water phase has been observed earlier in the season.30
Lake Geneva is fed by more than 60 rivers and streams. All tributaries longer than 10 km are listed in Table SI3.† Most incoming water (73%) enters the lake via the River Rhone to the east. The Rhone River has a glacial-nival regime with high flows in late spring and summer; water discharge values for the period 1935–2019 and for 2021 are shown in Fig. SI1.† Four other tributaries were sampled (Aubonne, Dranse, Venoge, and Versoix) in this study. Their regimes are pluvial with the highest discharge observed during winter and spring.
Sampling
Lake water samples were collected with Niskin bottles. Lake water samples at GE3 were transferred into 125 mL HDPE flasks (Nalgene) and stored refrigerated until further treatment. The flasks were cleaned with acid (1% HNO3) for at least one day, followed by incubation with Milli-Q water for at least another day. The flasks were further rinsed with lake water from the corresponding depth before water sampling. Lake water samples at SHL2 were transferred from the Niskin bottle into a glass beaker and immediately treated on site. In both cases, 15 mL was filtered using polyethersulfone (PES) filters of pore-size 0.20 μm and single-use syringes (HSW Henke-Ject). The samples were transferred into trace metal free centrifuge tubes (VWR) and 150 μL 0.2 M EDTA solution was added per 15 mL sample. The samples were refrigerated with cooling elements during the transfer from Geneva to Prague, where speciation analysis was carried out, and stored at 4 °C after arrival. Analyses for TMAO, MA(III) and iAs(III) were performed within 2–10 days from sampling. Analyses for iAs(III + V), MA(III + V) and DMA(III + V) were carried out within 3–13 days from sampling. Tests on preservation and matrix effects were performed in January 2021, before the first lake sampling campaign. The preservation of filtered samples (0.20 μm) stabilized by 2 mM EDTA and stored at 4 °C was verified by repeated analyses.
Previous sedimentological studies showed that surface sediments at this location consist mainly of silts, with 45% carbonates and 9% organic matter.31
Analysis
Concentrations of Astot,dis are available at site GE3 from 1997 and are used in this study to evaluate temporal trends. The concentrations were measured by ICP-MS on a Fisons PQ2+ before 2006, on a Thermo-Fisher X7 II from 2006 to 2015, and with an ICP-MS Agilent 7900 in high energy He mode since 2015 at SECOE. The analytical accuracy was followed by analysing the certified reference materials (CRM) SLRS-4, SLRS-5 and SLRS-6 (National Research Council Canada). The analytical laboratory is accredited for the analysis of the trace elements in freshwaters (accreditation number: STS245; ISO norm 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 025).
025).
For each series of As speciation analysis, a certified river water sample (SLRS-6) from the National Research Council of Canada was included as a quality control.
Other data
Monthly ancillary data (e.g., temperature, pH, conductivity, concentrations of oxygen, nutrients, and various metals, and phytoplankton counting) for location GE3 were provided by SECOE. They cover the period January 1997–December 2021. The analytical instruments used for metal analyses were changed twice since 1997. Analyses were performed by ICP-MS on a Fisons PQ2+ before 2006 and on a Thermo-Fisher X7 II from 2006 to 2015. Since 2015, concentrations are measured on an Agilent 7900. Ancillary data for sampling location SHL2 and ancillary data for sampling location SHL2 were provided by CARRTEL (Alpine Center for research on trophic networks and limnic ecosystems), France.Time series data statistical treatment
Non-parametric methods are required to analyse temporal trends of trace element concentrations in water because data do not typically follow a normal distribution.36 Thus, temporal trends can be studied by applying Mann–Kendall (MK) or seasonal Mann–Kendall (SMK) methods,37,38 depending on the presence or absence of seasonality in the data. The statistical significance of the trends is obtained in MK methods by applying the Z-statistic test of the sum of signs of the differences between every pair of values; Z shows a normal distribution, and therefore, the statistical significance of the temporal trend can be evaluated by using the p value. The Kruskall–Wallis test of equal medians for the data classified by depth and month is used to test for seasonality.Fourier power spectra of concentration time series have also been calculated to confirm seasonality. The REDFIT method used fits a first-order autoregressive process (AR(1)) to the concentration time series to calculate the Fourier spectrum.39 The 95% confidence limit of that spectrum is obtained with a Monte Carlo calculation. Spectral peaks above that confidence limit are considered as statistically significant.
Calculation programs used for MK and SMK tests are from https://www.mathworks.com/matlabcentral/fileexchange/11190-mann-kendalltau-b-with-sensmethod-enhanced/content/ktaub.m adapted by us. Kruskal–Wallis tests and REDFIT were performed with PAST.40
Results and discussion
Questions related to analytical methods
Rigorous tests on species stability and sample conservation were performed prior to analysis. Filtered samples (0.20 μm) stabilized by 2 mM EDTA addition and storage at 4 °C was proven a suitable strategy for all species including iAs(III) redox stability at tens of ng L−1 level for several months. This method was superior to acidification by HNO3 to pH 1 mainly due to minor method specific issues related to redox speciation based on pH selectivity of HG (see Section SI1†).
Great care was taken to verify that the small iAs(III) concentration of few tens of ng L−1 present in practically all samples was not due to an analytical artefact. However, no iAs(III) was observed in iAs(V) standards, nor in CRM SLRS-6 (acid conserved to pH 1.5). Additionally, less than 10 ng L−1 was found in SHL2 profile samples at depths higher than 100 m. All these facts makes us believe that the presence of low iAs(III) concentrations is real.
Due to the limited selectivity of the pH selective HG step, 1% of ‘apparent’ MA(III) and 6% of ‘apparent’ DMA(III) were generated from pentavalent forms, and assessed from the slopes of MA(V) and DMA(V) calibrations without prereduction. In all analysed samples, the proportion of DMA(III)/DMA(III + V) observed corresponded to this ratio and DMA(III) is believed to be absent in the samples, probably because this species is extremely unstable and easy to oxidize.13,41 On the other hand, a proportion of MA(III)/MA(III + V) higher than 1% and up to 10% was observed in samples except in January and February, sufficient to produce a distinctive profile. Caution should be, however, taken when interpreting MA(III) data, due to extremely low concentrations (2 ng L−1 As or below) and potentially limited redox stability (see Section SI1†).
The sum of the concentrations of all investigated As species (sumAs), i.e. the sum of iAs(III + V), MA(III + V), DMA(III + V) and TMAO, in G3 was always lower by 0.1–0.26 μg L−1 As (i.e. 8–24%) than Astot,dis (Tables SI7 and SI8†); a Kruskal–Wallis test performed on both sumAs and Astot,dis shows that the assumption of equal means among several months can be refused (p value < 0.0001). SumAs and Astot,dis however, closely followed the shape of the profiles along the season. Even an unexpected concentration drop in March 2021 was simultaneously observed in both, sumAs and Astot,dis. Similarly, sumAs concentrations were always lower than Astot,dis in the Grand Lac (Tables SI9 and SI10†). River samples showed the same behaviour (Tables 2 and 3) as those from the lake although differences seem to depend on the river with, for instance, values tending to agree more in the Rhone River than in other rivers. Astot,dis and sumAs values agreed well in sediments (Table SI11†). Both the certified and the measured values of sumAs were lower in SLRS-6 (Table SI6†) than the certified Astot,dis of 0.57 ± 0.08 μg L−1, value recently confirmed by a value of 0.58 ± 0.04 μg L−1.42 Three different laboratories contributed to the obtained values for Astot,dis and the deviation from sumAs was similar. Discrepancies between Astot,dis and sumAs had been attributed in the past to As organic compounds that are non-hydride reactive species,43–46 but it is unclear how such species could uniformly be present in all samples and also explain our observations. This discrepancy has no practical consequence for the conclusions reached in this study.
| River | Date | Time | Astot,dis | sumAs | iAs(V) | iAs(III) | MA(III + V) | DMA(III + V) | TMAO |
|---|---|---|---|---|---|---|---|---|---|
| Rhone | 16 April | 08:50 | 1190 | 1247 | 973 | 230 | 5.9 | 36,2 | 2.2 |
| 09:50 | 1151 | 1139 | 868 | 225 | 5.2 | 38.6 | 2.4 | ||
| 10:50 | 1200 | ||||||||
| 11:50 | 1166 | ||||||||
| 12:50 | 1204 | ||||||||
| Aubonne | 16 April | 16:20 | 187 | 161 | 110 | 37.2 | 0.9 | 11.7 | 1.1 |
| Dranse | 15 April | 09:00 | 134 | 116 | 62.2 | 42.7 | 0.9 | 9.3 | 0.5 |
| Venoge | 16 April | 15:20 | 336 | 207 | 118 | 58.5 | 3.9 | 23.1 | 3.1 |
| Versoix | 16 April | 17:20 | 219 | 158 | 106 | 25.7 | 1.6 | 23.0 | 1.4 |
| River | Date | Time | Astot,dis | sumAs | iAs(V) | iAs(III) | MA(III + V) | DMA(III + V) | TMAO |
|---|---|---|---|---|---|---|---|---|---|
| Rhone | 23 June | 08:50 | 908 | 879 | 742 | 68.2 | 2.4 | 64.8 | 1.2 |
| 09:50 | 767 | 752 | 668 | 58.6 | 1.6 | 22.4 | 1.2 | ||
| 10:50 | 781 | 743 | 661 | 56.4 | 1.5 | 22.4 | 1.3 | ||
| 11:50 | 721 | 713 | 625 | 52.3 | 1.5 | 24.5 | 1.2 | ||
| 12:50 | 762 | 717 | 629 | 52.5 | 1.7 | 32.7 | 1.2 | ||
| Aubonne | 22 June | 15:30 | 299 | 228 | 166 | 27.8 | 1.6 | 31.1 | 1.3 |
| Dranse | 22 June | 19:00 | 269 | 212 | 121 | 22.8 | 2.1 | 64.9 | 0.9 |
| Venoge | 22 June | 14:45 | 846 | 672 | 604 | 51.0 | 6.5 | 30.6 | 6.4 |
| Versoix | 22 June | 12:50 | 295 | 240 | 181 | 33.4 | 2.5 | 20.3 | 2.5 |
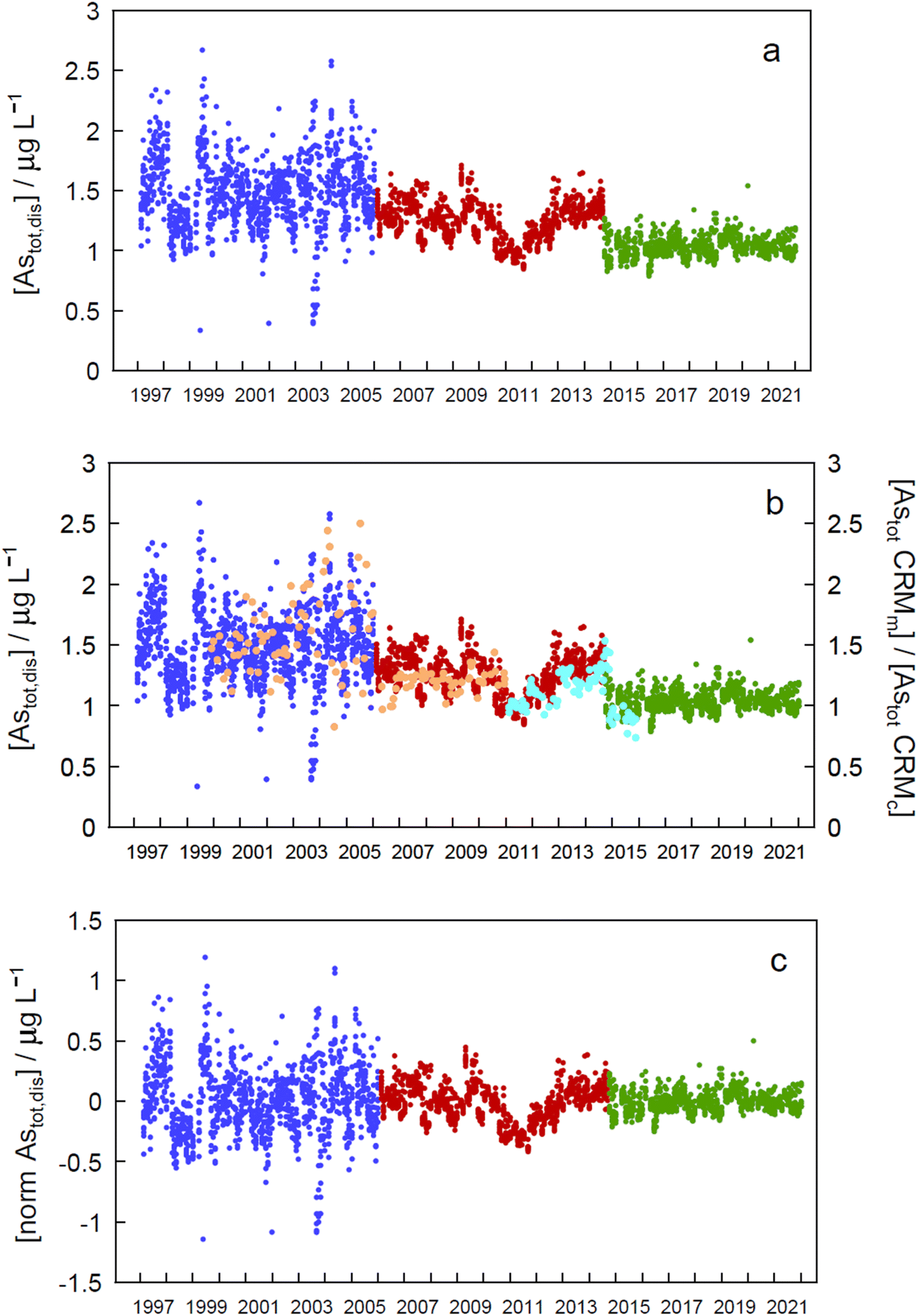 | ||
| Fig. 2 (a) Monthly total ‘dissolved’ arsenic concentrations (Astot,dis) at all sampling depths at sampling site GE3 between 1997 and 2021. Different colours correspond to different analytical instruments (blue, period one; red, period two; green, period three; see text for details). (b) Same data with values of the CRM simultaneously measured (Astot CRMm) and relative to their certified values (Astot CRMc) (orange: SLRS-4, certified 0.68 ± 0.06 μg L−1 As; turquoise: SLRS-5, certified 0.413 ± 0.039 μg L−1 As). (c) Same data after normalisation by using the median concentration of each period. | ||
Arsenic concentrations in the second period (in red in Fig. 2) show a marked decrease around 2011. Plotting As values in CRMs measured at the same time as the samples and normalized to their certified value (Fig. 2b, SLRS-4 in orange, and SLRS-5 in turquoise) clearly shows that the observed decrease is due to an analytical artefact and is not caused by lake processes. Thus, period 2 will be excluded in further analysis of the data.
Tracking arsenic along the spring productivity period
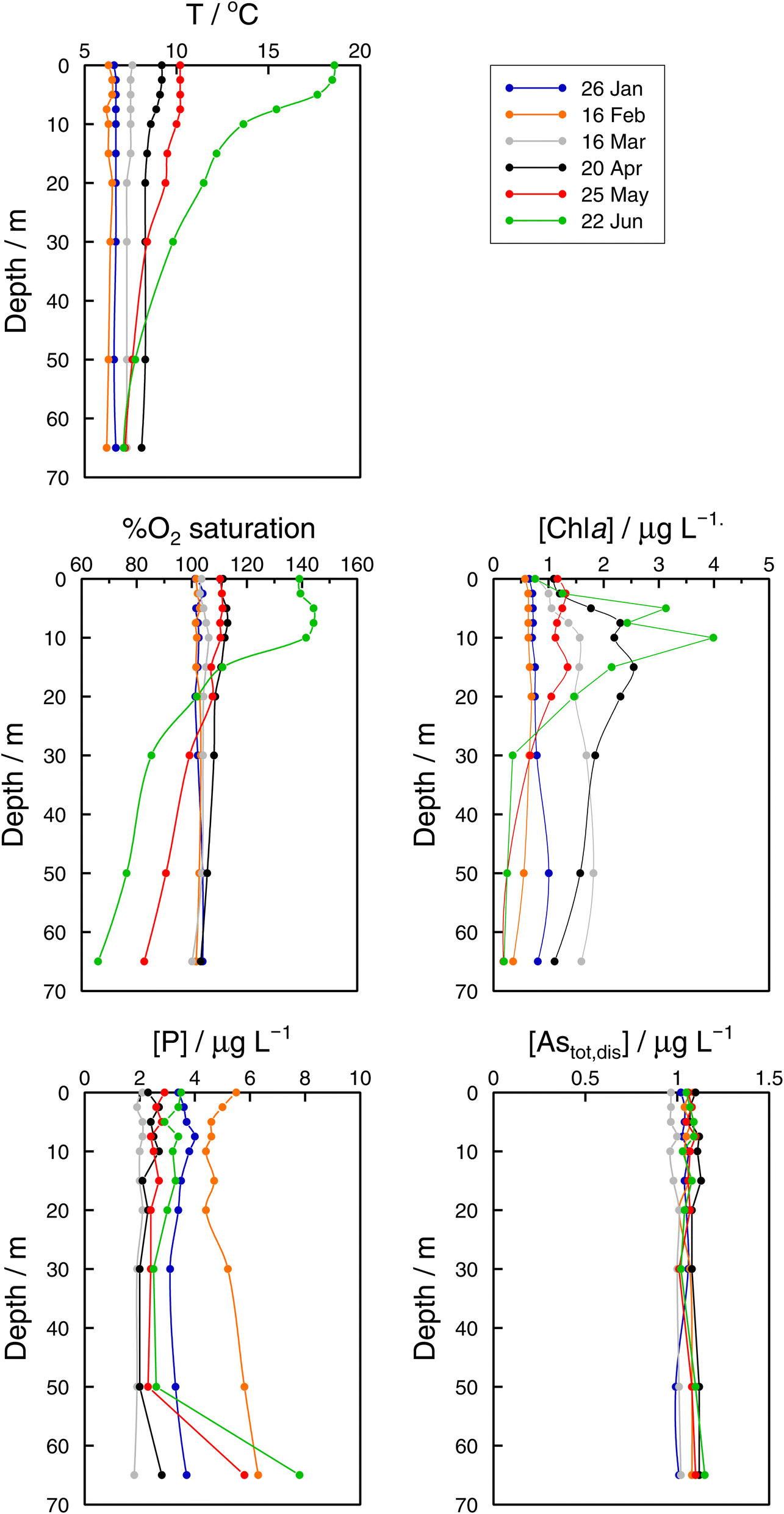 | ||
| Fig. 3 Vertical profiles of temperature, percentage of O2 saturation, chlorophyll a, dissolved reactive phosphorus (DRP), and total ‘dissolved’ arsenic, Astotal,dis, concentrations in the Petit Lac (point GE3) from January to June 2021. | ||
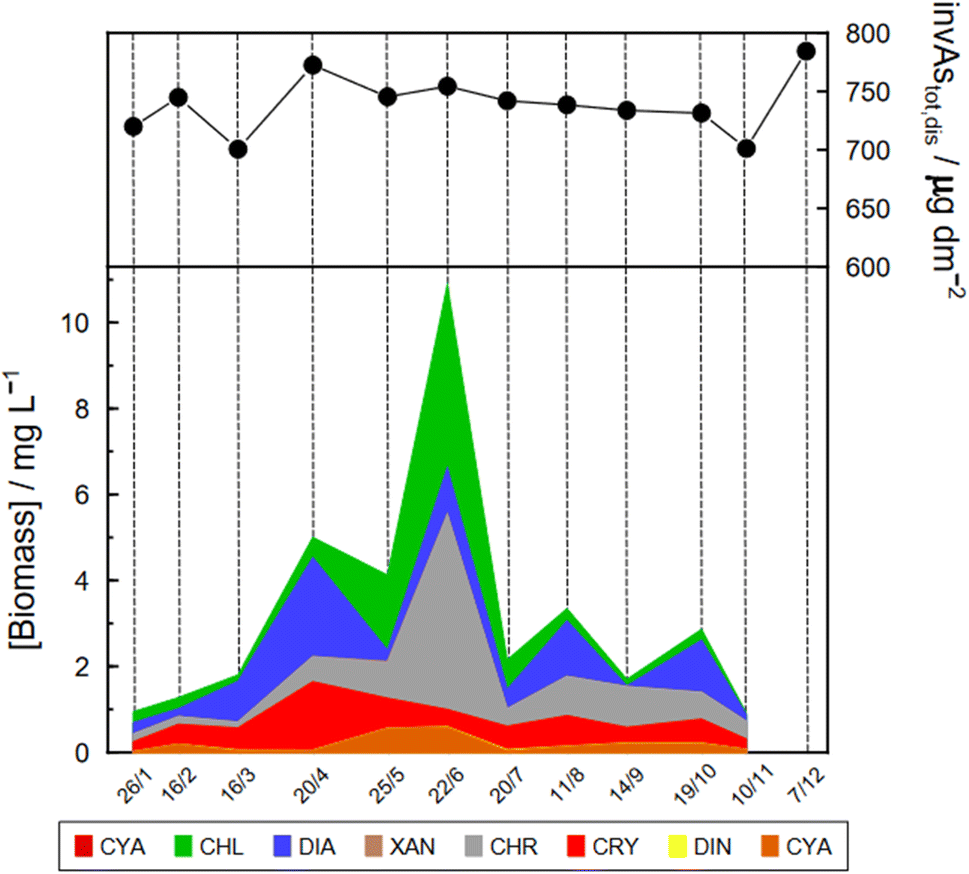 | ||
| Fig. 4 Monthly change in the biomass of phytoplankton groups at point G3 from January to November 2021 and corresponding change of invAstot,dis in the water column. Specific As mass inventories of Astot,dis were calculated by adding concentrations in layers of a virtual column of 1 dm2 section area and the height of the corresponding layer. CON: conjugatophyceae, CHL: chlorophyceae, DIA: diatoms; XAN: xantophyceae, CHR: chrysophyceae, CRY: cryptophyceae; DIN: dinophyceae, and CYA: cyanophyceae. | ||
In the Grand Lac, surface water temperatures were higher in June than in April (Fig. 5). This difference was limited to the surface layer, since below around 30 m, temperature profiles were reasonably similar between April and June. At the bottom of the lake, the water temperature was about 6 °C for both months. Chlorophyll a concentrations (Fig. 5) were higher in April than in June, with peaks observed at about the same depth (∼12 m). Surface water in June showed the higher oxygen saturation compared to that in April. Profiles of oxygen saturation were relatively similar below 50 m, with a minimum value of less than 30% reached at the bottom of the lake.
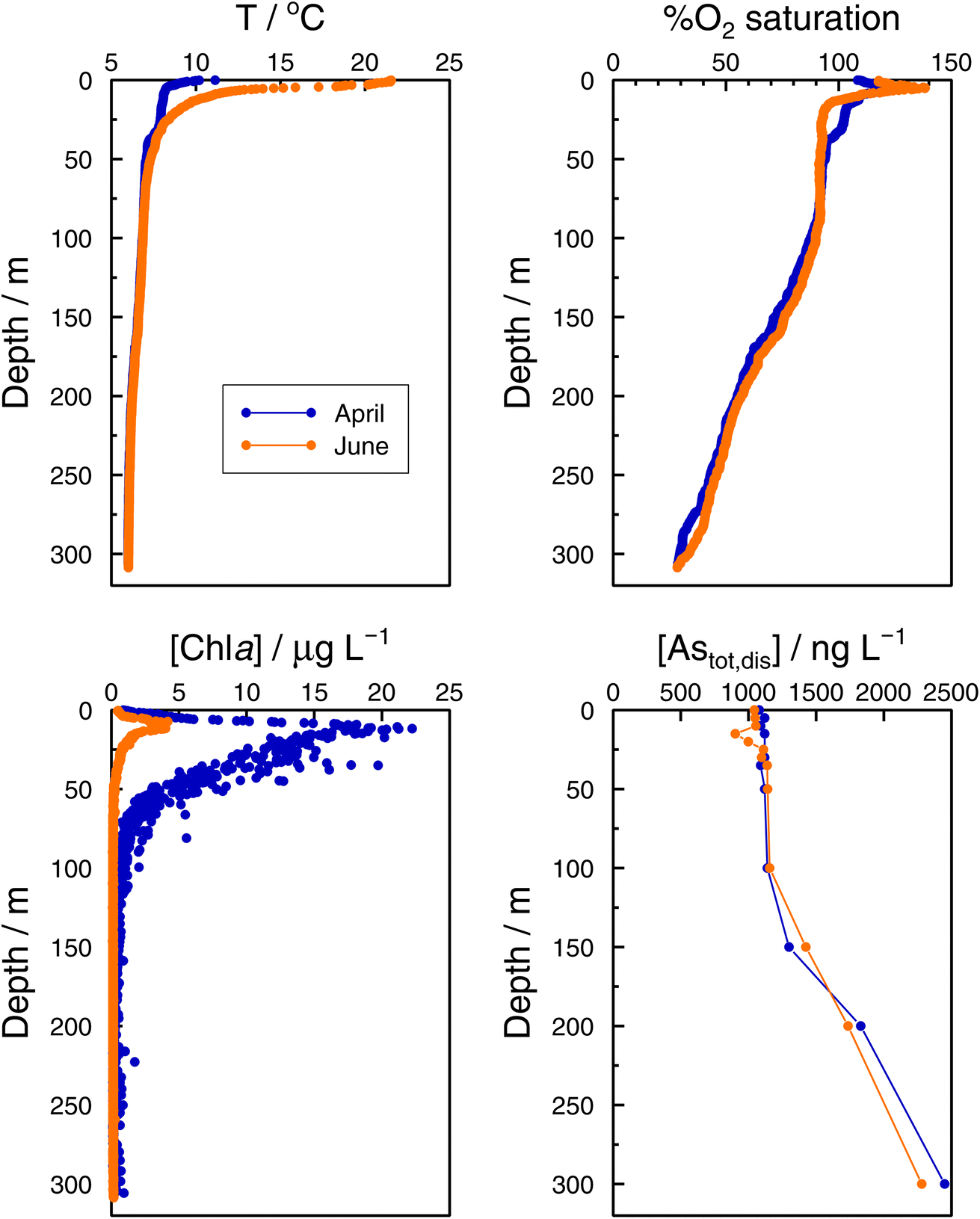 | ||
| Fig. 5 Vertical profiles of temperature, percentage of O2 saturation, chlorophyll a, and total dissolved arsenic, Astotal,dis in the Grand Lac (point SHL2) in April (blue) and June (orange) 2021. | ||
Specific As mass inventories, invAstot,dis, i.e. the mass of Astot,dis in an ideal water column of 1 dm2 section area were calculated by adding the mass of Astot,dis in the different layers resulting from the product of Astot,dis concentrations multiplied by the thickness of the corresponding layer. This parameter provides an indirect estimation of the incorporation of As into the ‘particulate’ phase, essentially phytoplankton, assuming that, at a given time, no sedimentation occurs. The results are shown in Fig. 4 for the Petit Lac where a clear decrease in As in March is observed, well correlated with an increase in algae growth. Specific As mass inventories for 2021 and the previous six years (all As ‘dissolved’ concentrations measured with the same instrument) are shown in Fig. SI3†, showing a high variability among the years. However, a Kruskal–Wallis test showed no seasonality of invAstot,dis along these years (p = 0.092). Specific As mass inventories in the Grand Lac, invAstot,dis, for the first 70 m gave very similar values to those of the Petit Lac: 778 μg dm−2vs. 772 in April and 764 μg dm−2vs. 754 in June, respectively.
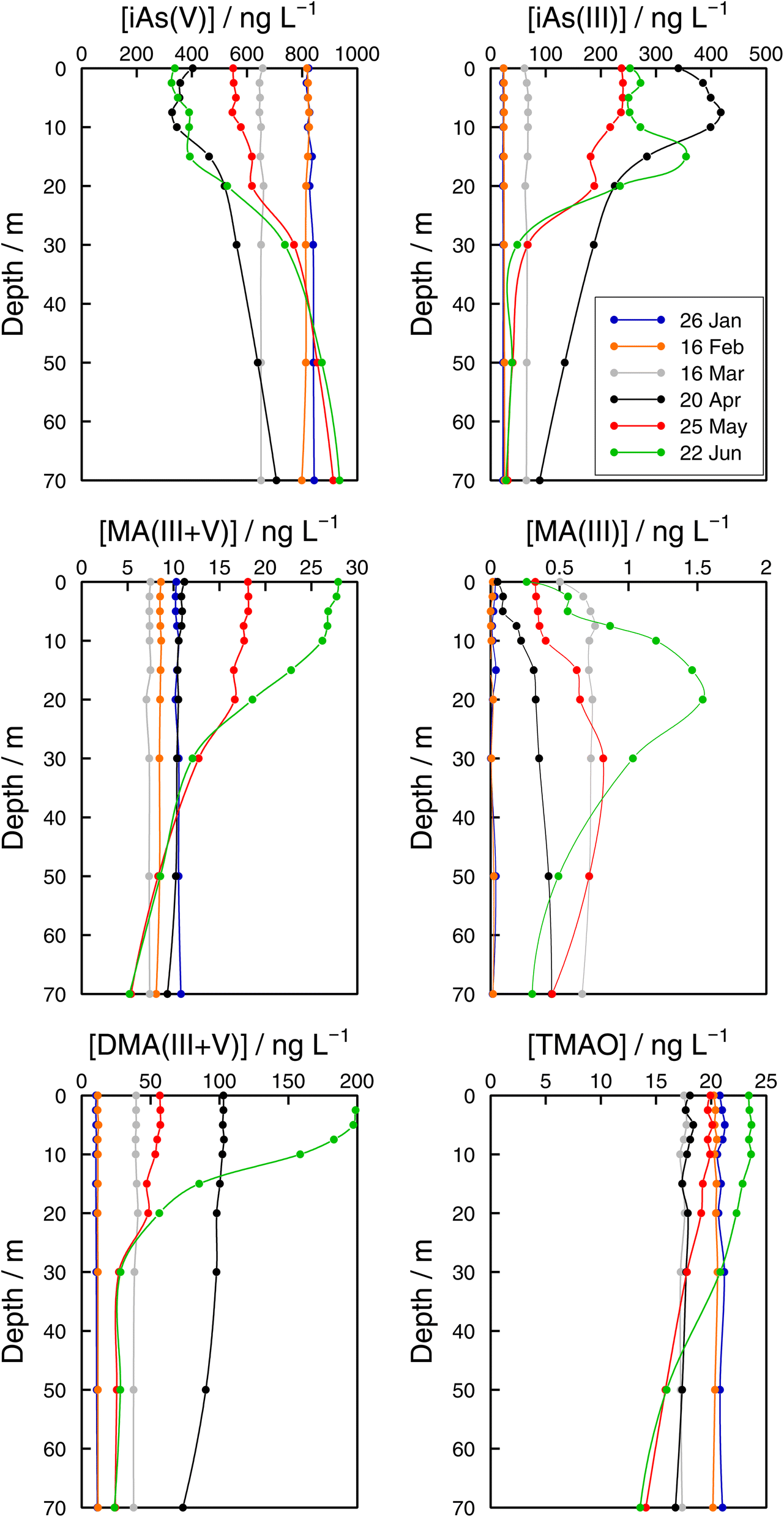 | ||
| Fig. 6 Vertical profiles of As species (iAs(V), iAs(III), MA(III + V), MA(III), DMA(III + V), and TMAO) concentrations in the Petit Lac (point GE3) from January to June 2021. | ||
Monthly follow-up in the Petit Lac. Solute concentration profiles are functions of the physical (mixing and stratification) and biogeochemical (production and degradation) processes taking place. When winter conditions prevailed at the beginning of the lake sampling campaign at GE3, the water column was well mixed and well oxygenated and with very low photosynthetic activity. As expected, concentration profiles were constant with depth, with a predominant presence of iAs(V) (820 ng L−1vs. < 25 ng L−1 iAs(III)). DMA(III + V) concentrations were slightly above 10 ng L−1. This corroborates previous observations (Table 1) that showed that, contrary to thermodynamic predictions, iAs(III) is present under oxic conditions either because it is very resilient to oxidation, or because of the presence of a source, or both. It is well known that iAs(III) oxidation by oxygen is extremely slow (e.g., only a few percent of As(III) is oxidised within seven days in the presence of air48) but As(III) is also subject to photo-oxidation49 which could result in relatively low As(III) residence times; for instance, 0.7 to 3 days under the conditions of the Pacific Ocean.10 However, fast photo-oxidation like that observed in boreal lakes50 requires the presence of high concentrations of specific types of dissolved organic matter – which is not the case in Lake Geneva.51 Possible sources of iAs(III), and also of methylated species even under winter conditions, could be produced by photosynthetic activity, even if very low, and minor diffusion from sediments.
The Petit Lac was still entirely vertically mixed in March with concentrations of iAs(V) being lower than in January–February (about 650 ng L−1) but higher for iAs(III) and DMA(III + V) (65 and 39 ng L−1, respectively). Phosphorous concentrations dropped, and chlorophyll a and phytoplankton biomass increased, all signs of biological activity. Weather conditions were favourable for algal growth because it was unusually warm at the beginning of March 2021 and insolation was above average (https://www.meteosuisse.admin.ch/home/service-et-publications/publications.html). A clear drop in sumAs occurred in March. This drop was also observed in Astot,dis, supporting that it was not due to an analytical error or artefact. The drop corresponded to a steep decrease in As(V) not compensated by the increase in other dissolved As-containing species. This suggests that, at this sampling date, As(V) had ‘moved’ from the ‘dissolved’ fraction to the ‘particulate’ one but with a very limited release of iAs(III) and DMA(III + V). Algal blooms could, thus, cause drops in Astot,dis by incorporation into algae, but these episodes can be easily missed in monthly sampling campaigns. This phenomenon has been observed in laboratory experiments with Closterium aciculare.52 At the current level of knowledge, it is not possible to relate the observed As retention inside algae to specific phytoplankton species in the lake. Patterns of As biotransformation differ across phytoplankton species53 with some algal species transforming iAs(V) efficiently into iAs(III) with rapid excretion into the culture medium, while others accumulate iAs(V) inside their cells or biotransform it into methylated and other organic As species. Those in the lake might belong to both categories.
In April, surface waters started to stratify at both sampling locations GE3 and SHL2 but mixing still occurred. Surface depletion of iAs(V) continued, and a minimum was observed at GE3 at 7.5 m, but this time a peak in iAs(III) appeared at the same depth. The first algal spring bloom seems to be dominated by a build-up of iAs(III), which agrees with observations in the literature.22,52 According to Hellweger et al. (2003),9 the build-up of iAs(III) is typical for non-P-depleted water bodies. The relationship As–P is further discussed below. The decrease in invAsdis,tot observed in March 2021 quickly recovered in April, further supporting the view that As had been retained inside algae in March. Although it is risky to draw many conclusions from invAsdis,tot values because they are a measure of both the amount retained in the floating algae at the moment of sampling and of any possible net losses of As by sedimentation of dead-algae and As in inorganic colloidal particles, in this case the quick recovery can safely be attributed to algae retention. It needs to be mentioned that the annual evolution of invAsdis,tot is very variable (Fig. SI3†) and important biological mechanisms can be missed if sampling is too widely spaced.
DMA(III + V) concentrations doubled from March to April, indicating strong production similar to iAs(III), but the two species showed a very different profile. While the iAs(III) profile was typical of a compound with a source at 7.5 m, the DMA(III + V) profile was close to vertical. Values observed in surface water at SHL2 of samples taken on the same day showed a similar tendency. This would suggest that water mixing was faster than the point production for DMA(III + V). Although mechanisms of methylation by microalgae remain controversial,14 DMA(V) is the final excreted product according to the classical Challenger mechanism54 and in possible alternative mechanisms. The Challenger mechanism includes reduction of iAs(V) into iAs(III) and successive oxidative and reductive methylation steps.
At the end of May, iAs(V) concentrations recovered, and iAs(III) and DMA(III + V) concentrations decreased compared with those in April. After the first algal bloom, the lake was in the clear water phase, a moment between the early spring bloom of algae and the second, summer algal bloom.30 This is an interesting situation where mechanisms at play are mostly decomposition reactions in the absence of an active source. Anderson and Bruland (1991) 16 showed that the degradation rate of DMA(V) spiked into waters collected before and after the lake's overturn is very different, the rate being much faster under oxic conditions. DMA(V) degraded into iAs(V). Probably, bacterial processes become important but they remain largely unexplored in oxic waters. For instance, Maki et al. (2005) 55 showed that the biomass of DMA(V) decomposing bacteria peaked approximately one month after the peak in chlorophyll a in Lake Kahokugata, Japan, but these bacteria, producing iAs(V), needed anaerobic, dark conditions.
The origin of MA(III + V) remains unclear. These species showed different dynamics from DMA(III + V). MA(III) and MA(V) are intermediate species formed inside algae but not the final products according to Challenger's and other mechanisms. MA(III + V) in lake waters could be either released directly from some algae or might be a microbial decomposition product of DMA(III + V) in the water. Microbial DMA demethylation had been demonstrated in soils, but not in aquatic systems until Giovannoni et al. (2019) 56 showed that the most abundant non-photosynthetic plankton in the oceans, SAR11 bacteria, are able to remove the methyl groups from dimethyl arsenate, releasing monomethyl As and iAs(V) back to the water. MA(III) has seldom been measured in freshwaters.20–22
In June, a new productivity period peaked (Fig. 4). iAs(V) concentrations in surface water were depleted and high DMA(III + V) and iAs(III) concentrations were established again. Observations at sampling site GE3 showed that the DMA(III + V) concentration was higher in June than in April, whereas iAs(III) concentrations showed the reverse trend (Fig. 6). This might be related to the molar iAs(V):P ratio, which was higher in April than in June (and higher in March than in May). Even if many studies have investigated the role of nutrient availability in the production of As species, it is uncertain how phosphate availability influences their production in different algae.57 Algae take up more phosphate than needed during the early phases of algal blooms (luxury uptake)9 and, simultaneously, large quantities of iAs(V) can enter the cells via the same pathway. In oceans, it has been postulated that a high iAs(V):P ratio causes toxic stress on phytoplankton, promoting fast transformation into iAs(III) and slow methylation.46 Kuhn and Sigg (1993) 17 proposed this hypothesis in eutrophic Lake Greifen (ratios < 0.001) to explain the absence of methylated species, but high DMA(III + V) concentrations were observed in Lake Biwa with high iAs(V):phosphate ratios.22 The molar iAs(V):P ratio at GE3 in Lake Geneva ranged between 0.07 and 0.25 during the sampling period. These ratios are far higher than the ratios covered by ocean studies.46 It is possible that a higher iAs(V) stress was exerted on algal communities during the first algal bloom in March and April compared with the second bloom in May–June, which could explain the higher concentration of DMA(III + V) in June in comparison with that in April. It is also possible, however, that differences in algae species present in the first and second blooms explain the observations.
TMAO has seldom been analysed in surface waters. It was included in none of the studies in Table 1. Like MA(III + V), TMAO showed a decreasing trend between January and March at sampling site GE3 (Fig. 6). Increases in surface water began in May. In addition to MA(V) and DMA(V), As(III) methylation products also include less toxic As species such as TMAO and volatile trimethylarsine (TMA).58–60 Savage et al. (2018) 61 observed that seawater samples spiked with DMA(V) led to an enhanced production and volatilisation of TMA into the gaseous phase, with TMAO as a probable intermediate in the aqueous phase. A similar process could lead to observed concentrations of TMAO in lakes.20 Concentrations of TMAO in January were higher than concentrations of MA(III + V) and DMA(III + V). This observation, considered in the context of the evolution of TMAO concentrations along the studied period, points to TMAO being a rather stable compound, resistant to the processes guiding the system back to the dominance of iAs(V), and whose main fate would be its transformation into volatile TMA. In addition, note that our analytical method cannot distinguish dissolved TMA and TMAO. TMAO is ubiquitous in atmospheric particles;62 a significant source being pesticides in soils.63
As speciation in lake bottom waters: lessons from the Grand Lac. The observed dynamics of As speciation at the bottom of Lake Geneva were distinct from the photic zone. In 2021, iAs(V) concentrations at GE3 at 65 m increased once lake stratification was established. Concentrations reached 936 ng L−1 in June, which represented 94% of sumAs. The results at the lake's deepest point (SHL2) confirmed iAs(V) dominance at sampling depths below the photic zone (Fig. 7). At both sampling dates (April and June), iAs(V) made up more than 95% of sumAs from 100 m towards the bottom of the lake. At 300 m, independent of the sampling date, the iAs(V) concentration reached almost 2000 ng L−1, representing 99% of sumAs. The predominance of iAs(V) agrees with what has been described in the literature: iAs(V) is the dominant species below the photic layer10,64 in oceans and in freshwaters. For instance, iAs(V) was the main species in the hypolimnion in the highly eutrophic Lake Greifen throughout the whole season,17 and the observed increase in sumAs with depth in both eutrophic and mesotrophic basins of Lake Biwa in summer was attributed to an increase in iAs(V).22 The predominance of iAs(V) in lake waters far from the productivity zone is in agreement with thermodynamic predictions and strongly supports the biological origin of iAs(III) when present. On the other hand, increasing iAs(V) concentrations with depth is a direct consequence of the lack of entire lake mixing. Winter mixing remained limited to 145 m in 2021, the depth was reached on 2 March 2021.29
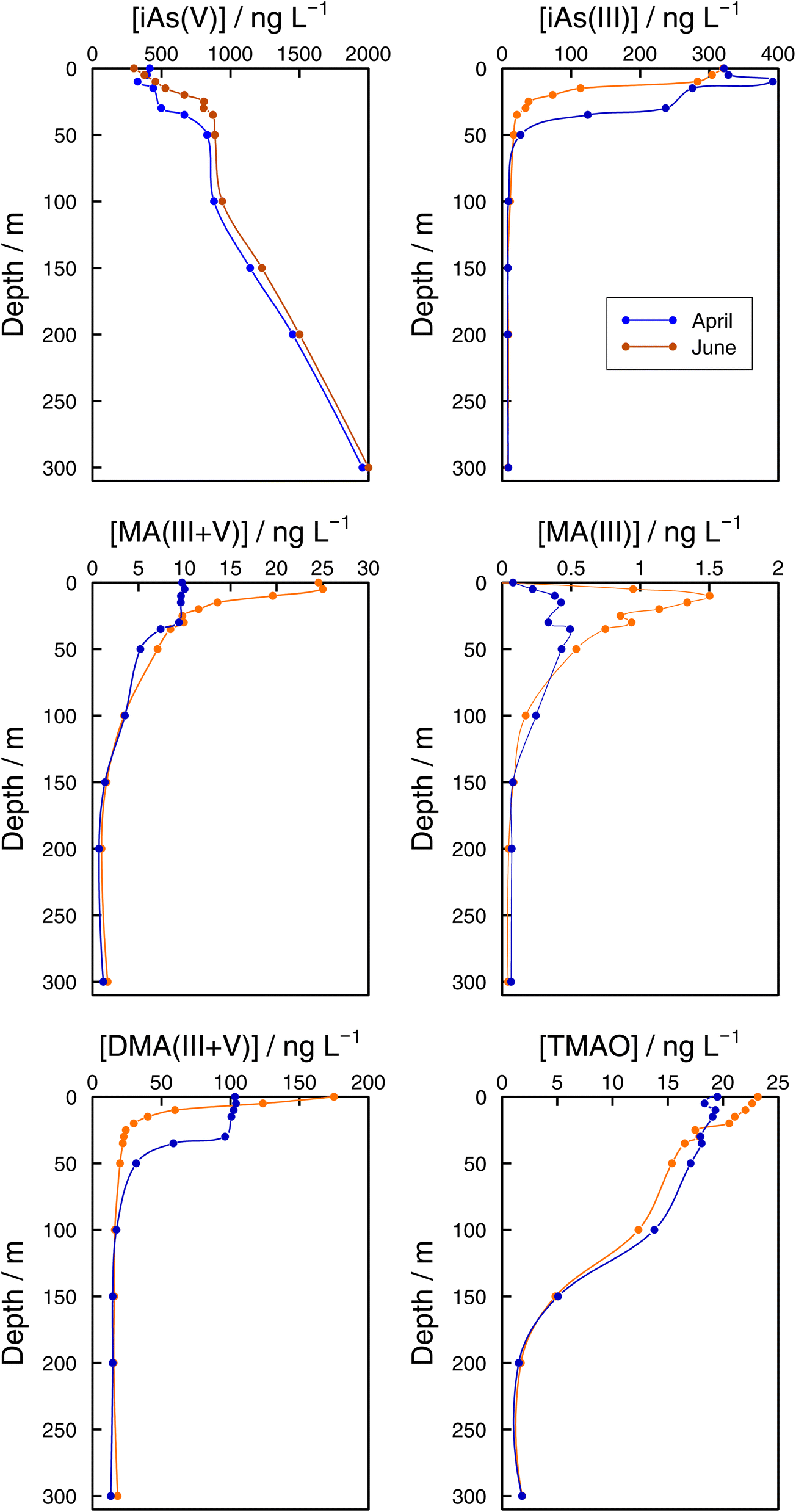 | ||
| Fig. 7 Vertical profiles of As species (iAs(V), iAs(III), MA(III,V), MA(III), DMA(III,V), and TMAO) concentrations in the Grand Lac (point SHL2) in April (blue) and June (orange) 2021. | ||
While methylated species were evenly distributed across the water column in winter months, differences between the surface water and the bottom layer were established as soon as the lake stratified (Fig. 6). Even if all methylated species showed profiles that resulted from a source in the epilimnion, their shape depended on the species considered. For instance, in June DMA(III + V) concentrations quickly approached a steady concentration with depth, whereas MA(III + V) and TMAO approached their minimum values more slowly at both sampling locations (Fig. 6 and 7). It can thus be assumed that methylated species are subject to decomposition at different rates, with rates for DMA(III + V) transformation being higher than for TMAO. It is interesting that DMA(III + V) values did not approach zero but remained around 15 ng L−1 in the deepest parts of the lake.
Exchange of arsenic species between sediment porewaters and the water column
Even if the number of samples was low and they reflected the unavoidable heterogeneity of lake sediments, concentration profiles of As species in the three sediment cores sampled at GE3 provide some interesting information. First, Astot,dis concentrations in the supernatant waters of the three cores were similar: 1.10 μg L−1 (C1), 1.14 μg L−1 (C2), and 1.38 μg L−1 (C3) versus 1.15 μg L−1 at 65 m depth in July, but they were always higher below the sediment water interface than in the supernatant water (for instance, 1.98 μg L−1 (C1), 5.95 μg L−1 (C2), and 9.52 μg L−1 (C3) at 2 cm depth) which implies a subsequent input of ‘dissolved’ As from the sediments to the water column. Secondly, this input is fed by iAs(III) and iAs(V) species since both species show higher concentrations in the interstitial waters compared with in the water column. The most probable source of ‘dissolved’ As is the release from the decomposition of sedimented algae, although a contribution from the release from settling iron oxyhydroxides cannot be excluded. A detailed discussion of these mechanisms is, however, entirely outside the scope of this study. Thirdly, Astot,dis in porewaters did not show similar profiles in the three sediment cores (Table SI11†), and profiles also differed for most of the As species measured among sediments (Fig. 8, Table SI15†) but some common patterns were observed: MA(III + V) profiles showed no diffusion or a very weak one and DMA(III + V) and TMAO clearly pointed to a net flux from the water column to the sediments, contrary to iAs(V) and iAs(III). This suggests that DMA(III, V) and TMAO are only – or predominantly – formed in algae.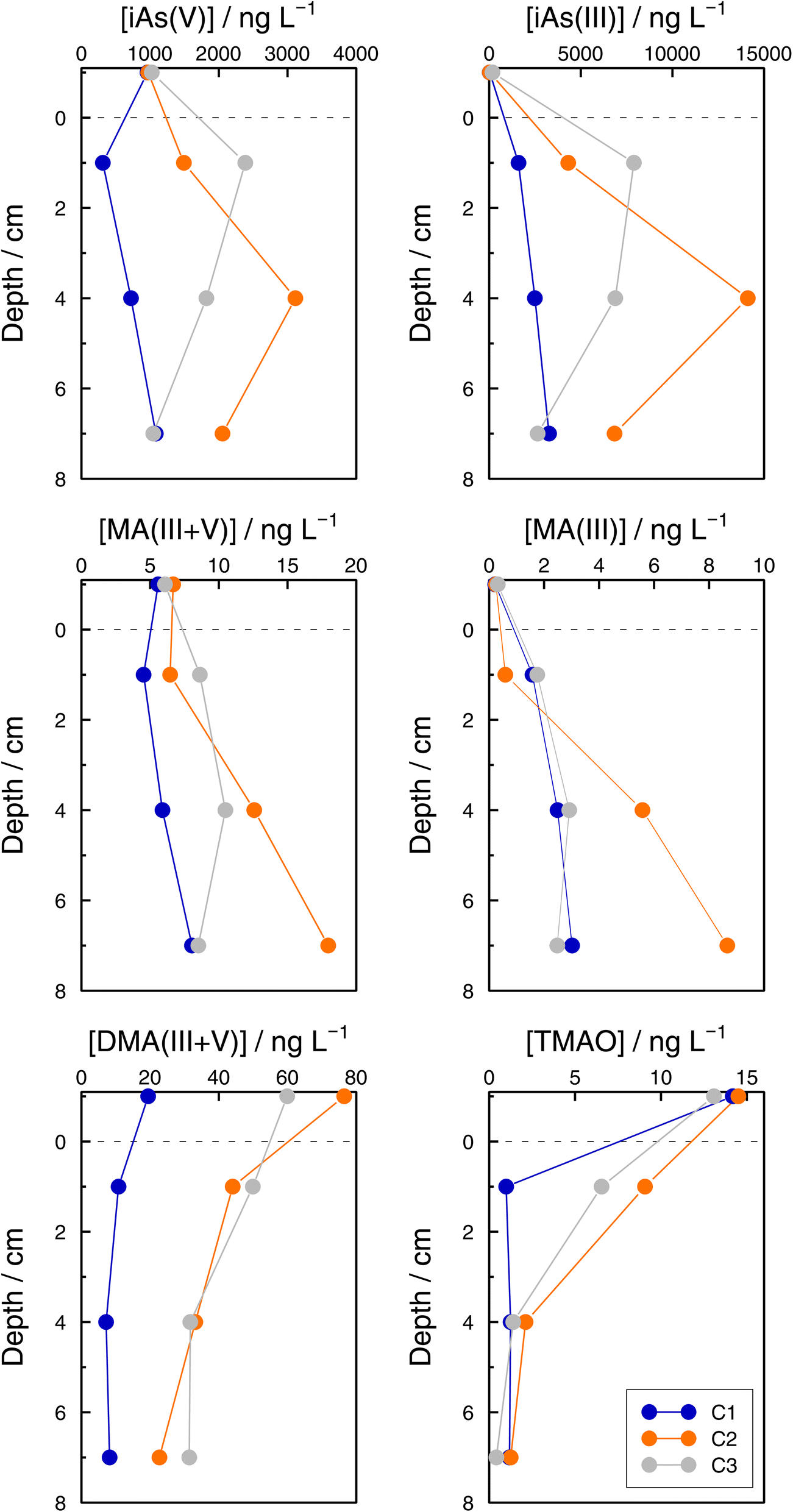 | ||
| Fig. 8 Concentrations of total dissolved arsenic (Astot,dis), iAs(V), iAs(III), MA(III + V), DMA(III + V), and TMAO concentrations in sediment pore waters and the overlying supernatant water in three cores (C1 (blue), C2 (orange), and C3 (grey)) in the Petit Lac, 12 July 2021. | ||
A rough calculation of diffusion fluxes has been attempted by applying Ficks's first law. In the steady state, the flux close to the sediment–water interface is equal to the product of the concentration gradient in the sediment pore waters and the diffusion coefficient of the substance considered. The concentration gradients were calculated from points at 1 and 4 cm depth in the sediments. The calculated fluxes are given in Table 4. Diffusion coefficients for As(OH)3 and HAsO42−, 11.6 × 10−6 cm2 s−1 and 7.27 × 10−6 cm2 s−1, respectively, are from ref. 65. No corrections for porosity or temperature dependence of the diffusion coefficients were considered since our calculations are only intended to provide a rough quantitative estimation. The mass of As in a 1 dm2 layer closer to the lake bottom (57.5 to 75 m) decreased from 136 (January) to 117 μg (March), and increased from March to June (150 μg in 98 days, which means 33 μg). This corresponds to a mean increase of 3.90 × 10−5 ng cm−2 s−1, which is in the same order of magnitude as the estimated diffusion fluxes of iAs using Fick's Law. This confirms that the order of magnitude of the estimated flux values is correct.
A complementary insight into arsenic in Lake Geneva tributaries and its possible contribution to lake observations
Astot,dis concentrations in the main tributaries to Lake Geneva were in the range 130–1200 ng L−1 (Tables 2 and 3) which is congruent with concentrations in stream waters in Europe; median value: 630 ng L−1.66 Concentrations in the Rhone River were higher than those in the lake in April but lower than those in June. Concentrations found in other tributaries were generally lower. The high Astot,dis detected in the Rhone River can be the result of catchment area of the Rhone including areas with elevated natural As concentrations.67 In June, concentrations in all rivers might have been strongly influenced by weather conditions before the sampling date: heavy thunderstorms occurred at the beginning of June (https://www.meteosuisse.admin.ch/home/service-et-publications/publications.html), and rivers were highly turbid and coloured brownish when sampled. The differences in Astot,dis between the two sampling dates in the Rhone River can be related to the differences in water discharge: 73–75 m3 s−1 in April and 504–541 m3 s−1 in June (points marked with arrows in Fig. SI1†) and could be attributed to a dilution effect, but the establishment of a sound concentration-discharge relationship – usually following a power law function68 – would require high frequency discharge data (available) and concentration data (outside current measuring device capabilities) over long periods of time. Knowledge of such relationships at least for the main tributaries would be essential, for instance, in order to establish a mass balance of the lake.| Core | iAs(III)/ng cm−2 s−1 | iAs(V)/ng cm−2 s−1 |
|---|---|---|
| C1 | –3.46 × 10−6 | –9.91 × 10−6 |
| C2 | –3.79 × 10−5 | –3.93 × 10−6 |
| C3 | 3.94 × 10−6 | 1.37 × 10−6 |
The major fraction of ‘dissolved’ As in the tributaries in April and June consisted of iAs(V) (Tables 2 and 3). The dominance of iAs(V) has been observed when rivers are oxygenated,69 as is in the case of most of the rivers considered (e.g., all sampling sites had an oxygen saturation higher than 90% in the Rhone River70). The Rhone carried a considerable amount of iAs(III) in April. Possible phytoplanktonic activity can be hypothesised as the cause since high densities of diatoms prevail in the Rhone even in winter months.70 As(III) concentrations were lower in June when high discharge and turbidity do not favour algal development. While MA(III + V) and TMAO constituted only minor contributions to the sum of As species, concentrations of DMA(III + V) were higher. In April, DMA(III + V) concentrations in all samples were lower than concentrations of iAs(III). In June, however, the situation depended on the river considered. The presence of organic species implies either a certain degree of photosynthetic activity in the water or soil or an anthropogenic origin. In this context, the River Venoge deserves a particular mention because it is known to be polluted by a diversity of substances.71 Despite organic As species being detected and measured in all rivers, given their concentration levels and lability, they do not seem to represent a significant input to the lake. Therefore, the presence of methylated species in the lake can be considered as the exclusive result of internal processes.
Temporal trends and seasonality of the total ‘dissolved’ arsenic, 1997–2021
Assessment of seasonality at each depth in each of the periods mentioned above (Fig. 2a) and in the whole data revealed significant differences (p < 0.05) between monthly concentration median values in different zones of the epilimnion in the three periods (Tables SI16a and SI16b†).To eliminate step changes between periods, which prevented all data from being treated together for temporal trend assessment, concentrations were normalized by subtracting the corresponding period median value in each datum (Fig. 2c). When the resulting time series was analysed for seasonality at each depth, statistically significant differences between months at every depth (except at 30 m) suggested seasonality (Table SI16a†). A Kruskall–Wallis test applied to all depths simultaneously (excluding or including period 2) gave a very significant value (p = 9.73 × 10−22 and 1.10 × 10−31, respectively), also indicating seasonality. Seasonality should also manifest by the presence of a significant annual peak in the Fourier series spectrum at a frequency of 0.00274 day−1, as was the case at 0 m depth (Fig. SI4†).
Both the normalized time series and the time series for each period showed no significant temporal trend (absolute SMK Z values less than 1.96, Tables SI17a and SI17b†), indicating that concentrations of As remained stationary in the Petit Lac between 1997 and 2021.
The lack of temporal trends of ‘dissolved’ As concentrations in the Petit Lac from 1997 to 2021 (with and without including 2006–2015 data) suggests that possible inputs from anthropogenic activities such as mining, use of As in agricultural chemicals, or atmospheric deposition are very minor in the lake watershed. No variation in geogenic inputs that could be derived, for instance, from changes in weathering rates linked to climate change seems to exist either.
Understanding seasonality results of total ‘dissolved’ As is not entirely straightforward. Methods applied to Astot,dis concentrations (Kruskall–Wallis with 12 ‘seasons’ and Fourier power spectra) over 1997–2021 point to a seasonal behaviour of As concentrations in the Petit Lac (i.e., understanding ‘seasonal’ behaviour here as differences between median values of monthly concentrations in the entire period). At first sight, this is not surprising for an element that is actively cycled through phytoplankton, which shows a clear seasonal succession. In Fig. 3, however, Astot,dis profiles look essentially vertical and not affected by this cycling. Moreover, specific As mass inventories for 2015–2021 (Fig. SI3†) show no seasonal character. The total ‘dissolved’ As remaining relatively constant, despite the presence and concentration levels of ‘dissolved’ As species being extremely dependent on algal cycling (as discussed in the previous sections), points to a strong internal species cycling, with exchanges between the ‘dissolved’ and ‘particulate’ fractions, but with few net As ‘losses’ and at a steady-state situation.
Specific As mass inventories for the first 70 m show similar values for the Petit Lac and Grand Lac, suggesting similar behaviours in this zone. The above situation does not apply, however, to the whole water column in the Grand Lac where the lack of water column mixing since 2012 has led to a steady increase in ‘dissolved’ As concentration with depth. Similar accumulation has been reported for other elements such as germanium34 and silica.29
Lessons learned
Arsenic behaviour in surface waters is very complex due to the redox equilibria involved and the formation of a myriad of alkylated species. The consequence is that our understanding heavily relies on the quality of the analytical methods applied, and on the spatial and temporal (frequency) of the sampling. This study clearly highlights some of these aspects, not always considered in previous studies:(1) The study of temporal trends of concentrations requires data over long periods of time to be available. In the case of trace elements, this is problematic because of the lack of such series and because of the often-ignored effect of analytical constraints in the acquisition of such data over the years. In this study, the possibility of having access to original analytical data including CRM results has allowed their evaluation, which is somewhat unusual. Adequate quality data tackling allows us to state that ‘dissolved’ As concentrations in Lake Geneva have remained stationary between 1997 and 2021.
(2) State-of-the-art analytical methods are needed in speciation measurements. In particular, great care needs to be taken at the analytical level to ensure the stability of the different species and the absence of measurement artefacts. The steps needed to ensure the preservation of samples are described in detail for application to future studies. Our approach has allowed us to reliably follow the dynamics of iAs(V), iAs(III), DMA(III + V), MA(III + V) and TMAO in the water column and measure them in river and sediment interstitial waters. We have confirmed the presence of MA(III) and TMAO in lake and river samples and the predominance of iAs(V) in most waters.
(3) ‘Dissolved’ vs. ‘particulate’ fractions. This study, as with most previous studies, deals with ‘dissolved’ As, meaning the As that is passed through a filter of a certain pore size. This implies that, if the ‘particulate’ compartment actively participates in the processes taking place (e.g., through algae in the lake), our interpretation of the results necessarily includes a ‘black-box’. This leads to an undesirable degree of speculation when discussing the information obtained.
(4) The role of algae in As speciation in oxic lakes is widely confirmed. Which are the algae involved, as well as the possible role of bacteria in oxic systems remain, however, open questions.
(5) Sampling frequency. In lakes, monthly sampling is not sufficient to adequately follow relevant physical, chemical and biological processes, many of them being much faster. It follows that studies with even lower sampling frequency, as for instance one sampling campaign in summer and one in winter (Table 1), will only be able to provide very limited information.
(6) Inter-year variability. Although, by definition, seasonal variations occur every year, the intensity and temporal location of some processes – particularly biological processes depending on climatic conditions – vary from year to year. This introduces a factor which hinders extrapolation of the results obtained in studies covering only one or a limited number of years. The effects of inter-year variability and modifications linked to climate change need to be considered in future studies.
Author contributions
M. Filella (conceptualization, data curation, resources, supervision, investigation, project administration, funding acquisition, visualization, validation, writing original draft, writing review and editing); S. Wey (conceptualization, data curation, investigation, visualization, writing original draft, writing review and editing); T. Matoušek (data curation, resources, formal analysis, investigation, validation, methodology, visualization, writing original draft, writing review and editing); M. Coster (resources, validation, funding acquisition, writing review and editing); J.C. Rodriguez-Murillo (data curation, software, formal analysis, validation, writing review and editing); J.-L. Loizeau (resources, supervision, investigation, funding acquisition, validation, writing review and editing).Conflicts of interest
There are no conflicts to declare.List of abbreviations
| AFS | Atomic fluorescence spectrometry |
| AR(1) | First-order autoregressive process |
| Astot,dis | Total ‘dissolved’ As concentrations |
| CON | Conjugatophyceae |
| CHL | Chlorophyceae |
| CHR | Chrysophyceae |
| CRM | Certified reference material |
| CRY | Cryptophyceae |
| CT | Cryotrapping |
| CYA | Cyanophyceae |
| DIA | Diatoms |
| DMA(V) | Dimethylarsinate |
| DIN | Dinophyceae |
| DMA(III) | Dimethylarsinite |
| DMA(III + V) | DMA(III) + DMA(V) |
| DRP | Dissolved reactive phosphorus |
| EDTA | Ethylenediaminetetraacetic acid |
| GE3 | Sampling point at the Petit Lac (6.2197° E/46.2994° N) |
| HG | Hydride generation |
| HPLC | High-performance liquid chromatography |
| iAs | Inorganic As |
| iAs(III) | Inorganic As(III) |
| iAs(V) | Inorganic As(V) |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| ICP-MS/MS | Inductively coupled plasma–tandem mass spectrometry |
| invAstot,dis | Specific As mass inventories |
| MA(III) | Methylarsonite |
| MA(V) | Methylarsonate |
| MA(III + V) | MA(III) + MA(V) |
| MK | Mann–Kendall trend test |
| REDFIT | Statistical method |
| SHL2 | Sampling point at the Grand Lac (6.5887° E/46.4527° N) |
| SMK | Seasonal Mann–Kendall trend test |
| sumAs | Sum of concentrations of all investigated As species |
| C1 | Sediment sampling point, Petit Lac (6.2276° E/46.3052° N) |
| C2 | Sediment sampling point, Petit Lac (6.2201° E/46.2971° N) |
| C3 | Sediment sampling point, Petit Lac (6.2123° E/46.2888° N) |
| TMA | Trimethylarsine |
| TMAO | Trimethylarsine oxide |
| XAN | Xantophyceae |
| Z | MK statistical parameter |
Acknowledgements
The Institute of Analytical Chemistry of the Czech Academy of Sciences [Institutional support RVO: 68081715] is gratefully acknowledged. We thank Philippe Arpagaus, Vincent Ebener, Emmanuel Farinoli, Jean-Christophe Hustache, Pascal Perney, and Inés Segovia. Without their help, this study would not have been possible.References
- J. O. Nriagu, Arsenic poisoning through the ages, in Environmental Chemistry of Arsenic, ed. W. T. Frankenberger Jr, Marcel Dekker, Inc., New York, 2002, pp. 1–26 Search PubMed.
- R. Nickson, J. McArthur, W. Burgess, K. Matin Ahmed, P. Ravenscroft and M. Rahmanñ, Arsenic poisoning of Bangladesh groundwater, Nature, 1998, 395, 338, DOI:10.1038/26387.
- A. H. Smith, E. O. Lingas and M. Rahman, Contamination of drinking-water by arsenic in Bangladesh: a public health emergency, Bull. W. H. O., 2000, 78, 1093–1103 CAS.
- P. Ravenscroft, H. Brammer and K. Richards, Arsenic Pollution: a Global Synthesis, Wiley-Blackwell, Chichester, 2009 Search PubMed.
- E. A. Woolson, Fate of arsenicals in different environmental substrates, Environ. Health Perspect., 1977, 19, 73–81, DOI:10.1289/ehp.771973.
- W. R. Cullen and K. J. Reimer, Arsenic speciation in the environment, Chem. Rev., 1989, 89, 713–764, DOI:10.1021/cr00094a002.
- R. S. Oremland and J. F. Stolz, The ecology of arsenic, Science, 2003, 300, 939–944, DOI:10.1126/science.1081903.
- S. Silver and L. T. Phung, Genes and enzymes involved in bacterial oxidation and reduction of inorganic arsenic, Appl. Environ. Microbiol., 2005, 71, 599–608, DOI:10.1128/AEM.71.2.599-608.2005.
- F. Hellweger, K. J. Farley, U. Lall and D. M. Di Toro, Greedy algae reduce arsenate, Limnol. Oceanogr., 2003, 48, 2275–2288, DOI:10.4319/lo.2003.48.6.2275.
- O. Wurl, R. U. Shelley, W. M. Landing and G. A. Cutter, Biogeochemistry of dissolved arsenic in the temperate to tropical North Atlantic Ocean, Deep Sea Res., Part II, 2015, 116, 240–250, DOI:10.1016/j.dsr2.2014.11.008.
- M. Vahter, Methylation of inorganic arsenic in different mammalian species and population groups, Sci. Prog., 1999, 82(Pt 1), 69–88, DOI:10.1177/003685049908200104.
- D. J. Thomas, Arsenic methylation - Lessons from three decades of research, Toxicology, 2021, 457, 152800, DOI:10.1016/j.tox.2021.152800.
- K. A. Francesconi and D. Kuehnelt, Determination of arsenic species: A critical review of methods and applications, 2000-2003, Analyst, 2004, 129, 373–395, 10.1039/B401321M.
- Y. Wang, S. Wang, P. Xu, C. Liu, M. Liu, Y. Wang, C. Wang, C. Zhang and Y. Ge, Review of arsenic speciation, toxicity and metabolism in microalgae, Rev. Environ. Sci. Bio/Technol., 2015, 14, 427–451, DOI:10.1007/s11157-015-9371-9.
- P. Seyler and J. M. Martin, Biogeochemical processes affecting arsenic species distribution in a permanently stratified lake, Environ. Sci. Technol., 1989, 23, 1258–1263, DOI:10.1021/es00068a012.
- C. D. Anderson and K. W. Bruland, Biogeochemistry of arsenic in natural waters: the importance of methylated species, Environ. Sci. Technol., 1991, 25, 420–427, DOI:10.1021/es00015a007.
- A. Kuhn and L. Sigg, Arsenic cycling in eutrophic Lake Greifen, Switzerland: Influence of seasonal redox processes, Limnol. Oceanogr., 1993, 38, 1052–1059, DOI:10.4319/lo.1993.38.5.1052.
- A. C. Aurillo, R. P. Mason and H. F. Hemond, Speciation and fate of arsenic in three lakes of the Aberjona watershed, Environ. Sci. Technol., 1994, 28, 577–585, DOI:10.1021/es00053a008.
- H. M. Spliethoff, R. P. Mason and H. F. Hemond, Interannual variability in the speciation and mobility of arsenic in a dimictic lake, Environ. Sci. Technol., 1995, 29, 2157–2161, DOI:10.1021/es00008a041.
- D. A. Bright, M. Dodd and K. J. Reimer, Arsenic in subarctic lakes influenced by gold mine effluent: The occurrence of organoarsenicals and “hidden” arsenic, Sci. Total Environ., 1996, 180, 165–182, DOI:10.1016/0048-9697(95)04940-1.
- H. Hasegawa, The behaviour of trivalent and pentavalent methylarsenicals in Lake Biwa, Appl. Organomet. Chem., 1997, 11, 305–311, DOI:10.1002/(SICI)1099-0739(199704)11:4<305::AID-AOC586>3.0.CO;2-6.
- Y. Sohrin, M. Matsui, M. Kawashima, M. Hojo and H. Hasegawa, Arsenic biogeochemistry affected by eutrophication in Lake Biwa, Japan, Environ. Sci. Technol., 1997, 31, 2712–2720, DOI:10.1021/es960846w.
- H. Hasegawa, M. A. Rahman, K. Kitahara, Y. Itaya, T. Maki and K. Ueda, Seasonal changes of arsenic speciation in lake waters in relation to eutrophication, Sci. Total Environ., 2010, 408, 1684–1690, DOI:10.1016/j.scitotenv.2009.11.062.
- J. L. Barringer, Z. Szabo, T. P. Wilson, J. L. Bonin, T. Kratzer, K. Cenno, T. Romagna, M. Alebus and B. Hirst, Distribution and seasonal dynamics of arsenic in a shallow lake in northwestern New Jersey, USA, Environ. Geochem. Health, 2011, 33, 1–22, DOI:10.1007/s10653-010-9289-7.
- C. Yan, F. Che, L. Zeng, Z. Wang, M. Du, Q. Wei, Z. Wang, D. Wang and Z. Zhen, Spatial and seasonal changes of arsenic species in Lake Taihu in relation to eutrophication, Sci. Total Environ., 2016, 563–564, 496–505, DOI:10.1016/j.scitotenv.2016.04.132.
- M. J. Palmer, J. Chételat, M. Richardson, H. E. Jamieson and J. M Galloway, Seasonal variation of arsenic and antimony in surface waters of small subarctic lakes impacted by legacy mining pollution near Yellowknife, NT, Canada, Sci. Total Environ., 2019, 684, 326–339, DOI:10.1016/j.scitotenv.2019.05.258.
- F. Che, X. Jiang, C. Yao, L. Zhao and K. Wang, Arsenic distribution and speciation in multiphase media of a lake basin, Tibet: The influences of environmental factors on arsenic biogeochemical behavior in the cold arid plateau lake, Sci. Total Environ., 2020, 714, 136772, DOI:10.1016/j.scitotenv.2020.136772.
- S. Ding, Y. Wang, M. Yang, R. Shi, T. Ma, G. Cui and X. Li, Distribution and speciation of arsenic in seasonally stratified reservoirs: Implications for biotransformation mechanisms governing interannual variability, Sci. Total Environ., 2022, 806, 150925, DOI:10.1016/j.scitotenv.2021.150925.
- CIPEL, Rapports sur les Études et Recherches Entreprises dans le Bassin Lémanique Campagne 2021, CIPEL: Nyon, Switzerland, 2022, https://www.cipel.org/wp-content/uploads/2022/11/rapport-scientifique-2021-2022-low.pdf, last accessed: 2 January 2023 Search PubMed.
- O. Anneville, M. Beniston, N. Gallina, C. Gillet, S. Jacquet, J. Lazzarotto and M. Perroud, L’empreinte du changement climatique sur le Léman, Arch. Sci., 2013, 66, 157–172, DOI:10.5169/seals-738482.
- J.-L. Loizeau, S. Makri, P. Arpagaus, B. Ferrari, C. Casado-Martinez, T. Benejam and P. Marchand, Micropolluants métalliques et organiques dans les sédiments superficiels du Léman. Rapport CIPEL contre Pollution, Campagne, 2016, 143–198, 2017 Search PubMed.
- T. Matoušek, J. M. Currier, N. Trojánková, J. R. Saunders, M. C. Ishida, C. Gonzalez-Horta, S. Musil, Z. Mester, M. Stýblo and J. Dědina, Selective hydride generation – cryotrapping – ICP-MS for arsenic speciation analysis at picogram levels: analysis of river and sea water reference materials and human bladder epithelial cells, J. Anal. At. Spectrom., 2013, 28, 1456–1465, 10.1039/c3ja50021g.
- A. García-Figueroa, M. Filella and T. Matoušek, Speciation of germanium in environmental water reference materials by Hydride Generation and Cryotrapping in combination with ICP-MS/MS, Talanta, 2021, 225, 121972, DOI:10.1016/j.talanta.2020.121972.
- M. Filella and T. Matoušek, Germanium in Lake Geneva (Switzerland/France) along the spring productivity period, Appl. Geochem., 2022, 143, 105352, DOI:10.1016/j.apgeochem.2022.105352.
- T. Matoušek, A. Hernandez-Zavala, M. Svoboda, L. Langrová, B. M. Adair, Z. Drobná, D. J. Thomas, M. Stýblo and J. Dědina, Oxidation state specific generation of arsines from methylated arsenicals based on L-cysteine treatment in buffered media for speciation analysis by hydride generation-automated cryotrapping-gas chromatography-atomic absorption spectrometry with the multiatomizer, Spectrochim. Acta, Part B, 2008, 63, 396–406, DOI:10.1016/j.sab.2007.11.037.
- C. Reimann and P. Filzmoser, Normal and lognormal distribution in geochemistry: death of a myth. Consequences for the statistical treatment of geochemical and environmental data, Environ. Geol., 2000, 39, 1001–1014, DOI:10.1007/s002549900081.
- R. M. Hirsch and J. R. Slack, A nonparametric trend test for seasonal data with serial dependence, Water Resour. Res., 1984, 20, 727–732, DOI:10.1029/WR020i006p00727.
- D. R. Helsel and R. M. Hirsch, Statistical Methods in Water Resources; Techniques of Water-Resources Investigations of the United States Geological Survey. Book 4, USGS, Reston, VA, 2002, Ch. A3 Search PubMed.
- M. Schulz and M. Mudelsee, REDFIT: estimating red-noise spectra directly from unevenly spaced paleoclimatic time series, Comput. Geosci., 2002, 28, 421–426, DOI:10.1016/S0098-3004(01)00044-9.
- Ø. Hammer, D. A. T. Harper and P. D. Ryan, PAST: Paleontological Statistics Software Package for Education and Data Analysis, Palaeontol. Electron., 2001, 4 Search PubMed , https://palaeo-electronica.org/2001_1/past/issue1_01.htm.
- J. M. Currier, M. Svoboda, T. Matoušek, J. Dědina and M. Stýblo, Direct analysis and stability of methylated trivalent arsenic metabolites in cells and tissues, Metallomics, 2011, 3, 1347–1354, 10.1039/c1mt00095k.
- D. Yeghicheyan, P. Grinberg, L. Y. Alleman, M. Belhadj, L. Causse, J. Chmeleff, L. Cordier, I. Djouraev, D. Dumoulin, J. Dumont, R. Freydier, H. Mariot, C. Cloquet, P. Kumkrong, B. Malet, C. Jeandel, A. Marquet, J. Riotte, M. Tharaud, G. Billon, G. Trommetter, F. Séby, A. Guihou, P. Deschamps and Z. Mester, Collaborative determination of trace element mass fractions and isotope ratios in AQUA-1 drinking water certified reference material, Anal. Bioanal. Chem., 2021, 413, 4959–4978, DOI:10.1007/s00216-021-03456-8.
- A. G. Howard and S. D. W. Comber, The discovery of hidden arsenic species in coastal waters, Appl. Organomet. Chem., 1989, 3, 509–514, DOI:10.1002/aoc.590030607.
- A. M. M. de Bettencourt and M. O. Andreae, Refractory arsenic species in estuarine waters, Appl. Organomet. Chem., 1991, 5, 111–116, DOI:10.1002/aoc.590050208.
- H. Hasegawa, M. Matsui, S. Okamura, M. Hojo, N. Iwasaki and Y. Sohrin, Arsenic speciation including ‘hidden’ arsenic in natural waters, Appl. Organomet. Chem., 1999, 13, 113–119, DOI:10.1002/(SICI)1099-0739(199902)13:2<113::AID-AOC837>3.0.CO;2-A.
- G. A. Cutter and L. S. Cutter, Biogeochemistry of arsenic and antimony in the North Pacific Ocean, Geochem., Geophys., Geosyst., 2006, 7, 1–12, DOI:10.1029/2005GC001159.
- J.-C. Rodríguez-Murillo, P. Nirel and M. Filella, Detecting trends in freshwater trace element concentrations: methodological issues and data treatment, H2Open J., 2018, 1, 87–98, DOI:10.2166/h2oj.2018.006.
- M. Jekel, Removal of arsenic in drinking water treatment, in Arsenic in the Environment Part I: Cycling and Characterization, ed. J. O. Nriagu, Wiley, New York, 1994, pp. 119–132 Search PubMed.
- J. Ryu, D. Monllor-Satoca, D. H. Kim, J. Yeo and W. Choi, Photooxidation of arsenite under 254 nm irradiation with a quantum yield higher than unity, Environ. Sci. Technol., 2013, 47, 9381–9387, DOI:10.1021/es402011g.
- M. Amyot, D. Bélanger, D. F. Simon, J. Chételat, M. Palmer and P. Ariya, Photooxidation of arsenic in pristine and mine-impacted Canadian subarctic freshwater systems, J. Hazard. Mater. Adv., 2021, 2, 100006, DOI:10.1016/j.hazadv.2021.100006.
- J.-C. Rodríguez-Murillo and M. Filella, Temporal evolution of organic carbon concentrations in Swiss lakes: Trends of allochthonous and autochthonous organic carbon, Sci. Total Environ., 2015, 520, 13–22, DOI:10.1016/j.scitotenv.2015.02.085.
- H. Hasegawa, Y. Sohrin, K. Seki, M. Sato, K. Norisuye, K. Naito and M. Matsui, Biosynthesis and release of methylarsenic compounds during the growth of freshwater algae, Chemosphere, 2001, 43, 265–272, DOI:10.1016/S0045-6535(00)00137-5.
- H. Hasegawa, R. I. Papry, E. Ikeda, Y. Omori, A. S. Mashio, T. Maki and M. A. Rahman, Freshwater phytoplankton: biotransformation of inorganic arsenic to methylarsenic and organoarsenic, Sci. Rep., 2019, 9, 1–13, DOI:10.1038/s41598-019-48477-7.
- F. Challenger, Biological methylation, Chem. Rev., 1945, 36, 315–361, DOI:10.1021/cr60115a003.
- T. Maki, H. Hasegawa and K. Ueda, Seasonal dynamics of dimethylarsinic-acid-decomposing bacteria dominating in Lake Kahokugata, Appl. Organomet. Chem., 2005, 19, 231–238, DOI:10.1002/aoc.696.
- S. J. Giovannoni, K. H. Halsey, J. Saw, O. Muslin, C. P. Suffridge, J. Sun, C.-P. Lee, E. R. Moore, B. Temperton and S. E. Noell, A parasitic arsenic cycle that shuttles energy from phytoplankton to heterotrophic bacterioplankton, mBio, 2019, 10, e00246-19, DOI:10.1128/mBio.00246-19.
- E. G. Duncan, W. A. Maher and S. D. Foster, Contribution of arsenic species in unicellular algae to the cycling of arsenic in marine ecosystems, Environ. Sci. Technol., 2015, 49, 33–50, DOI:10.1021/es504074z.
- S. Maeda, K. Kusadome, H. Arima, A. Ohki and K. Naka, Biomethylation of arsenic and its excretion by the alga Chlorella vulgaris, Appl. Organomet. Chem., 1992, 6, 407–413, DOI:10.1002/aoc.590060416.
- Suhendrayatna, A. Ohki, T. Kuroiwa and S. Maeda, Arsenic compounds in the freshwater green microalga Chlorella vulgaris after exposure to arsenite, Appl. Organomet. Chem., 1999, 13, 127–133, DOI:10.1002/(SICI)1099-0739(199902)13:2<127::AID-AOC810>3.0.CO;2-K.
- X. X. Yin, J. Chen, J. Qin, G. X. Sun, B. P. Rosen and Y. G. Zhu, Biotransformation and volatilization of arsenic by three photosynthetic cyanobacteria, Plant Physiol., 2011, 156, 1631–1638, DOI:10.1104/pp.111.178947.
- L. Savage, M. Carey, P. N. Williams and A. A. Meharg, Biovolatilization of arsenic as arsines from seawater, Environ. Sci. Technol., 2018, 52, 3968–3974, DOI:10.1021/acs.est.7b06456.
- T. Tziaras, S. A. Pergantis and E. G. Stephanou, Investigating the occurrence and environmental significance of methylated arsenic species in atmospheric particles by overcoming analytical method limitations, Environ. Sci. Technol., 2015, 49, 11640–11648, DOI:10.1021/acs.est.5b02328.
- H. Mukai and Y. Ambe, Detection of monomethylarsenic compounds originating from pesticide in airborne particulate matter sampled in an agricultural area in Japan, Atmos. Environ., 1987, 21, 185–189, DOI:10.1016/0004-6981(87)90284-8.
- M. O. Andreae, Arsenic speciation in seawater and interstitial waters: The influence of biological-chemical interactions on the chemistry of a trace element, Limnol. Oceanogr., 1979, 24, 440–452, DOI:10.4319/lo.1979.24.3.0440.
- M. Tanaka, Y. Takahashi, N. Yamaguchi, K.-W. Kim, G. Zheng and M. Sakamitsu, The difference of diffusion coefficients in water for arsenic compounds at various pH and its dominant factors implied by molecular simulations, Geochim. Cosmochim. Acta, 2013, 105, 360–371, DOI:10.1016/j.gca.2012.12.004.
- R. Salminen, M. J. Batista, M. Bidovec, A. Demetriades, B. De Vivo, W. De Vos, M. Duris, A. Gilucis, V. Gregorauskiene, J. Halamic, P. Heitzmann, A. Lima, G. Jordan, G. Klaver, P. Klein, J. Lis, J. Locutura, K. Marsina, A. Mazreku, P. J. O'Connor, S. A. Olsson, R.-T. Ottesen, V. Petersell, J. A. Plant, S. Reeder, I. Salpeteur, H. Sandström, U. Siewers, A. Steenfelt and T. Tarvainen, Geochemical Atlas of Europe. Part 1: Background Information, Methodology and Maps, Geological Survey of Finland, Espoo, 2005 Search PubMed.
- H.-R. Pfeifer, A. Häussermann, J. C. Lavanchy and W. Halter, Distribution and behavior of arsenic in soils and waters in the vicinity of the former gold-arsenic mine of Salanfe, Western Switzerland, J. Geochem. Explor., 2007, 93, 121–134, DOI:10.1016/j.gexplo.2007.01.001.
- S. E. Godsey, J. W. Kirchner and D. W. Clow, Concentration–discharge relationships reflect chemostatic characteristics of US catchments, Hydrol. Processes, 2009, 23, 1844–1864, DOI:10.1002/hyp.7315.
- W. Baeyens, A. de Brauwere, N. Brion, M. D. Gieter and M. Leermakers, Arsenic speciation in the River Zenne, Belgium, Sci. Total Environ., 2007, 384, 409–419, DOI:10.1016/j.scitotenv.2007.05.044.
- R. Bernard, F. Straub, L. Vuataz and M. Balet, Le Rhône – Observation de la qualitée des eaux de surface 2017. Canton du Valais: Département de la mobilité, du territoire et de l'environnement. Sevice de l'environnement, Sion, Switzerland, 2018, https://www.vs.ch/documents/19415/1603474/Qualit%E9deseauxduRh%F4neenavald%3FEvionnaz%2Ccampagne2017/acc4aecb-7ad8-46e3-be60-777eb16fc8ad?t=.now?long, last accessed: 31 December 2022 Search PubMed.
- C. M. Casado-Martinez, L. Molano-Leno, D. Grandjean, L. F. De Alencastro, I. Werner and B. J. D. Ferrari, Impact des séediments sur la qualitée de l’eau - Surveillance écotoxicologique de la qualitée de la rivière Venoge, Aqua Gas, 2016, 4, 56–63 Search PubMed , https://www.dora.lib4ri.ch/eawag/islandora/object/eawag:10561.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2em00431c |
| This journal is © The Royal Society of Chemistry 2023 |