
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron-rich pyrimidine rings enabling crystalline carbon nitride for high-efficiency photocatalytic hydrogen evolution coupled with benzyl alcohol selective oxidation†
Zhi
Lin
a,
Yiqing
Wang
a,
Ta
Thi Thuy Nga
b,
Jie
Zhang
c,
Ruizhe
Wang
a,
Zhengqi
Zhang
a,
Yufei
Xu
a,
Daming
Zhao
d,
Chung-Li
Dong
 b and
Shaohua
Shen
*a
b and
Shaohua
Shen
*a
aInternational Research Center for Renewable Energy, State Key Laboratory of Multiphase Flow in Power Engineering, Xi’an Jiaotong University, Shaanxi, 710049, China. E-mail: shshen_xjtu@mail.xjtu.edu.cn
bDepartment of Physics, Tamkang University, New Taipei City, 25137, Taiwan
cSchool of Civil and Architecture Engineering, Xi’an Technological University, Xi’an 710021, China
dSchool of Advanced Energy, Shenzhen Campus of Sun Yat-sen University, Shenzhen 518107, China
First published on 25th April 2023
Abstract
Photocatalytic water splitting over polymeric carbon nitride (PCN) has been seriously limited by the poor charge carrier transfer ability and sluggish four-electron water oxidation kinetics. Herein, crystalline carbon nitride (CCN-Pr) with electron-rich pyrimidine rings introduced in the molecular structure is synthesized by a two-step self-assembly and molten-salt annealing strategy for photocatalytic hydrogen evolution coupled with benzyl alcohol selective oxidation to benzaldehyde, instead of the kinetically sluggish water oxidation reaction. Owing to the synergistically tuned band and electronic structures, the obtained CCN-Pr exhibits excellent photocatalytic performances, with the highest hydrogen and benzaldehyde production rates reaching 149.39 μmol h−1 and 154.62 μmol h−1, respectively. The apparent quantum yield for hydrogen evolution is determined to be 20.27% at 420 nm, encouragingly standing at the highest level reported for simultaneous hydrogen and benzaldehyde production over PCN-based photocatalysts. It is well evidenced that the introduced electron-rich pyrimidine rings could finely tune the band structures for extended optical absorption and matched redox potentials for water reduction and benzyl alcohol oxidation. Theoretical calculation and experimental results reveal that the electronic structure engineered by pyrimidine rings alters the charge density distribution for promoted charge transport, and creates abundant reactive sites to accelerate the surface oxidation reaction kinetics. This work provides a reliable strategy to design efficient photocatalysts with band and electronic structures engineered by the tunable molecular structures, and also paves an alternative way to promote the economic benefits and the technology upgrading of solar energy conversion and utilization.
Broader contextPhotocatalytic water splitting for hydrogen production has been considered a promising technology to convert solar energy to chemical energy via zero-carbon cycle. However, the overall water splitting performance is still far from satisfactory, as it is mainly limited by the sluggish kinetics and endergonic thermodynamics of the water oxidation reaction. Moreover, oxygen production via photocatalytic water splitting is not economically and technically competitive, given the fact that oxygen could be easily produced on a large scale via simple air distillation process. Herein, crystalline PCN with electron-rich pyrimidine rings introduced in the molecular structure is used for photocatalytic hydrogen evolution coupled with benzyl alcohol selective oxidation to benzaldehyde, instead of the kinetically sluggish water oxidation reaction. It is well evidenced that the synergistic effects contributed from the tuned band and electronic structures give rise to the remarkable enhancement in photocatalytic benzyl alcohol oxidation and hydrogen evolution over PCN with molecular structures rationally engineered via the introduction of electron-rich pyrimidine rings. This work provides a reliable strategy to design efficient photocatalysts with band and electronic structures engineered by the tunable molecular structures, and also paves an alternative way to promote the economic benefits and technology upgrade of solar energy conversion and utilization. |
Introduction
Photocatalytic water splitting to produce hydrogen and oxygen is thought to be a promising and sustainable approach for solar energy conversion via zero-carbon cycle, which is expected to reduce the world's dependence on fossil fuels.1,2 The key to efficiently converting solar energy into hydrogen energy via photocatalytic water splitting is to develop stable, high-performance, and low-cost photocatalysts.3 Except for various metal-based semiconducting photocatalysts, polymeric carbon nitride (PCN), as a visible-responsive, inexpensive, nontoxic, and stable conjugated polymer semiconductor, has attracted much research attention and been extensively explored for photocatalysis in past years.4 Nevertheless, the fast charge carrier recombination, sluggish surface reaction kinetics, and the narrow visible photoresponse range largely limit its photocatalytic activity for water splitting.5 To overcome these drawbacks, many effective attempts have been proposed to improve the photocatalytic activity of PCN, including crystal structure engineering,6,7 element doping,8 morphology tuning,9,10 heterostructure construction,11–13 and single metal atom loading.14,15 Based on boron-doped and nitrogen-deficient PCN nanosheets, Shen et al. designed an all-PCN based 2D/2D Z-scheme heterojunction for efficient photocatalytic overall water splitting.16 Benefiting from the strong interfacial interaction, the matched band structure, and ultrathin nanostructures, this Z-scheme heterojunction received a solar-to-hydrogen (STH) efficiency as high as 1.16%. However, one should be aware that the overall water splitting performance is still far from satisfactory, as it is mainly limited by the water oxidation reaction with sluggish kinetics and endergonic thermodynamics.17 Moreover, oxygen production via photocatalytic water splitting is not economically and technically competitive, given the fact that oxygen could be easily produced on a large scale via simple air distillation process.18 Alternatively, oxidative synthesis of value-added organics that couples with hydrogen evolution could be expectedly more viable and valuable, with both photocatalytic efficiency and product value much improved, as compared to the photocatalytic water splitting system for simultaneous hydrogen and oxygen production.In the photoredox-catalyzed coupling reaction system, the organic substrate oxidation reaction would be much more kinetically favored than the four-electron process for oxygen production, accompanying the great enhancement in hydrogen evolution activity.19 Moreover, the value-added oxidative products generated by the coupled photocatalytic reactions could be widely used as fine chemicals with large economic benefits.20 As an essential industrial raw material, benzaldehyde obtained from the oxidation of benzyl alcohol is an important intermediate for the synthesis of herbicides, pharmaceuticals, and perfumes.21 However, the conventional oxidation method for benzaldehyde production requires hazardous oxidants (e.g., K2Cr2O7, V2O5, and KMnO4), with the oxidation reactions always occurring at high temperature and high pressure,22 which would inevitably increase production costs and cause environmental pollution. Thus, selective oxidation of benzyl alcohol to benzaldehyde by photocatalysis, along with efficient hydrogen production via water reduction, would be of great promise in industry and has made noticeable advances in recent years. For example, by introducing L-arginine into the molecular structure, Fu et al. prepared a PCN photocatalyst with adjustable bandgaps and achieved excellent photocatalytic performances for water reduction and benzyl alcohol oxidation to benzaldehyde,23 with the hydrogen evolution rate reaching 95.3 μmol h−1 and benzaldehyde production selectivity achieving 99.7%. Moreover, it has been proven to be effective to enhance the photocatalytic activity for hydrogen evolution coupled with benzyl alcohol selective oxidation, with the charge carrier separation and reaction kinetics accelerated by improving the crystallinity of PCN. By using NaCl as the ion-induction agent, Huo et al. prepared high crystallinity Na+-doped PCN nanosheets with altered charge density distribution,24 which exhibited facilitated charge transfer processes and rapid surface reaction kinetics, thus significantly improving the photocatalytic activity for hydrogen evolution and benzyl alcohol oxidation, with the apparent quantum yield reaching 10.5% at 420 nm. Based on these observations, it is expected that efficient photocatalysts for hydrogen evolution coupled with benzyl alcohol selective oxidation could be well-designed by tuning the band and electronic structures of crystalline PCN.
Herein, high crystalline PCN with electron-rich pyrimidine rings introduced into the molecular structure (CCN-Pr) was synthesized through a two-step self-assembly and molten-salt annealing strategy for the photocatalytic hydrogen evolution coupled with the selective oxidation of benzyl alcohol to benzaldehyde. With molecular structures altered by the introduced pyrimidine rings, excellent performances for the hydrogen evolution and selective oxidation of benzyl alcohol to benzaldehyde (conversion: 91%, selectivity: 99%, at 12 h) could be realized over the obtained CCN-Pr photocatalysts, with the hydrogen and benzaldehyde evolution rates reaching 149.39 and 154.62 μmol h−1, respectively, which stands at the highest level reported for PCN-based photocatalysts. Experimental and theoretical studies have shown that the introduced electron-rich pyrimidine rings could finely tune the band structures for the extended optical absorption and the matched redox potentials for water reduction and benzyl alcohol oxidation reactions, as well as modulate the electronic structures, thus altering the charge density distribution for the promoted charge carrier separation and transfer. Moreover, the C atoms in the introduced pyrimidine rings could serve as the reactive sites for the rapid adsorption of benzyl alcohol and desorption of benzaldehyde, and then accelerate the surface oxidation reaction kinetics.
Results and discussion
Herein, CCN-Pr with tunable molecular structures is synthesized through a two-step self-assembly and molten-salt annealing strategy (see Experimental section for details in ESI†). As illustrated in Fig. 1a, with 2,4,6-triaminepyrimidine (TAP), melamine and cyanuric acid self-assembled into the supramolecular precursors (MCA-x, x = 0, 20, 40, 60, 80), where x represents the feeding amounts (mg) of TAP in precursors, the CCN-Prx photocatalysts with pyrimidine rings introduced into the molecular structure could be obtained by annealing the MCA-x precursors in KCl/LiCl molten salts. The obtained CCN-Prx spheres (taking CCN-Pr40 as the example) with an average diameter of ∼1.5 μm (Fig. 1b and c) present high crystallinity, as identified by the clear lattice spacing of 0.33 nm (Fig. 1d), owing to the liquid-phase reaction environment provided by the KCl/LiCl molten salts that reduce the kinetic obstacles for the improved crystallinity.25 In comparison, without the KCl/LiCl molten salts during annealing, the obtained PCN sample (CN) with similar spherical morphology (Fig. S1a, ESI†) reveals an amorphous structure (Fig. S1b, ESI†). This demonstrates the necessity of molten salts for the crystallization of the annealing synthesized PCN, which is further supported by X-ray diffraction (XRD) analysis. As shown in Fig. 2a, the amorphous CN synthesized in the absence of KCl/LiCl molten salts during annealing presents two distinct XRD peaks at 2θ = 13.1° and 27.1°, assigned to the (100) and (002) planes of PCN, respectively. In comparison, all of the CCN-Prx samples synthesized in the presence of molten salts present very similar XRD patterns, with (100) and (002) peaks shifted to 2θ = 8.0° and 27.9°, respectively, indicating the reduced interlayer distance and the condensed in-plane conjugated skeleton for CCN-Prx with high crystallinity.25 The narrowed full width at half-maximum (FWHM) observed for the (002) peaks of CCN-Prx also evidences the improved crystallinity induced by molten-salt annealing (Table S1, ESI†).26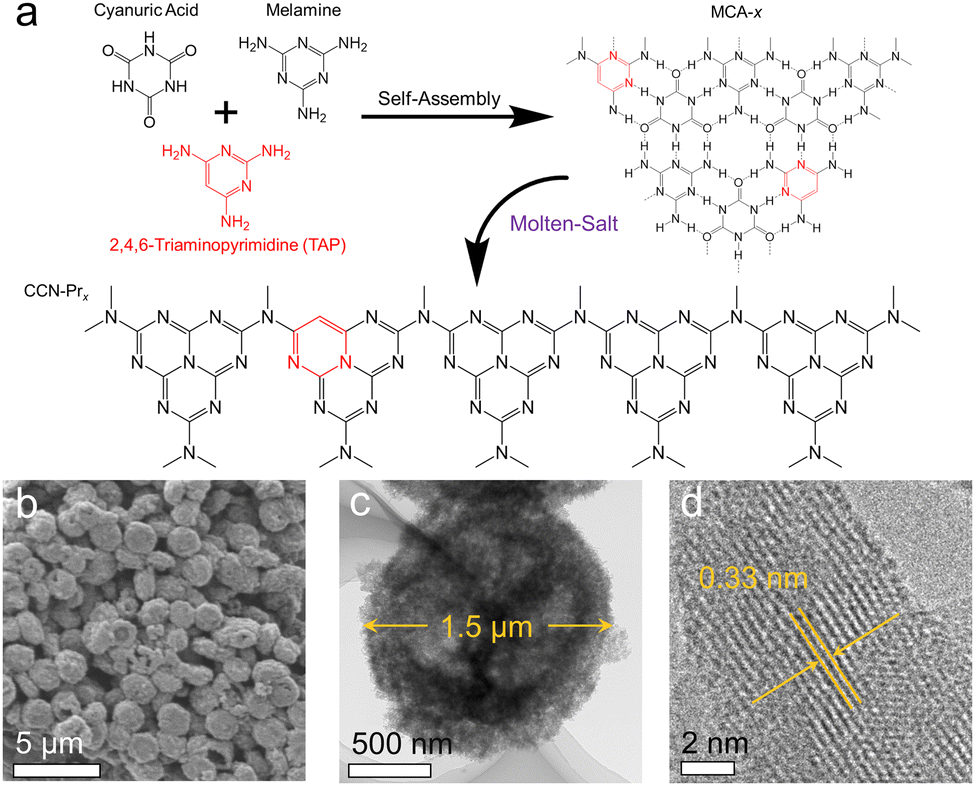 | ||
| Fig. 1 (a) The proposed preparation process of CCN-Prx. (b) Scanning electron microscopy (SEM) and (c and d) transmission electron microscopy (TEM) images of CCN-Pr40. | ||
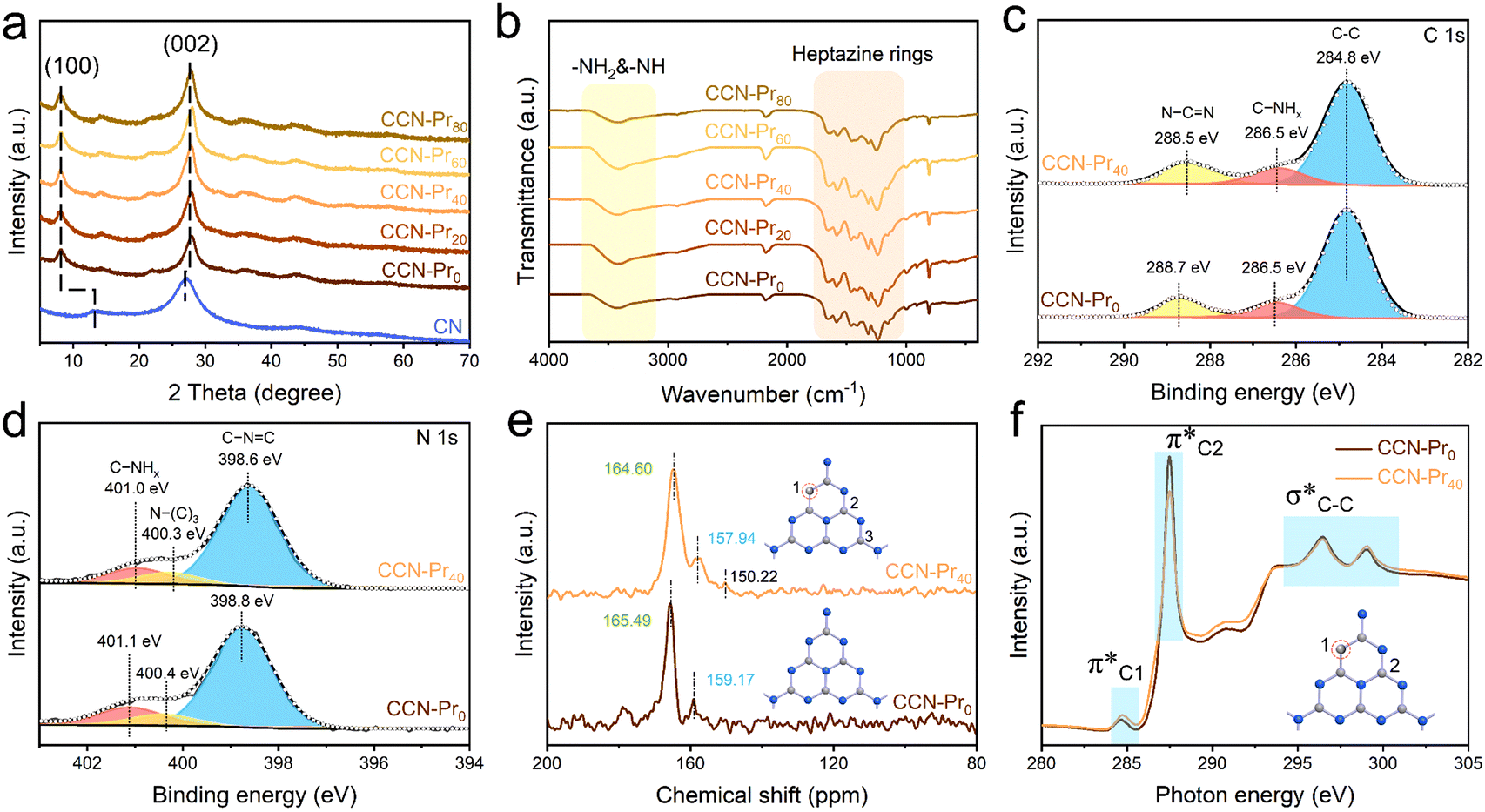 | ||
| Fig. 2 (a) XRD patterns of CN and CCN-Prx. (b) FT-IR spectra of CCN-Prx. High-resolution XPS spectra of (c) C 1s and (d) N 1s for CCN-Pr0 and CCN-Pr40. (e) Solid-state 13C CP-NMR and (f) C K-edge XANES spectra of CCN-Pr0 and CCN-Pr40. Gray: C, blue: N. | ||
Depending on the introduction of pyrimidine rings, the molecular structure evolution was then investigated over the obtained CCN-Prx photocatalysts. Elemental analysis reveals the gradually increased carbon contents in CCN-Prx with the increasing amounts of TAP introduced in the supramolecular precursors (Table S2, ESI†), suggesting the successful incorporation of pyrimidine rings into the PCN framework. Fourier transform infrared (FT-IR) spectra (Fig. 2b) show very similar fingerprint signals for all of the CCN-Prx photocatalysts, with the peaks at 1200–1700 cm−1, ∼810 cm−1 and 3000–3500 cm−1 attributed to the skeletal vibration of heptazine rings, the out-of-plane bending of heptazine rings, and the stretching vibrations of –NH2 and –NH–, respectively.27 These similar FT-IR fingerprints suggest that the introduction of pyrimidine rings would not remarkably change the original heptazine units in CCN-Prx.28 By further looking into the atomic structures monitored by X-ray photoelectron spectroscopy (XPS), one would note that the high-resolution XPS C 1s spectrum could be deconvoluted into three peaks at 284.8, 286.5, and 288.7 eV for CCN-Pr0 (Fig. 2c), corresponding to the adventitious carbon (C–C), the C–NHx groups, and the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N units in PCN framework, respectively.29 In comparison, the peak assigned to the N–CN units is slightly shifted to lower binding energy for CCN-Pr40, which verifies the possible replacement of N atoms in heptazine rings by C atoms with low electronegativity.30 It is also noted that the three distinct XPS N 1s peaks (Fig. 2d) corresponding to the C–NC, N–(C)3, and C–NHx groups31 are slightly shifted to lower binding energy for CCN-Pr40 relative to CCN-Pr0, suggesting that the introduced electron-rich pyrimidine rings would increase the electron density of N atoms.30 Solid-state 13C cross-polarization nuclear magnetic resonance (CP-NMR) spectra (Fig. 2e) further identify two strong peaks at 165.49 and 159.17 ppm for CCN-Pr0, which should be derived from the C atoms in the C–N3 and CN2–(NHx) groups, respectively, confirming the heptazine structures in CCN-Pr0.32 In comparison, these two peaks are slightly shifted to 164.60 and 157.94 ppm, respectively, for CCN-Pr40, indicating that CCN-Pr40 maintains the heptazine framework, but the electron density is disturbed by the introduced electron-rich pyrimidine rings. An additional peak could be observed at 150.22 ppm for CCN-Pr40, belonging to the C atoms in the sp2 CC electron-withdrawing group in pyrimidine rings,33 which evidences the introduction of pyrimidine rings into the PCN skeleton, as further confirmed by the C K-edge X-ray absorption near edge structure (XANES) spectra (Fig. 2f). The peaks at 284.7, 287.5 and 294.4–301.7 eV should be attributed to the orbital transition of π*CC (π*C1), π*C–NC (π*C2) and σ*C–C, respectively. The doublet peaks at 294.4–301.7 eV, featured for crystalline PCN, are observed in both CCN-Pr0 and CCN-Pr40 with high crystallinity.34 The π*C1 peak intensity is increased for CCN-Pr40, relative to CCN-Pr0, which is attributed to the successful introduction of pyrimidine rings with CC bonds into CCN-Pr40.35 Furthermore, the significantly reduced π*C2 peak intensity indicates that some of the empty orbitals in the heptazine units are filled with electrons provided by the electron-rich pyrimidine rings.36 These spectral variations evidence that the introduced pyrimidine rings would induce the charge density redistribution in the obtained CCN-Pr40.
N units in PCN framework, respectively.29 In comparison, the peak assigned to the N–CN units is slightly shifted to lower binding energy for CCN-Pr40, which verifies the possible replacement of N atoms in heptazine rings by C atoms with low electronegativity.30 It is also noted that the three distinct XPS N 1s peaks (Fig. 2d) corresponding to the C–NC, N–(C)3, and C–NHx groups31 are slightly shifted to lower binding energy for CCN-Pr40 relative to CCN-Pr0, suggesting that the introduced electron-rich pyrimidine rings would increase the electron density of N atoms.30 Solid-state 13C cross-polarization nuclear magnetic resonance (CP-NMR) spectra (Fig. 2e) further identify two strong peaks at 165.49 and 159.17 ppm for CCN-Pr0, which should be derived from the C atoms in the C–N3 and CN2–(NHx) groups, respectively, confirming the heptazine structures in CCN-Pr0.32 In comparison, these two peaks are slightly shifted to 164.60 and 157.94 ppm, respectively, for CCN-Pr40, indicating that CCN-Pr40 maintains the heptazine framework, but the electron density is disturbed by the introduced electron-rich pyrimidine rings. An additional peak could be observed at 150.22 ppm for CCN-Pr40, belonging to the C atoms in the sp2 CC electron-withdrawing group in pyrimidine rings,33 which evidences the introduction of pyrimidine rings into the PCN skeleton, as further confirmed by the C K-edge X-ray absorption near edge structure (XANES) spectra (Fig. 2f). The peaks at 284.7, 287.5 and 294.4–301.7 eV should be attributed to the orbital transition of π*CC (π*C1), π*C–NC (π*C2) and σ*C–C, respectively. The doublet peaks at 294.4–301.7 eV, featured for crystalline PCN, are observed in both CCN-Pr0 and CCN-Pr40 with high crystallinity.34 The π*C1 peak intensity is increased for CCN-Pr40, relative to CCN-Pr0, which is attributed to the successful introduction of pyrimidine rings with CC bonds into CCN-Pr40.35 Furthermore, the significantly reduced π*C2 peak intensity indicates that some of the empty orbitals in the heptazine units are filled with electrons provided by the electron-rich pyrimidine rings.36 These spectral variations evidence that the introduced pyrimidine rings would induce the charge density redistribution in the obtained CCN-Pr40.
All of the above molecular and atomic structural analyses confirm the successful introduction of electron-rich pyrimidine rings into CCN-Prx. Depending on the increasing TAP feeding amounts, the obtained CCN-Prx photocatalysts exhibit a progressive redshift in the optical absorption edges (Fig. 3a), with the bandgap (Eg) gradually narrowed from 2.43 eV for CCN-Pr0 to 1.49 eV for CCN-Pr80 (Fig. 3b). Given the very close conduction band (CB) and flat band potentials for a typical n-type semiconductor, the CB potentials (Ec) could be determined from the Mott–Schottky plots for CCN-Prx, cathodically shifted from −0.47 V (vs. reversible hydrogen electrode, RHE) for CCN-Pr0 to −0.73 V (vs. RHE) for CCN-Pr80 (Fig. 3c). Then, the valence band (VB) potentials (Ev), calculated by Eg and Ec (see Fig. S2 and related discussion in ESI†),37 are cathodically shifted from 1.96 V (vs. RHE) for CCN-Pr0 to 0.76 V (vs. RHE) for CCN-Pr80, depending on the increasing TAP feeding amounts (Fig. 3d). It is thus convincing that the introduction of pyrimidine rings could rationally engineer the band structures of CCN-Prx, with the CB and VB positions finely tuned to broaden the optical absorption region and match the redox potentials for the water reduction and benzyl alcohol oxidation reactions.38
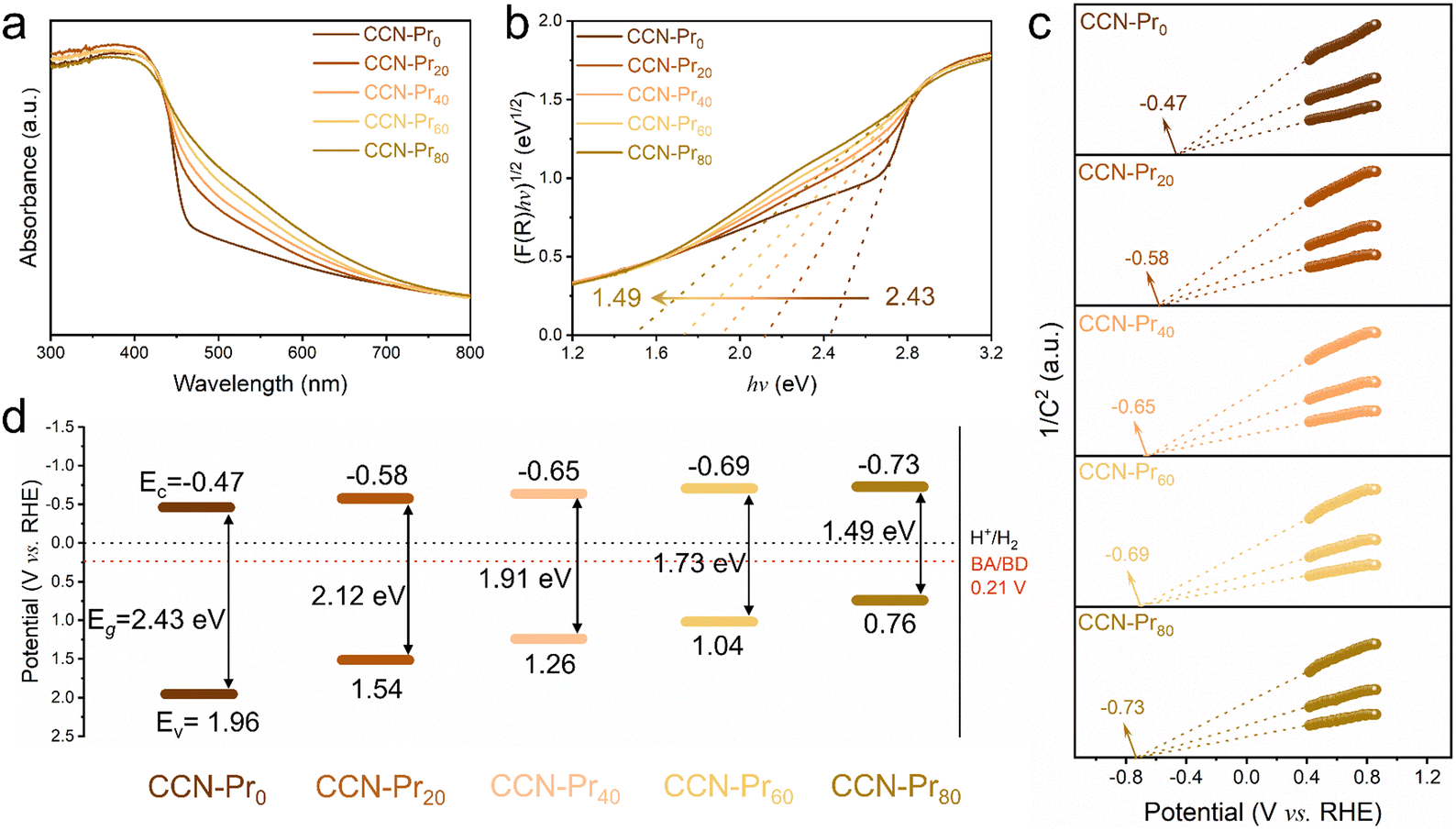 | ||
| Fig. 3 (a) Ultraviolet-visible-near infrared diffuse reflectance spectra, (b) plots of the transformed Kubelka–Munk function versus photon energy, (c) Mott–Schottky plots and (d) band structures of CCN-Prx (x = 0, 20, 40, 60, 80). BA and BD represent benzyl alcohol and benzaldehyde, respectively. | ||
To further understand the electronic structure evolution with the introduction of electron-rich pyrimidine rings, density-functional theory (DFT) calculations were carried out for CCN-Pr0 (Fig. 4a) and CCN-Pr40 (Fig. 4b). The band structures and density of states (DOS) calculation results demonstrate that both C 2p and N 2p orbitals contribute to the CB, while the VB is predominantly composed of N 2p orbitals for both CCN-Pr0 and CCN-Pr40 (Fig. 4c–f). With pyrimidine rings introduced, the C 2p orbitals of the newly introduced pyrimidine-carbon atoms would participate in the formation of VB and significantly elevate the VB position to narrow the bandgap.39 The theoretically calculated Eg is significantly decreased from 2.37 eV for CCN-Pr0 to 1.71 eV for CCN-Pr40, which agrees well with the extended optical absorption and measured Eg for CCN-Prx (Fig. 3a and b). Further investigation into the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) locations reveals that both LUMO and HOMO orbitals show symmetrical and overlapping distributions over heptazine rings (Fig. 4g). This is due to the high symmetry of the planar structure in CCN-Pr0. In contrast, the electron localization occurs near the electron-donating pyrimidine-carbon atoms in CCN-Pr40 (Fig. 4h), which leads to the redistribution of charge density, thus inducing the internal electric field to promote exciton dissociation and accelerate charge transport.40 Furthermore, this charge density redistribution would enable the separation of redox reactive sites, promoting charge carrier separation and suppressing back reactions, which is believed to benefit the photocatalytic activity.41
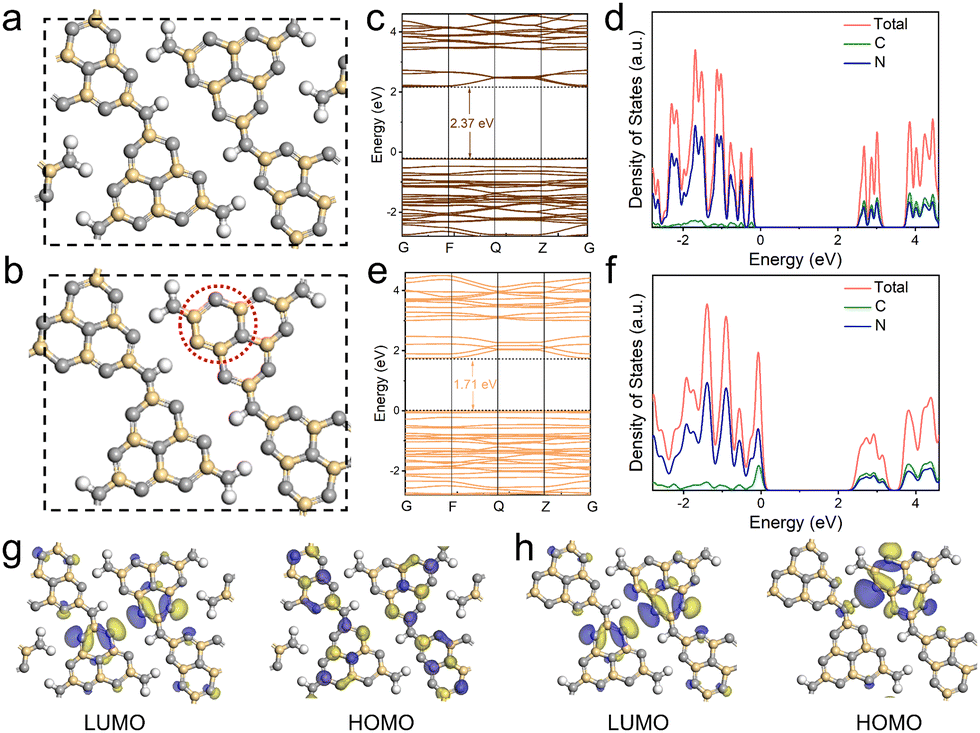 | ||
| Fig. 4 Structure models of (a) CCN-Pr0 and (b) CCN-Pr40. Calculated band structures and corresponding DOS of (c and d) CCN-Pr0 and (e and f) CCN-Pr40. LUMO and HOMO distributions of (g) CCN-Pr0 and (h) CCN-Pr40. The blue and yellow regions represent the net electron accumulation and depletion, respectively. The gray, yellow, and white spheres represent the N, C, and H atoms, respectively. | ||
The above theoretical calculation results suggest that the introduced electron-rich pyrimidine rings could promote exciton dissociation and charge separation by inducing the charge density redistribution in CCN-Prx. To strengthen this deduction, XANES spectra were further recorded in dark and under illumination at the C K-edge and N K-edge to monitor the photoexcited electronic transitions (Fig. 5a) in CCN-Pr0 and CCN-Pr40. With the introduction of pyrimidine rings, CCN-Pr40 exhibits more distinct dark-irradiation intensity variations in both C K-edge and N K-edge spectra than CCN-Pr0 (Fig. 5b). This demonstrates a more efficient electron transition from the occupied states to unoccupied states in CCN-Pr40.42 This observation could be explained by the introduction of electron-rich pyrimidine rings that could induce the internal electric field to accelerate the exciton dissociation by the charge density redistribution over the PCN networks.43 Moreover, the electron-rich pyrimidine rings would serve as the donor units to improve the charge separation and transfer by rapidly collecting photoexcited holes without recombination through the strong donor–acceptor interaction.39 To further support the above deduction, the charge transfer property was then systematically investigated by surface photovoltage (SPV), photoluminescence (PL), and electron paramagnetic resonance (EPR) spectra. As monitored by SPV spectra (Fig. 5c), the positive SPV signals identify both CN and CCN-Prx with n-type semiconducting characteristic.44 It should be noted that there are much stronger SPV signals observed for CCN-Prx than CN, meaning that the high crystallinity triggered by molten-salt annealing would favor photogenerated charge carrier separation. With electron-rich pyrimidine rings introduced, the SPV signals are gradually strengthened, reaching the maximum for CCN-Pr40. These signals are then weakened, depending on the increasing TAP feeding amounts. This phenomenon suggests that the introduction of pyrimidine rings at an appropriate level is important to facilitate the separation of photogenerated charge carriers in CCN-Prx, which could be further confirmed by steady-state PL emission spectra. As shown in Fig. 5d, CN shows a strong PL emission peak, indicating the rapid charge carrier recombination,17 while all the CCN-Prx samples exhibit significantly quenched PL emission, indicating the promoted charge carrier separation, as induced by the improved crystallinity of CCN-Prx. Specifically, the PL emission intensity first decreases and then increases, depending on the increasing TAP feeding amounts. The lowest PL intensity was attained for CCN-Pr40, revealing the accelerated separation of photogenerated charge carriers in CCN-Pr40. The improvement in charge carrier transfer ability could be also confirmed by the time-resolved transient PL decay spectra. As shown in Fig. 5e and Table S3 (ESI†), both CCN-Pr0 (3.92 ns) and CCN-Pr40 (2.19 ns) exhibit much shorter average lifetimes (τavg) of photogenerated charge carriers with respect to CN (8.31 ns), suggesting that the high crystallinity triggered by molten-salt annealing is beneficial to accelerate the charge carrier transfer process. Moreover, the shortest average lifetime of the charge carriers in CCN-Pr40 again evidences that the introduction of electron-rich pyrimidine rings could remarkably promote photogenerated charge carrier separation and transfer.15 These results are also supported by EPR analysis (Fig. S3, ESI†). It should be noted that all three samples, i.e., CN, CCN-Pr0, and CCN-Pr40, exhibit an obvious Lorentzian line with g factor of 2.003, which is caused by the unpaired electrons on the π-conjugated aromatic rings.17 In comparison to amorphous CN, CCN-Pr0 exhibits a remarkably increased EPR signal, indicating that the high crystallinity triggered by molten-salt annealing would favor the delocalization of electrons on the aromatic heptane rings. Moreover, the much stronger EPR signal observed for CCN-Pr40 than CCN-Pr0 means that the introduced electron-rich pyrimidine rings would provide more unpaired delocalized electrons in CCN-Pr40,17 which is beneficial to promote the charge carrier separation.
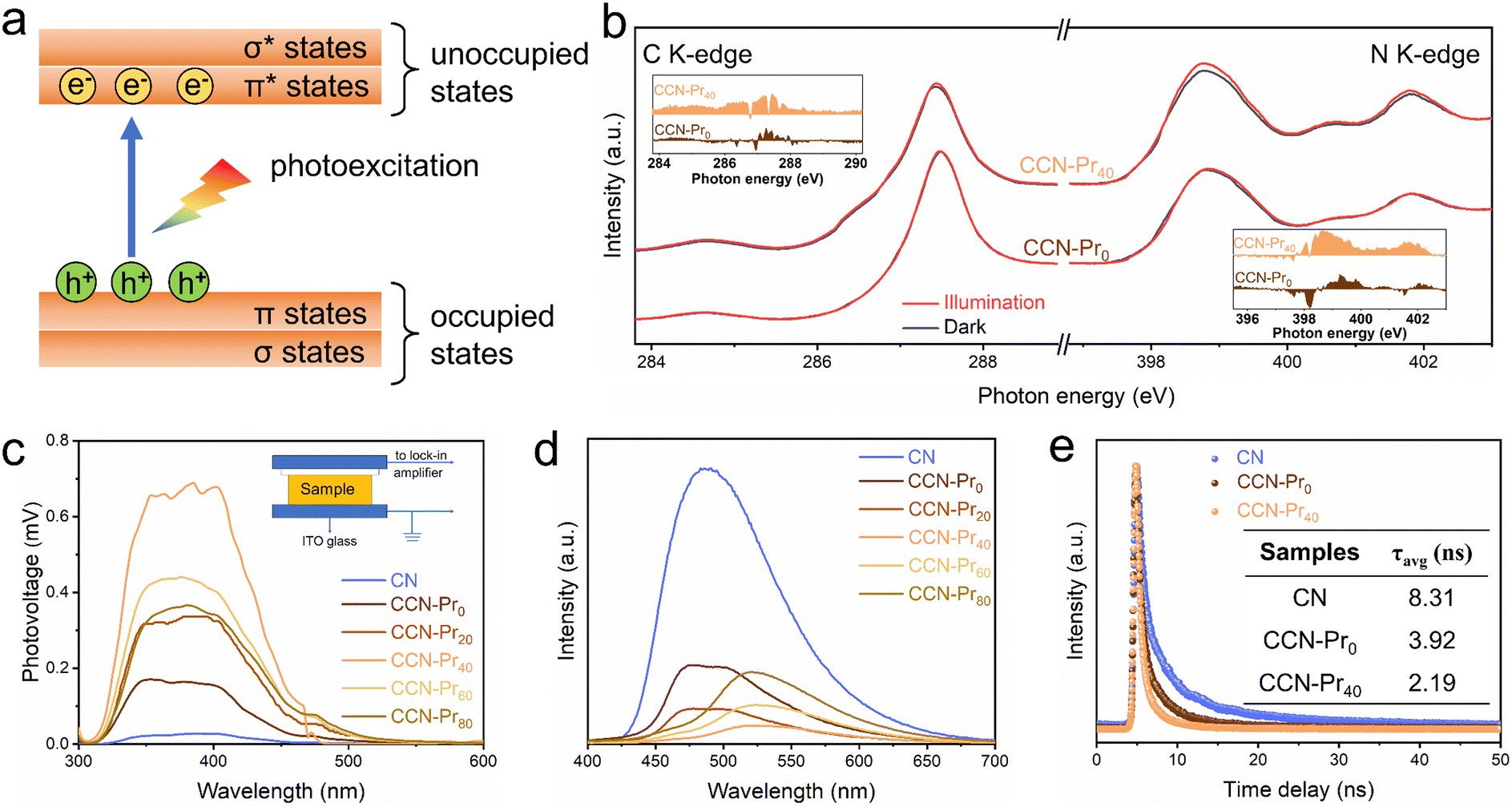 | ||
| Fig. 5 (a) Illustration of the electron transition processes from the occupied states to unoccupied states in PCN. (b) C K-edge and N K-edge XANES spectra of CCN-Pr0 and CCN-Pr40 with or without illumination. The inset in (b) exhibits the spectral difference between being in dark and illuminated conditions. (c) SPV and (d) steady-state PL emission spectra of CN and CCN-Prx. (e) Time-resolved transient PL decay spectra of CN, CCN-Pr0 and CCN-Pr40. | ||
The photocatalytic activity for hydrogen evolution over CCN-Prx was evaluated in an aqueous solution containing benzyl alcohol that was selectively oxidized to benzaldehyde. With the band and electronic structures well tuned by the introduction of electron-rich pyrimidine rings, the obtained CCN-Prx photocatalysts could effectively drive water reduction and benzyl alcohol oxidation relations for simultaneous and stoichiometric hydrogen and benzaldehyde evolution (Fig. 6a). All CCN-Prx (x = 0, 20, 40, 60, 80) samples with high crystallinity exhibit significantly increased photocatalytic hydrogen and benzaldehyde evolution rates, as compared to amorphous CN, which should be attributed to the improved light absorption (Fig. S4, ESI†) and the rapid charge carrier separation, benefiting from the high crystallinity triggered by molten salt annealing. Specifically, the photocatalytic activity increases first and then decreases for CCN-Prx, depending on the increasing contents of TAP fed in precursors. The highest hydrogen (149.39 μmol h−1) and benzaldehyde (154.62 μmol h−1) evolution rates are achieved over CCN-Pr40, which are about 6.7 times those of CN (22.36 μmol h−1 and 17.22 μmol h−1) and 2.2 times those of CCN-Pr0 (68.65 μmol h−1 and 64.18 μmol h−1), standing among the highest level reported for PCN-based photocatalysts (Table S4, ESI†). Considering that the introduction of pyrimidine rings does not significantly change the specific surface area (Fig. S5, ESI†), this volcano-shaped dependence of the photocatalytic activity on the TAP feeding amounts should be mainly attributed to the synergistically modulated band and electronic structures of CCN-Prx with introduced pyrimidine rings. With the introduced electron-rich pyrimidine rings, the VB position of CCN-Prx is gradually elevated to narrow the bandgap and extend the optical absorption (Fig. 3a and d). Meanwhile, the oxidation ability of the photogenerated holes gradually decreases with the elevating VB position,45 leading to the weakened driving force for the benzyl alcohol oxidation reaction. It should be further noted that with the electron-rich pyrimidine rings introduced into CCN-Prx at a moderate level, the triggered charge density redistribution would benefit the efficient electron excitation and the promoted charge transport. However, the excessive pyrimidine rings in CCN-Prx would serve as the recombination sites for charge carriers, and thus retard the charge transfer and separation processes.41 Therefore, CCN-Pr40 with synergistically engineered band and electronic structures exhibits the highest photocatalytic hydrogen and benzaldehyde evolution rates. This is due to the rapid charge carrier separation, the broadened optical absorption, and the well-tuned driving force for the benzyl alcohol oxidation reaction. CCN-Pr40 exhibits significantly improved conversion (91%) and selectivity (99%) for benzyl alcohol selective oxidation into benzaldehyde in a 12 hour reaction (Fig. 6b and Table S5, ESI†), with respect to CCN-Pr0 (conversion: 73%; selectivity: 89%) and CN (conversion: 36%; selectivity: 89%). Encouragingly, solar-to-hydrogen (STH) and apparent quantum yield (AQY) were calculated to be as high as 0.74% under one-sun illumination (Fig. 6c and Table S6, ESI†) and 20.27% at 420 nm (Fig. 6d), respectively, which even outperforms the most previously reported PCN-based photocatalysts for hydrogen evolution in sacrificial reagent solutions (Table S7, ESI†). The optical absorption profile closely dependent AQY values obtained for CCN-Pr40 suggest that this hydrogen evolution reaction is indeed driven by photoexcitation, with hydrogen mainly produced via water reduction as confirmed by the D2O isotope labeled photocatalytic measurement (Fig. S6, ESI†). A considerable activity could be still detected with the illumination wavelengths extended to be longer than 500 nm, indicating that the narrowed bandgap and the extended optical absorption greatly contribute to the improved photocatalytic activity of CCN-Pr40. The photocatalytic hydrogen evolution activity could be well recovered by supplementing benzyl alcohol after reacting for an extended time (Fig. S7, ESI†). Furthermore, the chemical structure and composition of CCN-Pr40 are well maintained before and after photocatalytic reaction (Fig. S8 and Table S8, ESI†), proving that CCN-Pr40 is stable for photocatalytic hydrogen evolution coupled with benzyl alcohol selective oxidation.
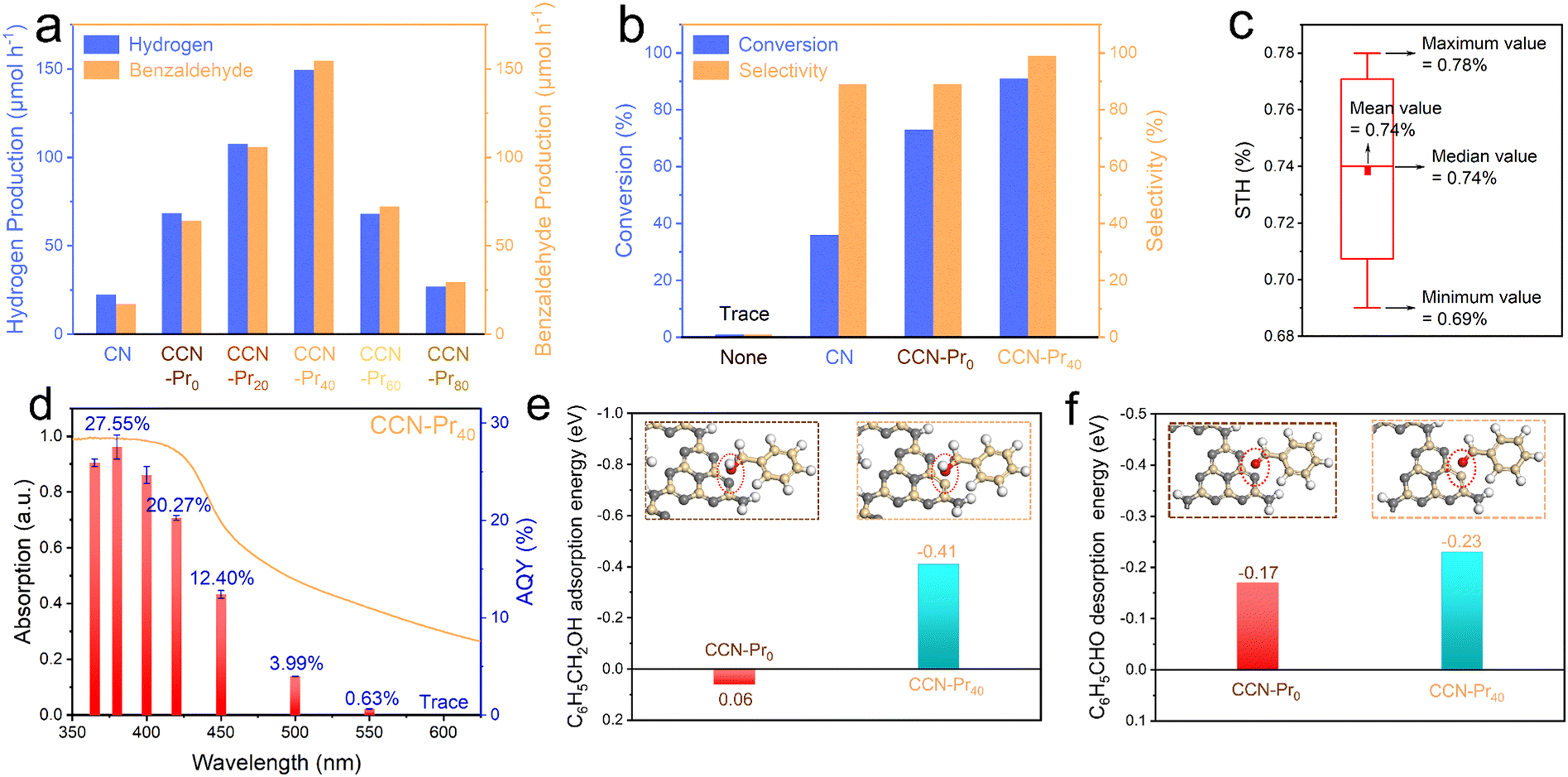 | ||
| Fig. 6 (a) Hydrogen and benzaldehyde evolution rates for CN and CCN-Prx. (b) The benzyl alcohol conversion and benzaldehyde selectivity of CN, CCN-Pr0 and CCN-Pr40. (c) The STH of CCN-Pr40 for photocatalytic hydrogen evolution in H2O and benzyl alcohol solution. The STH value was evaluated 11 times with separate samples. (d) The wavelength-dependent AQY of the hydrogen evolution reaction over CCN-Pr40. (e) Benzyl alcohol adsorption and (f) benzaldehyde desorption energies of CCN-Pr0 and CCN-Pr40. The gray, yellow, red and white spheres represent N, C, O and H atoms, respectively. | ||
By looking into the oxidation reaction for selective conversion of benzyl alcohol to benzaldehyde, the active sites were further theoretically identified to investigate the adsorption of benzyl alcohol molecules and the desorption of benzaldehyde molecules at the surface of CCN-Prx, which could be another determinant to the photocatalytic activity for simultaneous and stoichiometric hydrogen and benzaldehyde evolution. With computational configurations and adsorption energies of benzyl alcohol presented and calculated for CCN-Pr0 and CCN-Pr40 (Fig. 6e), one would note a positive adsorption energy (0.06 eV) required for the benzyl alcohol molecules to be adsorbed at N atoms in CCN-Pr0, indicating that the adsorption and activation of benzyl alcohol at the N atoms of heptazine rings is unfavored. In contrast, the adsorption of energy required for benzyl alcohol molecules at the pyrimidine-carbon atoms of the introduced pyrimidine rings in CCN-Pr40 is substantially reduced to −0.41 eV. Moreover, a more negative desorption energy of benzaldehyde is required for CCN-Pr40 (−0.23 eV) than CCN-Pr0 (−0.17 eV) (Fig. 6f), suggesting that the desorption of benzaldehyde on CCN-Pr40 is easier than on CCN-Pr0. Thus, the theoretical calculation results indicate that the pyrimidine-carbon atoms in pyrimidine rings could act as reactive sites that are superior to the N atoms in heptazine rings for benzyl alcohol adsorption and benzaldehyde desorption, thus accelerating the surface reaction kinetics for the selective oxidation of benzyl alcohol to benzaldehyde.46
Based on the above experimental characterization and theoretical calculation results, the significant improvement in the photocatalytic activity of CCN-Pr40 for hydrogen evolution coupled with benzyl alcohol selective oxidation can be explained as follows: (1) The introduced electron-rich pyrimidine rings could finely tune the CB and VB positions to extend the optical absorption, with the redox potentials well matched to the water reduction and benzyl alcohol selective oxidation reactions. (2) The introduced electron-rich pyrimidine rings would trigger charge density redistribution for efficient electron excitation and promoted charge transport. (3) The C atoms in the electron-rich pyrimidine rings could serve as the reactive sites for the rapid adsorption of benzyl alcohol and desorption of benzaldehyde, and then accelerate the surface oxidation reaction kinetics (Fig. 7). These synergistic effects contributed from the tuned band and electronic structures give rise to the remarkable enhancement in photocatalytic benzyl alcohol oxidation and hydrogen evolution over PCN with molecular structures rationally engineered via the introduction of electron-rich pyrimidine rings.
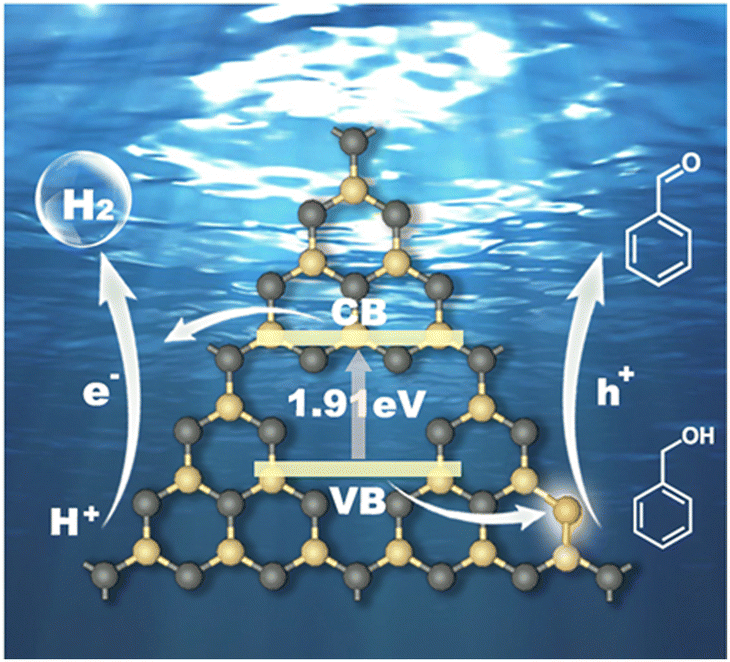 | ||
| Fig. 7 Reaction mechanism of the photocatalytic hydrogen evolution coupled with benzyl alcohol oxidation for CCN-Pr40. Yellow: C, gray: N. | ||
Conclusion
In summary, high crystalline CCN-Pr with electron-rich pyrimidine rings introduced into the molecular structure was synthesized via two-step self-assembly and molten-salt annealing strategy. With molecular structures altered by the introduced pyrimidine rings, the obtained CCN-Pr exhibits significant improvement in photocatalytic activities for hydrogen evolution and benzyl alcohol selective oxidation, with the optimized hydrogen and benzaldehyde evolution rates reaching 149.39 μmol h−1 and 154.62 μmol h−1. It is well evidenced that the band structures could be tuned successively via the introduction of electron-rich pyrimidine rings for the extended optical absorption and the matched redox potentials for water reduction and benzyl alcohol oxidation reactions. Furthermore, the introduced electron-rich pyrimidine rings would induce charge density redistribution for the promoted charge carrier transfer and separation. Meanwhile, the C atoms in the introduced pyrimidine rings could serve as the reactive sites for the rapid adsorption of benzyl alcohol and desorption of benzaldehyde, thus accelerating the surface oxidation reaction kinetics. This work provides a facile and reliable strategy to design novel and efficient PCN photocatalysts with band and electronic structures engineered by the tunable molecular structures, and also opens a new avenue for high performance and value-added solar energy conversion and utilization.Author contributions
For author contributions, Z. Lin carried out the sample preparations, characterizations and photocatalytic measurements. Y. Wang, Z. Zhang, and D. Zhao conducted the DFT calculations. Z. Lin, J. Zhang and S. Shen wrote the paper. T. T. Thuy Nga and C.-L. Dong carried out the XAS measurements. R. Wang and Y. Xu detected the benzyl alcohol and benzaldehyde concentrations. S. Shen conceived the idea and supervised the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (52225606, 51888103), the “Fundamental Research Funds for the Central Universities”, and “The Youth Innovation Team of Shaanxi Universities”.Notes and references
- X. Chen, S. Shen, L. Guo and S. S. Mao, Semiconductor-based Photocatalytic Hydrogen Generation, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- C. Bie, L. Wang and J. Yu, Challenges for photocatalytic overall water splitting, Chem, 2022, 8, 1567–1574 CAS.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Recent advances and perspectives for solar-driven water splitting using particulate photocatalysts, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Polymer photocatalysts for solar-to-chemical energy conversion, Nat. Rev. Mater., 2020, 6, 168–190 CrossRef.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability?, Chem. Rev., 2016, 116, 7159 CrossRef CAS PubMed.
- B. Zhai, H. Li, G. Gao, Y. Wang, P. Niu, S. Wang and L. Li, A Crystalline Carbon Nitride Based Near-Infrared Active Photocatalyst, Adv. Funct. Mater., 2022, 32, 2207375 CrossRef CAS.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Molecular-level insights on the reactive facet of carbon nitride single crystals photocatalysing overall water splitting, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- Y. Wang, D. Zhao, H. Deng, M. Li, J. Chen and S. Shen, Theoretical Insights into the Limitation of Photocatalytic Overall Water Splitting Performance of VIA Group Elements Doped Polymeric Carbon Nitride: A Density Functional Theory Calculation Predicting Solar-to-Hydrogen Efficiency, Solar RRL, 2021, 5, 2000630 CrossRef CAS.
- B. Fang, Z. Xing, D. Sun, Z. Li and W. Zhou, Hollow semiconductor photocatalysts for solar energy conversion, Adv. Powder Mater., 2022, 1, 100021 CrossRef.
- G. Zhang, G. Li, Z. A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Optimizing Optical Absorption, Exciton Dissociation, and Charge Transfer of a Polymeric Carbon Nitride with Ultrahigh Solar Hydrogen Production Activity, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- X. Chen, J. Wang, Y. Chai, Z. Zhang and Y. Zhu, Efficient Photocatalytic Overall Water Splitting Induced by the Giant Internal Electric Field of a g-C3N4/rGO/PDIP Z-Scheme Heterojunction, Adv. Mater., 2021, 33, 2007479 CrossRef CAS PubMed.
- D. Zhao, M. Wang, T. Kong, Y. Shang, X. Du, L. Guo and S. Shen, Electronic pump boosting photocatalytic hydrogen evolution over graphitic carbon nitride, Mater. Today Chem., 2019, 11, 296–302 CrossRef CAS.
- D. Zhao, X. Guan and S. Shen, Design of polymeric carbon nitride-based heterojunctions for photocatalytic water splitting: a review, Environ. Chem. Lett., 2022, 20, 3505–3523 CrossRef CAS.
- F. Chen, X. L. Wu, C. Shi, H. Lin, J. Chen, Y. Shi, S. Wang and X. Duan, Molecular Engineering toward Pyrrolic N-Rich M-N4 (M = Cr, Mn, Fe, Co, Cu) Single-Atom Sites for Enhanced Heterogeneous Fenton-Like Reaction, Adv. Funct. Mater., 2021, 31, 2007877 CrossRef CAS.
- Z. Lin, Z. Zhang, Y. Wang, Z. Peng, X. Wang, R. Wang, Y.-C. Huang, F. Meng, M. Li, C.-L. Dong, Q. Zhang, L. Gu and S. Shen, Anchoring Single Nickel Atoms on Carbon-vacant Carbon Nitride Nanosheets for Efficient Photocatalytic Hydrogen Evolution, Chem. Res. Chin. Univ., 2022, 38, 1243–1250 CrossRef CAS.
- D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, F. Xue, S. Shen and L. Guo, Boron-doped nitrogen-deficient carbon nitride-based Z-scheme heterostructures for photocatalytic overall water splitting, Nat. Energy, 2021, 6, 388–397 CrossRef CAS.
- Z. Lin, Y. Wang, Z. Peng, Y. C. Huang, F. Meng, J. L. Chen, C. L. Dong, Q. Zhang, R. Wang, D. Zhao, J. Chen, L. Gu and S. Shen, Single-Metal Atoms and Ultra-Small Clusters Manipulating Charge Carrier Migration in Polymeric Perylene Diimide for Efficient Photocatalytic Oxygen Production, Adv. Energy Mater., 2022, 12, 2200716 CrossRef CAS.
- A. Agosti, Y. Nakibli, L. Amirav and G. Bergamini, Photosynthetic H2 generation and organic transformations with CdSe@CdS-Pt nanorods for highly efficient solar-to-chemical energy conversion, Nano Energy, 2020, 70, 104510 CrossRef.
- M. Y. Qi, M. Conte, M. Anpo, Z. R. Tang and Y. J. Xu, Cooperative Coupling of Oxidative Organic Synthesis and Hydrogen Production over Semiconductor-Based Photocatalysts, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed.
- M. J. Lima, A. M. T. Silva, C. G. Silva, J. L. Faria and N. M. Reis, Selective photocatalytic synthesis of benzaldehyde in microcapillaries with immobilized carbon nitride, Chem. Eng. J., 2022, 430, 132643 CrossRef CAS.
- J. Li, M. Li, H. Sun, Z. Ao, S. Wang and S. Liu, Understanding of the Oxidation Behavior of Benzyl Alcohol by Peroxymonosulfate via Carbon Nanotubes Activation, ACS Catal., 2020, 10, 3516–3525 CrossRef CAS.
- M. Tayyab, Y. Liu, S. Min, R. Muhammad Irfan, Q. Zhu, L. Zhou, J. Lei and J. Zhang, Simultaneous hydrogen production with the selective oxidation of benzyl alcohol to benzaldehyde by a noble-metal-free photocatalyst VC/CdS nanowires, Chin. J. Catal., 2022, 43, 1165–1175 CrossRef CAS.
- B. Wu, L. Zhang, B. Jiang, Q. Li, C. Tian, Y. Xie, W. Li and H. Fu, Ultrathin Porous Carbon Nitride Bundles with an Adjustable Energy Band Structure toward Simultaneous Solar Photocatalytic Water Splitting and Selective Phenylcarbinol Oxidation, Angew. Chem., Int. Ed., 2021, 60, 4815–4822 CrossRef CAS PubMed.
- T. Huo, Q. Deng, F. Yu, G. Wang, Y. Xia, H. Li and W. Hou, Ion-Induced Synthesis of Crystalline Carbon Nitride Ultrathin Nanosheets from Mesoporous Melon for Efficient Photocatalytic Hydrogen Evolution with Synchronous Highly Selective Oxidation of Benzyl Alcohol, ACS Appl. Mater. Interfaces, 2022, 14, 13419–13430 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, Tri-s-triazine-Based Crystalline Graphitic Carbon Nitrides for Highly Efficient Hydrogen Evolution Photocatalysis, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- Y. Xu, X. He, H. Zhong, D. J. Singh, L. Zhang and R. Wang, Solid Salt Confinement Effect: An Effective Strategy to Fabricate High Crystalline Polymer Carbon Nitride for Enhanced Photocatalytic Hydrogen Evolution, Appl. Catal., B, 2019, 246, 349–355 CrossRef CAS.
- Y. Li, Y. Wang, C. L. Dong, Y. C. Huang, J. Chen, Z. Zhang, F. Meng, Q. Zhang, Y. Huangfu, D. Zhao, L. Gu and S. Shen, Single-atom nickel terminating sp2 and sp3 nitride in polymeric carbon nitride for visible-light photocatalytic overall water splitting, Chem. Sci., 2021, 12, 3633–3643 RSC.
- G. Zhang, Y. Xu, D. Yan, C. He, Y. Li, X. Ren, P. Zhang and H. Mi, Construction of K+ Ion Gradient in Crystalline Carbon Nitride to Accelerate Exciton Dissociation and Charge Separation for Visible Light H2 Production, ACS Catal., 2021, 11, 6995–7005 CrossRef CAS.
- Z. Yu, X. Yue, J. Fan and Q. Xiang, Crystalline Intramolecular Ternary Carbon Nitride Homojunction for Photocatalytic Hydrogen Evolution, ACS Catal., 2022, 12, 6345–6358 CrossRef CAS.
- X. Wang, J. Meng, X. Zhang, Y. Liu, M. Ren, Y. Yang and Y. Guo, Controllable Approach to Carbon-Deficient and Oxygen-Doped Graphitic Carbon Nitride: Robust Photocatalyst Against Recalcitrant Organic Pollutants and the Mechanism Insight, Adv. Funct. Mater., 2021, 31, 2010763 CrossRef CAS.
- X. Zhou, Y. Liu, Z. Jin, M. Huang, F. Zhou, J. Song, J. Qu, Y. J. Zeng, P. C. Qian and W. Y. Wong, Solar-Driven Hydrogen Generation Catalyzed by g-C3N4 with Poly(platinaynes) as Efficient Electron Donor at Low Platinum Content, Adv. Sci., 2021, 8, 2002465 CrossRef CAS PubMed.
- C. Qiu, Y. Xu, X. Fan, D. Xu, R. Tandiana, X. Ling, Y. Jiang, C. Liu, L. Yu, W. Chen and C. Su, Highly Crystalline K-Intercalated Polymeric Carbon Nitride for Visible-Light Photocatalytic Alkenes and Alkynes Deuterations, Adv. Sci., 2019, 6, 1801403 CrossRef PubMed.
- D. Liu, C. Li, C. Zhao, Q. Zhao, T. Niu, L. Pan, P. Xu, F. Zhang, W. Wu and T. Ni, Facile synthesis of three-dimensional hollow porous carbon doped polymeric carbon nitride with highly efficient photocatalytic performance, Chem. Eng. J., 2022, 438, 135623 CrossRef CAS.
- Y. Xu, M. Fan, W. Yang, Y. Xiao, L. Zeng, X. Wu, Q. Xu, C. Su and Q. He, Homogeneous Carbon/Potassium-Incorporation Strategy for Synthesizing Red Polymeric Carbon Nitride Capable of Near-Infrared Photocatalytic H2 Production, Adv. Mater., 2021, 33, 2101455 CrossRef CAS PubMed.
- Y. Jiang, S. Fang, C. Cao, E. Hong, L. Zeng, W. Yang, L. Huang and C. Yang, Enhanced light harvesting and charge separation of carbon and oxygen co-doped carbon nitride as excellent photocatalyst for hydrogen evolution reaction, J. Colloid Interface Sci., 2022, 612, 367–376 CrossRef CAS PubMed.
- C. Ehlert, W. E. Unger and P. Saalfrank, C K-edge NEXAFS spectra of graphene with physical and chemical defects: a study based on density functional theory, Phys. Chem. Chem. Phys., 2014, 16, 14083–14095 RSC.
- S. Chu, Y. Wang, Y. Guo, J. Feng, C. Wang, W. Luo, X. Fan and Z. Zou, Band Structure Engineering of Carbon Nitride: In Search of a Polymer Photocatalyst with High Photooxidation Property, ACS Catal., 2013, 3, 912–919 CrossRef CAS.
- Y. Wang, S. Gonell, U. R. Mathiyazhagan, Y. Liu, D. Wang, A. J. M. Miller and T. J. Meyer, Simultaneous Electrosynthesis of Syngas and an Aldehyde from CO2 and an Alcohol by Molecular Electrocatalysis, ACS Appl. Energy Mater., 2018, 2, 97–101 CrossRef.
- G. Zhang, Y. Xu, H. Mi, P. Zhang, H. Li and Y. Lu, Donor Bandgap Engineering without Sacrificing the Reduction Ability of Photogenerated Electrons in Crystalline Carbon Nitride, ChemSusChem, 2021, 14, 4516–4524 CrossRef CAS PubMed.
- G. Zhang, Y. Xu, G. Liu, Y. Li, C. He, X. Ren, P. Zhang and H. Mi, Pyrimidine donor induced built-in electric field between melon chains in crystalline carbon nitride to facilitate excitons dissociation, Chin. Chem. Lett., 2022, 34, 107383 CrossRef.
- D. Zhao, Y. Wang, C. L. Dong, F. Meng, Y. C. Huang, Q. Zhang, L. Gu, L. Liu and S. Shen, Electron-Deficient Zn-N6 Configuration Enabling Polymeric Carbon Nitride for Visible-Light Photocatalytic Overall Water Splitting, Nano-Micro Lett., 2022, 14, 223 CrossRef CAS PubMed.
- D. Zhao, C. L. Dong, B. Wang, C. Chen, Y. C. Huang, Z. Diao, S. Li, L. Guo and S. Shen, Synergy of Dopants and Defects in Graphitic Carbon Nitride with Exceptionally Modulated Band Structures for Efficient Photocatalytic Oxygen Evolution, Adv. Mater., 2019, 31, 1903545 CrossRef CAS PubMed.
- S. Gao, S. Wan, J. Yu and S. Cao, Donor–Acceptor Modification of Carbon Nitride for Enhanced Photocatalytic Hydrogen Evolution, Adv. Sustainable Syst., 2022, 7, 2200130 CrossRef.
- Z. Zhang, X. Chen, H. Zhang, W. Liu, W. Zhu and Y. Zhu, A Highly Crystalline Perylene Imide Polymer with the Robust Built-In Electric Field for Efficient Photocatalytic Water Oxidation, Adv. Mater., 2020, 32, 1907746 CrossRef CAS PubMed.
- J. Zhang, G. Zhang, X. Chen, S. Lin, L. Mohlmann, G. Dolega, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Co-monomer control of carbon nitride semiconductors to optimize hydrogen evolution with visible light, Angew. Chem., Int. Ed., 2012, 51, 3183–3187 CrossRef CAS PubMed.
- Q. Yang, T. Wang, Z. Zheng, B. Xing, C. Li and B. Li, Constructing interfacial active sites in Ru/g-C3N4−x photocatalyst for boosting H2 evolution coupled with selective benzyl-alcohol oxidation, Appl. Catal., B, 2022, 315, 121575 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00055a |
| This journal is © The Royal Society of Chemistry 2023 |