
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile fabrication of metal sulfide supraparticles by an in situ decomposition–assembly strategy†
Menglei
Wang
ab,
Fulin
Jia
bc,
Jianxiao
Gong
 *bc and
Yunsheng
Xia
*a
*bc and
Yunsheng
Xia
*a
aKey Laboratory of Functional Molecular Solids, Ministry of Education, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, P. R. China. E-mail: xiayuns@mail.ahnu.edu.cn
bCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, National Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: gongjx@nanoctr.cn
cSchool of Nanoscience and Technology, University of Chinese Academy of Sciences, Beijing 100149, China
First published on 22nd January 2023
Abstract
Supraparticles (SPs) are of great importance in both fundamental and applied studies due to their emerging collective properties, synergistic effects, and various applications. Metal sulfide nanomaterials are of vital importance in biomedicine, catalysis, battery materials, and other fields. Herein, an in situ decomposition–assembly strategy for the versatile fabrication of metal sulfide SPs is developed. In the fabrication, cysteine molecules and metal cations first react and form coordination polymers, which are then decomposed by heating to produce small-sized metal sulfide nanocrystals. Driven by elimination of the high surface energy of NCs generated by thermal decomposition and the van der Waals attraction, the resulting nanocrystals in situ self-assemble each other and form SP products. In addition to homogeneous Cu2S, CdS, and ZnS products, the proposed system can even be extended to fabricate hybrid Cu2S/Fe2O3 SPs. Furthermore, the SP size can be easily tuned from 10 to 100 nm by adjusting the proportion of cysteine and metal ions. The SPs not only exhibit various properties including photothermal conversion, fluorescence, and magnetism, depending on their composition, but can also combine these properties by the formation of hybrid structures.
Introduction
Supraparticles (SPs) are entities self-assembled from multiple building blocks, i.e., nanoparticles, driven by internal or external driving forces, which are endowed with specific morphology, definite size, and hierarchical topology.1–3 Compared with individual nanoparticles, there are several characteristics of SPs: firstly, due to more complex compositions and structures of SPs, the formation process involves more physical and chemical changes, which makes the preparation of SPs more challenging.4 Secondly, apart from the properties of nanoparticles, emerging physical and chemical properties are expected in SPs due to the opto-electronic coupling between the building blocks.5 What's more, SP properties could be accurately modulated by various parameters such as nanoparticle size,6 morphology,7,8 arrangement,9 composition,10,11etc. Finally, functional SPs with definite size, morphology, and hierarchical topology have demonstrated enormous application potentials in various fields, like energy, medicine, and biology.11–14Effective self-assembly strategies and high structural tunability of SPs are essential for developing novel properties inherent to nanoscale structural characteristics. Monodisperse SPs, not only demonstrated as important functional nanomaterials,15 but are also vital in investigating the self-assembly mechanism and serving as building blocks for advanced superstructures through hierarchical self-assembly. For instance, through the oil-in-water emulsion-assisted high-temperature drying method, Vanmaekelbergh et al.16 obtained three types of composite SPs with core@shell semiconductor nanocrystals (NCs), of which the emission can be feasibly tuned from pure to composite colors over the entire visible region by mixing the constituent NCs. Moreover, with penicillamine as the ligand, ZnS NCs can be assembled into monodisperse chiral SPs that can work as photocatalysts to selectively convert L- or D-tyrosine to dityrosine.17
Among these functional inorganic SPs, metal sulfide SPs are exceptionally useful due to their near-infrared region (NIR) absorbance,18 minimal toxicity,10 excellent catalytic/photocatalytic activity,19 robust and inexpensive nature. These characteristics make the synthesis methodology investigation of metal sulfide SPs important, especially meaningful for bio-applications. Our group20 proposed an in situ self-assembly strategy for the preparation of fluorescent and monodisperse CdS SPs. In addition to high stability, good biocompatibility, and high quantum efficiency, cationic surfactant modification not only well modulates the building blocks of CdS for self-assembly, but also endows the formed SPs with mitochondrial targeting ability. What's more, Xu et al.21 successfully synthesized CuxCoyS SPs with a self-limited self-assembly strategy producing the highest reactive oxygen species yield when illuminated with NIR light. Meanwhile, the CuxCoyS SPs can identify cancer cells by intracellular telomerase response fluorescence imaging in living cells. An eco-friendly and highly efficient assembly of metal sulfide SPs is strongly demanded to explore their potentials in biosensing, bioimaging, and biomedical fields. In our previous studies, we have found that, with the help of citrate as the capping ligand, amino acid molecules could facilitate the in situ one-step assembly of copper chalcogenide SPs in the aqueous phase, which has also been observed by other groups.22 This allows the possible design of a green and efficient assembly route with small amino acid molecules for metal sulfide SPs that are suitable for bio-applications.
Here, we developed an eco-friendly aqueous in situ decomposition–assembly strategy for a series of metal sulfide SPs, including CdS, Cu2S, and ZnS, which can even be extended to hybrid Cu2S/Fe2O3 SPs. As the only sulfur source and stabilizer, cysteine reacts with metal cations first to form coordination polymers in the fabrication and then decomposes on heating to generate small-size metal sulfide nanocrystals. Finally, driven by elimination of the high surface energy of NCs generated by thermal decomposition and the van der Waals attraction, the resulting nanocrystals self-assemble in situ and form SP products. It must be noted that this in situ decomposition–assembly strategy of preparing metal sulfide SPs has several advantages. (1) The method is eco-friendly. Cysteine was used instead of the traditional toxic sulfide as the sulfur source, and the SPs were prepared in aqueous phase. This versatile green fabrication approach greatly improves the biocompatibility of the metal sulfide SPs. (2) The obtained SPs have a narrow size distribution, which allows for exploration of hierarchically self-assembled superstructures and related novel properties. (3) The size of SPs can be effectively regulated in a wide range (10–100 nm, typically). This allowed the accurate tunability of their optical properties and benefits the application of these SPs in biosensing and medical imaging diagnosis and treatment. (4) This strategy could open up multifunctional hybrid nanomaterials. The photothermal conversion, fluorescence, and magnetic properties of a single SP are combined through the preparation of hybrid SPs, which could lead to a wider range of applications.
Experimental
Chemicals and materials
CuSO4·5H2O, CdSO4·8H2O, ZnSO4·7H2O, FeCl3·6H2O, and NaOH, were purchased from Innochem (Beijing). Cysteine was acquired from Aladdin (Shanghai, China). Deionized water from Millipore was used for all the experiments.In situ decomposition–assembly of Cu2S, CdS, and ZnS SPs
For the assembly of Cu2S SPs, 3 mL of 0.1 M cysteine aqueous solution and 125 μL of 0.4 M CuSO4·5H2O aqueous solution were mixed with 40 mL of water, followed by adjusting the pH value to 9.8 by addition of 0.5 M NaOH solution. The solution was heated to 80 °C and refluxed with N2 under vigorous stirring. The solution was kept at 80 °C for the reaction of about 12 h. At the end of the reaction, the supernatant was removed after centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RPM for 10 min. The SPs were collected and preserved under a vacuum seal. For the assembly of CdS SPs and ZnS SPs, the experimental procedures were the same as that of Cu2S SPs, except that CuSO4·5H2O was replaced by CdSO4·8H2O and ZnSO4·7H2O, respectively. All the deionized water used here was deoxidized with N2 bubbling.
000 RPM for 10 min. The SPs were collected and preserved under a vacuum seal. For the assembly of CdS SPs and ZnS SPs, the experimental procedures were the same as that of Cu2S SPs, except that CuSO4·5H2O was replaced by CdSO4·8H2O and ZnSO4·7H2O, respectively. All the deionized water used here was deoxidized with N2 bubbling.
In situ decomposition–assembly of hybrid Cu2S/Fe2O3 SPs
3 mL of 0.1 M cysteine aqueous solution, 62.5 μL of 0.4 M CuSO4·5H2O aqueous solution, and 62.5 μL of 0.4 M FeCl3·6H2O aqueous solution were dissolved in 40 mL of water, followed by adjusting the pH value to 9.8 by addition of 0.5 M NaOH. Under vigorous stirring, the solution was heated to 80 °C and refluxed with nitrogen. The solution was kept at 80 °C for the reaction of about 12 h. At the end of the reaction, the SPs were collected by centrifugation at 9000 RPM for 10 min and preserved under a vacuum seal. All the deionized water used here was deoxidized with N2 bubbling.Photothermal performance test of Cu2S and Cu2S/Fe2O3 SPs
The purified Cu2S SPs were first oxidized in air for 24 h to introduce copper deficiencies23 to enhance the photothermal conversion efficiency.24 The SP aqueous solutions with different copper concentrations of 0, 13.5, 27.5, 55, and 110 μg mL−1 were irradiated under an 808 nm laser beam (0.75 W cm−2) for 10 min at room temperature. The temperature change during the heating processes was recorded with an infrared thermal imager. To explore the photostability of the Cu2S SPs, 1 mL of 55 μg mL−1 Cu2S SP aqueous solution was irradiated with an 808 nm laser beam (0.75 W cm−2) for 10 min. Then the laser was turned off to cool the solution to the initial room temperature. The temperature change during the heating and cooling processes was recorded with an infrared thermal imager synchronously. Finally, the optical stability after 10 laser on/off cycles was evaluated.For the Cu2S/Fe2O3 SPs, procedures for photothermal performance tests were the same as that of the Cu2S SPs, except that the SP aqueous solution concentrations were 0, 12.5, 25, 50, and 100 μg mL−1, and a 1064 nm laser (1.0 W cm−2) was used instead of an 808 nm laser.
In vitro MRI test of Cu2S/Fe2O3 SPs
The purified Cu2S/Fe2O3 SP aqueous solutions with different Fe concentrations of 56, 28, 11.2, and 2.8 μg mL−1 were prepared and transferred to the test tube for the MRI test.Computational calculations
The bond energy calculations were performed by strongly constrained and appropriately normed (SCAN)25 meta-generalized-gradient approximation (Meta-GGA) functions as implemented in DMol3.26–28Characterization studies
Transmission electron microscopy (TEM) images and high-resolution TEM (HRTEM) images were obtained on a HT-7800 electron microscope (Hitachi, Japan) and a Tecnai F20 TEM (FEI, America). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and X-ray energy dispersive spectroscopy (X-EDS) were carried out using a Tecnai G2 F20 S-TWIN at 200 kV to characterize the elemental distribution of the SPs. Scanning electron microscopy (SEM) was carried out on a Hitachi SU8220 (Hitachi, Japan). The hydrodynamic particle sizes and ζ-potential values were examined on a Zeta-sizer Nano ZS series instrument (Malvern Instruments Co., Ltd., UK). Powder X-ray diffraction (XRD) patterns were recorded on a D/MAX-TTRIII (Cross Beam Optics) with Cu Kα radiation (λ = 1.542 Å) operating at 40 kV and 300 mA. X-ray photoelectron spectroscopy (XPS) measurements were carried out on an ESCALAB250Xi spectrometer at a pressure of ∼3 × 10−9 mbar using Al Kα as the excitation source (hν = 1486.6 eV) and operated at 15 kV and 20 mA. Absorption and steady-state fluorescence spectra were measured on a Hitachi U-3100 spectrophotometer and Hitachi F-4600 fluorescence spectrophotometer, respectively. Magnetic measurements were recorded on a PPMS-9 superconducting quantum interference device (SQUID) magnetometer (Quantum Design, USA) at −30000 to 30000 Oe and 300 K. Magnetic resonance imaging (MRI) was performed on a BioSpec70/20USR spectrometer (Bruker, Germany). An 808 nm laser with 0.75 W cm−2 (CNI, China) and a 1064 nm laser with 1.0 W cm−2 were used for in vitro photothermal irradiation and the temperature was measured via an infrared thermal imaging instrument (FLIR, A65).
Results and discussion
In situ decomposition–assembly of SPs
In this work, we systematically study a facile in situ decomposition–assembly method for metal sulfide SPs. As shown in Fig. 1, the aqueous reaction solution containing cysteine and metal coordination is adjusted to alkalinity with NaOH, followed by the decomposition of cysteine–Mx+ and assembly of SPs at 80 °C oil bath temperature.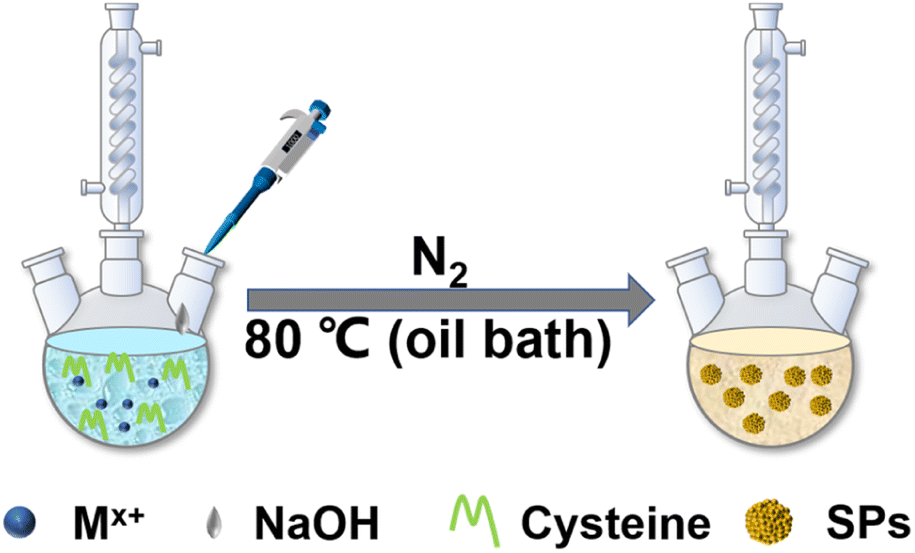 | ||
| Fig. 1 Scheme for the SP fabrication. | ||
It needs to be noted that, as a natural amino acid, cysteine is a non-toxic compound as compared with thioacetamide, thiourea, potassium thiocyanate, or other chemicals. Through the calculation, it is demonstrated that the instability of the C–S bonds from cysteine–Mx+ could enable the formation of functional inorganic metal sulfide SPs. As shown in Fig. 2, by comparing the bond length and bond energy of the C–S bonds in cysteine with that of cysteine–Mx+, it is found that the C–S bonds become less stable with the formation of cysteine–Mx+ during the heating process. As demonstrated by the calculation, the bond length of the C–S bonds in cysteine is 1.826 Å, whereas it becomes longer when cysteine is combined with metal ions, that is 1.849(5) Å (cysteine–Cd2+), 1.849 Å (cysteine–Cu+) (Cu2+ is reduced to Cu+) and 1.850 Å (cysteine–Zn2+), respectively. As consequence, the bond energy of the C–S bonds lowers after cysteine is bonded with metal ions. Specifically, the bond energy of the C–S bonds in pristine cysteine is 340.91 kJ mol−1, while the values decrease to 286.27 kJ mol−1 (cysteine–Cd2+), 302.04 kJ mol−1 (cysteine–Cu+) and 300.45 kJ mol−1 (cysteine–Zn2+), respectively. This implies that through forming cysteine–Mx+, cysteine could serve as a non-toxic sulfur source for constructing bio-compatible metal sulfide structures. In previous studies, cysteine was also used as a stabilizer in the preparation of nanomaterials, and metal sulfide nanomaterials were obtained by applying additional sulfur sources.22,29
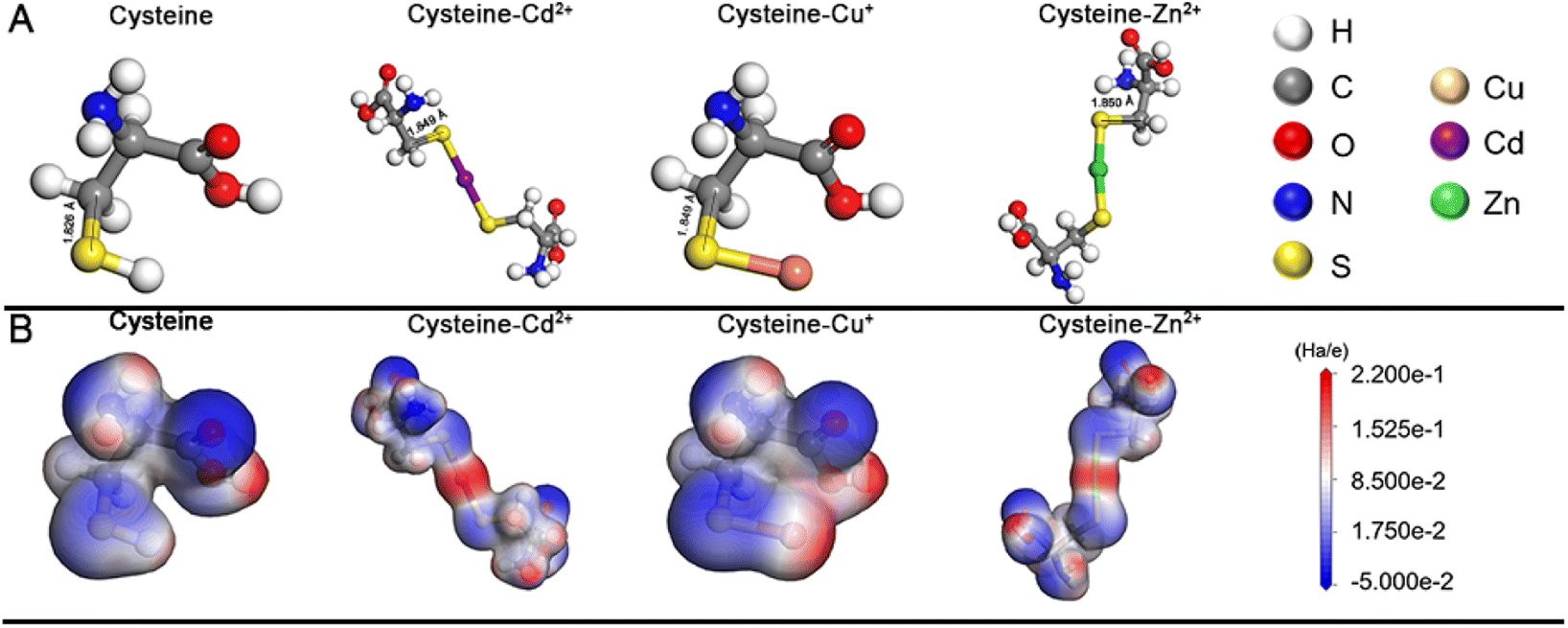 | ||
| Fig. 2 (A) Molecular models for cysteine, cysteine–Cd2+, cysteine–Cu+, and cysteine–Zn2+ after geometric optimization. (B) Molecular electrostatic potential maps for cysteine, cysteine–Cd2+, cysteine–Cu+, and cysteine–Zn2+ after geometric optimization. | ||
Herein, through our in situ decomposition–assembly strategy utilizing cysteine, four types of metal sulfide SPs are successfully obtained (Fig. 3). As shown in Fig. 3A–D, M–P, the obtained SPs are narrow size distributions of all four SPs around 7–11%, as measured by TEM, which is nearly comparable with high-quality NCs. It should be noted that such SPs are the prerequisite for developing a uniform device based on SPs and advanced superlattice through the hierarchical assembly. As observed in high magnification TEM images (Fig. 3E–H), the SPs are assembled by small nanoparticles and form flower-like spherical structures. The compositions of the SPs are characterized preliminarily by the elemental distribution mapping (Fig. 3I–L). The SPs are all composed of certain amount of 4–7 nm nanoparticles as subunits (marked with white circles in Fig. 4A–C), with the lattice fringes of interlayer spacings of 0.330 nm, 0.198 nm, and 0.270 nm, which correspond to the d-spacings of CdS (002), Cu2S (110), and ZnS (200) planes, respectively. Besides, the HRTEM images of the hybrid SPs in Fig. 4D and the corresponding FFT show that the diffraction spots of the two kinds of crystals correspond to the (220) plane of Cu2S and the (110) plane of Fe2O3, respectively. The crystal structures of the SPs are characterized by XRD (Fig. 4E–G), of which the peaks match well with those of the hexagonal Cu2S (JCPDS no. 46-1195), the hexagonal CdS (JCPDS no. 41-1049) and the hexagonal ZnS (JCPDS no. 80-0007), respectively. According to the Debye–Scherrer relation, the sizes of the subunits are 6.6 nm, 4.8 nm, and 3.6 nm, respectively, which correspond well with the TEM images (see ESI Table 1†). In Fig. 4H, the main peaks of 2θ = 31.8° and 45.5° correspond to the (200) and (220) crystal planes of cubic Cu2S (JCPDS no. 84-1770), and the peaks of 2θ = 35.6°, 40.8°, 54.0°, 62.4°, and 64.0° correspond to the (110), (113), (116), (214), and (300) crystal planes of hexagonal Fe2O3 (JCPDS no. 89-0596), respectively. This implies the formation of Cu2S/Fe2O3 hybrid SPs.
 | ||
| Fig. 3 The fabrication strategy used to synthesize SPs. TEM images of (A) CdS SPs, (B) Cu2S SPs, (C) ZnS SPs, and (D) Cu2S/Fe2O3 SPs at low magnification. TEM images of (E) CdS SPs, (F) Cu2S SPs, (G) ZnS SPs, and (H) Cu2S/Fe2O3 SPs at high magnification. The elemental distribution mapping images of the (I) CdS SPs, (J) Cu2S SPs, (K) ZnS SPs, and (L) Cu2S/Fe2O3 SPs. The corresponding size distribution of (M) CdS SPs, (N) Cu2S SPs, (O) ZnS SPs, and (P) Cu2S/Fe2O3 SPs. | ||
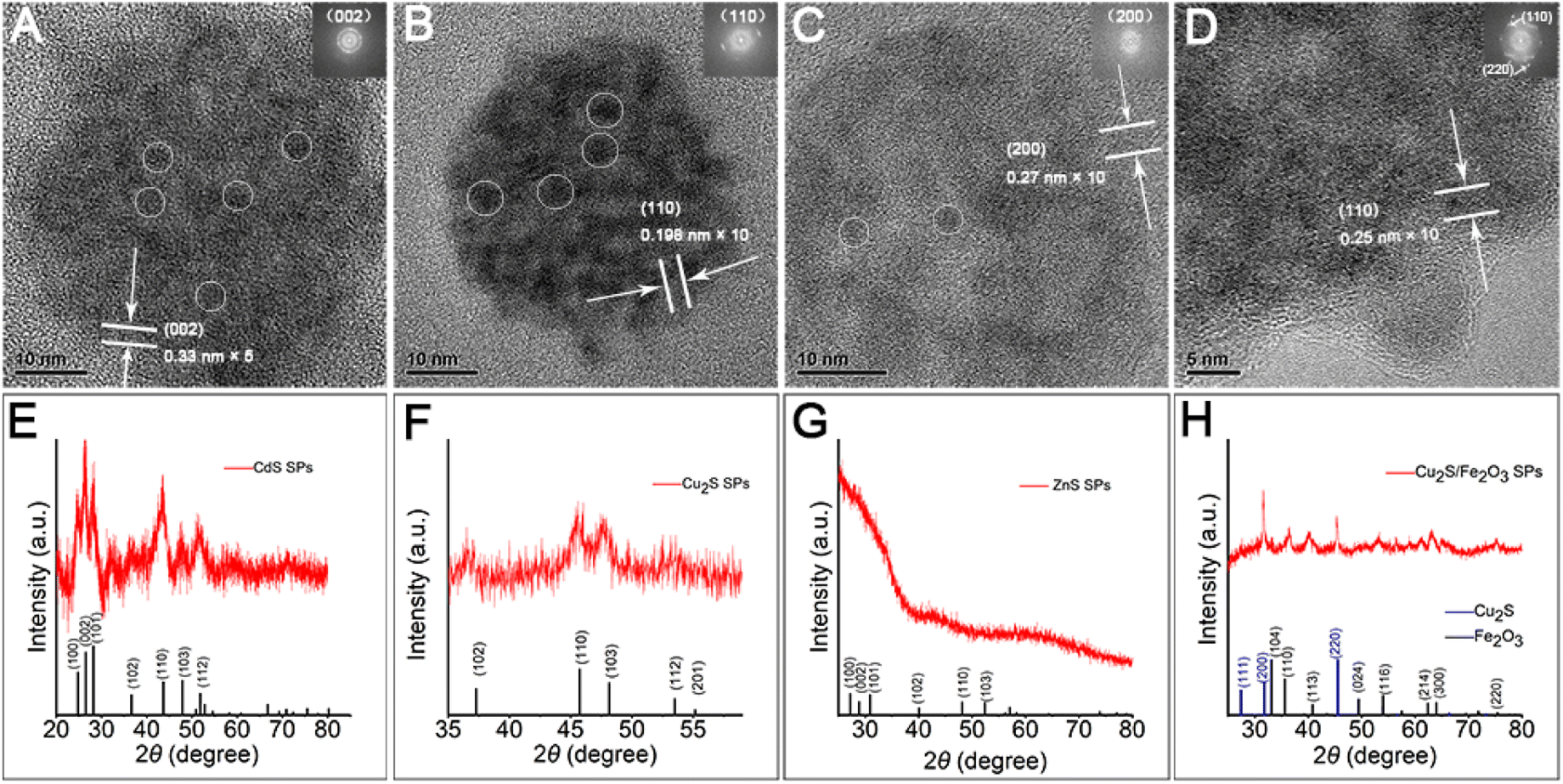 | ||
| Fig. 4 Structural characterization of SPs. HRTEM images of (A) CdS SPs, (B) Cu2S SPs, (C) ZnS SPs, and (D) Cu2S/Fe2O3 SPs. Individual NPs inside SPs are marked with white circles. XRD patterns of (E) CdS SPs, (F) Cu2S SPs, (G) ZnS SPs, and (H) Cu2S/Fe2O3 SPs. | ||
Formation mechanism of SPs
As a small biomolecule with three functional groups (–SH, –NH2, and –COOH), cysteine readily binds to metal cations to form complexes and the form of coordination varies.30 In our reaction systems, cysteine plays multiple roles: (1) as the sulfur source by forming a complex with metal ions, (2) reducing agent, and (3) stabilizer for SPs. With the addition of metal ions into the solution containing cysteine, milky white floc forms (Fig. 5A inset). We take CdS SPs as an example to investigate the formation of SPs. As shown in Fig. 5A and S1,† the white floc is a network composed of long strips. The existence of –NH2 and –COOH in cysteine and cysteine–Mx+ could lead to the formation of hydrogen bonds, thus making them crosslinked with each other to form the network. After adjusting the pH value to 9.8, the white floc disappears and the solution becomes clear and transparent (Fig. 5B inset). The network then decomposed to a smaller strip structure (Fig. 5B). This is due to the destruction of hydrogen bonds under alkaline conditions, which causes disconnection of the cross-links. The crystal plane spacing is 0.316 nm in HRTEM (Fig. S2†), which corresponds to the (101) crystal plane of hexagonal CdS. EDS mapping (Fig. S3†) of the strip structure shows that it is mainly composed of Cd and S, indicating that Cd is indeed bound to cysteine. To further explore the reaction process, samples were taken at different reaction times and analysed. As the reaction proceeds, the strip precursor (Fig. 5B) gradually decomposes into small crystallized particles (Fig. 5C) after heating for 1 h. In the first stage, under hydrothermal conditions, the ligands in the cysteine–Cd complexes were attacked by the strong nucleophilic OH− of NaOH molecules, leading to the weakening and breaking of the C–S bonds,31,32 forming preliminary CdS NCs. The size of these NCs is about 3–5 nm, which is consistent with the final size of the subunits of CdS SPs. This indicates that the CdS SPs are probably formed by the self-assembly of these NCs. In addition, the dynamic light scattering (DLS) data (Fig. 5G) show that the featured peak at about 1 μm gradually disappeared while a new peak around 10 nm (hydration diameter measured by DLS is larger than the actual NC size) grows. This supports the observation that the network precursor experiences consumption and the NCs form during the initial stage of the heating process. In this stage, driven by elimination of the high surface energy of NCs generated by thermal decomposition and the van der Waals attraction, the NCs are assembled to form SPs (Fig. 5D and E). After 12 hours (Fig. 5F), with the depletion of NCs in the solution, the negatively charged cysteine on the SP surface ensured the strong electrostatic repulsion (Fig. S4†) between SPs to prevent them from further growth and aggregation. Similar processes are also observed for other metal ions, such as Cu2S SPs in Fig. S5.† Therefore, the SP formation could be described by a “(I) S decomposition – (II) NC growth – (III) Self-assembly” three stage process as shown in the schematic diagram (Fig. 5H).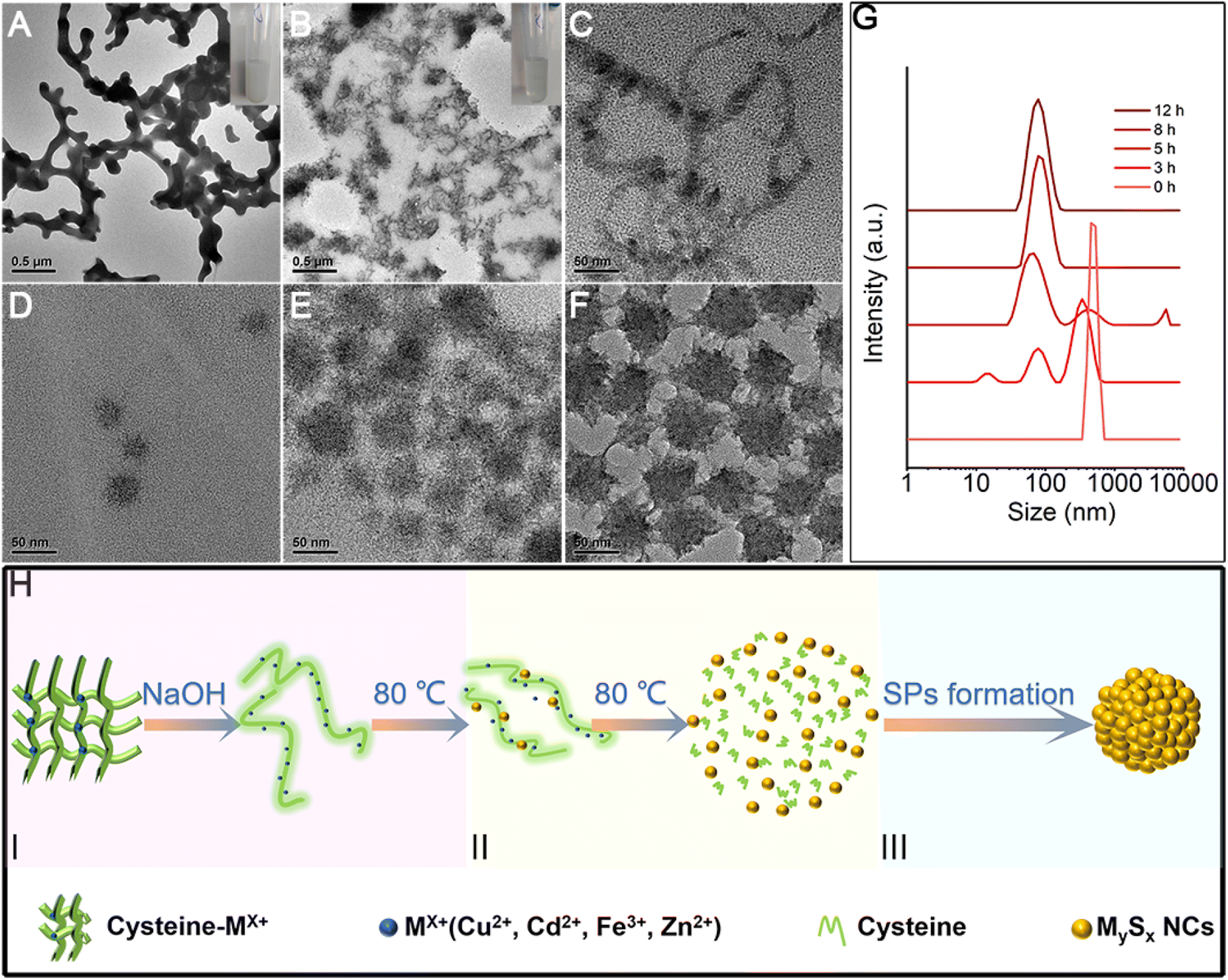 | ||
| Fig. 5 The self-assembly process of CdS SPs. (A) The TEM image of the precursor compound cysteine–Cd2+ before the addition of NaOH solution (the inset shows the photo picture of the corresponding reaction solution). (B–F) The TEM images of the products fabricated at (B) 0 h (the inset shows the photo picture of the corresponding reaction solution in 0 h), (C) 1 h, (D) 3 h, (E) 5 h, and (F) 12 h, respectively. (G) Corresponding DLS data of the reaction solution at different times. (H) Scheme of the SP formation process, I (red): S decomposition, II (yellow): NC growth, III (blue): self-assembly. | ||
As for the formation of the hybrid Cu2S/Fe2O3 SPs, the Ksp[FeS] is 6 × 10−18, which is much larger than Ksp[Cu2S] (2 × 10−48). So the –SH group in cysteine prefers to coordinate with Cu+ (cysteine reduces Cu2+ to Cu+) to form a precursor complex, while Fe3+ mainly combines with the carboxyl group in cysteine to form Fe2O3, which could be proved by the two new bands in the infrared spectra of the precursor complex (Fig. S12†), which correspond to the asymmetric νas (−COO−) and symmetric νs (–COO−) stretching vibrations, and are found at 1403 and 1627 cm−1, respectively. So the carboxyl groups in the ligands coordinate with Fe3+via monodentate bridging.33 Furthermore, the peaks at 667 and 599 cm−1 represent the vibration of the Fe–O lattice.
The size tunability of SPs
The size of the nanomaterials greatly influences their emerging physical and chemical properties,6,34 as well as their performances in related application areas, for instance, the uptake and resulting function of the cell, the mechanism of endocytosis, and the toxicity.35–37 Through tuning the proportion of cysteine and metal ions, the size of obtained SPs can be rationally designed. As the molar ratio of cysteine to Cd2+ in the system gradually increased, the size of obtained CdS SPs becomes larger and larger as shown in Fig. 6A–D, and their size is adjustable in the range of 40–110 nm. The results of in situ DLS detection in Fig. 6E indicate that this size modulation indeed happens in solution rather than on dry TEM grids. As shown in Fig. S6,† the ζ-potential values change from −46.1 to −33.0 mV as the ratio of cysteine to Cd2+ changes from 5:1 to 16:1. In other words, the less surface electrostatic potential causes weaker repulsive effects among the NPs (whether the SPs or their building blocks), which is beneficial for the self-assembly processes and leads to larger sized SPs. In addition to CdS SPs, Cu2S and Cu2S/Fe2O3 SPs of controlled sizes can also be effectively prepared by adjusting the ratio of ligands to metal ions (Fig. 6F–O). Taking Cu2S/Fe2O3 SPs as an example, the size of SPs increases from 29 nm to 93 nm when the ratio of Cu2+ and Fe3+ to cysteine increases from 1:1:12 to 1:1:32. For Cu2S SPs, the size can be accurately tuned from 10 nm to 100 nm by adjusting the ratio of Cu2+ to cysteine from 6:1 to 16:1.
 | ||
| Fig. 6 The size tunability of SPs. (A) TEM image of 33 nm CdS SPs was obtained by adjusting cysteine:Cd2+ = 5:1. (B) TEM image of 44 nm CdS SPs was obtained by adjusting cysteine:Cd2+ = 6:1. (C) TEM image of 75 nm CdS SPs was obtained by adjusting cysteine:Cd2+ = 8:1. (D) TEM image of 104 nm CdS SPs was obtained by adjusting cysteine:Cd2+ = 12:1. (E) Corresponding DLS data of CdS SPs were obtained with different ratios of cysteine to Cd. (F) TEM image of 20 nm Cu2S SPs was obtained by adjusting cysteine:Cu2+ = 6:1. (G) TEM image of 34 nm Cu2S SPs was obtained by adjusting cysteine:Cu2+ = 8:1. (H) TEM image of 39 nm Cu2S SPs was obtained by adjusting cysteine:Cu2+ = 12:1. (I) TEM image of 67 nm Cu2S SPs was obtained by adjusting cysteine:Cu2+ = 16:1. (J) Corresponding DLS data of Cu2S SPs were obtained with different ratios of cysteine to Cu2+. (K) TEM image of 29 nm Cu2S/Fe2O3 SPs with different sizes was obtained by adjusting cysteine:Cu2+:Fe3+ = 12:1:1. (L) TEM image of 39 nm Cu2S/Fe2O3 SPs with different sizes was obtained by adjusting cysteine:Cu2+:Fe3+ = 16:1:1. (M) TEM image of 52 nm Cu2S/Fe2O3 SPs with different sizes was obtained by adjusting cysteine:Cu2+:Fe3+ = 24:1:1. (N) TEM image of 93 nm Cu2S/Fe2O3 SPs with different sizes was obtained by adjusting cysteine:Cu2+:Fe3+ = 32:1:1. (O) Corresponding DLS data of Cu2S/Fe2O3 SPs were obtained with different ratios of cysteine to Cu and Fe. | ||
Related properties and potential applications of SPs
The successful in situ decomposition–assembly of the multiple SPs allows investigation of their properties and potential applications. The CdS SP solution exhibits a bright orange color under ultraviolet (UV) light (shown in the inset of Fig. 7A). Due to quantum confinement effects, CdS SPs exhibit a significant blue shift in the broad absorption band at 400–500 nm (Fig. S7†) compared to the bulk CdS material at 510 nm. As shown in Fig. 7A, the emission peak of the CdS SPs is symmetric and located at 590 nm. Because of the wide emission wavelength, the emission originates from the surface trap state rather than the intrinsic excitation recombination.20 Cysteine has been demonstrated to induce chirality in semiconductor NCs due to surface bonding. This is also observed in the cysteine-capped CdS SPs. As shown in Fig. 7B and S8,† CdS SPs with L- or D-cysteine as the surface ligand display circular dichroism (CD) peaks at 350 nm, which implies the induced chirality in the CdS SPs. These optical properties could be well applied in biological imaging, bio-probes, and photocatalytic asymmetric synthesis, which will be studied in future work. | ||
| Fig. 7 Properties and potential applications of SPs. (A) Fluorescence spectra of CdS SPs (the illustration shows the color changes before and after ultraviolet lamp irradiation). (B) CD spectra of CdS SPs. (C) Concentration-dependent extinction spectra after Cu2S SP oxidation for 24 h. (D) Temperature change of Cu2S SP solution with different concentrations of Cu2S SPs after laser irradiation for 10 min (808 nm, 0.75 W cm−2). (E) The photothermal image of Cu2S SPs varies with temperature. (F) The temperature change of Cu2S solution (55 μg mL−1) over ten cycles of heating/cooling. (G) Room temperature magnetization curves of Cu2S/Fe2O3 SPs (the inset shows the data of local amplification). (H) The transverse relaxivity r2 of Cu2S/Fe2O3 SPs (the inset shows the image of magnetic resonance imaging). (I) Temperature change of Cu2S/Fe2O3 SP solution with different concentrations of Cu2S/Fe2O3 SPs after laser irradiation (1064 nm, 1.0 W cm−2). (J) Photothermal image of Cu2S/Fe2O3 SPs varies with temperature. | ||
Cu vacancies, which are easily formed in Cu2S, can lead to localized surface plasmon resonance (LSPR) bands in Cu2S nanostructures.23 Thus, high photothermal conversion efficiency is expected by regulating the degree of copper vacancy, which can be dynamically tuned for copper chalcogenides by simple oxidation and reduction. The as-prepared Cu2S SPs were exposed to air for 24 h to generate strong and stable LSPR in the NIR (Fig. 7C), of which the absorption intensity increases linearly with the SP concentration due to the high dispersity of the SPs (Fig. S9†). In addition, the extinction coefficient was 10.8 L g−1 cm−1 by calculation; compared with the Cu2S prepared by other systems,38 the extinction coefficient is larger, which has stronger light absorption ability and can transform more heat. With 10 min of laser irradiation (808 nm, 0.75 W cm−2), the solution containing Cu2S SPs is effectively heated and the heating efficiency increases with the Cu2S SP concentration (Fig. 7D). Even when the concentration is as low as 13.5 μg mL−1, the temperature of the solution can be evidently increased. The photothermal conversion efficiency of the Cu2S SPs is as high as 72.2% (see ESI Section 1 and Fig. S10†). It is worth noting that the Cu2S SPs have an excellent photothermal imaging effect (Fig. 7E). After 10 heating/cooling cycles (Fig. 7F), the resulting heating temperature of the Cu2S solution only slightly changed (coefficient of variance < 1.8%), which proves the stable photothermal conversion of Cu2S SPs. In addition, the photothermal conversion efficiency of Cu2S SPs varies with the size of the SPs (see ESI Section 1†), of which the smaller size led to higher photothermal conversion efficiency. This could be attributed to the high absorption/scattering ratio of Cu2S SPs with small sizes. The high photothermal conversion efficiency and tunable size endow Cu2S SPs with great potential for therapy, bio-sensing, and related applications.
The hybrid Cu2S/Fe2O3 SPs, on the one hand, are superparamagnetic as shown in Fig. 7G, and can be used for T2 magnetic resonance imaging (Fig. 7H), and on the other hand, can be used for photothermal imaging (Fig. 7I and J) in the second NIR (1064 nm, 1.0 W cm−2), with the photothermal conversion efficiency as high as 45.2% (see ESI Section 1 and Fig. S11†), which makes the Cu2S/Fe2O3 SPs good candidates for biomedical combination therapy.
Conclusions
In summary, we have developed a versatile, efficient, and eco-friendly in situ decomposition–assembly route for metal sulfide SPs, including CdS, Cu2S, and ZnS, and even extended to hybrid Cu2S/Fe2O3 SPs. Various narrow-size distribution metal sulfide SPs are prepared by directly using cysteine as the sulfur source. The formation mechanism of the SPs is explored, of which the “decomposition–growth–assembly” process could benefit the structure and property design of functional SPs. By regulating the ratio of cysteine to metal ions, the size of the SPs can be precisely tuned in a broad range. Finally, the properties and potential applications of the prepared metal sulfide SPs are explored, which lays the foundation for practical applications of these SPs in the future.Author contributions
Menglei Wang: conceptualization; methodology; validation; formal analysis; writing – original draft preparation. Fulin Jia: software; writing – review & editing. Jianxiao Gong: writing – review & editing. Yunsheng Xia: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22274001) and Wanjiang Scholar program (Y. S. X.), the National Natural Science Foundation of China (No. 22272040) and the Hundred-Talent Program of Chinese Academy of Sciences (J. X. G.). The authors acknowledge Ge Guo for help in calculations and Hui Zhu for her help with the experiments.Notes and references
- S. Wintzheimer, T. Granath, M. Oppmann, T. Kister, T. Thai, T. Kraus, N. Vogel and K. Mandel, ACS Nano, 2018, 12, 5093–5120 CrossRef CAS PubMed.
- G. de Q. Silveira, N. S. Ramesar, T. D. Nguyen, J. H. Bahng, S. C. Glotzer and N. A. Kotov, Chem. Mater., 2019, 31, 7493–7500 CrossRef.
- Y. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580–587 CrossRef CAS PubMed.
- K. R. Deng, Z. S. Luo, L. Tan and Z. W. Quan, Chem. Soc. Rev., 2020, 49, 6002–6038 RSC.
- E. Piccinini, D. Pallarola, F. Battaglini and O. Azzaroni, Mol. Syst. Des. Eng., 2016, 1, 155–162 RSC.
- J. Ling, S. Gong and Y. Xia, Adv. Mater. Interfaces, 2020, 7, 2000804 CrossRef CAS.
- Y. Chen, G. Fu, Y. Li, Q. Gu, L. Xu, D. Sun and Y. Tang, J. Mater. Chem. A, 2017, 5, 3774–3779 RSC.
- H. Zhou, J.-P. Kim, J. H. Bahng, N. A. Kotov and J. Lee, Adv. Funct. Mater., 2014, 24, 1439–1448 CrossRef CAS.
- Z. Sun, W. Ni, Z. Yang, X. Kou, L. Li and J. Wang, Small, 2008, 4, 1287–1292 CrossRef CAS PubMed.
- H. Zhu, Y. Wang, C. Chen, M. Ma, J. Zeng, S. Li, Y. Xia and M. Gao, ACS Nano, 2017, 11, 8273–8281 CrossRef CAS PubMed.
- Y. Ling, D. Zhang, X. Cui, M. Wei, T. Zhang, J. Wang, L. Xiao and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 10542–10546 CrossRef CAS PubMed.
- Y. Yu, X. Yang, M. Liu, M. Nishikawa, T. Tei and E. Miyako, ACS Appl. Mater. Interfaces, 2019, 11, 18978–18987 CrossRef CAS PubMed.
- X. Ma, K. Zhao, H. Tang, Y. Chen, C. Lu, W. Liu, Y. Gao, H. Zhao and Z. Tang, Small, 2014, 10, 4664–4670 CrossRef CAS PubMed.
- J. Zhu, K. He, Z. Dai, L. Gong, T. Zhou, H. Liang and J. Liu, Anal. Chem., 2019, 91, 8237–8243 CrossRef CAS PubMed.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- F. Montanarella, T. Altantzis, D. Zanaga, F. T. Rabouw, S. Bals, P. Baesjou, D. Vanmaekelbergh and A. van Blaaderen, ACS Nano, 2017, 11, 9136–9142 CrossRef CAS PubMed.
- S. Li, J. Liu, N. S. Ramesar, H. Heinz, L. Xu, C. Xu and N. A. Kotov, Nat. Commun., 2019, 10, 4826 CrossRef PubMed.
- W. Zhang, J. Xiao, Q. Cao, W. Wang, X. Peng, G. Guan, Z. Cui, Y. Zhang, S. Wang, R. Zou, X. Wan, H. Qiu and J. Hu, Nanoscale, 2018, 10, 11430–11440 RSC.
- K. Hou, J. Han and Z. Tang, ACS Mater. Lett., 2019, 2, 95–106 CrossRef.
- R. Luo and Y. Xia, J. Anal. Test., 2021, 5, 30–39 CrossRef.
- S. Li, L. Xu, C. Hao, M. Sun, X. Wu, H. Kuang and C. Xu, Angew. Chem., Int. Ed., 2019, 58, 19067–19072 CrossRef CAS PubMed.
- H. Zhu, G. Guo and Y. Xia, Nano Res., 2022, 16, 1448–1458 CrossRef.
- T. A. Patel and E. Panda, Appl. Surf. Sci., 2019, 488, 477–484 CrossRef CAS.
- X. Chen, J. Yang, T. Wu, L. Li, W. Luo, W. Jiang and L. Wang, Nanoscale, 2018, 10, 15130–15163 RSC.
- F. Cavanna, R. Depalo, M. Aliotta, M. Anders, D. Bemmerer, A. Best, A. Boeltzig, C. Broggini, C. G. Bruno, A. Caciolli, P. Corvisiero, T. Davinson, A. di Leva, Z. Elekes, F. Ferraro, A. Formicola, Z. Fulop, G. Gervino, A. Guglielmetti, C. Gustavino, G. Gyurky, G. Imbriani, M. Junker, R. Menegazzo, V. Mossa, F. R. Pantaleo, P. Prati, D. A. Scott, E. Somorjai, O. Straniero, F. Strieder, T. Szucs, M. P. Takacs, D. Trezzi and L. Collaboration, Phys. Rev. Lett., 2018, 120, 239901 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- L. J. Singh and R. H. Singh, Int. J. Inorg. Chem., 2013, 2013, 1–9 Search PubMed.
- X. Mao, Z. Wang, D. Zeng, H. Cao, Y. Zhan, Y. Wang, Q. Li, Y. Shen and J. Wang, ACS Nano, 2019, 13, 2879–2887 CrossRef CAS PubMed.
- Y. Peng, L. Shang, Y. Cao, G. I. Waterhouse, C. Zhou, T. Bian, L. Z. Wu, C. H. Tung and T. Zhang, Chem. Commun., 2015, 51, 12556–12559 RSC.
- B. X. Li, Y. Xie and Y. Xue, J. Phys. Chem. C, 2007, 111, 12181–12187 CrossRef CAS.
- P. Roy and S. K. Srivastava, Cryst. Growth Des., 2006, 6, 1921–1926 CrossRef CAS.
- F. C. Nalle, R. Wahid, I. O. Wulandari and A. Sabarudin, Rasayan J. Chem., 2019, 12, 14–21 CrossRef CAS.
- S. Bhatt and M. Kumar, High Temp. - High Pressures, 2018, 47, 383–401 Search PubMed.
- M. Perde-Schrepler, A. Florea, I. Brie, P. Virag, E. Fischer-Fodor, A. Valcan, E. Gurzau, C. Lisencu and A. Maniu, J. Nanomater., 2019, 2019 Search PubMed.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed.
- M. Wu, H. Guo, L. Liu, Y. Liu and L. Xie, Int. J. Nanomed., 2019, 14, 4247–4259 CrossRef CAS PubMed.
- R. Hu, Y. Fang, M. Huo, H. Yao, C. Wang, Y. Chen and R. Wu, Biomaterials, 2019, 206, 101–114 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00747a |
| This journal is © The Royal Society of Chemistry 2023 |