
Bipotentiostatic tandem electrocatalysis of the CO2 reduction reaction yielding C2+ fuels†
Joo Yeon
Kim
,
Yeonsu
Kim
,
C. Hyun
Ryu
and
Hyun S.
Ahn
 *
*
Department of Chemistry, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea. E-mail: ahnhs@yonsei.ac.kr
First published on 31st May 2023
Abstract
Electrochemical CO2 conversion to fuel molecules is an attractive strategy towards atmospheric carbon attenuation. In the fuel conversion catalysis of carbon dioxide, surface adsorbed CO was often identified as a key intermediate, and the inherently low concentration of which has recently been highlighted as an important factor in the low yields of C2+ fuels. Here a bipotentiostatic tandem catalysis system was designed such that the loads for electrosynthesis of CO from CO2 and the subsequent C–C coupling reaction were split to two separate working electrodes with independent potential programming. Coulometric tracking of the reaction indicated efficient turnover of the electrosynthetic CO to fuel molecules at the second working electrode, achieving a high C2+ yield of 67.3% and a 6.5% faradaic efficiency towards 1-propanol. Importantly, the unique capability of the tandem catalysis system allowed control of the CO flux to the second working electrode, which enabled the direct quantification of the C–C coupling turnover frequency on a surface copper atom (0.43 ± 0.06 s−1), when the reaction current was electrode-kinetics controlled with sufficient CO mass transport. Furthermore, the bipotentiostatic reaction platform developed here exhibits modular tunability, such that the expansion of the platform to other sequential electrocatalyses is imaginable.
Introduction
Controlled emission of CO2 into the atmosphere is an international campaign due to the routine observation of the grim environmental impacts of anthropogenic greenhouse gas accumulation.1,2 Furthermore, front line research is taking the next step in the development of technologies for the reduction of atmospheric carbon by capture and utilization of CO2.3 A promising strategy in carbon attenuation is the electrochemically driven CO2 reduction reaction (CO2RR) to form value-added chemicals.4,5,48 Although electrochemical CO2RR is in its early stages of research, rapid progress in the understanding of the reaction has occurred in the last two decades. Regarding the CO2RR, the majority of research aims to produce fuel molecules containing more than two carbons in series. Thus far the only catalyst proven to facilitate C–C coupling from CO2 feedstock is copper.6–8 Numerous research studies strived to engineer copper (using thermal or redox stimuli and by alloying) to maximize C2+ selectivity,9–11,46 yet in most cases, other competing pathways leading to carbon monoxide and dihydrogen occupy significant portions of the reducing equivalents.Recent in situ and operando spectroscopic investigations revealed meaningful hints towards the mechanism behind the C–C coupling reaction on a catalytic surface;12–15,47 however, a firm understanding of the structural requirements for catalytic sites where C2+ products form and the key surface intermediates involved in the reaction remains elusive. The majority of reports in the literature concur on the importance of surface bound COads as a key intermediate in the C–C formation process (whether it is involved in homocoupling to another COads![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16,45 or reacts with a protonated COHads type species17–19), and a recent literature suggested that the C–C coupling efficiency remains low due to the inherently low surface concentration of COads.20–25 A collection of investigations on direct reduction of CO to C–C coupled fuel molecules26–29 further suggests the involvement of COads as an intermediate in the surface C–C formation. Electrochemical conversion of CO to fuels is an attractive vein of research; however, it has much less impact on CO2 attenuation.
16,45 or reacts with a protonated COHads type species17–19), and a recent literature suggested that the C–C coupling efficiency remains low due to the inherently low surface concentration of COads.20–25 A collection of investigations on direct reduction of CO to C–C coupled fuel molecules26–29 further suggests the involvement of COads as an intermediate in the surface C–C formation. Electrochemical conversion of CO to fuels is an attractive vein of research; however, it has much less impact on CO2 attenuation.
Collectively taking into consideration the importance of COads and its low surface concentration, a handful of researchers strived to design a bipotentiostatic system in which the first working electrode initiates selective conversion of CO2 to CO, and the second working electrode placed nearby attempts to proceed with the C–C coupling reaction of the electrosynthetic CO.30–34 Silver or gold electrodes were typically employed for CO production, with copper as the second electrode. Despite several reports on microfabrication of reactors,32–34 limited success in fuel formation compared to that with copper-only operation was achieved due to the challenges in fine independent control of the potentials on the two electrodes and their inefficient interplay.
In this work we designed a bipotentiostatic flow reactor for sequential conversion of CO2 to C–C coupled fuels, including C3 products such as 1-propanol. Coulometric tracking of the reaction suggested efficient fuel conversion of the CO produced from the first electrode. Although the reactor design prohibited direct spectroscopic identification of the COads surface intermediate, Multiphysics simulations revealed unprecedentedly high local concentration of CO near the second working electrode, approaching saturation concentration in the employed buffer. Fine control of the independent potential program on Ag–Cu electrodes allowed for their efficient tandem catalysis, resulting in overall 67.3% C2+ products from a CO2 feed, including 6.5% faradaic production of 1-propanol.
Results and discussion
A handful of literature precedents on bipotentiostatic CO2 conversion typically exhibit ill-controlled potentials and flawed geometry to support the interplay of the two electrodes.32–34 In order for the design to prevail, relative placement of the two electrodes for the optimized transport of the evolved CO along with careful independent potential control must be implemented for the minimization of side reactions and maximization of CO utilization for C–C coupling on the second working electrode. Ager and coworkers meticulously designed a CO mass transport model with micro-patterned Ag–Cu electrode arrays;34 however, due to the small spatial separation between the two electrodes, independent potential programming was difficult, resulting in minor improvements in C2+ yields. The flow reactor design employed in this work (Fig. 1 and see also Fig. S1–S5†) involved a typical planar Ag electrode as the first working electrode (WE1) and a mesh-type Cu electrode as the second (WE2) (see S11–S13†). WE2 integrated a central opening to maximize solution flux through the inter-electrode volume while encompassing the peripheries of WE1, where the rate of diffusion of the detaching CO species was at its maximum (see Fig. 1 and also Fig. S6, S6–S9, S16, S17 and S31†). This type of reactor and electrode geometry design were implemented to ensure efficient influx of the WE1 produced CO species into WE2, without interfering in the independent potential programming of both electrodes (see Fig. S13 and S51†). Additionally, inter-electrode spacing was considered as a control variable (see Fig. S28†). As a result, Multiphysics simulated CO concentration at Cu WE2 reached 0.63 mM (see Fig. 1 and also Fig. S6, S8, S9, and S31;† simulation details are given in the ESI†), approaching saturation concentration in buffered water.28,35 This type of efficient CO delivery allowed the operation of the bipotentiostatic system at inter-electrode distances of a few mm, much greater than the diffusion layer thickness (in the μm range),36 ensuring a negligible mutual field effect on the two electrodes. In our investigation of the correlation between the area of the WE2 central opening and the efficacy of CO mass transport from WE1 to WE2 (see Fig. S9 and S31†), the best results were obtained when the size of the central opening was equivalent to the area of WE1, in accordance with electrode mass transport theory.32,37–39,48 All subsequent discussions hereon regarding the bipotentiostatic CO2RR are on experiments performed with electrode configurations employing WE2 with a rectangular central opening mirroring WE1 dimensions.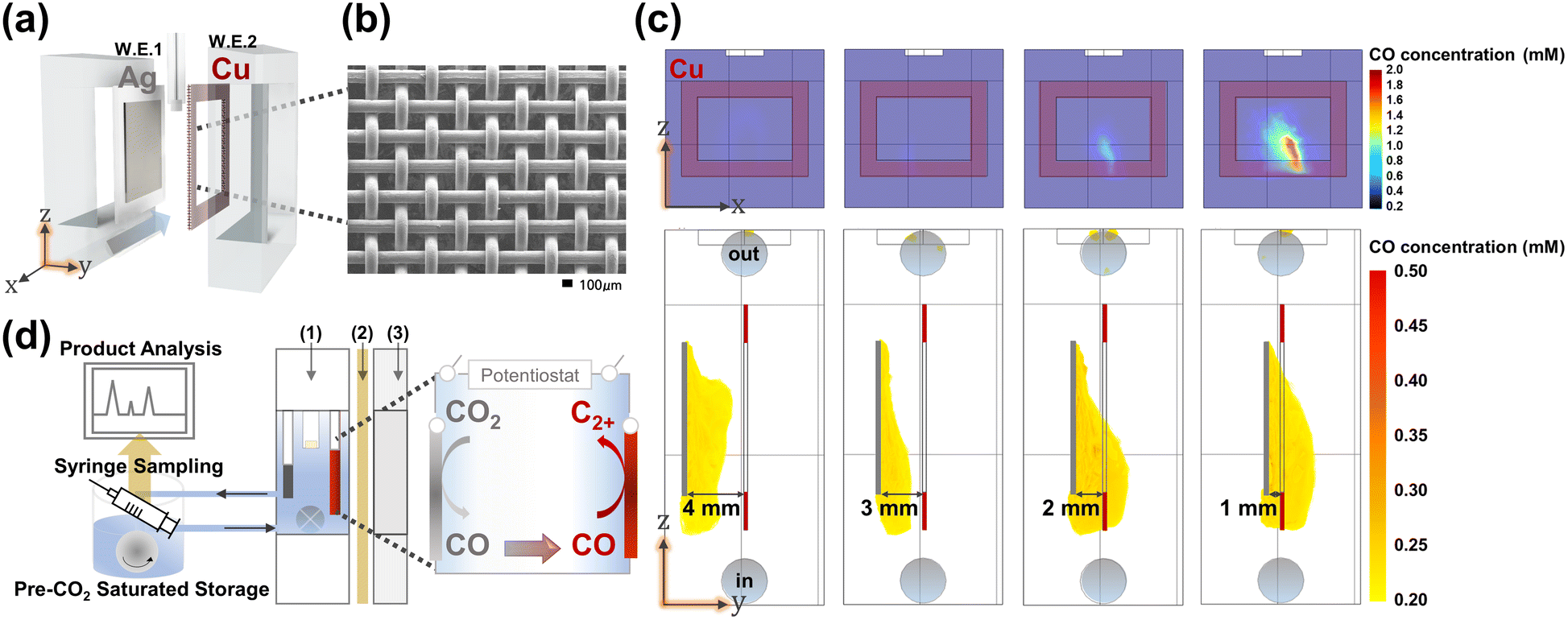 | ||
| Fig. 1 (a) Schematic representation of the Cu–Ag tandem CO2 reduction flow reactor, (b) SEM image of the Cu mesh WE2, and (c) COMSOL simulated map of the CO concentration at varying Ag–Cu distances. At inter-electrode distances smaller than 2 mm, efficient CO delivery to the Cu WE2 is visible. (d) Detailed scheme of the electrode configuration and the whole flow reactor used for the CO2RR. Compartments numbered (1), (2), and (3) represent the reference electrode (equipped with double junction), proton exchange membrane, and counter electrode part. Rather than using the synthesized Cu catalysts, we used the commercial Cu mesh as an electrocatalyst and a modulated potential application method (see S19†). The pulsed potential program we adopted here is a well-known method that possibly suppresses the HER through periodic anodic potential application with a small amount of charge.10,37 This experimental design provided better reproducibility in the CO2RR and excluded the biased comprehension of the specific catalysts. | ||
In the bipotentiostatic configuration shown in Fig. 1a, each electrode was operated independently to probe CO2RR product selectivity. Silver is well-known to selectively produce CO from CO2 (see Fig. S7†).8,40 Similarly, silver WE1 demonstrated selective CO production in our flow reactor at typical CO2RR potentials (−1.1 V vs. RHE, all potentials hereon are referenced to RHE; Fig. 2c). Copper WE2 yielded predominantly hydrogen with minor production of hydrocarbons when operated independently at −0.65 V (see Fig. S7 and S18†).
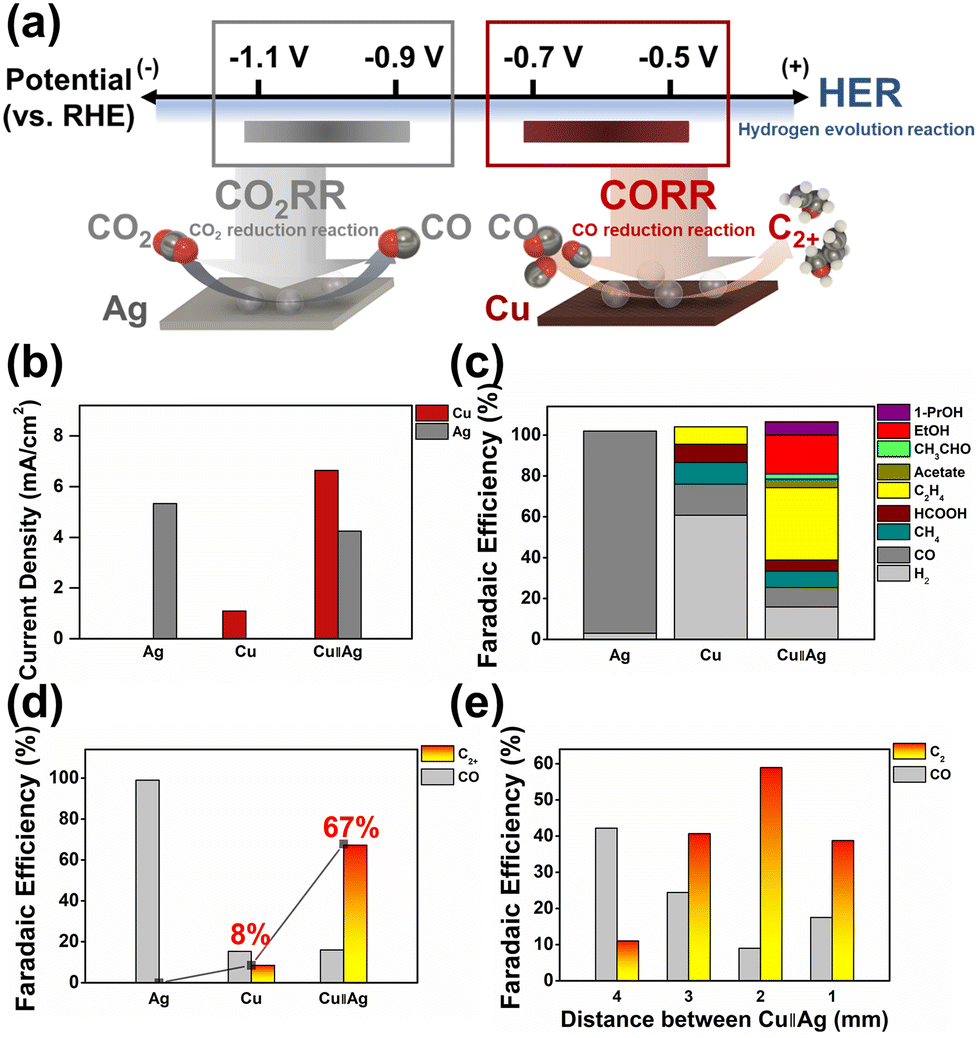 | ||
| Fig. 2 (a) A scheme of the potential space in the employed buffer system, and those applied at the two electrodes (Cu and Ag). Application of small overpotentials on Cu WE2 allowed for the selective reduction of the electrosynthetic CO with suppressed hydrogen evolution. (b) Average current densities, (c) total faradaic efficiencies, and (d) faradaic efficiencies for CO and C2+ in Ag-independent, Cu-independent, and Cu–Ag tandem catalytic operations. (e) Faradaic efficiencies toward CO and C2+ products at varying inter-electrode distances. Unless otherwise stated, the applied potentials on Ag and Cu were −1.1 V vs. RHE and −0.65 V/+0.45 V; 350 ms/350 ms, respectively. Reaction conditions for Cu–Ag tandem catalysis in (b–d) represent the most C2+ producing reaction conditions in the research, corresponding to constant CO2 supply with Cs+ cation incorporation. A CO2 saturated 0.05 M KHCO3 solution including 34 μM K2EDTA·2H2O was used as the electrolyte for (e). Products depicted as C2 and C2+ include C2H4, EtOH, acetate, and acetaldehyde in common, and 1-PrOH for C2+ (see S14–S25†). | ||
Typical onset potentials of the CO2RR on copper are reported in the range from −0.8 to −0.9 V,41,42 and therefore, dominant hydrogen production at less reducing potentials was expected (see Fig. S18†). Also, owing to the small overpotentials, current densities in copper-only operation were low, in the order of ca. 1 mA cm−2 (see Fig. 2b). In the bipotentiostatic mode (at an optimized inter-electrode distance of 2 mm and a flow rate of 100 cc min−1) (see Fig. S11 and S30†), WE2 current density was boosted to values exceeding 6 mA cm−2, suggesting that the CO molecules evolved from WE1 were efficiently being transported to WE2 and reduced (see Fig. 2b, S15 and S18†). Silver WE1 current density suffered ca. 20% decrease due to some competitive CO2RR at WE2 (despite the low potential at −0.65 V); however, it maintained a reasonable level at 4 to 5 mA cm−2 (see Fig. S15†). Significantly increased faradaic efficiencies toward fuel molecules such as ethylene, ethanol, and 1-propanol (Fig. 2c, d and S18†) indicated that the WE1 produced CO molecules were reductively and catalytically converted at WE2, yielding C–C bond formation. Notably, the C2+ faradaic yield reached 67.3% while that for hydrogen was suppressed to ca. 16%. Also, an impressively high yield of 1-propanol of 6.5% was recorded, comparable to the highest literature reported yields from a CO2 feed (Tables S55 and S56†). Inter-electrode distance was probed as a variable under set conditions (WE1 poised at −1.1 V and WE2 at −0.65 V), which revealed that the optimal fuel production efficiency is at 2 mm (see Fig. 2e and S8†). According to our Multiphysics calculations (see Fig. S8†), local CO concentration at WE2 was predicted to be the highest at an inter-electrode distance of 1 mm; however, it seems that beyond a certain flux of CO to copper, further improvement in C–C coupling efficiency was not feasible (also vide infra), presumably due to the limited catalytic rate at the copper electrode surface. All subsequently described experiments were performed at 2 mm distance between WE1 and WE2.
A handful of researchers attempted CO2 conversion via two sequential (albeit spatially separated) steps through the CO intermediate;30,31 however, the major difference and breakthrough in this work is that the tandem electrolyses are contained within the same reaction compartment and a short time of flight is allowed for the CO intermediate species between the two electrodes. An interesting observation occurred when we attempted direct reduction of CO (by saturating the buffered solution with CO gas purge) on copper, to compare the product distribution to that from the reduction of electrosynthetic CO (see Fig. S12, S14 and S29†). At copper potentials identical to those of the bipotentiostatic experiments (−0.65 V), a significantly lower C2+ yield was observed, along with pronounced selectivities towards hydrogen (Fig. S12†). The clear difference in the C2+ product selectivities between reductions of electrosynthetic and dissolved CO molecules further stresses that the bipotentiostatic tandem catalysis demonstrated here is a promising route for efficient fuel conversion of CO2.
Product selectivity distributions at increased overpotentials at WE1, and subsequently higher CO flux to WE2, were studied (see Fig. 3a and S19†). At WE1 potentials more anodic than −1.1 V, negatively biasing the potential systematically increased current densities at both WE1 and WE2, suggesting that increased CO production at WE1 translated into efficient conversion of the products at WE2 (see Fig. S15, S17 and S18†). However, at potentials of −1.2 and −1.3 V, WE2 current densities remained constant despite increased CO faradaic current at WE1 (see Fig. S16 and S17†). The excess CO produced was not further reduced at WE2 and was collected as products (see Fig. 3b and S15†). The overall amount of C2 products remained similar at WE1 potentials of −1.1 to −1.3 V; however, C2 faradaic yields diminished due to the dilution of products by unreacted CO at −1.2 and −1.3 V. From the trend displayed in Fig. 3a, we can deduce that the electrode reaction at WE2 transitioned from the CO mass-transfer-controlled mode at WE1 potentials more anodic than −1.1 V to electrode-kinetics-controlled mode at WE1 potentials from −1.1 to −1.3 V (see S51†).43,44
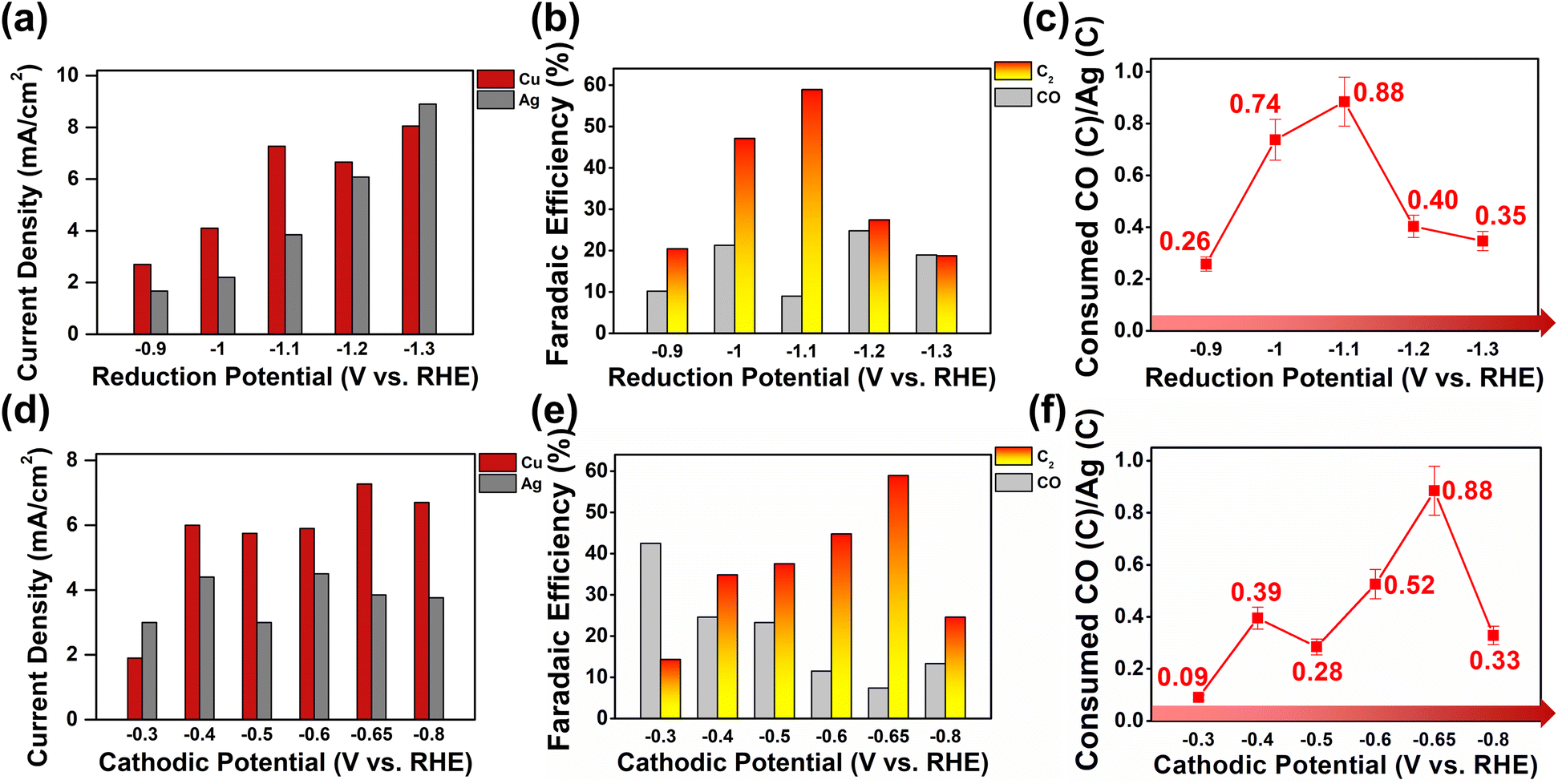 | ||
| Fig. 3 (a) Average current densities, (b) faradaic efficiencies for CO and C2, and (c) CO conversion efficiencies at varying applied potentials on Ag WE1 in Cu–Ag tandem catalysis. The applied potential on Cu was fixed to −0.65 V/+0.45 V (350 ms pulses). (d) Average current densities, (e) faradaic efficiencies for CO and C2, and (f) CO conversion efficiencies at varying applied potentials on Cu WE2. The applied potential on Ag was fixed at −1.1 V. All of the experiments were performed in a recirculating (100 cc min−1) 0.05 M KHCO3 buffer solution saturated with CO2. Details of the CO conversion efficiency calculation are provided in the ESI (Fig. S22 and S23†). | ||
This clear transition was observable due to the unique capability of the bipotentiostatic system, where CO production and delivery were systematically tunable by the control of WE1 potentials. The set of data with WE1 potentials more cathodic than −1.1 V (sufficient CO flux to WE2 and reaction current limited by electrode kinetics) allowed for the direct evaluation of the C–C coupling reaction kinetics at the copper surface (vide infra). Holding the WE1 potential constant at −1.1 V, the effect of WE2 potentials on bipotentiostatic tandem catalysis was observed (see Fig. 3d–f and S18†). Potentials from −0.3 V to −0.65 V clearly exhibited a systematic increase in faradaic efficiencies for C2 products while that for CO decreased proportionally. Potentials more cathodic than −0.65 V resulted in decreased C–C coupling and increased hydrogen evolution.
Optimal CO utilization (88 ± 9%; see the ESI for calculation details; S22 and S23†) was achieved with WE1 current densities of ca. 4 mA cm−2, corresponding to a CO flux of 9.9 mol s−1 cm−2. From the CO flux into WE2 and the amount of C–C coupled products formed, we can calculate the C–C coupling reaction turnover frequency of a copper surface atom to be 0.43 ± 0.06 s−1, under electrode-kinetics-controlled current conditions (see Fig. 3a and the ESI for detailed calculations; S32†). This information is particularly valuable because an electrocatalytic environment with a controlled flux of CO is difficult to achieve. To the best of our knowledge, this work marks the first quantification of the rate at which C–C coupling occurs at the copper surface when provided a sufficient influx of CO, and the current is limited by the electrode kinetics. Discussions on catalytic efficiency in the CO2RR typically revolve around product selectivity; however, as the field matures, defining lower-bound turnover frequencies as shown here towards a certain product is important in further designing and engineering catalysts.
In order to evaluate the stability of the bipotentiostatic flow reactor, continuous operation with real-time product monitoring was performed as shown in Fig. 4 (see also Fig. S25–S30†). For up to 4 hours, the C2+ product yield and CO utilization at copper remained stable, while methane and hydrogen selectivities were suppressed to low levels. The test system demonstrated good reproducibility and reasonable longevity for the systematic evaluation of Ag–Cu tandem electrocatalytic reduction of CO2. The flow reactor setup developed and optimized in this work can be modularly modified and applied to the study of other types of tandem electrocatalytic systems.
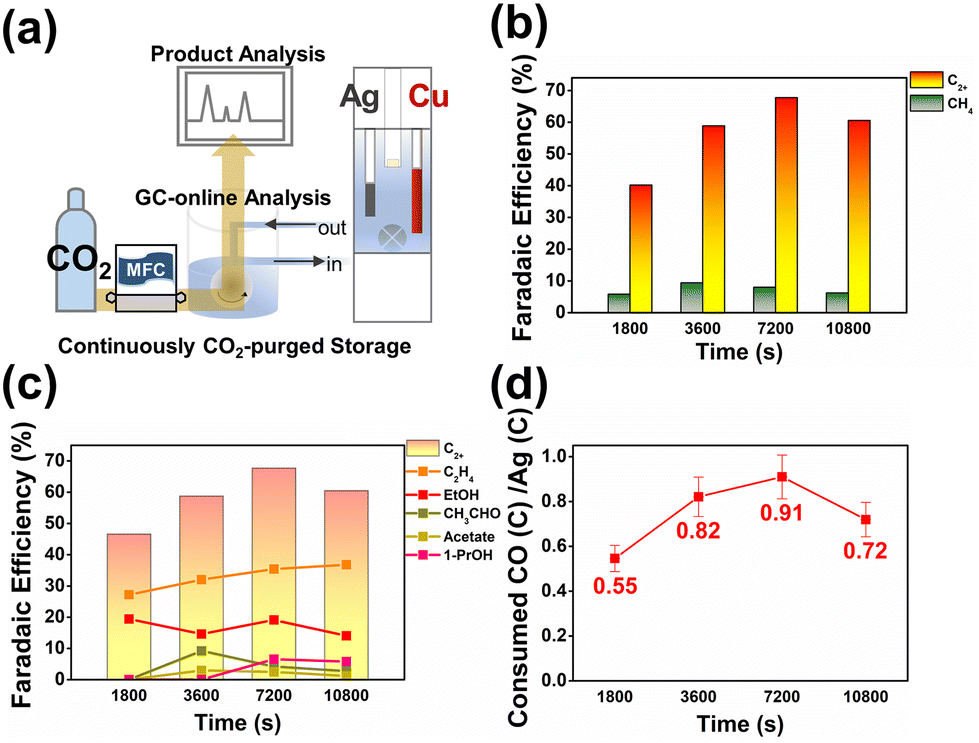 | ||
| Fig. 4 (a) Schematic diagram of the CO2RR experimental configuration with online-GC product quantification. (b) Faradaic efficiencies for CH4 and C2+ and (c) faradaic efficiencies towards each of the C2+ chemicals, namely C2H4, EtOH, acetate, acetaldehyde, and 1-PrOH, are plotted as a function of time. (d) CO conversion efficiencies of the Cu–Ag tandem electrocatalysis in various reactions. All experiments were performed in a recirculating (100 cc min−1) 0.05 M CsHCO3 buffer solution saturated with CO2. Except for the Cs cation, all other reaction parameters were fixed to those shown in Fig. 2 and 3. captions (see Fig. S20–S25†). | ||
Conclusion
In this work we demonstrated bipotentiostatic tandem electrocatalysis of the CO2 reduction reaction, leading to successful production of C2+ fuel molecules with high yields. Efficient sequential catalysis was feasible owing to the elaborate independent control of the potentials (subsequently rates) on the two electrodes without mutual interference. Coulometric tracking of the products indicated high utility of the electrosynthetic CO towards C–C coupled products. Notably, by separation of the electrosynthesis of CO and the subsequent reactions, electrode kinetics on the copper electrode during the C–C bond formation was evaluated. At a high enough CO flux into the second copper electrode where the current density was limited by the reaction kinetics, C–C coupling turnover frequency was found to be 0.43 ± 0.06 s−1 per surface copper atom. This type of evaluation was possible due to the unique capability of the tandem catalysis cell where the delivery of CO to the copper electrode was easily controllable by current density modulation on the silver electrode. The bipotentiostatic reaction platform developed here exhibits modular tunability over electrode materials, potential programming, and reactor geometry, such that the expansion of the platform to other sequential electrocatalyses is imaginable.Author contributions
Hyun S. Ahn conceived the concept of this research, and directed and supervised the overall project. Joo Yeon Kim performed the entire experiments (reactor design and CO2RR product distribution investigation). Yeonsu Kim and C. Hyun Ryu designed the COMSOL-based simulation model for the tandem CO2RR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea (NRF-2020R1C1C1007409 and NRF-2022K1A3A1A31092705).References
- K. Daehn, R. Basuhi, J. Gregory, M. Berlinger, V. Somjit and E. A. Olivetti, Nat. Rev. Mater., 2022, 7, 275–294 CrossRef CAS.
- H. D. Matthews and S. Wynes, Science, 2022, 376, 1404–1409 CrossRef CAS PubMed.
- K. M. G. Langie, K. Tak, C. Kim, H. W. Lee, K. Park, D. Kim, W. Jung, C. W. Lee, H.-S. Oh, D. K. Lee, J. H. Koh, B. K. Min, D. H. Won and U. Lee, Nat. Commun., 2022, 13, 7482 CrossRef CAS PubMed.
- T. N. Do, C. You and J. Kim, Energy Environ. Sci., 2022, 15, 169–184 RSC.
- J. Sisler, S. Khan, A. H. Ip, M. W. Schreiber, S. A. Jaffer, E. R. Bobicki, C.-T. Dinh and E. H. Sargent, ACS Energy Lett., 2021, 6, 997–1002 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- D. Cheng, Z.-J. Zhao, G. Zhang, P. Yang, L. Li, H. Gao, S. Liu, X. Chang, S. Chen, T. Wang, G. A. Ozin, Z. Liu and J. Gong, Nat. Commun., 2021, 12, 395 CrossRef CAS PubMed.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- M. Moradzaman and G. Mul, J. Phys. Chem. C, 2021, 125, 6546–6554 CrossRef CAS PubMed.
- Z.-Z. Wu, X.-L. Zhang, Z.-Z. Niu, F.-Y. Gao, P.-P. Yang, L.-P. Chi, L. Shi, W.-S. Wei, R. Liu, Z. Chen, S. Hu, X. Zheng and M.-R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 CrossRef CAS PubMed.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z.-J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed.
- Y. Kim, S. Park, S.-J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13, 4301–4311 RSC.
- X. Zhi, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem. Commun., 2021, 57, 9526–9529 RSC.
- X. Chang, J. Li, H. Xiong, H. Zhang, Y. Xu, H. Xiao, Q. Lu and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202111167 CAS.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed.
- K. Yao, J. Li, H. Wang, R. Lu, X. Yang, M. Luo, N. Wang, Z. Wang, C. Liu, T. Jing, S. Chen, E. Cortés, S. A. Maier, S. Zhang, T. Li, Y. Yu, Y. Liu, X. Kang and H. Liang, J. Am. Chem. Soc., 2022, 144, 14005–14011 CrossRef CAS PubMed.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. López and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- T. Akter, H. Pan and C. J. Barile, J. Phys. Chem. C, 2022, 126, 10045–10052 CrossRef CAS.
- L. Wang, D. C. Higgins, Y. Ji, C. G. Morales-Guio, K. Chan, C. Hahn and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12572–12575 CrossRef CAS PubMed.
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- L. Wang, S. A. Nitopi, E. Bertheussen, M. Orazov, C. G. Morales-Guio, X. Liu, D. C. Higgins, K. Chan, J. K. Nørskov, C. Hahn and T. F. Jaramillo, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- N. S. Romero Cuellar, C. Scherer, B. Kaçkar, W. Eisenreich, C. Huber, K. Wiesner-Fleischer, M. Fleischer and O. Hinrichsen, J. CO2 Util., 2020, 36, 263–275 CrossRef CAS.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5, 706–719 CrossRef CAS.
- D. Shu, M. Wang, F. Tian, H. Zhang and C. Peng, J. CO2 Util., 2021, 45, 101444 CrossRef CAS.
- Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef CAS.
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935–2944 RSC.
- J. Li, K. Chang, H. Zhang, M. He, W. A. Goddard, III, J. G. Chen, M.-J. Cheng and Q. Lu, ACS Catal., 2019, 9, 4709–4718 CrossRef CAS.
- J. C. Bui, C. Kim, A. J. King, O. Romiluyi, A. Kusoglu, A. Z. Weber and A. T. Bell, Acc. Chem. Res., 2022, 55, 484–494 CrossRef CAS PubMed.
- E. Pérez-Gallent, C. Sánchez-Martínez, L. F. G. Geers, S. Turk, R. Latsuzbaia and E. L. V. Goetheer, Ind. Eng. Chem. Res., 2020, 59, 5648–5656 CrossRef.
- K. Eckhard, X. Chen, F. Turcu and W. Schuhmann, Phys. Chem. Chem. Phys., 2006, 8, 5359–5365 RSC.
- S. Lu, Y. Wang, H. Xiang, H. Lei, B. B. Xu, L. Xing, E. H. Yu and T. X. Liu, J. Energy Storage, 2022, 52, 104764 CrossRef.
- Y.-W. Choi, F. Scholten, I. Sinev and B. Roldan Cuenya, J. Am. Chem. Soc., 2019, 141, 5261–5266 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- H. Xiao, T. Cheng, W. A. Goddard, III and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- J. Li, X. Chang, H. Zhang, A. S. Malkani, M.-j. Cheng, B. Xu and Q. Lu, Nat. Commun., 2021, 12, 3264 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2nd edn, 2001, pp. 534–579 Search PubMed.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed.
- L. Xu, J. Feng, L. Wu, X. Song, X. Tan, L. Zhang, X. Ma, S. Jia, J. Du, A. Chen, X. Sun and B. Han, Green Chem., 2023, 25, 1326–1331 RSC.
- L. Ma, N. Liu, B. Mei, K. Yang, B. Liu, K. Deng, Y. Zhang, H. Feng, D. Liu, J. Duan, Z. Jiang, H. Yang and Q. Li, ACS Catal., 2022, 12, 8601–8609 CrossRef CAS.
- S. Chang, Y. Xuan, J. Duan and K. Zhang, Appl. Catal., B, 2022, 306, 121135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, calculation of the TOF and CORR efficiency, and total product distribution of each electrolysis. See DOI: https://doi.org/10.1039/d3gc00974b |
| This journal is © The Royal Society of Chemistry 2023 |