
Facet-dependent electrocatalytic oxidation activity of Co3O4 nanocrystals for 5-hydroxymethylfurfural†
Zhenchuan
Zhang
a,
Zhaohui
Yang
a,
Chenyang
Wei
a,
Zhenghui
Liu
 b and
Tiancheng
Mu
*a
b and
Tiancheng
Mu
*a
aDepartment of Chemistry, Renmin University of China, Beijing 100872, China. E-mail: tcmu@ruc.edu.cn
bDepartment of Chemistry, Taizhou University, Taizhou 318000, Zhejiang, China
First published on 14th September 2023
Abstract
Metal oxide catalysts’ performance depends on their crystal structure, including the surface arrangement and coordination of metal cations and oxygen anions on the exposed facets. Here, we fabricated Co3O4 nanocrystals (NCs) with predominantly exposed (110), (111), (112), and (114) facets in the shapes of nanorod (Co-R), hexagonal nanoplate (Co-H), nanolaminar (Co-L), and nanoparticle (Co-P), respectively. The Co3O4 NC with exposed (114) high-index facet exhibits the highest 5-hydroxymethylfurfural (HMF) electrocatalytic activity, while the Co3O4 NC with exposed (112) shows the lowest activity. Both density functional theory (DFT) calculations and experiments reveal that the primary factor impacting the catalytic performance of Co3O4 NCs is the difference in HMF adsorption energy on various crystal facets. By doping Cr into Co3O4 (114), we tuned the adsorption energy of HMF to an optimal level, thereby achieving the best catalytic performance. Based on this, the adsorption behavior of other aldehyde–alcohol organic small molecules was also evaluated, and the linear relationship between adsorption strength and catalytic activity was confirmed. This work demonstrates that adjusting the exposed crystal facets of Co3O4 can alter the adsorption and catalytic capabilities of aldehyde–alcohol organic small molecules. It also provides explicit knowledge that could improve our understanding of facet-dependent reactions for other metal oxide catalysts.
Introduction
The development of modern society results in an increasing demand for energy and bulk chemicals. Unfortunately, this demand leads to the rapid depletion of finite fossil resources such as coal, crude oil, and natural gas.1,2 Moreover, increasing carbon emissions and associated global warming cause detrimental changes in climate, which becomes a global issue that urgently needs to be addressed.3 Developing renewable and earth-rich biomass (such as lignocellulose, fats, and starch crops) as a substitute for fossil resources to establish sustainable supply chains is a promising strategy.4,5 The United States ranked 5-hydroxymethylfurfural (HMF) as one of the top 10 platform molecules derived from biomass, which can be oxidized to pharmaceutical intermediates, monomers, and agricultural chemicals.6,7 2,5-Furan-dicarboxylic acid (FDCA), derived from HMF, serves as the foundational monomer for producing poly(2,5-furan-dicarboxylic acid ethylene glycol ester) as a replacement for polyethene terephthalate based on petroleum products.8HMF electrocatalytic oxidation (HMFOR) is a promising approach to achieving high yields of FDCA under ambient temperature and pressure conditions. This method also presents a potential solution to reduce the cell voltage and overcome the sluggish oxygen evolution reaction (OER) for efficient hydrogen production.9,10 The adsorption/desorption behavior11,12 of reactant molecules plays a crucial role in this catalytic process and is closely related to the electrooxidation activity of HMF on the catalyst surface. For instance, Li et al.13 discovered that Mn-doped NiS nanosheets directly grown on a 3D graphite felt substrate serve as efficient electrocatalysts for the electrooxidation of HMF to FDCA in H-cell systems at an industrial-level current density (500 mA cm−2). The enhanced catalytic activity can be attributed to the incorporation of Mn into the NiS material, which enhances adsorption and effectively promotes the electrooxidation performance of HMF. Lu et al.14 proposed a Co-doping strategy to modulate the surface electronic structure of β-Ni(OH)2 for HMFOR. Experimental and theoretical calculations demonstrated that 10% Co doping effectively facilitated the formation of active species Ni3+–O and increased the adsorption energy of HMF and OH−, thereby enhancing the catalytic activity. Our group's research15 further indicated that adjusting the HMF adsorption energy is crucial for achieving optimal catalytic activity, especially given the competitive adsorption between OH− and HMF molecules at the limited reaction sites of HMFOR. Therefore, HMF adsorption of different strengths on catalysts can influence HMFOR activity.
In general, the performance of heterogeneous catalysts is determined by their composition, shape and structure on the surface.16 Exposed facets are a crucial factor of surface structure that play a decisive role in determining the physicochemical properties of catalysts and their performance. For example, Li et al.17 developed a two-step redox strategy to reconstruct the surface atomic arrangement of copper foam. By combining various quasi-in situ/in situ techniques and DFT calculations, it was demonstrated that the key factor controlling the reaction is the surface effect of the metal Cu crystal: the Cu (110) surface is the most favorable surface for enhancing the chemical adsorption of reactants and selectively electrochemically hydrogenating HMF into 2,5-bis(hydroxymethy)furan, while the Cu (100) surface may lead to the accumulation of a byproduct, 5,50-bis(hydroxymethyl) furan. Metal oxides can expose different crystal facets due to their various morphologies, and the exposed facets primarily determine their performance.18,19 For example, Co3O4 nanorods are more active than Co3O4 nanoparticles in the oxidation of CO and NO. Researchers attributed the excellent activity of the Co3O4 nanorods to the abundant Co3+ sites active on the predominantly exposed {110} facet.20,21 Xiao et al.22 have reported that the (111) facet of Co3O4 has better electrochemical properties than other facets. The underlying mechanisms behind the different catalytic activities of various crystal facets remain elusive. Moreover, the role of crystal facets in modulating the electrocatalytic performance of aldehyde–alcohol organic small molecules has been less investigated. Hence, unraveling the effects of crystal facets at the micro level is crucial for designing and optimizing nanocrystal catalysts.
In this study, we employed HMF as a model substrate of aldehyde–alcohol organic small molecules, and investigated the role of crystal facets in HMFOR by using Co-R, Co-H, Co-L, and Co-P as catalysts. By combining experimental observations with DFT calculations, diverse crystal facets exhibit different HMF adsorption energies, which is a crucial factor affecting catalytic activity. To further confirm the dominance of adsorption energy on crystal facets, we applied minor Cr-doping of Co-P to adjust the HMF adsorption strength on the (114) facet of Co3O4. Our results show that on an undamaged (114) facet of Co3O4, the HMF adsorption strength and catalytic activity of Co3O4 NCs can be described by an approximate volcano curve, with 1.2 wt% Cr-doped Co-P exhibiting the highest catalytic activity, consistent with adsorption energy as an activity descriptor.15 Based on this, we employed open-circuit potential (OCP) tests to evaluate the adsorption of small organic molecules that possess aldehyde and alcohol groups. The results reveal a positive correlation between adsorption strength and catalytic activity. Thus, the decisive influence of the adsorption strength of aldehyde–alcohol organic small molecules on Co3O4 crystal facets on catalytic activity is demonstrated. This study provides valuable insights into understanding the catalytic mechanism of crystal facets and offers guidance for designing catalysts with highly active exposed facets in the future.
Results and discussion
Characterization of Co3O4 NCs with different shapes
Uniform Co3O4 nanorods (Co-R), hexagonal nanoplates (Co-H), and nanolaminars (Co-L) were prepared by hydrothermal synthesis with predominantly exposed (110), (111) and (112) facets, respectively. Co3O4 nanoparticles (Co-P) with mainly exposed (114) were synthesized by the one-step precipitation approach. The micromorphologies of prepared catalysts were investigated using scanning electron microscope (SEM) and transmission electron microscope (TEM), as shown in Fig. 1. Measurement of the lattice spacing and the lattice angle by TEM allows us to determine the exposed facets of all the catalysts using the reported method of crystal facet determination.23 Co-R's average diameter and length are around 10–20 and 300 nm, respectively (Fig. 1a, a1). The lattice fringe spaces over Co-R are 0.243 and 0.233 nm in the TEM image in Fig. 1a2, which corresponds to the (31−1) and (2−22) facets of the Co3O4, respectively. It is evident from this that the Co-R sample mainly exposes {110} facets, which are perpendicular to both (31−1) and (2−22) facets. Co-H material has an average edge length and thickness of around 150–250 and 15–20 nm, respectively (Fig. 1b and b1). According to the TEM image, the Co-H sample primarily exposes {111} facets that are perpendicular to both (022) and (220) facets (Fig. 1b2). Co-L has a layered structure with a length of about 1–6 μm. The thickness is approximately 10–20 nm, and two exposed {112} facets are perpendicular to two groups of orthogonal (2−20) and (222) facets (Fig. c, c1, and c2). The Co-P material has a uniform size of about 10–15 nm (Fig. 1d and d1). It can be determined from Fig. 1d2 by TEM that Co-P primarily exposes {114} high-index facet (perpendicular to (2−20) and (311) facets). Fig. 1a3–d3 shows the surface atomic structure model of (110), (111), (112), and (114) facets, respectively. The success in controlling the synthesis of Co3O4 with the various morphologies allowed us to obtain different exposed facets. More information on the shape can be found in Fig. S1.† In contrast to the other two catalysts, Co-R and Co-P exhibit a wide diffraction ring in their selected area electron diffraction (SAED) pattern, which is indicative of low crystallinity.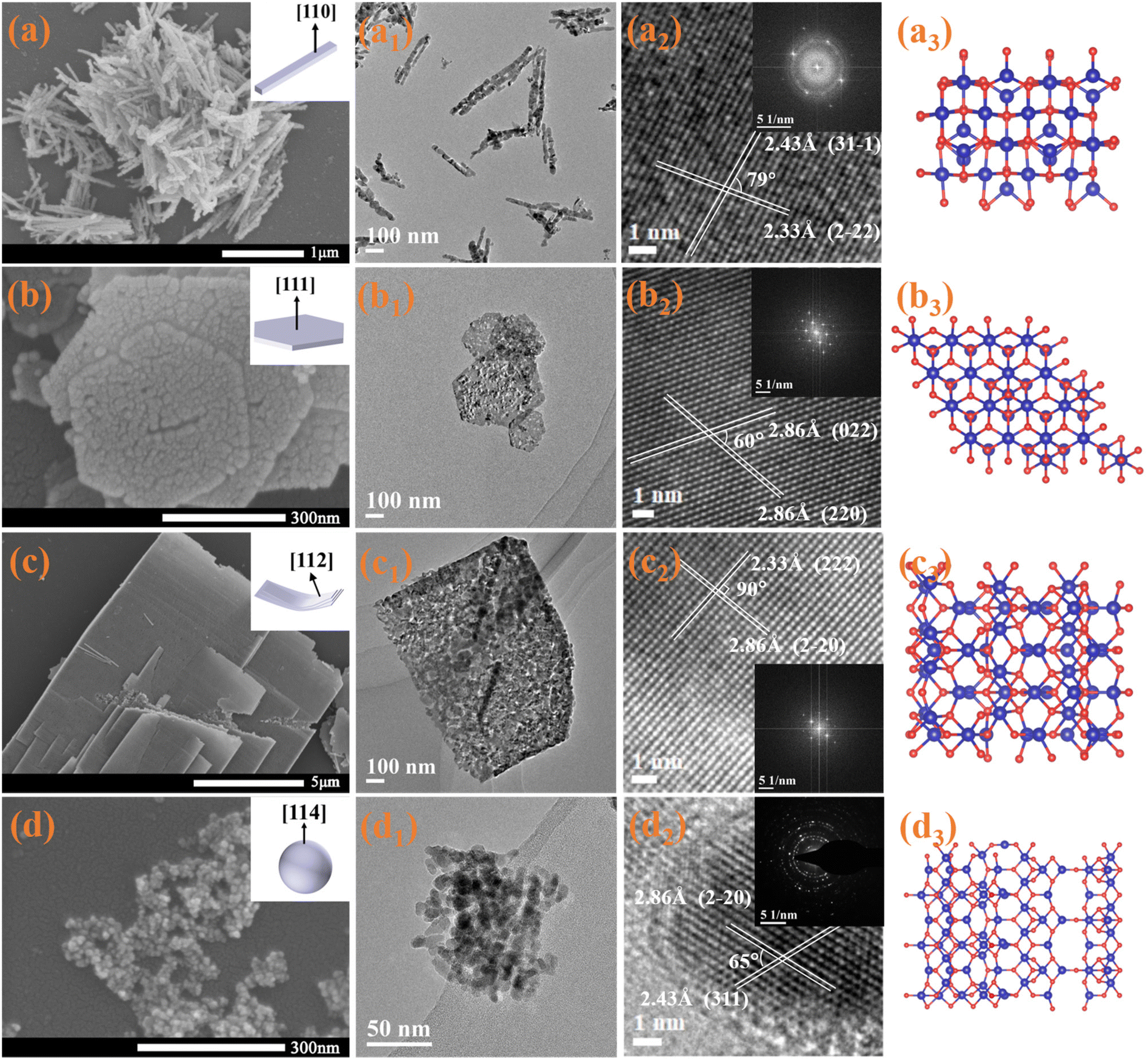 | ||
| Fig. 1 SEM and TEM images of Co-R (a and a1), Co-H (b and b1), Co-L (c and c1) and Co-P (d and d1) (insets show the corresponding 3D models of Co3O4). a2, b2, c2 and d2 show the corresponding high-resolution images and SAED patterns. Surface atomic structure models of (110), (111), (112) and (114) facets (a3, b3, c3 and d3). | ||
The four catalysts exhibit comparable X-ray diffraction (XRD) patterns (Fig. S1e†) and are indexed as face-centered-cubic Co3O4 structures (space group Fd3m). For all samples, the (111), (220), (311), (222), (400), (422), (511), and (440) planes attributable to Co3O4 (PDF #42-1467) can be seen. Co-P's relatively large peak width indicates its comparatively small grain size. The XRD patterns show no other crystalline impurities or intermediates. X-ray photoelectron spectroscopy (XPS) analysis shows the detailed states of elements on the surface of the Co3O4 samples. Only Co, C, and O can be seen in the entire XPS survey spectra, as shown in Fig. S1f,† proving that all catalysts are pure cobalt oxides. According to Fig. S2a,† the Co 2p bonding states of Co-R, Co-H, Co-L, and Co-P are almost identical. The strong peaks observed at approximately 795.2 eV and 779.7 eV correspond to the Co 2p3/2 and Co 2p1/2 spin–orbit peaks of Co3O4, respectively. Co3+ and Co2+ component peaks at approximately 779.9 eV and 781.2 eV, respectively, can be further deconvoluted from the Co 2p3/2 spectra. The coexistence of Co3+ and Co2+ species is shown, with no significant shakeup satellite peaks observed in Co 2p, indicating the formation of Co3O4 phase.24
Electrochemical measurements of different Co3O4 NCs on HMF oxidation
The performance of the synthetic materials as an electrocatalyst for the oxidation of HMF was tested in a typical three-electrode device. The material was applied to a carbon cloth working electrode with a loading of 1 mg cm−2. Linear scanning voltammetry curves (LSV) were obtained by scanning at a rate of 5 mV s−1 with a 90% current compensation to minimize the effects of resistance. Fig. 2a shows the LSV curves of four Co3O4 NCs (Co-P, Co-H, Co-R, and Co-L) in a 1.0 M KOH solution with 10 mM HMF. The results show that the onset potential of Co-P is approximately 1.30 V, while the other Co3O4 NCs are around 1.35 V. Co-P exhibits the highest catalytic activity for the electrooxidation of HMF, with the catalytic activity order as follows: Co-P > Co-H > Co-R > Co-L. However, the density of electrochemically active sites for HMF electrooxidation varies due to differences in the morphologies/structures of the four Co3O4 NCs. To compare the intrinsic activities of Co3O4 NCs accurately, the geometric current densities were normalized by the electrochemically active surface area (ECSA). The ECSA represents the density of accessible sites, including those with defects.25 The results show that Co-P exhibited the largest Cdl of 13.44 mF cm−2, indicating its relatively high electrochemically active surface area (Fig. 2b). After normalization by ECSA, the intrinsic current density of Co3O4 NCs follows the same trend (Fig. S3†), indicating that the inherent catalytic activity for HMF electrooxidation follows the order of Co-P > Co-H > Co-R > Co-L. Accordingly, Co-P with exposed (114) facets possesses the highest intrinsic activity for the electrooxidation of HMF. The electron transfer rate corresponding to the Tafel slope was also calculated for a more detailed analysis. The results are shown in Fig. 2c. The Tafel slopes of Co-P, Co-H, Co-R, and Co-L for the HMFOR are calculated to be 126, 146, 160, and 258.2 mV dec−1, respectively. This finding suggests that Co-P is the most efficient catalyst in electron transfer rate, with a lower Tafel slope and a lower potential required to increase the HMF oxidation rate.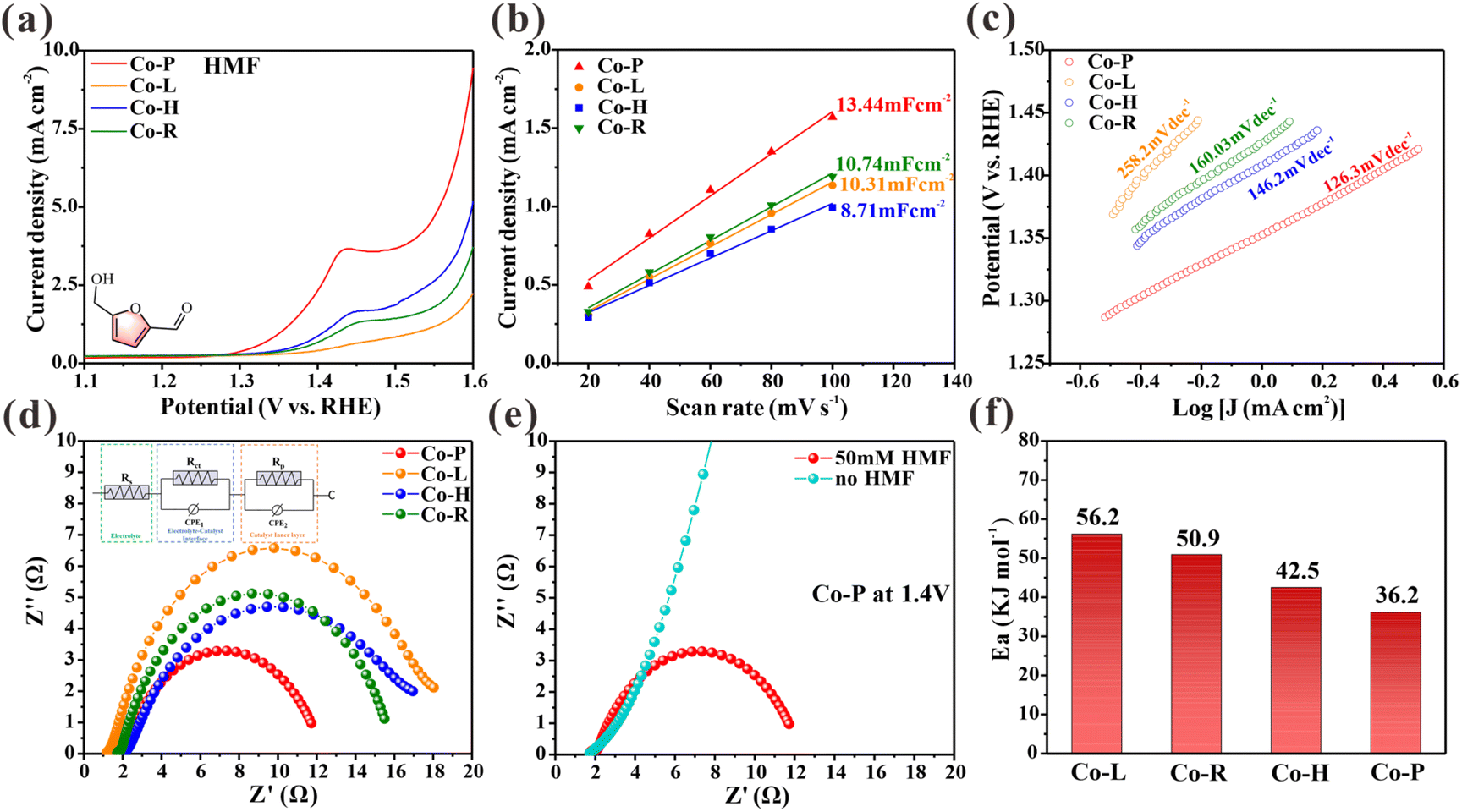 | ||
| Fig. 2 (a) LSV curves of Co-R, Co-L, Co-H and Co-P. (b) Charging current differences (Δj = janode − jcathode) plotted against scan rates for Co-R, Co-L, Co-H and Co-P. (c) Tafel slopes derived from LSVs. (d) Nyquist plots for Co-R, Co-L, Co-H and Co-P in 1.0 M KOH with 50 mM HMF at 1.4 V vs. RHE. (e) Nyquist plots for Co-P in 1.0 M KOH with and without 50 mM HMF at 1.4 V vs. RHE. (f) The average activation energy of Co-R, Co-L, Co-H and Co-P. | ||
In situ electrochemical impedance spectroscopy (EIS) was conducted at different potentials to analyze the catalytic kinetics of HMF electrochemical oxidation on the obtained electrode. An equivalent circuit was presented by fitting the Nyquist plots (Fig. S4†). Fig. 2d compares the Nyquist plots of the four Co3O4 NCs at 1.4 V. The electronic conductivity and charge-transfer rate of Co-P are better than other materials, as indicated by its low Rt value.26 In 1.0 M KOH at 1.4 V, Nyquist plots of Co-P with and without HMF were compared in Fig. 2e. Without HMF, a steep line is observed, indicating high charge transfer resistance and no occurrence of OER. With HMF, a semicircle is observed, indicating oxidation at 1.4 V. In addition, Fig. S5† compares the Nyquist plots of the four Co3O4 NCs electrodes at different voltages in 1.0 M KOH with 50 mM HMF. Nyquist plots exhibit approximated vertical lines at the low voltage of 1.0–1.15 V, indicating high charge transfer resistance. When the applied voltage increases to 1.3 V, there is a significant change in the Nyquist plots, with a complete semicircle appearing, indicating the start of a faradaic reaction. This suggests that HMF begins to oxidize at this potential. Additionally, the in situ Bode phase plots in Fig. S6† show a low-frequency peak (100 Hz–101 Hz), which is commonly associated with nonhomogeneous charge distribution,26 such as the formation of oxidizing species on the electrode surface. All catalysts examined exhibit an apparent transition peak at a potential of 1.6 V in the absence of HMF, suggesting the occurrence of OER. Upon adding HMF to the electrolyte, an earlier transition peak at 1.3 V is observed on the Co-P electrode, indicating its high catalytic activity, consistent with the onset potential obtained from the LSV curve (Fig. 2a).
Electrochemical reactions’ activation energy (Ea) provides valuable insight into the intrinsic activity of electrodes, as it depends solely on material composition and is not influenced by extrinsic factors such as surface area.27,28 The activation energy of the HMFOR was determined for Co3O4 NCs, as shown in Fig. 2f and Fig. S7–10.† The average Ea value of Co-P is found to be 36.17 kJ mol−1, which is lower compared to the Ea value of Co-H (42.52 kJ mol−1), Co-R (50.93 kJ mol−1) and Co-L (56.17 kJ mol−1). These results further confirm the exceptional intrinsic activity of the Co-P nanomaterial. The observed electrocatalytic activity order for the electrooxidation of HMF may be applicable to other organic small molecules containing aldehydes and alcohols, as the electrooxidation of HMF can be extended to such molecules.7,29 To test this hypothesis, we used furfural (FF), furfuryl alcohol (FA), benzaldehyde (BzH), benzyl alcohol (BnOH), and ethanol (EtOH) as representative small organic molecules containing both aldehydes and alcohols. The electrooxidation activities of these molecules were evaluated on four Co3O4 NCs using LSV curves (Fig. S11†). As expected, our results demonstrate that catalytic activity followed the sequence of Co-P > Co-H > Co-R > Co-L. Therefore, prepared Co3O4 NCs may exhibit a reaction mechanism similar to that of HMFOR for the electrocatalytic oxidation of small molecules containing aldehydes and alcohols.
Investigating the mechanism of HMF catalytic activities in different Co3O4 nanocatalysts
It has been previously reported that oxygen vacancies (OV) play a role in the electrooxidation of HMF in Co3O4 crystalline materials, as OH− ions can fill the vacancies through the lattice oxygen oxidation process, thereby accelerating the dehydrogenation of 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) intermediates.30 To gain insight into the causes of the varying catalytic properties of Co3O4 crystals with different crystal facets, X-ray photoelectron spectroscopy (XPS) data was analyzed. The O 1s XPS spectra of the prepared material are shown in Fig. S2b.† The peak, centered at 529.8 eV, corresponds to lattice oxygen (OLatt). The peak, centered at 531 eV, is attributed to low-coordination oxygen or oxygen that is associated with oxygen vacancies, indicating the presence of vacancies on the material.31 The OV/OLatt ratios for Co-P, Co-H, Co-R, and Co-L are determined to be 0.40, 0.33, 0.36, and 0.40, respectively (Fig. 3b and Table S1†). Only minor differences in the OV/OLatt ratios are found, and no correlation is found with the order of catalyst activity. To further investigate, electron paramagnetic resonance (EPR) tests were performed, which revealed a representative EPR signal of g = 2.003 in all Co3O4 crystalline materials, indicating the presence of electrons trapped in oxygen vacancies (Fig. 3a).32,33 The results of the XPS and EPR show that the synthesized Co3O4 NCs with different exposed facets have similar oxygen vacancy contents and do not correlate with their catalytic activity order. Therefore, oxygen vacancies are not the main factor causing differences in catalytic activity among Co3O4 nanocrystals. Further investigation is needed to elucidate the underlying mechanism responsible for their varying activity.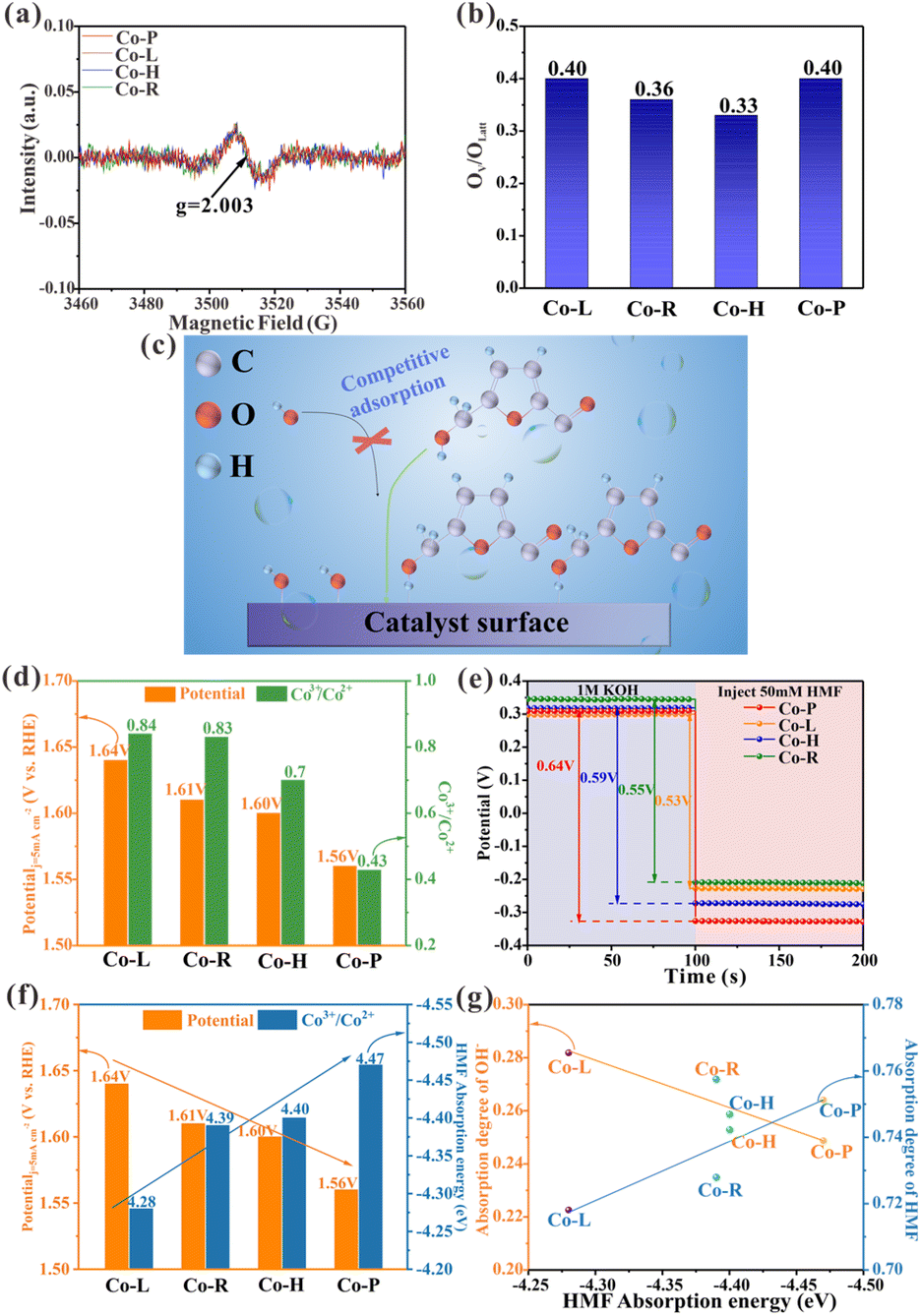 | ||
| Fig. 3 (a) EPR spectra of Co-R, Co-L, Co-H and Co-P. (b) The OV/OLatt ratio of Co-R, Co-L, Co-H and Co-P from XPS spectra. (c) Competitive adsorption between HMF and OH− in the schematic. (d) Histogram showing the correlation between Co3+/Co2+ ratio from XPS spectra and HMF electrooxidation activity. (e) OCP curves of Co3O4 NCs in 1.0 M KOH and 50 mM HMF was injected subsequently. (f) Histogram showing the correlation between adsorption energy on Co3O4 NCs and HMF electrooxidation activity. (g) Adsorption degree of OH− and HMF on Co3O4 NCs as HMF adsorption energy rises. | ||
Adsorption/desorption behavior is a critical step in catalytic reactions. Electrooxidation of aldehyde–alcohol molecules typically employs OHads as a significant intermediate, similar to OER.29,34 OH− and HMF molecules are adsorbed and combined on the surface of the electrode during oxidation.31 OHads extract H+ from HMFads, activating and breaking C–H/O–H bonds to facilitate dehydrogenation during the process of oxidation.7,35 Weak HMF adsorption on the catalyst surface results in higher free energy required for dissociating C–H/O–H bonds, unfavorable for its oxidation.36,37 Transition metal catalysts typically utilize metal sites on their surface for the adsorption of HMF and OH−.34,38 HMF and OH− compete for adsorption at a limited number of reaction sites (Fig. 3c). Strong HMF adsorption crowds out OH− from these sites and limits OHads formation, hindering HMF oxidation.39 The adsorption equilibrium of HMF and OH− on the electrode surface is crucial for the electrooxidation of HMF since they are competitive adsorption species.15 A recent study by Lu et al. suggests that Co2+ in Co3O4 can chemisorb on HMF, while Co3+ plays a catalytic oxidation role in the HMF oxidation process.8 The diverse levels of Co2+ and Co3+ on the surface may be the key to the problem due to the different surface atomic ordering of Co3O4 nanocrystals with different crystalline exposed facets. The Co 2p XPS spectra analysis (Fig. S2a†) reveals that the surface Co3+/Co2+ ratios of four Co3O4 nanocrystals (i.e., Co-L, Co-R, Co-H and Co-P) were 0.84, 0.83, 0.7 and 0.34, respectively, which correlated with the observed catalytic activity trend (Fig. 3d and Table S1†). The lower Co3+/Co2+ ratios are attributed to having more Co2+ capable of chemisorption to HMF molecules or playing less Co3+ to catalytic oxidation in HMF oxidation. Based on the analysis of the Co3+/Co2+ ratios of the four Co3O4 NCs, it is clear that the better catalytic activity of Co-P with exposed (114) crystal facet compared to the other Co3O4 NCs comes from the fact that the catalyst surface contains more Co2+. This leads to a more substantial adsorption capacity and thus increased catalytic activity. Notably, relatively abundant Co3+ catalytic sites were found on all catalysts. This is because the catalytic activity increased gradually with decreasing Co3+/Co2+ ratio. Therefore, there is no case where the catalytic activity decreases due to insufficient Co3+ catalytic sites. In summary, we proposed a hypothesis that the difference in surface adsorption capacity is the key to the varying HMF catalytic activities of the four Co3O4 NCs with distinct exposed crystal facets.
The open-circuit potential (OCP) measurements were measured to correlate the adsorption strength of HMF on Co3O4 NCs with varying crystal facets to their catalytic activity. Reduced OCP values can suggest changes in adsorbates within the Helmholtz layer, with a greater decrease indicating more robust adsorption.40 As shown in Fig. 3e, we injected 50 mM HMF and measured OCP, which decreased as follows: Co-P (0.64 V) > Co-H (0.59 V) > Co-R (0.55 V) > Co-L (0.53 V). This indicates that HMF adsorption strength on these four Co3O4 NCs varies in the same order: Co-P > Co-H > Co-R > Co-L. However, OCP measurements are only a rough indicator of adsorption behavior. To provide a theoretical insight into the mechanism, DFT calculations were carried out by adopting (110), (111), (112) and (114) facets of Co3O4 as model slabs (Fig. S12†). Fig. S13† illustrates the adsorption configuration of HMF and its associated adsorption energy (Eads). The calculated HMF adsorption energies on the surface of Co3O4 (112), Co3O4 (110), Co3O4 (111), and Co3O4 (114) are −4.28 eV, −4.39 eV, −4.4 eV, and −4.47 eV, respectively, in agreement with the OCP tests. When arranging the Co3O4 NCs based on increasing HMF adsorption energies, their ability to produce a current density of 5 mA cm−2 decreases, which aligns with the catalytic activity order shown in Fig. 3f. The adsorption of OH− was also studied on these four Co3O4 NCs by DFT calculations (Fig. S14†). We calculated the ratio of HMF or OH− adsorption energy to their sum [Eads, HMF/(Eads, HMF + Eads, OH−)] for HMF and [Eads, OH−/(Eads, HMF + Eads, OH−)] for OH− to compare their adsorption degree on the catalyst surface.15Fig. 3g shows that the HMF adsorption degree increases and the OH− adsorption degree decreases as HMF adsorption energy increases. This reveals a competition between HMF and OH− for adsorption sites on the catalyst surface. HMF oxidation is inefficient when HMF adsorption is weak, even with a high OH− adsorption ratio. This suggests that HMF adsorption is more important than OH− adsorption in most cases. Based on previous reports15 and the above analysis, if the adsorption energy is the key factor for the activity difference of Co3O4 electrocatalysis HMF on different exposed facets, then the formation of OHads intermediates will be affected as the HMF adsorption energy increases further, leading to a decrease in catalytic activity (Fig. 3c). This became the direction of our subsequent study.
Doping is a simple strategy that can be used to adjust the adsorption strength of substrate molecules on catalysts through electronic interactions.41 It has been reported that doping Cr into cobalt phosphide can affect the electronic structure of cobalt phosphide and thus regulate the free energy of hydrogen adsorption, thereby improving the efficiency of hydrogen evolution reaction (HER).42 Ali et al.43 found that 6at% Cr-doped Co3O4 exhibited good electrochemical performance. In both studies, Cr doping enhances the stability of the materials, and the Cr3+ ion radius is close to that of Co3+, which can better maintain the stability of the structure. Therefore, Cr doping is beneficial to adjust the adsorption strength of HMF while maintaining the same crystal facet.
Modulating the adsorption strength of HMF on Co3O4 through Cr doping
Fig. 4a and b shows the nanoparticles structure of 1.2 wt% Cr-doped Co-P. As shown in Fig. S15–17,† the spherical shape can be maintained up to a Cr content of 2.1 wt% by adjusting the amount of Cr during the nanoparticle synthesis. For higher Cr loadings, more plate-like structures are observed, which affects the exposure of the Co3O4 (114) crystal facet and therefore are not considered for further discussion. XRD patterns indicate the formation of cubic Co3O4 (PDF#42-1467) (Fig. S18†), and the incorporation of Cr atoms did not alter the crystal structure of Co3O4. Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis reveals the synthesis of Co-P doped with 0.6 wt%, 1.2 wt%, and 2.1 wt% Cr (denoted as 0.6% Cr-Co-P, 1.2% Cr-Co-P, and 2.1% Cr-Co-P, respectively) (Table S2†). Energy-dispersive X-ray spectroscopy (EDS) mapping shows the uniform distribution of all elements, including Co, Cr, and O (Fig. 4d–d3). The corresponding high-resolution transmission electron microscopy (HRTEM) image in Fig. 4c displays easily distinguishable lattice fringes with a spacing of 0.243 nm and 0.286 nm, and a lattice angle of 65°, which corresponds to the Co3O4 (114) crystal facet. In fact, due to the small amount of Cr doping, the lattice spacing of M% Cr-Co-P only slightly changed, and the exposed (114) crystal facet remained undisturbed. The relevant information of 0.6% Cr-Co-P and 2.1% Cr-Co-P is shown in Fig. S15 and 16.†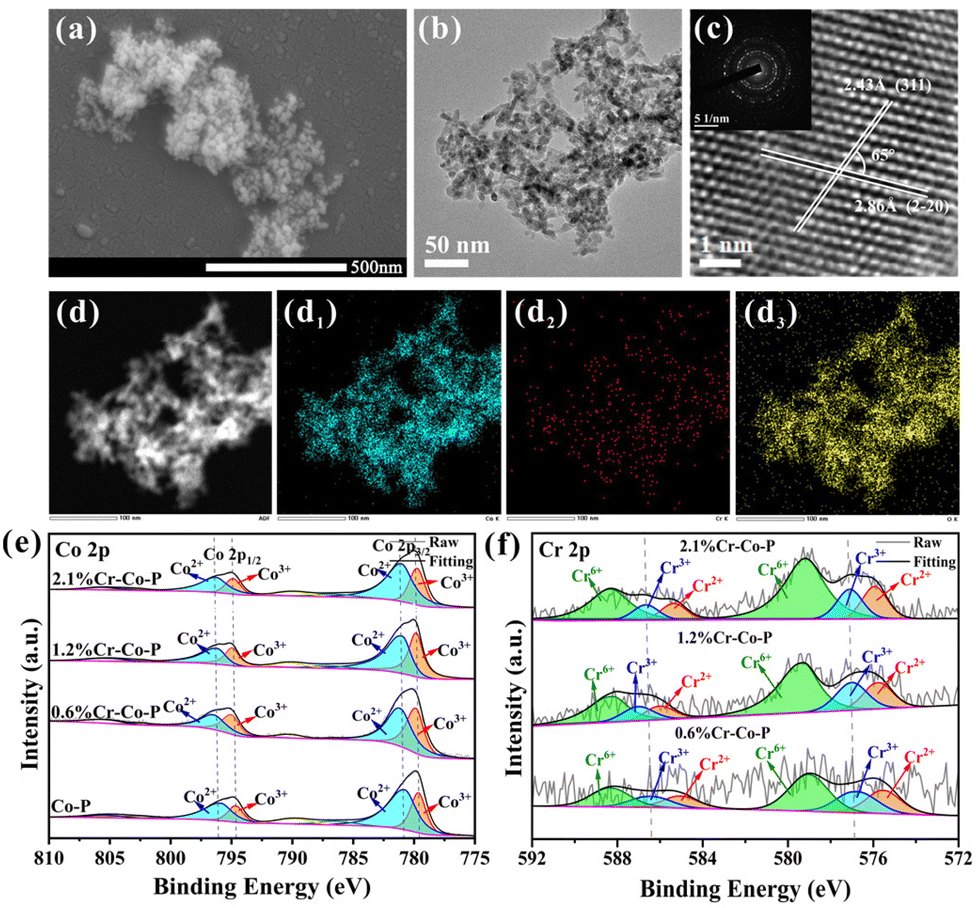 | ||
| Fig. 4 SEM and TEM images of 1.2% Cr-Co-P (a and b). (c and inset c) the corresponding high-resolution images and SAED patterns. (d–d3) Corresponding elemental mappings of 1.2% Cr-Co-P with Co (d1), Cr (d2) and O (d3). (e) Co 2p and (f) Cr 1s XPS spectra of M% Cr-Co-P. | ||
The XPS measurements were conducted to analyze the chemical makeup and electronic interactions of the surface in M% Cr-Co-P (Fig. 4e and f and Fig. S19†). Our primary objective was to use XPS data to understand how doping impacts the electronic structure of Co3O4. Fig. 4e and f show that Cr doping results in peak shifts in the XPS spectra, which become more pronounced with increasing Cr content, indicating more electron transfer and stronger electronic interactions.41 Due to the lower electronegativity of Cr than Co, Co has a more vital electron-attracting ability. Thus, electrons are more likely to transfer from Cr to Co, which explains why the XPS peaks of Co and Cr shifted to higher binding energies. Electronic interactions are crucial for adjusting the electronic environment of metal centers, which is necessary to tune the HMF adsorption energy on the surface of Co3O4 following Cr doping.41
LSV evaluated the electrocatalytic performance of HMF on doped and undoped Co3O4 with Cr in 1.0 M KOH with 10 mM HMF. Fig. 5a shows that Cr-doped Co3O4 has higher activity than undoped Co3O4, suggesting that modifying the electronic configuration of Co3O4 through Cr doping has a beneficial impact on HMF oxidation. With increased Cr content, the catalytic performance first increased and then decreased, with the 1.2% Cr-Co-P exhibiting the best catalytic activity. Oxygen evolution reaction (OER) is a primary rival reaction for HMF electrooxidation in the aqueous solution. As shown in Fig. 5b, the LSV curve of 1.2% Cr-Co-P can achieve a catalytic current density of 10 mA cm−2 at 1.43 V, which is about 184 mV lower than the voltage when OER reaches the same current density. Cdl was evaluated by CV measurement (Fig. S20†) to study ECSA. 1.2% Cr-Co-P shows the smallest Cdl of 9.75 mF cm−2 (Fig. 5c), indicating its smaller ECSA value. The EIS measurement results are shown in Fig. 5d. The fitted charge transfer resistance (Rct) of 1.2% Cr-Co-P is the lowest among the tested catalysts. In summary, 1.2% Cr-Co-P has the smallest active site density and impedance similar to other M% Cr-Co-P, but it exhibits the highest activity for HMF electrooxidation. This implies that there are additional factors that positively impact the electrooxidation of HMF.
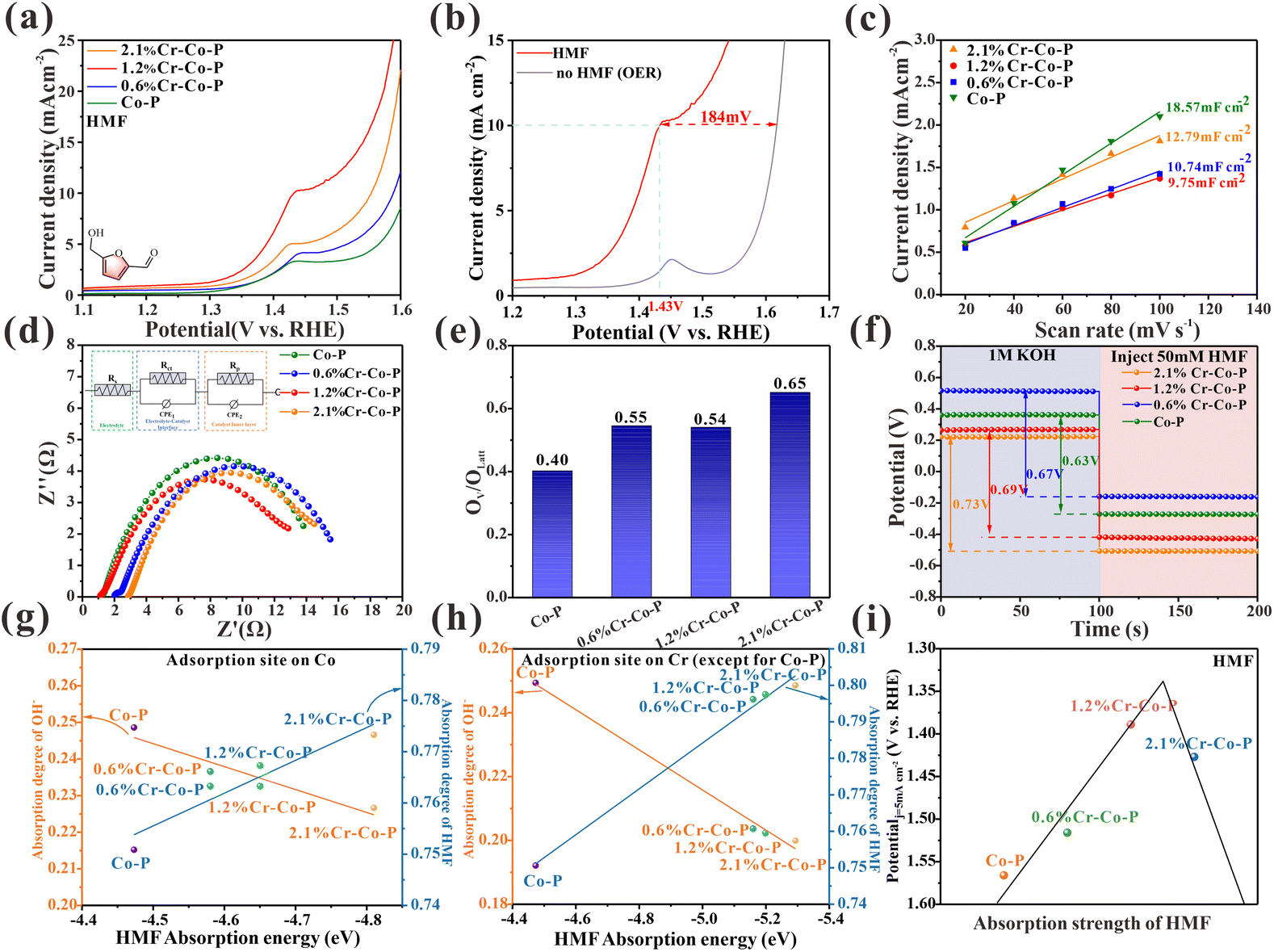 | ||
| Fig. 5 (a) LSV curves of M% Cr-Co-P. (b) The LSV curves of 1.2% Cr-Co-P in 1.0 M KOH with and without 10 mM HMF at a scan rate of 5 mV s−1. (c) Scanning rates for M% Cr-Co-P displayed versus charging current differences (j = janode − jcathode). (d) Nyquist plots for M% Cr-Co-P in 1.0 M KOH with 50 mM HMF at 1.4 V vs. RHE. (e) The OV/OLatt ratio of M% Cr-Co-P from XPS spectra. (f) OCP curves of Co3O4 NCs in 1.0 M KOH and 50 mM HMF was injected subsequently. (g) Adsorption degree of OH− and HMF on M% Cr-Co-P with the increase of HMF adsorption energy (adsorption site only on Co). (h) Adsorption degree of OH− and HMF on M% Cr-Co-P with the increase of HMF adsorption energy (adsorption site on Cr). (i) Plotting the electrooxidation of HMF on M% Cr-Co-P using a volcanic plot. | ||
Given the assumptions presented earlier, the discussion in this section focuses on the adsorption energy of HMF. To rule out the effect of oxygen vacancies, we examined the O 1s XPS spectra (Fig. S19) of M% Cr-Co-P samples. As shown in Fig. 5e and Table S3,† The OV/OLatt ratios for Co-P, 0.6% Cr-Co-H, 1.2% Cr-Co-R, and 2.1% Cr-Co-L are determined to be 0.40, 0.55, 0.54, and 0.65, respectively (Fig. 5e and Table S3†). Cr doping enhances the oxygen vacancy formation, but there is no clear relationship between the OV/OLatt ratio and the catalytic performance. Moreover, 2.1% Cr-Co-P, which exhibits the highest OV/OLatt ratio, do not show superior activity among all catalysts. Hence, oxygen vacancy concentration is not a key factor determining catalytic activity. We measured OCP to study the HMF adsorption strength on M% Cr-Co-P. As shown in Fig. 5f, when 50 mM HMF was injected, the OCP of the electrochemical cell decreased more significantly with increasing Cr content, indicating that the surface adsorption of HMF on Co3O4 gradually strengthened. Density functional theory (DFT) calculations were used to verify our findings theoretically. There are two scenarios that need to be considered due to the small amount of doping. In the first scenario, the adsorption site is a Co2+ atom, and its chemical state is affected by the Cr3+ doping around it, which influences its adsorption strength for HMF. In the second scenario, the adsorption site is the Cr3+ atom, which loses electrons due to the Cr doping on the surface of Co3O4, resulting in a positive charge relative to the surrounding Co atoms and a stronger adsorption ability for HMF. Fig. S21 and S23† show that the HMF adsorption energy decreases with increasing Cr doping for both scenarios, indicating enhanced HMF adsorption ability. The OH− adsorption energy shows little variation with Cr doping for both scenarios (Fig. S22, and S24†). Fig. 5g and h illustrate the correlation between HMF adsorption energies and adsorption degree of HMF and OH− in two different scenarios. It is evident that both scenarios exhibit a similar trend, and HMF and OH− exhibit competitive adsorption. Therefore, more Cr doping on the Co3O4 (114) crystal facet within a certain range of doping amounts will lead to stronger surface adsorption of HMF. The difference in charge density (Fig. S25†) reveals that electrons transferred from Cr to Co atoms, which affects the electronic distribution of the entire surface. This indicates that Cr doping effectively adjusts the electronic structure of the adsorption site, thereby enhancing its adsorption of HMF. However, over-doping can cause HMF to be strongly adsorbed, leading to reduced performance. This is due to a decrease in the adsorption of OH− and insufficient formation of OHads, as shown in Fig. 5g and h. The relationship between adsorption energy and catalytic activity in M% Cr-Co-P is an approximate volcano curve (Fig. 5i). This activity trend still exists even after ECSA normalization (Fig. S26†). Based on previous reports, experiments and theoretical calculations, we confirmed that the difference in HMF catalytic performances is mainly responsible for the diverse surface adsorption strength of Co3O4 with exposed different crystal facets. Based on this conclusion, we measured the overall adsorption strength of FF, FA, BzH, BnOH and EtOH in four Co3O4 NCs using OCP tests. The results show that the adsorption energy is proportional to the catalytic activity (Fig. S11, and S27†). This indicates that the adsorption strength of organic small molecules containing aldehydes and alcohols plays a dominant role in the crystal facet-dependent catalytic reaction of Co3O4.
Conclusions
In short, we synthesized four Co3O4 nanocrystals (NCs) with similar oxygen vacancy concentrations but different exposed facets. Combining density functional theory (DFT) calculations with experiments, we found that the catalytic activity of 5-hydroxymethylfurfural (HMF) on Co3O4 exposed facets was proportional to the surface adsorption energy. By doping trace amounts of Cr, we tuned the adsorption energy of the catalyst surface while maintaining the same (114) facet. Based on the previously reported active descriptor of HMF, we demonstrate that the HMF adsorption strength causes the difference in catalytic activity on different facets. By selectively exposing specific facets of Co3O4, the catalytic performance of the HMF electrocatalytic oxidation reaction (HMFOR) can be improved, and further enhancement can be achieved by adjusting the surface adsorption energy of facets. In addition, the conclusion of this work can be extended to more electrocatalytic reactions of organic small molecules containing aldehydes and alcohols. This work provides new insights into the effect of facet on electrocatalysis and is helpful for designing high-performance electrocatalysts.Experimental sections
Chemicals
5-Hydroxymethylfurfural (C6H6O3, 99%, Aladdin Biochemical Technology Co., Ltd), 5-hydroxymethyl-2-furancarboxylic acid (C6H6O4, 98%, Innochem Co., Ltd), 2,5-diformylfuran (C6H4O3, 98%, Innochem Co., Ltd), 5-formylfuran-2-carboxylic acid (C6H4O4, 98%, Innochem Co., Ltd), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 98%, Innochem Co., Ltd), ethyl alcohol (C2H6O, 99%, Innochem Co., Ltd), ethylene glycol (C2H6O2, 99%, Innochem Co., Ltd), ammonium hydroxide (NH3·H2O, AR, Macklin Biochemical Co., Ltd), polyvinylpyrrolidone (K30, TCL Co., Ltd), sodium hydroxide (NaOH, AR, Macklin Biochemical Co., Ltd), urea (CH4N2O, 99%, Innochem Co., Ltd), hexadecyl trimethyl ammonium Bromide (C19H42BrN, AR, SCR Co., Ltd), glycerol (C3H8O3, 99.5%, Innochem Co., Ltd), sodium carbonate anhydrous (Na2CO3, AR, SCR Co., Ltd), KOH (95%, Macklin Biochemical Co., Ltd), furfural alcohol (C5H6O2, 99%, SCR Co., Ltd), furfural (C5H4O2, 99%, SCR Co., Ltd), benzaldehyde (C7H6O, AR, SCR Co., Ltd), benzyl alcohol (C7H8O, AR, SCR Co., Ltd). Chromium(III) chloride hexahydrate (CrCl3·6H2O, AR, SCR Co., Ltd). Deionized water (DI, resistivity: ∼18 MΩ cm) was used in all experiments.Synthesis of Co3O4-110 nanorods
A typical synthesis44 involved combining 10 mL of NH3 (aq) and 25 mL of C2H6O2. The solution was then treated with 1.5 mL of 1 M Na2CO3 (aq) and stirred magnetically for several minutes. Next, 5 mL of 1 M Co(NO3)2 (aq) was added to the mixture while stirring continuously for 20 min. The sample was placed into a Teflon-lined stainless steel autoclave (50 mL) and heated to 170 °C, remaining for 17 h. After centrifuging the cooled suspension, the precipitate was repeatedly washed with deionized water and ethanol and then dried at 60 °C under vacuum to produce Co(CO3)0.5(OH)0.11·H2O nanorods. Upon calcination in air at 300 °C for 3 h, Co3O4 nanorods were obtained.Synthesis of Co3O4-111 hexagonal nanoplatelets
A typical procedure45 involved dissolving 1.2 g of Co(NO3)2·6H2O in 5 mL of deionized water and 5 mL of ethanol. Then, 1 g of polyvinylpyrrolidone was added as a surfactant, and the mixture was stirred for 30 min. Next, 25 mL of 0.4 M NaOH (aq) was slowly added dropwise, taking about 90 min for the solution to change from light red to dark green. The suspension was then transferred into a 50 mL Teflon-lined autoclave and heated to 120 °C for 10 h. After cooling, the precipitate was centrifuged and rinsed with ethanol and deionized water to obtain β-Co(OH)2. By annealing the β-Co(OH)2 precursor at 450 °C for 2 h in air, the final Co3O4 nanocrystals were produced.Synthesis of Co3O4-112 nanolaminars
Using a facile hydrothermal approach,46 we synthesized Co3O4 nanolaminars with highly-exposed {112} facets by calcining Co2(OH)2CO3 precursor. Briefly, 8 mmol of urea (CO(NH2)2), 2 mmol of Co(NO3)2·6H2O, and 0.4 g of hexadecyl trimethyl ammonium bromide as a soft template were dissolved in 40 mL of deionized water and stirred for 30 min. The mixture was loaded into a Teflon-lined autoclave (50 mL) and heated to 140 °C for 12 h. The suspension was cooled to room temperature and then centrifuged. The resulting solid was washed multiple times with deionized water and ethanol. The final mesoporous Co3O4 nanolaminars were obtained by calcining the Co2(OH)2CO3 precursor in air at 450 °C for 2 h.Synthesis of Co3O4-114 nanoparticles
A one-step precipitation method47 was used to prepare the catalyst. Firstly, a 1 M Co(NO3)2 aqueous solution was created by dissolving Co(NO3)2·6H2O in 20 ml of deionized water. Glycerin was then added to the solution at a molar ratio of n (C3H8O3) to n (Co) of 1, and the solution was stirred until complete dissolution. Next, a 1 M Na2CO3 solution was added to the resulting suspension to adjust the pH to 9, after which the mixture was stirred at room temperature for 1 h. The mixture was filtered and rinsed with ethanol and deionized water. Finally, the sample was dried for 12 h at 70 °C in an oven to obtain Co3O4 nanoparticles.To create Cr-doped Co3O4 catalysts, the same synthesis method was used, with the addition of Chromic chloride hexahydrate (CrCl3·6H2O, Aldrich) in solution at an adjusted molar concentration. The resulting mixture was then processed similarly to the Co3O4 nanoparticles.
Electrochemical measurements
Electrochemical experiments were conducted using a CHI 660E electrochemical analyzer, with a single three-electrode cell used unless otherwise specified. To ensure HMF molecules were fully adsorbed onto the electrode surface before electrochemical testing, stir the dissolved electrolyte with HMF for a few minutes in the electrochemical cell with a working electrode. The counter and reference electrodes used were Pt foil and Hg/HgO (1.0 M KOH). As for all catalysts, the working electrode was a carbon cloth electrode loading catalyst with a surface area of 1 cm × 1 cm. Typically, 3 mg of catalyst was dissolved in 400 μL deionized water, 480 μL ethanol, and 20 μL of Nafion solution (5 wt%) to create homogenous ink with the aid of ultrasound. The ink was applied to the carbon cloth surface using a micropipette and left to dry at room temperature. The total catalyst loading on the working electrode was 1 mg cm−2 for all catalysts. Linear sweep voltammetry (LSV) was conducted at room temperature with stirring in 1.0 M KOH, with or without 10 mM HMF at a rate of 5 mV s−1. A 90% iR compensation was used to record all polarization curves. ERHE = EHg/HgO + 0.059 pH + 0.098 V was used to convert the reported potentials to the RHE scale. To determine the double-layer capacitance (Cdl), cyclic voltammetry (CV) was used in 1.0 M KOH with 10 mM HMF. Various scan rates of 20, 40, 60, 80, and 100 mV s−1 were employed within a potential window that did not involve faradaic processes. Using the formula ECSA = Cdl/Cs, where Cs = 118 μF cm−2, the Cdl obtained was converted to an electrochemically active surface area (ECSA), and the intrinsic current density of each NC was calculated. With an AC amplitude of 10 mV, Operando electrochemical impedance spectroscopy (EIS) experiments were conducted over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz to 0.01 Hz. The open-circuit potential (OCP) was measured in 1.0 M KOH with and without 50 mM HMF. To obtain the Arrhenius activation energy (Ea) of each catalyst for the HMFOR, LSV curves of Co-R, Co-H, Co-L and Co-P were obtained at different temperatures from 25 °C to 45 °C with an interval of 5 °C. The Ea values were calculated according to Arrhenius equation: Inj = InA − (Ea/R)/T, where A is pre-exponential factor, R is gas constant and T is the temperature (kelvin).
000 Hz to 0.01 Hz. The open-circuit potential (OCP) was measured in 1.0 M KOH with and without 50 mM HMF. To obtain the Arrhenius activation energy (Ea) of each catalyst for the HMFOR, LSV curves of Co-R, Co-H, Co-L and Co-P were obtained at different temperatures from 25 °C to 45 °C with an interval of 5 °C. The Ea values were calculated according to Arrhenius equation: Inj = InA − (Ea/R)/T, where A is pre-exponential factor, R is gas constant and T is the temperature (kelvin).
Density functional theory (DFT) calculation
All density functional theory calculations were performed using the Vienna ab initio Simulation Program (VASP).48,49 The generalized gradient approximation (GGA) in the Perdew–Burke–Ernzerhof (PBE) form and cutoff energy of 450 eV for the plane-wave basis set was adopted.50 A 3 × 3 × 1 Monkhorst–Pack grid was used to sample the brillouin zones at structure optimization.51 The ion-electron interactions were described by the projector-augmented wave (PAW) method.52 The convergence criteria of structure optimization were chosen as the maximum force on each atom less than 0.02 eV Å−1 with an energy change of less than 1 × 10−5 eV. The DFT-D3 semiempirical correction was described via Grimme's scheme method.53 Hubbard-U Correction method was applied to improve the description of localized Co d-electrons with U = 3.3 for Co. The adsorption energy change is defined as:| ΔEads = E (*ads) − E(*) − E (ads) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22238011, 22073112).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- P. Prabhu, Y. Wan and J. M. Lee, Matter, 2020, 3, 1162–1177 CrossRef.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- Y. Lu, C. L. Dong, Y. C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- Y. Song, W. Xie, Y. Song, H. Li, S. Li, S. Jiang, J. Y. Lee and M. Shao, Appl. Catal., B, 2022, 312, 121400 CrossRef CAS.
- Y. Sun, J. Wang, Y. Qi, W. Li and C. Wang, Adv. Sci., 2022, 9, 2200957 CrossRef CAS PubMed.
- J. Hulva, M. Meier, R. Bliem, Z. Jakub, F. Kraushofer, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Science, 2021, 371, 375–379 CrossRef CAS PubMed.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li, X. Zhong and J. Wang, Adv. Funct. Mater., 2023, 33, 2214488 CrossRef CAS.
- L. Lu, C. Wen, H. Wang, Y. Li, J. Wu and C. Wang, J. Catal., 2023, 424, 1–8 CrossRef CAS.
- Y. Yang, D. Xu, B. Zhang, Z. Xue and T. Mu, Chem. Eng. J., 2022, 433, 133842 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- M. Li, T. Zheng, D. Lu, S. Dai, X. Chen, X. Pan, D. Dong, R. Weng, G. Xu and F. Wang, J. Energy Chem., 2023, 84, 101–111 CrossRef CAS.
- M. H. Huang, S. Rej and S. C. Hsu, Chem. Commun., 2014, 50, 1634–1644 RSC.
- S. Rong, P. Zhang, F. Liu and Y. Yang, ACS Catal., 2018, 8, 3435–3446 CrossRef CAS.
- X. W. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- L. Ma, W. Zhang, Y. G. Wang, X. Chen, W. Yu, K. Sun, H. Sun, J. Li and J. W. Schwank, Appl. Catal., B, 2020, 267, 118371 CrossRef CAS.
- X. Xiao, X. Liu, H. Zhao, D. Chen, F. Liu, J. Xiang, Z. Hu and Y. Li, Adv. Mater., 2012, 24, 5762–5766 CrossRef CAS PubMed.
- J. Qu, Y. Wang, X. Mu, J. Hu, B. Zeng, Y. Lu, M. Sui, R. Li and C. Li, Adv. Mater., 2022, 34, 2203320 CrossRef CAS PubMed.
- P. W. Menezes, A. Indra, D. González-Flores, N. R. Sahraie, I. Zaharieva, M. Schwarze, P. Strasser, H. Dau and M. Driess, ACS Catal., 2015, 5, 2017–2027 CrossRef CAS.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W. X. Li, J. Greeley and P. Strasser, Angew. Chem., 2021, 133, 14567–14578 CrossRef.
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- S. Ibraheem, G. Yasin, A. Kumar, M. A. Mushtaq, S. Ibrahim, R. Iqbal, M. Tabish, S. Ali and A. Saad, Appl. Catal., B, 2022, 304, 120987 CrossRef CAS.
- Y. Shi, J. Wang, C. Wang, T. T. Zhai, W. J. Bao, J. J. Xu, X. H. Xia and H. Y. Chen, J. Am. Chem. Soc., 2015, 137, 7365–7370 CrossRef CAS PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K. S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- Y. Lu, Y. T. Liu, C. L. Dong, C. Yang, L. Zhou, Y. C. Huang, Y. Li, B. Zhou, Y. Zou and S. Wang, Adv. Mater., 2022, 34, 2107185 CrossRef CAS PubMed.
- W. Li, Y. Wang, X. Y. Cui, S. Yu, Y. Li, Y. Hu, M. Zhu, R. Zheng and S. P. Ringer, ACS Appl. Mater. Interfaces, 2018, 10, 19235–19247 CrossRef CAS PubMed.
- K. Zhang, G. Zhang, J. Qu and H. Liu, Small, 2018, 14, 1802760 CrossRef PubMed.
- J. Zheng, Y. Lyu, C. Xie, R. Wang, L. Tao, H. Wu, H. Zhou, S. Jiang and S. Wang, Adv. Mater., 2018, 30, 1801773 CrossRef PubMed.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C. L. Dong, Y. C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- X. Peng, X. Nie, L. Zhang, T. Liang, Y. Liu, P. Liu, Y. L. Men, L. Niu, J. Zhou, D. Cui and Y. X. Pan, ACS Appl. Mater. Interfaces, 2020, 12, 56943–56953 CrossRef CAS PubMed.
- M. H. Mahyuddin, A. Staykov, Y. Shiota and K. Yoshizawa, ACS Catal., 2016, 6, 8321–8331 CrossRef CAS.
- X. Lu, K. H. Wu, B. Zhang, J. Chen, F. Li, B. J. Su, P. Yan, J. M. Chen and W. Qi, Angew. Chem., 2021, 133, 14649–14656 CrossRef.
- N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806 RSC.
- Y. Lu, T. Liu, C. L. Dong, Y. C. Huang, Y. Li, J. Chen, Y. Zou and S. Wang, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed.
- Y. Sun, K. Xu, Z. Wei, H. Li, T. Zhang, X. Li, W. Cai, J. Ma, H. J. Fan and Y. Li, Adv. Mater., 2018, 30, 1802121 CrossRef PubMed.
- L. Zhang, J. Zhang, J. Fang, X. Y. Wang, L. Yin, W. Zhu and Z. Zhuang, Small, 2021, 17, 2100832 CrossRef CAS PubMed.
- F. Ali and N. R. Khalid, Appl. NanoSci., 2020, 10, 1481–1488 CrossRef CAS.
- Y. Wang, Z. Zhong, Y. Chen, C. T. Ng and J. Lin, Nano Res., 2011, 4, 695–704 CrossRef CAS.
- D. Su, S. Dou and G. Wang, Sci. Rep., 2014, 4, 5767 CrossRef PubMed.
- L. Cui, J. Li and X. G. Zhang, J. Appl. Electrochem., 2009, 39, 1871–1876 CrossRef CAS.
- D. He, K. Zhao, L. Zhang, J. Yuan, L. Yang, T. Cai, B. Xiao and C. Jin, China CN107857306B, 2019.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02398b |
| This journal is © The Royal Society of Chemistry 2023 |