
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
S-Adenosylmethionine: more than just a methyl donor
Yu-Hsuan
Lee†
 a,
Daan
Ren†
a,
Byungsun
Jeon‡
a and
Hung-wen
Liu
*ab
a,
Daan
Ren†
a,
Byungsun
Jeon‡
a and
Hung-wen
Liu
*ab
aDepartment of Chemistry, University of Texas at Austin, Austin, TX 78712, USA. E-mail: H.w.liu@mail.utexas.edu
bDivision of Chemical Biology & Medicinal Chemistry, College of Pharmacy, University of Texas at Austin, Austin, TX 78712, USA
First published on 9th March 2023
Abstract
Covering: from 2000 up to the very early part of 2023
S-Adenosyl-L-methionine (SAM) is a naturally occurring trialkyl sulfonium molecule that is typically associated with biological methyltransfer reactions. However, SAM is also known to donate methylene, aminocarboxypropyl, adenosyl and amino moieties during natural product biosynthetic reactions. The reaction scope is further expanded as SAM itself can be modified prior to the group transfer such that a SAM-derived carboxymethyl or aminopropyl moiety can also be transferred. Moreover, the sulfonium cation in SAM has itself been found to be critical for several other enzymatic transformations. Thus, while many SAM-dependent enzymes are characterized by a methyltransferase fold, not all of them are necessarily methyltransferases. Furthermore, other SAM-dependent enzymes do not possess such a structural feature suggesting diversification along different evolutionary lineages. Despite the biological versatility of SAM, it nevertheless parallels the chemistry of sulfonium compounds used in organic synthesis. The question thus becomes how enzymes catalyze distinct transformations via subtle differences in their active sites. This review summarizes recent advances in the discovery of novel SAM utilizing enzymes that rely on Lewis acid/base chemistry as opposed to radical mechanisms of catalysis. The examples are categorized based on the presence of a methyltransferase fold and the role played by SAM within the context of known sulfonium chemistry.
 Yu-Hsuan Lee | Yu-Hsuan Lee was born in Taipei (Taiwan) and received her bachelor's degree from National Taiwan University in 2016, where she spent the last year as an exchange student at the Technical University of Munich. She then joined the graduate program in chemistry at the University of Texas at Austin. In the research group led by Prof. Hung-wen (Ben) Liu, she has studied the mechanisms of radical SAM enzymes involved in the biosynthesis of nucleoside natural products. |
 Daan Ren | Daan Ren was born in Nanjing, China. He received his bachelor's degree from Nanjing University in 2016. He then joined Prof. Hung-wen Liu's group at the University of Texas at Austin as a graduate student. His graduate research has focused on the biosynthesis of C-nucleoside natural products and elucidating the mechanisms of enzymatic reactions. |
 Byungsun Jeon | Byungsun Jeon received his PhD from the University of Texas at Austin, where he studied the enzymes and biosynthetic pathway of spinosyn A using kinetic analysis and organic synthesis under the guidance of Prof. Hung-wen (Ben) Liu. Upon completing his studies in 2017, he worked with Prof. Christopher J. Chang at University of California, Berkeley as a postdoctoral researcher. In 2019, he started his independent career at Korea Institute of Science and Technology (KIST). His current research focuses on organic synthesis of natural products and biased-ligand development for G-protein coupled receptors. |
 Hung-wen Liu | Hung-wen (Ben) Liu was born in Taipei (Taiwan). He completed his PhD degree with Prof. Koji Nakanishi from Columbia University in natural product chemistry and continued his postdoctoral research with Prof. Christopher Walsh at MIT. He started his independent career in the chemistry department at the University of Minnesota in 1984 and moved to University of Texas at Austin in 2000. His most recent research focuses on atypical sugar biosynthesis, radical SAM enzymology and mechanistic investigations of enzyme catalyzed transformations during natural product biosynthesis. |
1 Introduction
S-Adenosyl-L-methionine (SAM or AdoMet, 1) is a sulfonium-containing primary metabolite found in both prokaryotic and eukaryotic cells.1 SAM is best known for the role it plays in one-carbon metabolism where it serves as a methyl donor in many biological reactions,2–4 and a search in InterPro reveals that approximately 3 million protein sequences have been annotated as SAM-dependent methyltransferases.5 The molecular mechanism of SAM-dependent methylation generally involves a nucleophilic substitution at the sulfonium methyl carbon of SAM to generate a variety of C-, O-, N- and S-methylated products.6Not all enzymes, however, utilize SAM to catalyze methylation reactions. In particular, a significant number of enzymes also bind an [Fe4S4] cluster typically via a highly conserved CX3CX2C motif in the active site that serves to reduce SAM leading to the homolytic cleavage of the C5′–S linkage.7,8 The resulting 5′-deoxyadenosyl radical equivalent (2) can then initiate a broad range of radical-mediated transformations. Radical SAM enzymes thus constitute a protein superfamily with more than 700 thousand protein sequences9 arguably becoming one of the focal points of modern enzymological study as summarized in many recent reviews.10–17
A number of other enzymes are also dependent on SAM for their activity; however, they are not easily classified as either SAM-dependent transferases or radical SAM enzymes.18–21 Among these enzymes, SAM may serve as an alkyl donor as opposed to a strict methyl donor or stabilize reaction intermediate(s) via the positive charge of its cationic sulfonium moiety. Alternatively, SAM may only play a structural role helping to maintain the integrity of the enzyme and its active site and thus remain intact throughout the catalytic cycle. While many of these enzymes are structurally related to the SAM-dependent methyltransferases,22–24 this is not universally the case.
This review emphasizes the varied enzymology of SAM in biological transformations that are not expected to involve radical intermediates (Fig. 1). The enzymes will therefore be discussed based on whether they are structurally related to the SAM-dependent methyltransferases (Section 2) or appear to represent different classes of enzymes altogether (Section 3). As will become evident, however, even structurally related enzymes can exhibit unexpected behavior thereby complicating bioinformatics efforts to infer their activities and placing an emphasis on experimental investigation. Research has nevertheless made steady progress, and since 2010 there has been a considerable expansion in the recognized scope of SAM enzymology observed in nature. Furthermore, continued study in the future is certain to fill in many of the gaps in the current understanding of these enzymes as well as identify new and potentially unforeseen biological reactions involving SAM.
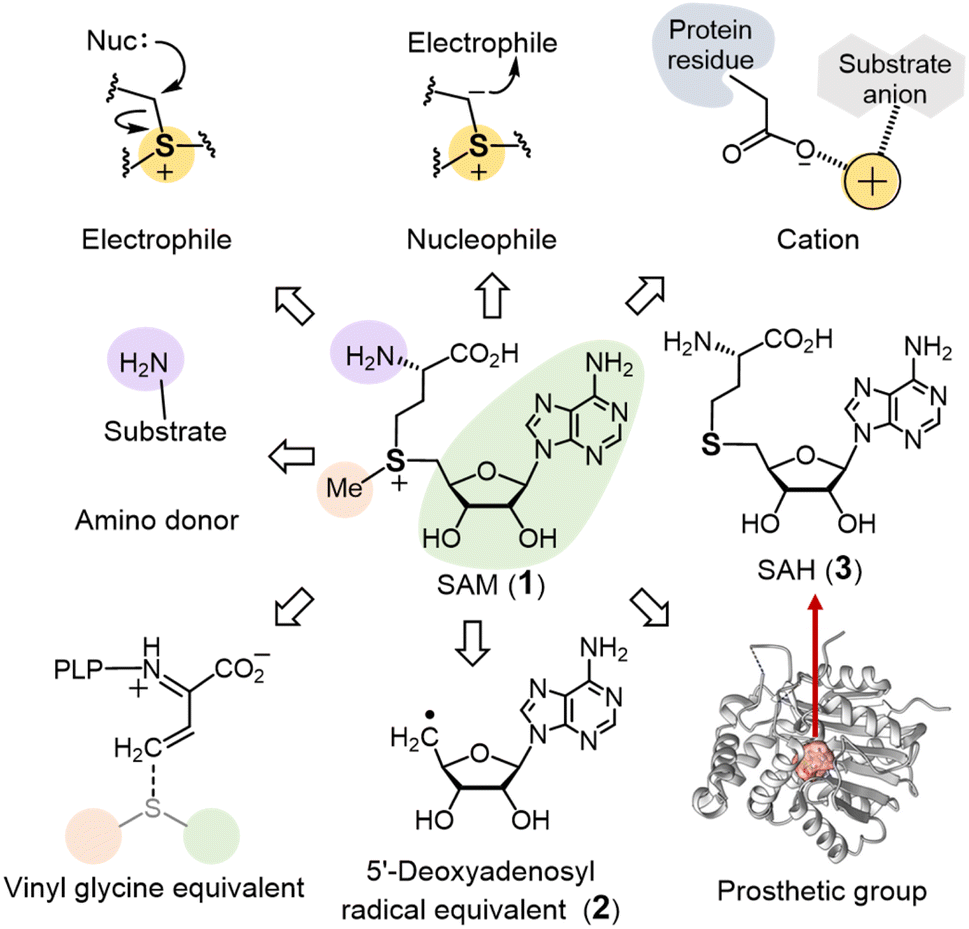 | ||
| Fig. 1 Distinct uses of SAM in enzymatic reactions. | ||
2 Enzymes with a methyltransferase (MT) fold
The electrophilicity of the SAM sulfonium moiety is illustrated by its involvement in a number of different biological alkylation reactions (Fig. 2). However, the regioselectivity of nonenzymatic alkyl transfer from sulfonium ions bearing three different primary alkyl groups cannot be precisely controlled by tuning either the steric bulkiness of the substituents or other reaction conditions such as the solvent, counter ion or polarizability.25–28 Consequently, the regioselectivity of enzyme catalyzed alkyl transfer from SAM appears to originate in variations in the enzyme active sites that tune the relative positioning between the alkyl donor and the nominal substrate, which serves as the alkyl acceptor. Moreover, a number of enzymes have also evolved active site arrangements that preferentially recognize derivatives of SAM thereby further increasing the scope of biological alkylation reactions. Protein crystallography has thus become a critical tool in understanding the details of SAM-dependent alkylation reactions. The following section is focused mainly on more recent findings regarding transfer of the 3-amino-3-carboxypropyl group and the modified methyl group from SAM, each catalyzed by an enzyme adopting a methyltransferase fold. Finally, a brief discussion is included of the adenosyl transfer enzymes, which lack a methyltransferase fold yet utilize SAM as the adenosyl donor.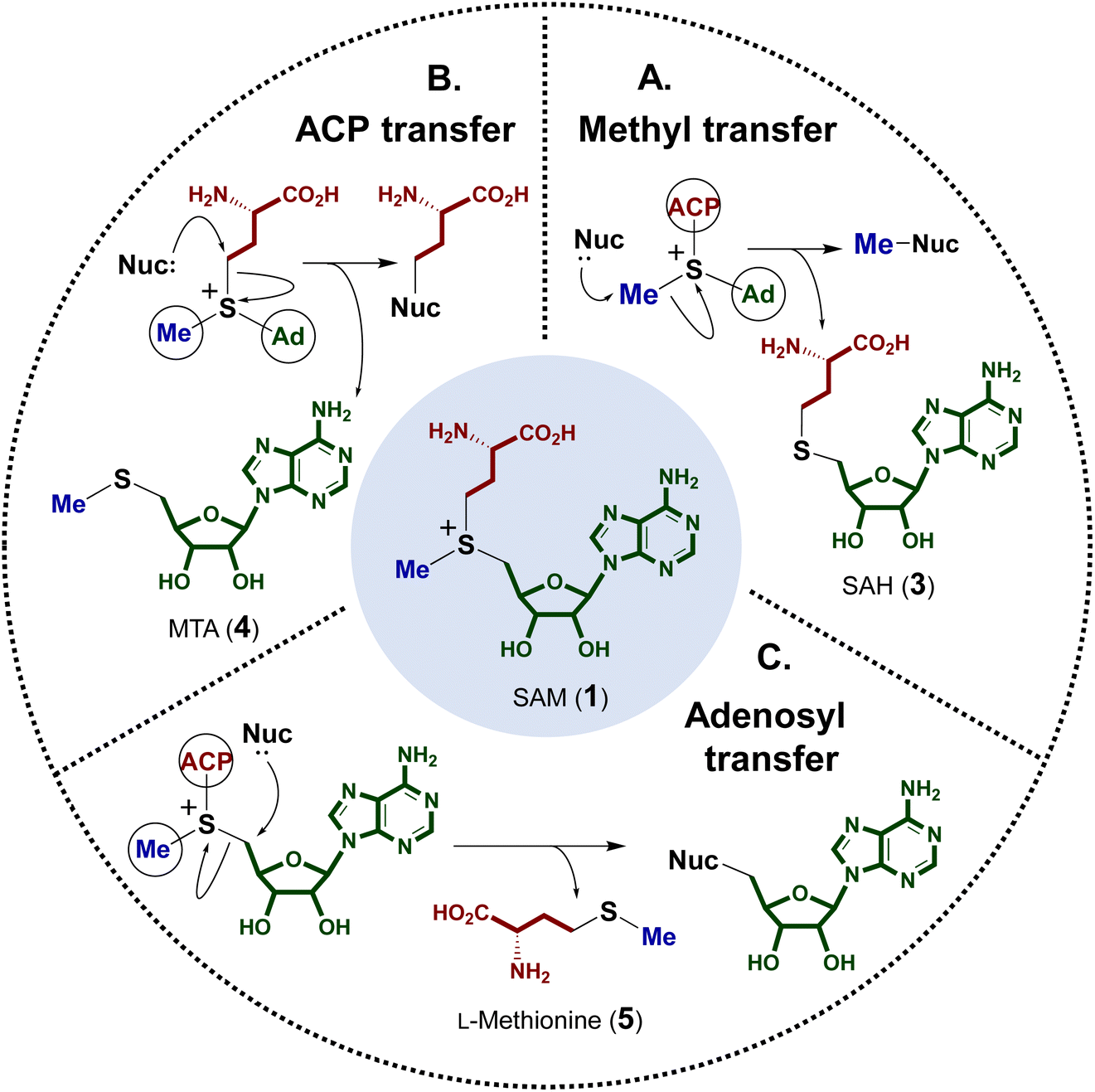 | ||
| Fig. 2 Summary of SAM-dependent alkylation reactions, showing the by-product generated following each group transfer. (A) Methyl transfer yields S-adenosyl homocysteine (SAH, 3), (B) ACP (3-amino-3-carboxypropyl) transfer yields 5′-methylthioladenosine (MTA, 4) and (C) adenosyl transfer yields L-methionine (5). | ||
2.1 SAM-dependent alkylation reactions
A number of enzymes have been discovered to selectively catalyze transfer of the 3-amino-3-carboxypropyl (ACP) group from the SAM sulfonium to an acceptor nucleophile expelling methylthioadenosine (MTA) as the leaving group. This type of chemistry has been described in the biosynthesis of several natural products as well as modification of tRNA and rRNA.29–31 Moreover, analogous reactions have also been reported for derivatives of SAM and in particular decarboxy-SAM (dc-SAM, 6) leading to aminopropyl transfer reactions observed during the biosynthesis of spermidine (7) (Fig. 3).32–34 Catalysis of both ACP transfer and aminopropyl transfer involves an inversion of the stereocenter consistent with a direct nucleophilic displacement reaction.35–37 Substrate binding is of particular importance for regioselective alkyl transfer, and thus the emphasis of the following discussion will be placed on how SAM binding allows for selective C–S bond cleavage.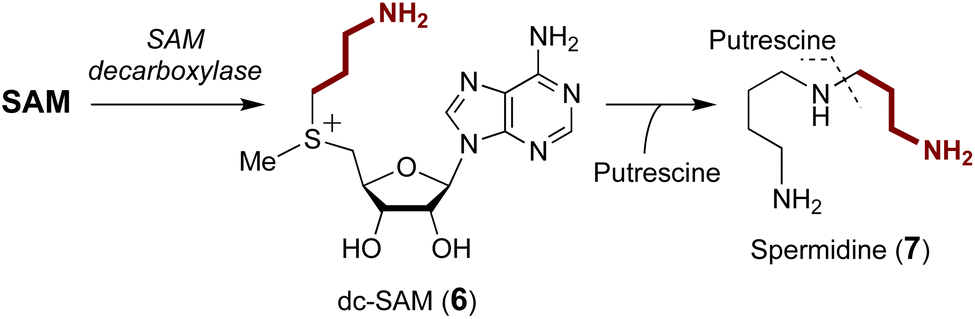 | ||
| Fig. 3 Aminopropyl group transfer during the biosynthesis of spermidine. SAM is first decarboxylated to form decarboxy-SAM (dc-SAM, 6) before the 3-aminopropyl group is transferred to putrescine to yield spermidine (7). | ||
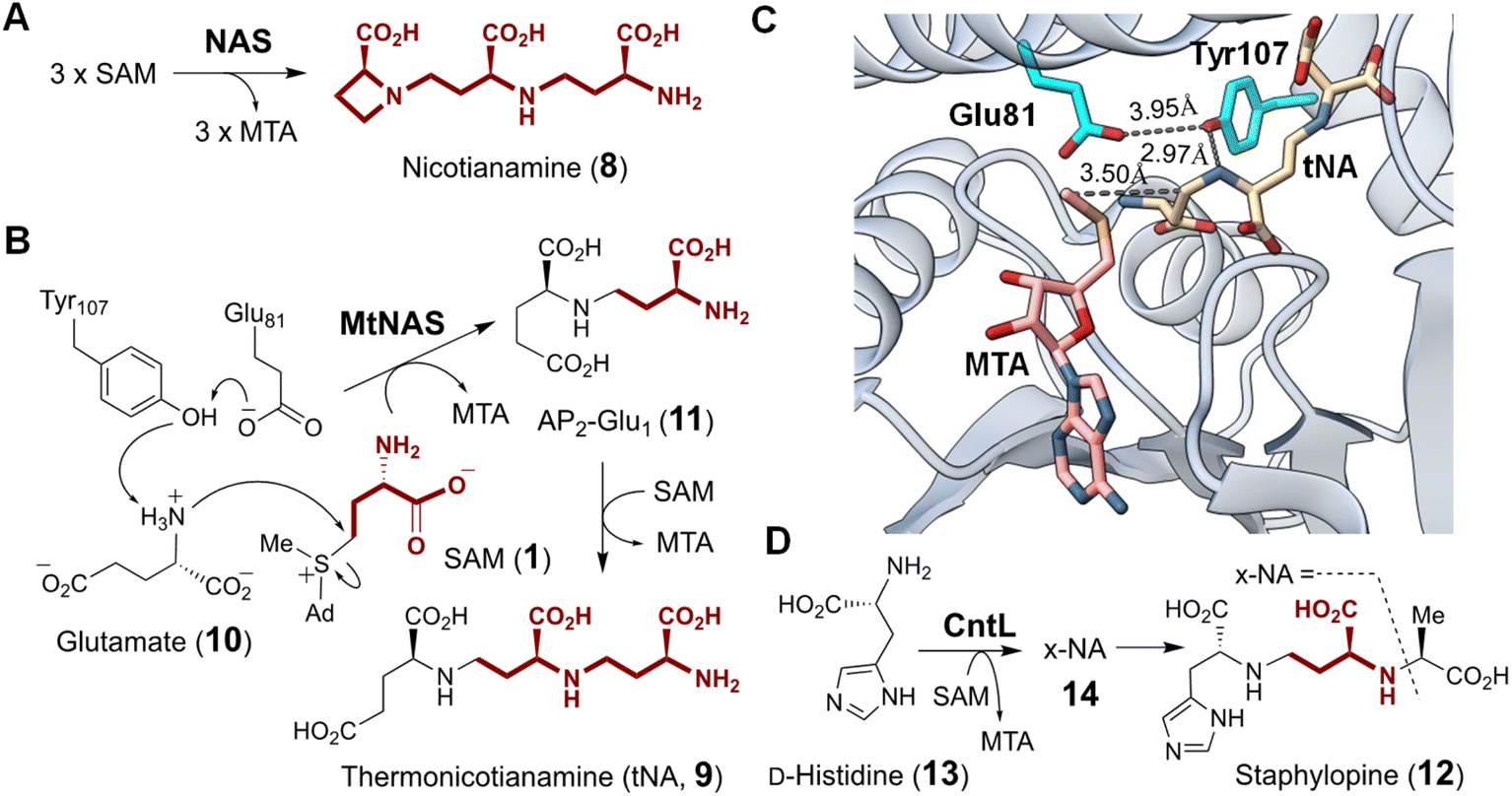 | ||
| Fig. 4 (A) Nicotianamine synthase (NAS) catalyzes the formation of nicotianamine (8) from three molecules of SAM. (B) Molecular mechanism proposed for MtNAS during the biosynthesis of thermonicotianamine (tNA, 9). (C) Crystallographic snapshot of MtNAS (PDB ID: 3FPF) complexed with MTA (pink) and tNA (tan). Nucleophilic displacement was facilitated by deprotonation of the substrate nitrogen by Tyr107 and Glu81 (cyan). (D) The NAS-like enzyme CntL catalyzes transfer of one ACP to D-His (13) providing x-NA (14) as the precursor of bacterial metallophore, staphylopine (12). | ||
Archaeal NAS from Methanothermobacter thermautotrophicus (MtNAS) was found to copurify with thermonicotianamine (tNA, 9), which is structurally distinct from nicotianamine in that the first condensation partner is a glutamate (10) rather than an ACP moiety (Fig. 4B).45 Various crystallographic snapshots of MtNAS in complex with its substrate, a proposed intermediate and its product have led to a mechanistic proposal involving two rounds of ACP transfer to glutamate leading to tNA.45,47 In particular, the active site of MtNAS extends deep into the protein interior from a solvent exposed entrance allowing for sequential binding of glutamate and SAM.45 As SAM binds, glutamate translocates from the donor site at the entrance of the reaction chamber to the buried acceptor site. Transfer of the ACP moiety from SAM to glutamate or the intermediate AP2-Glu1 (11) is facilitated by Glu81 and Tyr107, which reside in the middle of the active site and deprotonate the nucleophilic amine on the acceptor substrate (i.e., Glu or AP2-Glu1) (Fig. 4C).45
CntL is a bacterial NAS-like enzyme that participates in the biosynthesis of staphylopine (12),46 a metallophore in the pathogenic bacterium Staphylococcus aureus.48–50 Mutant strains that do not produce CntL show reduced virulence and decreased intracellular metal content.46 CntL catalyzes the transfer of ACP from SAM to D-histidine (13) forming xNA (14) (Fig. 4D).46 The structure of CntL was solved at 1.81 Å and found to be similar to that of MtNAS.51 Both MtNAS and CntL possess a Rossmann fold MT C-terminal domain with a characteristic glycine-rich motif that binds SAM as the ACP donor.45,51 An open to closed conformational change upon substrate binding was noted for CntL and involves the partially disordered interdomain linker in the CntL–SAM binary complex becoming an ordered α-helix in the CntL–MTA–xNA ternary complex.51 The mechanism of CntL catalyzed ACP transfer is otherwise analogous to that of MtNAS. In addition to NAS and CntL, many other NAS-like enzymes have been identified or proposed in the biosynthesis of structurally diverse metallophores with an amino group serving as the acceptor nucleophile, which underscores the importance of SAM-dependent ACP modification of metallophores in bacteria.52–54
Microcin C (15) produced from E. coli is a potent inhibitor of aspartyl-tRNA synthetase.55 The biosynthesis of microcin C involves aminopropylation of the phosphate which enhances the inhibitory effect by approximately 10-fold.56 Two enzymes have been proposed to catalyze aminopropylation including a SAM-dependent methyltransferase MccD (PDB ID: 5FCD) and the N-terminal pyridoxal 5′-phosphate (PLP)-dependent decarboxylase domain of MccE (Fig. 5, route a).56In vitro characterization of the two enzymes initially suggested that aminopropylation may proceed with ACP transfer to McC1120 (16) catalyzed by MccD followed by decarboxylation of the alkylated intermediate McC1221 (17) catalyzed by MccE.57 However, an alternative route similar to polyamine biosynthesis, where decarboxy-SAM (6) is the aminopropyl donor such that MccE and MccD operate in the reversed order (Fig. 5, route b), has not been completely ruled out. Moreover, the activity of MccD can be significantly enhanced by the nucleosidase Mtn, which can hydrolyze the MTA by-product and presumably alleviate product inhibition.57
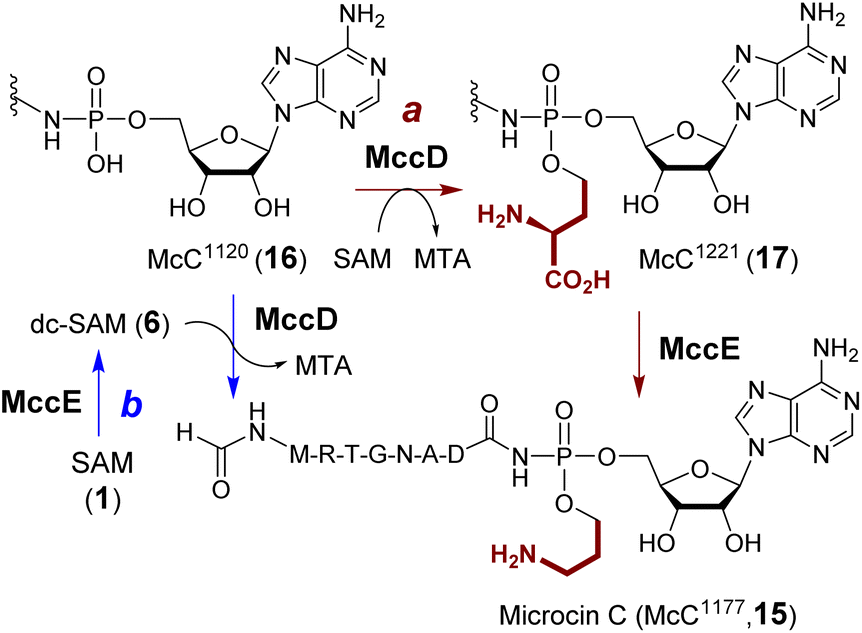 | ||
| Fig. 5 Aminopropylation during maturation of microcin C (15). MccD can catalyze transfer of an ACP group onto McC1120 (16), which can further be decarboxylated in a reaction catalyzed by MccE (route a). An alternative pathway starts with the formation of dc-SAM catalyzed by MccE followed by MccD mediated aminopropyl transfer (route b). | ||
Additional ACP transferases with small molecule substrates are also involved in the biosynthesis of isonocardicin C (18),58,59 betaine lipid (19),60 discadenine (20)61,62 and 2-(3-amino-3-carboxypropyl)-isoxazolin-5-one (ACI) (21) (Fig. 6);63,64 however, none of these enzymes appear to be homologous to the aforementioned ACP transferases NAS, MtNAS, CntL and MccD. With the exception of ACI, ACP transfer from SAM is respectively catalyzed in these cases by NAT, BtaA and discadenine synthase. However, while the in vitro formation of ACI is known to require a partially purified enzyme fraction from sweet pea seedlings as well as the presence of ATP and magnesium,63,64 the responsible enzyme as well as the role of ATP remains elusive.
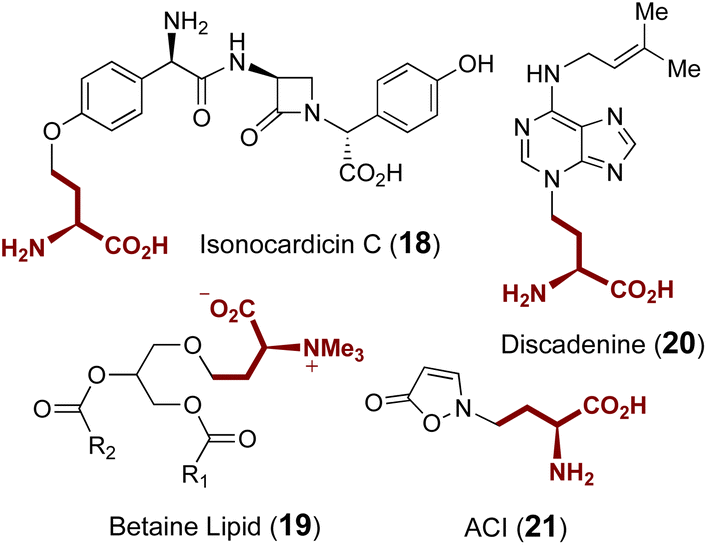 | ||
| Fig. 6 Other natural products with ACP groups derived from SAM. | ||
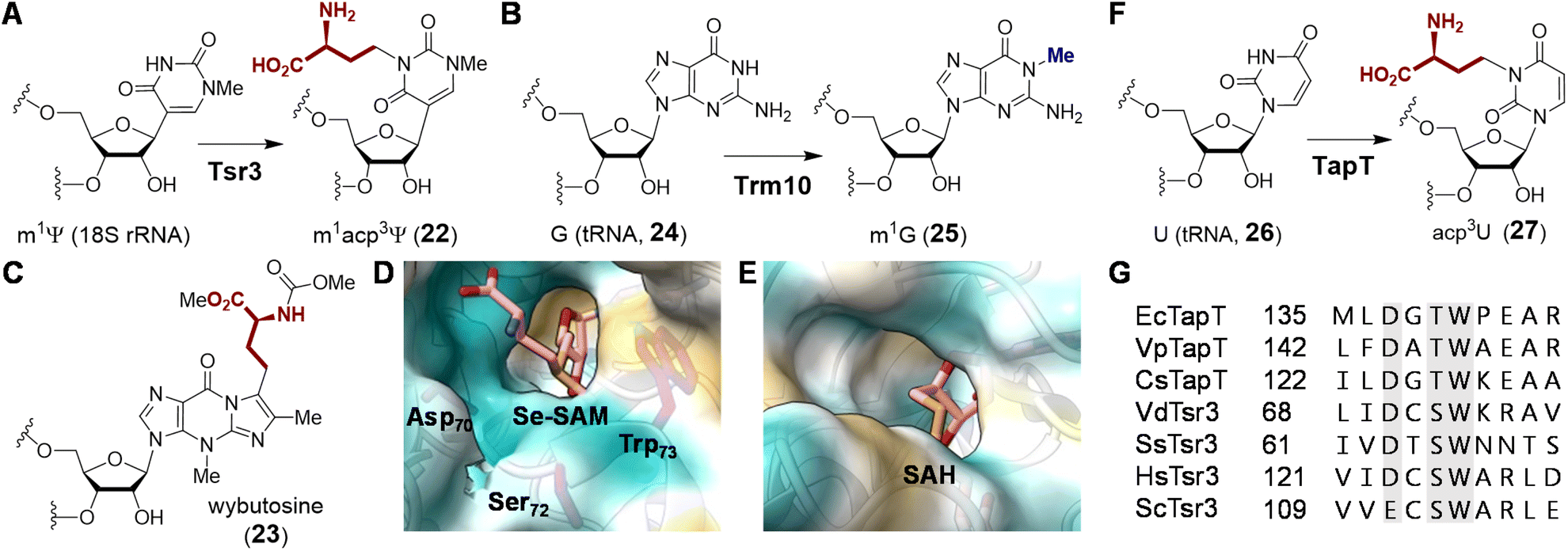 | ||
| Fig. 7 Enzymatic functions of (A) Tsr3 and (B) its closest homolog Trm10. Tsr3 catalyzes ACP modification of a pseudouridine residue whereas Trm10 catalyzes methylation of a guanosine or adenosine residue. (C) Structure of wybutosine highlighting the ACP moiety transferred under the action of Tyw2. Solvent accessible area of (D) Tsr3 (PDB ID: 5APG) and (E) Trm10 (PDB ID: 4JWF) is shown colored with the most hydrophilic residues in cyan to the most hydrophobic in gold. In Tsr3, the ACP side chain of Se-SAM (pink) is exposed to solvent and stabilized by hydrophilic interactions. Asp70, Ser72 and Trp73 in the DTW domain are in purple. In contrast, the ACP side chain of SAH (pink) in Trm10 is buried, and the entrance to the binding cavity is lined with hydrophobic residues. (F) Enzymatic function of TapT. (G) Sequence alignment of Tsr3 and TapT at the DTW domain (gray) (Ec: Escherichia coli; Vp: Variovorax paradoxus; Cs: Clostridium saccharoperbutylacetonicum; Vd: Vulcanisaeta distributa; Ss: Sulfolobus solfataricus; Hs: Homo sapiens; Sc: Saccharomyces cerevisiae). | ||
Comparison of the crystal structures of VdTsr3 and Trm10 with SAM bound reveals overall similarity at the interface of the N- and C-terminal domains.66 This is where SAM binds with the adenine moiety buried in a hydrophobic pocket and the ribose moiety hydrogen-bonded to the protein backbone. However, closer inspection of the VdTsr3 active site shows the carboxyl moiety of ACP positioned towards the entrance of the substrate binding pocket surrounded by hydrophilic residues, while the methyl group is directed into a hydrophobic pocket consisting of an unconserved aliphatic residue and a conserved tryptophan (Trp73 in VdTsr3, see Fig. 7D).66 Mutation of the Trp to Ala results in both diminished SAM affinity and ACP transferase activity in SsTsr3.66 Furthermore, Tsr3 employs a conserved residue (Asp70 in VdTsr3) that serves as a general base, whereas the equivalent Asp210 in Trm10 is not essential for methyl transfer.74 The above catalytically important residues in Tsr3 constitute the DTW motif with the conserved primary sequence (D/E)X(T/S)W (Fig. 7G),75 in which Asp is the possible catalytic base and Trp assists in methyl group positioning.76 Proteins with this domain are classified into the TDD superfamily, which is named after the three representative protein members Tsr3, DTWD1 and DTWD2.75
Although ACP transfer catalyzed by Tsr3 is the final step in m1acp3Ψ modification during 18S rRNA maturation, studies of yeast mutants lacking enzymes for pseudouridylation and methylation have suggested that in vivo ACP transfer activity is independent of either modification such that the corresponding mutants can produce acp3U (27) or acp3Ψ, respectively.77,78 These results suggest that Tsr3 and its homologs can catalyze modification of unmodified uridine residues in RNA. Two recent studies involving comparative genomic and ribonucleome analysis have supported this hypothesis by showing that the bacterial Tsr3 homolog TapT from E. coli catalyzes acp3U (27) modification at U47 (26) in the V-loop of several bacterial tRNAs (Fig. 7F).76,79 While a structure of TapT is not yet available, sequence analyses and mutational studies imply that TapT harbors the catalytically important DTW motif similar to that of archaeal Tsr3 despite overall low sequence identity (Fig. 7G).76
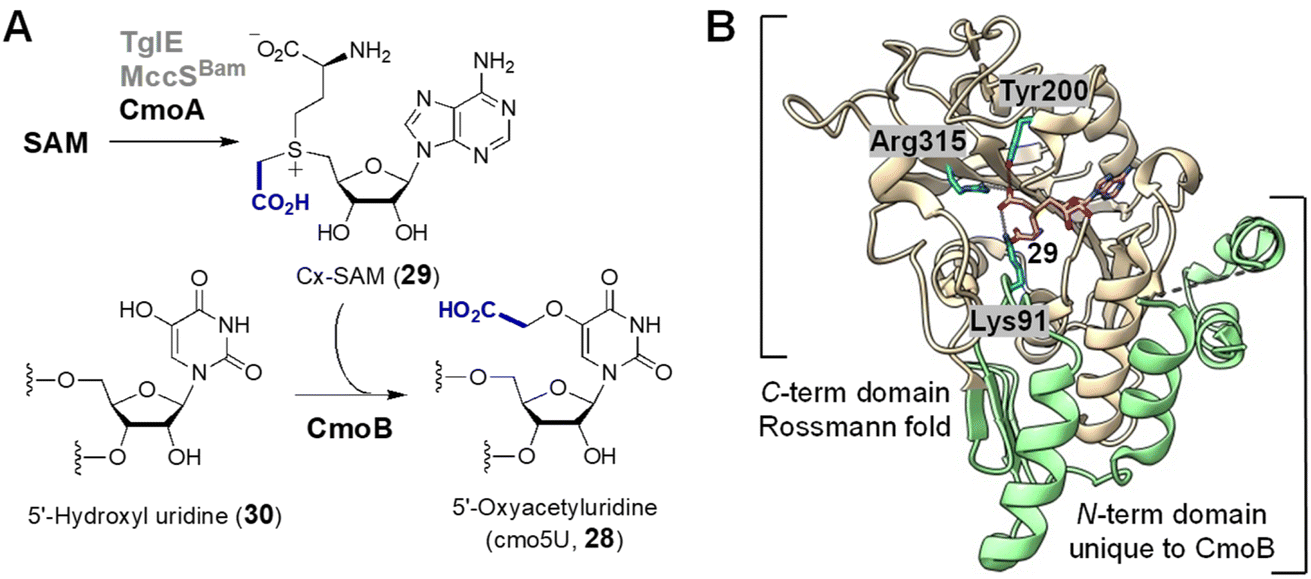 | ||
| Fig. 8 (A) Enzymatic functions of CmoA and CmoB. Two CmoA homologs TglE and MccSBam also catalyze the formation of 29. (B) Crystal structure of Cx-SAM bound to CmoB (PDB ID: 4QNV). The carboxymethyl moiety of Cx-SAM is coordinated by Tyr200, Lys91 and Arg315. The unique 100 residues at the N-terminus of CmoB are shown in green. | ||
While CmoB can catalyze alkyl transfer from both SAM and Cx-SAM, it exhibits a strong preference for Cx-SAM.88 In order to identify the structural determinants of this selectivity, the crystal structure of CmoB was solved by Kim and co-workers (Fig. 8B).88,89 It was found that the C-terminal domain of CmoB adopts a Rossmann fold shared among typical methyltransferases, whereas the structure of the N-terminal domain, which spans around 100 amino acid residues, is unique to CmoB.88 Lys91 was found adjacent to the anionic carboxymethyl moiety and mutation of Lys to Ala completely abolished the production of cmo5U (28) in vivo, the effect of which is attributed to a 19-fold increase in affinity towards SAM and a 275-fold reduction in affinity towards Cx-SAM.88 Consequently, Lys91 likely serves to stabilize the carboxymethyl moiety in the active site via electrostatic interactions thereby enhancing the specificity of CmoB towards Cx-SAM (29) versus SAM. Furthermore, unlike typical SAM methyltransferases, CmoB does not appear to employ general base catalysis to increase the nucleophilicity of the acceptor oxygen,88 because the substrate hydroxyl has an apparent pKa of 7.68 which is sufficient for specific acid/base catalysis.90
Following discovery of the CmoA/CmoB carboxymethylation cascade, a homologous enzyme pair was found in the biosynthetic pathway of 3-thiaglutamate (31), a nonproteinogenic amino acid with unknown function.91 TglE/F show high sequence identity with CmoA/B, and TglF has been shown to catalyze the trans-carboxymethylation of a norcysteine thiol in a ribosomally synthesized and post-translationally modified peptide (32 → 33) prior to amide hydrolysis by the protease TglG to release 3-thiaglutamate (31) (Fig. 9A).91 Primary sequence alignment between CmoB and TglF showed that the essential residues including Lys91 for selective binding of Cx-SAM in CmoB are all conserved in TglF. Furthermore, the biosynthetic gene cluster (MccBam) of the Trojan horse peptide-cytidylate antibiotic microcin C (34) encodes MccBBam which has been shown to catalyze transfer of the carboxymethyl moiety from Cx-SAM synthesized by MccSBam to its tRNA substrate 35 (Fig. 9B).92 The C-terminal domain of MccBBam (MccBBamCTD) adopts a methyltransferase fold that does not show significant similarity with CmoB and is strictly specific for Cx-SAM (29).92 Although the site of carboxymethylation in 34 has yet to be determined, this finding implies that the two-enzyme cascade for this modification is operative in both Gram-negative and Gram-positive bacteria.92
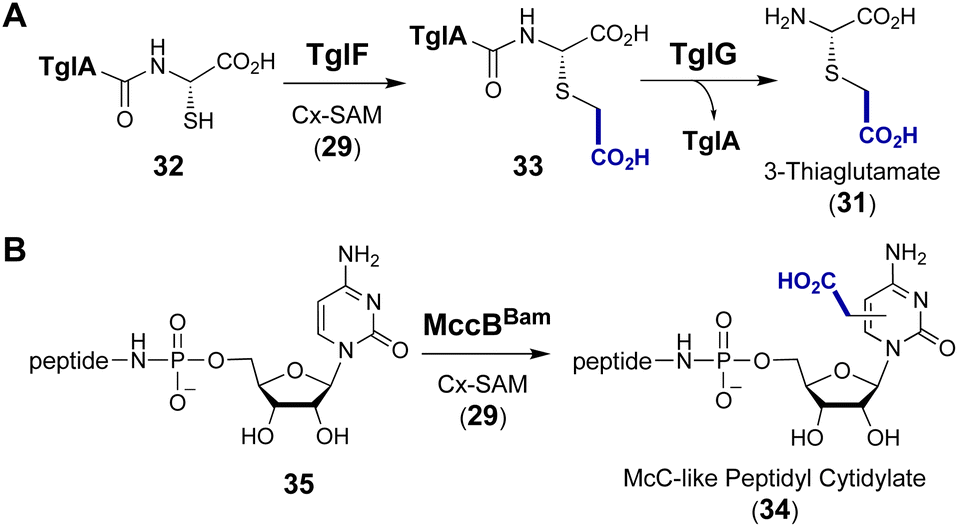 | ||
| Fig. 9 SAM-dependent carboxymethylation during the biosynthesis of (A) 3-thiaglutamate (31) and (B) McC-like peptidyl cytidylate (34). | ||
The discovery of Cx-SAM demonstrates that SAM can be naturally modified leading to additional diversity among the available types of biological SAM-dependent alkyl transfer reactions. Given the high intracellular concentration of SAM versus its modified counterparts such as Cx-SAM,88,93 enzymes must employ recognition mechanisms in order to differentiate between the SAM congeners and thereby avoid inhibition or nonproductive shunt reactions. The structures of CmoB demonstrate that interactions between specific residues and the bound SAM analog at least play a part in this specificity. Consequently, it may be possible to engineer new alkyl transferases via optimized point mutations to facilitate the selective binding of other non-natural SAM analogs and thereby further expand the range of biocatalytic alkylation reactions for biotechnological purposes.94,95
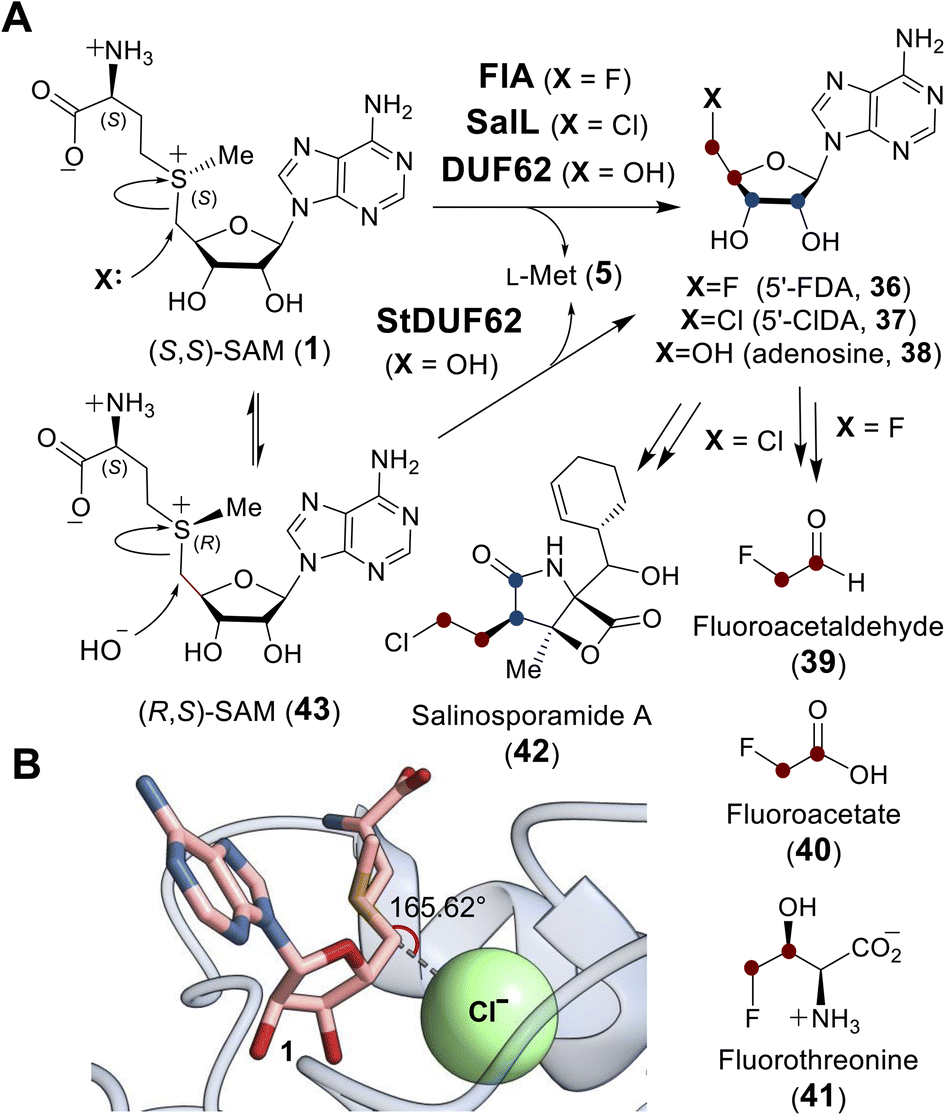 | ||
| Fig. 10 (A) Adenosyl transfer reactions catalyzed by FlA, SalL and DUF proteins. The resulting 5′-deoxy-5′-haloadenosine (36, 37) is a precursor to halogen containing natural products (39–42). (B) In the active site of SalL-Y70T mutant (PDB ID: 2Q6O), SAM (1, pink) and chloride (green) are aligned and poised for SN2 reaction. | ||
Crystallographic studies of both FlA and SalL revealed an overall structure distinct from SAM-dependent methyltransferases. The structures of FlA and SalL instead resemble those of proteins belonging to the DUF62 superfamily despite none of the members having been functionally characterized at the time (Fig. 11). Subsequently, one protein from the DUF62 superfamily, SaDUF62, was shown to catalyze hydrolysis of SAM, which is effectively adenosyl transfer from SAM to a hydroxide ion thereby producing adenosine (38) and methionine (5).103,104 Moreover, a strictly conserved His-Arg-Asp triad is only present in DUF62 proteins (Fig. 11C), which activates one of the two H-bonded waters as the hydrolytic nucleophile necessary for the displacement reaction.105 In contrast, both halogenases lack the catalytic triad and waters are instead expelled from the vicinity of the sulfonium of SAM and the adjacently trapped halide nucleophile.
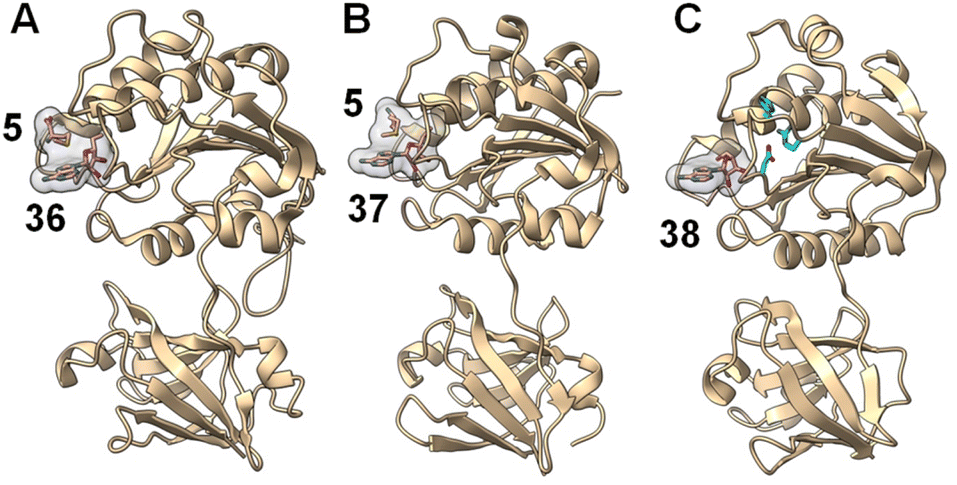 | ||
| Fig. 11 A similar overall structure was observed for three adenosyl transfer enzymes. (A) The product complex between fluorinase FlA/36/5 (PDB ID: 1RQR). (B) The product complex between chlorinase SalL/37/5 (PDB ID: 2Q6I). (C) The product complex of a DUF62 protein from Pyrococcus horikoshii OT3 with 38 (PDB ID: 1WU8). SAM-derived products (i.e., 5 and 36–38) have a gray surface and the conserved His-Arg-Asp triad in DUF62 proteins is colored cyan. | ||
Comparison of the binary FlA/SAM substrate complex with the ternary FlA/5′-FDA/Met product complex has suggested that the C5′ of SAM is positioned between the fluoride and the sulfur of SAM during the substitution reaction indicative of an SN2-type mechanism.100 A catalytically inactive SalL mutant forms a ternary complex with chloride and SAM revealing a colinear arrangement between the chloride ion, C5′ and the sulfur center of SAM (Fig. 10B).101 Although the biological function of hydroxide adenosyltransferases is not as obvious as their halogenase counterparts, recent research identified a DUF62-containing protein, StDUF62, catalyzing the stereoselective hydrolysis of the non-native (R,S)-SAM (43), which is a stereoisomer that equilibrates with biological (S,S)-SAM (1) upon racemization of the sulfonium.106 This hints at a role played by DUF62 proteins in preventing in vivo accumulation of the nonbiological SAM diastereomer.
2.2 Intramolecular cyclization of SAM
Transformations that utilize SAM as the alkylating agent take advantage of the inherent electrophilicity of the SAM sulfonium group107 This property renders SAM susceptible to nucleophilic attack not only intermolecularly but also intramolecularly. In fact, the major decomposition pathway of SAM under neutral or slightly acidic conditions is the intramolecular cyclization that yields homoserine lactone (44) and MTA (4) (Fig. 12).108,109 According to Baldwin's rules,110 this reaction is a favored 5-exo-tet ring closure and likely proceeds through direct nucleophilic attack of the carboxylate oxygen at the C-γ carbon. Although other non-enzymatic intramolecular cyclizations of SAM have not been reported, enzyme catalyzed cyclizations of the aminocarboxypropyl moiety to yield three-, four- and five-membered rings have been described in various natural product biosynthetic pathways (Fig. 12).111–113 However, not all of these biosynthetic enzymes adopt a SAM-dependent methyltransferase (MT) fold despite each utilizing SAM as the substrate. Therefore, only MT-like cyclases are included in the following section, and the rest will be discussed in Section 3.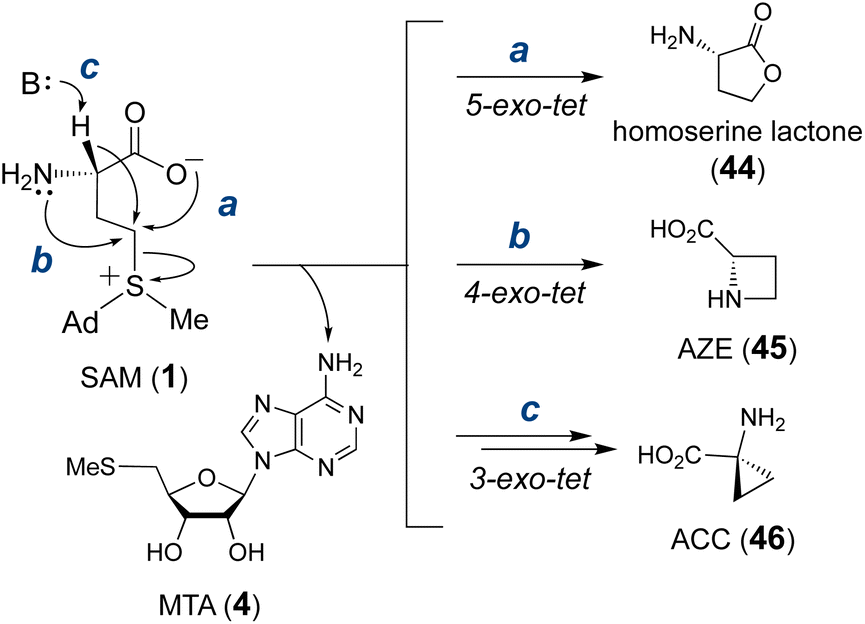 | ||
| Fig. 12 Intramolecular cyclization of the SAM aminocarboxypropyl moiety into homoserine lactone (44, via route a), azetidine 2-carboxylic acid (AZE, 45, via route b) and 1-aminocyclopropane-1-carboxylic acid (ACC, 46, via route c), each with elimination of MTA (4). | ||
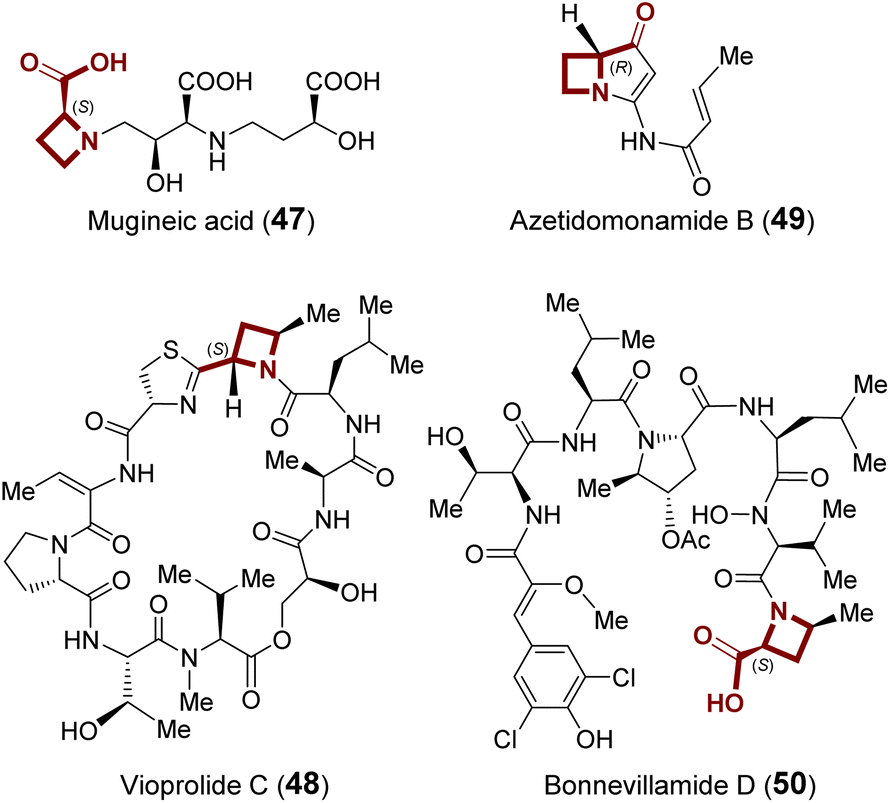 | ||
| Fig. 13 Examples of natural products that contain an azetidine 2-carboxylate (AZE) moiety. | ||
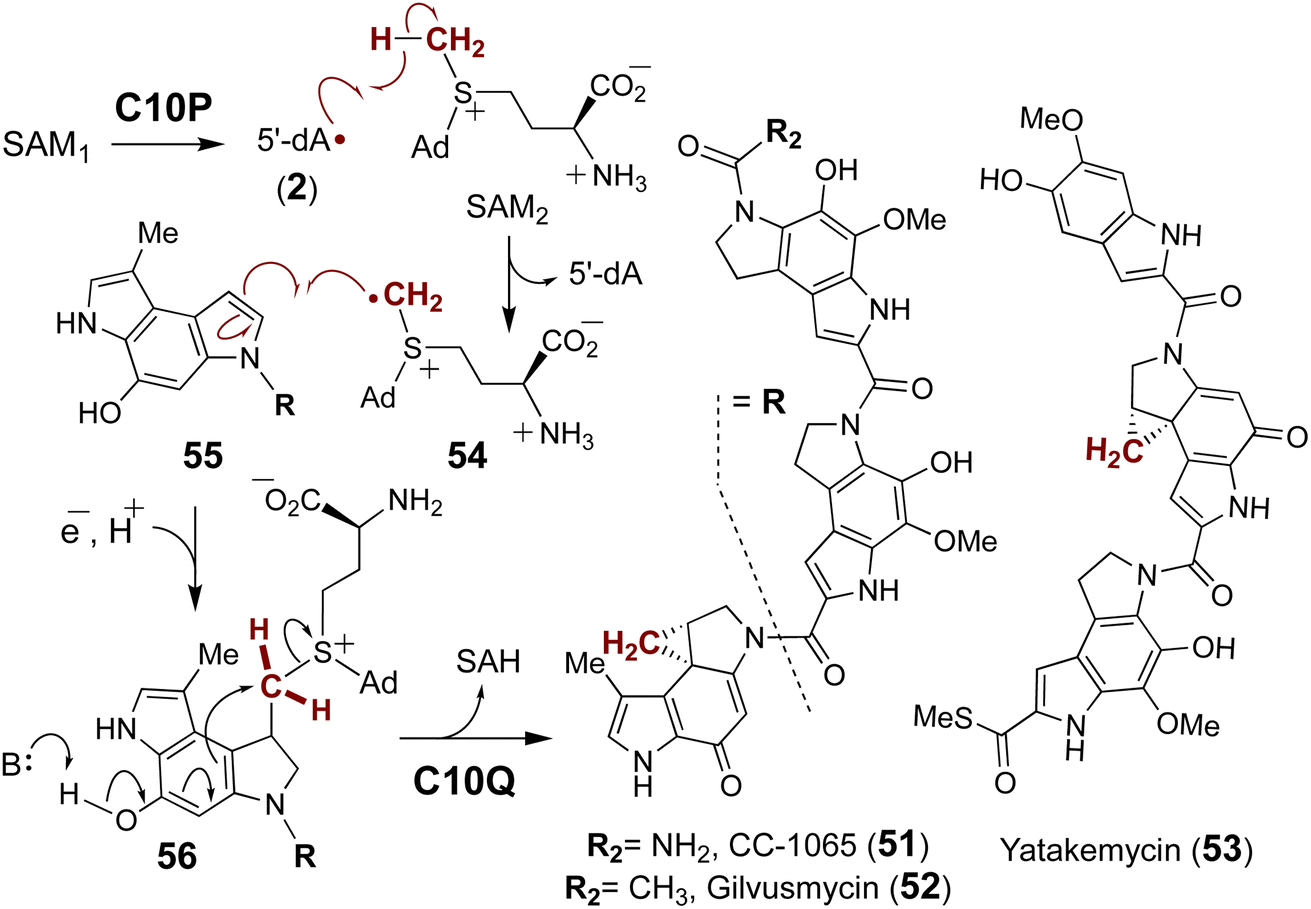 | ||
| Fig. 14 The proposed two enzyme cascade for C10P and C10Q catalyzed formation of the cyclopropane ring during biosynthesis of CC-1065 (51) and related compounds (52 & 53). | ||
2.3 Methylation induced cascade reaction
SAM is also involved in enzyme catalyzed cyclization reactions that proceed in tandem with alkylation.128,129 The majority of examples of this chemistry characterized to date come from the study of terpene biosynthesis. Terpenoid lipids are characterized by extremely diverse cyclic scaffolds constructed from the two unsaturated precursors isopentenyl-pyrophosphate and dimethylallyl-pyrophosphate.130,131 The structural diversity is attributed to the formation of carbocation intermediates such as 57 that can rearrange in a number of ways within the active sites of terpene cyclases.132,133 The activated carbocation intermediates are typically generated via departure of pyrophosphate (58), which may still be associated with the carbocation species as an ionic complex, from the respective precursors (59) (Fig. 15A); however, synthetic methodologies have been developed that employ a Lewis acid to induce carbocation formation including epoxide ring opening and alkene protonation.134 This type of chemistry has also been found in natural product biosynthesis where SAM serves as the Lewis acid catalyst by essentially donating a methyl cation to the electron rich π-system of the terpenoid precursor. Cyclization of the resulting carbocation thus proceeds in tandem with alkylation before deprotonation, which in the case of cyclopropyl fatty acid synthases is performed by an active site bicarbonate ion (60) (Fig. 15B).135,136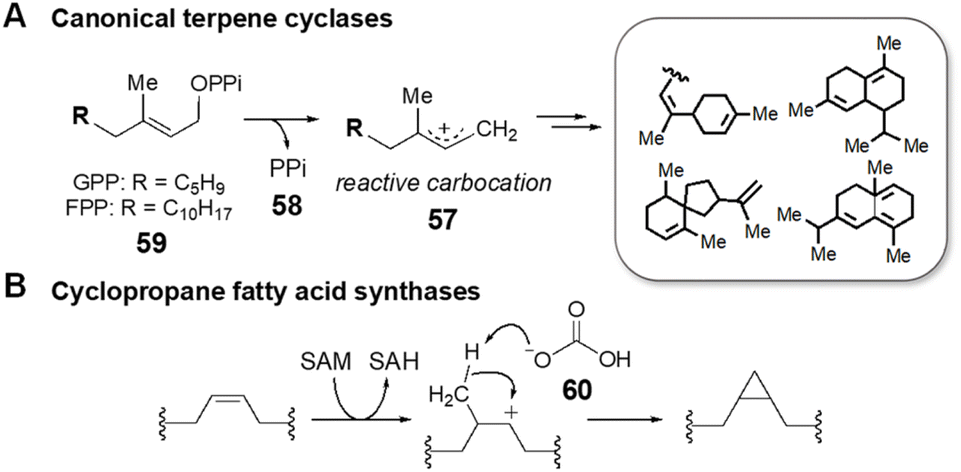 | ||
| Fig. 15 Cation-induced cyclizations catalyzed by (A) canonical terpene cyclases and by (B) cyclopropane fatty acid synthases. | ||
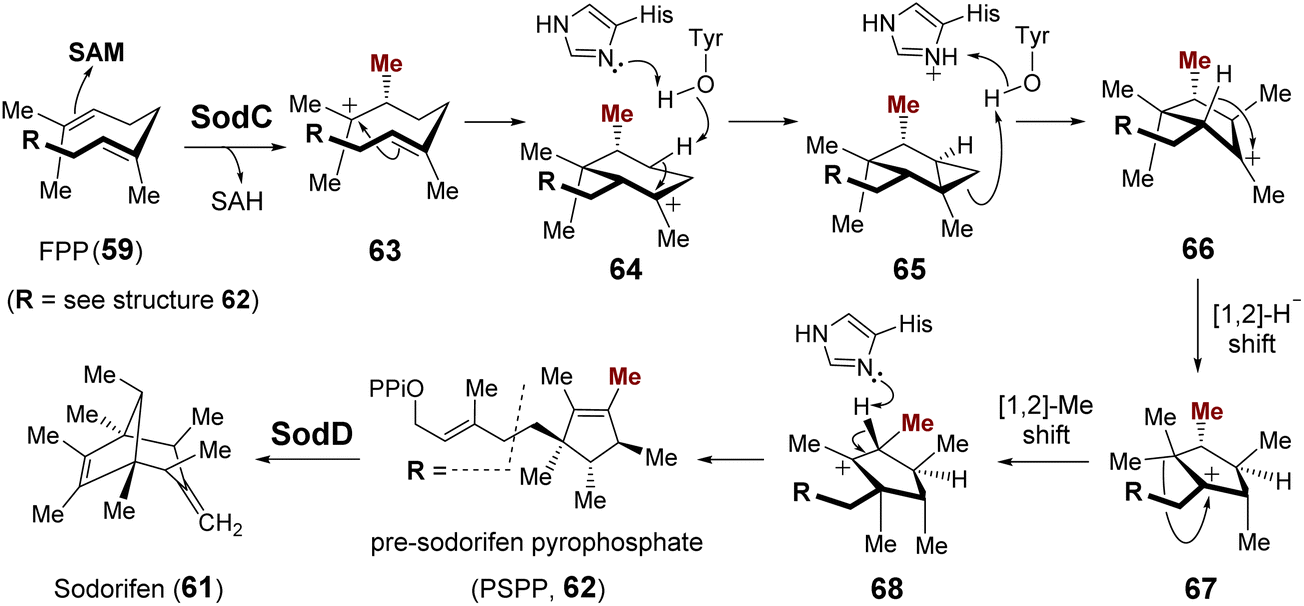 | ||
| Fig. 16 Mechanism proposed for the SodC catalyzed cyclization of FPP. | ||
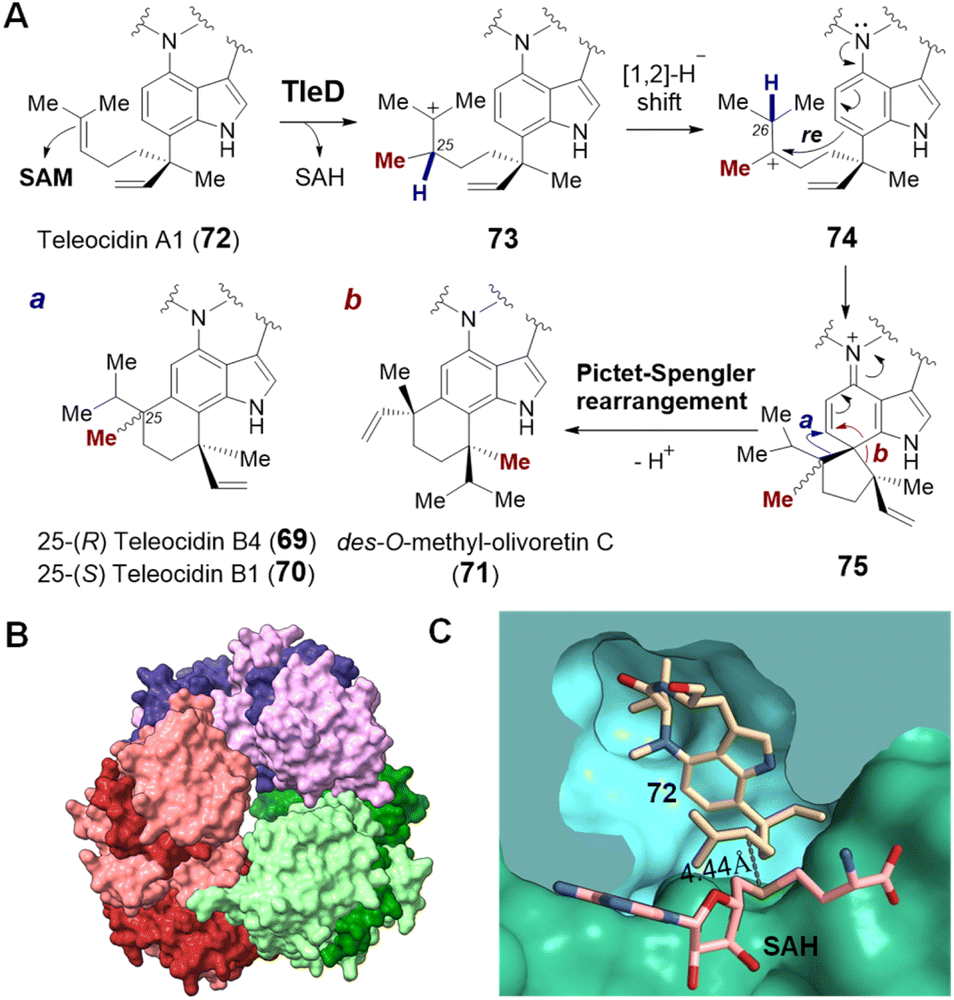 | ||
| Fig. 17 (A) Methylation induced cyclization catalyzed by TleD in teleocidin biosynthesis. Crystal structures of (B) TleD hexamer (PDB ID: 5GM2) and (C) TleD active site formed at the interface of two monomers (green & blue) with bound teleocidin A1 (72, tan) and product SAH (pink). | ||
Crystal structures of TleD have been solved with SAH bound (2.5 Å) as well as both SAH and substrate bound (2.8 Å) in the active site.147 These structures place the sulfur of SAH approximately 4.4 Å away from C25 of the teleocidin A1 (72), which is consistent with the proposed methyltransfer mechanism.147 Unlike typical SAM methyltransferases, TleD appears to be active as a hexamer with each active site formed at the interface of two TleD monomers in a domain-swapped fashion (Fig. 17B and C).147 The active site of TleD is tightly lined with hydrophobic residues and thus water is excluded and prevented from quenching the putative intermediate carbocations.147 The geranyl moiety of the substrate appears to be flexible and assumes two different conformations in the crystal structure; however, molecular dynamics analysis suggests that only one of them is likely to account for the observed product distribution.147
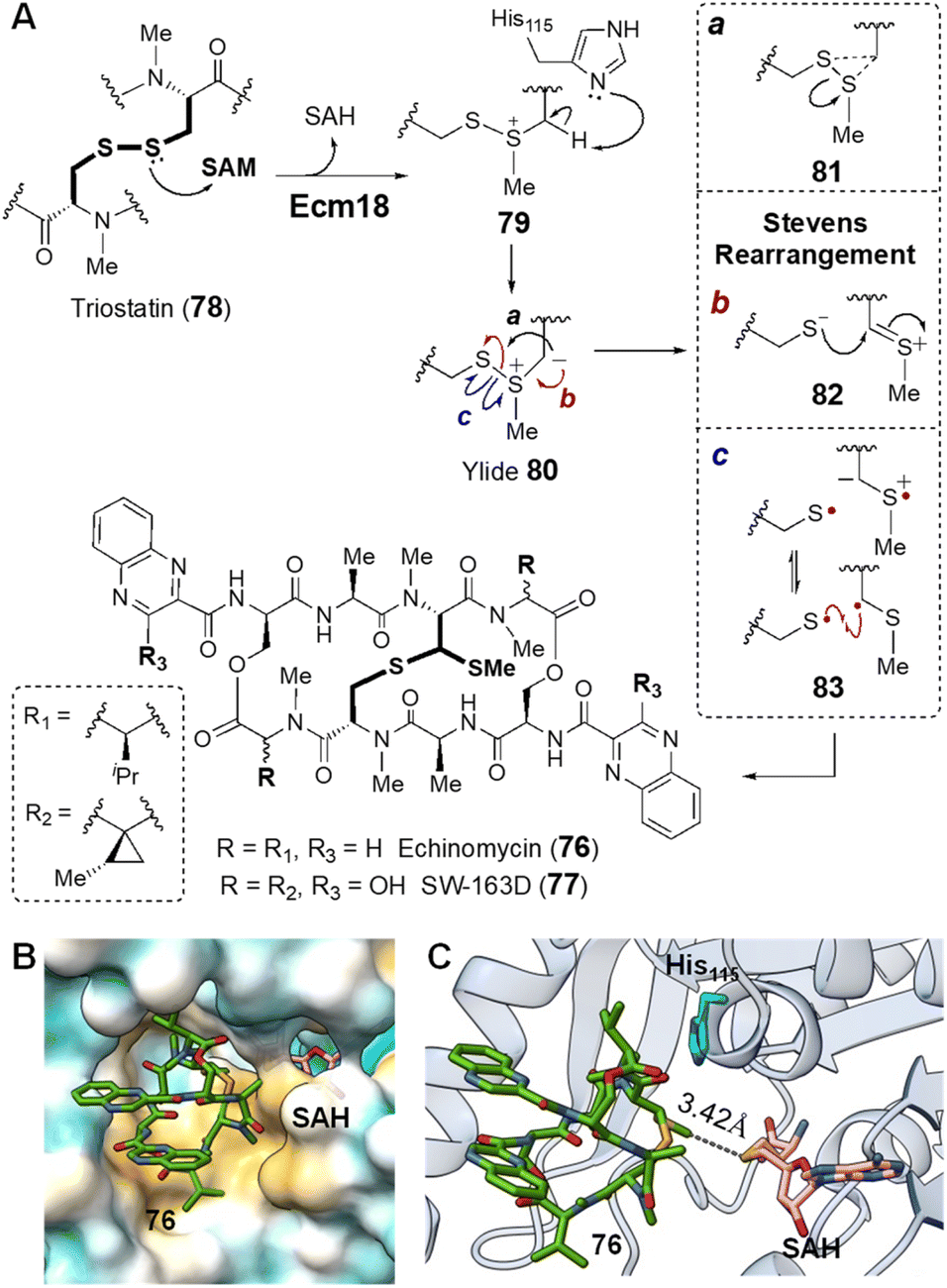 | ||
| Fig. 18 (A) Mechanism of Ecm18 catalyzed rearrangement of a disulfide bridge to a thioacetal. (B) Hydrophobic surface of the Ecm18 substrate binding pocket (PDB ID: 4NEC) colored according to most hydrophobic (gold) and most hydrophilic (cyan) residues. (C) Ecm18/76/SAH ternary complex. The distance between the sulfur of SAH and the methyl group in 76 is consistent with the proposed methyltransfer reaction. The basic nitrogen on His115 (cyan) is near the disulfide bridge and could facilitate ylide 80 formation via deprotonation of the methylated sulfonium intermediate 79. | ||
The catalytic cycle of Ecm18 is hypothesized to proceed through an intermediary sulfonium ion generated via SAM-dependent methylation of one of the two sulfur atoms of the disulfide bridge in triostatin (78 → 79).154 The sulfonium ion can then undergo α-deprotonation in the presence of a general base to yield an ylide (80), which is capable of a Stevens rearrangement to yield the methylthioacetal linkage (80 → 76). To probe the mechanistic details of Ecm18 catalysis, Hotta and co-workers solved the 1.5 Å crystal structure of Ecm18 with the reaction products SAH and echinomycin (76) bound in the active site.159 Ecm18 was thus found to adopt a characteristic SAM binding Rossmann fold featuring a compact hydrophobic substrate/product binding site (Fig. 18B).159 The proposed methylation reaction is consistent with the linear arrangement of the sulfur center of SAH, the transferred methyl group and the sulfur center on the product. A polar histidine residue resides sufficiently close to the thioacetal and could serve as a general base for α-deprotonation of the substrate sulfonium to yield the putative sulfur ylide 80 (Fig. 18C).159 Rearrangement of the ylide may then proceed via a 3-endo-tet cyclization and ring-opening to yield the methyl thioacetal in 76 (Fig. 18A, mechanism a).159 However, two other mechanistic hypotheses for the rearrangement have also been proposed that do not require intermediary cyclization via81 to effect the Stevens rearrangement as shown in Fig. 18A.107,160,161 One involves heterolytic cleavage of the S–S bond followed by thiolate addition to the resulting thionium 82 (mechanism b). The other starts with homolytic cleavage of the S–S linkage and a subsequent radical–radical recombination to construct the C–S bond in 76 (mechanism c).
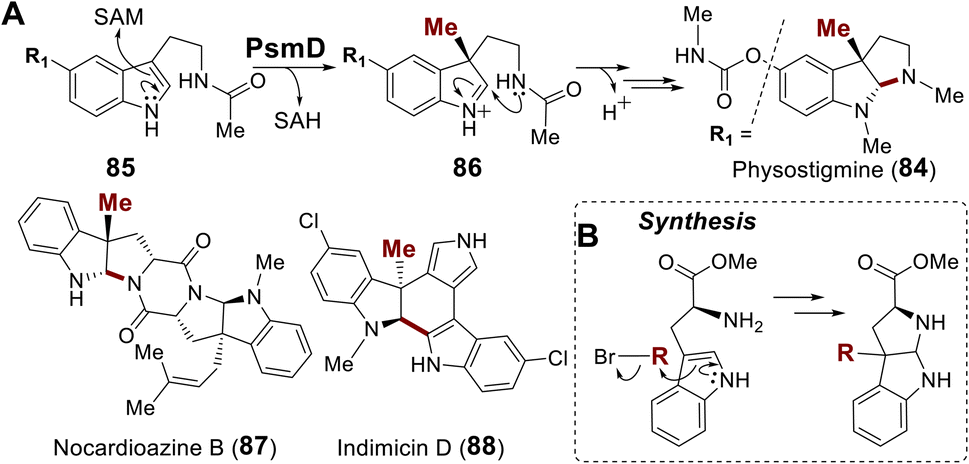 | ||
| Fig. 19 (A) Methylation induced cyclization in pyrroloindoline alkaloid biosynthesis. (B) Synthesis of pyrroloindolines via alkylation–cyclization cascade. | ||
2.4 SAM as a prosthetic group
Apart from direct participation in the group transfer reactions, SAM can also serve as a prosthetic group for many enzymes that are annotated as SAM-dependent methyltransferases. Among these examples, SAM does not undergo any covalent changes in its structure during the reaction.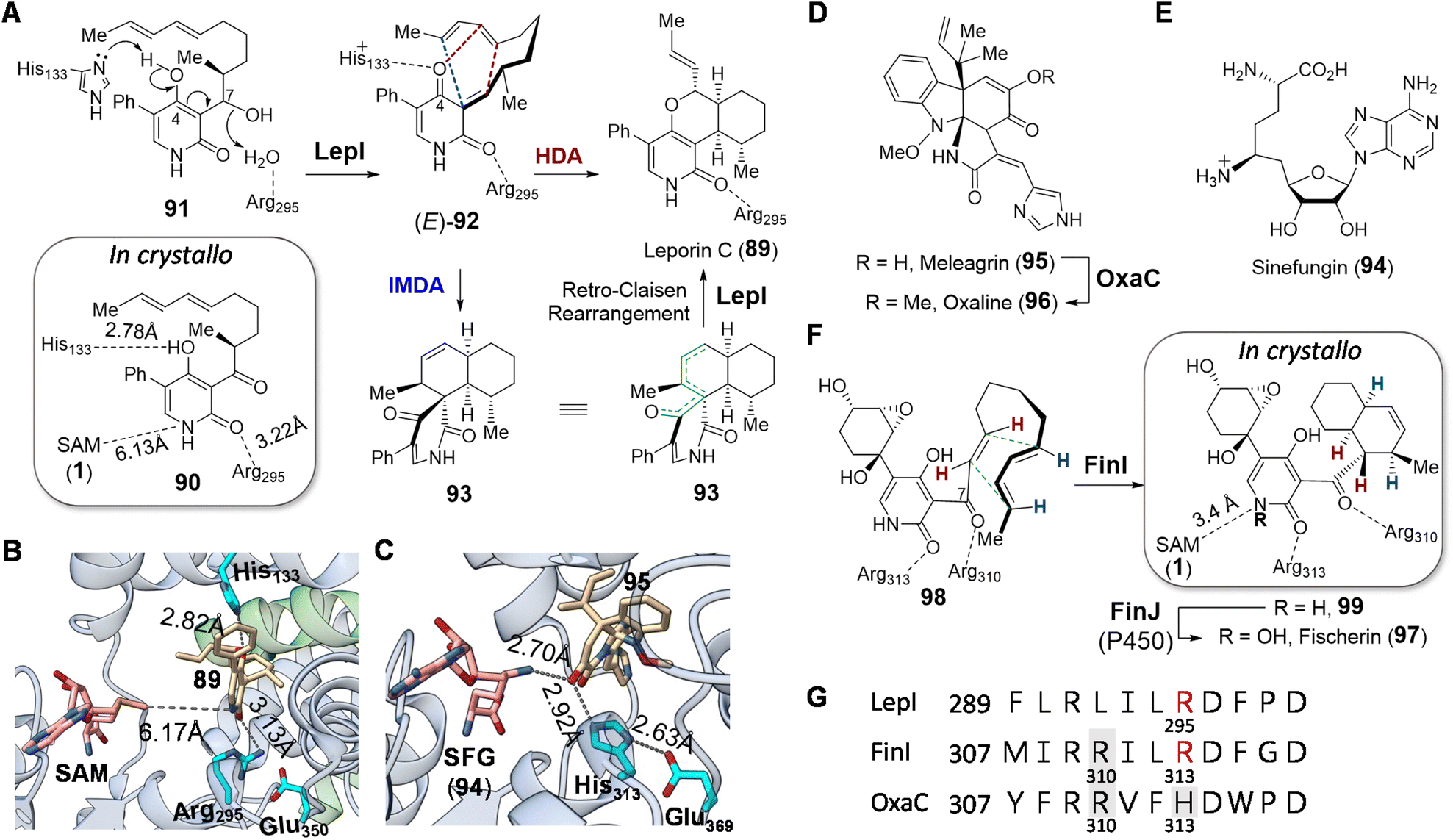 | ||
| Fig. 20 (A) LepI catalyzed dehydration–cyclization cascade during leporin C biosynthesis. Although the stereochemistry at C7 in 91 is currently unclear, it has been shown that the diastereomer of 91 is not accepted by LepI. Comparison of crystal structures of (B) LepI/SAM/89 complex (PDB ID: 6IX9) and (C) OxaC/SFG/95 complex (PDB ID: 5W7S). In LepI, SAM is too far from the product (89, in tan) for methyl transfer; however, in the methyltransferase OxaC, the substrate meleagrin (95) (tan) is located in between SFG and the His313-Glu369 dyad. In contrast, the catalytic histidine is substituted with Arg295 in LepI such that deprotonation cannot occur. Green ribbons in LepI are residues from the second monomeric peptide. (D) OxaC catalyzed methylation during formation of oxaline (96). (E) Structure of sinefungin (94). (F) Cyclization of 98 catalyzed by FinI. (G) Sequence alignment of LepI, FinI and OxaC highlighting the conserved SAM binding arginine residue (Arg310) that interacts with SAM, the catalytic base (His313) in OxaC (in gray boxes) and the essential arginine (Arg295/Arg313) in the pericyclases in red. | ||
The positive charge on SAM and sinefungin was originally proposed to play an electrostatic role in catalysis, since coordination of the substrate to a sulfonium moiety is suggested by computational simulations to accelerate the cyclization and drive the periselectivity towards the favored product.170 However, the crystal structure of LepI complexed with SAM and an unreactive substrate analog (90) revealed that the distance between the two ligands is too remote for coordination (Fig. 20A and 21),171 whereas a product complex between LepI/SAM/89 indicated that the guanidinium group of Arg295 forms a hydrogen bond with the amide carbonyl of the LepI product (89).171 In addition, the His133 residue essential for LepI catalysis is near the 4-O of 89, implying its role in deprotonation of 91 (Fig. 20B). The resulting imidazolium of His133, along with the guanidinium of Arg295, is thus hypothesized to stabilize the transition-state during cycloaddition via92.171 This conclusion has also been corroborated by three other independent crystallography studies.172–174 Sequence and structural comparison between LepI and its closest homolog OxaC, which is an O-methyltransferase catalyzing the methyltransfer of meleagrin (95) to yield oxaline (96) (Fig. 20D),175 revealed that Arg295 in LepI aligns with an essential histidine (His313) in OxaC (Fig. 20G).171 His313, along with Glu369, is conserved among functional O-methyltransferases for substrate deprotonation, and the substitution of the former in LepI may provide an evolutionary perspective on the mechanistic shift from performing methylation to dehydration and cyclization.
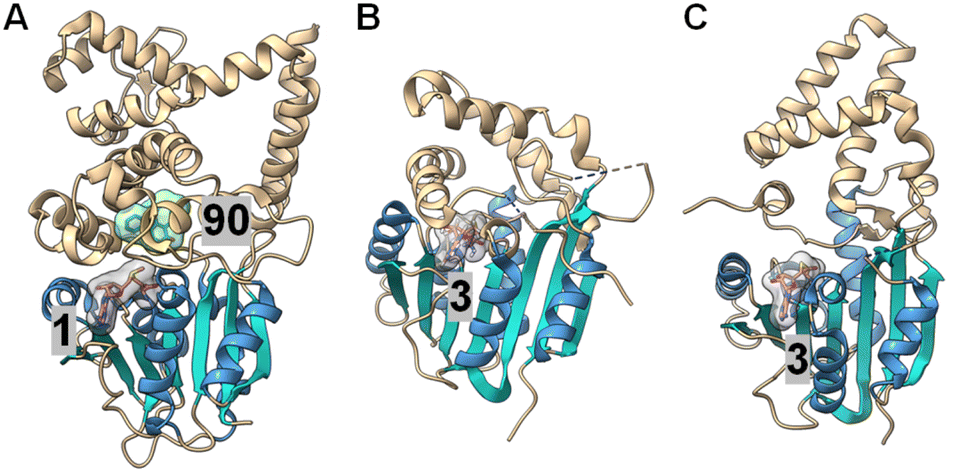 | ||
| Fig. 21 Crystal structures of (A) LepI (PDB ID: 6IX5), (B) SpnF (PDB ID: 4PNE) and (C) SpnL (PDB ID: 7V6H). Each enzyme contains a C-terminal Rossmann fold catalytic domain containing seven β-strands (cyan) surrounded by five α-helices (blue), while the N-terminal domains differ between each protein. The cofactor, SAM (1) for LepI and SAH (3) for SpnF and SpnL (pink with a gray surface), binds similarly at the C-terminal domain of all three enzymes. The substrate analog (90) bound in the LepI active site is shown with a green surface. | ||
Such repurposed methyltransferases catalyzing cyclization reactions have also been found to participate in other biosynthetic pathways. For example, fischerin (97) features a cis-decalin moiety, the biosynthesis of which is not immediately obvious. The dedicated cyclase FinI shows 25% sequence identity and structural homology with LepI and also co-purifies with SAM as does LepI.170,176 Moreover, FinI catalyzed cyclization of 98 to 99 is believed to involve an exo-selective DA reaction (Fig. 20F), which is rare among known pericyclases.176 Unlike the case of LepI, however, SAM and the substrate 98 bind the FinI active site so as to place the sulfonium methyl of SAM close to the amide nitrogen of 98 at a distance of 3.4 Å suggesting an electrostatic interaction and a possible role for SAM in substrate recognition. Whereas in the case of LepI Arg295 can act as a Lewis acid to activate the diene for cyclization,171 the homologous residue Arg313 in FinI is unable to do so given the structure of 98 (Fig. 20F). Instead, Arg310 may play a similar role serving to activate the dienophile in the FinI catalyzed reaction by forming a hydrogen bond with the carbonyl group at C7 of 98. Although Arg310 in FinI is conserved in homologous methyltransferases such as OxaC, where it interacts with SAM, this residue appears to have been evolutionarily repurposed in the FinI active site to interact and likely activate the substrate for cyclization (Fig. 20G).
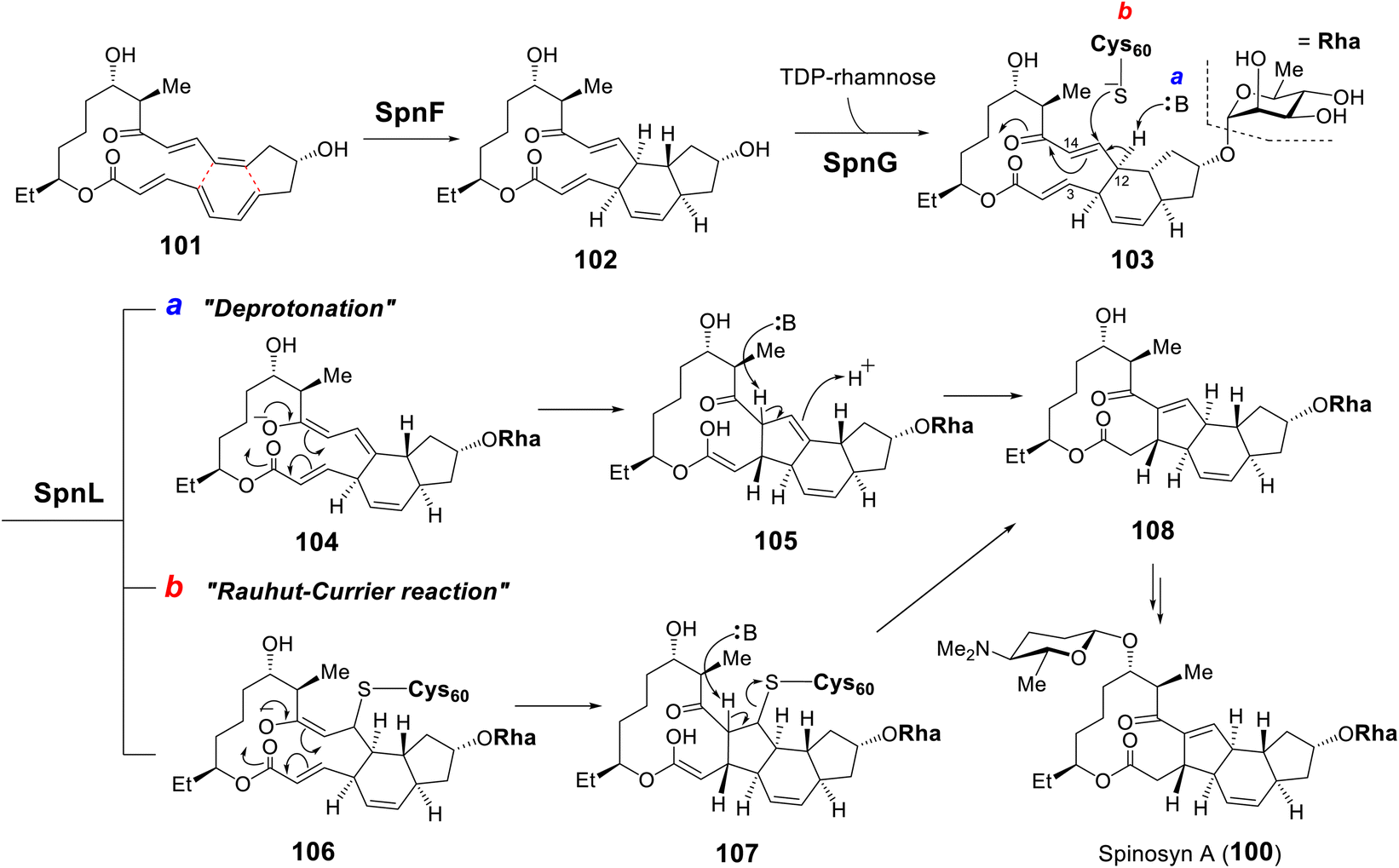 | ||
| Fig. 22 Biosynthesis of spinosyn A highlighting the [4 + 2] cycloaddition catalyzed by SpnF and two proposed mechanisms for the transannulation catalyzed by SpnL. | ||
Following SpnF catalyzed [4 + 2] cycloaddition and SpnG catalyzed glycosylation,182 SpnL catalyzes the transannular carbon–carbon bond formation between C3 and C14 of 103 to generate 108 (Fig. 22).177 Two possible mechanisms have been postulated, the first involving deprotonation at C12 to form an enolate (104) poised for Michael addition at C3 giving 105. Alternatively, a nucleophilic residue in the active site may first add to C13 to yield intermediate 106, which may then cyclize into 107 prior to elimination of the assisting nucleophile. Involvement of a nucleophilic residue during modification of the β-carbon of an enone moiety is indicative of a Rauhut–Currier (RC) reaction146,183 and is reminiscent of thymidylate synthase chemistry.184 When the SpnL reaction was conducted in buffered D2O, only a single solvent deuterium was incorporated into the product; however, no significant primary KIE was observed with the C12-deuterated substrate.185 Moreover, a C13-fluorinated substrate analog 109 led to inactivation of SpnL via the formation of a covalent enzyme–substrate adduct (112) demonstrated by mass spectrometry (Fig. 23A).185 Thus, all the biochemical evidence is consistent with an RC mechanism. SpnL shares 35% sequence identity with SpnF and also copurifies with bound SAH.185 The binary complex of SpnL with SAH bound was solved at 3.05 Å (Fig. 21C) and demonstrated that Cys60 resides nearby the bound SAH with its thiol moiety buried and apparently inaccessible from the active site cavity (Fig. 23B).185 Consequently, a conformational change upon substrate binding was proposed to allow for Cys60 exposure to the substrate thereby facilitating catalysis.
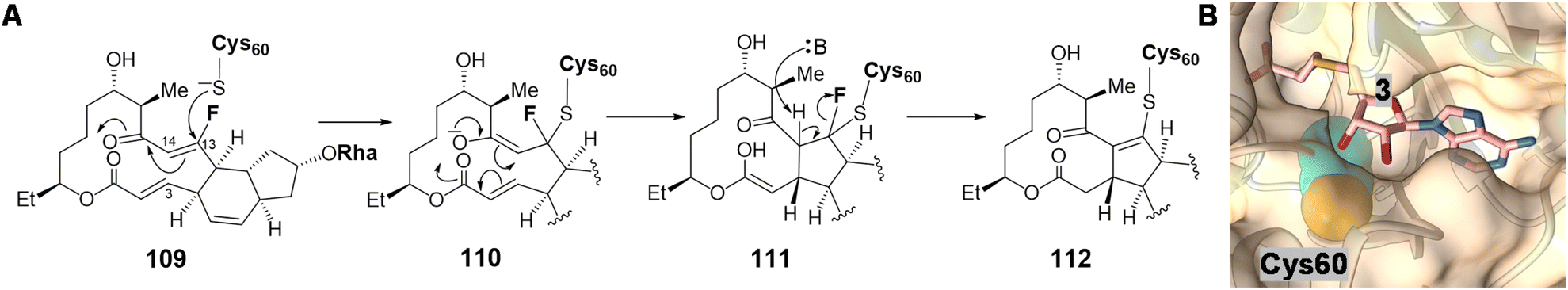 | ||
| Fig. 23 (A) C13-fluorinated substrate (109) leads to covalent modification of SpnL. (B) Crystal structure of SpnL/SAH binary complex (PDB ID: 7V6H) highlighting the SAH (3) binding site and the catalytic Cys60 (carbons as cyan and sulfur as yellow spheres), which is buried in the protein interior. | ||
During biosynthesis of the fungal secondary metabolite ilicicolin H (113), the cyclohexene moiety is believed to be constructed via an inverse-electron demand Diels–Alder (DA) reaction catalyzed by IccD, which harbors an intact SAM-binding motif yet does not copurify with SAM (Fig. 24A).186 Moreover, two additional groups of methyltransferase-like proteins were also found to catalyze pericyclic reactions. EpiI, UpiI and HpiI are proposed to catalyze a hetero-DA reaction yielding 119 on the quinone methide substrate (117) generated upon dehydration of 116, while PdxI, AdxI and ModxI catalyze conversion of the same substrate to 121via a proposed Alder-ene reaction (Fig. 24B).187 The catalytic cycles of these pericyclases, however, do not depend on either SAM or SAH indicating they are mechanistically distinct from those of LepI or SpnF and SpnL.186,187 Pericyclases characterized by a SAM-binding fold appear both evolutionarily and catalytically distinct.188–190
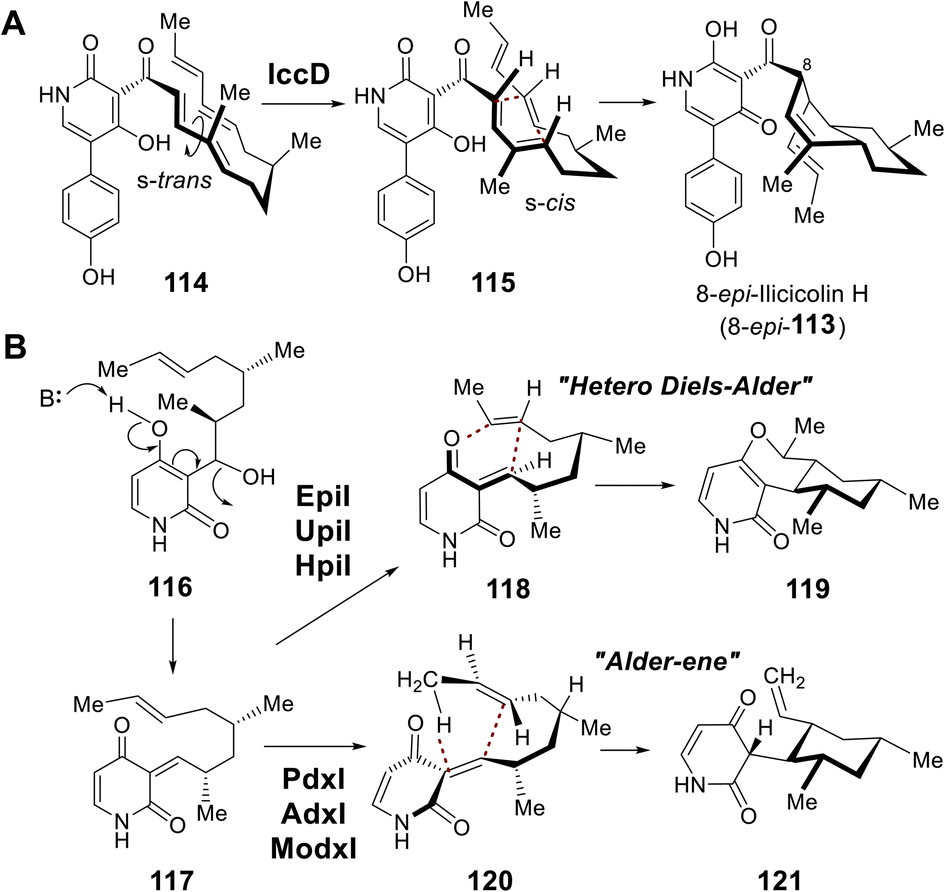 | ||
| Fig. 24 (A) Cyclization catalyzed by IccD during the biosynthesis of ilicicolin H. (B) Dehydration–cyclization reactions catalyzed by SAM-independent pericyclases that each adopt a SAM-binding fold. | ||
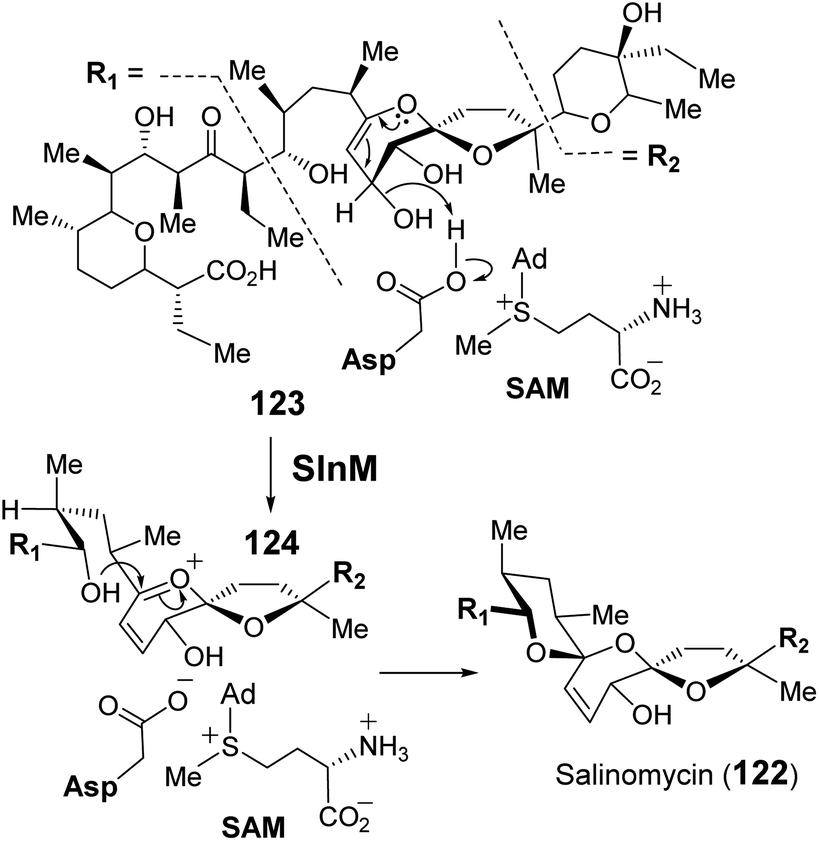 | ||
| Fig. 25 SlnM catalyzes spirocyclization during salinomycin biosynthesis. SAM is proposed to stabilize an anionic aspartate residue via electrostatic interactions to facilitate substrate protonation. | ||
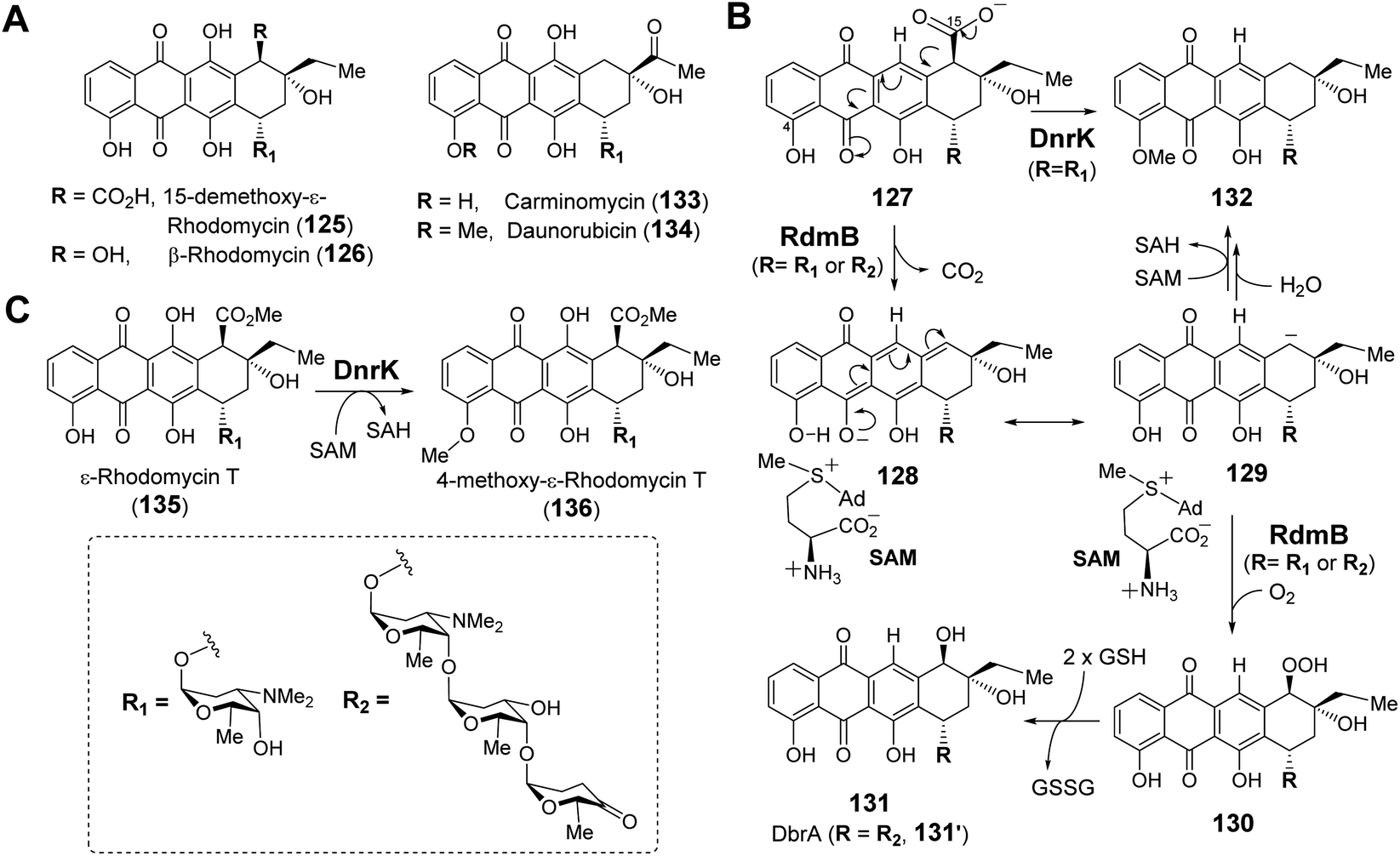 | ||
| Fig. 26 (A) Selected anthracycline natural products generated via RdmB or DnrK catalysis. (B) Proposed mechanism for RdmB/DnrK reaction with 127. (C) Activity of methyltransferase DnrK with 135. | ||
There is reason to believe that RdmB may have diverged from an evolutionary lineage of SAM-dependent O-methyltransferases upon losing methyltransferase activity. DnrK is a homolog of RdmB (52% sequence identity) that catalyzes methylation at O-4 of carminomycin (133) yielding daunorubicin (134),202 which is a powerful chemotherapeutic agent and close structural analog of β-rhodomycin (126) (Fig. 26A).203 Comparison of the crystal structures of RdmB and DnrK demonstrated subtle changes consistent with the divergence in catalytic activities. Thus, the crystal structure of DnrK bound with the methylation product 4-methoxy-ε-rhodomycin T (136, Fig. 26C) and SAH shows alignment between the acceptor hydroxyl group, the transferred methyl and the sulfur of SAH as expected for methyltransferase activity (Fig. 27A).204 In contrast, the ternary complex of RdmB bound with SAM and the hydroxylation product 11-deoxy-β-rhodomycin A (DbrA, 131′) revealed an O–C–S angle of 122.9° between the SAM sulfonium and the corresponding hydroxyl oxygen of 131′ suggesting misalignment for efficient nucleophilic displacement (Fig. 27A).200
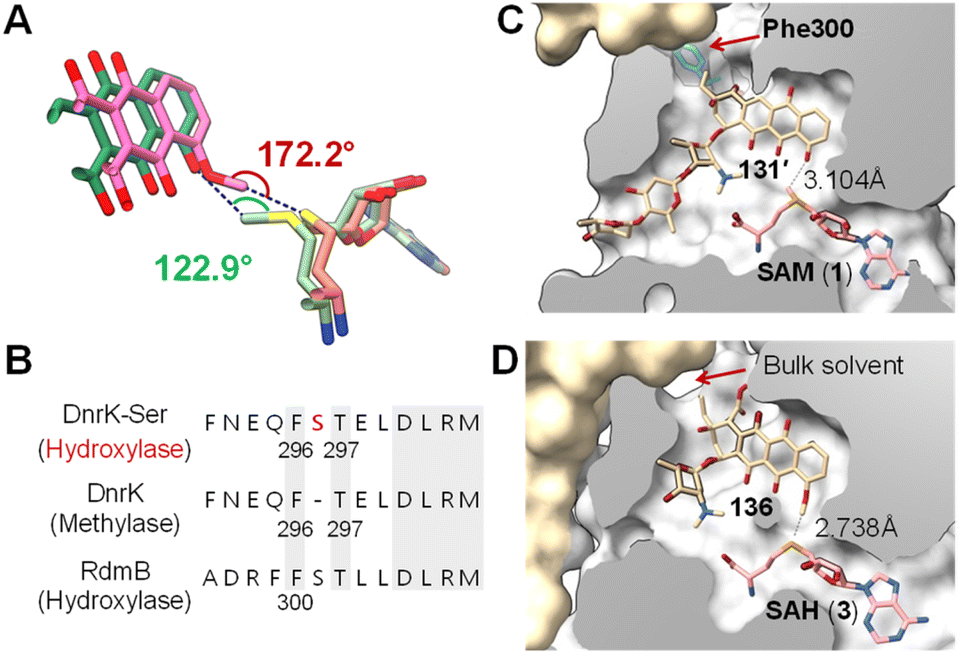 | ||
| Fig. 27 (A) Comparison of the binding of SAM (light green)/131′ (green) in RdmB and SAH (salmon)/136 (pink) in DnrK. (B) Sequence alignment of DnrK and RdmB at the α-helix that contains the aromatic residue, highlighting the position of serine insertion in DnrK-Ser. The orientation of Phe296 in DnrK or Phe300 in RdmB dictates the catalytic activity being methylation or hydroxylation. (C) Clip view of the RdmB/SAM/131′ complex (PDB ID: 1XDS). (D) Clip view of the DnrK/SAH/136 complex (PDB ID: 1TW2). The channel to the bulk solvent is blocked in RdmB due to the presence of Phe300 (cyan), whereas it is opened in DnrK. | ||
Recent structural comparisons of RdmB and DnrK coupled with extensive mutational studies by Grocholski and co-workers have provided even more insight into this question.205 The Michaelis complex of RdmB is solvent inaccessible,200 which prevents protonation of the anionic intermediate 129 by water. Closure of the active site is facilitated by a loop that includes Phe300 and blocks the channel connecting the substrate binding site to the external media (Fig. 27C).205 In contrast, the corresponding Phe296 in DnrK faces away from the channel leaving the active site of DnrK solvent accessible (Fig. 27D). Notably, DnrK catalyzes the conversion of 127 to 132in vitro (Fig. 26B).205 Although the timing of methylation at C4–OH is unclear, this observation is consistent with the hypothesis that upon decarboxylation at C15, the resulting anion (i.e., 129) is quenched by water in the DnrK active site.205 Moreover, the different orientation of Phe300 in RdmB and Phe296 in DnrK is due to Ser301 in RdmB, which is missing in the primary sequence of DnrK205 (Fig. 27B); however, Phe296 in DnrK can be rotated towards the channel thus blocking solvent access by insertion of a serine residue between Phe296 and Thr297, which mimics the Ser301 in RdmB (Fig. 27B).205 As expected, this DnrK insertion mutant (DnrK-Ser) did exhibit hydroxylase activity, implying the importance of solvent extrusion to support hydroxylation activity.205
Human complex I is a respiratory chain protein that drives proton pumping for ATP synthesis by mediating electron transfer between NADH and ubiquinone.206,207 NDUFAF5 is an essential protein in the biogenesis of human complex I and is characterized by a conserved SAM-binding motif at the C-terminus. Suppression of NDUFAF5 expression results in reduced hydroxylation of an arginine residue in the protein NDUFS7, which is a subunit in human complex I.208 Thus, NDUFAF5 has been hypothesized to be an arginine hydroxylase targeting NDUFS7 (137 → 138) (Fig. 28). The secondary structure of NDUFAF5 is predicted to be similar to that of RdmB in both the substrate and SAM binding regions indicating that NDUFAF5 might adopt an active site structure similar to that of RdmB.208 While the in vivo activity of NDUFAF5 has been correlated with arginine hydroxylation, the in vitro activity and mechanistic details regarding whether the hydroxylase activity is SAM or SAH dependent and how the arginine is activated for oxidation remains unclear.
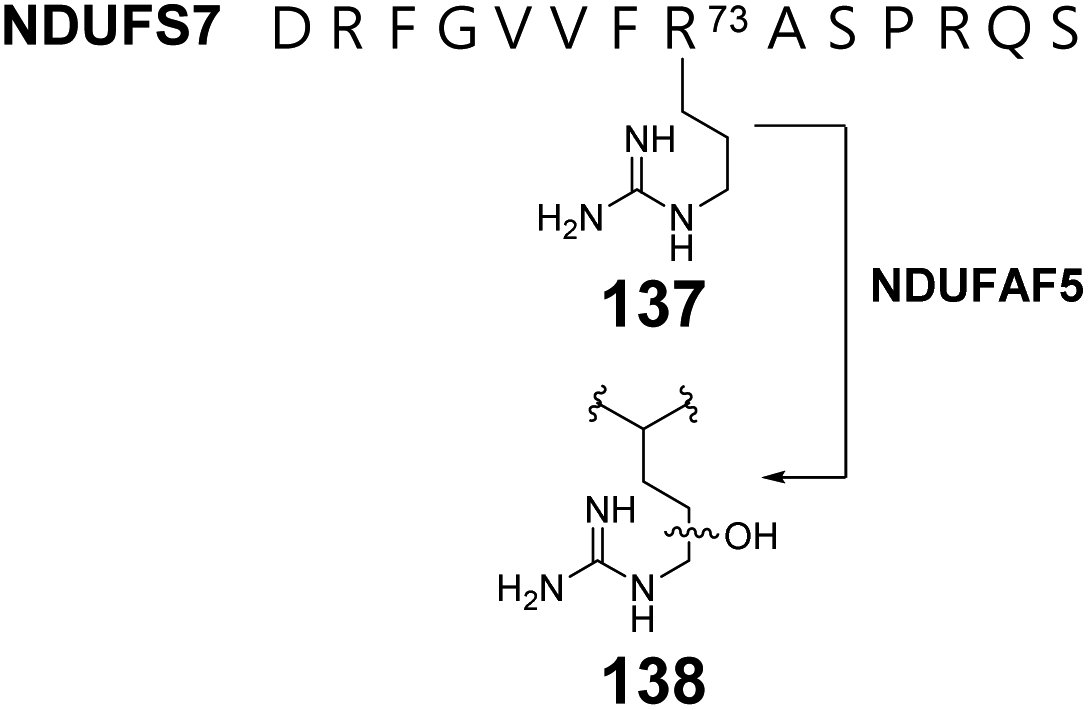 | ||
| Fig. 28 NDUFAF5 catalyzes modification of the substrate protein NDUFS7 at Arg73. The precise site of hydroxylation is unknown. | ||
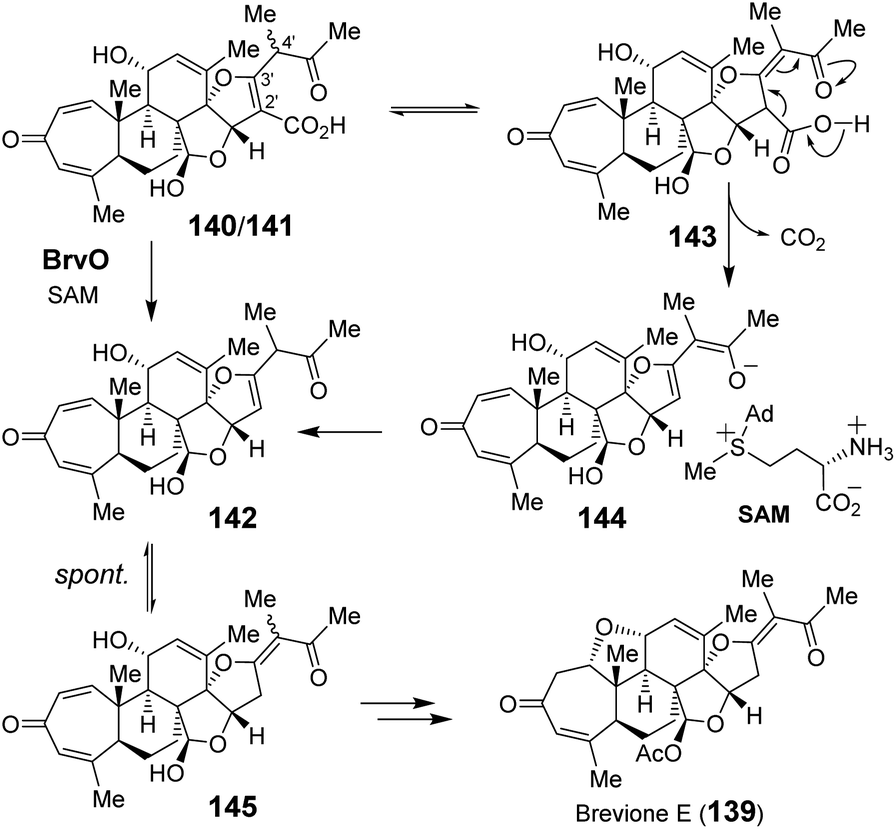 | ||
| Fig. 29 Proposed mechanism for BrvO catalyzed decarboxylation. | ||
2.5 SAM as a nucleophile
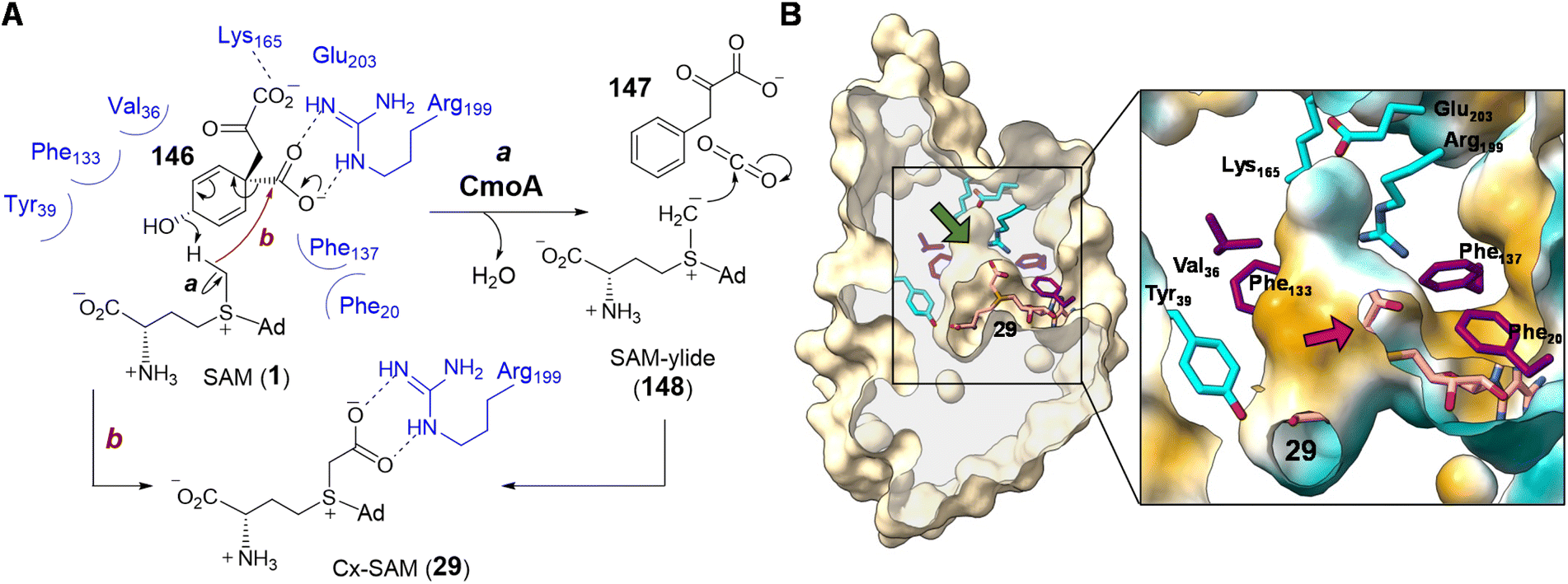 | ||
| Fig. 30 (A) Proposed mechanism for CmoA catalysis. (B) Clip view of Cx-SAM/CmoA complex structure (PDB ID: 4GEK) highlighting the inner cavity for Cx-SAM binding. Computational substrate docking models have suggested that SAM adopts a similar binding configuration as that of the product Cx-SAM. Prephenate (146) is predicted to bind in the hydrophobic pocket adjacent to SAM (green arrow). In the zoomed in region, the surface coloring indicates hydrophobicity (hydrophobic in gold and hydrophilic in cyan). The red arrow indicates the position of the methyl group of SAM, which is surrounded by a hydrophobic environment. | ||
The first half of the CmoA catalyzed reaction is hypothesized to proceed analogous to that of prephenate dehydratase, where decarboxylation of prephenate facilitates elimination of a hydroxide ion thereby generating CO2 and phenylpyruvate (147).211 In the catalytic cycle of prephenate dehydratase, the eliminated hydroxide is believed to abstract a proton from an active site threonine residue thereby completing the reaction.211 As the pKa of the threonine hydroxyl is close to that measured for the trimethylsulfonium ion,212 the same hydroxide is expected to be competent for deprotonating the methyl of SAM. The resulting sulfonium methylide (148) can then add to the eliminated molecule of CO2 to yield Cx-SAM (29). Moreover, the X-ray crystal structure of Cx-SAM-bound CmoA from E. coli revealed an overall Rossmann fold86,87 and a cavity adjacent to the Cx-SAM binding site that was predicted to be the prephenate (146) binding site by molecular docking.86 Notably, the region of the active site adjacent to the eliminated prephenate hydroxide is lined primarily with hydrophobic residues leaving the methyl group of SAM the only available proton source nearby (Fig. 30B).86
Computational substrate docking has also suggested an arrangement of the prephenate hydroxyl and SAM methyl that disallows a typical methylation reaction.86 This is consistent with the observation that CmoA does not catalyze O-methylation of prephenate. The docking results instead predicted a 3.4 Å O⋯H–C hydrogen bond between the prephenate hydroxyl group and the SAM methyl, which is more consistent with the proposed deprotonation reaction.86 Formation of the putative sulfonium methylide (148) was also supported by the observed exchange of only a single deuterium from CD3-labelled SAM with solvent under turnover conditions. Finally, the low dielectric constant of the hydrophobic active site may enhance the basicity of the eliminated hydroxide while increasing the acidity of the SAM methyl.213–216 This would address the large difference in pKa between water and sulfonium α-carbons measured in polar solvents.212,217 Likewise, a hydrophobic substrate binding site has also been proposed in the case of Ecm18 to lower the pKa value of a sulfonium intermediate in assisting the formation of a sulfur ylide (80) (Section 2.3.3).159 While current evidence points to an intermediary sulfonium ylide (148), the reaction could also occur in a concerted manner avoiding the formation of a discrete ylide intermediate (Fig. 30A, route b).
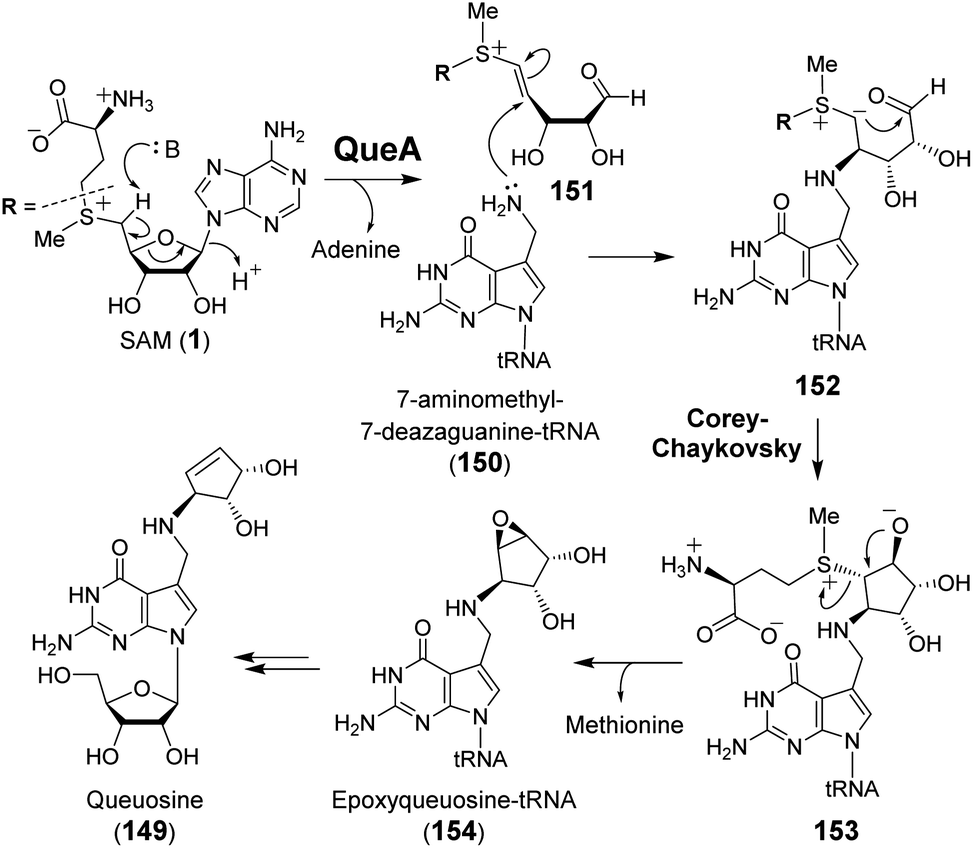 | ||
| Fig. 31 Proposed mechanism of QueA catalyzed ribose transfer reaction. | ||
2.6 SAM as an amino donor
SAM is also recognized as an amino donor; however, only two instances of such enzymology have been reported. BioA depends on PLP and catalyzes the oxidative deamination of SAM to its α-keto acid congener while reductively aminating 7-keto-8-aminopelargonic acid (KAPA, 156) to 7,8-diaminopelargonic acid (DAPA, 157) during the biosynthesis of biotin (155) (Fig. 32A).223,224 The mechanism of BioA is thus no different from other PLP transaminases,225 despite SAM serving as the amine donor rather than a free amino acid. In contrast, the second example appears to be unrelated to PLP chemistry and contributes to the biosynthesis of rhodoquinone in Rhodospirillum rubrum.226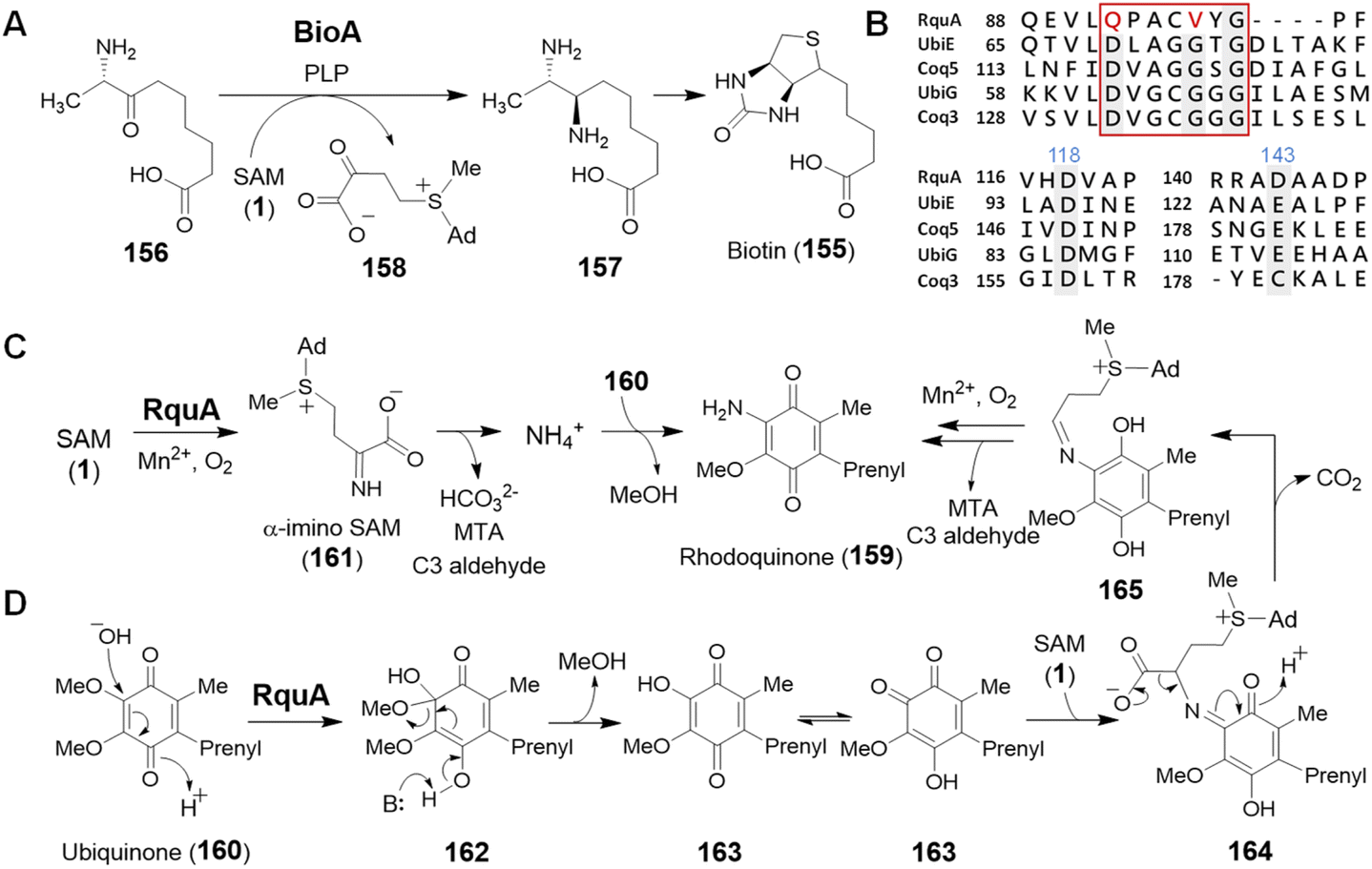 | ||
| Fig. 32 (A) Transamination of SAM catalyzed by BioA. (B) Sequence alignment of RquA with related functional methyltransferases. RquA has two essential aspartate residues (Asp118 and Asp143, numbers in blue) for SAM binding whereas the DXXXGXG (red box) motif typically found in Rossmann fold MTs is missing in RquA. (C and D) Two proposed mechanisms for RquA catalyzed transamination during the biosynthesis of rhodoquinone (159). | ||
Rhodoquinone (159) differs structurally from ubiquinone (160) by replacing an O-methoxy group with an amine in a reaction that requires the enzyme RquA.227,228 RquA was recently characterized as a manganese-dependent enzyme that catalyzes transfer of the α-amino group from SAM to ubiquinone (160) to yield rhodoquinone (159).229 While RquA resembles SAM-dependent methyltransferases, only a subset of the conserved residues in the predicted Rossmann fold generally associated with SAM binding are required for activity (Fig. 32B).227,229 In addition to a divalent metal ion, RquA catalysis also depends on aerobic conditions as well and is expected to involve a redox component. Indeed, isotope labelling experiments have suggested the oxidative decomposition of SAM to CO2, MTA and an aldehyde fragment during the transformation.229 Mechanistic hypotheses for this transformation are shown in Fig. 32C and D; however, much work is still left to be done before the chemistry of this unique enzyme is fully understood.
3 Enzymes without a typical MT-fold
As discussed in the previous sections, evolutionary diversification of SAM-dependent methyltransferases has led to the emergence of several different types of chemistry. However, there also exist groups of SAM utilizing enzymes that lack the MT-fold making their evolutionary relationship with the SAM-dependent methyltransferases less clear. These enzymes also show a broad range of different activities again emphasizing the mechanistic versatility and intrinsic reactivity of SAM as a biological substrate.3.1 Pyridoxal 5′-phosphate-dependent enzymes
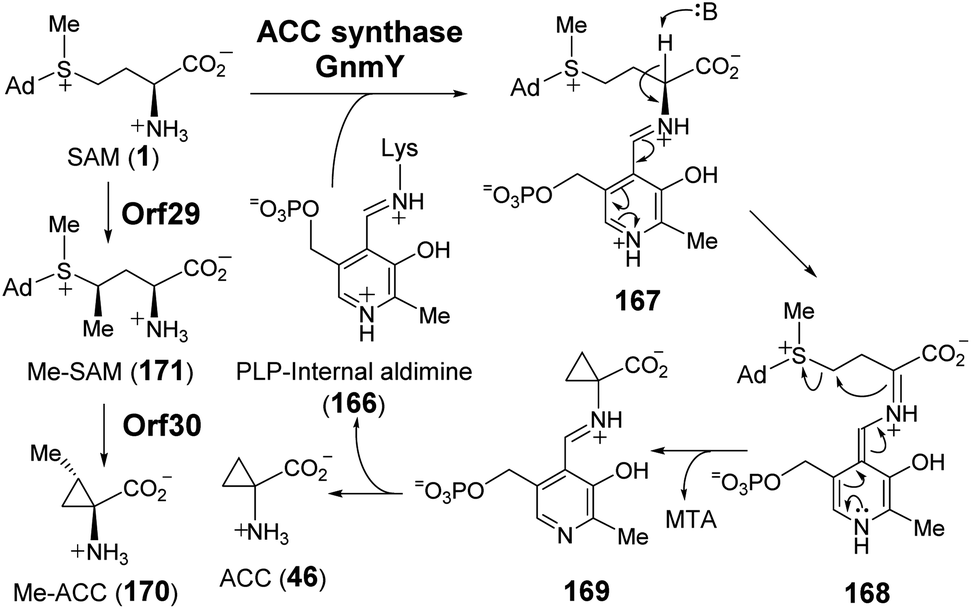 | ||
| Fig. 33 Proposed mechanism for the transformation of SAM to ACC (46) and Me-ACC (170) catalyzed by GnmY and Orf29/Orf30, respectively. | ||
Recently, guangnanmycin (GNM, 172), which is a structural homolog of the antitumor antibiotic leinamycin (LNM), was discovered through genome mining.239 Apart from a hybrid peptide–polyketide backbone, GNM also features an unusual ACC moiety (Fig. 34).239 Comparative analysis of lnm-type biosynthetic gene clusters between guangnanmycin producers and non-producers revealed that the gene product of gnmY may catalyze the formation of ACC from SAM. This functional assignment was then validated by in vivo inactivation of gnmY, which rendered the mutant strain unable to produce guangnanmycin unless exogenous ACC is supplemented.240 Sequence analysis revealed that GnmY belongs to the aspartate aminotransferase superfamily, and subsequent in vitro characterizations confirmed that GnmY catalyzes the conversion of SAM to ACC in a PLP-dependent manner.240 GnmY also exhibits steady-state kinetic parameters comparable to those of plant ACC synthases and utilizes a catalytic lysine residue (Lys251) to form an internal aldimine (166) with PLP during catalysis.240 Nevertheless, bioinformatics analyses have suggested that GnmY is phylogenetically distinct from the family of plant ACC synthases despite sharing a similar mechanism of catalysis.240
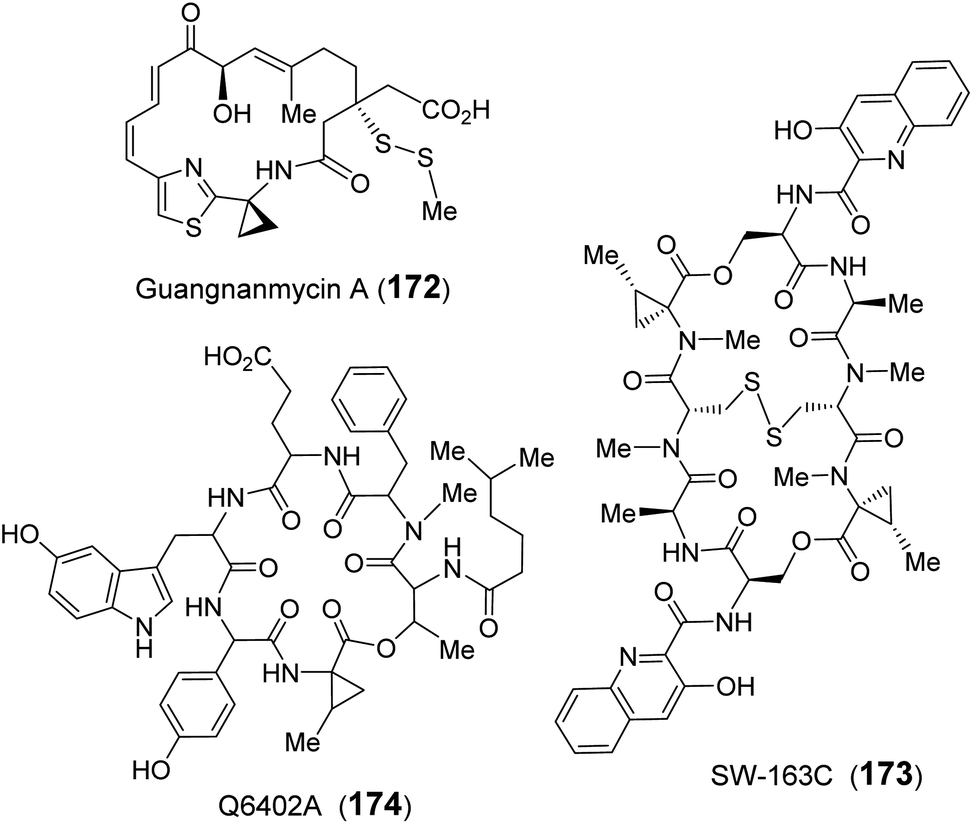 | ||
| Fig. 34 Natural products that contain SAM-derived cyclopropane moieties. | ||
Homologs of GnmY have also been found in the putative biosynthetic gene clusters for two other peptide antibiotics, SW-163C (173) and Q6402A (174),157,241 which are each decorated with an unusual 2-methyl-ACC (MeACC, 170) moiety (Fig. 34).242,243 In the biosynthetic gene cluster of Q6402A, the gene product of orf30 shows homology with GnmY suggesting that its activity may involve the formation of MeACC (170).241 Initial characterization of purified Orf30 revealed its ability to catalyze the conversion of SAM to ACC.241 However, Orf30 exhibits a 1900-fold lower value of kcat compared to GnmY such that SAM may not be the native substrate of Orf30.241 The B12-dependent radical SAM methylase Orf29 encoded by a gene fragment upstream of Orf30 was later demonstrated to catalyze methylation of SAM yielding (4′′R)-4′′-methyl-SAM (171).244 This SAM derivative (171) can be efficiently cyclized to give MeACC (170) under the action of Orf30 (Fig. 33).244 It remains to be established how Orf30 distinguishes between SAM and methylated-SAM.
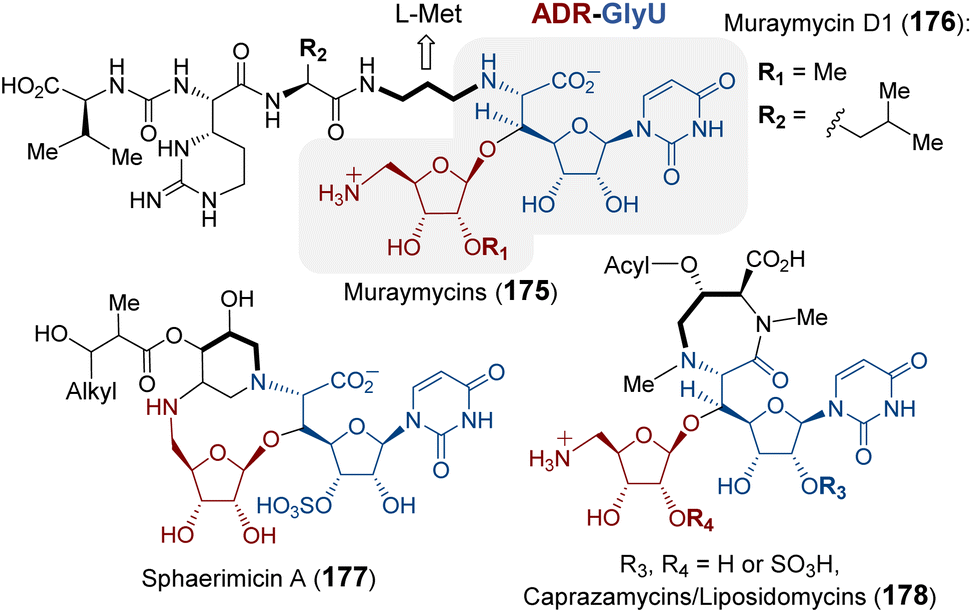 | ||
| Fig. 35 Natural products that contain the ADR-GlyU disaccharide. The bold alkyl chain indicates the putative methionine-derived aminopropyl moiety. | ||
Purification and in vitro characterization of Mur24 and its homologs LipJ250 and SphL252 indicated that all three enzymes do indeed catalyze ACP transfer from SAM to the substrate (182) amine coupled with the elimination of MTA (Fig. 36).248 To investigate the role of PLP in this reaction, the conserved lysine residue necessary for internal aldimine formation with PLP was mutated leading to a complete loss of ACP transfer activity.248 The catalytic mechanism of Mur24 was further studied using SAM isotopologs, the results of which provided evidence that two protons are washed out in the product (183) including the C-α proton and either the C-β or C-γ proton.248 Furthermore, reactions run in D2O led to a mixture of both single and double solvent deuterium incorporation into the product 183.248 These observations implied sequential and reversible deprotonation at C-α and C-β leading to elimination of MTA following SAM–PLP aldimine (167) formation. The unsaturated iminium intermediate, or vinylglycine–PLP aldamine (181), thereby generated then undergoes aza-Michael addition to yield the N-alkylated product 183 (Fig. 36).
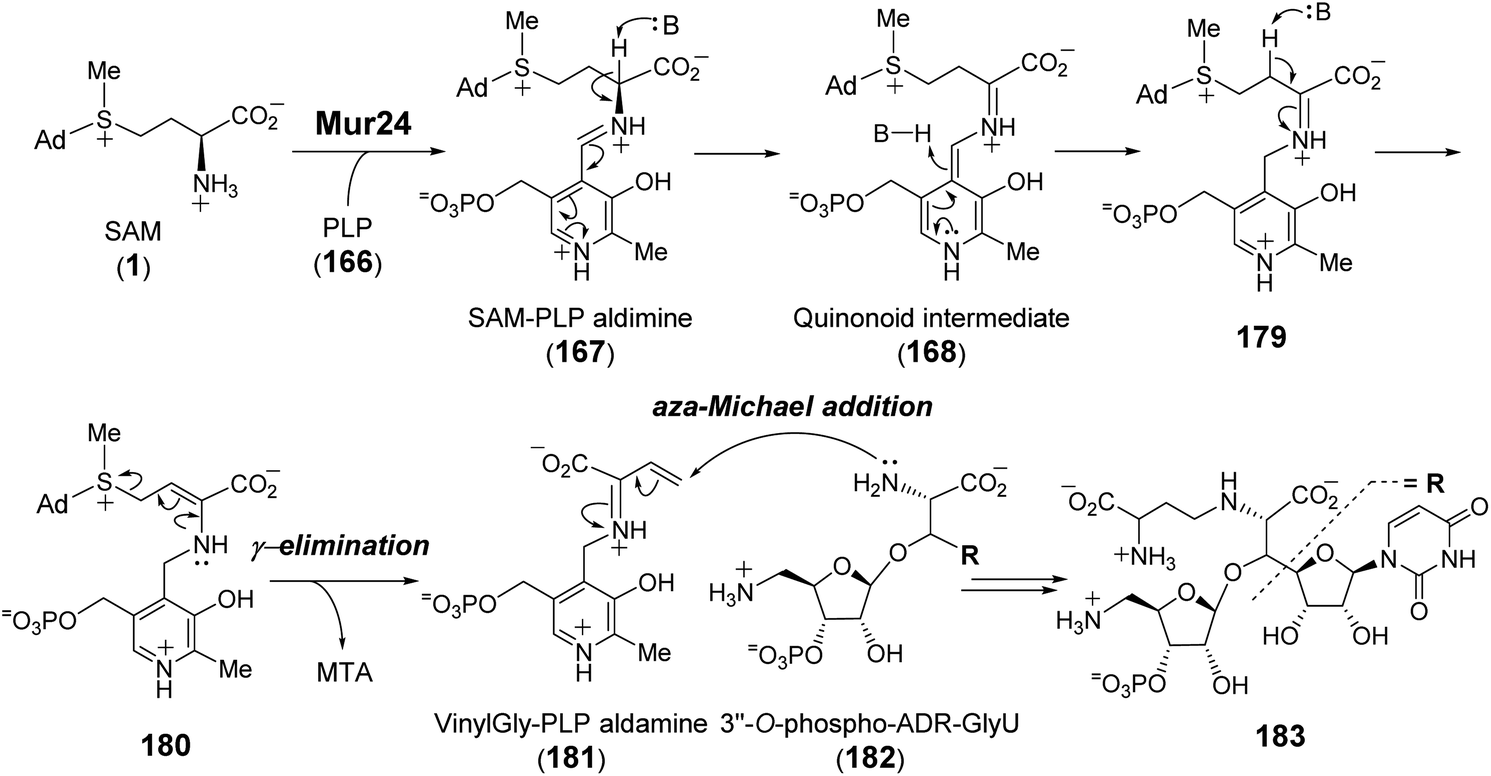 | ||
| Fig. 36 Proposed mechanism for Mur24 catalyzed reaction. | ||
More recently, the PLP-dependent enzyme SbzP was found to catalyze formation of the 6,5-bicyclic core of 6-azatetrahydroindane containing anticancer natural products (184–186).253–255 Feeding experiments established that the biosynthetic origin of the C4 moiety on the five-member ring is likely derived from aspartate and the six-member ring from a nicotinamide containing nucleotide.256 Extensive substrate screening corroborated SAM and β-nicotinamide adenine dinucleotide (β-NAD, 187) as the native substrates of SbzP (Fig. 37).256 Steady-state kinetic analysis implied a ping-pong Bi–Bi mechanism. Upon incubation of SbzP with SAM, MTA is released with concomitant formation of an enzyme-bound intermediate prior to NAD binding and subsequent formation of the annulation product.256
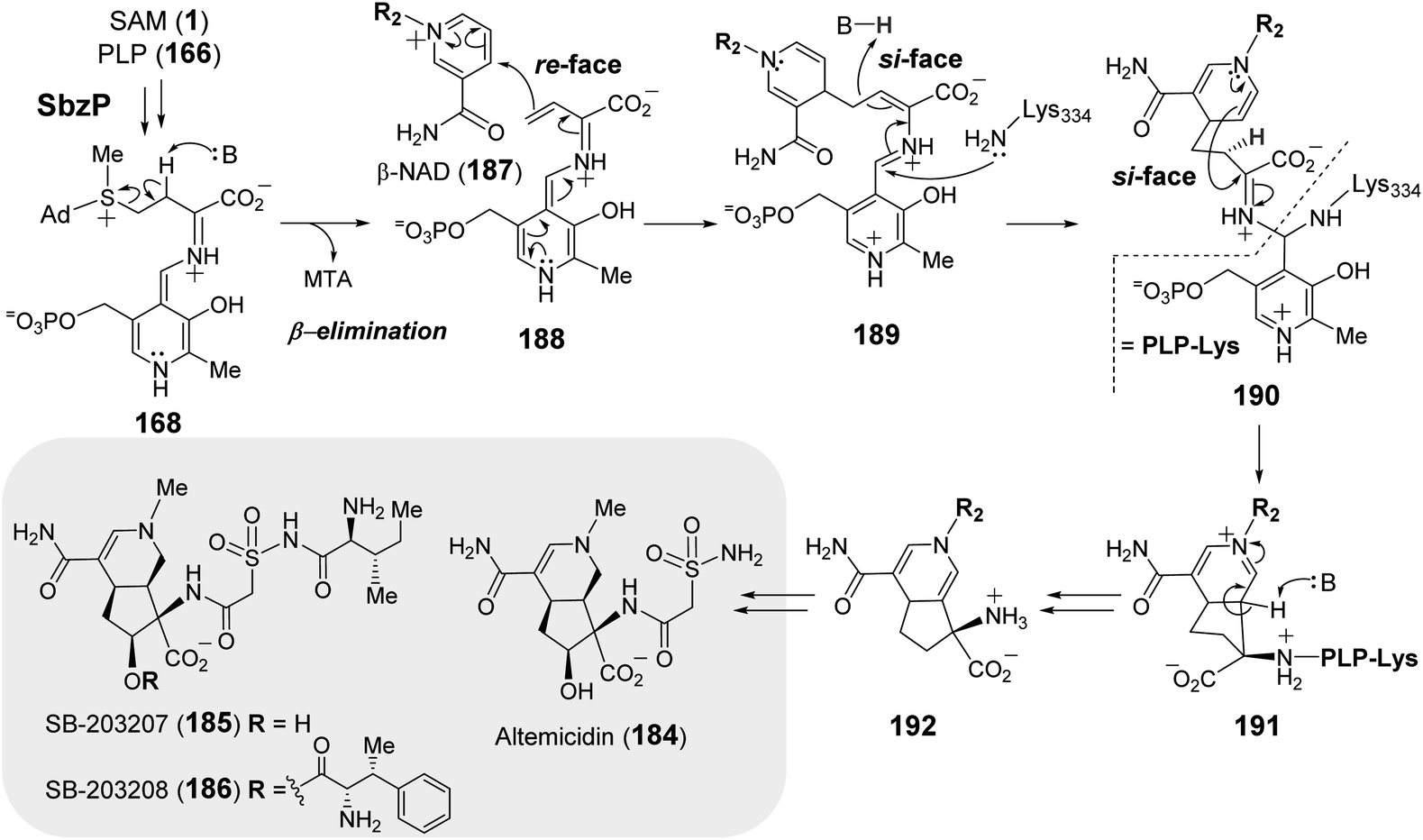 | ||
| Fig. 37 Proposed mechanism of SbzP catalyzed transformation of SAM (1) and β-NAD (187) into 192, which serves as the precursor to 6-azatetrahydroindane containing natural products (184–186). | ||
Although the SbzP catalyzed fragmentation of SAM appears similar to that of Mur24 at the point of generating 168, the second half reactions catalyzed by Mur24 and SbzP are distinct. While the unsaturated iminium (181) generated during the Mur24 catalytic cycle is connected to PLP as an aldamine and undergoes alkylation by an incoming nucleophile, during SbzP catalysis, the vinyl glycine iminium 188 instead links to a quinonoid form of PLP and serves to increase the nucleophilicity of C-γ facilitating its addition to the electrophilic nicotinamide ring of NAD (188 → 189) (Fig. 37). Addition of the active site lysine residue covalently links the intermediate 189 with the enzyme as a gem-diamine 190 and facilitates rearrangement to the bicyclic structure 192. Spectroscopic analysis of SbzP revealed that addition of SAM results in a unique absorption at 520 nm consistent with the formation of a β,γ-unsaturated quinonoid (188) with an extended π-conjugation as predicted.256 An analogous spectroscopic characterization of Mur24 is presently unavailable; however, it would also help provide additional mechanistic insight into how the PLP cofactor participates in reactions with opposite electronic demands.
The enzymes described above employ a SAM–PLP quinonoid intermediate (168) that can react via several different pathways eliminating MTA to either yield an unsaturated iminium or induce an intramolecular cyclization. In the former case, the iminium is susceptible to both electrophilic and nucleophilic addition. Moreover, electrophilic addition can occur at either C-α or C-γ, with the latter exemplified by SbzP. In contrast, attack at C-α is operative in the enzyme CqsA. Initial study of CqsA (PDB ID: 2WK8) revealed its structural resemblance with other PLP-dependent enzymes257,258 and its activity towards different amino acids. However, SAM was later identified as the biosynthetic CqsA substrate to yield EA-CAI-1 (194), which is the precursor to the quorum sensing autoinducer CAI-1 (193) (Fig. 38).257 CAI-1-type autoinducers are produced by Vibrio cholerae, which is the bacterium that causes the life-threatening disease cholera.259 The net transformation of SAM to EA-CAI-1 (194) catalyzed by CqsA involves C–S bond cleavage at C-γ, C–C bond formation at C-α with a fatty acyl-CoA (195) and decarboxylation. The catalytic cycle appears to begin with formation of a PLP–SAM adduct that is converted to a quinonoid intermediate (188) between vinyl glycine and PLP following elimination of MTA as in the case of SbzP. CqsA catalysis then proceeds with nucleophilic addition of the α-carbon to the incoming acyl-CoA substrate (195) before subsequent decarboxylation (196 → 197). The resulting EA-CAI-1 (194) is then converted to CAI-1 (193) by subsequent enzymes in vivo.260 While the aforementioned transformations exploit the nucleofugality of the sulfonium moiety of SAM, they are not direct SN2-type substitution reactions and may also be considered within the context of exotic PLP chemistry, which has been reviewed recently.261
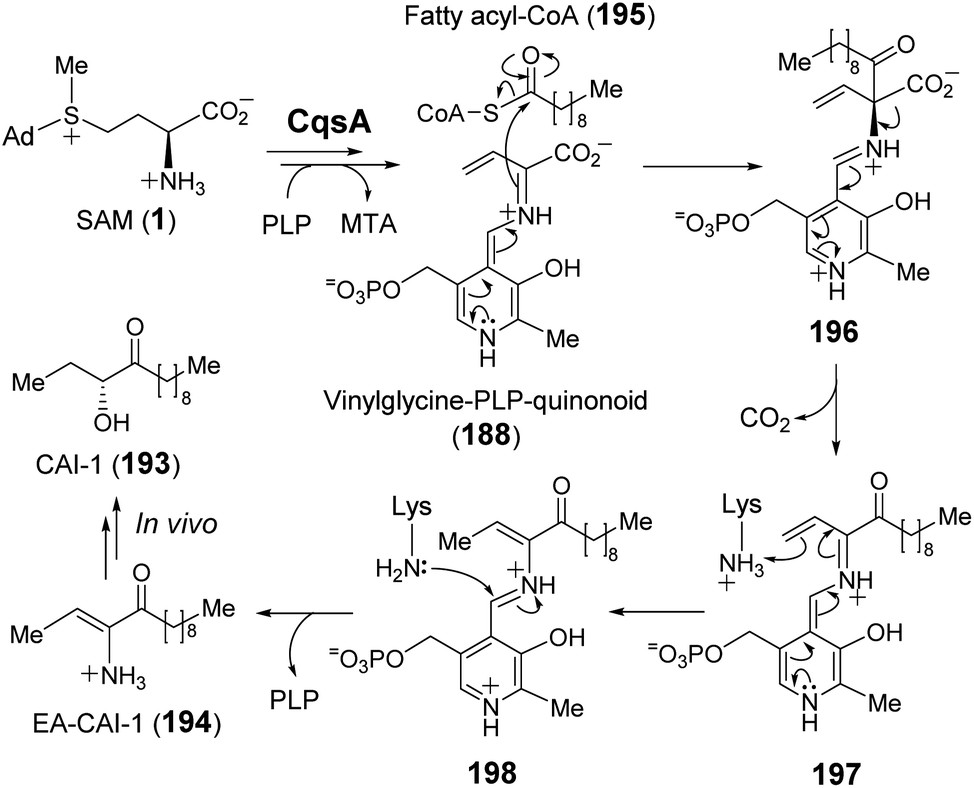 | ||
| Fig. 38 Proposed mechanism of CqsA reaction. | ||
3.2 Polyketide synthase
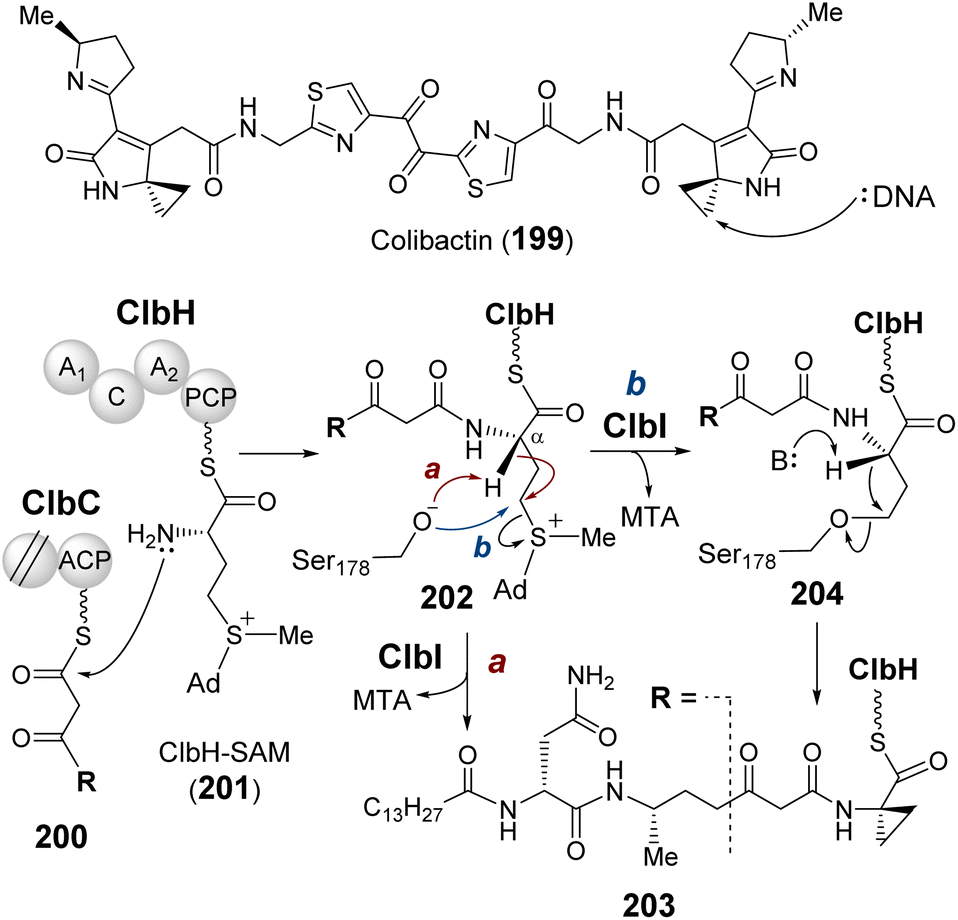 | ||
| Fig. 39 Proposed mechanism for the ClbI catalyzed cyclopropylation during the formation of colibactin biosynthetic intermediate 203. | ||
4 Conclusions
SAM is ranked among the most frequently observed substrates in enzymatic transformations.18 The majority of known SAM dependent biological reactions involve methylation; however, regioselective transfer of all three alkyl substituents of the SAM sulfonium have been reported (Sections 2.1 and 2.2) and may represent components of more complex biosynthetic cascades (Section 2.3). In addition to alkyl transfer, several other types of SAM-dependent reactions catalyzed by enzymes have been established including α-deprotonation with the potential for ylide formation (Section 2.5) as well as β-elimination concomitant with elimination of a neutral disulfide (Section 3).In all cases, these enzyme catalyzed reactions conform to the well-known chemistry of the sulfonium cation, such that the role of the enzyme is to selectively stabilize those transition states representing one mode of reaction over another. Structural investigations of these enzymes have been critical in defining these constraints thereby explaining both the modes of catalysis as well as the observed regiochemistry underlying these various enzyme catalyzed reactions. Moreover, the principles underlying sulfonium enzymology are not limited to SAM-dependent enzymes alone and have also been appreciated among the dimethylsulfoniopropionate (DMSP) lyases found in marine bacteria266 as well as BurG during the biosynthesis of malleicyprols.267,268
Many aspects of SAM enzymology, however, remain to be explored. In particular, several enzymes as described in Section 2.4 require SAM even though the catalytic cycles of these enzymes do not appear to involve any sort of covalent rearrangement or fragmentation of SAM itself. In these instances, SAM may be required for proper folding and structural integrity of the enzyme,269 or it may play a more direct role in catalysis by modifying the structural and electrostatic features of the active site.270,271
Many of the SAM-dependent enzymes also exhibit structural features that imply homology with the SAM methyltransferases, whereas others do not, suggesting different evolutionary lineages. Consequently, how these enzymes diversified in some cases from a common evolutionary origin to exhibit the broad range of biochemical activities and unique catalytic roles of SAM seen today is yet another question that remains open to investigation. In any case, SAM has become a focal point of modern enzymology that will likely bring many new discoveries and surprises in the years to come.
5 Conflicts of interest
The authors declare no conflicts of interest.6 Acknowledgements
We thank Dr Mark W. Ruszczycky for his help with the English and his comments on the accuracy of the content. This work was supported by grants from the National Institutes of Health (GM035906 and GM040541) and the Welch Foundation (F-1511).7 References
- S. C. Lu, Int. J. Biochem. Cell Biol., 2000, 32, 391–395 CrossRef CAS PubMed.
- P. K. Chiang, R. K. Gordon, J. Tal, G. C. Zeng, B. P. Doctor, K. Pardhasaradhi and P. P. McCann, FASEB J., 1996, 10, 471–480 CrossRef CAS.
- W. A. M. Loenen, Biochem. Soc. Trans., 2006, 34, 330–333 CrossRef CAS PubMed.
- D. K. Liscombe, G. V. Louie and J. P. Noel, Nat. Prod. Rep., 2012, 29, 1238–1250 RSC.
- M. Blum, H.-Y. Chang, S. Chuguransky, T. Grego, S. Kandasaamy, A. Mitchell, G. Nuka, T. Paysan-Lafosse, M. Qureshi, S. Raj, L. Richardson, G. A. Salazar, L. Williams, P. Bork, A. Bridge, J. Gough, D. H. Haft, I. Letunic, A. Marchler-Bauer, H. Mi, D. A. Natale, M. Necci, C. A. Orengo, A. P. Pandurangan, C. Rivoire, C. J. A. Sigrist, I. Sillitoe, N. Thanki, P. D. Thomas, S. C. E. Tosatto, C. H. Wu, A. Bateman and R. D. Finn, Nucleic Acids Res., 2021, 49, D344–D354 CrossRef CAS.
- A.-W. Struck, M. L. Thompson, L. S. Wong and J. Micklefield, ChemBioChem, 2012, 13, 2642–2655 CrossRef CAS.
- H. J. Sofia, G. Chen, B. G. Hetzler, J. F. Reyes-Spindola and N. E. Miller, Nucleic Acids Res., 2001, 29, 1097–1106 CrossRef CAS.
- P. A. Frey and O. T. Magnusson, Chem. Rev., 2003, 103, 2129–2148 CrossRef CAS PubMed.
- N. Oberg, T. W. Precord, D. A. Mitchell and J. A. Gerlt, ACS Bio Med Chem Au, 2022, 2, 22–35 CrossRef CAS PubMed.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS PubMed.
- W. E. Broderick and J. B. Broderick, J. Biol. Inorg Chem., 2019, 24, 769–776 CrossRef CAS.
- M. W. Ruszczycky, Y. Ogasawara and H.-w. Liu, Biochim. Biophys. Acta, 2012, 1824, 1231–1244 CrossRef CAS.
- J. L. Vey and C. L. Drennan, Chem. Rev., 2011, 111, 2487–2506 CrossRef CAS PubMed.
- M. R. Challand, R. C. Driesener and P. L. Roach, Nat. Prod. Rep., 2011, 28, 1696–1721 RSC.
- K. Boswinkle, J. McKinney and K. D. Allen, J. Bacteriol., 2022, 204, e0019722 CrossRef.
- Y. Nicolet, Nat. Catal., 2020, 3, 337–350 CrossRef CAS.
- M. W. Ruszczycky, A. Zhong and H.-w. Liu, Nat. Prod. Rep., 2018, 35, 615–621 RSC.
- M. Fontecave, M. Atta and E. Mulliez, Trends Biochem. Sci., 2004, 29, 243–249 CrossRef CAS.
- S. Roje, Phytochemistry, 2006, 67, 1686–1698 CrossRef CAS PubMed.
- Q. Sun, M. Huang and Y. Wei, Acta Pharm. Sin. B, 2021, 11, 632–650 CrossRef CAS PubMed.
- L. Barra, T. Awakawa and I. Abe, JACS Au, 2022, 2, 1950–1963 CrossRef CAS PubMed.
- H. L. Schubert, R. M. Blumenthal and X. Cheng, Trends Biochem. Sci., 2003, 28, 329–335 CrossRef CAS PubMed.
- P. Z. Kozbial and A. R. Mushegian, BMC Struct. Biol., 2005, 5, 19 CrossRef PubMed.
- J. L. Martin and F. M. McMillan, Curr. Opin. Struct. Biol., 2002, 12, 783–793 CrossRef CAS PubMed.
- K. Umemura, H. Matsuyama and N. Kamigata, Bull. Chem. Soc. Jpn., 1990, 63, 2593–2600 CrossRef CAS.
- M. Kobayashi, K. Umemura, N. Watanabe and H. Matsuyama, Chem. Lett., 1985, 14, 1067–1070 CrossRef.
- H. M. R. Hoffmann, J. Chem. Soc., 1965, 823 CAS.
- H. Matsuyama, H. Minato and M. Kobayashi, Bull. Chem. Soc. Jpn., 1975, 48, 3287–3292 CrossRef CAS.
- H. Lin, Bioorg. Chem., 2011, 39, 161–170 CrossRef CAS.
- A. Noma, Y. Kirino, Y. Ikeuchi and T. Suzuki, EMBO J., 2006, 25, 2142–2154 CrossRef CAS.
- K. E. Sloan, A. S. Warda, S. Sharma, K. D. Entian, D. L. J. Lafontaine and M. T. Bohnsack, RNA Biol., 2017, 14, 1138–1152 CrossRef PubMed.
- H. Tabor and C. W. Tabor, Adv. Enzymol. Relat. Areas Mol. Biol., 1972, 36, 203–268 CAS.
- Y. Ikeguchi, M. C. Bewley and A. E. Pegg, J. Biochem., 2006, 139, 1–9 CrossRef CAS PubMed.
- A. E. Pegg and A. J. Michael, Cell. Mol. Life Sci., 2010, 67, 113–121 CrossRef CAS.
- B. T. Golding and I. K. Nassereddin, J. Am. Chem. Soc., 1982, 104, 5815–5817 CrossRef CAS.
- B. T. Golding and I. K. Nassereddin, J. Chem. Soc., Perkin Trans. 1, 1985, 2017–2024 RSC.
- G. R. Orr, D. W. Danz, G. Pontoni, P. C. Prabhakaran, S. J. Gould and J. K. Coward, J. Am. Chem. Soc., 1988, 110, 5791–5799 CrossRef CAS.
- N. von Wiren, S. Klair, S. Bansal, J. F. Briat, H. Khodr, T. Shioiri, R. A. Leigh and R. C. Hider, Plant Physiol., 1999, 119, 1107–1114 CrossRef CAS PubMed.
- C. Curie, G. Cassin, D. Couch, F. Divol, K. Higuchi, M. Le Jean, J. Misson, A. Schikora, P. Czernic and S. Mari, Ann. Bot., 2009, 103, 1–11 CrossRef CAS PubMed.
- J. S. McFarlane and A. L. Lamb, in Comprehensive Natural Products III: Chemistry and Biology, ed. H.-w. Liu and T. Begley, Elsevier, Amsterdam, 3rd edn, 2020, ch. 16, vol. 5, pp. 395–414 Search PubMed.
- K. Higuchi, K. Kanazawa, N.-K. Nishizawa, M. Chino and S. Mori, Plant Soil, 1994, 165, 173–179 CrossRef CAS.
- A. Herbik, G. Koch, H. P. Mock, D. Dushkov, A. Czihal, J. Thielmann, U. W. Stephan and H. Bäumlein, Eur. J. Biochem., 1999, 265, 231–239 CrossRef CAS.
- K. Higuchi, K. Suzuki, H. Nakanishi, H. Yamaguchi, N. K. Nishizawa and S. Mori, Plant Physiol., 1999, 119, 471–480 CrossRef CAS PubMed.
- H. Q. Ling, G. Koch, H. Bäumlein and M. W. Ganal, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7098–7103 CrossRef CAS PubMed.
- C. Dreyfus, D. Lemaire, S. Mari, D. Pignol and P. Arnoux, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16180–16184 CrossRef CAS PubMed.
- G. Ghssein, C. Brutesco, L. Ouerdane, C. Fojcik, A. Izaute, S. Wang, C. Hajjar, R. Lobinski, D. Lemaire, P. Richaud, R. Voulhoux, A. Espaillat, F. Cava, D. Pignol, E. Borezée-Durant and P. Arnoux, Science, 2016, 352, 1105–1109 CrossRef CAS PubMed.
- C. Dreyfus, M. Larrouy, F. Cavelier, J. Martinez, D. Pignol and P. Arnoux, Chem. Commun., 2011, 47, 5825–5827 RSC.
- L. Remy, M. Carrière, A. Derré-Bobillot, C. Martini, M. Sanguinetti and E. Borezée-Durant, Mol. Microbiol., 2013, 87, 730–743 CrossRef CAS PubMed.
- K. P. Grim, B. San Francisco, J. N. Radin, E. B. Brazel, J. L. Kelliher, P. K. Párraga Solórzano, P. C. Kim, C. A. McDevitt and T. E. Kehl-Fie, mBio, 2017, 8 DOI:10.1128/mbio.01281-17.
- L. Song, Y. Zhang, W. Chen, T. Gu, S.-Y. Zhang and Q. Ji, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3942–3947 CrossRef CAS PubMed.
- Z. Luo, S. Luo, Y. Ju, P. Ding, J. Xu, Q. Gu and H. Zhou, FASEB J., 2021, 35, e21575 CAS.
- S. Lhospice, N. O. Gomez, L. Ouerdane, C. Brutesco, G. Ghssein, C. Hajjar, A. Liratni, S. Wang, P. Richaud, S. Bleves, G. Ball, E. Borezée-Durant, R. Lobinski, D. Pignol, P. Arnoux and R. Voulhoux, Sci. Rep., 2017, 7, 17132 CrossRef.
- J. S. McFarlane and A. L. Lamb, Biochemistry, 2017, 56, 5967–5971 CrossRef CAS PubMed.
- M. C. Mastropasqua, M. D'Orazio, M. Cerasi, F. Pacello, A. Gismondi, A. Canini, L. Canuti, A. Consalvo, D. Ciavardelli, B. Chirullo, P. Pasquali and A. Battistoni, Mol. Microbiol., 2017, 106, 543–561 CrossRef CAS.
- K. Severinov, E. Semenova, A. Kazakov, T. Kazakov and M. S. Gelfand, Mol. Microbiol., 2007, 65, 1380–1394 CrossRef CAS.
- A. Metlitskaya, T. Kazakov, G. H. Vondenhoff, M. Novikova, A. Shashkov, T. Zatsepin, E. Semenova, N. Zaitseva, V. Ramensky, A. Van Aerschot and K. Severinov, J. Bacteriol., 2009, 191, 2380–2387 CrossRef CAS PubMed.
- A. Kulikovsky, M. Serebryakova, O. Bantysh, A. Metlitskaya, S. Borukhov, K. Severinov and S. Dubiley, J. Am. Chem. Soc., 2014, 136, 11168–11175 CrossRef CAS PubMed.
- B. A. Wilson, S. Bantia, G. M. Salituro, A. M. Reeve and C. A. Townsend, J. Am. Chem. Soc., 1988, 110, 8238–8239 CrossRef CAS.
- A. M. Reeve, S. D. Breazeale and C. A. Townsend, J. Biol. Chem., 1998, 273, 30695–30703 CrossRef CAS PubMed.
- R. M. Klug and C. Benning, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5910–5915 CrossRef CAS PubMed.
- M. Ihara, Y. Tanaka, K. Yanagisawa, Y. Taya and S. Nishimura, Biochim. Biophys. Acta, 1986, 881, 135–140 CrossRef CAS.
- Y. Taya, Y. Tanaka and S. Nishimura, FEBS Lett., 1978, 89, 326–328 CrossRef CAS PubMed.
- F. Ikegami, R. Sakai, T. Ishikawa, Y.-H. Kuo, F. Lambein and l. Murakoshi, Biol. Pharm. Bull., 1993, 16, 732–734 CrossRef CAS.
- A. Callebaut, F. Lambein and R. Van Parijs, Arch. Int. Physiol. Biochim., 1977, 85, 157–158 CAS.
- A. G. Saponara and M. D. Enger, Biochim. Biophys. Acta, 1974, 349, 61–77 CrossRef CAS PubMed.
- B. Meyer, J. P. Wurm, S. Sharma, C. Immer, D. Pogoryelov, P. Kötter, D. L. J. Lafontaine, J. Wöhnert and K. D. Entian, Nucleic Acids Res., 2016, 44, 4304–4316 CrossRef CAS PubMed.
- K. L. Tkaczuk, S. Dunin-Horkawicz, E. Purta and J. M. Bujnicki, BMC Bioinf., 2007, 8, 73 CrossRef PubMed.
- S. E. Strassler, I. E. Bowles, D. Dey, J. E. Jackman and G. L. Conn, J. Biol. Chem., 2022, 298, 102393 CrossRef CAS PubMed.
- M. Umitsu, H. Nishimasu, A. Noma, T. Suzuki, R. Ishitani and O. Nureki, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15616–15621 CrossRef CAS PubMed.
- Z. Shao, W. Yan, J. Peng, X. Zuo, Y. Zou, F. Li, D. Gong, R. Ma, J. Wu, Y. Shi, Z. Zhang, M. Teng, X. Li and Q. Gong, Nucleic Acids Res., 2014, 42, 509–525 CrossRef CAS PubMed.
- B. Van Laer, M. Roovers, L. Wauters, J. M. Kasprzak, M. Dyzma, E. Deyaert, R. Kumar Singh, A. Feller, J. M. Bujnicki, L. Droogmans and W. Versées, Nucleic Acids Res., 2016, 44, 940–953 CrossRef CAS.
- J. E. Jackman, R. K. Montange, H. S. Malik and E. M. Phizicky, RNA, 2003, 9, 574–585 CrossRef CAS.
- K. L. Tkaczuk, S. Dunin-Horkawicz, E. Purta and J. M. Bujnicki, BMC Bioinf., 2007, 8, 73 CrossRef PubMed.
- A. Krishnamohan and J. E. Jackman, Nucleic Acids Res., 2017, 45, 9019–9029 CrossRef CAS.
- A. M. Burroughs and L. Aravind, Front. Genet., 2014, 5, 424 Search PubMed.
- B. Meyer, C. Immer, S. Kaiser, S. Sharma, J. Yang, P. Watzinger, L. Weiß, A. Kotter, M. Helm, H.-M. Seitz, P. Kötter, S. Kellner, K.-D. Entian and J. Wöhnert, Nucleic Acids Res., 2020, 48, 1435–1450 CrossRef CAS PubMed.
- X.-H. Liang, Q. Liu and M. J. Fournier, RNA, 2009, 15, 1716–1728 CrossRef CAS PubMed.
- B. Meyer, J. P. Wurm, P. Kötter, M. S. Leisegang, V. Schilling, M. Buchhaupt, M. Held, U. Bahr, M. Karas, A. Heckel, M. T. Bohnsack, J. Wöhnert and K.-D. Entian, Nucleic Acids Res., 2011, 39, 1526–1537 CrossRef CAS PubMed.
- M. Takakura, K. Ishiguro, S. Akichika, K. Miyauchi and T. Suzuki, Nat. Commun., 2019, 10, 5542 CrossRef CAS.
- A. Babaian, K. Rothe, D. Girodat, I. Minia, S. Djondovic, M. Milek, S. E. Spencer Miko, H.-J. Wieden, M. Landthaler, G. B. Morin and D. L. Mager, Cell Rep., 2020, 31, 107611 CrossRef CAS PubMed.
- P. F. Agris, F. A. P. Vendeix and W. D. Graham, J. Mol. Biol., 2007, 366, 1–13 CrossRef CAS PubMed.
- A. Weixlbaumer, F. V. Murphy IV, A. Dziergowska, A. Malkiewicz, F. A. Vendeix, P. F. Agris and V. Ramakrishnan, Nat. Struct. Mol. Biol., 2007, 14, 498–502 CrossRef CAS PubMed.
- A. M. Edwards, M. A. Addo and P. C. Dos Santos, Genes, 2020, 11, 907 CrossRef CAS.
- P. Boccaletto, F. Stefaniak, A. Ray, A. Cappannini, S. Mukherjee, E. Purta, M. Kurkowska, N. Shirvanizadeh, E. Destefanis, P. Groza, G. Avşar, A. Romitelli, P. Pir, E. Dassi, S. G. Conticello, F. Aguilo and J. M. Bujnicki, Nucleic Acids Res., 2022, 50, D231–D235 CrossRef CAS PubMed.
- S. J. Nasvall, P. Chen and G. R. Bjork, RNA, 2004, 10, 1662–1673 CrossRef.
- J. Kim, H. Xiao, J. B. Bonanno, C. Kalyanaraman, S. Brown, X. Tang, N. F. Al-Obaidi, Y. Patskovsky, P. C. Babbitt, M. P. Jacobson, Y.-S. Lee and S. C. Almo, Nature, 2013, 498, 123–126 CrossRef CAS.
- R. T. Byrne, F. Whelan, P. Aller, L. E. Bird, A. Dowle, C. M. Lobley, Y. Reddivari, J. E. Nettleship, R. J. Owens, A. A. Antson and D. G. Waterman, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1090–1098 CrossRef CAS PubMed.
- J. Kim, H. Xiao, J. Koh, Y. Wang, J. B. Bonanno, K. Thomas, P. C. Babbitt, S. Brown, Y. S. Lee and S. C. Almo, Nucleic Acids Res., 2015, 43, 4602–4613 CrossRef CAS PubMed.
- S. Jeong and J. Kim, Biochem. Biophys. Res. Commun., 2021, 534, 604–609 CrossRef CAS.
- C. J. La Francois, Y. H. Jang, T. Cagin, W. A. Goddard III and L. C. Sowers, Chem. Res. Toxicol., 2000, 13, 462–470 Search PubMed.
- C. P. Ting, M. A. Funk, S. L. Halaby, Z. Zhang, T. Gonen and W. A. van der Donk, Science, 2019, 365, 280–284 CrossRef CAS PubMed.
- M. Serebryakova, D. Tsibulskaya, O. Mokina, A. Kulikovsky, M. Nautiyal, A. Van Aerschot, K. Severinov and S. Dubiley, J. Am. Chem. Soc., 2016, 138, 15690–15698 CrossRef CAS PubMed.
- B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Chem. Biol., 2009, 5, 593–599 CAS.
- R. Wang and M. Luo, Curr. Opin. Chem. Biol., 2013, 17, 729–737 CrossRef CAS PubMed.
- A. Y. Rudenko, S. S. Mariasina, P. V. Sergiev and V. I. Polshakov, Mol. Biol., 2022, 56, 229–250 CrossRef CAS.
- H. Deng and D. O'Hagan, Curr. Opin. Chem. Biol., 2008, 12, 582–592 CrossRef CAS PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef.
- D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed.
- C. Schaffrath, H. Deng and D. O'Hagan, FEBS Lett., 2003, 547, 111–114 CrossRef CAS PubMed.
- C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561–565 CrossRef CAS PubMed.
- A. S. Eustáquio, F. Pojer, J. P. Noel and B. S. Moore, Nat. Chem. Biol., 2008, 4, 69–74 CrossRef.
- D. B. Harper and D. O'Hagan, Nat. Prod. Rep., 1994, 11, 123–133 RSC.
- H. Deng, C. H. Botting, J. T. Hamilton, R. J. Russell and D. O'Hagan, Angew. Chem., Int. Ed., 2008, 47, 5357–5361 CrossRef CAS PubMed.
- A. S. Eustáquio, J. Härle, J. P. Noel and B. S. Moore, ChemBioChem, 2008, 9, 2215–2219 CrossRef.
- H. Deng, S. A. McMahon, A. S. Eustáquio, B. S. Moore, J. H. Naismith and D. O'Hagan, ChemBioChem, 2009, 10, 2455–2459 CrossRef CAS PubMed.
- T. Kornfuehrer, S. Romanowski, V. de Crécy-Lagard, A. D. Hanson and A. S. Eustáquio, ChemBioChem, 2020, 21, 3495–3499 CrossRef CAS.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS.
- J. L. Hoffman, Biochemistry, 1986, 25, 4444–4449 CrossRef CAS.
- D. F. Iwig and S. J. Booker, Biochemistry, 2004, 43, 13496–13509 CrossRef CAS PubMed.
- J. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC.
- S. Ma, D. Mandalapu, S. Wang and Q. Zhang, Nat. Prod. Rep., 2022, 39, 926–945 RSC.
- P. Diana and G. Cirrincione, Biosynthesis of Heterocycles: From Isolation to Gene Cluster, Wiley, Hoboken, 2015 Search PubMed.
- M. E. Churchill and L. Chen, Chem. Rev., 2011, 111, 68–85 CrossRef CAS.
- L. Fowden, Nature, 1955, 176, 347–348 CrossRef CAS.
- E. Leete, J. Am. Chem. Soc., 1964, 86, 3162 CrossRef CAS.
- E. Leete, G. E. Davis, C. R. Hutchinson, K. W. Woo and M. R. Chedekel, Phytochemistry, 1974, 13, 427–433 CrossRef CAS.
- E. Leete, L. L. Louters and H. S. Prakash Rao, Phytochemistry, 1986, 25, 2753–2758 CrossRef CAS.
- T. Takemoto, K. Nomoto, S. Fushiya, R. Ouchi, G. Kusano, H. Hikino, S. Takagi, Y. Matsuura and M. Kakudo, Proc. Jpn. Acad., Ser. B, 1978, 54, 469 CrossRef CAS.
- S. Mori and N. Nishizawa, Plant Cell Physiol., 1987, 28, 1081–1092 CAS.
- S. Shojima, N. K. Nishizawa, S. Fushiya, S. Nozoe, T. Irifune and S. Mori, Plant Physiol., 1990, 93, 1497–1503 CrossRef CAS PubMed.
- F. Yan, D. Auerbach, Y. Chai, L. Keller, Q. Tu, S. Hüttel, A. Glemser, H. A. Grab, T. Bach, Y. Zhang and R. Müller, Angew. Chem., Int. Ed., 2018, 57, 8754–8759 CrossRef CAS PubMed.
- F. Yan and R. Müller, ACS Chem. Biol., 2019, 14, 99–105 CrossRef CAS PubMed.
- Z. Hong, A. Bolard, C. Giraud, S. Prévost, G. Genta-Jouve, C. Deregnaucourt, S. Häussler, K. Jeannot and Y. Li, Angew. Chem., Int. Ed., 2019, 58, 3178–3182 CrossRef CAS PubMed.
- Y.-H. Shin, Y. H. Ban, J. Shin, I. W. Park, S. Yoon, K. Ko, J. Shin, S.-J. Nam, J. M. Winter, Y. Kim, Y. J. Yoon and D.-C. Oh, J. Org. Chem., 2021, 86, 11149–11159 CrossRef CAS PubMed.
- W. B. Jin, S. Wu, X.-H. Jian, H. Yuan and G.-L. Tang, Nat. Commun., 2018, 9, 2771 CrossRef PubMed.
- X. Wang, S. Wu, W. Jin, B. Xu, G. Tang and H. Yuan, Acta Biochim. Biophys. Sin., 2018, 50, 516–518 CrossRef CAS PubMed.
- W. Huang, H. Xu, Y. Li, F. Zhang, X.-Y. Chen, Q.-L. He, Y. Igarashi and G.-L. Tang, J. Am. Chem. Soc., 2012, 134, 8831–8840 CrossRef CAS PubMed.
- I. Abe, J. Antibiot., 2018, 71, 763–768 CrossRef CAS.
- J. D. Rudolf and C. Y. Chang, Nat. Prod. Rep., 2020, 37, 425–463 RSC.
- J. S. Dickschat, Nat. Prod. Rep., 2016, 33, 87–110 RSC.
- C. Schmidt-Dannert, Adv. Biochem. Eng./Biotechnol., 2015, 148, 19–61 CrossRef CAS PubMed.
- D. J. Tantillo, Chem. Soc. Rev., 2010, 39, 2847–2854 RSC.
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS PubMed.
- J. F. Quílez del Moral, Á. Pérez and A. F. Barrero, Phytochem. Rev., 2020, 19, 559–576 CrossRef.
- D. F. Iwig, A. Uchida, J. A. Stromberg and S. J. Booker, J. Am. Chem. Soc., 2005, 127, 11612–11613 CrossRef CAS.
- J. E. Cronan and T. Luk, Microbiol. Mol. Biol. Rev., 2022, 86, e0001322 CrossRef PubMed.
- M. Kai, E. Crespo, S. M. Cristescu, F. J. M. Harren, W. Francke and B. Piechulla, Appl. Microbiol. Biotechnol., 2010, 88, 965–976 CrossRef CAS PubMed.
- R. Schmidt, V. de Jager, D. Zühlke, C. Wolff, J. Bernhardt, K. Cankar, J. Beekwilder, W. van Ijcken, F. Sleutels, W. de Boer, K. Riedel and P. Garbeva, Sci. Rep., 2017, 7, 862 CrossRef PubMed.
- S. von Reuss, D. Domik, M. C. Lemfack, N. Magnus, M. Kai, T. Weise and B. Piechulla, J. Am. Chem. Soc., 2018, 140, 11855–11862 CrossRef CAS PubMed.
- M. C. Lemfack, W. Brandt, K. Krüger, A. Gurowietz, J. Djifack, J.-P. Jung, M. Hopf, H. Noack, B. Junker, S. von Reuß and B. Piechulla, Sci. Rep., 2021, 11, 3182 CrossRef CAS PubMed.
- E. R. Duell, P. M. D'Agostino, N. Shapiro, T. Woyke, T. M. Fuchs and T. A. M. Gulder, Microb. Cell Fact., 2019, 18, 32 CrossRef.
- T. Awakawa and I. Abe, Org. Biomol. Chem., 2018, 16, 4746–4752 RSC.
- T. Awakawa, J. Nat. Med., 2021, 75, 467–474 CrossRef CAS PubMed.
- T. Awakawa, L. Zhang, T. Wakimoto, S. Hoshino, T. Mori, T. Ito, J. Ishikawa, M. E. Tanner and I. Abe, J. Am. Chem. Soc., 2014, 136, 9910–9913 CrossRef CAS.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef PubMed.
- C.-I. Lin, R. M. McCarty and H.-w. Liu, Angew. Chem., Int. Ed., 2017, 56, 3446–3489 CrossRef CAS.
- F. Yu, M. Li, C. Xu, B. Sun, H. Zhou, Z. Wang, Q. Xu, M. Xie, G. Zuo, P. Huang, H. Guo, Q. Wang and J. He, Biochem. J., 2016, 473, 4385–4397 CrossRef CAS PubMed.
- K. Katagiri, J. Shoji and T. Yoshisa, J. Antibiot., 1962, 15, 273 CAS.
- M. J. Waring and L. P. Wakelin, Nature, 1974, 252, 653–657 CrossRef CAS PubMed.
- D. Kong, E. J. Park, A. G. Stephen, M. Calvani, J. H. Cardellina, A. Monks, R. J. Fisher, R. H. Shoemaker and G. Melillo, Cancer Res., 2005, 65, 9047–9055 CrossRef CAS PubMed.
- A. Cornish, M. J. Waring and R. D. Nolan, J. Antibiot., 1983, 36, 1664–1670 CrossRef CAS PubMed.
- J. S. Lee and M. J. Waring, Biochem. J., 1978, 173, 115–128 CrossRef CAS PubMed.
- K. R. Fox, N. L. Harrison and M. J. Waring, FEBS Lett., 1981, 133, 305–310 CrossRef CAS PubMed.
- K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS PubMed.
- C. Zhang, L. Kong, Q. Liu, X. Lei, T. Zhu, J. Yin, B. Lin, Z. Deng and D. You, PLoS One, 2013, 8, e56772 CrossRef CAS PubMed.
- M. Sato, T. Nakazawa, Y. Tsunematsu, K. Hotta and K. Watanabe, Curr. Opin. Chem. Biol., 2013, 17, 537–545 CrossRef CAS PubMed.
- K. Watanabe, K. Hotta, M. Nakaya, A. P. Praseuth, C. C. C. Wang, D. Inada, K. Takahashi, E. Fukushi, H. Oguri and H. Oikawa, J. Am. Chem. Soc., 2009, 131, 9347–9353 CrossRef CAS PubMed.
- C. L. Lim, T. Nogawa, M. Uramoto, A. Okano, Y. Hongo, T. Nakamura, H. Koshino, S. Takahashi, D. Ibrahim and H. Osada, J. Antibiot., 2014, 67, 323–329 CrossRef CAS PubMed.
- K. Hotta, R. M. Keegan, S. Ranganathan, M. Fang, J. Bibby, M. D. Winn, M. Sato, M. Lian, K. Watanabe, D. J. Rigden and C. Y. Kim, Angew. Chem., Int. Ed., 2014, 53, 824–828 CrossRef CAS PubMed.
- S. H. Pine, B. A. Catto and F. G. Yamagishi, J. Org. Chem., 1970, 35, 3663–3666 CrossRef CAS.
- J. B. Sweeney, Chem. Soc. Rev., 2009, 38, 1027–1038 RSC.
- C. Sun, W. Tian, Z. Lin and X. Qu, Nat. Prod. Rep., 2022, 39, 1721–1765 RSC.
- S. Funayama and G. A. Cordell, in Alkaloids, ed. S. Funayama and G. A. Cordell, Academic Press, Massachusetts, 1st edn, 2015, ch. 2, pp. 63–102 Search PubMed.
- J. Liu, T. Ng, Z. Rui, O. Ad and W. Zhang, Angew. Chem., Int. Ed., 2014, 53, 136–139 CrossRef CAS PubMed.
- H. Li, Y. Qiu, C. Guo, M. Han, Y. Zhou, Y. Feng, S. Luo, Y. Tong, G. Zheng and S. Zhu, Chem. Commun., 2019, 55, 8390–8393 RSC.
- L. Ma, W. Zhang, Y. Zhu, G. Zhang, H. Zhang, Q. Zhang, L. Zhang, C. Yuan and C. Zhang, Appl. Microbiol. Biotechnol., 2017, 101, 6123–6136 CrossRef CAS PubMed.
- N. Alqahtani, S. K. Porwal, E. D. James, D. M. Bis, J. A. Karty, A. L. Lane and R. Viswanathan, Org. Biomol. Chem., 2015, 13, 7177–7192 RSC.
- T. M. Khopade, K. Ajayan, S. S. Joshi, A. L. Lane and R. Viswanathan, ACS Omega, 2021, 6, 10840–10858 CrossRef CAS PubMed.
- M. R. TePaske, J. B. Gloer, D. T. Wicklow and P. F. Dowd, Tetrahedron Lett., 1991, 32, 5687–5690 CrossRef CAS.
- M. Ohashi, F. Liu, Y. Hai, M. Chen, M. C. Tang, Z. Yang, M. Sato, K. Watanabe, K. N. Houk and Y. Tang, Nature, 2017, 549, 502–506 CrossRef PubMed.
- Y. Cai, Y. Hai, M. Ohashi, C. S. Jamieson, M. Garcia-Borras, K. N. Houk, J. Zhou and Y. Tang, Nat. Chem., 2019, 11, 812–820 CrossRef CAS PubMed.
- Z. Chang, T. Ansbacher, L. Zhang, Y. Yang, T.-P. Ko, G. Zhang, W. Liu, J.-W. Huang, L. Dai, R.-T. Guo, D. T. Major and C.-C. Chen, Org. Biomol. Chem., 2019, 17, 2070–2076 RSC.
- M. Chang, Y. Zhou, H. Wang, Z. Liu, Y. Zhang and Y. Feng, Biochem. Biophys. Res. Commun., 2019, 515, 255–260 CrossRef CAS PubMed.
- Q. Sun, Y. Hu, Y. Gu, J. Huang, J. He, L. Luo, Y. Yang, S. Yin, C. Dou, T. Wang, X. Fu, L. He, S. Qi, X. Zhu, S. Yang, X. Wei and W. Cheng, Signal Transduction Targeted Ther., 2019, 4, 17 CrossRef.
- S. A. Newmister, S. Romminger, J. J. Schmidt, R. M. Williams, J. L. Smith, R. G. S. Berlinck and D. H. Sherman, Org. Biomol. Chem., 2018, 16, 6450–6459 RSC.
- M. Ohashi, D. Tan, J. Lu, C. S. Jamieson, D. Kanayama, J. Zhou, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2023, 145, 3301–3305 CrossRef CAS PubMed.
- H. J. Kim, M. W. Ruszczycky, S.-h. Choi, Y.-n. Liu and H.-w. Liu, Nature, 2011, 473, 109–112 CrossRef CAS PubMed.
- B.-S. Jeon, M. W. Ruszczycky, W. K. Russell, G.-M. Lin, N. Kim, S.-h. Choi, S.-A. Wang, Y.-n. Liu, J. W. Patrick, D. H. Russell and H.-w. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10408–10413 CrossRef CAS PubMed.
- A. Patel, Z. Chen, Z. Yang, O. Gutiérrez, H.-w. Liu, K. N. Houk and D. A. Singleton, J. Am. Chem. Soc., 2016, 138, 3631–3634 CrossRef CAS PubMed.
- Z. Yang, S. Yang, P. Yu, Y. Li, C. Doubleday, J. Park, A. Patel, B.-s. Jeon, W. K. Russell, H.-w. Liu, D. H. Russell and K. N. Houk, Proc. Natl. Acad. Sci. U. S. A., 2017, 115, E848–E855 Search PubMed.
- C. D. Fage, E. A. Isiorho, Y.-n. Liu, D. T. Wagner, H.-w. Liu and A. T. Keatinge-Clay, Nat. Chem. Biol., 2015, 11, 256–258 CrossRef CAS.
- E. A. Isiorho, H.-w. Liu and A. T. Keatinge-Clay, Biochemistry, 2012, 51, 1213–1222 CrossRef CAS PubMed.
- C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069–4084 CrossRef CAS.
- C. W. Carreras and D. V. Santi, Annu. Rev. Biochem., 1995, 64, 721–762 CrossRef CAS PubMed.
- S.-h. Choi, B. Jeon, N. Kim, H.-H. Wu, T.-P. Ko, M. W. Ruszczycky, E. A. Isiorho, Y.-n. Liu, A. T. Keatinge-Clay, M.-D. Tsai and H.-w. Liu, J. Am. Chem. Soc., 2021, 143, 20291–20295 CrossRef CAS PubMed.
- Z. Zhang, C. S. Jamieson, Y.-L. Zhao, D. Li, M. Ohashi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2019, 141, 5659–5663 CrossRef CAS PubMed.
- M. Ohashi, C. S. Jamieson, Y. Cai, D. Tan, D. Kanayama, M.-C. Tang, S. M. Anthony, J. V. Chari, J. S. Barber, E. Picazo, T. B. Kakule, S. Cao, N. K. Garg, J. Zhou, K. N. Houk and Y. Tang, Nature, 2020, 586, 64–69 CrossRef CAS PubMed.
- B.-S. Jeon, S.-A. Wang, M. W. Ruszczycky and H.-w. Liu, Chem. Rev., 2017, 117, 5367–5388 CrossRef CAS PubMed.
- C. S. Jamieson, M. Ohashi, F. Liu, Y. Tang and K. N. Houk, Nat. Prod. Rep., 2019, 36, 698–713 RSC.
- K. Watanabe, J. Nat. Med., 2021, 75, 434–447 CrossRef CAS PubMed.
- Y. Miyazaki, M. Shibuya, H. Sugawara, O. Kawaguchi and C. Hirsoe, J. Antibiot., 1974, 27, 814–821 CrossRef CAS PubMed.
- S. Zhou, F. Wang, E. T. Wong, E. Fonkem, T.-C. Hsieh, J. M. Wu and E. Wu, Curr. Med. Chem., 2013, 20, 4095–4101 CrossRef CAS PubMed.
- J. Dewangan, S. Srivastava and S. K. Rath, Tumor Biol., 2017, 39 DOI:10.1177/1010428317695035.
- C. Jiang, H. Wang, Q. Kang, J. Liu and L. Bai, Appl. Environ. Microbiol., 2012, 78, 994–1003 CrossRef CAS PubMed.
- M. E. Yurkovich, P. A. Tyrakis, H. Hong, Y. Sun, M. Samborskyy, K. Kamiya and P. F. Leadlay, ChemBioChem, 2012, 13, 66–71 CrossRef CAS PubMed.
- C. Jiang, Z. Qi, Q. Kang, J. Liu, M. Jiang and L. Bai, Angew. Chem., Int. Ed., 2015, 54, 9097–9100 CrossRef CAS PubMed.
- A. Jansson, J. Niemi, Y. Lindqvist, P. Mäntsälä and G. Schneider, J. Mol. Biol., 2003, 334, 269–280 CrossRef CAS PubMed.
- Y. Wang, J. Niemi, K. Airas, K. Ylihonko, J. Hakala and P. Mäntsälä, Biochim. Biophys. Acta, 2000, 1480, 191–200 CrossRef CAS PubMed.
- Y. Wang, J. Niemi and P. Mäntsälä, FEMS Microbiol. Lett., 2002, 208, 117–122 CrossRef CAS PubMed.
- A. Jansson, H. Koskiniemi, A. Erola, J. Wang, P. Mäntsälä, G. Schneider and J. Niemi, J. Biol. Chem., 2005, 280, 3636–3644 CrossRef CAS PubMed.
- C. Gui, X. Mo, Y.-C. Gu and J. Ju, Org. Lett., 2017, 19, 5617–5620 CrossRef CAS PubMed.
- K. Madduri, F. Torti, A. L. Colombo and C. R. Hutchinson, J. Bacteriol., 1993, 175, 3900–3904 CrossRef CAS PubMed.
- H. A. Blair, Drugs, 2018, 78, 1903–1910 CrossRef PubMed.
- A. Jansson, H. Koskiniemi, P. Mäntsälä, J. Niemi and G. Schneider, J. Biol. Chem., 2004, 279, 41149–41156 CrossRef CAS PubMed.
- T. Grocholski, P. Dinis, L. Niiranen, J. Niemi and M. Metsä-Ketelä, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9866–9871 CrossRef CAS PubMed.
- J. Hirst, Annu. Rev. Biochem., 2013, 82, 551–575 CrossRef CAS PubMed.
- L. A. Sazanov, Nat. Rev. Mol. Cell Biol., 2015, 16, 375–388 CrossRef CAS PubMed.
- V. F. Rhein, J. Carroll, S. Ding, I. M. Fearnley and J. E. Walker, J. Biol. Chem., 2016, 291, 14851–14860 CrossRef CAS PubMed.
- F. A. Macías, R. M. Varela, A. M. Simonet, H. G. Cutler, S. J. Cutler, F. M. Dugan and R. A. Hill, J. Org. Chem., 2000, 65, 9039–9046 CrossRef PubMed.
- D. Yan and Y. Matsuda, Angew. Chem., Int. Ed., 2022, 61, e202210938 CAS.
- J. Van Vleet, A. Kleeb, P. Kast, D. Hilvert and W. W. Cleland, Biochim. Biophys. Acta, 2010, 1804, 752–754 CrossRef CAS PubMed.
- J. Crosby and C. J. M. Stirling, J. Chem. Soc. B, 1970, 671–679 RSC.
- W. E. Stites, A. G. Gittis, E. E. Lattman and D. Shortle, J. Mol. Biol., 1991, 221, 7–14 CAS.
- F. Jordan, H. Li and A. Brown, Biochemistry, 1999, 38, 6369–6373 CrossRef CAS PubMed.
- J. J. Dwyer, A. G. Gittis, D. A. Karp, E. E. Lattman, D. S. Spencer, W. E. Stites and B. García-Moreno, Biophys. J., 2000, 79, 1610–1620 CrossRef CAS PubMed.
- D. G. Isom, C. A. Castañeda, B. R. Cannon and B. García-Moreno, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260–5265 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- S. D. Kinzie, B. Thern and D. Iwata-Reuyl, Org. Lett., 2000, 2, 1307–1310 CrossRef CAS PubMed.
- R. M. McCarty and V. Bandarian, Bioorg. Chem., 2012, 43, 15–25 CrossRef CAS PubMed.
- S. G. Van Lanen and D. Iwata-Reuyl, Biochemistry, 2003, 42, 5312–5320 CrossRef CAS PubMed.
- I. Mathews, R. Schwarzenbacher, D. McMullan, P. Abdubek, E. Ambing, H. Axelrod, T. Biorac, J. M. Canaves, H.-J. Chiu, A. M. Deacon, M. DiDonato, M.-A. Elsliger, A. Godzik, C. Grittini, S. K. Grzechnik, J. Hale, E. Hampton, G. W. Han, J. Haugen, M. Hornsby, L. Jaroszewski, H. E. Klock, E. Koesema, A. Kreusch, P. Kuhn, S. A. Lesley, I. Levin, M. D. Miller, K. Moy, E. Nigoghossian, J. Ouyang, J. Paulsen, K. Quijano, R. Reyes, G. Spraggon, R. C. Stevens, H. van den Bedem, J. Velasquez, J. Vincent, A. White, G. Wolf, Q. Xu, K. O. Hodgson, J. Wooley and I. A. Wilson, Proteins, 2005, 59, 869–874 CrossRef CAS PubMed.
- C. Grimm, R. Ficner, T. Sgraja, P. Haebel, G. Klebe and K. Reuter, Biochem. Biophys. Res. Commun., 2006, 351, 695–701 CrossRef CAS PubMed.
- G. L. Stoner and M. A. Eisenberg, J. Biol. Chem., 1975, 250, 4029–4036 CrossRef CAS PubMed.
- S. Mann and O. Ploux, Biochim. Biophys. Acta, 2011, 1814, 1459–1466 CrossRef CAS PubMed.
- R. S. Breen, D. J. Campopiano, S. Webster, M. Brunton, R. Watt and R. L. Baxter, Org. Biomol. Chem., 2003, 1, 3498–3499 RSC.
- B. C. Brajcich, A. L. Iarocci, L. A. G. Johnstone, R. K. Morgan, Z. T. Lonjers, M. J. Hotchko, J. D. Muhs, A. Kieffer, B. J. Reynolds, S. M. Mandel, B. N. Marbois, C. F. Clarke and J. N. Shepherd, J. Bacteriol., 2010, 192, 436–445 CrossRef CAS PubMed.
- Z. T. Lonjers, E. L. Dickson, T.-P. Chu, J. E. Kreutz, F. A. Neacsu, K. R. Anders and J. N. Shepherd, J. Bacteriol., 2012, 194, 965–971 CrossRef CAS PubMed.
- A. C. Bernert, E. J. Jacobs, S. R. Reinl, C. C. Y. Choi, P. M. Roberts Buceta, J. C. Culver, C. R. Goodspeed, M. C. Bradley, C. F. Clarke, G. J. Basset and J. N. Shepherd, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2019, 1864, 1226–1234 CrossRef CAS PubMed.
- T. Neupane, L. R. Chambers, A. J. Godfrey, M. M. Monlux, E. J. Jacobs, S. Whitworth, J. E. Spawn, S. H. K. Clingman, K. L. Vergunst, F. M. Niven, J. J. Townley, I. W. Orion, C. R. Goodspeed, K. A. Cooper, J. D. Cronk, J. N. Shepherd and D. N. Langelaan, Commun. Chem., 2022, 5, 89 CrossRef CAS PubMed.
- T. L. Amyes and J. P. Richard, Synlett, 2017, 28, 2407–2421 CrossRef.
- G. D. Peiser, T.-T. Wang, N. E. Hoffman, S. F. Yang, H.-w. Liu and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 3059–3063 CrossRef CAS PubMed.
- S. F. Yang and N. E. Hoffmann, Annu. Rev. Plant Physiol., 1984, 35, 155–189 CrossRef CAS.
- F. X. Nascimento, M. J. Rossi and B. R. Glick, Front. Plant Sci., 2018, 9, 114 CrossRef PubMed.
- Y. Li, L. Feng and J. F. Kirsch, Biochemistry, 1997, 36, 15477–15488 CrossRef CAS PubMed.
- M. Jakubowicz, Acta Biochim. Pol., 2002, 49, 757–774 CrossRef CAS PubMed.
- H.-P. Zhang, H. Kakeya and H. Osada, Tetrahedron Lett., 1998, 39, 6947–6948 CrossRef CAS.
- C. J. Thibodeaux, W.-c. Chang and H.-w. Liu, Chem. Rev., 2012, 112, 1681–1709 CrossRef CAS PubMed.
- Y. Y. Fan, X. H. Gao and J. M. Yue, Sci. China: Chem., 2016, 59, 1126–1141 CrossRef CAS.
- G. Pan, Z. Xu, Z. Guo, Hindra, M. Ma, D. Yang, H. Zhou, Y. Gansemans, X. Zhu, Y. Huang, L.-X. Zhao, Y. Jiang, J. Cheng, F. Van Nieuwerburgh, J.-W. Suh, Y. Duan and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E11131–E11140 CAS.
- Z. Xu, G. Pan, H. Zhou and B. Shen, J. Am. Chem. Soc., 2018, 140, 16957–16961 CrossRef CAS PubMed.
- C. Maruyama, Y. Chinone, S. Sato, F. Kudo, K. Ohsawa, J. Kubota, J. Hashimoto, I. Kozone, T. Doi, K. Shin-Ya, T. Eguchi and Y. Hamano, Biomolecules, 2020, 10, 775 CrossRef CAS PubMed.
- M. Hiramoto, A. Niwa, A. Miyake, H. Yamamoto, Y. Takebayashi, T. Nishikawa, M. Shibazaki and K. Nagai, Tennen Yuki Kagobutsu Toronkai Koen Yoshishu, 1993, 35, 678–684 Search PubMed.
- K. Kurosawa, K. Takahashi and E. Tsuda, J. Antibiot., 2001, 54, 615–621 CrossRef CAS PubMed.
- F. Kudo, A. Minato, S. Sato, N. Nagano, C. Maruyama, Y. Hamano, J. Hashimoto, I. Kozone, K. Shin-Ya and T. Eguchi, Org. Lett., 2022, 24, 8975–8979 CrossRef CAS PubMed.
- C. T. Walsh and W. Zhang, ACS Chem. Biol., 2011, 6, 1000–1007 CrossRef CAS PubMed.
- M. McErlean, X. Liu, Z. Cui, B. Gust and S. G. Van Lanen, Nat. Prod. Rep., 2021, 38, 1362–1407 RSC.
- X. Chi, P. Pahari, K. Nonaka and S. G. Van Lanen, J. Am. Chem. Soc., 2011, 133, 14452–14459 CrossRef CAS PubMed.
- Z. Cui, J. Overbay, X. Wang, X. Liu, Y. Zhang, M. Bhardwaj, A. Lemke, D. Wiegmann, G. Niro, J. S. Thorson, C. Ducho and S. G. Van Lanen, Nat. Chem. Biol., 2020, 16, 904–911 CrossRef PubMed.
- L. Kaysser, L. Lutsch, S. Siebenberg, E. Wemakor, B. Kammerer and B. Gust, J. Biol. Chem., 2009, 284, 14987–14996 CrossRef CAS PubMed.
- M. Funabashi, S. Baba, K. Nonaka, M. Hosobuchi, Y. Fujita, T. Shibata and S. G. Van Lanen, Chembiochem, 2010, 11, 184–190 CrossRef CAS PubMed.
- L. Cheng, W. Chen, L. Zhai, D. Xu, T. Huang, S. Lin, X. Zhou and Z. Deng, Mol. BioSyst., 2011, 7, 920–927 RSC.
- M. Funabashi, S. Baba, T. Takatsu, M. Kizuka, Y. Ohata, M. Tanaka, K. Nonaka, A. P. Spork, C. Ducho, W.-C. L. Chen and S. G. Van Lanen, Angew. Chem., Int. Ed., 2013, 52, 11607–11611 CrossRef CAS PubMed.
- A. Takahashi, S. Kurasawa, D. Ikeda, Y. Okami and T. Takeuchi, J. Antibiot., 1989, 42, 1556–1561 CrossRef CAS PubMed.
- A. L. Stefanska, R. Cassels, S. J. Ready and S. R. Warr, J. Antibiot., 2000, 53, 357–363 CrossRef CAS PubMed.
- C. S. Houge-Frydrych, M. L. Gilpin, P. W. Skett and J. W. Tyler, J. Antibiot., 2000, 53, 364–372 CrossRef CAS PubMed.
- L. Barra, T. Awakawa, K. Shirai, Z. Hu, G. Bashiri and I. Abe, Nature, 2021, 600, 754–758 CrossRef CAS PubMed.
- R. C. Kelly, M. E. Bolitho, D. A. Higgins, W. Lu, W.-L. Ng, P. D. Jeffrey, J. D. Rabinowitz, M. F. Semmelhack, F. M. Hughson and B. L. Bassler, Nat. Chem. Biol., 2009, 5, 891–895 CrossRef CAS PubMed.
- N. Jahan, J. A. Potter, M. A. Sheikh, C. H. Botting, S. L. Shirran, N. J. Westwood and G. L. Taylor, J. Mol. Biol., 2009, 392, 763–773 CrossRef CAS PubMed.
- D. A. Higgins, M. E. Pomianek, C. M. Kraml, R. K. Taylor, M. F. Semmelhack and B. L. Bassler, Nature, 2007, 450, 883–886 CrossRef CAS PubMed.
- Y. Wei, L. J. Perez, W.-L. Ng, M. F. Semmelhack and B. L. Bassler, ACS Chem. Biol., 2011, 6, 356–365 CrossRef CAS.
- Y. L. Du and K. S. Ryan, Nat. Prod. Rep., 2019, 36, 430–457 RSC.
- A. R. Healy, H. Nikolayevskiy, J. R. Patel, J. M. Crawford and S. B. Herzon, J. Am. Chem. Soc., 2016, 138, 15563–15570 CrossRef CAS PubMed.
- M. Xue, C. S. Kim, A. R. Healy, K. M. Wernke, Z. Wang, M. C. Frischling, E. E. Shine, W. Wang, S. B. Herzon and J. M. Crawford, Science, 2019, 365, eaax2685 CrossRef CAS PubMed.
- M. R. Wilson, Y. Jiang, P. W. Villalta, A. Stornetta, P. D. Boudreau, A. Carrá, C. A. Brennan, E. Chun, L. Ngo, L. D. Samson, B. P. Engelward, W. S. Garrett, S. Balbo and E. P. Balskus, Science, 2019, 363, eaar7785 CrossRef CAS PubMed.
- L. Zha, Y. Jiang, M. T. Henke, M. R. Wilson, J. X. Wang, N. L. Kelleher and E. P. Balskus, Nat. Chem. Biol., 2017, 13, 1063–1065 CrossRef CAS PubMed.
- J. S. Dickschat, P. Rabe and C. A. Citron, Org. Biomol. Chem., 2015, 13, 1954–1968 RSC.
- F. Trottmann, K. Ishida, J. Franke, A. Stanišić, M. Ishida-Ito, H. Kries, G. Pohnert and C. Hertweck, Angew. Chem., Int. Ed., 2020, 59, 13511–13515 CrossRef CAS PubMed.
- F. Trottmann, K. Ishida, M. Ishida-Ito, H. Kries, M. Groll and C. Hertweck, Nat. Chem., 2022, 14, 884–890 CrossRef CAS PubMed.
- P. Wittung-Stafshede, Acc. Chem. Res., 2002, 35, 201–208 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- S. D. Fried and S. G. Boxer, Annu. Rev. Biochem., 2017, 86, 387–415 CrossRef CAS PubMed.
Footnotes |
| † These authors contributed equally. |
| ‡ Current affiliation: Center for Neuro-Medicine, Korea Institute of Science & Technology, Seoul, Korea 02792. |
| This journal is © The Royal Society of Chemistry 2023 |