
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding and controlling lithium morphology in solid polymer and gel polymer systems: mechanisms, strategies, and gaps†
Kyra D.
Owensby
 ab,
Ritu
Sahore
a,
Wan-Yu
Tsai
a and
X. Chelsea
Chen
*a
ab,
Ritu
Sahore
a,
Wan-Yu
Tsai
a and
X. Chelsea
Chen
*a
aOak Ridge National Laboratory, Chemical Sciences Division, Oak Ridge, Tennessee 37831, USA. E-mail: chenx@ornl.gov
bThe Bredesen Center for Interdisciplinary Research and Graduate Education, The University of Tennessee Knoxville, Knoxville, Tennessee 37996, USA
First published on 19th October 2023
Abstract
Lithium metal anode promises the highest theoretical energy density and may enable high energy designs such as lithium–sulfur and lithium–air batteries. However, stable lithium plating and stripping remains a challenge in all electrolyte systems including liquids, polymers, and ceramic electrolytes. In this perspective, we examine literature studies of lithium morphologies in solid polymer and gel polymer systems and compare that with well-studied liquid electrolytes. In solid polymer electrolytes, current density and mechanical properties are both governing parameters for lithium morphology, differing from conventional liquid electrolytes. Stable lithium electrodeposition may be accomplished by a polymer electrolyte with good stiffness operating at significantly lower current densities than its limiting current density, which is defined by the Sand equation. In gel polymer electrolytes, the reported lithium morphology is more similar to that in liquid electrolytes, suggesting similar nucleation and growth mechanisms. Based on experimental evidence and theoretical guidance, current strategies to control lithium morphology in solid polymer and gel polymer electrolytes are summarized. The limitations of these strategies are discussed. In particular, we note the knowledge gap in understanding the solid electrolyte interphase in solid polymer systems and the critical role it can play in regulating lithium morphologies.
 Kyra D. Owensby | Kyra D. Owensby is a PhD student at the University of Tennessee – Knoxville in the Bredesen Center for Research and Graduate Education studying Energy Science and Engineering. She is currently working under Dr X. Chelsea Chen in the Chemical Sciences Division at Oak Ridge National Laboratory. She received her BE in Chemical Engineering from Vanderbilt University. Her research interests currently include investigating the interfaces of lithium metal–polymer electrolyte batteries. |
 Ritu Sahore | Ritu Sahore is a R&D Associate in the Chemical Sciences Division at Oak Ridge National Laboratory. She received her PhD in Materials Science and Engineering from Cornell University in 2015, followed by two postdoctoral appointments at Argonne National Laboratory and Oak Ridge National Laboratory. She has over 10 years of research experience in the field of batteries with chemistries ranging from lithium–sulfur, lithium-ion, and solid-state batteries, with over 30 peer-reviewed publications. Her research interests include understanding fundamental cell degradation mechanisms, with a current focus on high-voltage lithium-ion and solid-state battery chemistries. |
 Wan-Yu Tsai | Wan-Yu Tsai is an R&D Associate at Oak Ridge National Laboratory (ORNL). She obtained her PhD in Materials Science at Paul Sabatier University (Toulouse, France) in 2015, and later joined ORNL as a postdoc before becoming an ORNL staff in 2020. Her research focuses on interfacial properties and their impact on charge storage mechanisms in energy storage materials using in situ electrochemical atomic force microscopy and other techniques. |
 X. Chelsea Chen | Xi (Chelsea) Chen is an R&D Staff and Polymer Scientist at Oak Ridge National Laboratory (ORNL). She has extensive experience in polymers, composites, interfaces, electron microscopy and neutron/X-ray scattering. She leads programs in solid state batteries and polymer electrolytes at ORNL. She earned her PhD in 2011 from the University of Michigan, Ann Arbor and pursued postdoctoral research at Lawrence Berkeley National Laboratory. She worked as a senior chemist at Dow Electronic Materials before joining ORNL in 2017. She has 60 peer-reviewed publications and 1 issued patent. She received the 2022 American Chemical Society PMSE Division's Young Investigator Award. |
1. Introduction
Solid-state, lithium (Li) metal batteries are promising candidates for developing safe, energy-dense devices needed to transition to an electrified economy. Lithium metal's high theoretical specific capacity (3860 mA h g−1) and low redox potential (−3.04 V vs. standard hydrogen electrode) make it especially attractive. However, lithium is highly reactive, making it thermodynamically unstable when in contact with many electrolyte materials. This reactivity can lead to continuous consumption of lithium and the electrolyte, significantly decreasing battery life.1–3 Aside from reactivity, another significant challenge inhibiting the commercialization of lithium metal anode is non-uniform lithium stripping and plating during cycling, which leads to cell shorting. Controlling lithium morphology during cycling is the key to enabling lithium anode.Although liquid electrolytes are currently the most viable options for battery energy storage, they have several drawbacks. Organic liquid electrolytes are highly flammable and prone to leaking and gassing, leading to safety concerns. Most liquid electrolytes are unstable with lithium metal, and the side reactions decrease cycling Coulombic efficiency and battery life. Furthermore, due to unregulated lithium-ion flux, achieving uniform lithium plating and stripping in a liquid system is difficult. Solid-state batteries are a potential solution to these common issues in liquid batteries. Most solid-state electrolytes are nonflammable as they don’t utilize flammable organic solvents, and they have a wider voltage window with thermal stability, making them safer than traditional liquid electrolytes.4 Polymer electrolytes, oxide ceramics, sulfides, and halides are the major types of solid electrolytes. This perspective focuses on lithium morphology in polymer-based systems.
Solid polymer electrolytes (SPEs) have many advantages over liquid electrolytes. These benefits include lithium-ion flux regulation, improved safety features (as they are more electrochemically and thermally stable than liquid electrolytes), and mechanical flexibility and strength.5–7 However, solid polymer electrolytes generally do not have sufficient room temperature ionic conductivity. Adding a liquid component to a polymer host, making it a gel polymer electrolyte (GPE), is an effective way to improve ionic conductivity.8,9
Lithium plating and stripping are inseparably linked to the formation of the solid–electrolyte interphase (SEI), a passivation layer created by the decomposition of the electrolyte and the anode.10–12 The importance of SEI in controlling lithium morphology has been recognized in liquid systems and has been under intense investigation in recent years. In contrast, there are fewer reports about the SEI at the polymer–Li interface, as it is buried and challenging to characterize.
Herein, we provide a perspective of the current understanding of lithium morphology in solid polymer and gel polymer systems, comparing that to typical liquid systems. We summarize the theoretical foundation of the current strategies to control lithium morphology. The limitations of these strategies will be discussed. This perspective consists of five sections. In Section 2, a brief description of lithium morphology control in liquid systems will be presented. In Sections 3 and 4, we examine studies on solid polymer and gel polymer systems, respectively. In Section 5, a comparison of the three systems will be provided.
2. Liquid systems
Lithium plating and stripping in liquid systems have been extensively studied. There are some excellent reviews on this topic.13 Lithium morphology is affected by both the plating and stripping processes during cycling. During plating, lithium first goes through nucleation, followed by growth. Nuclei density is governed by the nucleation overpotential and surface energy of lithium interfaces.14 Although lithium deposits begin with nucleation, growth can occur at both the base and tip of the deposit.3,12,15 The SEI formation rate affects root vs. surface growth modes.12 Lithium is often not deposited uniformly during plating because of crystalline defects and grain boundaries on the electrode or inhomogeneous ion conductivity of the interphase layer.3There have been several attempts to classify the various geometries of lithium deposits. For example, Wood et al. attempted to be concise with just three types: needle, mossy, and fractal dendrites.16 In the review paper by Horstmann et al., two morphologies are distinguished: moss and fractal dendrites.13 Below the limiting current density, lithium metal electrochemically plates as nanosized whiskers, which quickly get entangled and form mossy porous lithium. Above the limiting current density, fractal-like micron-sized dendrites form. Most liquid systems perform below the limiting current density, which is defined by the Sand equation:17
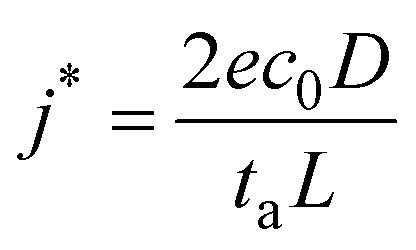 | (1) |
Much of the current research on lithium morphology focuses on plating rather than stripping, primarily because stripping is a subsurface process, making it difficult to characterize. However, understanding stripping is equally important for lithium morphology control. During the stripping process, the inhomogeneous dissolution of Li metal can lead to nanovoids in the anode18–21 and dead lithium.3,19–22 Dead lithium most commonly occurs during the dissolution phase of inhomogeneous stripping. Jiang et al. have proposed three modes of lithium stripping based on dissolution sites, similar to the plating growth modes: the tip-, the base-, and the tip-/base-stripping models.19,23 In the tip-stripping model, dissolution begins at the top of a lithium deposit and works its way down. The lithium maintains contact with the anode throughout the process and will not form dead lithium, making it the most desirable outcome. In the base-stripping model, dissolution begins at the root of the lithium deposit, which becomes electrically isolated from the anode, leading to the formation of dead lithium. This is the most likely outcome, as the roots of Li deposits always exhibit a higher current density than the tip. In the tip-/base-stripping model, the root and tip are active dissolution sites. If the stripping rate at the tip is larger than that at the root, it will reduce the amount of dead lithium produced.
Lithium morphology in liquid systems is intrinsically linked to the SEI as SEI composition and nanostructure affect the plating and stripping of lithium metal.10–12 The properties of this passivation layer significantly affect cell performance. An ideal SEI or interphase layer requires the following features:
(1) High lithium-ion conductivity. If lithium ions cannot efficiently pass through the SEI, cell impedance will increase.24
(2) Little to no electronic conductivity. If electrons are not blocked, there will be continuous lithium and electrolyte decomposition, causing the SEI to grow.25 This growth is related to the next point:
(3) Appropriate thickness with a compact/dense structure. The resistance of the SEI layer is reported to be proportional to its thickness.9,26 Therefore, continuous growth will ultimately reduce the coulombic efficiency and battery capacity.
(4) High elastic strength (high Young's modulus).9,26,27 This is mechanically necessary to suppress dendrite growth; however, the SEI should still be flexible enough to accommodate non-uniform electrochemical behavior.9,26
Unfortunately, most interphase layers are not ideal.
It is believed that fluorination improves the performance of lithium metal batteries by introducing lithium fluoride (LiF) to the SEI, typically via fluoroethylene carbonate (FEC). Lithium is the metal with the lowest redox potential, and fluorine is the strongest oxidizing agent. Because of this, LiF is believed to have more extreme properties than other compounds.28 LiF is an excellent electronic insulator with high interfacial and low diffusing energy, promoting higher interphase stability, limiting corrosive side reactions, and promoting more uniform lithium deposition.28–32,37 Bulk LiF is a poor Li ion conductor, but it has been shown to interface with other crystalline materials in the SEI, improving Li+ conductivity overall.28 Furthermore, incorporating inorganic materials improves the mechanical stability of the SEI, as LiF has a very high Young's modulus (65 GPa) and shear modulus (49 GPa).28 This can help suppress protruding lithium deposits and contribute to the nanostructure.33
There are two established models of SEI nanostructures in liquid electrolytes: the mosaic model and the multilayer model. The mosaic model, initially proposed by Peled et al., suggests a heterogeneous distribution of inorganic and organic components that forms a mosaic of crystalline and amorphous microphases.34 This is the most common nanostructure observed. The multilayer model, initially proposed by Aurbach et al., on the other hand, suggests an ordered structure with separate organic and inorganic layers.35
Currently, the best technique to visualize SEI nanostructures is cryo-transmission electron microscopy (cryo-TEM). To our knowledge, in 2017, Li et al. became the first group to utilize cryo-TEM to characterize lithium metal batteries.36 They investigated the fundamental role of SEI nanostructure on battery performance, as shown in Fig. 1.22 For the mosaic model, which was observed in an unmodified carbonate-based electrolyte (ethylene carbonate/diethyl carbonate, EC/DEC), the primary failure mode was found to be isolated and inactive lithium due to lithium deposits being surrounded by electrically insulating SEI and disconnected from the current collector. This disconnection occurred on one of the branches of the dendrites, leaving behind empty SEI unable to collapse because of dead lithium. In the multilayer model, which was observed in the same base electrolyte with added 10 v% FEC, uniform stripping and plating were observed until the Li–metal was fully stripped. The lithium deposits developed a wavy and curved morphology on the surface of the anode rather than protruding morphologies that could pierce the SEI or become isolated. The uniform morphology seems to suggest that a multilayer SEI is preferred.
 | ||
| Fig. 1 Li metal deposition and stripping morphology with mosaic and multilayer SEI. The top row shows (A) a schematic of the Mosaic SEI nanostructure noted, (B) the top-view SEM of deposited Li, and (C) top-view SEM of stripped Li in an EC/DEC electrolyte. The second row shows (D) a schematic of the multilayer SEI nanostructure noted, (E) the top-view SEM of deposited Li, and (F) top-view SEM of stripped Li in an modified EC/DEC electrolyte with 10 vol% FEC. Reproduced from ref. 22 with permission from Elsevier, copyright 2018. | ||
It is worth mentioning that Li et al. also discovered that contrary to the commonly held belief that the crystalline structure of LiF improves battery performance, there was no LiF lattice after adding FEC.36 Although FEC does not seem to guarantee crystalline LiF, several other studies have reported that using FEC as an additive results in multilayer SEIs.15,37–39 Incorporating more inorganic materials, especially LiF, is known to improve the performance of Li–metal batteries. Due to the recent advances in cryo-TEM technology, the importance of SEI nanostructure is becoming better understood.
3. Dry solid polymer electrolytes
Lithium electrodeposition morphology and the lithium-polymer interface have not been extensively studied in dry SPEs compared with liquid electrolytes. This is due primarily to the difficulty of imaging the Li/SPE interface. Poly(ethylene oxide) (PEO) is the best and most commonly used polymer host to form dry polymer electrolytes.5,40–42 The addition of lithium salt, like lithium bis(trifluoroethane sulfonyl)imide (LiTFSI) or lithium bis(fluoro sulfonyl)imide (LiFSI), has been shown to form a low resistance passivation layer on lithium electrode.43 PEO-based SPEs operate above their melting temperatures (∼60 °C) and create intimate contact with the electrode at melt conditions. This intimate contact is advantageous in battery performance but causes challenges in characterizing the buried Li/SPE interface.3.1 Lithium morphology in dry SPEs
In a pioneering study by Brissot and Rosso et al., direct in situ observation of dendritic electrodeposition of lithium was performed in lithium symmetric cells using the standard polymer electrolyte PEO–LiTFSI.44 They recorded lithium electrodeposition morphology using a microscope and a CCD camera. At low current density regimes (below the limiting current density), whisker morphology was observed; at high current density regimes (above the limiting current density), tree-like morphology was observed. Above the limiting current density, the onset of growth and growth velocity agree with Chazalviel's model.3,10 In this model, if the limiting current density, where the current density exceeds the diffusion limit of lithium, is exceeded, then a large ion concentration gradient is formed, and lithium growth occurs at the tip of the lithium, leading to bulk dendrite growth.In a widely cited study from the Balsara group, synchrotron X-ray microtomography was used to monitor lithium morphology evolution in the lithium symmetric cells sandwiching a PEO–polystyrene block copolymer electrolyte.45 Globule-like morphologies emerged from subsurface structures located within the lithium electrode. This study introduced a powerful tool – X-ray tomography – as a non-destructive way to study buried interfaces between SPE and Li–metal.
In a later review by the same group, Frenck et al. summarized lithium morphology observed in PEO-based electrolytes and compared the data with liquid electrolytes and a few representative ceramic and sulfide solid electrolytes (Fig. 2).46 In liquid systems, lithium growth morphology is mainly determined by the applied current density, as discussed in the previous session. In contrast, in polymer systems, lithium morphology is not only affected by the current density but also governed by the mechanical properties of the polymer electrolyte.47
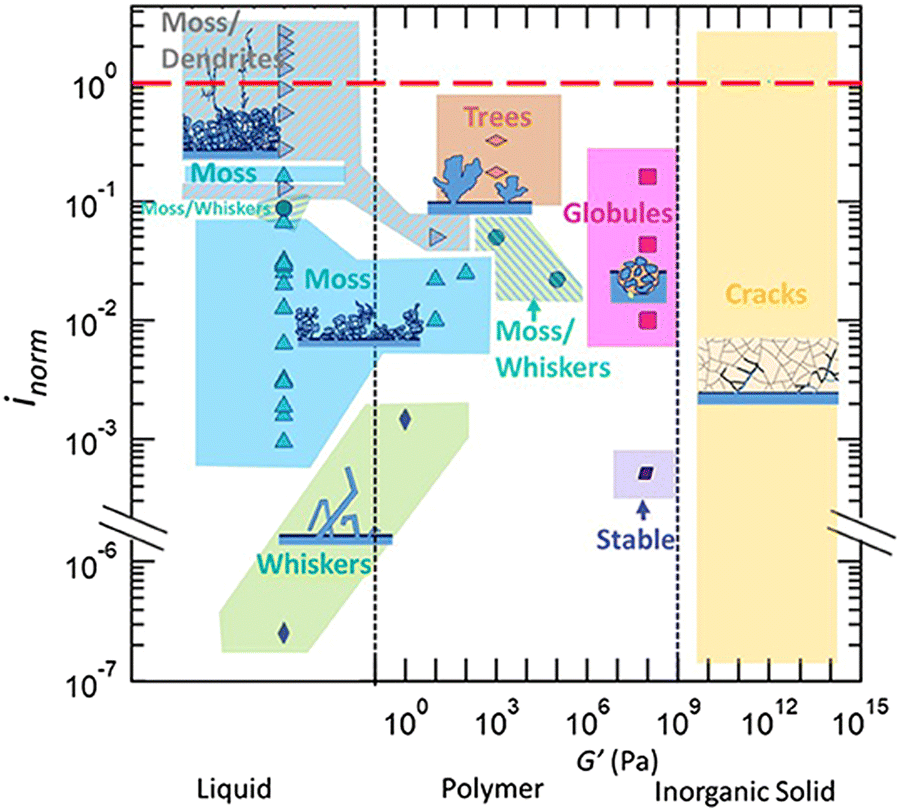 | ||
| Fig. 2 Morphology data of lithium protrusions plotted as a function of the current density normalized by the limiting current density, inorm, vs. the electrolyte type and storage shear modulus, G′. Reproduced from ref. 46 with permission from frontiers, copyright 2019. | ||
In Fig. 2, lithium morphology is plotted as a function of inorm (current density normalized by the limiting current density) and G′ (shear modulus) of the polymer electrolyte. With low modulus polymers and at low inorm values (0.001–0.04), similar lithium morphologies to liquid electrolytes are observed (whiskers and moss). For polymers with higher shear moduli (G′ = 103 Pa), coexisting moss and whisker morphology are seen in the range where inorm = 0.02–0.05. At inorm above 0.2, tree lithium deposits are observed.
Importantly, in this master plot, one polymer electrolyte with a shear modulus of 108 Pa resulted in stable lithium deposition at inorm = 0.005. Although this current is too low for practical applications, note that in typical carbonate-based liquid electrolytes, no stable lithium electrodeposition has been observed, regardless of the applied current density. In the same high shear modulus range, an inorm of 0.01–0.2 results in the formation of globular deposits. Globular deposits progress slower than trees and dendrites. Fig. 2 suggests that SPEs with higher shear moduli lead to Li morphologies that have not been observed in liquid electrolytes. Besides shear modulus, which is related to elastic deformation, a more recent work by the Balsara group showed that SPEs with high yield stress, which is related to plastic deformation, produced slower growing globular protrusions and had better cycle life than those with lower yield stress, which produced dendrites.47 These results give scientists more strategies to tackle this problem, differing from liquid electrolytes.
3.2 Theoretical considerations for lithium dendrite suppression in polymer electrolytes
In 2005, Monroe and Newman published a report, “The Impact of Elastic Deformation on Deposition Kinetics at Lithium/Polymer Interfaces” in the Journal of Electrochemical Society.48 This work laid the theoretical foundation for using the mechanical stress of a polymer electrolyte to suppress lithium dendrite growth. This report concludes that the SPE's shear modulus needs to be approximately twice that of lithium metal to mechanically suppress dendrite growth. This work has been viewed by many as guidance for designing polymer electrolytes to stabilize lithium anode. However, the shear modulus of bulk lithium is 3.4 GPa (3.4 × 109 Pa), at least an order of magnitude higher than a typical polymer electrolyte can achieve, as shown in Fig. 2. Monroe and Newman's prediction has made it a daunting challenge to the polymer community to suppress lithium dendrite growth.As experimental evidence accumulates, apparent discrepancies between experimental data and Monroe and Newman's work are observed. For example, as Fig. 2 shows, polymer electrolytes can exhibit stable lithium plating and stripping under a particular combination of current density and mechanical stiffness, and their mechanical moduli are much lower than twice that of lithium. In contrast, based on Monroe and Newman's model, dendrite should immediately grow in polymer electrolytes with insufficient shear moduli, regardless of the applied current density.
In 2017, Barai and Srinivasan et al. reexamined Monroe and Newman's model and modified some assumptions.49 They discovered that Monroe and Newman's prediction could be voided entirely by changing the force lithium anode experiences from tensile stress (pre-stressed lithium) to compressive stress (relaxed lithium). In the latter case, where lithium experiences compressive stress, stable lithium stripping and plating occur in all electrolytes regardless of their shear moduli. A relatively low applied current density of 0.1 mA cm−2 has been used in this report. This modified calculation result also seems to contradict experimental data as PEO-based electrolytes often short at this current density with extended cycling.
The same group then published another report and further improved their models to include elastic–plastic deformation of both the electrolyte and lithium.50 In Monroe and Newman's model, only elastic deformations were considered. Using this improved methodology, a phase map of the applied current with respect to the shear modulus of the polymer electrolyte is developed to demarcate conditions where stable lithium cycling can be achieved, as shown in Fig. 3.
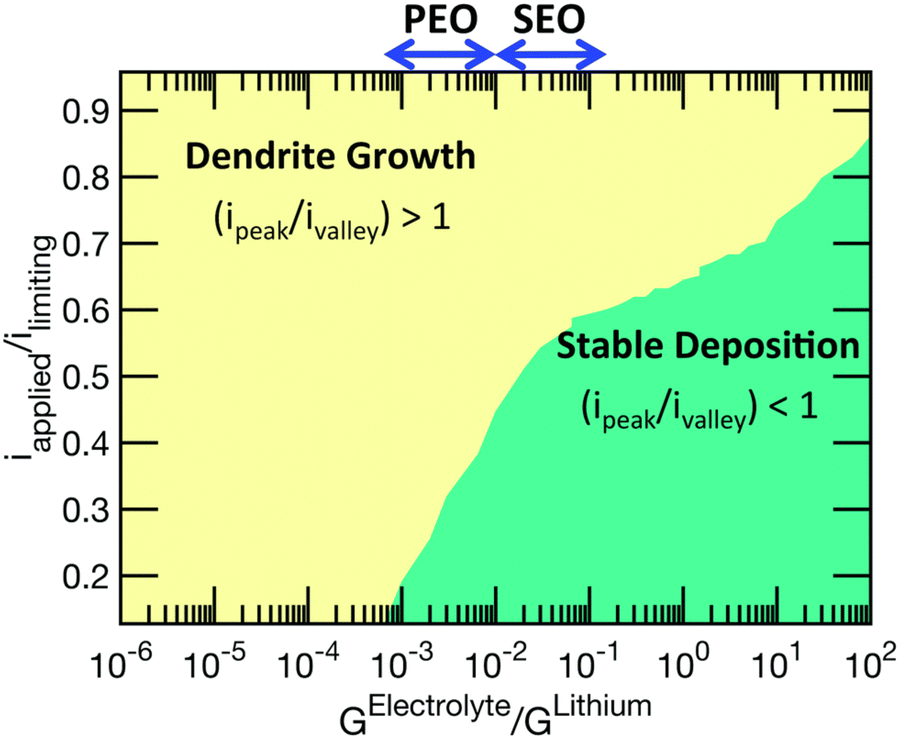 | ||
| Fig. 3 A phase map of the applied current with respect to the shear modulus of the polymer electrolyte phase. Reproduced from ref. 50 with permission from the Royal Society of Chemistry, copyright 2017. | ||
In this phase map, stable lithium electrodeposition can be achieved if the applied current density is less than 40% of the limiting current density for PEO-based electrolytes. The stiffer (larger shear modulus) the polymer electrolyte, the higher the current density (with respect to its limiting current density) it can sustain before dendrite growth can occur. The authors also noted that increasing the yield strength of the electrolyte can effectively suppress dendrite growth, even with a shear modulus two orders of magnitude smaller than that of lithium. This map is more aligned with the experimental data summarized by Frenck et al. in Fig. 2.
More recently, Ganser et al. pointed out that ion transport and mechanics in the electrolyte cannot be treated as decoupled and adopted a coupled electro-chemo-mechanical transport model to examine the effect of transport properties and mechanical stiffness of PEO-based polymer electrolytes on the morphological stability lithium.51 Their results indicate a trade-off relationship between transport properties and mechanical stiffness requirement: i.e., the better the ion transport properties, the lower the Young's modulus required to suppress dendrite growth. Additionally, a higher lithium transference number will also lower the required Young's modulus to stabilize lithium electrodeposition.
It is worth pointing out that recent studies on lithium indicate that the mechanical properties of lithium at interfaces may significantly deviate from its bulk properties.52,53 This may cause a discrepancy between experimental results and theoretical predictions. Further, the SEI layer has not been considered in the theory work cited above. The vital role SEI plays in regulating lithium plating/stripping morphology has been recognized from liquid systems. Yet, it is unclear how the SEI will affect lithium morphology in polymer systems, as it has not been extensively studied and, therefore, is not well understood.
3.3 SEI in dry SPE systems
As mentioned in the previous section, the SEI layer of dry SPEs is not well understood. A few studies investigating the chemical composition of the SEI show that it comprises similar components as liquid electrolytes.54,55 The most prominent inorganic components appear to be Li2O, LiF, Li2CO3, and LiOH. The importance of these components has not been extensively investigated. Unlike liquid electrolyte systems, the SEI in SPEs can take several days to form, depending on the electrolyte's composition, cell assembly, and electrode surface morphology.55,56Similarly, there has been little investigation into the nanostructure of polymer SEI since Peled's initial suggestion of a mosaic model until recently.34 Much, if not all, of the work on imaging the Li/SPE interface using cryo-TEM has been completed by O. Sheng and coworkers. This technique has allowed for a more thorough investigation of the SEI. They found that the interface of unmodified linear PEO–LiTFSI was a mosaic containing only crystalline Li, Li2O, and LiOH randomly distributed in the amorphous component.57,58 The SEI was described as fluffy and porous. The lithium morphology was noted to be blocky or irregular bulk, which differs from the morphologies previously described. Sheng et al. also examined how various modifications to PEO affected the interface. These modifications included adding a platinum nano-layer, fluorination, and iodination. They noted that the lithium grew in dense, almost cube-like bulks for all modifications. The platinum nano-layer resulted in a primarily amorphous mosaic SEI structure with evenly distributed alloys of crystalline Li, Li2O, LiOH, Pt/Li–Pt.57 Similarly, the PEO doped with I2 resulted in a mosaic structure SEI with Li, Li2O, LiI, and LiIO3 nanocrystals embedded in an amorphous layer primarily comprised of polymeric Li salts.59 The inorganic products here differ from what has been observed in SPEs because of the differences in additives. However, when Sheng et al. added Li2S to promote LiF growth, they noted two distinct layers: the surface layer was composed of SPE, Li metal, LiF, and Li2O, while the bottom layer on deposited lithium mainly consisted of Li, LiF, and Li2O.58 Though they called it a mosaic structure, the presence of distinct layers is indicative of the multilayer nanostructure. This combination supports a newly theorized model from Nanda et al. called the nanomosaic–multilayer hybrid SEI model.60 This model combines the two commonly accepted nanostructures into a more complex model. It has thus far only been explored in gel electrolytes; however, Sheng et al.'s description of their fluorinated SEI appears to fit this model best. Nanda et al.'s work was completed with tip-enhanced Raman spectroscopy (TERS), adding another viable technique to analyze nanostructure (Fig. 4).
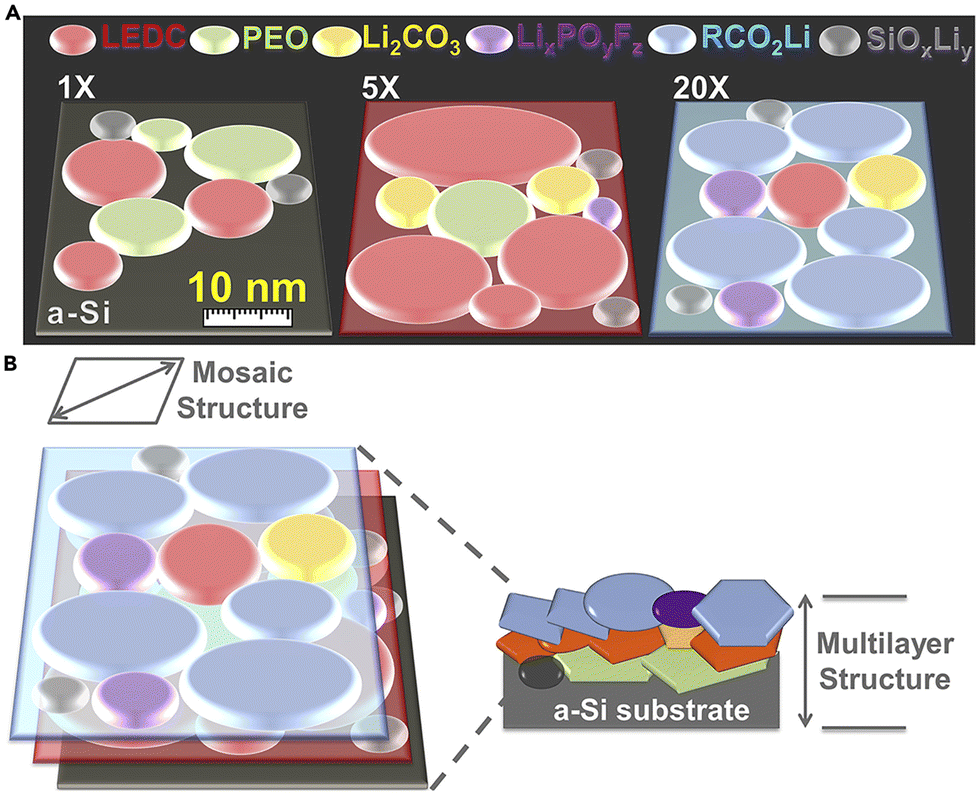 | ||
| Fig. 4 (A) Schematic of the SEI tomography evolution correlated to increasing cycle number probed by tip-enhanced Raman spectroscopy (TERS). (B) Schematic Illustration of the Nanomosaic–Multilayer Hybrid SEI Model on a cycled a-Si surface. Reproduced from ref. 60 with permission from Elsevier, copyright 2019. | ||
4. Gel polymer electrolytes
To decrease operating temperatures, plasticizers are often infused into a polymer electrolyte to form GPEs. We use a separate section to discuss GPEs because this subset of polymer electrolytes is not entirely the same as the dry SPEs. GPEs are semi-solid, and the small molecule plasticizers facilitate lithium-ion transport.8,9,61,62 The presence of small molecule plasticizers also modifies the interface with lithium. There were concerns that adding a liquid phase could compromise thermal stability and safety and deteriorate mechanical strength.8,9 However, it has been shown that the liquid content's effect on flammability and thermal stability can be mitigated because the liquid is hosted in the polymer matrix.634.1 Lithium morphology in GPEs
The lithium morphologies observed in GPEs align with those in liquid electrolytes. Whisker,31,64 mossy,33,61,65 dendrite,33 and spherical deposits33,38,61,64 are the most commonly reported morphologies. More ideal, dense, dome-like morphologies have been reported in some of the fluorinated GPEs,31,61 although not all of them. Other noted morphologies included what was described as “flaky deposits” by Lu et al.,64 and “bush-like” by He et al.66 To our knowledge, the only globular lithium deposit in a GPE was observed in a single-ion conducting gel electrolyte with a PVDF-HFP host.67 Thus far, it seems that globular, tree, and bulk deposits have not been noted in gel electrolytes with PEO backbones.In our own recent work, lithium morphology evolution during initial cycling in a cross-linked PEO-based gel composite electrolyte full cell is monitored via post-mortem photographs and scanning electron microscopy (SEM), shown in Fig. 5.68 Due to the cross-linked molecular structure of the membrane, cross-linked PEO doesn’t go through a melting transition and can be more easily separated from the lithium anode post-mortem, unlike linear PEO, which melts onto the lithium anode and it is very difficult to expose the interface. Our results show that lithium electrodeposition in the initial cycle is homogeneous and planar. However, severe surface pitting occurs as early as the second stripping cycle. The leading cause of the Li surface roughening and dendrite growth mechanism in the model gel composite electrolyte is pit formation and continuous dissolution during stripping. This dendrite growth mechanism resembles that of the liquid electrolyte. This report substantiated the hypothesis that since a gel electrolyte is a polymeric framework swelled with liquid electrolytes, the underlying growth mechanisms for SEI growth and lithium morphology are similar to liquid electrolytes.11
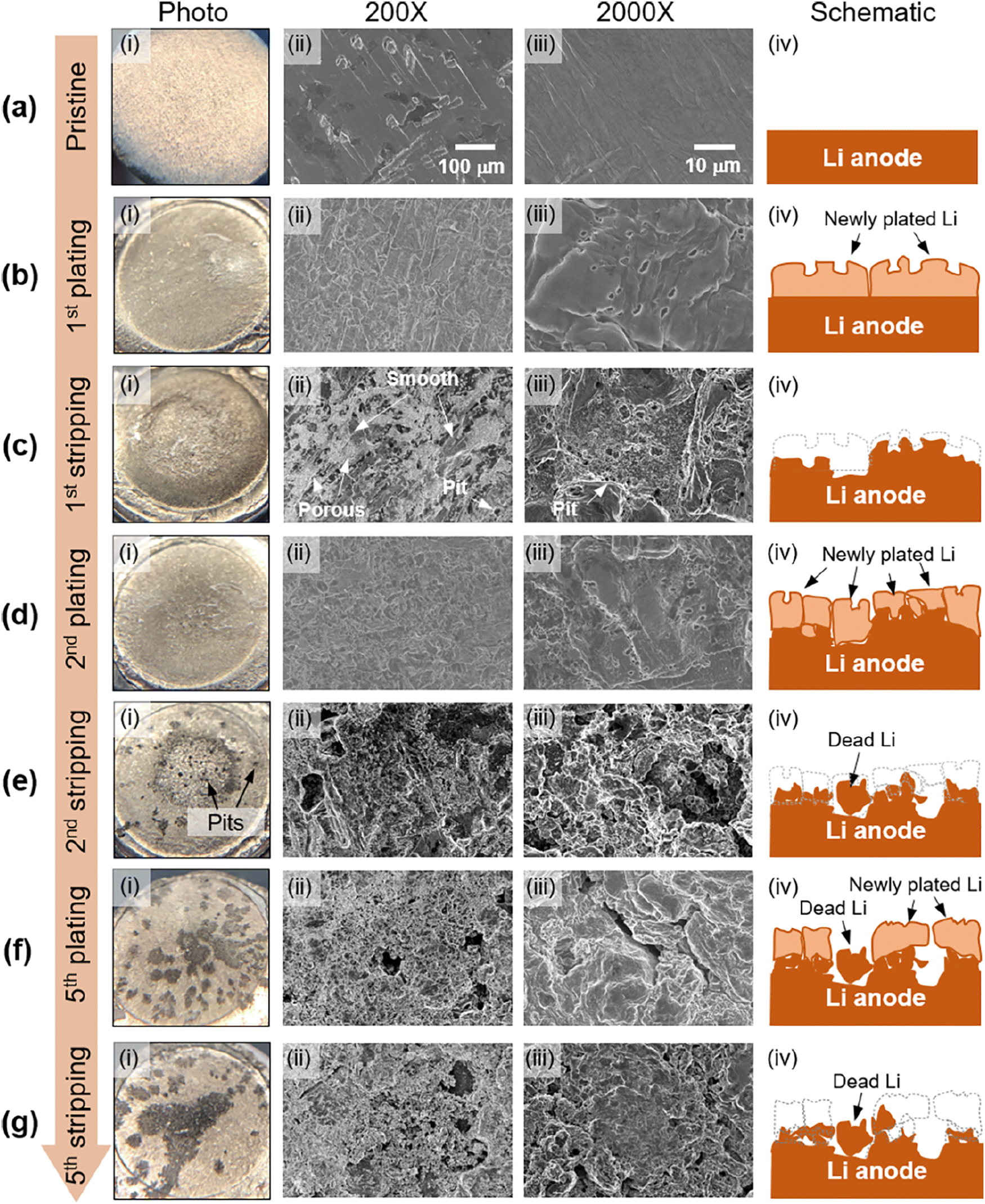 | ||
| Fig. 5 Li anode morphology evolution at early stages of full cell cycling: (i) Li anodes photographs, (ii) 200× and (iii) 2000× SEM images, and (iv) schematics of Li morphology evolution at (a) the pristine stage, (b) end of the first plating, (c) end of the first stripping, (d) end of the second plating, (e) end of the second stripping, (f) end of the fifth plating, and (g) end of the fifth stripping. Reproduced from ref. 68 with permission from the American Chemical Society, copyright 2022. | ||
These similarities to liquid electrolytes do not mean that the polymer does not play a role in lithium deposition in gel electrolytes. In studies where a comparison was made to liquid electrolytes, the lithium morphology of the GPE was far more uniform.31,61 The Archer group has reported at a polymer content of above 40%, the plasticizer molecules are fully associated with the polymer network.69 This allows for regulation of the microscale thermodynamics of the GPE while also controlling macroscale electrokinetics.
Both the mechanical properties and the applied current density affect lithium deposition morphology in GPEs. Y. Shi et al. compared a PEO–LiTFSI plasticized with TEGDME (with a Derjaguin–Müller–Toporov (DMT) modulus of 10.5 MPa) with a high-modulus GPE electrolyte (9.6 GPa).33 At low current densities, uniform plating of spherical lithium deposits was observed. As current density increases, mossy and branched lithium form. Increasing the electrolyte's modulus can partially inhibit lithium dendrite formation. However, the dendrites were eventually able to penetrate the polymer because of poor interfacial wettability. The dependence on current density demonstrates the diffusion-limited Chazalviel model of lithium growth. It also follows the idea of Barai's predictive model (Fig. 3) when combined with the high modulus GPEs’ ability to further suppress dendrites.
H. Wu et al. finds that operating voltage may change lithium morphology.70 For their PEO-based, salt-rich GPE, at 4.3 V, there is homogenous lithium deposition, but increasing to 4.5 V produces uneven lithium growth thickness that varies between 37 and 58 μm but appears to be mossy; the paper does not clarify. Wu's group also noticed that at the higher potential of 4.5 V, there are more organic species and lower lithium content in the SEI than at 4.3 V, where the SEI has higher inorganic and lower organic content. The continued growth and decrease of inorganic components suggest that the SEI at 4.5 V is unstable. Therefore, voltage can affect the composition of the SEI in addition to the rate of growth.70
4.2 SEI in gel polymer electrolyte systems
The SEI composition and nanostructure of GPE systems have been more widely studied and better understood than dry SPEs. In GPEs, lithium salts play a significant role in the formation of the SEI, similar to liquid systems. The decomposition of LiTFSI or LiFSI contributes to the formation of LiF, improving the inorganic components.31,33,70 In Y. Shi et al.'s work, they compared GPE made with LiFSI versus GPE made with LiTFSI and discovered that LiFSI is a more favorable ionic salt for inducing LiF formation.33 In the cells made with LiFSI, dendrites did not form when run at the same potentials as those made with LiTFSI. The new LiFSI gel polymer's SEI had a DMT modulus 5× higher than the original, and the SEI shells were more uniform and richer in LiF. This suggests that the additional mechanical stiffness from the inorganic component played a role in the suppression of the dendrites.Y. Shi's group was also able to visualize the stripping process of their gel electrolyte using in situ optical microscopy, as shown in Fig. 6. Longer stripping caused the volume of mossy dendrites to contract continuously, leaving behind dead lithium surrounded by a hollow SEI shell. During cycling, mossy dendrites spread along boundary sites of the electrode and on the remaining SEI shell. Lithium deposition is not inclined to occur where a hollow SEI shell exists because of the insulating nature of the interphase. This suggests that an SEI shell that does not shrink contributes to the discontinuity and steric hindrance of the gel electrolyte. Dead lithium's role in this issue is emphasized because the SEI shell they investigated maintained the same spherical geometry as the initial uniform lithium deposits.
 | ||
Fig. 6
In situ optical microscopy investigations of Li plating/stripping processes in a GPE-based electrolyte under galvanostatic control, after (a) plating at a current density of −1 mA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−2, (b) stripping at 1 mAcm−2, (c) plating at −5 mAcm−2, (d) stripping at 5 mAcm−2, (e) plating at −8 mAcm−2, and (f) stripping at 8 mAcm−2. Reproduced from ref. 33 with permission from JOHN/WILEY & SONS, INC., copyright 2020. cm−2, (b) stripping at 1 mAcm−2, (c) plating at −5 mAcm−2, (d) stripping at 5 mAcm−2, (e) plating at −8 mAcm−2, and (f) stripping at 8 mAcm−2. Reproduced from ref. 33 with permission from JOHN/WILEY & SONS, INC., copyright 2020. | ||
The stripping process in GPE, observed by Y. Shi et al., reminds one of the stripping process in liquid electrolytes. When lithium deposits are fully dissolved, the SEI shell is able to collapse.15,20,22 However, when dead lithium forms, it becomes trapped in the SEI shell, and the SEI shell cannot collapse or be dissolved further. The isolated lithium and its associated lithium shell separate into the SEI, reducing the cell's capacity as uniform plating is no longer able to occur. Forming an ideal SEI layer is important to reduce the likelihood of this occurring.
The production of hollow SEI shells seems to lend itself to the mosaic model of SEI growth; however, the paper, like many GPE studies, does not investigate the structure of the SEI. Still, layered,64,71 mosaic,31,34,61 and nanomosaic–multilayer hybrid60 structures have been noted in gel electrolytes thanks largely to the incorporation of cryo-TEM. Like in liquid electrolyte systems, mosaic structures are the most common nanostructure in GPEs. However, they typically contain more crystalline components than their liquid counterparts. The higher inorganic content and, therefore, higher stability of the SEI layer seen in gel electrolytes seems to indicate that SEI formation is closer to that of polymers than liquid electrolytes.
Similar to liquid electrolytes, it has been shown that adding fluorination can produce a multilayer SEI in GPEs. Fluorination with FEC and LiBF4 seems to have an advantage over LiPF6, as the latter resulted in a mosaic structure rather than the desired multilayer.64,72,73 However, there is not enough data to draw any conclusions, as only a few studies have investigated the fluorination of GPEs. In a recent study by Lin et al., they examined electrodeposited lithium using a cross-linked PEO-based electrolyte containing a succinonitrile solid plasticizer and FEC additive.31 Using cryo-TEM, they discovered a mosaic structure of the SEI. The FEC additive resulted in a fluorine-rich SEI containing amorphous F-species with inorganic Li2O crystals dispersed within (Fig. 7). This seems to break the trend in liquid systems where FEC is known to cause multilayer structured SEIs.
 | ||
| Fig. 7 (a) Cryogenic HAADF-STEM images and EDS maps of lithium domes using PEO-based electrolyte containing SN and FEC plasticizers. (b) Cryogenic atomic-resolution TEM image of the mosaic SEI's nano-sized domain's different crystallographic orientations. Reproduced from ref. 31 with permission from Springer Nature, copyright 2022. | ||
5. Discussion
We now compare the lithium morphologies in liquid electrolytes, SPEs, and GPEs. We compared 22 papers (9 on GPEs, 9 on liquids, and 4 on SPEs) with 26 samples to note differences in their lithium morphology, SEI composition, and nanostructure. This table was created using the given data and author descriptions. Because of this, there may be inconsistencies due to differing terminology, characterization techniques that cannot detect certain features or chemical compounds, or because a detail was not integral to the paper's argument.5.1 Lithium morphology
As discussed, current density is the governing parameter for Li morphology in liquid electrolytes. As shown by Table 1, no stable cycling can be found in conventional liquid electrolytes; however, improvements have been made with the use of additives to modify the SEI composition. This will be discussed further in the next section.| Study | Electrolyte composition | Li morphology | SEI nanostructure | Inorganic materials | |
|---|---|---|---|---|---|
| Gel polymer | 133 | PEO + TEGDME + LiTFSI + silica | (1) Spherical, (2) mossy, (3) branched dendrites | — | LiF |
| 133 | PEO + TEGDME + LiFSI + silica | Uniform spheres | — | LiF | |
| 231 | PolyEA + TFSI + SN | (1) Whiskers + domes, cracks in the whiskers | — | LiF, Li3N | |
| 231 | PolyEA + TFSI + SN + 5% FEC | Dense domes | Mosaic | Li2O, LiF, Li2CO3, Li3N | |
| 364 | PVDF-HFP + LiBF4 + PC | (1) Domes, (2) flaky Li | Bilayer | Li2O, LiF, LiBF4 | |
| 364 | PVDF-HFP + LiTFSI + PC | Whiskers | Mostly amorphous | Li2O | |
| 761 | PEO + LiPF6 + methacrylate + DCM + EC/DEC | Dense domes | Mosaic | LiOH, Li2O, LiF and Li2CO3 | |
| 1174 | LiTFSI/LiDFOB + FEC + PTFEMA | Flat, dense, mossy | — | LiF, LiBO2 | |
| 1460 | PEO + LEDC | — | Nanomosaic–multilayer hybrid SEI model | Li2CO3, LiOH, Li2O | |
| 1573 | LiFSI + TMP/FEC + PEGMA | Flat, mossy | Mosaic | Li2CO3, LiF, Li2O, Li3N | |
| 1663 | PVDF-HFP + PEGDME | Dense, mossy | — | LiF | |
| 2275 | Random PAN/PVDF-LiFSI + LiPF6 in EC/PC + 2.0 wt% VC | Loose cracks | — | Li3PO4, Li2O, LiF | |
| 2275 | Patterned PAN/PVDF-LiFSI + LiPF6 in EC/PC + 2.0 wt% VC | Dense lithium | — | Li3PO4, Li2O, LiF, Li2CO3 | |
| Liquid | 465 | LiFSI/DME + TFETFE | Flat, dense, mossy | — | LiF, Li2S, Li2O |
| 761 | LiPF6 + EC/DEC | (1) Mossy, (2) whisker | Mosaic, mostly amorphous | LiOH, Li2O and Li2CO3 | |
| 836 | EC/DEC + LiPF6 | (1) Mossy, (2) whisker | Mosaic, mostly amorphous | Li2O, Li2CO3 | |
| 836 | EC/DEC +LiPF6 + 10 v% FEC | (1) Mossy, (2) whisker | Layered | Li2O, Li2CO3 | |
| 1012 | LiTFSI + DOL–DME + 5% LiNO3 | Even spheres | — | LiF, Li2CO3, Li2O, Li2S, Li3N | |
| 1012 | LiPF6 + PC + 2w% LiAsF6 + 2w% VC | Whiskers | — | LiF | |
| 1174 | LiTFSI/LiDFOB + FEC | Protruding, dense moss | — | — | |
| 1938 | LiPF6 in PC:FEC:ethyl 2,2,2-trifluoroethyl carbonate with 10 wt% BCF, 5 wt% LiBF4, and 0.5 wt% LiNO3 | — | Multilayer | LiF, Li2CO3, Li2O | |
| 2039 | LiTFSI in DOL/DME with 1% LiNO3 | — | Multilayer at 60 °C, amorphous at 20 °C | Li2O | |
| 2137 | LiPF6 in DMC and EC + FEC | — | Multilayer | Li2CO3, LiF | |
| Solid polymer | 558 | PEO + LiTFSI + Li2S | Dense bulks | Mosaic | Li2O, LiOH, Li2CO3, Li2S and LiF |
| 558 | PEO + LiTFSI | Irregular, loose | Mosaic | LiOH, Li2O, Li2S, Li2CO3 | |
| 659 | PEO + LiTFSI + 1 wt% I2 | Block structure | Mosaic | Li2O, LiI, LiF, and LiIO3 | |
| 976 | PEO–LiN(CF3SO2)2 | — | — | Li2O, Li | |
| 1854 | PEO + LiTFSI | — | — | Li2O, LiF, Li2CO3 |
In SPEs, on the other hand, current density and mechanical property are both governing parameters for lithium morphology. Theory suggests that the two go hand in hand; a higher mechanical modulus can sustain a higher current density for stable lithium cycling, at least for the plating mechanism. Little research has been done on the stripping mechanism of SPEs. Void formation due to uneven stripping has not been reported in SPE systems. The only instance of void formation we noted in the literature has been attributed to a lack of lithium deposition on top of an impurity particle.77 Their hypothesis that lithium will not deposit on the impurity particle because of its insulating nature follows observations of lithium not depositing on SEI shells in liquid22 and gel systems.33
Lithium morphology in GPEs has a lot of similarities with LEs. As far as we know, there have not been studies that describe the bulks, blocks, or globules that are seen in dry polymer systems. Furthermore, void formation and its resulting pitting have been shown in GPE systems.68 However, there are some differences, as a planar morphology seems more likely to be achieved in GPEs. There are three possible explanations from the literature that we have gathered: (1) participation of TFSI leads to a more crystalline SEI layer, which will be discussed further in the next section; (2) the polymer network allows for regulation of the microscale thermodynamics of the GPE while also controlling macroscale electrokinetics;69 and (3) higher mechanical modulus. Despite the morphological similarities to liquid electrolytes, GPE systems are believed to depend on mechanical moduli. As mentioned previously, Y. Shi's group showed that increasing the electrolyte's DMT modulus could partially inhibit dendrite formation.33 However, as previously discussed with the SPE systems, the modulus of the electrolyte is not the only factor in suppressing dendrite formation.33
5.2 SEI
To better understand the chemical composition of the various SEIs in these studies, Table 2 below summarizes the top 5 inorganic species noted in the SEI of these 26 samples.| LiF | Li2O | Li2CO3 | LiOH | Li2S | # of samples | |
|---|---|---|---|---|---|---|
| Liquid | 6 | 7 | 6 | 1 | 2 | 10 |
| Solid polymer | 3 | 5 | 3 | 2 | 2 | 5 |
| Gel polymer | 9 | 6 | 4 | 2 | 0 | 11 |
| Total | 18 | 18 | 13 | 5 | 4 | 26 |
The only uniform Li morphology noted in this set of studies for liquid electrolytes was in those containing Li2S. The presence of LiF and Li2O did not seem to always prevent the growth of Li whiskers. In polymer systems, introducing or increasing the amount of LiF and/or Li2O in the SEI promoted more uniform lithium morphology. LiF can be promoted directly (i.e., FEC31,73,74 or LiPF664) or indirectly (i.e., Li2S58). There are inconsistencies in whether the TFSI participates in SEI formation in the polymer systems. There are studies where LiTFSI is utilized, but no LiF is reported. Again, the lack of noted LiF could be because of the technique used.
There is no clear link between these inorganic species and the nanostructure of the SEI. Most of the studies did not investigate whether the inorganic species were in a crystalline or amorphous phase. These studies also did not investigate the overall nanostructure of the SEI. From the studies that did utilize cryo-TEM, there was a variety of amorphous and crystalline combinations of the above inorganic species. Still, only six samples were officially classified as having a layered nanostructure, four of which were liquid,22,37–39 and two GPEs.64,73 This, however, makes sense as the mosaic nanostructure is the most common.
Liquid systems, overall, tend to have primarily amorphous SEI layers, with only a small portion of their inorganic species being crystalline.61,78 GPEs, on the other hand, seem to have more significant amounts of crystalline species in their nanostructure. Few studies have looked into the SEI of SPEs, and even fewer have investigated the nanostructure of the SEI. Those conducted by Sheng et al. have classified all their SPEs’ SEI nanostructures as mosaics. A better understanding of how the chemical composition and nanostructure of the SEI affect lithium morphology and what additives impact this is imperative to improving the viability of lithium metal batteries.
5.3 Strategies to stabilize lithium anode
As discussed in earlier sections, SEI plays a critical role in regulating lithium morphologies in liquid systems. SEI design and engineering is the main strategy to stabilize lithium electrodeposition in liquid systems. Recently, new type of liquid electrolytes, localized high-concentration electrolytes (LHCE), are under intense investigation. LHCE changes the lithium-ion solvation structure and the associated SEI formation process. Ren et al. developed an LHCE formulation that promotes an effective protection interphase enriched in LiF. The full cell can cycle stably for 150 cycles under practical conditions.79Guided by the theory work from Newman, Srinivasan, and Ganser, in dry SPE systems, we can generalize two strategies for stabilizing lithium metal anode: (1) increase the mechanical stiffness of the polymer electrolyte. However, this is challenging and may compromise transport and interfacial properties. (2) Maintain some mechanical stiffness and significantly increase the limiting current density of the polymer electrolytes. According to eqn (1), to increase limiting current density, one could design a polymer electrolyte with improved diffusion coefficient, reduced anion transference number, or reduced thickness. Particularly, a class of polymer electrolyte with covalently immobilized anions, the single-ion-conducting polymers, have a cation transference number greater than 0.9.80 The high cation transference number may lead to greatly improved limiting current densities. However, single-ion-conducting polymers fail at current densities way below the theoretical values.81 The failure mechanisms are not entirely understood.67 One possible mechanism is that impurities in the lithium metal itself may contribute to the premature cell failure.
Several methods have been proposed to improve the performance of polymer electrolytes by improving ionic conductivity, Li+ transference number, and mechanical properties. They have been discussed in detail in several great review papers.82–84 For example, introducing active and nonactive additives can improve both ionic conductivity and Li+ transference number.82,83 Another method to improve performance is synthesizing block copolymer, graft copolymer, or star polymer electrolytes to achieve high ionic conductivity and desired mechanical properties.83 Reinforcing polymers through electrospinning has been shown to improve the mechanical strength of both SPEs and GPEs and reduce their thickness.75,82
GPEs generally have a much lower mechanical modulus than SPEs due to the presence of plasticizers. Based on the theoretical guidance detailed in Section 3.2, GPEs will require even higher limiting current densities to enable stable lithium deposition. In this sense, a combination of strategies, SEI engineering employed in LEs and polymer designs in SPEs, may be used to stabilize Li anode in GPE systems.
5.4 Gaps
One of the most significant gaps in this work is the lack of understanding of SEI in SPE systems. This gap makes sense as there are several challenges when investigating the interfaces of solid-state batteries. One of the most pervasive is the fact that the interface is buried.18,68,85 This makes it difficult to probe and characterize with many techniques, especially in situ techniques popular in liquid electrolytes. As a result, when methods to improve the polymer are explored, the lithium morphology and composition and nanostructure of the SEI layer are often not investigated. In addition, most investigations are done post-mortem; unfortunately, normal disassembly can disrupt the morphology of the lithium deposits and the SEI. Both mechanically by pulling the layers apart and chemically by introducing new solvents to dissolve residual polymer.58 This can limit the accuracy of the measurements. Cryo-TEM as a technique is maturing, as evidenced by the recent studies involving cyro-TEM to investigate the Li/electrolyte interface of batteries. This new use of the technology has enhanced our ability to investigate SEI.22 Room temperature TEM damages the samples as many battery components are sensitive to electronic-beam irradiation. Furthermore, Li metal doesn’t react with nitrogen at cryogenic temperatures, so the structural and chemical properties of the Li–electrolyte interface are preserved. This reduction in reactivity allows for the visualization of the nanostructures and lithium morphology and the identification of chemical compounds via their crystal orientation. Although challenges still remain with sample preparation and the representativeness of real battery systems, cryo-TEM is one of the best techniques to characterize SEI nanostructures. As this technique matures for lithium metal batteries, it will be important to find ways to better replicate the environment of a full cell battery.A second gap is that lithium dissolution/stripping behavior is not well understood in SPEs and GPEs. The difference in physical nature of the SPEs and GPEs versus liquid electrolytes also affects the lithium dissolution behavior and, therefore, the coulombic efficiencies (CEs) measured in anode-free and thin Li//thick Li cell configurations. Liquid electrolytes can flow into the pits created during stripping and maintain the Li/electrolyte contact. SPEs, on the other hand, are non-flowable (barring linear PEO-based SPEs above their molten temperature) and, therefore, can lose contact with the electrolyte upon stripping and become inactive in the absence of high external pressures. GPEs are in between the two with a flowable component and, as discussed in our recent work,68 show a porous stripped lithium morphology like liquid electrolytes. We recently reported the C.E. of a GPE in thin Li//thick Li cell configuration to be 60–70%,86 which is even lower than the carbonate-based liquid electrolytes without any special additives, suggesting the contact-loss element to be also partially contributing to the low CEs.
Another significant gap in the current research is our understanding of how the properties of the lithium anode itself may affect the SEI layer and, therefore the lithium morphology.67 The mechanical properties of lithium at interfaces deviate from its bulk properties. A better understanding of lithium may feed into more accurate theoretical predictions of polymer electrolyte properties to stabilize lithium anode. For anode-free designs, the surface chemistry of the current collector can affect lithium morphology just as much.38 Research on anode-free designs is still in its infancy.
6. Conclusion
In this work, we examine literature studies of lithium morphologies in solid polymer and gel polymer systems and compare that with liquid systems. In solid polymer electrolytes, lithium morphology is not only a function of the applied current density but also governed by the mechanical properties. Stable lithium electrodeposition may be accomplished by a polymer electrolyte with good stiffness operating at significantly lower current densities than its limiting current density, defined by the Sand equation. In gel polymer electrolytes, the reported lithium morphology is more similar to that in liquid electrolytes, suggesting similar nucleation and growth mechanisms.Based on experimental evidence and theoretical guidance, two strategies can be adopted to promote stable lithium electrodeposition in SPEs: greatly increase the mechanical modulus of the SPE, or greatly increase its limiting current density while maintaining some mechanical stiffness. The caveat of the former is that it is challenging to achieve mechanical modulus in the GPa range and improvement in mechanical modulus often leads to compromised ion transport properties and interfacial contact. The limitation of the latter is that polymer electrolytes with high theoretical limiting current densities such as single-ion-conducting polymers, may still fail at current densities much lower than the theoretical values. Given the similarities between gel polymer electrolytes and liquids, a combination of SEI engineering and polymer design may be adopted to enable lithium anode in GPE systems. To achieve revolutionary advancements in polymer lithium batteries, a primary focus should be on enhancing our understanding of how to better control Li+ flux.
Finally, there are some significant gaps in the understanding of lithium morphology control in SPE and GPE systems: the need to understand the role of SEIs in SPEs, the lack of study on the dissolution/stripping behavior of SPEs, and the need to better understand lithium itself. With this perspective, we hope to help readers from outside the polymer community to quickly grasp the current status of lithium morphology studies in polymer-based batteries and provoke thoughts from readers inside the community to generate new ideas towards designing polymer systems for stable lithium plating and stripping.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was sponsored by the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy for the Vehicle Technologies Office's Advanced Battery Materials Research Program (Simon Thompson and Tien Duong, Program Manager).References
- D. Ren, K. Smith, D. Guo, X. Han, X. Feng, L. Lu, M. Ouyang and J. Li, J. Electrochem. Soc., 2018, 165, A2167–A2178 CrossRef CAS.
- S. Lou, F. Zhang, C. Fu, M. Chen, Y. Ma, G. Yin and J. Wang, Adv. Mater., 2021, 33, 2000721 CrossRef CAS PubMed.
- J. H. Um, S.-H. Yu, J. H. Um and S.-H. Yu, Adv. Energy Mater., 2021, 11, 2003004 CrossRef CAS.
- T. Inoue and K. Mukai, ACS Appl. Mater. Interfaces, 2017, 9, 1507–1515 CrossRef CAS PubMed.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- N. Riphaus, B. Stiaszny, H. Beyer, S. Indris, H. A. Gasteiger and S. J. Sedlmaier, J. Electrochem. Soc., 2019, 166, A975–A983 CrossRef CAS.
- A. A. Rojas, S. Inceoglu, N. G. Mackay, J. L. Thelen, D. Devaux, G. M. Stone and N. P. Balsara, Macromolecules, 2015, 48, 6589–6595 CrossRef CAS.
- M. Que, Y. Tong, G. Wei, K. Yuan, J. Wei, Y. Jiang, H. Zhu and Y. Chen, J. Mater. Chem. A, 2016, 4, 14132–14140 RSC.
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Chem. Rev., 2020, 120, 6820–6877 CrossRef CAS PubMed.
- J. A. Lewis, F. Javier Quintero Cortes, M. G. Boebinger, J. Tippens, T. S. Marchese, N. Kondekar, X. Liu, M. Chi, M. T. McDowell and G. W. Woodruff, ACS Energy Lett., 2019, 4, 591–599 CrossRef CAS.
- B. Wu, S. Wang, J. Lochala, D. Desrochers, B. Liu, W. Zhang, J. Yang and J. Xiao, Energy Environ. Sci., 2018, 11, 1803–1810 RSC.
- Y. He, W. Fan, Y. Zhang, Z. Wang, X. Li, Z. Liu and Z. Lü, ACS Appl. Mater. Interfaces, 2020, 12, 22268–22277 CrossRef CAS PubMed.
- B. Horstmann, J. Shi, R. Amine, M. Werres, X. He, H. Jia, F. Hausen, I. Cekic-Laskovic, S. Wiemers-Meyer, J. Lopez, D. Galvez-Aranda, F. Baakes, D. Bresser, C. C. Su, Y. Xu, W. Xu, P. Jakes, R. A. Eichel, E. Figgemeier, U. Krewer, J. M. Seminario, P. B. Balbuena, C. Wang, S. Passerini, Y. Shao-Horn, M. Winter, K. Amine, R. Kostecki and A. Latz, Energy Environ. Sci., 2021, 14, 5289–5314 RSC.
- J. I. Yamaki, S. I. Tobishima, K. Hayashi, K. Saito, Y. Nemoto and M. Arakawa, J. Power Sources, 1998, 74, 219–227 CrossRef CAS.
- A. Kushima, K. P. So, C. Su, P. Bai, N. Kuriyama, T. Maebashi, Y. Fujiwara, M. Z. Bazant and J. Li, Nano Energy, 2017, 32, 271–279 CrossRef CAS.
- K. N. Wood, M. Noked and N. P. Dasgupta, ACS Energy Lett., 2017, 2, 664–672 CrossRef CAS.
- K. Borzutzki, J. R. Nair, M. Winter and G. Brunklaus, ACS Appl. Mater. Interfaces, 2022, 14, 5211–5222 CrossRef CAS PubMed.
- F. Shi, A. Pei, D. T. Boyle, J. Xie, X. Yu, X. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8529–8534 CrossRef CAS PubMed.
- F.-N. Jiang, S.-J. Yang, H. Liu, X.-B. Cheng, L. Liu, R. Xiang, Q. Zhang, S. Kaskel and J.-Q. Huang, SusMat, 2021, 1, 506–536 CrossRef CAS.
- S. Y. Lee, J. Shangguan, S. Betzler, S. J. Harris, M. M. Doeff and H. Zheng, Nano Energy, 2022, 102, 107641 CrossRef CAS.
- D. Tewari, S. P. Rangarajan, P. B. Balbuena, Y. Barsukov and P. P. Mukherjee, J. Phys. Chem. C, 2020, 124, 6502–6511 CrossRef CAS.
- Y. Li, W. Huang, Y. Li, A. Pei, D. T. Boyle and Y. Cui, Joule, 2018, 2, 2167–2177 CrossRef CAS.
- A. Jana and R. E. García, J. Electrochem. Soc., 2023, 170, 030533 CrossRef CAS.
- P. Lu, C. Li, E. W. Schneider and S. J. Harris, J. Phys. Chem. C, 2014, 118, 896–903 CrossRef CAS.
- F. Ding, W. Xu, X. Chen, J. Zhang, M. H. Engelhard, Y. Zhang, B. R. Johnson, J. V. Crum, T. A. Blake, X. Liu and J.-G. Zhang, J. Electrochem. Soc., 2013, 160, A1894–A1901 CrossRef CAS.
- X. B. Cheng, R. Zhang, C. Z. Zhao, F. Wei, J. G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef PubMed.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- Y. Wang, J. Liang, X. Song and Z. Jin, Energy Storage Mater., 2023, 54, 732–775 CrossRef.
- Y. Yuan, F. Wu, Y. Bai, Y. Li, G. Chen, Z. Wang and C. Wu, Energy Storage Mater., 2019, 16, 411–418 CrossRef.
- J. Pan, Y. T. Cheng and Y. Qi, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 134116 CrossRef.
- R. Lin, Y. He, C. Wang, P. Zou, E. Hu, X. Q. Yang, K. Xu and H. L. Xin, Nat. Nanotechnol., 2022, 17, 768–776 CrossRef CAS PubMed.
- X.-Q. Zhang, X.-B. Cheng, X. Chen, C. Yan, Q. Zhang, X. Zhang, X. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef.
- Y. Shi, J. Wan, G.-X. Liu, T.-T. Zuo, Y.-X. Song, B. Liu, Y.-G. Guo, R. Wen, L.-J. Wan, Y. Shi, J. Wan, G. Liu, T. Zuo, Y. Song, Y. Guo, R. Wen, L. Wan and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 18120–18125 CrossRef CAS PubMed.
- E. Peled, D. Golodnitsky and G. Ardel, J. Electrochem. Soc., 1997, 144, L208–L210 CrossRef CAS.
- D. Aurbach, E. Zinigrad, Y. Cohen and H. Teller, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS.
- Y. Li, Y. Li, A. Pei, K. Yan, Y. Sun, C. L. Wu, L. M. Joubert, R. Chin, A. L. Koh, Y. Yu, J. Perrino, B. Butz, S. Chu and Y. Cui, Science, 2017, 358, 506–510 CrossRef CAS PubMed.
- X. Zhou, P. Li, Z. Tang, J. Liu, S. Zhang, Y. Zhou and X. Tian, Energies, 2021, 14, 7467 CrossRef CAS.
- Y. Gao, T. Rojas, K. Wang, S. Liu, D. Wang, T. Chen, H. Wang, A. T. Ngo and D. Wang, Nat. Energy, 2020, 5, 534–542 CrossRef CAS.
- J. Wang, W. Huang, A. Pei, Y. Li, F. Shi, X. Yu and Y. Cui, Nat. Energy, 2019, 4, 664–670 CrossRef CAS.
- C. Liu, X. Tang, Y. Wang, R. L. Sacci, W. Bras, J. K. Keum and X. C. Chen, ACS Macro Lett., 2022, 11, 595–602 CrossRef CAS PubMed.
- Y. Zhao, R. Tao and T. Fujinami, Electrochim. Acta, 2006, 51, 6451–6455 CrossRef CAS.
- S. Tang, W. Guo and Y. Fu, Adv. Energy Mater., 2021, 11, 2000802 CrossRef CAS.
- L. Edman, M. M. Doeff, A. Ferry, J. Kerr and L. C. De Jonghe, J. Phys. Chem. B, 2000, 104, 3476–3480 CrossRef CAS.
- C. Brissot, M. Rosso, J. N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81–82, 925–929 CrossRef CAS.
- K. J. Harry, D. T. Hallinan, D. Y. Parkinson, A. A. MacDowell and N. P. Balsara, Nat. Mater., 2013, 13, 69–73 CrossRef PubMed.
- L. Frenck, G. K. Sethi, J. A. Maslyn and N. P. Balsara, Front. Energy Res., 2019, 7, 115 CrossRef.
- S. Chakraborty, G. K. Sethi, L. Frenck, A. S. Ho, I. Villaluenga, H. Wantanabe and N. P. Balsara, ACS Appl. Energy Mater., 2022, 5, 852–861 CrossRef CAS.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, A396–A404 CrossRef CAS.
- P. Barai, K. Higa and V. Srinivasan, J. Electrochem. Soc., 2017, 164, A180–A189 CrossRef CAS.
- P. Barai, K. Higa and V. Srinivasan, Phys. Chem. Chem. Phys., 2017, 19, 20493–20505 RSC.
- M. Ganser, F. E. Hildebrand, R. M. McMeeking and M. Kamlah, J. Electrochem. Soc., 2020, 167, 130525 CrossRef CAS.
- E. G. Herbert, N. J. Dudney, M. Rochow, V. Thole and S. A. Hackney, J. Mater. Res., 2019, 34, 3593–3616 CrossRef CAS.
- E. G. Herbert, S. A. Hackney, V. Thole, N. J. Dudney and P. Sudharshan Phani, J. Mater. Res., 2018, 33, 1347–1360 CrossRef CAS.
- C. Xu, B. Sun, T. Gustafsson, K. Edström, D. Brandell and M. Hahlin, J. Mater. Chem. A, 2014, 2, 7256–7264 RSC.
- E. E. Ushakova, A. Frolov, A. A. Reveguk, D. Y. Usachov, D. M. Itkis and L. V. Yashina, Appl. Surf. Sci., 2022, 589, 153014 CrossRef CAS.
- I. Ismail, A. Noda, A. Nishimoto and M. Watanabe, Electrochim. Acta, 2001, 46, 1595–1603 CrossRef CAS.
- O. Sheng, C. Jin, M. Chen, Z. Ju, Y. Liu, Y. Wang, J. Nai, T. Liu, W. Zhang and X. Tao, J. Mater. Chem. A, 2020, 8, 13541–13547 RSC.
- O. Sheng, J. Zheng, Z. Ju, C. Jin, Y. Wang, M. Chen, J. Nai, T. Liu, W. Zhang, Y. Liu, X. Tao, O. Sheng, J. Zheng, Z. Ju, C. Jin, Y. Wang, M. Chen, J. Nai, T. Liu, W. Zhang, Y. Liu and X. Tao, Adv. Mater., 2020, 32, 2000223 CrossRef CAS PubMed.
- O. Sheng, H. Hu, T. Liu, Z. Ju, G. Lu, Y. Liu, J. Nai, Y. Wang, W. Zhang, X. Tao, O. Sheng, H. Hu, T. Liu, Z. Ju, G. Lu, Y. Liu, J. Nai, Y. Wang, W. Zhang and X. Tao, Adv. Funct. Mater., 2022, 32, 2111026 CrossRef CAS.
- J. Nanda, G. Yang, T. Hou, D. N. Voylov, X. Li, R. E. Ruther, M. Naguib, K. Persson, G. M. Veith and A. P. Sokolov, Joule, 2019, 3, 2001–2019 CrossRef CAS.
- V. Jabbari, V. Yurkiv, M. G. Rasul, M. T. Saray, R. Rojaee, F. Mashayek and R. Shahbazian-Yassar, Energy Storage Mater., 2022, 46, 352–365 CrossRef.
- C. Liu, R. L. Sacci, R. Sahore, G. M. Veith, N. J. Dudney and X. C. Chen, J. Power Sources, 2022, 527, 231165 CrossRef CAS.
- J. Castillo, A. Santiago, X. Judez, I. Garbayo, J. A. Coca Clemente, M. C. Morant-Miñana, A. Villaverde, J. A. González-Marcos, H. Zhang, M. Armand and C. Li, Chem. Mater., 2021, 33, 8812–8821 CrossRef CAS.
- X. Lu, Y. Cheng, M. Li, Y. Zou, C. Zhen, D. Wu, X. Wei, X. Li, X. Yang, M. Gu, X. Lu, Y. Cheng, M. Li, Y. Zou, C. Zhen, D. Wu, X. Wei, X. Li, X. Yang and M. Gu, Adv. Funct. Mater., 2023, 2212847 CrossRef CAS.
- J. F. Ding, R. Xu, N. Yao, X. Chen, Y. Xiao, Y. X. Yao, C. Yan, J. Xie and J. Q. Huang, Angew. Chem., Int. Ed., 2021, 60, 11442–11447 CrossRef CAS PubMed.
- X. He, S. Schmohl and H. D. Wiemhöfer, ChemElectroChem, 2019, 6, 1166–1176 CrossRef CAS.
- L. Frenck, P. Lennartz, D. Y. Parkinson, M. Winter, N. P. Balsara and G. Brunklaus, ACS Appl. Mater. Interfaces, 2022, 14, 53893–53903 CrossRef CAS PubMed.
- W. Y. Tsai, X. C. Chen, S. Kalnaus, R. Sahore, Z. Du and A. S. Westover, ACS Appl. Energy Mater., 2022, 5, 11362–11369 CrossRef CAS.
- S. Choudhury, S. Stalin, D. Vu, A. Warren, Y. Deng, P. Biswal and L. A. Archer, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- H. Wu, P. Gao, H. Jia, L. Zou, L. Zhang, X. Cao, M. H. Engelhard, M. E. Bowden, M. S. Ding, J. Hu, D. Hu, S. D. Burton, K. Xu, C. Wang, J. G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 31583–31593 CrossRef CAS PubMed.
- Z. Li, R. Yu, S. Weng, Q. Zhang, X. Wang and X. Guo, Nat. Commun., 2023, 14, 1–12 CAS.
- Y. Wang, F. Liu, G. Fan, X. Qiu, J. Liu, Z. Yan, K. Zhang, F. Cheng and J. Chen, J. Am. Chem. Soc., 2021, 143, 2829–2837 CrossRef CAS PubMed.
- Z. Wen, Z. Zhao, T. Zhang, Y. Wang, J. Zhang, Z. Sun, L. Li, Y. Li, F. Wu and R. Chen, J. Mater. Chem. A, 2022, 10, 21905–21911 RSC.
- G. Zhou, J. Yu and F. Ciucci, Energy Storage Mater., 2023, 55, 642–651 CrossRef.
- Q. Kang, Z. Zhuang, Y. Liu, Z. Liu, Y. Li, F. Pei, H. Zhu, H. Li, P. Li, Y. Lin, K. Shi, Y. Zhu, J. Chen, C. Shi, Y. Zhao, P. Jiang, Y. Xia, D. Wang, X. Huang, Q. Kang, Y. Liu, H. Li, P. Li, Y. Lin, K. Shi, Y. Zhu, J. Chen, P. Jiang, X. Huang, Z. Zhuang and D. Wang, Adv. Mater., 2023, 2303460 CrossRef CAS PubMed.
- F. Kong and F. McLarnon, J. Power Sources, 2000, 89, 180–189 CrossRef CAS.
- K. J. Harry, X. Liao, D. Y. Parkinson, A. M. Minor and N. P. Balsara, J. Electrochem. Soc., 2015, 162, A2699–A2706 CrossRef CAS.
- Y. Xu, H. Wu, H. Jia, J. G. Zhang, W. Xu and C. Wang, ACS Nano, 2020, 14, 8766–8775 CrossRef CAS PubMed.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- J. Zhu, Z. Zhang, S. Zhao, A. S. Westover, I. Belharouak and P.-F. Cao, Adv. Energy Mater., 2021, 11, 2003836 CrossRef CAS.
- K. Borzutzki, J. R. Nair, M. Winter and G. Brunklaus, ACS Appl. Mater. Interfaces, 2022, 14, 5211–5222 CrossRef CAS PubMed.
- Y. Zhao, L. Wang, Y. Zhou, Z. Liang, N. Tavajohi, B. Li, T. Li, Y. Zhao, Y. Zhou, B. Li, L. Wang, Z. Liang, N. Tavajohi and T. Li, Adv. Science, 2021, 8, 2003675 CrossRef CAS PubMed.
- K. S. Ngai, S. Ramesh, K. Ramesh and J. C. Juan, Ionics, 2016, 22, 1259–1279 CrossRef CAS.
- L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- S. Wenzel, T. Leichtweiss, D. Krüger, J. Sann and J. Janek, Solid State Ionics, 2015, 278, 98–105 CrossRef CAS.
- R. Sahore, Z. Du, X. C. Chen, W. B. Hawley, A. S. Westover and N. J. Dudney, ACS Energy Lett., 2021, 6, 2240–2247 CrossRef CAS.
Footnote |
| † This paper was authored by UT-Battelle, LLC, under Contract No. DE-AC05-00OR22725 with the U.S. DOE. The US government retained and the publisher, by accepting the article for publication, acknowledged that the US government retained a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this paper, or allow others to do so, for US government purposes. DOE would provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan). |
| This journal is © The Royal Society of Chemistry 2023 |