
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic device combining hydrodynamic and dielectrophoretic trapping for the controlled contact between single micro-sized objects and application to adhesion assays†
Clémentine
Lipp
 a,
Laure
Koebel
b,
Romain
Loyon
c,
Aude
Bolopion
b,
Laurie
Spehner
c,
Michaël
Gauthier
b,
Christophe
Borg
c,
Arnaud
Bertsch
a and
Philippe
Renaud
*a
a,
Laure
Koebel
b,
Romain
Loyon
c,
Aude
Bolopion
b,
Laurie
Spehner
c,
Michaël
Gauthier
b,
Christophe
Borg
c,
Arnaud
Bertsch
a and
Philippe
Renaud
*a
aLaboratory of Microsystems LMIS4, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland. E-mail: philippe.renaud@epfl.ch
bInstitut FEMTO-ST, Département AS2M, Univ. Bourgogne Franche-Comté, CNRS, Besançon, France
cUnité RIGHT, UMR INSERM 1098, Établissement Français du Sang Bourgogne Franche-Comté, Besançon, France
First published on 14th July 2023
Abstract
The understanding of cell–cell and cell–matrix interactions via receptor and ligand binding relies on our ability to study the very first events of their contact. Of particular interest is the interaction between a T cell receptor and its cognate peptide–major histocompatibility complex. Indeed, analyzing their binding kinetics and cellular avidity in large-scale low-cost and fast cell sorting would largely facilitate the access to cell-based cancer immunotherapies. We thus propose a microfluidic tool able to independently control two types of micro-sized objects, put them in contact for a defined time and probe their adhesion state. The device consists of hydrodynamic traps holding the first type of cell from below against the fluid flow, and a dielectrophoretic system to force the second type of object to remain in contact with the first one. First, the device is validated by performing an adhesion frequency assay between fibroblasts and fibronectin coated beads. Then, a study is conducted on the modification of the cellular environment to match the dielectrophoretic technology requirements without modifying the cell viability and interaction functionalities. Finally, we demonstrate the capability of the developed device to put cancer cells and a population of T cells in contact and show the discrimination between specific and non-specific interactions based on the pair lifetime. This proof-of-concept device lays the foundations for the development of next generation fast cell–cell interaction technologies.
Multicellular organisms require a very well organized and finely balanced cell–cell communication, adhesion, coordination and programmed cell death to ensure the organism's homeostasis. These functions rely on specialized receptors placed at the cell membrane whose binding to their ligand triggers a defined function. Receptor–ligand interaction is thus a cornerstone of multicellularity, not only maintaining the cells physically adhered to one another, but also enabling communication to ensure function and organism homeostasis. Malfunction of the receptors to properly trigger the appropriate response leads to imbalances in the organism and failure to exert specific functions, and our understanding of these pathologies thus relies on the capacity to study such interactions. Because they characterize the very first instance of the reaction chain induced by a binding event, the binding kinetic parameters defining the rate of bond formation and dissociation are of particular interest. Receptors whose ligands are in solution benefit from well characterized tools to study their binding kinetics, such as surface plasmon resonance.1 In contrast, tools available to study receptors whose ligands are anchored on a cell or on a surface remain relatively inaccessible.
Measuring the binding kinetics of two surface anchored receptors and ligands demands the independent control of the spatial position of two micrometer sized objects with high temporal resolution, which is technically challenging.2 State-of-the-art methods for such assays comprise atomic force microscopy (AFM), which is a precision tool of choice to measure the force characteristics. Typically, a cell is picked and attached to the tip of a soft microfabricated cantilever using either surface functionalization3 or an embedded fluidic system4 and used to make contact with another cell adhered to a substrate. The time of contact can be controlled before probing the adhesion state and measuring the force resistance of the formed bond(s) through the deformation of the cantilever. Single bond detachment can be resolved thanks to the high force sensitivity of the probe. Alternatively, the dual pipette assay (DPA) makes use of two micropipettes to attach one cell per pipette via the aspiration of the cells. The pipettes can then be guided using micromanipulators coupled to high resolution high speed imaging to probe the adhesion state as a function of time of contact.5 Variations of this method comprise the attachment of a red blood cell coated with a specific ligand to one of the cells. Indeed red blood cells are soft and a precise force measurement of the bond can be deduced from their deformation. Alternatively, a bead coated with the ligand can be attached to an aspirated red blood cell and put in contact with the receptor presenting cell attached to the other pipette. In that case, the thermal fluctuations of the bead are measurable because of the high deformability of the red blood cells and indicate the adhesion state of the pair.6 A direct measurement of the on and off rates from the adhesion state timeline is then made possible. Optical tweezers (OT) are another tool that makes possible the precise manipulation of objects thanks to the attraction of particles towards the waist of a highly focused laser beam. OT are thus able to both push an object trapped in the waist towards a cell adhered to a surface or trapped in another OT and pull on it with known force to probe the adhesion state and force of adhesion.7,8 These three methods rely on a very soft spring linked to one of the cells to push and pull it to and from another cell to allow a controlled force and time of interaction. They are thus highly precise in force and position control but are also of very low throughput and require highly skilled staff to perform the assay.
Thanks to their ability to control the position of micro-sized objects through various forces, microtechnologies have potential to control the contact between two objects at larger throughput than the above mentioned methods. The strategy of confining two objects in the same micro-sized environment was largely used to study downstream effects after the contact,9–11 but lacks the possibility of independently controlling the two objects to study binding kinetics. Systems for the dynamic and serial cell–cell and cell–bead interaction were developed in microfluidic chips by immobilizing in a first step the first type of particle using monolayer adhesion,12,13 hydrodynamic traps14,15 and hydrodynamic traps combined with sedimentation.16 The second type of cell was flowed on top and the reduction in speed upon contact between the two objects, pair lifetime, resistance to flow shear stress or downstream signalling molecules was monitored to characterize the interaction. These systems are powerful tools to probe receptor–ligand interactions at higher throughput than the macrosized methods presented above, but they lack the possibility of controlling both force and contact time between the two objects.
Controlled contact between cells is of specific interest for cell-based cancer immunotherapies. Indeed the process of selecting patient T cells with high antitumor activity is currently a long and very expensive process, limiting the generalization of such an approach despite its proven efficacy.17 In this context, a tool able to reliably pair T lymphocytes with antigen presenting cells (APCs) or tumor cells and rapidly assess the specificity of the interaction could simplify the long process of T cell selection and facilitate in the long term the access to this kind of therapy.
In this context, we propose a microfluidic tool to control cell–cell and cell–bead contact in a microfluidic chip using forces derived from a different phenomenon for each object. The effects are thus orthogonal for an independent manipulation of the two micro-sized particles. The method relies on the combination of two trapping methods based on planar hydrodynamic trapping of cells18 and dielectrophoretic (DEP) trapping,19 both physical principles being widely used to precisely manipulate and trap single cells.20,21 The fabrication processes were successfully implemented sequentially to provide a novel device with both capabilities. We first present the working principle of the microfluidic device and its capabilities (section 2.1). The functionality of the tool is then validated by performing an adhesion frequency assay with fibroblast cells and fibronectin coated beads, and the extracted binding kinetic parameters are compared to the literature (section 2.2). In order to demonstrate the potential of such a tool in immunotherapy applications, the pairing of T cells with cancer cells is then performed and the effect of the binding of T cell receptors (TCRs) to their cognate peptide–major histocompatibility complex (pMHC) is studied (section 2.3). The pair lifetime is measured and the effect of TCR–pMHC bonds on this parameter is demonstrated. A measurement of cellular avidity based on these measurements is then proposed by assigning a pair lumped off rate as a metric. The characteristics of the device are finally discussed in section 3 as well as future developments and perspectives. The throughput of this device has the potential for scale up by parallelization and combination of the shown principles with computer vision control.
1 Experimental
1.1 Microfabrication
Buried microchannels were fabricated as detailed by ref. 18 for the planar hydrodynamic trapping. Shortly, 500 nm Al2O3 was sputtered on a fused silica substrate and access holes as well as traps and outlets were defined by photolithography and etched using ion beam etching. The channels were defined by exposure to a vapor phase of hydrofluoric acid (HF) that selectively underetched the fused silica substrate without attacking the Al2O3 layer. Access holes were then sealed by depositing a 2.5 μm thick layer of low temperature oxide doped with boron and phosphate (BPSG) leading to 3 μm diameter traps. Electrodes for the dielectrophoretic actuation were then defined by a lift-off process. The negative resist AZ nLOF (MicroChemicals) was coated (ACS 200, Süss), exposed (MLA 150, Heidelberg Instruments) and developed (ACS 200, Süss) on top of the structured fused silica substrate. 20 nm of titanium and 200 nm of platinum were evaporated (EVA 760, Alliance-Concept) and lifted off in a remover bath (MICROPOSIT Remover 1165). The photoresist residues remaining in the buried channels were then cleaned using oxygen plasma (GIGAbatch, PVA TePla). The top PDMS channels were then molded, aligned and permanently bonded to the electrodes and glass channels as described by Lipp et al.19 The full fabrication process is shown in Fig. S1,† and pictures of the fabricated combined traps and the whole final chip in Fig. S2 and S3A,† respectively.1.2 Experimental platform and protocols
The chips were primed with Pierce protein-free (PBS) blocking buffer for 2 hours to prevent proteins from adhering to the surfaces. The cells or beads were placed in a chromatography vial connected to the punched PDMS with 360 μm outer diameter tubing for tight sealing. Pressure was applied to the vials using Fluigent Flow-EZ pressure controllers. The chip was mounted on and electrically connected to a custom PCB placed on the stage of a Leica DMI3000 B inverted microscope and observed using a uEye (IDS) camera. All the electric signals for the DEP manipulation of particles were sent through a home-made PCB multiplying an AC signal at 100 kHz by DC signals whose amplitudes are controlled by the computer with an adapted C++ program through an analog output generator (Mccdaq USB-3100). The full setup is shown in Fig. S4† and the chip and PCB mounting in Fig. S3 in the ESI.† The maximum peak value for deviation voltages V1 and V2 was set to 10 V whereas the peak trapping voltages Vsync and Vcontact were set to 8 V. Pressures during contact experiments were set to 15 mbar to ensure constant drag force exerted on the dielectrophoretically manipulated particles.1.3 Cell culture and preparation
The adherent mouse fibroblast cell line NIH-3T3 was cultured in medium (DMEM, high glucose, GlutaMAX Supplement) supplemented with 10% FBS and 1% penicillin–streptomycin at 37 °C in a 5% CO2 atmosphere. The semi-adherent cancer cell line Colo205 was cultured in medium (RPMI) supplemented with 10% FBS and 1% penicillin–streptomycin at 37 °C in a 5% CO2 atmosphere. Colo205 tumor cells were pulsed with 10 μg mL−1 specific or irrelevant peptide, and incubated for 1 h at 37 °C in 5% CO2 in RPMI medium supplemented with 10% FBS and 1% penicillin–streptomycin. The specificity of CD8+ T cells towards the peptide was previously assessed by co-culture of 5 × 104 T cells with pulsed tumor cell lines in the presence of Golgi Plug (BD Biosciences) in a 96-well plate at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E:T cell ratio.
:1 E:T cell ratio.
Staining of the cells was performed by incubating the cells in PBS with 4 μm calcein UltraBlue AM (Cayman Chemical) or with 1 μm calcein AM (Invitrogen™) for 1 h. The working solution is composed of 40% RPMI and 60% deionized water. The solution was compensated for osmolarity by the addition of dextrose (Sigma-Aldrich) and cleaned through a 0.22 μm filter. After staining, the cells were resuspended in the working solution and passed through a 40 μm cell strainer before the experiment. All the reagents were from Gibco unless specified.
1.4 Medium compatibility protocols
For the cell viability assay, cells were immersed for 5 h in complete medium diluted in different percentages of DI water with the corresponding amount of dextrose to compensate for osmolarity at 37 °C in a 5% CO2 atmosphere. After 5 h, the cells were resuspended in standard complete medium and cultivated at 37 °C in a 5% CO2 atmosphere for 48 h. Cell viability was assessed using trypan blue after the 5 h incubation in custom medium and 24 h and 48 h after resuspension in standard medium.For the cell activation assay, CD8+ T cells were stimulated with specific or irrelevant peptides at 5 μg mL−1 (JPT Technologies) in the presence of Golgi Plug (BD Biosciences) in complete medium (RPMI medium supplemented with 10% FBS and 1% penicillin–streptomycin) or in complete medium diluted in different percentages of DI water with the corresponding amount of dextrose to compensate for osmolarity. CD8+ T cells were incubated at 37 °C in 5% CO2 for 5 h for cell viability and cytokine production analysis. Cytokine production was analyzed by flow cytometry upon cell staining performed according to the manufacturer's instructions (BD). Briefly, cells were stained for 30 min with Live/Dead fixable dead cell stain (eBioscience™ Fixable Viability Dye eFluo™ 506 Invitrogen), anti-CD3 (PB, BD Pharmingen), anti-CD4 (APC-H7, BD Pharmingen) and anti-CD8 (PE, Diaclone). For intracellular staining, cells were incubated with Cytofix/Cytoperm™ (BD Biosciences) for 30 min and then stained with anti IFNγ (APC, BD Pharmingen) and anti TNFα (FITC, BD Pharmingen). Cells were acquired on a BD FACS- Canto II (BD Biosciences) flow cytometer and data were analyzed using FlowJo Software.
1.5 Bead coating
5 μm in diameter polystyrene beads coated with streptavidin were purchased from Spherotech. The beads were cleaned 3 times in PBS and resuspended in a solution of PBS with 20 μg ml−1 biotinylated fibronectin (cytoskeleton) or biotinylated BSA (Pierce) and left to incubate for 1 h at room temperature and with gentle rotation. The beads were then washed three times in PBS supplemented with 7.5% BSA to prevent the beads from adhering to each other and resuspended in the working solution.2 Results
2.1 Principle of operation: combining hydrodynamic trapping and dielectrophoresis in a microfluidic chip
We present a microfluidic system combining fluidic and dielectrophoretic actuation capable of performing in flow cell–particle interaction. The device is shown in Fig. 1a and comprises two inlets for medium perfusion controlled by pressures Pin,1 and Pin,2 followed by serpentine channels to increase the flow resistance. The two channels merge before entering the interaction chamber. This latter is composed of an upstream DEP actuated deviation system capable of steering the incoming particles to specific streamlines as detailed by Demierre et al.22 through the modulation of the voltage ratio V1/V2. The deviation system is followed by an expansion of the channel and two lines of DEP traps. Each line is made of coplanar electrodes and comprises four distinct trapping units capable of focusing the particles to one point in three dimensions as described by Lipp et al.,19 as well as a bypass zone to which unwanted particles can be deviated to leave the chamber. The upstream line is named the synchronization line because its role is to synchronize the arrival of particles to the second line once the amount of particles per trap is reached. The second line is named the contact line and features hydrodynamic traps placed along the center of each one of the DEP traps. The hydrodynamic traps are precisely described by Lipp et al.18 and feature holes, placed at the bottom of the main PDMS channel and connected to a second lower level of channels, named buried channels. The buried channels connect the traps to a pressure control channel, where pressure Pc defines the flow direction in the buried channel. Particles of larger diameter than the trap and lying in the streamlines passing through the traps and buried channels clog the traps due to their larger dimensions, stopping the flow in the buried channels and immobilizing the particle thanks to the pressure difference built across it. Fig. 1b is an optical microscopy image of the synchronization (top) and contact (bottom) lines with transparent buried channels and hydrodynamic traps. Fig. 1d is a scanning electron microscopy image with two hydrodynamic traps surrounded by coplanar electrodes (highlighted in yellow for better visualization).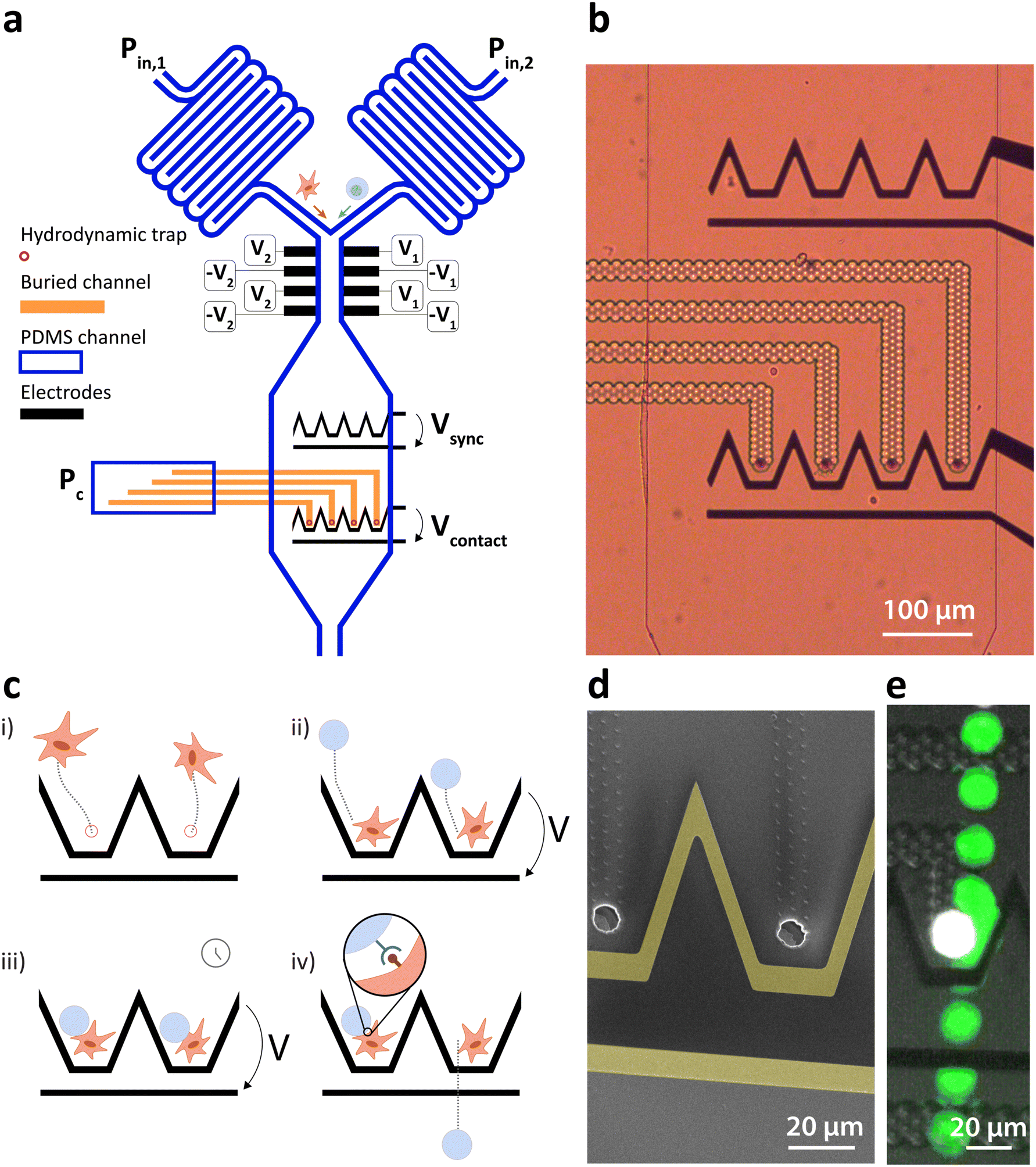 | ||
| Fig. 1 (a) Schematic representation of the microfluidic chip and electrodes with controlled voltages and pressures. The orange part represent the buried channel embedded in the glass substrate and running at a lower level under the PDMS channel. (b) Optical microscopy picture of the interaction zone with the synchronization line (top) and contact line (bottom). Each funnel shaped electrode feature generates a DEP force field with a single equilibrium position against the flow at its center when a voltage is applied. The contact line comprises hydrodynamic traps at the center of the DEP traps. (c) Workflow steps for the controlled interaction between objects with emphasis on two traps of the synchronization line: i) Hydrodynamic trapping of the first type of cell. ii) DEP trapping of the second type of cell. iii) The contact is maintained for the desired duration. iv) The voltage is turned off and the second type of cell is dragged by the flow detaching the pairs, unless a bond was formed between them. (d) SEM picture of two units of the synchronization line. The hydrodynamic trap is visible at the center of the funnel shaped electrodes. (e) Timelapse image of the controlled contact between two cells. The hydrodynamically trapped cell is coloured in white and the dielectrophoretically manipulated cell in green by image processing for clarity. | ||
The interaction zone thus comprises DEP and hydrodynamic traps, whose roles are to exert independent effects on the particles. Once a particle is immobilized in the hydrodynamic trap, the DEP force does not affect it because it is weaker than the force exerted by the pressure difference across the hydrodynamic trap. This particle is hereafter named the hydrodynamically trapped particle (HTP). A second particle, however, is affected by the DEP induced force when a potential difference is applied across the contact electrodes. Because the hydrodynamic trap is placed at the equilibrium position of the DEP trap, its effect directs the second particle, hereafter named dielectrophoretically manipulated particle (DMP), towards the HTP and immobilizes it in close contact with the HTP against the flow. The workflow steps for the controlled interaction are depicted in Fig. 1c with emphasis on two traps of the contact line. The first step consists of the introduction of the first type of particle in the interaction chamber by increasing Pin,1 and setting Pin,2 to zero. The particles are then immobilized in the hydrodynamic traps by setting a negative value to Pc (Fig. 1ci)). Once all the traps are filled with HTPs, Pc can be increased to a value close to zero to minimize the pressure difference across them and associated deformation. The second type of particle is then introduced in the chamber by increasing Pin,2 and setting Pin,1 to zero. The arriving particles are first steered towards each of the trapping units of the synchronization line (not shown) using the deviation system and trapped against the flow drag force under the effect of the DEP force generated by Vsync. The destination of the particles is defined by the ratio of voltage applied to each side of the channel V1/V2 as described by Demierre et al.22 Once the synchronization line is filled by one particle per trap, Vsync can be turned off and the DMPs are dragged downstream by the flow and immobilized in close contact with the HTP by turning on Vcontact (Fig. 1cii)). The voltage is maintained for the desired contact duration (Fig. 1ciii)). Once this duration is elapsed, Vcontact is turned off and the DMPs are dragged away from the HTP by the constant flow. Adhesion events mediated through receptor–ligand bonds can be observed and related parameters such as the state of adhesion or lifetime of the pairs can then be assessed and measured (Fig. 1civ)).
Fig. 1e is a timelapse image of the contact between two cells demonstrating the device's capability, the HTP is coloured in white and the DMP in green by image post-processing for clarity.
2.2 Validation of the device via the interaction between fibronectin and fibroblasts
In order to validate the device, a well-known and characterized pair of receptor–ligand interaction was studied using the developed microfluidic chip. Integrins present on the surface of mouse fibroblasts were shown to bind specifically to fibronectin coated on a surface.7 For the specific case of a species outnumbering the other one, the mathematical expression describing the probability of adhesion of one bond after contact time t was derived:5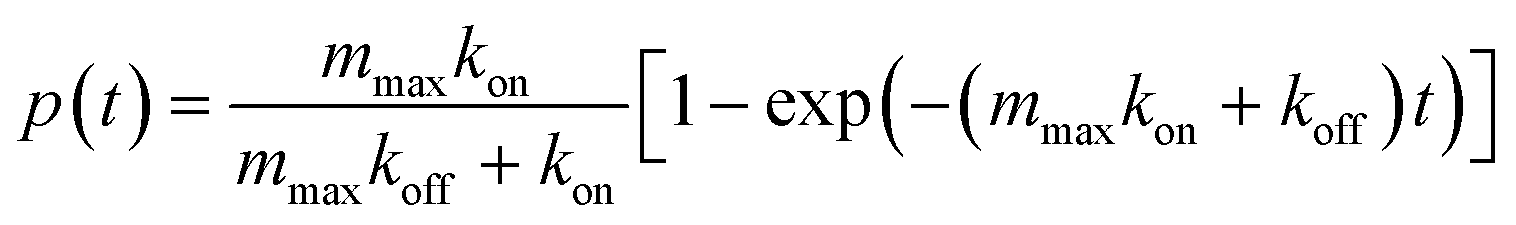 | (1) |
 | (2) |
 the binomial coefficient. The probability of adhesion Pa mediated by minimum nmin bonds is then given by:
the binomial coefficient. The probability of adhesion Pa mediated by minimum nmin bonds is then given by: | (3) |
| Pa = 1 − [1 − p(t)]Acmmin | (4) |
 | ||
| Fig. 2 (a) Timelapse image of a fibronectin bead placed in contact with a mouse fibroblast during 3 seconds. No adhesion was observed after the contact and the bead was dragged downstream after the release. (b) Percentage of adhesion events as a function of contact duration for fibronectin coated beads as DMPs and mouse fibroblasts as HTPs. BSA coated beads as DMPs are used as a control. We demonstrate that the adhesion is mediated by integrin receptors at the surface of the fibroblasts binding specifically to fibronectin and extract the binding parameters from the fitting eqn (4) to the data. | ||
Integrins were taken as the less abundant species and one bond was considered as sufficient to mediate an adhesion as deduced by the similar work performed by Thoumine et al.7 As expected from eqn (4), the percentage of adhesion of fibronectin coated beads increases as a function of contact duration until reaching a plateau. The control data display a significantly lower percentage of adhesion with no adhesion event reported for the lower range of contact duration. Experimentally, the strength of adhesion was significantly lower for BSA as the adhered beads would detach from the cell at a very low flow rate, contrary to most fibronectin beads which remained attached even when increasing the flow rate until the maximum value allowed by the pump. Eqn (4) was fitted to the data and is represented as the continuous line in the graph of Fig. 2b. The parameters extracted from the best fit are kr = 0.17 s−1 and Acmminmmaxkf = 0.76 s−1. This latter parameter is constant for a given adhesion curve and was found to be in the range reported by the similar adhesion experiment by Thoumine et al.7
2.3 Application to the measurement of T cell–cancer cell pair lifetime
The device was used to test more complex receptor–ligand pairs such as TCR–pMHC. An HLA-A2-restricted clone of human CD8+ T cells was thus used as DMPs and cancer cells pulsed with the peptide to which the TCRs of the T cell clone are specific were used as HTPs. The negative control consisted of the same cancer cells pulsed with an irrelevant peptide. | ||
| Fig. 3 Intracellular IFN-γ and TNF-α contents after 5 h co-incubation of T cell clones with the specific peptide in medium composed of different percentages of RPMI supplemented with FBS and diluted in DI water with the adopted amount of dextrose to compensate for osmolarity. A minimum content of 40% complete medium was found to be necessary to ensure T cell clone activation and viability. | ||
 | ||
| Fig. 4 (a) Pair lifetime measurements after 22 seconds of forced contact between Colo205 and peptide-specific T cell clones in the case where the HLA-A2+ Colo205 cells were pulsed with the TCR cognate peptide (specific) or irrelevant peptide (non-specific). (b) A lumped off rate can be fitted to the data indicating a faster dissociation of pairs without the specific peptide than that of the pairs with the specific peptide. | ||
The sample distributions were tested for normality using the Kolmogorov–Smirnov test and the null hypothesis of a normal distribution was rejected in both cases. The samples were thus tested for equal median using the Mann–Whitney U test and a p-value of 0.03 was obtained, rejecting at 95% confidence interval the null hypothesis and indicating a larger median lifetime in the presence of the specific peptide on the cancer cells. The TCR–pMHC interaction thus tends to increase the pair lifetime.
The lifetime of single bonds following first order dissociation kinetics can be described by an exponential decay and the probability Pi of a bond formed at time t = 0 to remain intact at time tb follows the law:
| Pi = exp(−kofftb) | (5) |
3 Discussion
We demonstrated the development of a novel microfluidic chip capable of performing in flow interaction assays based on two distinct phenomena for the independent manipulation of two micro-sized objects. The orthogonal manipulation of the two objects allows spatial and time control over the contact, which unlocks for the first time different adhesion assays on chip. Two different assays were performed as a proof of concept to demonstrate the possibilities achievable with this method. First a biophysical experiment was performed in which fibroblasts were put in contact with fibronectin coated beads during different contact times. The adhesion mediated by the binding of integrin receptors on the surface of the fibroblasts to fibronectin was assessed, and the binding kinetics of the receptor ligand pair were extracted by fitting the theoretical curve to the data. Second, human T cell clones were put in contact with cancer cells pulsed with a peptide to which the TCRs are specific. The pair lifetime was measured after a contact time of 22 seconds, indicating a longer adhesion of T cells to cancer cells pulsed with the specific peptide than to the cancer cells not pulsed with the specific peptide. The data were approximated by a single bond dissociation to extract a pair lumped off rate describing the avidity of the interaction. This second assay opens the door to application in cancer immunotherapy for specific T cell selection via avidity evaluation.13,28 The combination of lifetime measurement together with another criteria indicating a specific activation of the T cells, for example an intracellular calcium increase10,13 or a change of the cell's electrical impedance,29 could allow a precise assay for specific T cell recognition. Future development will comprise the addition of a dielectrophoresis actuated cell sorter (DACS) downstream of the contact chamber to sort and retrieve cells of interest.30 However, the largest improvement will come from computer vision automation of the actuation for the reasons described below.Reverse binding constants are known to be dependent on the force applied to disrupt the bonds. First order forward and backward kinetics were described by a single energy barrier in the potential landscape along the distance between the receptor and the ligand. The effect of a force F applied on the complex can be understood as a lowering in the energy barrier and an increase in the off rate, as described by the Bell model:31
| koff = k0off exp (F/Fb) | (6) |
Variation of the compressive force pushing the two objects together changes the area of contact between them and has an impact on the parameter Acmminmmaxkon. The compressive force is defined by the difference between drag and DEP force and the latter is dependent not only on the volume of the DMPs, but also on the position of the HTP surface. Large variation in both HTP and DMP size thus has an impact on both the compressive force and the disruptive force experienced by the DMPs and alters the extracted binding kinetics, which should be taken into account when working with samples of large size distribution. Using computer vision control, the voltage used to compress the DMPs in contact with the HTPs could thus be dynamically adapted to both sizes to reach a constant compressive force.
Precise control of the time of contact is not possible when defining the time of contact as tVsync = 0 – tVcontact = 0, indeed particles have a distribution in velocity due to differences in size and position along the channel height and do not stabilize in contact simultaneously. Control of the time of contact for less than 6 seconds thus made a single trap at a time, but the minimum contact time was limited to 1 second due to human limitation in reactivity. This aspect would also be solved using automated control of the interaction, together with identification and sorting of events of interest.
4 Conclusion
In summary, we proposed and developed a new microfluidic chip combining two types of actuation for the controlled contact and separation of two micro-sized objects, in particular single cells, and their independent controlled motions. Hydrodynamic traps were designed to first trap and arrange single cells in the chip, and the dielectrophoresis phenomenon is used on the second type of particle, either beads or cells, to bring them towards the first cells and create a forced monitored interaction. This developed tool is specially designed to guarantee manipulation methods that preserve cell integrity and receptor functions and paves the way to a second generation of larger throughput devices as it can easily be combined with automation. We performed two different assays to demonstrate the capability of the device to generate repeatable cell–bead and cell–cell interactions, first on the fibronectin–integrin bond to fit the experimental data to the well-known binding kinetic model and validate the device. Finally, we tackled the challenge of the TCR–pMHC bond and succeeded in discriminating between specific and non-specific interactions, which shows the potential of the device in cell-based cancer immunotherapy development once combined with automation for faster and less expensive T cell screening and sorting. This novel method thus opens new perspectives for applications in biophysical experiments and adoptive cell therapy developments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EIPHI Graduate School under Contract ANR-17-EURE-0002, by the French Agence Nationale de la Recherche and the Swiss National Science Foundation through the CoDiCell project (contract “ANR-17-CE33-0009” and “FNS 00021E_175,592/1”, respectively), and by the French ROBOTEX network and its Micro and Nanorobotics center under Grant ANR-10-EQPX-44-01. The authors thank Olivier Thoumine for the fruitful discussions, Laura Collodel for her help with the experiments, Niccolò Piacentini for the technical advice and support with the analysis, François Marionnet for the valuable technical advice, and the French RENATECH network and its FEMTO-ST technological facility as well as the EPFL Center of MicroNanoTechnology (CMi) for the support with the microfabrication.References
- P. Pattnaik, Appl. Biochem. Biotechnol., 2005, 126, 079–092 CrossRef CAS PubMed.
- X. Hang, S. He, Z. Dong, G. Minnick, J. Rosenbohm, Z. Chen, R. Yang and L. Chang, Biosens. Bioelectron., 2021, 179, 113086 CrossRef CAS PubMed.
- J. Helenius, C.-P. Heisenberg, H. E. Gaub and D. J. Muller, J. Cell Sci., 2008, 121, 1785–1791 CrossRef CAS PubMed.
- E. Potthoff, O. Guillaume-Gentil, D. Ossola, J. Polesel-Maris, S. LeibundGut-Landmann, T. Zambelli and J. A. Vorholt, PLoS One, 2012, 7, e52712 CrossRef CAS PubMed.
- S. E. Chesla, P. Selvaraj and C. Zhu, Biophys. J., 1998, 75, 1553–1572 CrossRef CAS PubMed.
- W. Chen, E. A. Evans, R. P. McEver and C. Zhu, Biophys. J., 2008, 94, 694–701 CrossRef CAS PubMed.
- O. Thoumine, P. Kocian, A. Kottelat and J.-J. Meister, Eur. Biophys. J., 2000, 29, 398–408 CrossRef CAS PubMed.
- S. Dai, J. Mi, J. Dou, H. Lu, C. Dong, L. Ren, R. Zhao, W. Shi, N. Zhang, Y. Zhou, J. Zhang, J. Di and J. Zhao, Biosens. Bioelectron., 2022, 206, 114131 CrossRef CAS PubMed.
- M. Kirschbaum, M. S. Jaeger, T. Schenkel, T. Breinig, A. Meyerhans and C. Duschl, J. Chromatogr. A, 2008, 1202, 83–89 CrossRef CAS PubMed.
- B. Dura, M. M. Servos, R. M. Barry, H. L. Ploegh, S. K. Dougan and J. Voldman, Proc. Natl. Acad. Sci., 2016, 113, year CrossRef PubMed.
- Y. Zhou, N. Shao, R. Bessa de Castro, P. Zhang, Y. Ma, X. Liu, F. Huang, R.-F. Wang and L. Qin, Cell Rep., 2020, 31, 107574 CrossRef CAS PubMed.
- P. Moura Rosa, N. Gopalakrishnan, H. Ibrahim, M. Haug and O. Halaas, Lab Chip, 2016, 16, 3728–3740 RSC.
- J. F. Ashby, J. Schmidt, N. KC, A. Kurum, C. Koch, A. Harari, L. Tang and S. H. Au, Adv. Healthcare Mater., 2022, 11, 2200169 CrossRef CAS PubMed.
- M. Duchamp, T. Dahoun, C. Vaillier, M. Arnaud, S. Bobisse, G. Coukos, A. Harari and P. Renaud, RSC Adv., 2019, 9, 41066–41073 RSC.
- M. A. Stockslager, J. S. Bagnall, V. C. Hecht, K. Hu, E. Aranda-Michel, K. Payer, R. J. Kimmerling and S. R. Manalis, Biomicrofluidics, 2017, 11, 064103 CrossRef PubMed.
- H. Ide, W. V. Espulgar, M. Saito, T. Aoshi, S. Koyama, H. Takamatsu and E. Tamiya, Sens. Actuators, B, 2020, 330, 129306 CrossRef.
- S. A. Rosenberg and N. P. Restifo, Science, 2015, 348, 62–68 CrossRef CAS PubMed.
- C. Lipp, K. Uning, J. Cottet, D. Migliozzi, A. Bertsch and P. Renaud, Lab Chip, 2021, 21, 3686–3694 RSC.
- C. Lipp, L. Koebel, A. Bertsch, M. Gauthier, A. Bolopion and P. Renaud, Front. Bioeng. Biotechnol., 2022, 10, year Search PubMed.
- Y. Zhou, S. Basu, E. Laue and A. A. Seshia, Biosens. Bioelectron., 2016, 81, 249–258 CrossRef CAS PubMed.
- S. V. Puttaswamy, N. Bhalla, C. Kelsey, G. Lubarsky, C. Lee and J. McLaughlin, Biosens. Bioelectron., 2020, 170, 112661 CrossRef CAS PubMed.
- N. Demierre, T. Braschler, P. Linderholm, U. Seger, H. van Lintel and P. Renaud, Lab Chip, 2007, 7, 355–365 RSC.
- A. Ramos, H. Morgan, N. G. Green and A. Castellanos, J. Phys. D: Appl. Phys., 1998, 31, 2338–2353 CrossRef CAS.
- J. Cottet, A. Kehren, S. Lasli, H. Lintel, F. Buret, M. Frenea-Robin and P. Renaud, Electrophoresis, 2019, 40, 1498–1509 CrossRef CAS PubMed.
- J.-R. Hwang, Y. Byeon, D. Kim and S.-G. Park, Exp. Mol. Med., 2020, 52, 750–761 CrossRef CAS PubMed.
- J. B. Huppa and M. M. Davis, Nat. Rev. Immunol., 2003, 3, 973–983 CrossRef CAS PubMed.
- D. Campillo-Davo, D. Flumens and E. Lion, Cell, 2020, 9, 1720 CrossRef CAS PubMed.
- T. Yerbury, z-Movi Cell Avidity Analyzer - LUMICKS, 2022, https://lumicks.com/products/z-movi-cell-avidity-analyzer/ Search PubMed.
- E. Rollo, E. Tenaglia, R. Genolet, E. Bianchi, A. Harari, G. Coukos and C. Guiducci, Biosens. Bioelectron., 2017, 94, 193–199 CrossRef CAS PubMed.
- D. Lee, B. Hwang and B. Kim, Micro Nano Syst. Lett., 2016, 4, 2 CrossRef.
- G. I. Bell, Science, 1978, 200, 618–627 CrossRef CAS PubMed.
- B. Liu, W. Chen and C. Zhu, Annu. Rev. Phys. Chem., 2015, 66, 427–451 CrossRef CAS PubMed.
- F. Kong, A. J. Garcia, A. P. Mould, M. J. Humphries and C. Zhu, J. Cell Biol., 2009, 185, 1275–1284 CrossRef CAS PubMed.
- S. Kobel, A. Valero, J. Latt, P. Renaud and M. Lutolf, Lab Chip, 2010, 10, 857 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00400g |
| This journal is © The Royal Society of Chemistry 2023 |