
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing C≥2 product selectivity in electrochemical CO2 reduction by controlling the microstructure of gas diffusion electrodes†
Francesco
Bernasconi
 ab,
Alessandro
Senocrate
*ac,
Peter
Kraus
a and
Corsin
Battaglia
abc
ab,
Alessandro
Senocrate
*ac,
Peter
Kraus
a and
Corsin
Battaglia
abc
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, 8600 Dübendorf, Switzerland. E-mail: alessandro.senocrate@empa.ch
bETH Zürich, Department of Materials, 8093 Zürich, Switzerland
cETH Zürich, Department of Information Technology and Electrical Engineering, 8093 Zürich, Switzerland
First published on 29th August 2023
Abstract
We fabricate polymer-based gas diffusion electrodes with controllable microstructure for the electrochemical reduction of CO2, by means of electrospinning and physical vapor deposition. We show that the microstructure of the electrospun substrate is affecting the selectivity of a Cu catalyst, steering it from H2 to C2H4 and other multicarbon products. Specifically, we demonstrate that gas diffusion electrodes with small pores (e.g. mean pore size 0.2 μm) and strong hydrophobicity (e.g. water entry pressure >1 bar) are necessary for achieving a remarkable faradaic efficiency of ∼50% for C2H4 and ∼75% for C≥2 products in neutral 1M KCl electrolyte at 200 mA cm−2. We observe a gradual shift from C2H4 to CH4 to H2 during long-term electrochemical reduction of CO2, which we ascribe to hygroscopic carbonate precipitation in the gas diffusion electrode resulting in flooding of the Cu catalyst by the electrolyte. We demonstrate that even with minimal electrolyte overpressure of 50 mbar, gas diffusion electrodes with large pores (mean pore size 1.1 μm) lose selectivity to carbon products completely, suddenly, and irreversibly in favor of H2. In contrast, we find that gas diffusion electrodes with small pore size (mean pore size 0.2 μm) and strong hydrophobicity (water entry pressure ∼5 bar) are capable of resisting up to 1 bar of electrolyte overpressure during CO2RR without loss of selectivity. We rationalize these experimental results in the context of a double phase boundary reactivity, where an electrolyte layer covers the Cu catalyst and thus governs local CO2 availability. Our results emphasize the pivotal role of microstructure and hydrophobicity in promoting high C≥2 product selectivity and long-term stability in CO2RR flow cells.
Broader contextThe electrochemical CO2 reduction reaction offers the unique possibility to power the conversion of CO2 with electricity from renewable energy technologies, which is a necessary condition to close the carbon loop. An important technological goal is to maximize the selectivity towards one single carbon product while avoiding the parasitic H2 evolution reaction, to avoid costly and energy-intensive separation methods. Amongst carbon products, C2H4 is one of the most sought after, as it is a suitable platform chemical for the synthesis of long-chain hydrocarbons. Presently, Cu is the only known electrocatalyst capable of converting CO2 to C2H4 at technological relevant current densities, albeit with limited selectivity. To improve the selectivity towards C2H4 selectivity and suppress the H2 revolution reaction we purposefully modify the morphology of hydrophobic polymer-based gas diffusion electrodes made in-house by electrospinning. We find that gas diffusion electrodes substrates with small pore sizes and strong hydrophobicity grant high C2H4 and C≥2 products selectivity (∼50% and ∼75%, respectively) in neutral electrolyte, as well as resilience to electrolyte overpressures (>1 bar) during CO2RR. These findings highlight the potential of substrate control as a scalable strategy to improve the selectivity of the electrochemical CO2 reduction reaction. |
Introduction
The conversion of CO2 into sustainable fuels and chemicals has been identified as a key technology to achieve carbon neutrality by closing the carbon loop.1,2 Converting CO2 electrochemically via the electrochemical CO2 reduction reaction (CO2RR) offers the unique possibility to power this conversion with electricity from renewable energy technologies.1 Significant scientific effort has been devoted to improving CO2RR activity and selectivity, as well as to understanding its mechanisms, starting from the studies of Hori, who pioneered the reduction of CO2 dissolved in aqueous electrolytes in the 1980s.3–5 More recent developments employ gas diffusion electrodes (GDEs) to circumvent CO2 mass transport limitations to the catalyst resulting from the low solubility of CO2 in water (35 mM at 25 °C),6 enabling CO2RR at technologically relevant current densities ≥ 200 mA cm−2.7,8An important technological goal to avoid costly product separation is to achieve high selectivity towards one single carbon product, while suppressing the parasitic hydrogen evolution reaction (HER). However, depending on the catalyst material,9 pH and composition of the electrolyte,5,10–17 temperature,18,19 pressure,20,21 and GDE type,22–25 the product selectivity of the CO2RR can vary significantly. For example Cu, the most studied catalyst for CO2RR, can produce both single carbon (CH4, CO and HCOOH, collectively labeled C1) and multicarbon products (e.g. C2H4, CH3CH2OH, etc., collectively labeled C≥2), yielding a complex product distribution. Amongst multicarbon products, C2H4 is particularly desirable as it can be used as a precursor to obtain long-chain hydrocarbons. An emerging strategy to enhance C2H4 selectivity and suppress HER consists in controlling the local environment of the catalyst,23,24,26–28 for example by creating an abrupt interface between a hydrophobic polymer substrate and a hydrophilic Cu catalyst yielding high C2H4 selectivity in strongly basic electrolytes.29
In this work, we purposely control the pore size of hydrophobic polymer substrates made in-house by electrospinning. We find that Cu GDEs based on substrates with small pore size and strong hydrophobicity achieve high C2H4 and C≥2 products selectivity in neutral electrolyte as well as resilience to electrolyte overpressures (>1 bar) during CO2RR. Through mass transport considerations and the comparison between CO2 and CO reduction experiments we provide evidence that the Cu active sites are covered by a layer of electrolyte, forcing the reaction to take place mainly at double phase boundaries. Our work highlights the necessity of controlling GDE substrate microstructure to minimize the parasitic HER and offers a scalable strategy towards high C≥2 products selectivity.
Methods
GDE substrates were prepared by electrospinning solutions of 15, 30, and 32 wt% poly-(vinyliden-fluoride-co-hexafluorpropylene) (PVDF-HFP) in a 1 wt% solution of tetraethylammonium bromide (TEABr) in dimethylformamide (DMF) at 30 kV. These substrates were used to prepare GDEs by single-sided deposition of a Cu catalyst layer with a nominal thickness of 500 nm employing physical vapor deposition (Cu-GDE). Pore size and water entry pressure measurements on both substrates and GDEs are carried out by capillary flow porometry. Details on the fabrication and structural characterization are provided in the ESI† and in Fig. S1–S4.Cu-GDE product selectivity and activity during CO2RR was assessed in a three-compartment electrochemical cell. Details of the cell assembly are shown in Fig. S5 (ESI†). Briefly, the GDE sits between a gas chamber where the CO2 reactant flows, and a liquid chamber, which contains the catholyte (1M KCl) and a Ag/AgCl reference electrode. The anolyte (1M KHCO3) is separated from the catholyte by a cation exchange membrane (Nafion 117). A Ti grid coated with IrOx serves as anode for the oxygen evolution reaction, oversizing the anode to avoid limiting the cathodic performance. Gaseous and liquid reaction products are quantified using gas and liquid chromatography, respectively (details are reported in the ESI†).
Results and discussion
Fig. 1a–c shows cross-sectional scanning electron microscopy (SEM) images of Cu-GDEs obtained by electrospinning solutions with increasing polymer concentration. The pore size distribution shown in Fig. 1d, from which we extract the mean pore sizes of the GDEs was determined by capillary flow porometry (see Fig. S3 and S4 for more details, ESI†). With increasing polymer concentration in the electrospinning solution, the mean pore size increases from 0.2 to 0.7 to 1.1 μm. To get information on the GDEs' wetting behavior, we also measure their water entry pressure, which is the pressure of water required to penetrate a dry, porous, hydrophobic substrate. The water entry pressure is related to the polymer's hydrophobicity, and thus to its chemistry, but also depends on the substrate pore size. As the GDE substrates are made of the same polymer, their different water entry pressures are mainly dictated by their different mean pore size, as described by the Young–Laplace equation. Fig. 1e shows the correlation between these morphological descriptors, with GDEs with smaller mean pore size showing higher water entry pressures. | ||
| Fig. 1 SEM images of Cu-GDEs with mean pore size of (a) 0.2 μm (15% PDVF-HFP), (b) 0.7 μm (30% PDVF-HFP), and (c) 1.1 μm (32% PDVF-HFP), and (d) corresponding pore size distribution, and (e) water entry pressure vs. mean pore size. Note the different scale bars in the SEM images. The obtained fibers (darker contrast) are coated with Cu (lighter contrast) close to the top surface of the GDE. | ||
Next, we analyze the product selectivity and activity of the GDEs during CO2RR. Fig. 2 displays the faradaic efficiencies (FEs) towards the main gaseous products (H2, C2H4, CH4 and CO) and liquid products (formic acid, acetic acid, EtOH, and n-PrOH), and the total FE as a function of applied current density for the Cu-GDEs with mean pore sizes 0.2 μm, 0.7 μm, and 1.1 μm. The total FE also accounts for minor gaseous products such as allyl alcohol, propane, propene, propionaldehyde, acetaldehyde, and acetone, each accounting for FE < 1%. Inspection of Fig. 2 reveals that when increasing the current density from 10 to 50 mA cm−2, all GDEs show a clear decrease of FE towards H2, CO, and formic acid, a strong increase towards C2H4, ethanol, and acetic acid, as well as a mild increase in FE towards CH4 irrespective of their mean pore size. In this current density range, the only difference between the GDEs is a lower FE towards CO and formic acid and a higher FE towards H2 for the GDE with 1.1 μm mean pore size. When increasing the current density further from 50 to 200 mA cm−2, the FE of the GDE with 1.1 μm mean pore size shifts distinctly towards H2. In comparison, for the GDEs with 0.2 and 0.7 μm mean pore size, the FE for C2H4 continues to increase up to 200 mA cm−2, where a remarkable value of ∼50% is achieved. For n-PrOH, an initial increase followed by a decrease at high current densities is observed. In addition, the FE towards CH4 decreases for the 0.2 μm and 0.7 μm GDEs with increasing current density, while it stays almost constant for the GDE with 1.1 μm mean pore size. The total FE shown in Fig. 2i falls between 90 and 110% considering all GDEs and all current densities applied. These relative variations, however, do not affect the observed trends with GDE pore size, as these are also present after normalization of the total FEs to 100% (Fig. S6, ESI†). The statistical error associated to the FE was assessed by evaluating the performance of nominally identical GDEs with 0.2 um pore size. This analysis is given in Fig. S10 (ESI†), from which we extract a relative standard deviation of ±5% for the FE of the main gaseous products, averaged over a 1 h measurement at 200 mA cm−2. We also provide the current vs. voltage slopes in Fig. S7 (ESI†).
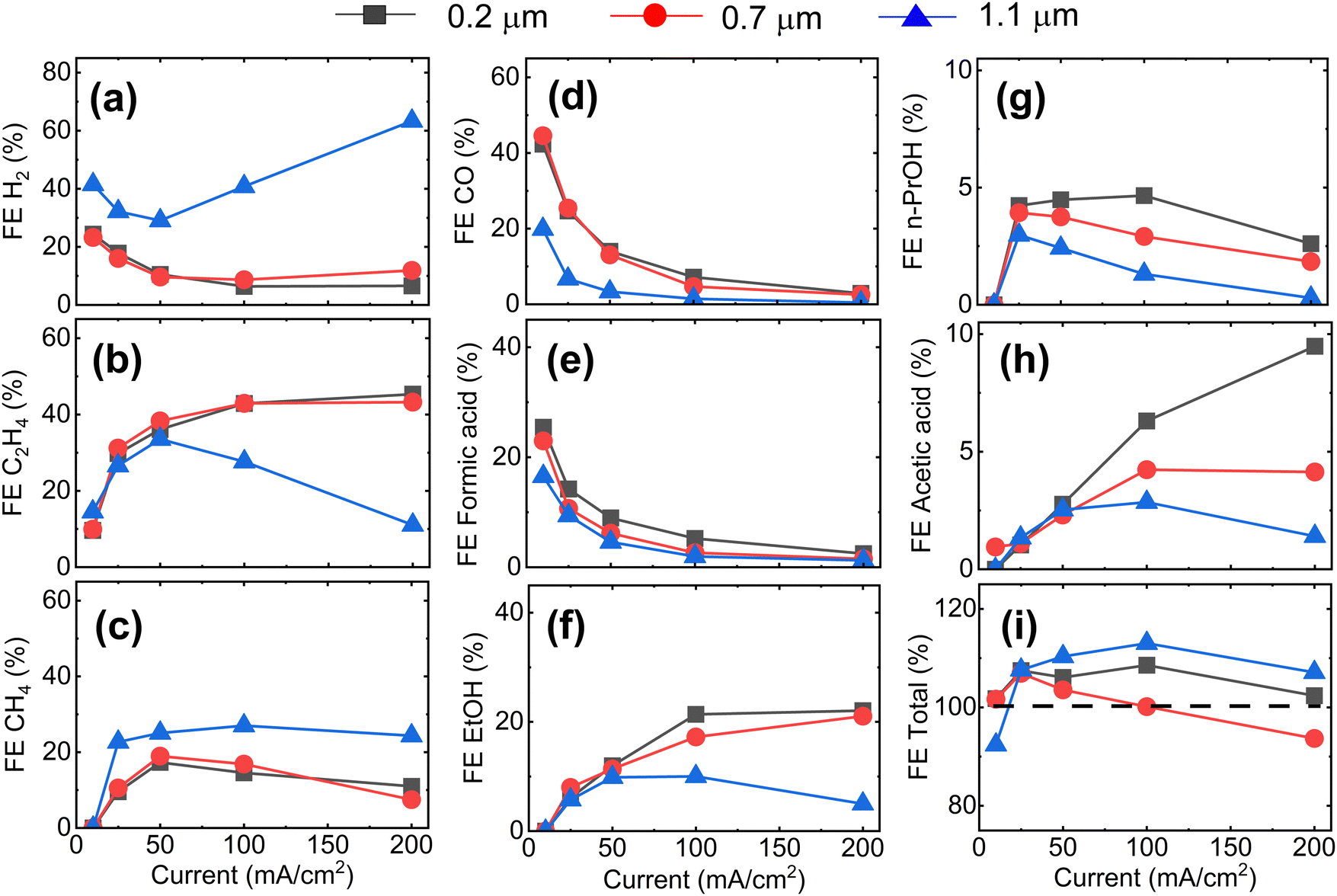 | ||
| Fig. 2 Faradaic efficiency of Cu-GDEs with different mean pore size as a function of current density towards (a) H2 (b) C2H4, (c) CH4, (d) CO, (e) formic acid (f) EtOH (g) n-PrOH (h) acetic acid and (i) total FE. Additional trace compounds (FE < 1%) including allyl alcohol, propane, propene, propionaldehyde, acetaldehyde, and acetone were detected in the gas products and are accounted for in the total FE. The FE towards gaseous products is assessed every five minutes and it is averaged over one hour of reaction, while the FE towards liquid products is determined at the end of the reaction. | ||
The plateau in FE towards C≥2 product observed in Fig. 2b is a first evidence for CO2RR taking place at double-phase boundaries between the solid Cu catalyst and liquid electrolyte with dissolved CO2,30 rather than at triple phase boundaries (between catalyst, electrolyte, and gaseous CO2). As direct gaseous access is unlikely to be restricted in our highly porous GDEs (∼80% porosity by liquid pycnometry), these results suggest that the active sites of the Cu catalyst are covered by an electrolyte layer, which indirectly limits the availability of CO2. The uniform behaviour of the GDEs below 50 mA cm−2 is consistent with this hypothesis.
To gather additional evidence for CO2RR taking place at double-phase boundaries, we extend the CO2RR experiment to higher current densities up to 600 mA cm−2. In parallel, we assess the GDE performance during the electrochemical reduction of CO (CORR). Fig. 3a–c displays the FE for the main gaseous products (H2, C2H4, and CH4) during CO2RR at current densities up to 600 mA cm−2. Results are consistent with Fig. 2, even showing a higher maximum FE of ∼55% for C2H4 at 200 mA cm−2 for the GDE with the smallest mean pore size of 0.2 μm. Maxima in FE are also observed for CH4 shifting from 400 mA cm−2 to 200 mA cm−2 with increasing GDE mean pore size followed by a rapid rise in FE for H2 at higher current densities. For CORR shown in Fig. 3d–f, we observe that the highest FE towards C2H4 is achieved at a ten-fold lower current density compared to CO2RR for the GDE with the mean pore size of 0.2 μm, which is attributed to the lower solubility of CO in water (1 mM at 25 °C).31 At higher current densities, the FE for C2H4 falls rapidly. A similar behavior is observed for the GDEs with 0.7 μm and 1.1 μm mean pore size with the maxima shifted towards lower current densities.
 | ||
| Fig. 3 Faradaic efficiency for H2, C2H4, and CH4 as function of applied current for GDEs with different mean pore size during CO2RR (a)–(c) and CORR (d)–(f). To avoid resistive heating at current densities >200 mA cm−2, we reduce the exposed area of the catalyst from 1 cm2 to 0.2 cm2 for CO2RR by masking the GDEs with a polytetrafluoroethylene tape. The voltage of the reactions is reported in Fig. S8 (ESI†). Note the difference in the scale of the x-axis for CO2RR and for CORR plots. | ||
These findings show that a maximum FE towards C2H4 is obtained at an optimum value of the current density which depends on the GDE's mean pore size. The selectivity towards C1 products is promoted when the availability of protons is substantial (indicated by high parasitic HER), while the selectivity towards C≥2 products is maximized when proton availability is limited (low HER). In addition, the CORR results provide a strong evidence for the double phase boundary hypothesis, as the much more severe mass transport limitations observed there are consistent with the 35-fold lower solubility of CO in water compared with CO2. As the thickness of the electrolyte layer covering the catalyst is the key parameter governing CO/CO2 availability, the results reveal a correlation between GDE pore size and electrolyte layer thickness, with smaller pores yielding thinner electrolyte layers.
Having shown that the GDE microstructure dictates CO2RR product selectivity, we now study the impact of GDE microstructure on CO2RR stability. In Fig. 4, we present the evolution of the CO2RR FEs during 300 min operation of the Cu-GDEs at 200 mA cm−2 (see Fig. S9 for other products, ESI†). During the first 180 min, the FE toward H2 remains below 5% for the GDEs with mean pore size of 0.2 μm and 0.7 μm, then progressively increases to 40% after 300 min. For the GDE with the largest mean pore size of 1.1 μm, the FE for H2 starts at 60% and increases to 100% after 300 min. The FE for C2H4 follows an opposite trend and starts at 50% for the GDEs with 0.2 μm and 0.7 μm mean pore size, but then gradually decays over time. A more complicated evolution is observed for the FE for CH4: for the GDEs with 0.2 μm and 0.7 μm mean pore size, it slowly increases and reaches a maximum after several hours, while for the GDE with 1.1 μm mean pore size, the FE for CH4 reaches a maximum very early and then decreases continuously.
 | ||
| Fig. 4 Faradaic efficiency for (a) H2, (b) C2H4, and (c) CH4 at 200 mA cm−2 as a function of time for GDEs with mean pore size 0.2 μm, 0.7 μm, and 1.1 μm. | ||
Overall, we observe a gradual shift from C2H4 to CH4 to H2 during long-term CO2RR, which we ascribe to precipitation of carbonates in the GDE resulting in flooding of the Cu catalyst by the electrolyte, as described previously.32–34 According to the reported mechanism, high local pH values in the electrolyte near the Cu catalyst, resulting from localized proton depletion, lead to the precipitation of carbonates or bicarbonates, which are highly hygroscopic. Over time, the accumulation of these salts in the GDE causes a loss of hydrophobicity, and promotes electrolyte penetration into the GDE. As a result, catalyst flooding worsens CO2 mass transport and favors parasitic HER by increasing the diffusion pathway length of CO2. As we are employing strongly hydrophobic GDE substrates, we have never observed complete, macroscopic flooding of the GDE. However, it is still likely that local precipitation of salts close to the Cu catalyst takes place, reducing local hydrophobicity and causing an increase in the thickness of the electrolyte layer covering the catalyst. One strategy to mitigate or reduce the decay of performance over time is to periodically refresh the electrolyte and/or to periodically interrupt the reaction.35 As we focus here on the effect of the morphology of the GDE on their performance, we refrain from using these strategies in this study.
An additional parameter for controlling local wetting of the GDE is provided by the pressure difference between the liquid electrolyte and the reactant gas inside the electrochemical cell. A negative pressure difference (ΔP < 0), i.e. a gas overpressure, can hinder electrolyte penetration into the GDE and consequently prevent catalyst flooding, while a positive pressure difference (ΔP > 0), i.e. an electrolyte overpressure, can promote it. Fig. 5 displays the results of a CO2RR experiment in which we purposefully vary the pressure difference by using needle valves, monitoring the change with pressure sensors. As can be seen in Fig. 5a and b, switching the pressure difference from negative to slightly positive (only 50 mbar) immediately causes the C2H4 selectivity to drop to zero while H2 selectivity dominates the product distribution for the GDEs with 0.7 μm and 1.1 μm mean pore size. Only the GDE with 0.2 μm mean pore size is capable of maintaining high C2H4 selectivity despite the pressure change, highlighting again the importance of small mean pore sizes. Note that 50 mbar liquid overpressure is insufficient to push the liquid electrolyte through the GDE, as even the water entry pressure for the GDE with the largest pore size is 0.6 bar. Further, this loss of selectivity is irreversible even if the pressure difference is set back to negative values. Notably, the GDE with smallest mean pore size of 0.2 μm is unaffected by the change in pressure difference even up to ΔP > 1000 mbar, as shown in Fig. 5c. From an engineering point of view, GDEs with small pores thus offer the attractive advantage of withstanding pressure spikes both in gas and in liquid overpressure, which could take place, for example during CO2RR startup.
 | ||
| Fig. 5 Faradaic efficiency for (a) H2 and (b) C2H4 at 200 mA cm−2 as a function of time and pressure for GDEs with mean pore size 0.2 μm, 0.7 μm, and 1.1 μm. In the first 60 minutes, the pressure difference between liquid and gas chamber is negative (ΔP < 0), then switched to positive (ΔP = 50 mbar, liquid overpressure) for 60 minutes, finally the pressure is reverted back to negative values (ΔP < 0). (c) Faradaic efficiency towards C2H4 at 200 mA cm−2 as a function of time and with stepwise increase of pressure from ΔP ∼ −200 mbar to ΔP ∼ 1000 mbar for the GDE with mean pore size 0.2 μm. | ||
Our findings provide multiple evidence that, for all mean pore sizes considered, the active sites of the Cu GDE are covered by an electrolyte layer, and that the thickness of this layer is governed by GDE pore size, with smaller pores yielding thinner electrolyte layers. This means that, in these polymer-based GDEs, double-phase boundaries dominate the CO2RR or CORR reactivity over triple phase boundaries. We also note that, while thinner electrolyte layers cause enhanced CO2 transport, they also yield a more limited transport of H+/OH− to/from the catalyst active sites. The H+/OH− transport limitation will make the pH shift more strongly to higher values during CO2RR for small pore size GDEs, potentially also influencing the product selectivity.
Considering 200 mA cm−2 as a technologically relevant threshold value for the current density, our results show that the upper limit for the GDE's mean pore size is certainly below 1.1 μm, likely below 0.7 μm. In addition, we can expect a lower limit dictated by the pore size at which the mass transport of CO2 to the catalyst transitions from pressure driven to Knudsen diffusion, i.e. when the mean free path of the gas molecule is comparable with the diameter of the pore, which is ∼0.05 μm for CO2 gas at room pressure.36 These mass transport considerations, in agreement with our experimental results, indicate an optimal pore size for GDE substrates between ∼0.05 and ∼0.7 μm to maximize C2H4 selectivity on Cu catalysts.
Nonetheless, we note that C2H4 is always synthetized alongside a variety of other C≥2 products, highlighting the need for strategies able to steer the selectivity toward a single C≥2 product. Several different approaches in this direction have been recently assessed, from the development of engineered pathways for CO2 and H+/OH− transport,37 to the insertion of hydrophobic zones near the active sites,23 to the use of complex electrolyte solutions.30,38 As these approaches have yielded important – but not sufficient – selectivity improvements, we expect that a higher degree of control of the local catalyst environment, for example achieved by the targeted introduction of hydrophobic – hydrophilic zones within the GDE, may be necessary to obtain a single C≥2 product during CO2RR.
Conclusions
In conclusion, we employed electrospinning and physical vapor deposition to prepare Cu GDEs with tunable substrate pore sizes and water entry pressures. We demonstrated that GDE mean pore size dictates product selectivity and were able to achieve a remarkable FE of ∼50% towards C2H4 and ∼75% towards C≥2 products with the GDE with smallest mean pore size of 0.2 μm. Based on mass transport considerations, we estimate the optimal pore size of the substrate to be between ∼0.05 and ∼0.7 μm to maximize C≥2 product selectivity. Finally, we assessed the capability of GDEs to sustain liquid electrolyte overpressures and found that GDEs with the smallest mean pore size are able to withstand up to 1 bar overpressure, while GDEs with larger mean pore size (above 0.7 μm) suddenly and irreversibly lose their selectivity towards carbon products in favor of HER with just 50 mbar of electrolyte overpressure. The dependence of the highest FE towards C2H4 on the pore size for both CO2RR and CORR, the progressive increase of FE towards H2 over time and the increased sensitivity of GDEs with large pore size to liquid overpressure provide evidence for the presence of an electrolyte layer covering the active site of the Cu catalyst, and thus of the dominance of double phase boundary reactivity. To complement the picture emerging from electrochemical measurements with direct experimental in-formation on the local catalyst environment, future studies will focus on in situ methods.Data availability statement
Data used in this work can be found on the ZENODO repository using the following link: https://zenodo.org/record/8211440 (DOI: 10.5281/zenodo.8211440).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation (grant number 180544). This work also received funding from the ETH Board in the framework of the Joint Strategic Initiative “Synthetic Fuels from Renewable Resources”. The authors acknowledge the support of the Scientific Center for Optical and Electron Microscopy (ScopeM) of the ETH Zurich and of Dr. Peng Zeng of the ScopeM for the FIB-SEM results. Additionally, the authors are grateful to Dr. Fabian Itel from Empa's Laboratory for Biometic Membranes and Textiles for fruitful discussions on the electrospinning process.References
- I. E. A. Exploring, Clean Energy Pathways, Explor. clean energy pathways, 2019 DOI:10.1787/c76b829e-en.
- D. W. Keith, Why Capture CO2 from the Atmosphere?, Science, 2009, 325(5948), 1654–1655, DOI:10.1126/science.1175680.
- Y. Hori, K. Kikuchi and S. Suzuki, Production of CO and CH4 in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Hydrocarbonate Solution, Chem. Lett., 1985, 1695–1698, DOI:10.1246/cl.1985.1695.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Production of Methane and Ethylene in Electrochemical Reduction of Carbon Dioxide at Copper Electrodes in Aqueous Hydrogencarbonate Solution, Chem. Lett., 1986, 897–898, DOI:10.1246/cl.1986.897.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, Electrochemical Reduction of CO at a Copper Electrode, J. Phys. Chem. B, 1997, 101(36), 7075–7081, DOI:10.1021/jp970284i.
- P. Scharlin, Carbon Dioxide in Water and Aqueous Electrolyte Solutions. IUPAC, Solubility Data Ser., 1996, 62(7), 21 Search PubMed.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, High Rate Gas Phase CO2 Reduction to Ethylene and Methane Using Gas Diffusion Electrodes, J. Electrochem. Soc., 1990, 137(2), 607–608, DOI:10.1149/1.2086515.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, CO2 Electrolysis to Multicarbon Products in Strong Acid, Science, 2021, 372(6546), 1074–1078, DOI:10.1126/science.abg6582.
- A. Senocrate and C. Battaglia, Electrochemical CO2 Reduction at Room Temperature: Status and Perspectives, J. Energy Storage, 2021, 36, 102373, DOI:10.1016/j.est.2021.102373.
- N. Gupta, M. Gattrell and B. MacDougall, Calculation for the Cathode Surface Concentrations in the Electrochemical Reduction of CO2 in KHCO3 Solutions, J. Appl. Electrochem., 2006, 36(2), 161–172, DOI:10.1007/s10800-005-9058-y.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, Theoretical Insights into a CO Dimerization Mechanism in CO2 Electroreduction, J. Phys. Chem. Lett., 2015, 6(11), 2032–2037, DOI:10.1021/acs.jpclett.5b00722.
- A. Seifitokaldani, C. M. Gabardo, T. Burdyny, C. T. Dinh, J. P. Edwards, M. G. Kibria, O. S. Bushuyev, S. O. Kelley, D. Sinton and E. H. Sargent, Hydronium-Induced Switching between CO2 Electroreduction Pathways, J. Am. Chem. Soc., 2018, 140(11), 3833–3837, DOI:10.1021/jacs.7b13542.
- C. T. Dinh, F. P. García De Arquer, D. Sinton and E. H. Sargent, High Rate, Selective, and Stable Electroreduction of CO2 to CO in Basic and Neutral Media, ACS Energy Lett., 2018, 3(11), 2835–2840, DOI:10.1021/acsenergylett.8b01734.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and Cu, J. Am. Chem. Soc., 2016, 138(39), 13006–13012, DOI:10.1021/jacs.6b07612.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119(12), 7610–7672, DOI:10.1021/acs.chemrev.8b00705.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, The Effect of Electrolyte Composition on the Electroreduction of CO2 to CO on Ag Based Gas Diffusion Electrodes, Phys. Chem. Chem. Phys., 2016, 18(10), 7075–7084, 10.1039/c5cp05665a.
- Q. Zhang, X. Shao, J. Yi, Y. Liu and J. Zhang, An Experimental Study of Electroreduction of CO2 to HCOOH on SnO2/C in Presence of Alkali Metal Cations (Li+, Na+, K+, Rb+ and Cs+) and Anions (HCO3−, Cl−, Br− and I−), Chinese J. Chem. Eng., 2020, 28(10), 2549–2554, DOI:10.1016/j.cjche.2020.04.015.
- S. T. Ahn, I. Abu-Baker and G. T. R. Palmore, Electroreduction of CO2 on Polycrystalline Copper: Effect of Temperature on Product Selectivity, Catal. Today, 2017, 288, 24–29, DOI:10.1016/j.cattod.2016.09.028.
- B. Yang, K. Liu, H. J. W. Li, C. Liu, J. Fu, H. Li, J. E. Huang, P. Ou, T. Alkayyali, C. Cai, Y. Duan, H. Liu, P. An, N. Zhang, W. Li, X. Qiu, C. Jia, J. Hu, L. Chai, Z. Lin, Y. Gao, M. Miyauchi, E. Cortés, S. A. Maier and M. Liu, Accelerating CO2 Electroreduction to Multicarbon Products via Synergistic Electric-Thermal Field on Copper Nanoneedles, J. Am. Chem. Soc., 2022, 3039–3049, DOI:10.1021/jacs.1c11253.
- J. Li, Y. Kuang, Y. Meng, X. Tian, W.-H. Hung, X. Zhang, A. Li, M. Xu, W. Zhou, C.-S. Ku, C.-Y. Chiang, G. Zhu, J. Guo, X. Sun and H. Dai, Electroreduction of CO2 to Formate on a Copper-Based Electrocatalyst at High Pressures with High Energy Conversion Efficiency, J. Am. Chem. Soc., 2020, 142(16), 7276–7282, DOI:10.1021/jacs.0c00122.
- B. Sahin, J. J. Leung, E. Magori, S. Laumen, A. Tawil, E. Simon and O. Hinrichsen, Controlling Product Distribution of CO2 Reduction on CuO-Based Gas Diffusion Electrodes by Manipulating Back Pressure, Energy Technol., 2022, 10(12), 2200972, DOI:10.1002/ente.202200972.
- J. Wicks, M. L. Jue, V. A. Beck, J. S. Oakdale, N. A. Dudukovic, A. L. Clemens, S. Liang, M. E. Ellis, G. Lee, S. E. Baker, E. B. Duoss and E. H. Sargent, 3D-Printable Fluoropolymer Gas Diffusion Layers for CO2 Electroreduction, Adv. Mater., 2021, 33(7), 1–8, DOI:10.1002/adma.202003855.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Enhancing Carbon Dioxide Gas-Diffusion Electrolysis by Creating a Hydrophobic Catalyst Microenvironment, Nat. Commun., 2021, 12(1), 1–11, DOI:10.1038/s41467-020-20397-5.
- A. Senocrate, F. Bernasconi, D. Rentsch, K. Kraft, M. Trottmann, A. Wichser, D. Bleiner and C. Battaglia, Importance of Substrate Pore Size and Wetting Behavior in Gas Diffusion Electrodes for CO2 Reduction, ACS Appl. Energy Mater., 2022, 5(11), 14504–14512, DOI:10.1021/acsaem.2c03054.
- W. Ju, F. Jiang, H. Ma, Z. Pan, Y. B. Zhao, F. Pagani, D. Rentsch, J. Wang and C. Battaglia, Electrocatalytic Reduction of Gaseous CO2 to CO on Sn/Cu-Nanofiber-Based Gas Diffusion Electrodes, Adv. Energy Mater., 2019, 9(32), 1901514, DOI:10.1002/aenm.201901514.
- A. Inoue, T. Harada, S. Nakanishi and K. Kamiya, Ultra-High-Rate CO2 Reduction Reactions to Multicarbon Products with a Current Density of 1.7 A cm−2 in Neutral Electrolytes, EES Catal., 2023, 1(1), 9–16, 10.1039/d2ey00035k.
- Z. Wang, Y. Li, X. Zhao, S. Chen, Q. Nian, X. Luo, J. Fan, D. Ruan, B. Q. Xiong and X. Ren, Localized Alkaline Environment via In Situ Electrostatic Confinement for Enhanced CO2-to-Ethylene Conversion in Neutral Medium, J. Am. Chem. Soc., 2022, 145(11), 6339–6348, DOI:10.1021/jacs.2c13384.
- H. Y. Erbil, Precursor Film Formation on Catalyst-Electrolyte-Gas Boundaries during CO2 Electroreduction with Gas Diffusion Electrodes, Catal. Sci. Technol., 2022, 106(26), 6933–6944, 10.1039/d2cy01576e.
- C. T. Dinh, T. Burdyny, G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, CO2 Electroreduction to Ethylene via Hydroxide-Mediated Copper Catalysis at an Abrupt Interface, Science, 2018, 360(6390), 783–787, DOI:10.1126/science.aas9100.
- N. T. Nesbitt, T. Burdyny, H. Simonson, D. Salvatore, D. Bohra, R. Kas and W. A. Smith, Liquid − Solid Boundaries Dominate Activity of CO2 Reduction on Gas Diffusion Electrodes, ACS Catal., 2020, 10(23), 14093–14106, DOI:10.1021/acscatal.0c03319.
- W. Robert Cargill. Solubility Data Series Volume 43 Carbon Monoxide. IUPAC Solubility Data Series. 1990, 43, 4.
- Y. Kong, H. Hu, M. Liu, Y. Hou, V. Kolivoška, S. Vesztergom and P. Broekmann, Visualisation and Quantification of Flooding Phenomena in Gas Diffusion Electrodes Used for Electrochemical CO2 Reduction: A Combined EDX/ICP–MS Approach, J. Catal., 2022, 408, 1–8, DOI:10.1016/j.jcat.2022.02.014.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, Investigating Electrode Flooding in a Flowing Electrolyte, Gas-Fed Carbon Dioxide Electrolyzer, ChemSusChem, 2020, 13(2), 400–411, DOI:10.1002/cssc.201902547.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, A New Mechanism for the Selectivity to C1 and C2 Species in the Electrochemical Reduction of Carbon Dioxide on Copper Electrodes, Chem. Sci., 2011, 2(10), 1902, 10.1039/c1sc00277e.
- R. Casebolt, K. Levine, J. Suntivich and T. Hanrath, Pulse Check: Potential Opportunities in Pulsed Electrochemical CO2 Reduction, Joule, 2021, 5(8), 1987–2026, DOI:10.1016/j.joule.2021.05.014.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Hoboken, 95th edn, 2014 Search PubMed.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Modulating Local CO2 Concentration as a General Strategy for Enhancing C−C Coupling in CO2 Electroreduction, Joule, 2020, 4(5), 1104–1120, DOI:10.1016/j.joule.2020.03.013.
- X. H. Yang, M. Papasizza, A. Cuesta and J. Cheng, Water-In-Salt Environment Reduces the Overpotential for Reduction of CO2 to CO2− in Ionic Liquid/Water Mixtures, ACS Catal., 2022, 6770–6780, DOI:10.1021/acscatal.2c00395.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00140g |
| This journal is © The Royal Society of Chemistry 2023 |