
Enhanced electroreduction of CO2 to ethanol via enriched intermediates at high CO2 pressures†
Rongxing
Qiu
a,
Jun
Jia
 a,
Li
Peng
*a,
Ruiqing
Li
a,
Sen
Yan
a,
Jiaran
Li
a,
Jie
Zhang
b,
Daniel T.
Sun
cd,
Zhipeng
Lan
a,
Tianwei
Xue
a,
Guangkuo
Xu
a,
Linxiao
Cui
a,
Zeyu
Lv
a,
Cheng
Li
a,
Yanzhen
Hong
a,
Yuzheng
Guo
e,
Bin
Ren
a,
Shuliang
Yang
*f,
Jun
Li
*a and
Buxing
Han
g
a,
Li
Peng
*a,
Ruiqing
Li
a,
Sen
Yan
a,
Jiaran
Li
a,
Jie
Zhang
b,
Daniel T.
Sun
cd,
Zhipeng
Lan
a,
Tianwei
Xue
a,
Guangkuo
Xu
a,
Linxiao
Cui
a,
Zeyu
Lv
a,
Cheng
Li
a,
Yanzhen
Hong
a,
Yuzheng
Guo
e,
Bin
Ren
a,
Shuliang
Yang
*f,
Jun
Li
*a and
Buxing
Han
g
aCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, Fujian, PR China. E-mail: li.peng@xmu.edu.cn; junnyxm@xmu.edu.cn
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore
cSunchem LLC, 395 South Van Ness Ave., San Francisco, CA 94103, USA
dThe Molecular Foundry, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, California 94720, USA
eThe Institute of Technological Sciences, Wuhan University, Wuhan 430072, Hubei, PR China
fCollege of Energy, Xiamen University, Xiamen 361102, Fujian, PR China. E-mail: ysl@xmu.edu.cn
gBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, PR China
First published on 14th December 2022
Abstract
Electrochemical conversion of CO2 into liquid fuels such as ethanol, powered by renewable electricity, is an efficient strategy for CO2 utilization to produce high value-added products. In this work, we discovered that the primary C2+ product could be switched from gaseous ethylene to liquid ethanol by directly increasing the CO2 pressure when Cu2O@Cu with a hollow sphere morphology was adopted as the catalyst. The faradaic efficiency (FE) of ethanol reached as high as 36.6% at a low overpotential of −1.0 V vs. Ag/AgCl (−0.48 V vs. RHE) at 100 bar, which was 4.6 times higher than that of 1 bar. Moreover, faster kinetics and lower overpotential for ethanol formation were obtained at high CO2 pressures. In situ Raman spectroscopy studies at different pressures in combination with density functional theory calculations demonstrated that the *CO surface coverage was increased significantly at increased CO2 pressure, which is responsible for facilitating ethanol formation during the electrochemical CO2RR. This study provides a novel and promising strategy for the selective production of ethanol on Cu-based catalysts by facilely adjusting the CO2 pressure.
Introduction
Carbon capture and conversion has received considerable attention as a potential means of reducing anthropogenic CO2 emissions.1 The electrochemical CO2 reduction reaction (CO2RR) into high value-added chemicals and fuels has great potential for closing the carbon cycle and alleviating the energy shortage crisis.2,3 Tremendous effort has been devoted to developing a series of novel heterogeneous catalysts for the electrochemical conversion of CO2 into C1 products (i.e., CO4–7 and HCOO−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8,9) using renewable energy. However, further reduction of these chemicals into more valuable chemicals and fuels (C2+ compounds, i.e., ethylene,10–13 ethanol14–18 and n-propanol19,20) is highly desired. Ethylene and ethanol, as vital C2+ chemicals with high energy density, share many intermediates in the 12-electron transfer process. Obtaining ethanol from electrochemical CO2 reduction is an extremely attractive approach owing to its high energy density and economic value. However, there are still numerous challenges that exist in selectively producing ethanol due to the C–C coupling limitation and the presence of the competitive reaction for ethylene.21,22
8,9) using renewable energy. However, further reduction of these chemicals into more valuable chemicals and fuels (C2+ compounds, i.e., ethylene,10–13 ethanol14–18 and n-propanol19,20) is highly desired. Ethylene and ethanol, as vital C2+ chemicals with high energy density, share many intermediates in the 12-electron transfer process. Obtaining ethanol from electrochemical CO2 reduction is an extremely attractive approach owing to its high energy density and economic value. However, there are still numerous challenges that exist in selectively producing ethanol due to the C–C coupling limitation and the presence of the competitive reaction for ethylene.21,22
At present, Cu-based materials have been regarded as the most effective catalysts to generate C2+ products by the electroreduction of CO2.23,24 Many tactics for designing catalysts have been applied to CO2 (CO) electroreduction to generate C2+ products, including oxide-derived copper,25–27 selective facet exposure,28–30 morphological control,31,32 heteroatom doping/bimetallic strategies,33–35 vacancy effect,36 molecular modifications,15,37,38 introducing compressive strain,39 and free-copper based catalysts.40,41 Other experimental parameters including local pH environment,42 temperature43 and pressure44,45 have also been considered, which would greatly affect the selectivity of products in the CO2RR.
It has been recognized that the low solubility of CO2 in the aqueous electrolyte at ambient pressure often forms limited active carbon species, which results in lower current density and inferior selectivity.46,47 Thus, adjusting the type of electrolyte is a prevalent approach to improve CO2 solubility such as using ionic liquid-based electrolytes.48–50 Furthermore, some research groups have reported that increasing the CO2 concentration by increasing CO2 pressure can effectively accelerate mass transfer and thus enhance the current density, along with suppressing the hydrogen evolution reaction (HER) to improve CO2RR selectivity.51,52 For example, Fontecave et al. have reported that an Ag alloyed Zn electrode can drastically increase the partial current density of CO production from −21 mA cm−2 at 1 bar to −286 mA cm−2 at 9.5 bar. Meanwhile, FECO maintained an average of 90% over 40 h and an average of 85% over 100 h at 9.5 bar.53 Dai et al. have reported that square-wave-Cu2O/Cu can reduce CO2 electrochemically to nearly pure formate at 45 bar, while the HER was the dominant reaction at ambient pressure.46 Typically, C1 chemicals are the primary products in the electrochemical CO2RR at high pressures in previous studies. However, there are very rare studies realizing CO2–ethanol conversion using electrical energy at high pressures especially above the supercritical pressure as far as we know, which highly deserves further exploration.
Herein, a Cu2O@Cu hollow sphere catalyst (denoted as HS-Cu) was prepared successfully and its performance for CO2 electrocatalysis at different CO2 pressures was studied comprehensively. We surprisingly discovered that the product distribution could be regulated remarkably with an increase in the CO2 pressure, accompanied by increasing the current density. In particular, at ambient pressure, a broad distribution of products (H2, CO, CH4, HCOOH, C2H4, CH3CH2OH and n-propanol) was observed, and ethylene was the main C2+ product. Interestingly, ethylene production was suppressed and the production of ethanol was improved greatly by increasing the CO2 pressure. The FE of ethanol could reach as high as 36.6% at a low overpotential of −1.0 V vs. Ag/AgCl (−0.48 V vs. RHE) at 100 bar, which was 4.6 times higher than that of 1 bar. Moreover, in situ Raman spectroscopy measurements at different CO2 pressures coupled with density-functional theory (DFT) calculation studies demonstrate that denser *CO surface coverage at a higher CO2 pressure is responsible for the promotion of ethanol formation during the CO2 electrocatalysis process. To the best of our knowledge, this is the first report using a Cu catalyst to mainly produce liquid ethanol from the CO2RR only with the assistance of a high CO2 pressure. The primary CO2RR products could be easily switched from gaseous ethylene at ambient pressure into liquid ethanol at a high CO2 pressure.
Experimental
Materials and chemicals
Copper(II) chloride dihydrate (CuCl2·2H2O, AR), sodium hydroxide (NaOH, AR), D-glucose (AR), potassium bicarbonate (AR), dimethyl sulfoxide (DMSO, AR) and isopropanol (AR) were produced by Sinopharm Chemical Reagent Co., Ltd. Deuterium oxide (D2O, 99.9 atom% D) was provided by Energy Chemical. Nafion solution (5 wt% in water and isopropanol) was obtained from Meryer (Shanghai) Chemical Technology Co., Ltd. Conductive carbon paper (hydrophobic) was purchased from Suzhou Sinero Technology Co., Ltd. CO2 (99.999%) was provided by Fuzhou Xinhang Industrial Gases Co., Ltd. Unless otherwise noted, deionized water (18 MΩ cm−2) was used throughout this work.Catalyst preparation
The Cu2O@Cu hollow sphere catalyst was prepared according to the reported literature with some modifications.54 CuCl2·2H2O (0.3410 g) was added to a mixed solution of 20 mL of ethylene glycol and 10 mL of deionized water, and the solution was stirred for 10 min and transferred into a 100 mL two-necked flask immersed in a 60 °C water bath. Whereafter, 10 mL of NaOH (2 g) aqueous solution was added drop by drop. After 5 min, 10 mL of D-glucose (2 g) solution was added rapidly and kept at 60 °C for 30 min. The resulting product was centrifuged, and rinsed with distilled water several times. Finally, the Cu2O@Cu hollow sphere catalyst was obtained by drying it in a vacuum oven at 70 °C for 12 h.Characterization of materials
Powder XRD was recorded using a Rigaku SmartLab-SE powder X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å) at room temperature. SEM images were acquired using a field emission scanning electron microscope from ZEISS Sigma. The morphology of the products was measured using an FEI Tecnai F30 transmission electron microscope at an acceleration voltage of 300 kV. XPS measurements were implemented with a Thermo Escalab 250Xi spectrometer using a photon energy of 461 eV at an energy resolution of 0.1 eV. 1H NMR spectra were operated using a Bruker AVANCE III 500 MHz nuclear magnetic resonance spectrometer. In situ Raman measurements were conducted with a Renishaw invia instrument using a 633 nm excitation laser and signals were recorded using 20 s integration in a home-made high pressure in situ Raman cell setup with a three-electrode, the laser power was 0.4 mW, and the objective (Nikon, ×10, NA 0.25) was used for exciting and collecting the Raman signals.Preparation of the working electrode
The as-prepared Cu2O@Cu hollow sphere catalyst (10 mg) was dispersed in 1980 μL of isopropanol and 20 μL of Nafion solution (5 wt% in water and isopropanol), followed by ultrasonication for 1 h at room temperature. Then 200 μL of catalyst ink was drop-coated onto hydrophobic carbon paper (geometric area: 1 × 1 cm2), the catalyst loading was about 1 mg cm−2, and the electrode was dried in a vacuum oven at 70 °C for 12 h. HS-Cu was then obtained by in situ electroreduction of the Cu2O@Cu hollow sphere catalyst.Results and discussion
The HS-Cu catalyst was first synthesized through a simple wet-chemical method. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of the Cu2O@Cu catalyst exhibited a hollow sphere morphology (Fig. 1a and b). High-resolution TEM (HR-TEM) images (Fig. 1c) revealed that the two different sets of lattice fringes in Cu2O@Cu correspond to the (111) facet of Cu2O and the (111) facet of Cu, respectively. The X-ray diffraction (XRD) pattern (Fig. 1d) showed three peaks located at 43.4°, 50.5° and 74.1°, corresponding to the diffraction from the Cu(111), (200) and (220) facets, respectively. Furthermore, the peak at 36.6° could be assigned to the diffraction from the Cu2O(111) facet, which was consistent with the HR-TEM characterization results. Two peaks at 953.8 eV and 933.9 eV were observed in the Cu 2p X-ray photoelectron spectroscopy (XPS) spectra (Fig. 1e), corresponding to Cu2+, which could be caused by the rapid and unavoidable surface oxidation of Cu+/Cu0 when exposed to the air.55,56 Auger electron spectroscopy (AES) of Cu LMM showed two peaks located at 574.1 eV and 569.2 eV, corresponding to Cu+ and Cu0, respectively (Fig. 1f). Taken together, the HS-Cu catalyst containing Cu+ and Cu0 was synthesized successfully.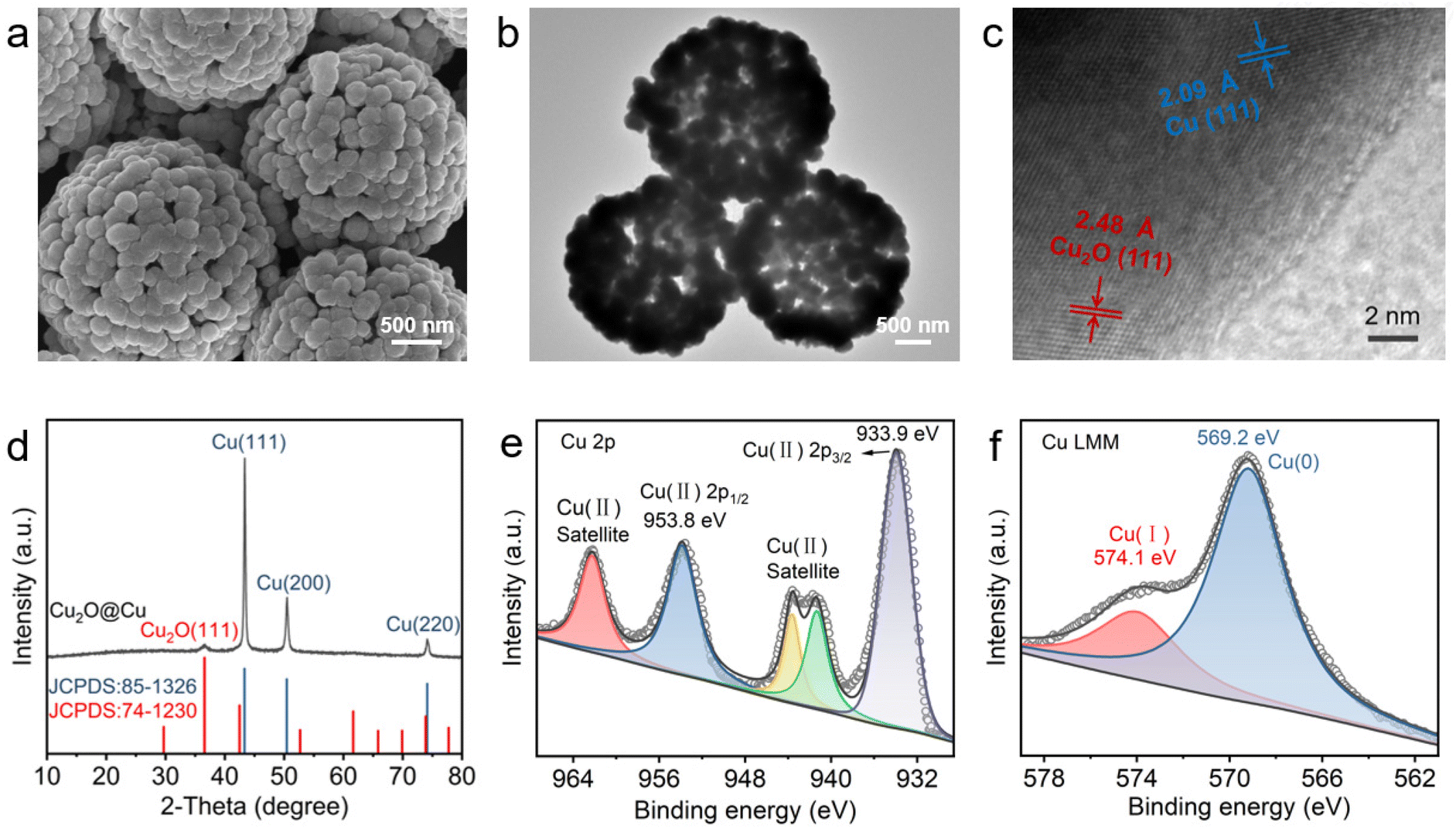 | ||
| Fig. 1 Characterization of the as-prepared HS-Cu catalyst. (a) SEM image, (b and c) HRTEM image, (d) XRD pattern, (e and f) XPS Cu 2p and Cu LMM Auger electron spectra of Cu2O@Cu. | ||
HS-Cu was then obtained by in situ electroreduction of the Cu2O@Cu hollow sphere catalyst via five runs of linear sweep voltammetry (LSV) with a scan speed of 20 mV s−1 over a potential range from −0.3 V to −2.1 V vs. Ag/AgCl. The XRD pattern of HS-Cu displayed only metallic Cu facets (Cu(111), Cu(200) and Cu(220)) as shown in Fig. S1.† Whereafter, the CO2RR performance over HS-Cu at ambient and high pressure was inspected carefully. For the electrochemical measurement at ambient and high CO2 pressures, the CO2 electroreduction reaction was performed in a high-pressure electrolytic cell with a two-compartment PEEK lining separated by a proton exchange membrane to prevent the crossover of liquid products (Fig. 2a and Fig. S2†). The concentration of active CO2 (aq.) and H2CO3 species in the electrolyte increased with an increase in CO2 pressure (Fig. S3†).44 The detailed calculation of the concentrations of carbon species and protons in 0.1 M KHCO3 at various CO2 partial pressures is shown in the experimental section of the ESI.†
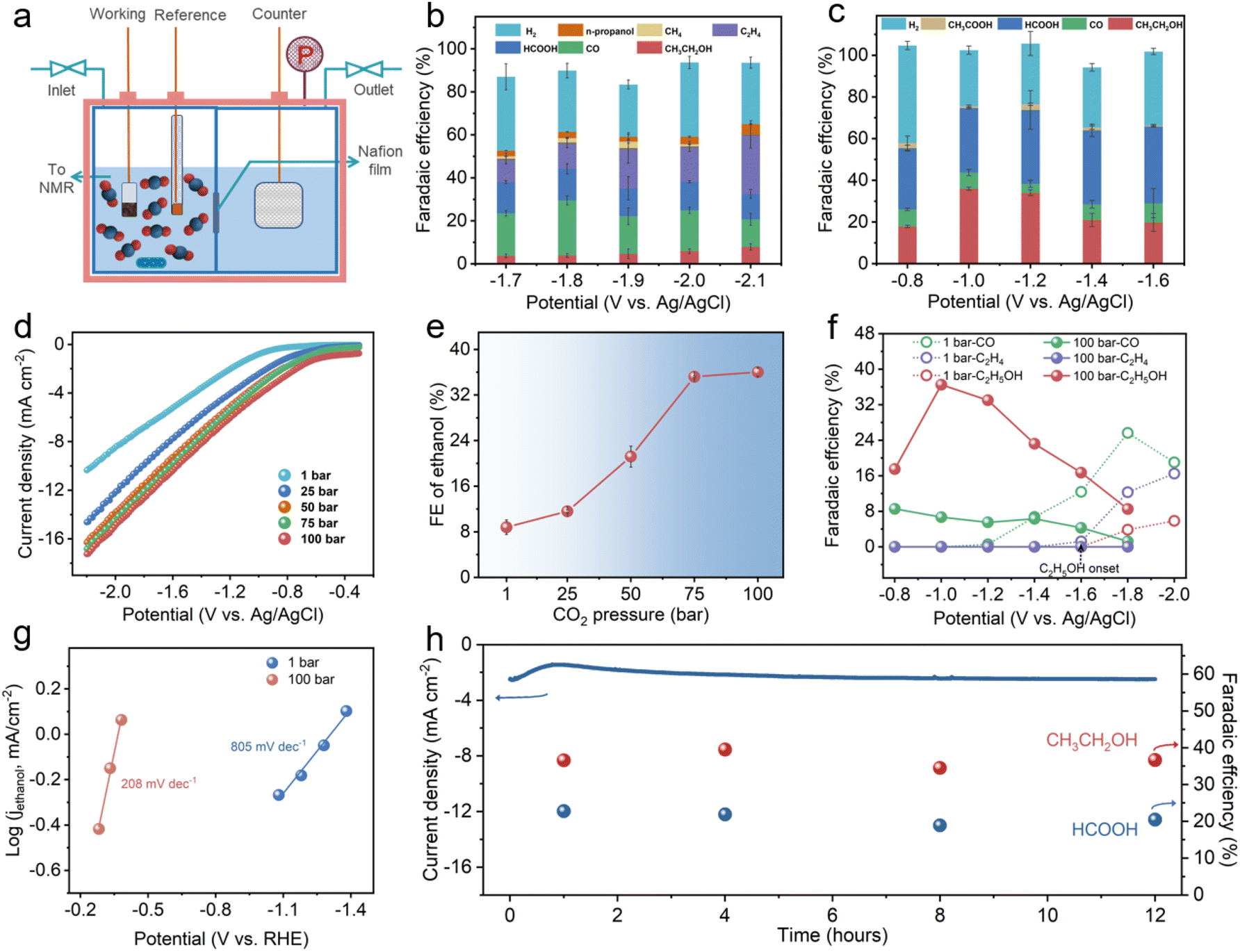 | ||
| Fig. 2 CO2RR performance at different CO2 pressures. (a) Schematic description of the high-pressure electrolytic cell. (b) Faradaic efficiencies of different products on the HS-Cu electrode at 1 bar and various potentials for one hour test. (c) Faradaic efficiencies of different products on the HS-Cu electrode at 100 bar and various potentials. (d) LSV of HS-Cu in 0.1 M KHCO3 aqueous solution at different CO2 pressures. (e) Correlation of FEethanol and CO2 pressure. (f) A comparison of the potential-dependent FECO, FEethylene and FEethanol at 1 bar and 100 bar CO2 pressure. (g) Tafel slopes for the ethanol partial current density (jethanol) at 1 and 100 bar. (h) Stability test of the HS-Cu catalyst on a glassy carbon electrode at 100 bar CO2 pressures at a potential of −1.0 V vs. Ag/AgCl (non-iR corrected. The red and purple balls represent the corresponding FEs of ethanol and formate, respectively). | ||
The CO2RR product distribution at ambient and high pressures (100 bar) is shown in Fig. 2b and c. A linear sweep voltammetry (LSV) test was conducted with a sweep rate of 20 mV s−1 at different CO2 pressures. It should be noted that the LSV curves emerged with a decreasing trend of the overpotential and an increase in current density (Fig. 2d and Fig. S4†), suggesting the immense impact of the different CO2 pressures in the CO2RR. Product analysis at a low overpotential of 1 bar revealed that the reaction was dominant by the competitive HER (ranging from −1.2 V vs. Ag/AgCl to −1.6 V vs. Ag/AgCl, FEH2 > 60%) (Fig. 2b and Fig. S5†). Though the HER was suppressed at a high overpotential (ranging from −1.7 V vs. Ag/AgCl to −2.1 V vs. Ag/AgCl, FEH2 < 40%), a wide CO2RR product distribution was observed. It could be seen that CO was the dominant C1 product (FECO = 25.7% at −1.8 V vs. Ag/AgCl) and ethylene was the main C2+ product (FEethylene = 27.8% at −2.1 V vs. Ag/AgCl). In addition, the maximum FEethanol was only 7.9% at −2.1 V vs. Ag/AgCl.
However, the product selectivity of the CO2RR at different high pressures (25 bar, 50 bar, 75 bar and 100 bar) showed a significant difference. In particular, analysis of the liquid products illustrated that the selectivity of ethanol was obviously improved with increasing CO2 pressure. The FEethanol values at 25 bar, 50 bar and 75 bar were 11.8%, 22.8% and 35.8%, respectively (Fig. S6–S8†). Remarkably, a FEethanol of 36.6% was achieved at a higher CO2 pressure of 100 bar, which was 4.6 times higher than that obtained at 1 bar. Also, only −1.0 V vs. Ag/AgCl (−0.48 V vs. RHE) was required for the cathodic reduction, which is a low reduction overpotential for the CO2RR to ethanol (Fig. 2c). To reveal the variation tendency of FEethanol influenced by different high CO2 pressures, the correlation between FEethanol and CO2 pressure was studied (Fig. 2e). The FE of ethanol was rapidly enhanced upon increasing the CO2 pressure from 1 bar to 75 bar. When the pressure continued to rise to 100 bar, the FEethanol reached a plateau. This indicates that the FEethanol is mainly affected by the CO2 pressure up to supercritical CO2 conditions. Surprisingly, a significant decrease in FEethylene was observed accordingly. A very low FEethylene of 3.5% was attained at 25 bar and no ethylene was detected at 50 bar, 75 bar and 100 bar as shown in Fig. 2c and Fig. S6–S8.† These results manifest that the ethylene product, as the competitive product of ethanol, was restrained by suppressing at the same potentials at 100 bar, which we surmise is due to the higher CO2 solubility at the high pressure (Fig. 2c and Fig. S5†).46,52,53 For C1 products, as displayed in Fig. 2c and Fig. S5–S8,† FECO was decreased and FEHCOOH was improved at a high CO2 pressure. Moreover, FECO, FEethylene and FEethanol in the potential range of −0.8 to −2.0 V vs. Ag/AgCl at 1 bar and 100 bar are presented in Fig. 2f, which revealed a super-low ethanol overpotential (<−0.8 V vs. Ag/AgCl (<−0.3 V vs. RHE)) at 100 bar in contrast to that at 1 bar (−1.6 V vs. Ag/AgCl).20 The FEethanol was increased accompanied by the decrease of FECO at 100 bar, which strongly implies that CO is a vital intermediate for the CO2RR to ethanol.
To investigate whether the product distribution is caused by CO2 pressure directly or pH changes at high CO2 pressure, the CO2RR experiments at 1 bar used 0.1 M KHCO3 + H2SO4 aqueous solutions of pH ∼ 5.0 as the electrolyte was operated. As shown in Fig. S10,† a similar product distribution was obtained in the CO2RR at 1 bar in different local pH environments, which indicates that the FEC2+ switching with the increase of CO2 pressure is due to the CO2 pressure inherently. For the CORR of the HS-Cu catalyst, a similar trend for FEethylene has been observed in the CORR at different CO pressures shown in Fig. S11,† which indicated that FEethylene was decreased with the CO concentration increase. There is no noticeable change in FEethanol at 1 bar and 3 bar, which is probably due to the other C2+ competitive products.
In order to compare the reaction kinetics of the CO2RR at ambient and high pressures, we obtained Tafel slopes for the CO2RR current density attributed to ethanol production (jethanol). The slopes were found to be 208 mV dec−1 and 805 mV dec−1 for 100 bar and 1 bar, respectively (Fig. 2g). The decreased Tafel slope value with increased CO2 pressure suggested that a faster reaction kinetics could be achieved at a higher CO2 pressure.44 In addition, the continuous CO2RR at 100 bar was performed at −1.0 V vs. Ag/AgCl for 12 h to illuminate the long-term stability of the HS-Cu. As displayed in Fig. 2h, there were no obvious changes in both the current density and FE of the ethanol and formate, illustrating the excellent stability of the prepared HS-Cu catalyst at a high CO2 pressure.52 Furthermore, XRD, XPS and SEM of the electrode after the CO2RR at 100 bar were characterized. Only characteristic peaks of bare Cu could be observed from the XRD pattern (Fig. S12†), but a minor signal of the Cu+ peak was detected from XPS analysis (Fig. S13†), which could be ascribed to the reoxidation of Cu0 after exposure to air during the sample transfer process. The SEM image showed that the sample still exhibited the hollow sphere morphology (Fig. S14†), which further indicated the good stability of the catalyst.
To gain insight into the reaction mechanism on HS-Cu for the CO2RR at different CO2 pressures, in situ Raman measurement was conducted at various applied potentials in a custom-made high pressure in situ Raman cell (Fig. S15†). As shown in Fig. 3 and Fig. S16,† the spectra at different potentials were collected to uncover the reaction intermediates at different CO2 pressures. The C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching vibration of adsorbed *CO at ca. 2091 cm−1 and the C–H stretching vibration of adsorbed CH3 at ca. 2861 and 2930 cm−1 were clearly observed at negative applied potentials from −0.8 V to −1.3 V vs. Ag/AgCl at 80 bar (Fig. 3a), which could be assigned to the key intermediates for producing ethanol.57–59 Moreover, the peaks at ca. 278 cm−1 and 352 cm−1 assigned to the Cu–CO stretching vibration modes could be unambiguously detected. In addition, a broad peak emerging at ca. 2091 cm−1 corresponds to the *CO vibrational mode. With the applied potential being increased to −1.3 V vs. Ag/AgCl, the peak intensity increased continuously, which indicates that the *CO coverage on the catalyst was improved (Fig. 3a).60 Furthermore, in situ Raman spectra at different CO2 pressures were compared at a controlled potential (−1.0 V vs. Ag/AgCl). Due to the weak adsorption of *CO on the catalyst surface at ambient pressure, no obvious peak could be observed at different potentials at 1 bar CO2 pressure. The elevated peak intensity of the Cu–CO and *CO stretching vibrations with the increased CO2 pressure from 1 bar to 80 bar is due to the effective enrichment of the adsorbed *CO intermediates on the surface of the catalyst under the high-pressure conditions (Fig. 3b). In situ Raman data collected at various applied potentials and CO2 pressures thus directly and clearly demonstrates that the high CO2 pressure contributes to improving the *CO coverage on the catalyst,60,61 which effectively promotes C–C coupling and enhances ethanol selectivity. Besides, the characteristic Cu2O peaks at around 520 cm−1 and 620 cm−1 disappeared with the overpotential from −0.1 to −1.3 V vs. Ag/AgCl, further indicating that only Cu(0) existed stably during the CO2RR at high pressure (Fig. S16†).
O stretching vibration of adsorbed *CO at ca. 2091 cm−1 and the C–H stretching vibration of adsorbed CH3 at ca. 2861 and 2930 cm−1 were clearly observed at negative applied potentials from −0.8 V to −1.3 V vs. Ag/AgCl at 80 bar (Fig. 3a), which could be assigned to the key intermediates for producing ethanol.57–59 Moreover, the peaks at ca. 278 cm−1 and 352 cm−1 assigned to the Cu–CO stretching vibration modes could be unambiguously detected. In addition, a broad peak emerging at ca. 2091 cm−1 corresponds to the *CO vibrational mode. With the applied potential being increased to −1.3 V vs. Ag/AgCl, the peak intensity increased continuously, which indicates that the *CO coverage on the catalyst was improved (Fig. 3a).60 Furthermore, in situ Raman spectra at different CO2 pressures were compared at a controlled potential (−1.0 V vs. Ag/AgCl). Due to the weak adsorption of *CO on the catalyst surface at ambient pressure, no obvious peak could be observed at different potentials at 1 bar CO2 pressure. The elevated peak intensity of the Cu–CO and *CO stretching vibrations with the increased CO2 pressure from 1 bar to 80 bar is due to the effective enrichment of the adsorbed *CO intermediates on the surface of the catalyst under the high-pressure conditions (Fig. 3b). In situ Raman data collected at various applied potentials and CO2 pressures thus directly and clearly demonstrates that the high CO2 pressure contributes to improving the *CO coverage on the catalyst,60,61 which effectively promotes C–C coupling and enhances ethanol selectivity. Besides, the characteristic Cu2O peaks at around 520 cm−1 and 620 cm−1 disappeared with the overpotential from −0.1 to −1.3 V vs. Ag/AgCl, further indicating that only Cu(0) existed stably during the CO2RR at high pressure (Fig. S16†).
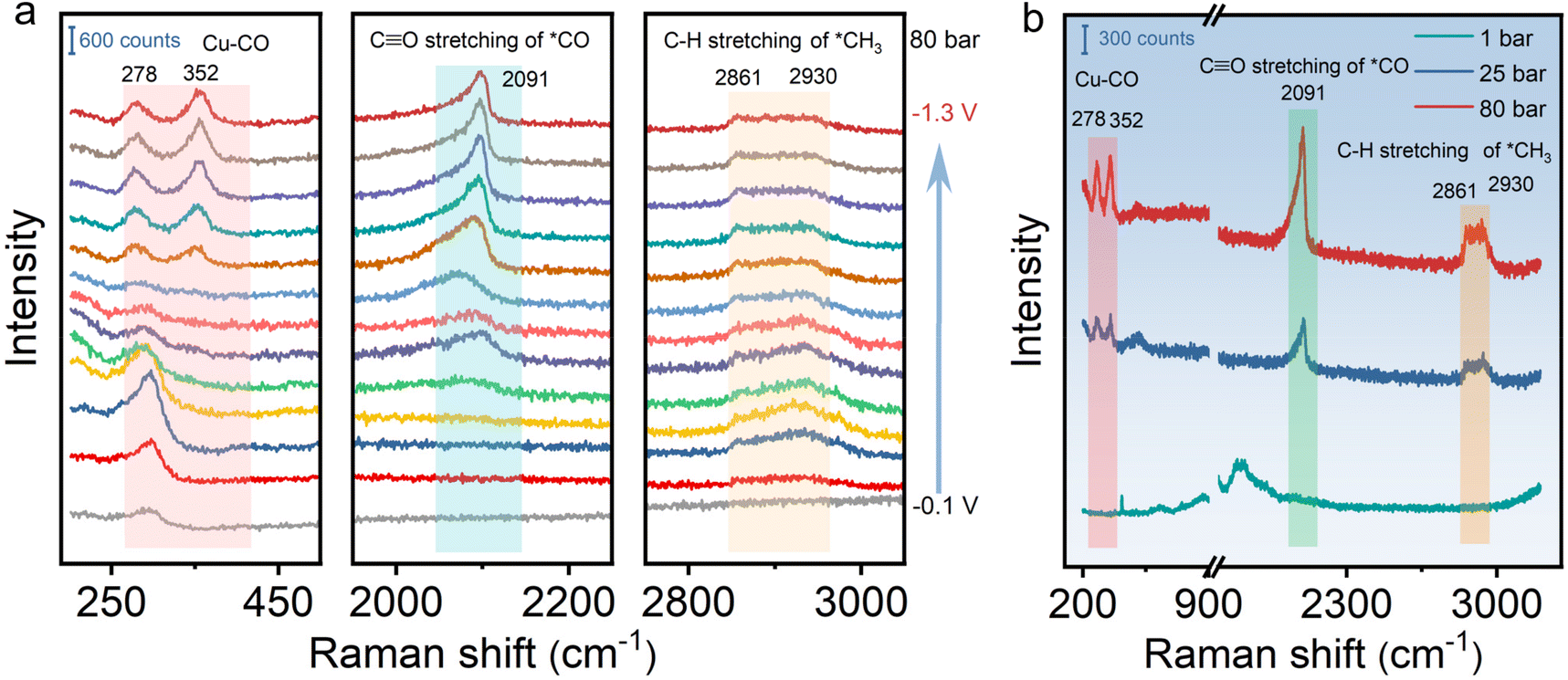 | ||
| Fig. 3 In situ Raman spectra of HS-Cu obtained (a) in the potential range of −0.1 to −1.3 V vs. Ag/AgCl during the CO2RR at 80 bar CO2 pressure, (b) at different CO2 pressures during the CO2RR at a controlled potential of −1.0 V vs. Ag/AgCl. | ||
On the basis of the observed pressure-dependent activity for CO2 conversion to ethanol, DFT calculations were further conducted to gain a more in-depth understanding of the underlying relationship between the production pathways of ethylene, ethanol and the local *CO concentration on the catalyst surface (Fig. 4 and S17†). Previous reports proposed that the formation of the intermediate *CHCOH is a decisive step in generating C2+ products during the process of the CO2RR on the Cu(111) surface model systems.60,62 The *CHCOH intermediate is further hydrogenated to form *CCH and *CHCHOH, which are the critical intermediates for ethylene and ethanol formation, respectively (Fig. 4a). We first calculated the binding energy of *CHCHOH and *CCH intermediates (Fig. 4b). We found that the binding energy decreases more for the formation of *CHCHOH (ethanol path) compared with that of *CCH (ethylene path) in the presence of *CO. With the increase of *CO coverage from 1/9–4/9 ML, the binding energy of *CHCHOH intermediates also reduces continuously, which indicates that more heat is released during the formation of *CHCHOH intermediates. Therefore, we discovered that higher *CO coverage on the catalyst surface effectively facilitates the selective formation of ethanol rather than ethylene. Besides, it is reasonable to calculate the required free energy for generating the intermediates *CCH and *CHCHOH to evaluate the possible reaction paths (Fig. S18†). The free energy of *CHCHOH decreases more than that of *CCH with *CO coverage reaching from 1/9 to 4/9. This phenomenon, which allows for high *CO coverage on the catalyst surface, is beneficial for generating *CHCHOH, thus promoting the production of ethanol. The good agreement with experimental results and the DFT calculations suggests that the selectivity for C2+ products in the CO2RR could be effectively controlled by modulating the *CO coverage on the surface of Cu-based catalysts.
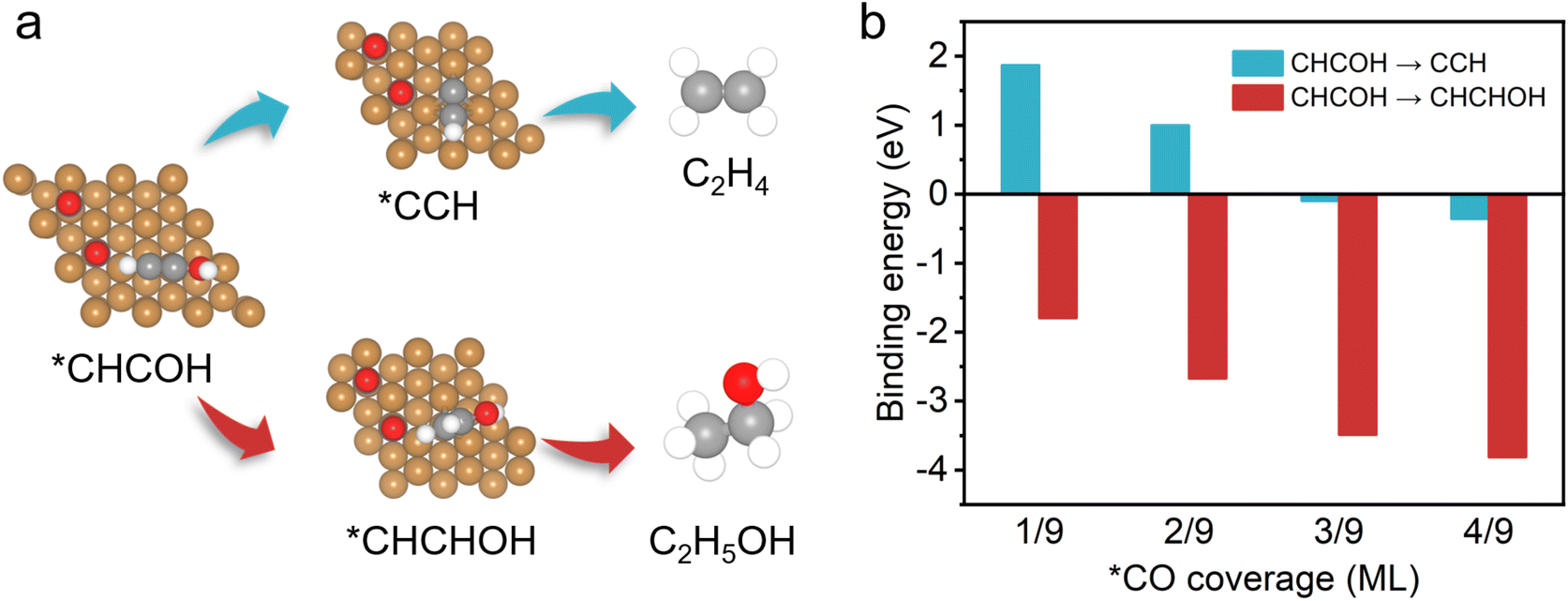 | ||
| Fig. 4 DFT calculation results on the effects of *CO coverage. (a) Key reaction pathways for the CO2RR to ethylene and ethanol on the Cu(111) surface. Yellow, copper; grey, carbon; red, oxygen; and white, hydrogen. (b) Binding energies of *CCH (cyan) and *CHCHOH (red) intermediates at different *CO coverages on the Cu(111) surface. | ||
Furthermore, we have also conducted techno-economic analysis (TEA) of the electrocatalytic CO2 conversion to ethanol at high pressures. From the technology and its impact on society, the process of electrochemical CO2 reduction to produce ethanol is the green pathway for CO2 conversion relevant to carbon reduction/utilization. Meanwhile, the high-pressure strategy does not obviously depend on the pressurization/CO2 supply cost but mainly on the electrolysis energy cost (as displayed in the ESI† in detail).
Conclusions
In summary, different CO2 pressures from 1 to 100 bar were facilely introduced into an electrochemical CO2RR system to regulate the product selectivity, and it was interesting to find that the main C2+ products could be switched from ethylene at ambient pressure to ethanol under high pressure conditions over the HS-Cu catalyst. In our work, the FEethanol could reach as high as 36.6% at 100 bar CO2 pressure, which is 4.6 times higher than that of 1 bar accompanied by faster kinetics and lower overpotential for ethanol formation. In situ Raman spectra at different pressures prove that higher *CO coverage on the surface of the HS-Cu catalyst could be detected at higher CO2 pressures. DFT calculations further confirmed that higher *CO coverage at a high CO2 pressure on the Cu surface effectively facilitates the selective formation of ethanol rather than ethylene. This work provides a facile strategy for regulating CO2 conversion into ethanol by using renewable electricity with the assistance of high CO2 pressure. In particular, to further enhance the FEethanol, the CO2RR performance of other Cu-based catalysts which have already been demonstrated with good FEethylene or FEethanol under ambient conditions will be evaluated with the assistance of high CO2 pressures in the near future. Moreover, we believe that the interesting observation under a high pressure CO2 regulation system reported here could also be extended to plenty of other novel catalysts for the efficient conversion of CO2 to high value-added chemicals and fuels.Author contributions
R. X. Q., L. P., S. L. Y., and J. L. proposed the project, designed the experiments, and wrote the manuscript. R. X. Q. performed the whole experiment. J. J., and Y. Z. G. conducted the DFT calculations. J. R. L., J. Z., D. T. S., Z. Y. L., and C. L. performed the analysis of experimental data. R. Q. L., S. Y., and B. R. conducted a part of the characterization studies. Z. P. L., T. W. X., G. K. X., L. X. C., and Y. Z. H. participated in the discussions. L. P., S. L. Y., J. L., and B. X. H. supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (21903066 and 22078274) and the President Fund of Xiamen University (No. 20720210046). The authors are also grateful for the help of Yanan Xu, Wenli Hao, Zhihong Gao, Jia Zhao, Guang Li, Wanfeng Xiong and Prof. Dr Jinqing Lin.Notes and references
- R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 RSC.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- G. Wen, B. Ren, Y. Zheng, M. Li, C. Silva, S. Song, Z. Zhang, H. Dou, L. Zhao, D. Luo, A. Yu and Z. Chen, Adv. Energy Mater., 2021, 12, 2103289 CrossRef.
- R. G. Mariano, K. McKelvey, H. S. White and M. W. Kanan, Science, 2017, 358, 1187–1192 CrossRef CAS.
- W. Guo, X. Tan, J. Bi, L. Xu, D. Yang, C. Chen, Q. Zhu, J. Ma, A. Tayal, J. Ma, Y. Huang, X. Sun, S. Liu and B. Han, J. Am. Chem. Soc., 2021, 143, 6877–6885 CrossRef CAS PubMed.
- L. Jiao, W. Yang, G. Wan, R. Zhang, X. Zheng, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 20589–20595 CrossRef CAS.
- X. Suo, F. Zhang, Z. Yang, H. Chen, T. Wang, Z. Wang, T. Kobayashi, C. L. Do-Thanh, D. Maltsev, Z. Liu and S. Dai, Angew. Chem., Int. Ed., 2021, 60, 25688–25694 CrossRef CAS PubMed.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C. W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS.
- Z. Wang, Y. Zhou, D. Liu, R. Qi, C. Xia, M. Li, B. You and B. Y. Xia, Angew. Chem., Int. Ed., 2022, 61, e202200552 CAS.
- D. L. Meng, M. D. Zhang, D. H. Si, M. J. Mao, Y. Hou, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 25485–25492 CrossRef CAS PubMed.
- X. F. Qiu, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS.
- P. Shao, W. Zhou, Q. L. Hong, L. Yi, L. Zheng, W. Wang, H. X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687–16692 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- C. Chen, X. Yan, S. Liu, Y. Wu, Q. Wan, X. Sun, Q. Zhu, H. Liu, J. Ma, L. Zheng, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 16459–16464 CrossRef CAS PubMed.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- X. Su, Z. Jiang, J. Zhou, H. Liu, D. Zhou, H. Shang, X. Ni, Z. Peng, F. Yang, W. Chen, Z. Qi, D. Wang and Y. Wang, Nat. Commun., 2022, 13, 1322 CrossRef CAS.
- H. Han, Y. Noh, Y. Kim, S. Park, W. Yoon, D. Jang, S. M. Choi and W. B. Kim, Green Chem., 2020, 22, 71–84 RSC.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T. K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed.
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 CrossRef CAS PubMed.
- D. Karapinar, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2021, 6, 694–706 CrossRef CAS.
- J. Santatiwongchai, K. Faungnawakij and P. Hirunsit, ACS Catal., 2021, 11, 9688–9701 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- C. Kim, K. M. Cho, K. Park, J. Y. Kim, G. T. Yun, F. M. Toma, I. Gereige and H. T. Jung, Adv. Funct. Mater., 2021, 31, 2102142 CrossRef CAS.
- J. Wang, H. Y. Tan, Y. Zhu, H. Chu and H. M. Chen, Angew. Chem., Int. Ed., 2021, 60, 17254–17267 CrossRef CAS.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef.
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z. J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS PubMed.
- O. Piqué, F. Viñes, F. Illas and F. Calle-Vallejo, ACS Catal., 2020, 10, 10488–10494 CrossRef.
- C. Zhu, Z. Zhang, L. Zhong, C.-S. Hsu, X. Xu, Y. Li, S. Zhao, S. Chen, J. Yu, S. Chen, M. Wu, P. Gao, S. Li, H. M. Chen, K. Liu and L. Zhang, Chem, 2021, 7, 406–420 CAS.
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, e1803111 CrossRef PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS.
- A. N. Kuhn, H. Zhao, U. O. Nwabara, X. Lu, M. Liu, Y. T. Pan, W. Zhu, P. J. A. Kenis and H. Yang, Adv. Funct. Mater., 2021, 31, 2101668 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- R. Feng, Q. Zhu, M. Chu, S. Jia, J. Zhai, H. Wu, P. Wu and B. Han, Green Chem., 2020, 22, 7560–7565 RSC.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- Z. Z. Niu, F. Y. Gao, X. L. Zhang, P. P. Yang, R. Liu, L. P. Chi, Z. Z. Wu, S. Qin, X. Yu and M. R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed.
- S. Jia, Q. Zhu, M. Chu, S. Han, R. Feng, J. Zhai, W. Xia, M. He, H. Wu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 10977–10982 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS.
- Y. Song, W. Chen, C. Zhao, S. Li, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840–10844 CrossRef CAS.
- N. Sikdar, J. R. C. Junqueira, S. Dieckhofer, T. Quast, M. Braun, Y. Song, H. B. Aiyappa, S. Seisel, J. Weidner, D. Ohl, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 23427–23434 CrossRef CAS.
- S. Kaneco, N.-h. Hiei, Y. Xing, H. Katsumata, H. Ohnishi, T. Suzuki and K. Ohta, J. Solid State Electrochem., 2003, 7, 152–156 CrossRef CAS.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O'Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- C. I. Shaughnessy, D. J. Sconyers, H.-J. Lee, B. Subramaniam, J. D. Blakemore and K. C. Leonard, Green Chem., 2020, 22, 2434–2442 RSC.
- J. Li, Y. Kuang, Y. Meng, X. Tian, W. H. Hung, X. Zhang, A. Li, M. Xu, W. Zhou, C. S. Ku, C. Y. Chiang, G. Zhu, J. Guo, X. Sun and H. Dai, J. Am. Chem. Soc., 2020, 142, 7276–7282 CrossRef CAS PubMed.
- J. Li, J. Guo and H. Dai, Sci. Adv., 2022, 8, eabo0399 CrossRef CAS.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- X. Kang, L. Li, A. Sheveleva, X. Han, J. Li, L. Liu, F. Tuna, E. J. L. McInnes, B. Han, S. Yang and M. Schroder, Nat. Commun., 2020, 11, 5464 CrossRef CAS PubMed.
- D. V. Vasilyev and P. J. Dyson, ACS Catal., 2021, 11, 1392–1405 CrossRef CAS.
- C. I. Shaughnessy, D. J. Sconyers, T. A. Kerr, H. J. Lee, B. Subramaniam, K. C. Leonard and J. D. Blakemore, ChemSusChem, 2019, 12, 3761–3768 CrossRef CAS PubMed.
- K. j. Puring, O. Evers, M. Prokein, D. Siegmund, F. Scholten, N. Mölders, M. Renner, B. Roldan Cuenya, M. Petermann, E. Weidner and U.-P. Apfel, ACS Catal., 2020, 10, 12783–12789 CrossRef.
- S. Lamaison, D. Wakerley, J. Blanchard, D. Montero, G. Rousse, D. Mercier, P. Marcus, D. Taverna, D. Giaume, V. Mougel and M. Fontecave, Joule, 2020, 4, 395–406 CrossRef CAS.
- J. Kou, A. Saha, C. Bennett-Stamper and R. S. Varma, Chem. Commun., 2012, 48, 5862–5864 RSC.
- T. C. Chou, C. C. Chang, H. L. Yu, W. Y. Yu, C. L. Dong, J. J. Velasco-Velez, C. H. Chuang, L. C. Chen, J. F. Lee, J. M. Chen and H. L. Wu, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed.
- M. Li, Y. Ma, J. Chen, R. Lawrence, W. Luo, M. Sacchi, W. Jiang and J. Yang, Angew. Chem., Int. Ed., 2021, 60, 11487–11493 CrossRef CAS PubMed.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Gratzel, Angew. Chem., Int. Ed., 2019, 58, 15036–15040 CrossRef CAS.
- F. Hu, L. Yang, Y. Jiang, C. Duan, X. Wang, L. Zeng, X. Lv, D. Duan, Q. Liu, T. Kong, J. Jiang, R. Long and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 26122–26127 CrossRef CAS PubMed.
- A. D. Handoko, K. W. Chan and B. S. Yeo, ACS Energy Lett., 2017, 2, 2103–2109 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2019, 3, 75–82 CrossRef.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- Y. W. Lum, T. Cheng, W. A. Goddard and J. W. Ager, J. Am. Chem. Soc., 2018, 140, 9337–9340 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03343g |
| This journal is © The Royal Society of Chemistry 2023 |