
Advances and status of anode catalysts for proton exchange membrane water electrolysis technology
Qiannan
Wu†
,
Yuannan
Wang†
,
Kexin
Zhang
,
Zhoubing
Xie
,
Ke
Sun
,
Wei
An
,
Xiao
Liang
* and
Xiaoxin
Zou
 *
*
State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: liangxiao@jlu.edu.cn; xxzou@jlu.edu.cn
First published on 27th January 2023
Abstract
Coupling renewable electricity with proton exchange membrane water electrolysis (PEMWE) technology to generate decarbonized “green hydrogen” is a key route for the international “carbon neutrality” target. Large-scale applications of PEMWE hydrogen production technology urgently demands low-cost, efficient, and robust anode catalysts, which are competent to industrial requirements. In this review, the developments and status of anode catalysts for PEMWE are systematically summarized. We start by introducing the stack structure and principle of the PEM electrolyzer, mechanism of oxygen evolution reaction (OER) in acid, and activity–stability trade-off of anode catalysts. Then, an overview of reported anode materials, such as Ir-based catalysts, Ru-based catalysts, and noble-metal-free catalysts, is provided. Considering the fact that the superior activity and stability of anode catalysts evaluated in the laboratory are rarely rendered to high performance under industrial conditions, we discuss the inherent reasons for the discrepancy in performance between the two test systems, i.e., the three-electrode cell set-up and PEM electrolyzer. Subsequently, the strategies for designing viable anode catalysts as well as intermediate assessment methods for narrowing the gap between efficient anode materials and high-performance PEM electrolyzers are provided. Finally, we explore the future direction of developing viable anode catalysts for PEMWE hydrogen production technology.
 Qiannan Wu | Qiannan Wu has received her master's degree in physical chemistry from Jilin University in 2020. She is currently studying for her PhD under the supervision of Prof. Xiaoxin Zou at the State Key Laboratory of Inorganic Synthesis and Preparative Chemistry in Jilin University. Her research interests focus on the design of water splitting electrocatalysts, especially the iridium-based and low iridium-based electrocatalysts in proton exchange membrane water electrolyzer applications. |
 Yuannan Wang | Yuannan Wang has received his bachelor's degree in material chemistry from the College of Chemistry, Jilin University in 2018. He is currently studying for his PhD under the supervision of Prof. Xiaoxin Zou at the State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University. His research interests focus on the design and synthesis of iridium-based oxygen evolution catalysts, especially the application of membrane electrode assemblies with low iridium loading in proton exchange membrane water electrolyzers. |
 Xiao Liang | Xiao Liang received her PhD in inorganic chemistry from Jilin University in 2021. She is currently a postdoctoral researcher at State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University. Her research interests focus on the design of water splitting electrocatalysts, especially the acidic water oxidation electrocatalysts in proton exchange membrane water electrolysis application. |
 Xiaoxin Zou | Xiaoxin Zou has received his PhD in inorganic chemistry from Jilin University in June 2011, and then moved to the University of California, Riverside, and Rutgers, The State University of New Jersey, as a postdoctoral scholar from July 2011 to October 2013. He is currently a professor at the State Key Laboratory of Inorganic Synthesis and Preparative Chemistry in Jilin University. His research interests are in hydrogen energy materials chemistry, comprising the elucidation of the atomic basis for water splitting electrocatalysts, prediction and searching of efficient catalysts with novel crystal structures and preparative technology of industrial water splitting catalysts. |
1 Introduction
In recent years, the consumption of fossil energy and emission of greenhouse gases (e.g., CO2) have aggravated energy crisis, causing climate change. Obviously, the fossil fuel-oriented energy system no longer meets the demands of sustainable development. Thus, more than 130 countries and regions in the world have developed the “carbon neutrality” consensus with the mission to achieve conversion from carbon-based fossil fuels to clean energy.1,2 Hydrogen (H2), benefiting from its high calorific value, high energy density, abundant resources, and environmental friendliness, is considered as the hopeful energy to alleviate the energy and climate issues.3–5 However, global hydrogen energy is currently dominated by “gray hydrogen” produced from fossil fuels, which diverges from the original intention of net-zero emissions.6,7 A real clean energy system can be built by coupling renewable electricity with water electrolysis to produce decarbonized “green hydrogen”, which is recognized as the future direction of the global hydrogen energy industry.8,9Among the mainstream water electrolysis technologies, proton exchange membrane water electrolysis (PEMWE) emerges as the focus due to its technological superiority, such as large operating current density (1–2 A cm−2), high purity of the product (≥99.99%), and compact construction. Especially, the rapid response to power inputs enables PEMWE to the most suitable hydrogen production technology for integrating with renewable electricity.10–12 The PEMWE hydrogen/oxygen production technology has a history of half a century (Fig. 1), dating back to 1973, when General Electric Company developed the promising PEM electrolyzer.11 Later, the U.S. space program first applied it to generate oxygen (O2) as the breathing support on the aircraft.13 Having established reliability, PEMWE was employed again for military applications on nuclear submarines.14 In 1980 s, PEMWE's first commercial utility was founded for producing high-purity hydrogen to replace the cylinder in gas chromatographs and cooling gas in electrical generators, which relieved the economic pressure for gas-handling.15 The generalization of PEMWE was not achieved until Proton OnSite commercially manufactured the PEMWE hydrogen production devices in 1996. Owing to the simplified manufacturing technique and advanced material components, PEMWE's investment cost has been gradually reduced over the years.16–18 Recently, with the surge in global hydrogen demand, the MW-scale PEMWE hydrogen production facilities have been built in multiple countries.19 In addition, Plug Power will provide the 1 GW hydrogen production installation system, the world's largest PEM electrolyzer, in Denmark.
 | ||
| Fig. 1 Development timeline infographic of PEMWE. | ||
Even though PEMWE hydrogen production technology has developed rapidly over the past 50 years, it remains rudimentary for large-scale industrial applications and faces numerous challenges. A major challenge is a desideration to exploit low-cost, efficient, and robust anode catalysts, which are competent for the PEMWE's operating environment.20–24 The commercial anode catalyst in PEMWE is iridium dioxide (IrO2), which is also the only known stable material under harsh conditions. Unfortunately, there are two problems for IrO2: (i) iridium is scarce (annual capacity <7.5 tons) and expensive ($157 per gram in December 2022).25 Global iridium capacity can only support 10–12 GW hydrogen production installation per year in the present state. (ii) The catalytic activity of IrO2 is not desired, which makes it difficult to improve the electrolysis efficiency of PEMWE.16 With the purpose of developing novel anode catalysts to replace IrO2, many efforts have been made by researchers. However, the emergent catalysts are often susceptible to suffering adverse structural evolution during catalysis, such as cation leaching, irreversible oxidation, and surface reconstruction. These lead to crystal structure collapse and ultimately catalyst degradation.26–28 More importantly, the configuration and operating conditions of the three-electrode cell and PEM electrolyzer are substantially different, resulting in the failure to translate the excellent activity and stability of the anode catalysts into the high performance of PEM electrolyzers. This gap between active anode catalysts and high-performance PEM electrolyzers renders that, the majority of emergent anode catalysts cannot meet the application demands in PEMWE.29 Therefore, in order to explore the design strategy of new anode catalysts for PEMWE, it is necessary to summarize the potential anode catalysts and discuss the gap between the catalyst and membrane electrode assembly (MEA).
In this review, we present a comprehensive summary of the advances and application status of the anode catalysts for PEMWE. The stack structure and principle of the PEM electrolyzer are first introduced. Then, we highlight the catalytic/degradation mechanism during anode catalysis, followed by a discussion on the inverse activity–stability relationship. The great efforts to develop anode catalysts for PEMWE are looked back upon by summarizing the representative candidates, including Ir-based catalysts, Ru-based catalysts, and noble-metal-free catalysts. With regard to the disparity between efficient anode catalysts and superb PEM electrolyzers, we compare the three-electrode cell set-up with the PEM electrolyzer, which is employed to assess the performance in the laboratory and industry. The inherent reasons for the discrepancy in the performance between the two test systems are clarified. In order to bridge this gap, we put forward the design strategies for viable anode catalysts and outline the effective intermediate assessment methods. Finally, an outlook on the remaining challenges and relevant suggestions in the progress of the anode materials is provided, which benefits promoting PEMWE hydrogen production technology.
2 Mechanisms in PEMWE
2.1 Structure and principle of PEM electrolyzers
Significantly different from conventional aqueous electrolysis cells (e.g., AWE), the PEM electrolyzer uses PEM as a solid electrolyte and separator for the bipolar chambers.30,31 Therefore, the structure of the PEM electrolyzer needs to be specially designed to accommodate this change. The main components of PEMWE include PEM, catalyst layers (CL), gas diffusion layers (GDL), bipolar plates (BPs), and end plates (Fig. 2a). As a core configuration of the PEM electrolyzer, the MEA, consisting of PEM, CL and/or GDL, provides a place for electrochemical reactions and largely determines the overall performance of the PEM electrolyzer.32 In addition to the basic conductivity and heat transfer, the BPs are mainly used to distribute water evenly within the cell.33 Finally, the end plates act as mechanical supports and fixtures for all the components. Obviously, the contact and interface structure between multiple components are the key indicators that determine the efficiency of the PEM electrolyzer at higher current densities. | ||
| Fig. 2 (a) Structure and (b) operating principles of the PEM electrolyzer. | ||
As the basis of the water electrolyzer, OER at the anode and hydrogen evolution reaction (HER) occur at the cathode of three-phase interface on the catalyst surface. The typical operating principles of a PEM electrolyzer are as follows (Fig. 2b): pure water supplied from the anode port flows through the BPs and GDL to the surface of the anode catalyst, where it is further dissociated into protons and oxygen. The evolved oxygen is released along the water flow, while the protons pass through the PEM to the surface of the cathode catalyst and are immediately reduced to hydrogen.
2.2 OER Mechanisms in acid
OER at the anode is sluggish because it involves multiple proton–electron transfers, resulting in a higher oxidation potential and lower voltage efficiency. Although only pure water needs to be supplied to the PEMWE, the anode catalysts are in a somewhat acidic environment due to the enormous number of protons produced by the dissociation of water. However, acidic conditions require the stability of the OER catalyst more strictly, that is, on the basis of acid corrosion resistance, more electrochemical corrosion resistance.34,35 Electrochemical corrosion is often considered to be closely related to the reaction mechanism of OER. Therefore, it is of great significance to understand the mechanism of OER in acidic conditions to improve the energy conversion efficiency and operating life of PEMWE. Early OER reaction paths were mostly inferred from kinetic parameters obtained by electrochemical methods, and it was generally believed that O*, HO*, and HOO* intermediates were partially or completely involved in OER, whether in acidic or alkaline conditions. However, the complex OER dynamic processes make the mechanism analysis challenging because dynamic characteristics are affected by multiple factors, which may lead to similar responses to different mechanisms.36 Consequently, it is necessary to explore the OER mechanism in conjunction with thermodynamics.With the development of density functional theory (DFT), Rossmeisl et al. proposed an adsorbate evolution mechanism (AEM) involving four concerted proto–electron transfer (CPET) reactions (Fig. 3a).37,38 In this way, the reaction free energy (ΔG) can be quantified, and the step with the greatest ΔG is regarded as the rate-determining step, which controlls the overall OER activity.39 Based on extensive experimental and computational data from various catalysts, the adsorption energy of the HO* and HOO* intermediates had been proved to have a near-constant gap of 3.2 eV, that is, the so-called scaling relationship (Fig. 3b).40 Thus, a theoretical OER overpotential volcano appears and limits the minimum overpotential that a catalyst can achieve to 0.37 V rather than 0 V (Fig. 3c). Although the scaling relationship has guided the selection of catalysts with appreciable activity, there is an urgent need to break this relationship to meet the quest for active catalysts with greater benefits. Since the scaling relationship is rooted in the similar adsorption patterns of HO* and HOO* on the catalyst surface,41 optimizing the adsorption of intermediates separately through appropriate pathways is a feasible route to break the scaling relationship (Fig. 3d).42
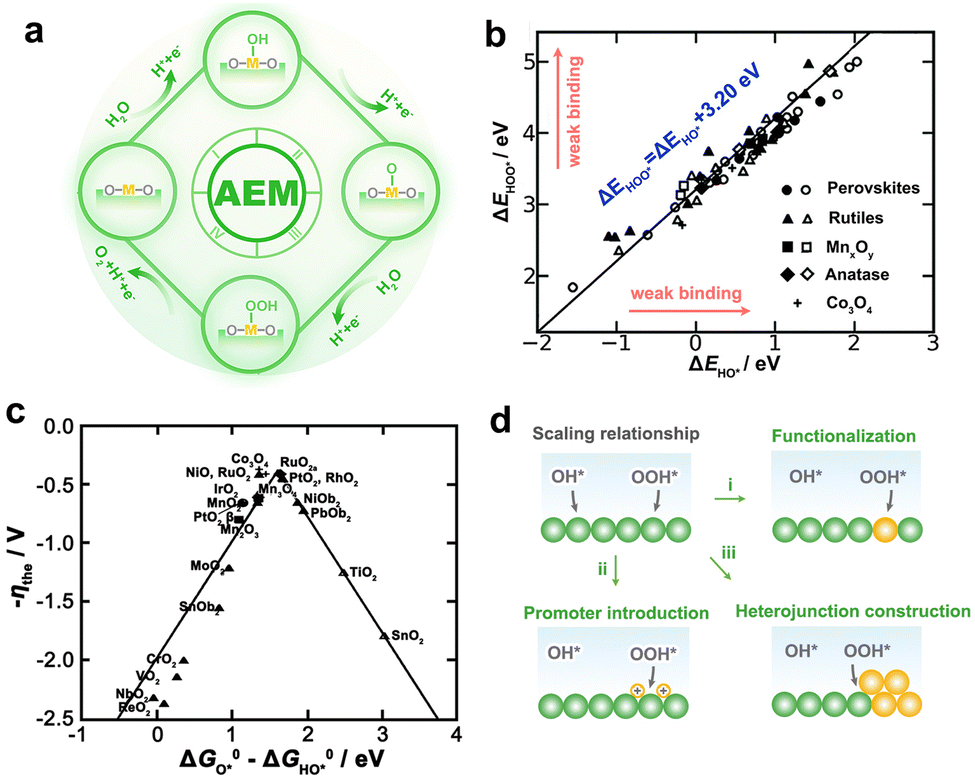 | ||
| Fig. 3 (a) Schematic illustration of the AEM. (b) The function of the adsorption energies (ΔE) of HOO* with HO* on various catalysts. (c) The volcano plot for the theoretical overpotential as a function of the adsorption-free energy difference between O* and HO*. (b and c) Reproduced with permission from ref. 40. Copyright 2011, Wiley-VCH. (d) Schematic of strategies for breaking the scaling relationship in OER. | ||
In recent years, lattice oxygen-mediated mechanism (LOM) is expected to break the scaling relationship due to the unique existence of non-concerted proton–electron transfer reactions.43,44 Although the pioneering work of Grimaud et al. targeted the perovskite system under alkaline conditions,44 the LOM under acidic conditions was also observed experimentally,45 as such, LOM in acid could be reasonably deduced from that under alkaline conditions. In the LOM, lattice oxygen is a reactive electrophilic oxygen capable of coupling with water molecules via nucleophilic attack, which subsequently releases oxygen and creates oxygen vacancies (Vo). Vo can be filled by water or bulk oxygen migration to restart the catalytic cycle (Fig. 4a). Based on the understanding of AEM, LOM with the CPET process seems to be possible, but this contradicts the experimental result that the LOM is accompanied by pH dependence. The pH dependence indicates the decoupling of CPET in the reaction steps, especially in the rate-determining step (Fig. 4b). Essentially, this is because the oxygen-containing species on the surface have either high electron or proton affinity, and thus interact strongly with the electrolyte.46,47 It is worth noting that the pH dependence does not necessarily result in LOM, such as in the case where the electron transfer rate does not match the affinity of the intermediate.48,49 For further insight, acidic LOM can be enriched by studies of alkaline LOM, such as the electron structure analysis of the p-band center and the charge transfer energy.46,50
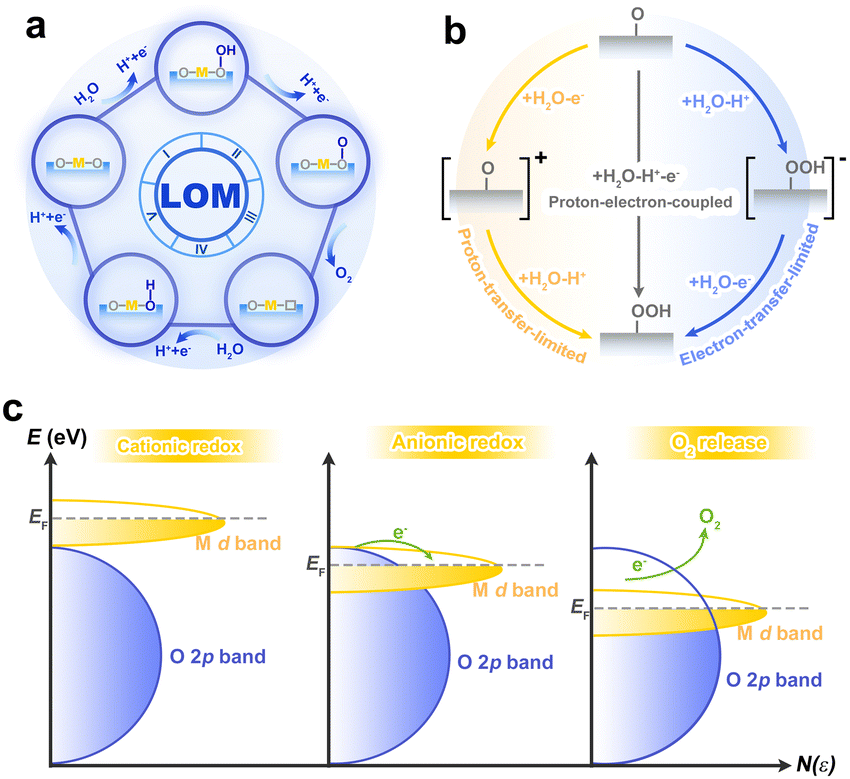 | ||
| Fig. 4 (a) Schematic illustration of the LOM. (b) Schematic diagram of decoupled proton–electron transfer of a classical rate-determining step under acidic conditions. (c) Description of O 2p bands falling into M d orbitals and achieving an anionic redox process. Reproduced with permission from ref. 52. Copyright 2016, Springer Nature. | ||
Generally speaking, the differences between LOM and AEM can be summarized in the following three aspects: (i) source of oxygen. For LOM, oxygen is not only derived from water molecules but also partly or completely from the catalyst lattice. This can be experimentally detected by isotope labelled online electrochemical mass spectrometry.51 (ii) Redox process. For AEM, the metal is the only active site on which water undergoes a continuous four-step proton–electron coupling reaction. Essentially, this is a cationic redox process, as the metal ions take on different valence states to accommodate changes in the oxygen-containing intermediates during the reaction. While in the LOM, the oxygen in the lattice connected to the metal is the catalytic active O− species. This process can be viewed as an anionic redox process, similar to anion-driven capacity storage in lithium-ion batteries (Fig. 4c).52 (iii) Activity and stability. LOM is able to bypass the general scaling relationship of intermediates in AEM, which led to the discovery of highly active catalysts. However, since lattice oxygen in LOM mediated the reaction, the instability of the Vo and the inevitable metal dissolution will cause a certain degree of irreversible damage to the structure of the catalyst, although the generated Vo can be filled.20,53 Therefore, catalysts dominated by LOM appear to be more active but less stable than those of AEM. In order to obtain high performance catalysts, reasonable strategies are required to balance this activity–stability relationship.
For the practical application of PEMWE, the catalyst should first satisfy the high stability, and then the high activity.54 The instability, or deactivation of the catalyst is caused by both external (e.g., catalyst detachment and surface blocking) and internal (e.g., catalyst dissolution and surface reconstruction) factors.55,56 Defining the dissolution path is particularly important because other factors can be appropriately modified by process adjustment. On the other hand, it helps to determine the main parameters affecting the stability of OER electrocatalysts.26
In the case of Ir-based oxide catalysts commonly used in acid OER, more recently, Ir dissolution has been found to be highly correlated with OER.57 At different potentials, the iridium electrodes mainly complete the OER catalytic cycle through IrIII or IrVI, which leads to different dissolution paths (Fig. 5). (i) For the less active heat-treated iridium oxide, the higher applied potential is required, so that the core IrV intermediate is further oxidized into volatile IrO3, which can be decomposed into IrO2 or become soluble IrO42−. (ii) For the highly active metallic Ir and reactively sputtered Ir oxide, low overpotential can drive an appreciable reaction rate, but the potential is not enough to oxidize IrV. Subsequently, IrV intermediates decompose into HIrO2 species, and then become soluble IrIII ions or deprotonate to form IrO2. As indicated above, OER and Ir dissolution are closely related reactions sharing an identical unstable intermediate. Therefore, inhibiting the dissolution of intermediates is a necessary route to obtain high stability catalyst. However, the competition between different paths of dissolution or deposition is controlled by the relevant reaction kinetics, which requires more in-depth kinetic simulation and advanced characterization techniques to explore the complex activity–stability relationship of actual catalysts.
 | ||
| Fig. 5 Universal mechanism linking the OER to possible Ir dissolution pathways. Reproduced with permission from ref. 57. Copyright 2018, Wiley-VCH. | ||
3 Advances in anode catalysts for PEMWE
3.1 Ir-based anode catalysts
Morphology regulation of Ir black catalyst is an effective way to improve its catalytic activity and iridium utilization.10 There have been a variety of synthesis methods for developing Ir-based catalysts with different morphologies, such as the wet means (e.g., sol–gel method, polyol method, aqueous method) and thermal decomposition methods (e.g., Adams fusion method and Pechini-thermal treatment method).58–60 Lee et al. reported the synthesis of Ir nanodendrites with an extremely high OER activity than that of commercial Ir black.61 Fu et al. reported the Ir wavy nanowires with a small size of 1.7 nm. The Ir wavy nanowires exhibiting enhanced activity and durability for OER can be ascribed to theirspecial morphology.62 Subsequently, they also reported ultrasmall Ir nanoparticles with narrow size distribution by a colloidal method for the first time, which yielded excellent OER activity under acidic media.63 Pi et al. reported a new class of three-dimensional (3D) Ir superstructure catalysts, which showed a low onset overpotential (Fig. 6a and b).64 Although Ir black catalysts exhibit good OER activity, to make it a potential material to be applied in PEMWE, the irreversible oxidation of iridium metal to iridium oxide during the catalytic process has to be taken into account.
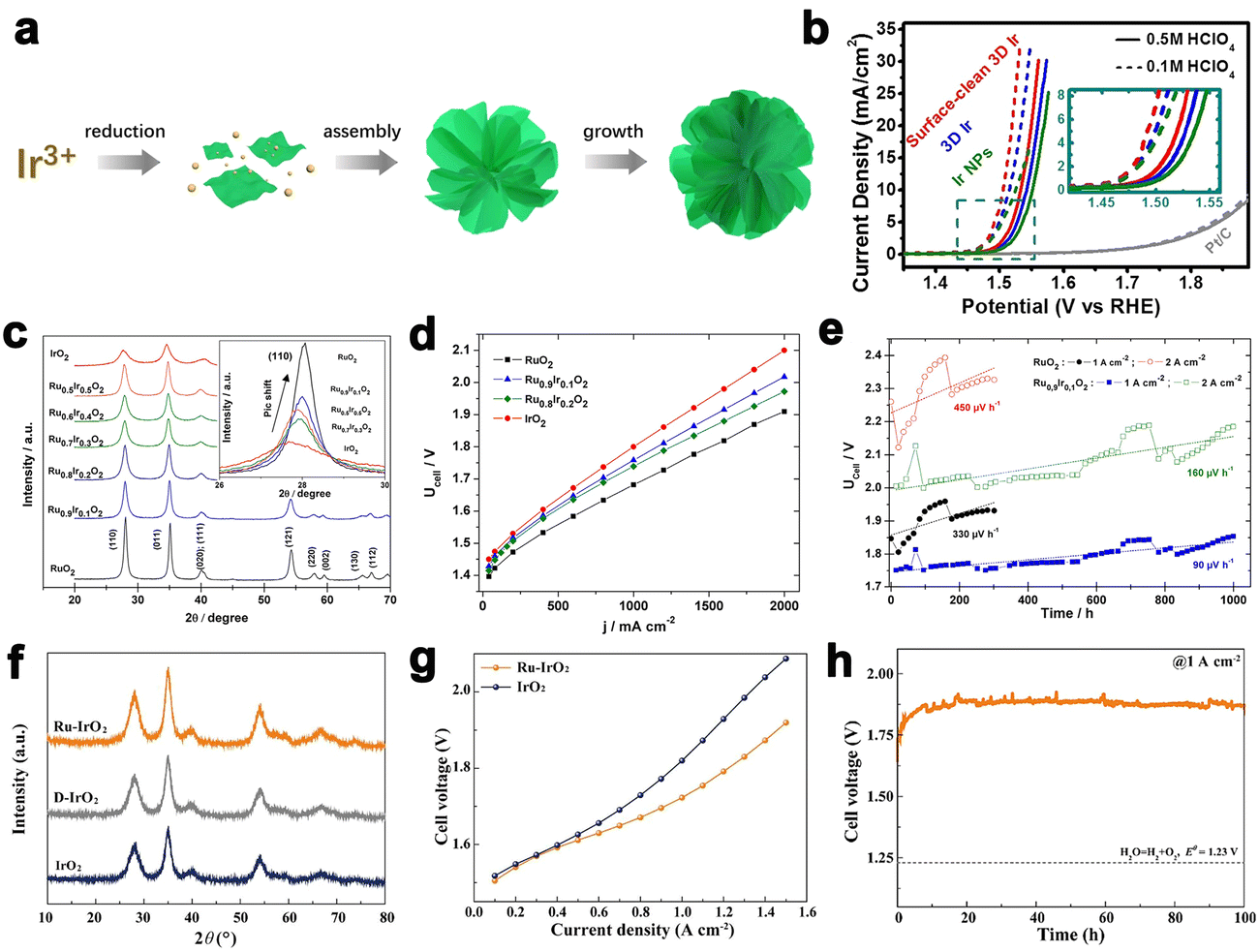 | ||
Fig. 6 (a) Schematic illustration of the synthesis process of the 3D Ir superstructures. (b) The OER performance of catalysts in acidic conditions. (a and b) Reprinted with permission from ref. 64. Copyright 2016, American Chemical Society. (c) The XRD pattern of Ir–Ru mixed oxides. (d) The polarization curves of Ir–Ru mixed oxides at 80 °C. (e) The stability test for Ru0.9Ir0.1O2 at 1 A cm−2 and 2 A cm−2. (c–e) Reprinted with permission from ref. 71. Copyright 2014, Elsevier. (f) The XRD pattern of Ru–IrO2. (g) Cell voltage–current density curves of Ru–IrO2 with iR-free. (h) Durability tests of Ru–IrO2 at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A cm−2. (f–h) Reprinted with permission from ref. 77. Copyright 2022, Elsevier. A cm−2. (f–h) Reprinted with permission from ref. 77. Copyright 2022, Elsevier. | ||
IrO2 with a rutile phase is currently the commercial anode catalyst for PEMWE. The IrO2 loading on MEA is usually 2–2.5 mg cm−2, and it is expected to be reduced by improving the catalytic activity of IrO2. For instance, Lim et al. developed a molten salt method to synthesize ultrathin IrO2 nanoneedles (diameter ≈ 2 nm), which is facile and scalable. The OER performance of IrO2 nanoneedles was significantly enhanced compared to that of the conventional nanoparticles.65 Especially, the low loading of IrO2 about 0.38 mg cm−2 on MEA can achieve a high performance of 2 A cm−2 at 1.633 V and had no obvious degradation after 122 h.66 On the other hand, iridium oxide with poor crystallinity, namely, IrOx, tends to show better OER activity than crystalline IrO2. Its excellent activity can be attributed to a large number of active sites and oxygen defects. But the existential fact is the poor structure stability of IrOx due to Ir dissolution during the catalytic process. Even though the catalyst with poor structural stability may show good catalytic stability during the short-term electrochemical test in the laboratory, the loss of active sites and the collapse of structure will shorten the lifetime of PEMWE. Therefore, it is very significant to balance the catalytic activity and structural stability of the catalyst. Jiang et al. developed an amorphous IrOx catalyst through a sophisticated soft-templating method. This mesoporous IrOx shows superior OER activity and good stability.67 Yu et al. fabricated the IrOx dispersed in the Nafion® catalyst layer. The IrOx/Nafion catalyst showed good stability in PEMWE, and especially, the catalysts layer was used with a low Ir loading of about 0.08 mg cm−2. They thought three-points can influence the total performance: (i) the content of iridium(IV) to iridium(III) species was high; (ii) a well-dispersed catalyst; (iii) the homogeneous distribution of the catalyst layer in the PEM.68 Therefore, it will have great potential for optimized IrOx to apply in the PEMWE.
:Ru was 0.7:0.3) by a thermal treatment method, the IrRuOx displayed higher OER activity compared to IrOx in PEMWE.70 Audichon et al. developed Ru0.9Ir0.1O2 mixed nanocatalysts with a stability loss rate of 90 μV h−1 (in 1000 h) at 1 A cm−2 in the operation potential of 1.806 V (Fig. 6c–e).71 Furthermore, coating more stable IrO2 on the surface of RuO2 is another effective strategy to obtain highly active and stable electrocatalysts. For instance, Lv et al. designed self-assembled RuO2@IrOx nanocomposite electrocatalysts with mediate OER activity and sufficient durability, which exhibited a lower cell voltage (1.683 V@1 A cm−2) than RuO2 (1.715 V@1 A cm−2) and IrO2 (1.748 V@1 A cm−2).72 In another case, Du et al. chose IrO2 as a stable matrix, and the Ru atoms were in the IrO2 matrix. In PEMWE, the catalysts displayed a current density of 1 A cm−2 at 1.72 V. (Fig. 6f–h). The superior activity and stability were due to the improved water affinity of the surface and the broken of IrO6 octahedrons in the Ir sites. In addition, the change in the mechanism about catalytic process will make the change in the OER activity. From the view of dynamics vapor sorption (DVS) measurements and X-ray absorption spectroscopy (XAS) results, the oxygen intermediates (such as *OH, *O, and *OOH) of Ru–IrO2 were stronger than those on IrO2. The OER process for Ru–IrO2 was dominated by LOM, which was also different from single IrO2.73
 | ||
| Fig. 7 (a) Synthesis of GB-Ta0.1Tm0.1Ir0.8O2−δ catalyst. (b) Chronopotentiometry curve at 1.5 A cm−2. (a and b) Reprinted with permission from ref. 81. Copyright 2021, Nature Publishing Group. (c) The TEM image of the 20% IrOx/30%W-TiO2. (d) The polarization curves in PEMWE with various MEAs at 80 °C. (e) The stability test in PEMWE under 1 A cm−2. (c–e) Reprinted with permission from ref. 85. Copyright 2021, Elsevier. (f) The HAADF-STEM image of Ir/Nb2O5−x. (g) Chronopotentiometry curves using Ir/Nb2O5−x catalyst at a continuous current density. (f and g) Reprinted with permission from ref. 89. Copyright 2022, Wiley-VCH. | ||
Perovskite-type SrIrO3 is the first reported iridate anode catalyst. Seitz et al. synthesized orthorhombic SrIrO3 by pulsed laser deposition (PLD) and found that the Sr leaching behavior during the first 30 hours of OER catalysis.90 The high activity of the resulting IrOx/SrIrO3 structure was confirmed by experiments and theoretical calculations (Fig. 8a–i). Similar results have also been found in the study of pyrochlore-type anode catalysts (e.g., Pr2Ir2O7, Y2Ir2O7, Bi2Ir2O7).91 A′ element on the surface will also dissolve into the electrolyte, generating amorphous IrOx, which is the actual active phase. In fact, the structural evolution of iridate during OER catalysis involves not only the A (or A′)-site leaching but also the Ir dissolution triggered by it. Especially for some doped iridate anode catalysts (e.g., Ba2PrIrO6, Sr2CoIrO6, La2LiIrO6, Sr2YIrO6), the severe leaching of the dopant and A (or A′)-site element results in the surface (even the bulk) reconstruction to amorphous IrOx.22,35,92,93 These behaviors are likely to aggravate the dissolving of Ir active sites and finally lead to catalyst degradation. It is important to note that these structural evolutions might not even be the results of electrochemical corrosion, but may also be caused by transient acid treatment (Fig. 8j and k).34 This structural instability has raised questions about whether these catalysts are actual low-iridium catalysts and has also forced researchers to focus on developing iridate anode catalysts with excellent structural stability.
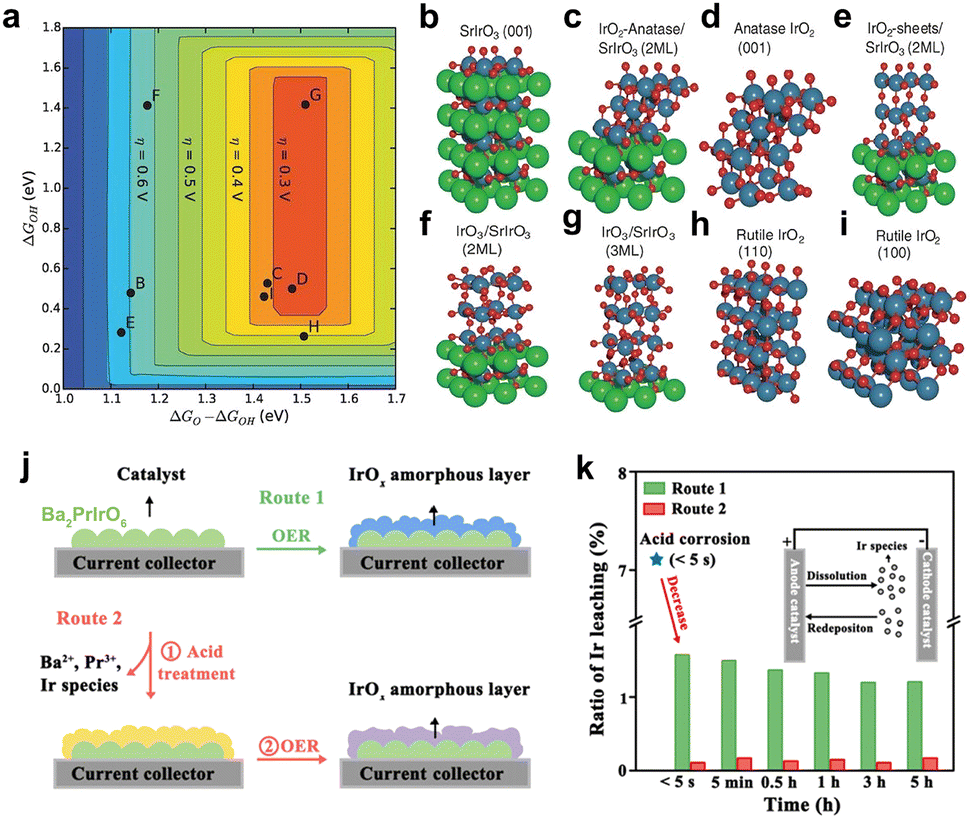 | ||
| Fig. 8 (a) Theoretical overpotential volcanic plot based on the scaling relation of 3C-SrIrO3 system. (b–i) Surface models of IrOx and 3C-SrIrO3, where (b–i) correspond to (b–i) in the volcano plot in (a). (a–i) Reprinted with permission from ref. 90. Copyright 2016, American Association for the Advancement of Science. (j) Schematic of two experimental routes for Ba2PrIrO6 in the catalytic corrosion test. (k) Comparison of the percentage of Ir leaching from Ba2PrIrO6 in Route 1 and 2 ratios during the OER test. (j and k) Reprinted with permission from ref. 34. Copyright 2022, Elsevier. | ||
Due to the diverse crystal structure of iridate, their structural stability can be optimized by phase engineering. Our group reported monoclinic SrIrO3 (6H-SrIrO3), which is a polymorph of orthorhombic SrIrO3 (3C-SrIrO3).94 Although the two have the same elemental composition, the mass activity of 6H-SrIrO3 was twice that of 3C-SrIrO3 (Fig. 9a). In addition, the amount of Sr and Ir leaching of 6H-SrIrO3 was much less than that of 3C-SrIrO3, and this enhanced structural stability can be attributed to the unique face-sharing IrO6 octahedral dimers in the crystal structure (Fig. 9b). Considering the well-maintained crystal structure of 6H-SrIrO3, we investigated its theoretical activity according to the AEM. The DFT calculations revealed that the excellent activity of 6H-SrIrO3 was attributed to the moderate adsorption energy of the oxygen-containing intermediate. In the later work, we further developed 6H-SrIrO3 by facet regulation. The two-dimensional ultrathin {001}-faceted 6H-SrIrO3 (2D-SIO) was prepared, and the nanosheets displayed higher activity and less Sr leaching than their bulk counterparts (Fig. 9c).95 Combining the results of the experimental and theoretical calculations, we found the exposed {001} facet can not only create the beneficial O* intermediates but also prevent the subsequent leaching of Sr (Fig. 9d). These effects enabled it possible to simultaneously accelerate the O–O bond coupling and maintain the structural integrity, thus endowing 2D-SIO with superior catalytic activity and structural stability.
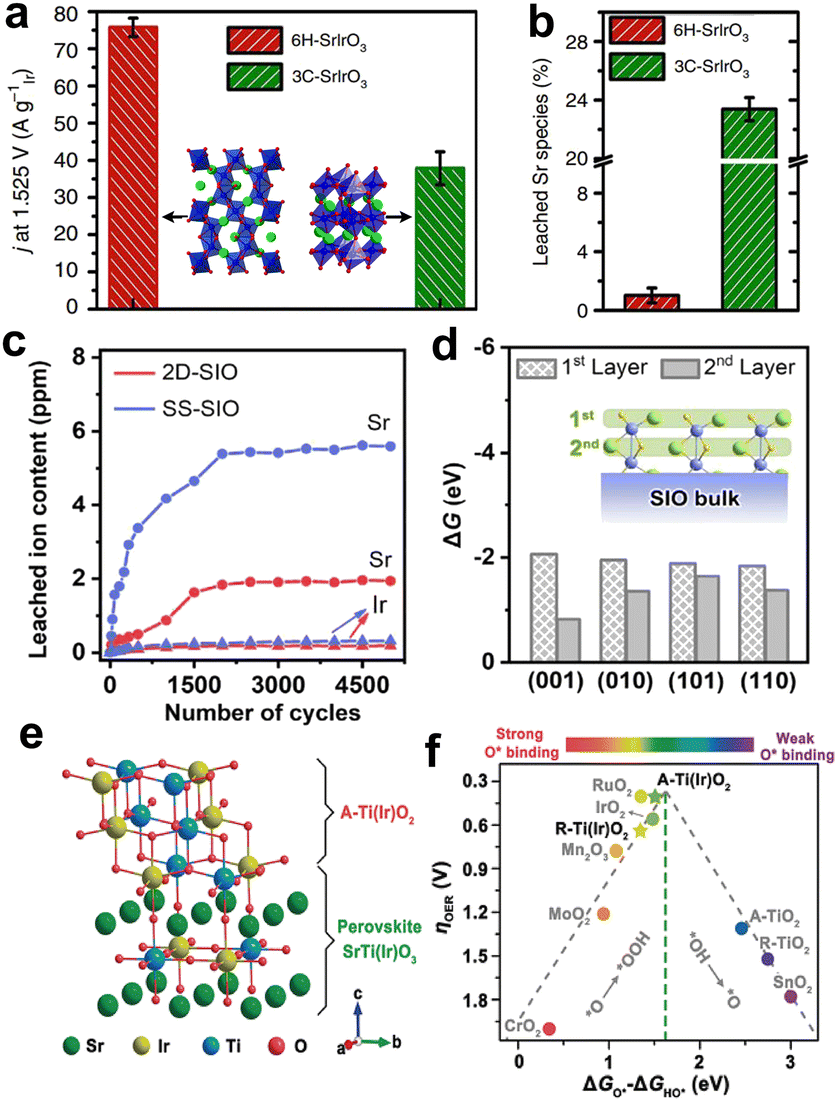 | ||
| Fig. 9 (a) Mass activity comparison of 6H-SrIrO3 and 3C-SrIrO3, normalized by the Ir mass at 1.525 V vs. RHE. (b) Percentage of Sr leaching from 6H-SrIrO3 and 3C-SrIrO3 after 30 h of the catalytic stability test. (a and b) Reprinted with permission from ref. 94. Copyright 2018, Springer Nature. (c) Contents of Sr and Ir leaching from 2D-SIO and SS-SIO during 5000-cycle measurements. (d) The thermodynamic driving forces of Sr leaching from the first and second atomic layers at different SrIrO3 surfaces. (c and d) Reprinted with permission from ref. 95. Copyright 2021, Elsevier. (e) Surface model of SrTiO3–SrIrO3 after little Sr leaching and forming anatase Ti(Ir)O2. (f) Theoretical overpotential volcanic plot of different oxide materials. (e and f) Reprinted with permission from ref. 98. Copyright 2020, Wiley. | ||
Another effective approach to improve the structural stability of iridate is to stabilize Ir active sites in the parent materials with strong corrosion resistant (e.g., SrTiO3, SrZrO3, BaTiO3). Different from the above-mentioned-doped iridate, which introduces the element dissolving easily during catalysis, the robust non-noble metals in these iridates hardly oxidize and dissolve during the acid treatment and electrocatalysis. On the contrary, the stable crystal framework constructed by them can inhibit the leaching of A-site elements and iridium, which can effectively prevent the crystal structure of the anode catalysts. For example, our group reported several perovskite-type solid solutions, such as SrTiO3–SrIrO3 and SrZrO3–SrIrO3.96–98 The introduction of Ti or Zr adjusted the hybridization of Ir 5d and O 2p orbitals, which relieved the excessively strong Ir–O bond covalency, ensuring the excellent structural stability. After further comparing the thermodynamic stability of key oxygen-absorbed structures involved in AEM and LOM, we found that OER proceeds preferably via AEM rather than LOM in these catalysts. The highly active phase is the anatase solid solutions (i.e., TiO2–IrO2, ZrO2–IrO2) formed on the surface of the perovskite-type solid solution after slight Sr leaching (Fig. 9e). The stable Ti and Zr can optimize the adsorption of the Ir active site to oxygen-containing intermediates, thus improving the activity of the anode catalysts (Fig. 9f).
3.2 Ru-based anode catalysts
Ru-based catalysts are deemed to be the alternate candidates for Ir-based anode catalysts for PEMWE. On the one hand, Ru-based anode catalysts usually exhibit better catalytic activity than Ir-based counterparts, contributing to the favorable binding ability for oxygen-containing intermediates (e.g., *O, *OH, and *OOH) of Ru sites.99 On the other hand, ruthenium costs nine times less than iridium (i.e., $17 per gram in December 2022), which can effectively provide saving on the catalyst investment. Unfortunately, despite the advantages in cost and activity, Ru-based anode catalysts face a fatal problem, which is the rather poor stability during OER catalysis. Their poor stability can be attributed to two points: (i) Ru-based materials are prone to be excessively oxidized to soluble Rux (X > +4), e.g., soluble RuO4 derivatives, when the applied voltage exceeds 1.4 V during OER catalysis. The definitive reaction is RuO2 + 2H2O → RuO4 (aq) + 4 H+ + 4e−. (ii) The catalysis on Ru-based anode catalysts involves LOM, which releases lattice oxygen and generates oxygen vacancies, accelerating the leaching of the Ru site, as well as the destruction of the crystal structure.44,100,101 Therefore, the stabilization of the active sites is the top priority for Ru-based anode catalysts.Elemental doping is a successful strategy for improving the stability of Ru-based anode catalysts. It has been reported that with the introduction of foreign elements, such as V, Sn, Co, Ni, Zn, Fe, W, and La, the electronic structure of the Ru site can be modulated and prevent the formation of oxygen vacancies, and thus is effective to improve the stability of Ru-based anode catalysts.102,103 Recently, Hao et al. synthesized the W0.2Er0.1Ru0.7O2−δ anode catalyst by bimetallic doping, enabling this catalyst to exhibit long-term stability for 500 h.104 They found that the W and Er dopants were capable of downshifting the O 2p-band center to the Fermi level, which increased the oxygen vacancy formation energy. The authors believed that the increase of the oxygen vacancy formation energy inhibits the direct O–O coupling together from the lattice of W0.2Er0.1Ru0.7O2−δ, which makes the OER inclined to the AEM and reduces the tendency of Ru dissolution and oxidation (Fig. 10a–c). Besides, the authors concluded that the calculated ΔG for the potential determining steps between OOH* and O* followed the order of W0.2Er0.1Ru0.7O2−δ (0.53 eV) < W0.2Ru0.8O2−δ (0.6 eV) < W0.1Ru0.9O2−δ (0.72 eV) < RuO2 (0.79 eV), which suggested that these effects simultaneously boosted the OER activities and stabilities of RuO2 (Fig. 10d and e).
 | ||
| Fig. 10 (a) DOS plots of Ru 4d and O 2p states in RuO2 and W0.2Er0.1Ru0.7O2−δ−1. (b) Schematic representation of rigid band models for RuO2 and W0.2Er0.1Ru0.7O2−δ−1 toward acidic OER. (c) The formation energy of VO at different positions. (d) Diagram of the energy barrier for W0.2Er0.1Ru0.7O2−δ−1. (e) The long-term stability (500 h) of W0.2Er0.1Ru0.7O2−δ nanosheets in 0.5 M H2SO4. (a–e) Reprinted with permission from ref. 104. Copyright 2020, Springer Nature. (f) Calculated PDOS plots of Ru 4d and O 2p orbitals for Y2Ru2O7 and RuO2. (g) Comparison of the central energies of the overlapping bands of Ru 4d and O 2p orbital and retained current density at 1.50 V for Y2Ru2O7 and RuO2, respectively. (f and g) Reprinted with permission from ref. 108. Copyright 2017, American Chemical Society. | ||
Apart from RuO2, Ru-based complex oxides with diverse crystal structures (e.g., perovskite-type and pyrochlore-type) were also studied to exploit the robust Ru-based anode catalysts.22,105 Among these, the Ru-based complex oxide, perovskite-type SrRuO3 is identified as a classical model, whose catalytic/degradation mechanism has been systematically explored. The most effective stabilization approach for SrRuO3 is also proven to be element doping. For example, Na and Ca substitutions for Sr were investigated by Retuerto et al. and Hirai et al., respectively.106,107 It has been shown that Na or Ca substitution at the A site can improve catalytic stability by adjusting the electronic structure of the Ru site and alleviating the structural distortion of RuO6 octahedra. In addition to the influence of elements, the unique crystal structure of the complex oxide is also conducive to the stability enhancement of the Ru-based anode catalysts. For example, Kim et al. reported a pyrochlore-type Y2Ru2O7−δ anode catalyst, which possesses good activity and stability.108 As suggested from the theoretical calculation results, the overlapped Ru 4d-orbital and O 2p-orbital band center energies of Y2Ru2O7−δ is less than that of RuO2, which makes the Ru–O bond of Y2Ru2O7−δ more stable (Fig. 10f and g).
All the above discussion showed that substantial progress has been made in the stabilization of Ru-based anode catalysts. However, the harsh truth is that they are far from PEMWE's technical targets. Although several Ru-based anode catalysts have been evaluated in the PEM electrolyzer, the operating current is not up to the amperage level for PEMWE. For example, the W0.2Er0.1Ru0.7O2−δ anode catalyst discussed above can operate at a current density of 100 mA cm−2 for 120 hours in a PEM electrolyzer. Wu et al. tested the Ni-doped RuO2 anode catalyst in a PEM electrolyzer, which could maintain the current density of 200 mA cm−2 for 1000 hours.109 Admittedly, it is great progress for Ru-based anode catalysts to achieve such a degree of durability in the PEM electrolyzer. However, once the operating voltage raises higher (e.g., 2 V), there is a strong possibility of catalyst degradation. In the short term, the more promising strategy is to combine Ru-based anode catalysts with iridium-based counterparts.
3.3 Noble-metal-free anode catalysts
Although Ir-based and Ru-based materials exhibit excellent performance for OER, their price and scarceness heavily hinder the scale-up development of PEMWEs. Thus, it is urgently required to explore acid-stable noble-metal-free materials. The 3d transition metal-based oxides have excellent adsorption energy of oxygen intermediates, and thus show better catalytic activity. Nevertheless, there is a shortcoming for transition metal-based oxides: poor stability not enough to catalyze the water splitting in an acidic medium. The Pourbaix diagrams, as an effective tool for theoretical prediction, can reflect the stable redox species ranging with potential and pH in aqueous solutions, and thereby screen out the metal elements with good acidic stability.110 The theoretical and experimental results show that some metal elements (e. g., Ti, Mo, Nb, Ta, W, Pb, etc.) can be stable under the pH and potential of acidic OER.111 However, these metals and their oxides have poor OER activity, therefore, coupling with some stabilizing structural elements into transition metal-based oxides with good OER activity can improve the stability of unstable metal oxide catalysts to a certain extent. For example, Huynh et al. selected Pb as a stabilizing structural element in CoFePbOx films.112 CoFePbOx showed high OER activity at pH = 2.5 and long-term stability for over 50 h at 1 mA cm−2. Frydendal et al. incorporated TiO2 and MnO2 to obtain the catalyst that led to the improvement of stability.113 Apart from introducing metal elements, introducing non-metallic elements can bring amazing results. Patel et al. developed F-doped Cu1.5Mn1.5O4 catalysts with unchanged current density for 24 h testing of constant potential (1.55 V vs. RHE).114 Pan et al. successfully designed low-cost and high-performance OER catalysts in acidic medium, named Mn7.5O10Br3.115 The obtained Mn7.5O10Br3 can maintain excellent stability about 500 h at 10 mA cm−2 and 300 h at 100 mA cm−2 in PEMWE, which can be attributed to the formation of a tightly-packed oxide on the surface of the catalysts. This work also confirmed that the introduction of halogen ions with excellent electron transport capacity can further enhance the OER activity (Fig. 11a–c). Moreover, the interface engineering has been considered to enhance stability. Li et al. chose to deposit γ-MnO2 on the fluorine-doped tin oxide support, which had no obvious decay during galvanostatic electrolysis at 10 mA cm−2 over 8000 h measurement.116 Huang et al. mixed CeO2 and Co3O4 to construct Co3O4/CeO2 nanocomposite, with the overpotential of 423 mV (FTO substrate) and 347 mV (C substrate) at 10 mA cm−2.117 The closely bonded interface between CeO2 and Co3O4 expanded the electronic regulatory effect at the interface, changed its local structural characteristics, and promoted the catalytic performance of acid OER. (Fig. 11d and e) All kinds of strategies can be implemented to obtain efficient acid OER catalysts using the abundant transition metal oxides, which are expected to replace noble-metal materials one day.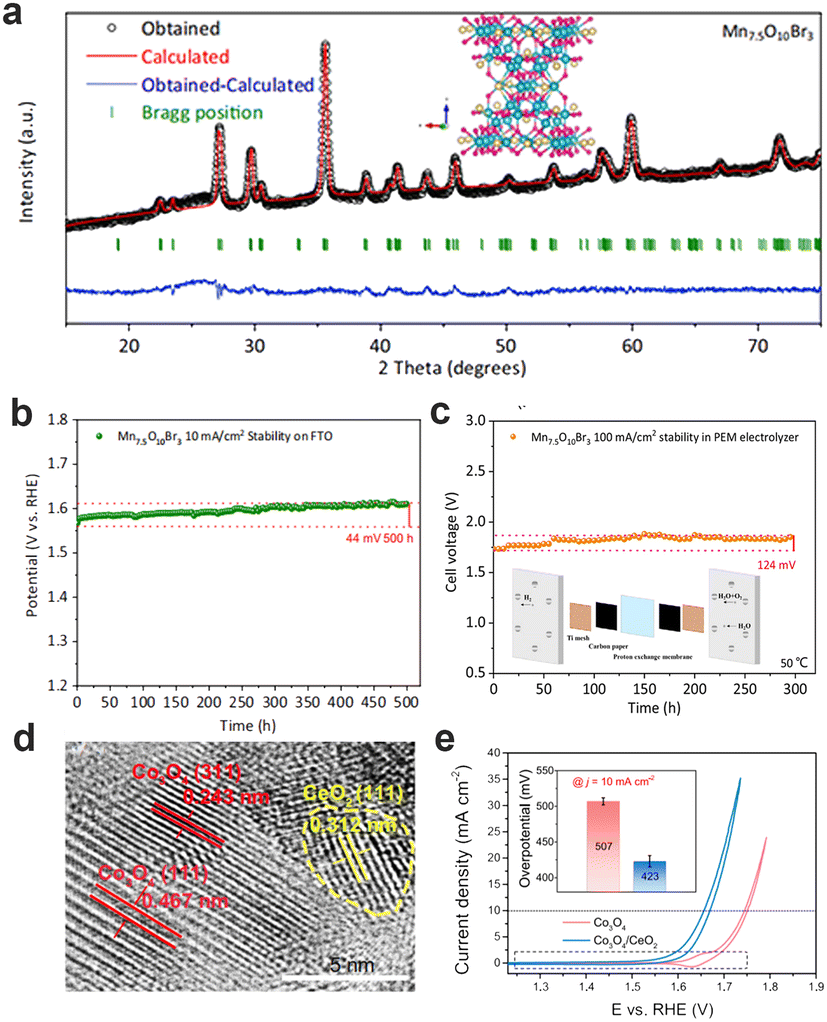 | ||
| Fig. 11 (a) XRD patterns of Mn7.5O10Br3. (b) Chronopotentiometry curves (on FTO) of Mn7.5O10Br3 at 10 mA cm−2 (25 °C). (c) Stability tests of Mn7.5O10Br3 at 100 mA cm−2 in PEMWE. (a–c) Reprinted with permission from ref. 115. Copyright 2022, Springer Nature. (d) HRTEM images of Co3O4/CeO2 samples. (e) The iR-corrected CV curves of Co3O4 and Co3O4/CeO2 in 0.5 M H2SO4 solution. (d and e) Reprinted with permission from ref. 117. Copyright 2021, Springer Nature. | ||
The other non-noble metal materials, including metal sulfides, metal borides, and metal phosphides, (e.g., MoS2, TaS2, TiB2, CoPi, etc.) also have great appeal in the field of the acidic medium. For example, Wu et al. exfoliated 2D metal sulfides (1T-MoS2, 1T-TaS2, 2H-MoS2, 2H-TaS2) nanosheets through a liquid phase and lithium intercalation, which showed poor activity and stability.118 MoS2/Co9S8/Ni3S2/Ni, was also demonstrated an excellent electrocatalyst in a wide pH range (alkaline, acidic and neutral electrolytes).119 In addition, some intermetallic compounds were examined for OER in the acidic medium. For instance, the intermetallic Ni2Ta can produce the current for OER in the acidic electrolyte.120 The corrosion rates of Ni2Ta was 2 orders lower than that of pure Ni. TiB2 showed the high overpotential of about 560 ± 20 mV. Although poor activity was found for TiB2, it had moderate stability in acidic conditions.
Although some works have been reported about the application of non-precious metal-based catalysts, these catalysts are much less active than Ir-based and Ru-based catalysts. The differences are shown as follows: (i) the high overpotential (>400 mV) at 10 mA cm−2; (ii) the leaching of nonprecious-metal-based electrocatalysts in acid was serious, and the catalysts had no time to react and thereby resulted in poor stability. Taking into account strongly acidic conditions and the high performance of the PEMWE, the activity and stability of these materials cannot meet the industrial demand for PEMWE. Therefore, how to enhance the activity and stability of nonprecious-metal-based electrocatalysts, making them potential to be applied in PEMWE by substituting precious metals (e.g., Ir and Ru), will be very challenging work.
4 Keys for translating efficient anode catalysts into superb PEM electrolyzers
Because PEMWE is considered to be one of the most promising green hydrogen production routes, long-term development plans have been announced by many countries.121 For example, the U.S. Department of Energy (DOE) has established the technology goals before 2025 for PEM electrolyzer stacks as follows:122 the operating current density of more than 3 A cm−2@1.9 V, a lifetime of more than 80 khr with a degradation rate less than 2.25 mV khr−1, and a load of platinum-group metals (PGM) mass on MEA less than 0.5 mg cm−2. Similarly, China's “hydrogen energy technology” key project in the National Key Research and Development Plan also states a series of goals: operating current density of more than 2 A cm−2@1.8 V@80 °C, lifetime more than 3 khr with an overpotential increase rate of less than 2%, Ir mass loading less than 0.3 mg cm−2. To achieve these accomplishments, the durability and activity of the anode catalyst must be evaluated in an actual PEM electrolyzer. Over the past decade, many advances have been made in the development of anode catalysts for PEMWE. The activity and stability of some novel anode catalysts have been proven to be significantly better than those of the commercial catalyst IrO2.123,124 However, their performances were evaluated in a three-electrode test system rather than in a PEM electrolyzer. Once these novel anode catalysts are evaluated in a PEM electrolyzer, most of them can hardly reproduce their excellent performance.125 This fact can be attributed to the great difference between the three-electrode cell and PEM electrolyzer.4.1 Comparison of three-electrode cell and PEM electrolyzer
Three-electrode cell (Fig. 12a) has been extensively used in the laboratory to evaluate the intrinsic activity of electrocatalysts.126 Generally, a classic three-electrode cell includes one working electrode (e.g., rotating disk electrode, RDE), one counter electrode (e.g., platinum wire or mesh), and one reference electrode (e.g., Hg/Hg2Cl2 or Hg/HgSO4 electrode). The diluted acid solution (e.g., 0.1 M HClO4) is used as a liquid electrolyte. The catalyst loading on the working electrode is fairly low (a few to tens of micrograms), benefiting from the less dosage of PGM. And the simple fabrication of a three-electrode cell makes it a powerful tool to quickly and conveniently measure the inherent activity of the anode catalysts.127 | ||
| Fig. 12 (a) Schematic illustration of three-electrode cell set-up (left). The enlarged model of the catalyst layer on RDE and the advantages and disadvantages of three-electrode cell test (right). (b) Schematic illustration of PEM electrolyzer (left). The enlarged model of the catalyst layer on MEA and the advantages and disadvantages of PEM electrolyzer (right). | ||
In contrast, the performance of the catalyst in PEMWE is based on MEA, the core component of the PEM electrolyzer (Fig. 12b). More PGM catalysts (e.g., Pt load: 0.5 mg cm−2, Ir load: 2 mg cm−2) tend to be consumed during the preparation of MEA. Coupled with the complex components introduced above, assembling a PEM electrolyzer is much more expensive and time-consuming. Even so, the PEM electrolyzer test reflects the actual activity and stability of the electrocatalyst under the actual industrial operating conditions.
The catalytic activities of the anode catalysts exhibited in three-electrode cells (i.e., RDE-level activity) and in the PEM electrolyzer (i.e., MEA-level activity) hinge on different determinants. In a three-electrode cell, the RDE-level activity is commonly tested with a narrow potential window of 1.2–1.6 V vs. RHE, which generates a relatively low current density (i.e., several to tens mA cm−2), resulting in the reaction kinetics (related to intrinsic activity) playing the crucial role in RDE-level activity.29 With respect to the PEM electrolyzer, PEM shortens the proton transmission distance, so that the operating current density of the PEM electrolyzer can reach tens to hundreds of times than that of the three-electrode test system, accompanied by a large amount of gas release. Under the conditions of frequent gas–liquid transfer and high current density (>1000 mA cm−2), factors affecting the actual performance include not only the reaction kinetics but also the mass transfer loss and ohmic resistance of the catalyst layer,128 which are not reflected in the three-electrode test system. Therefore, the discrepancy in mass transport and charge transport behaviors provides the different catalytic performances in the two test systems, especially at high current density.
In addition, the RDE-level stability and MEA-level stability of the same anode catalysts behave differently.129 For example, Knöppel et al. stated the underestimated RDE-level stability in a three-electrode cell for anode catalysts.130 They found that, for the same catalyst, the RDE-level lifetime (i.e., in units of the day) was several orders of magnitude lower than the MEA-level lifetime (i.e., in units of the year). They proposed a dimensionless descriptor, stability number (S-number), which is the ratio of oxygen evolved to iridium dissolved during the OER catalysis.35 By evaluating the effects of different system parameters (e.g., pH of the electrolyte, ionomer content of the catalyst layer, and catalyst loading) on S-number, they revealed that the main reason for different Ir dissolution behaviors was the pH discrepancy between the three-electrode cell and PEM electrolyzer, which will become more apparent over time. In another case, the RDE-level stability is also closely related to the properties of the electrode materials covered by the electrocatalyst. The common working electrodes (e.g., glass carbon, ATO) are often passivated, which leads to a continuous increase in contact resistance, eventually resulting in a sudden potential change.131 This result cannot simply be attributed to catalyst degradation. It suggested using a gold electrode or boron-doped diamond electrode as the working electrode because they are less pron to be passivated.129
In a word, the RDE-level test has the advantages of convenience, universality, and minimum catalyst consumption, which is conducive to the rapid screening of promising catalysts with high intrinsic activity. However, the usability of anode catalysts demands to be assessed in the MEA-level test system, which is superior to providing real activity and stability for PEMWE. In order to directly understand the performance differences assessed by the two test systems, we have listed the performance parameters of the mentioned anode catalysts in the review in Table 1.
| Catalyst | Electrolyte | RDE-level activity | MEA-level activity | Stability | Ref. |
|---|---|---|---|---|---|
| Ir dendrites | 0.5 M H2SO4 | 1.58 V vs. SHE@0.005 A | 61 | ||
| Ir nanowires | 0.5 M HClO4 | 283 mV@10 mA cm−2 | 25000 s@5 mA cm−2 |
62 | |
| Ir nanoparticles | 0.5 M HClO4 | 290 mV@10 mA cm−2 | 25000 s@10 mA cm−2 |
63 | |
| 3D Ir superstructures | 0.1 M HClO4 | 270 mV@10 mA cm−2 | 8 h@2.5 mA cm−2 | 64 | |
| IrO2 nanoneedles | 1 M H2SO4 | 313 mV@10 mA cm−2 | 2.1 V@2 A cm−2@80 °C | 200 h@2 A cm−2 | 65 |
| IrO2 | 2 V@2 A cm−2@80 °C | 122 h@1 A cm−2 | 66 | ||
| Amorphous IrOx | 0.5 M H2SO4 | 320 mV@10 mA cm−2 | 12 h@2.5 mA cm−2 | 67 | |
| Nano-size IrOx | 0.1 M HClO4 | 1.88 V@1.8 A cm−2@80 °C | 4500 h@1.8 A cm−2 | 68 | |
| Ir0.7Ru0.3Ox | 1.85 V@3.2 A cm−2@90 °C | 70 | |||
| Ir0.1Ru0.9O2 | 0.5 M H2SO4 | 1.806 V@1 A cm−2@80 °C | 1000 h@1.0 A cm−2 | 71 | |
| RuO2@IrOx | 0.5 M H2SO4 | 215 mV@10 mA cm−2 | 1.683 V@1 A cm−2@80 °C | 300 h@1.0 A cm−2 | 72 |
| Ru–IrO2 | 0.5 M H2SO4 | 266 mV@10 mA cm−2 | 1.722 V@1 A cm−2@80 °C | 100 h@1.0 A cm−2 | 73 |
| Ir0.7W0.2Sn0.1Ox | 0.5 M H2SO4 | 236 mV@10 mA cm−2 | 220 h@1.0 A cm−2 | 80 | |
| GB-Ta0.1Tm0.1Ir0.8O2−δ | 0.5 M H2SO4 | 198 mV@10 mA cm−2 | 1.935 V@2 A cm−2@50 °C | 500 h@1.0 A cm−2 | 81 |
| IrO2/TiO2 | 1.57 V@1 A cm−2@80 °C | 84 | |||
| IrOx/W-TiO2 | 0.5 M H2SO4 | 1607 A g−1@1.4 V vs. Ag/AgCl | 1.602 V@1 A cm−2@80 °C | 450 h@1.0 A cm−2 | 85 |
| Ir/Nb2O5−x | 0.5 M H2SO4 | 218 mV@10 mA cm−2 | 1.839 V@3 A cm−2@80 °C | 2000 h@2.0 A cm−2 | 89 |
| IrOx/SrIrO3 | 0.5 M H2SO4 | 270–290 mV@10 mA cm−2 | 30 h@10 mA cm−2 | 90 | |
| 6H-SrIrO3 | 0.5 M H2SO4 | 248 mV@10 mA cm−2 | 30 h@10 mA cm−2 | 94 | |
| 2D-SIO | 0.05 M H2SO4 | 243 mV@10 mA cm−2 | 5000 CV cycles (1.2–1.6 V vs. RHE) | 95 | |
| SrTiO3–SrIrO3 | 0.1 M HClO4 | 247 mV@10 mA cm−2 | 20 h@10 mA cm−2 | 96 | |
| SrZrO3–SrIrO3 | 0.1 M HClO4 | 240 mV@10 mA cm−2 | 30 h@10 mA cm−2 | 97 | |
| W0.2Er0.1Ru0.7O2−δ | 0.5 M H2SO4 | 168 mV@10 mA cm−2 | 500 h@10 mA cm−2 | 100 | |
| Y2Ru2O7−δ | 0.1 M HClO4 | 190 mV@10 mA cm−2 | 8 h@1 mA cm−2 | 104 | |
| Ni-RuO2 | 0.1 M HClO4 | 214 mV@10 mA cm−2 | 1.95 V@1 A cm−2@80 °C | 200 h@10 mA cm−2 | 105 |
| CoFePbOx | 0.1 M Pi + 1 M KNO3 | >50 h@1 mA cm−2 | 112 | ||
| Ti-MnO2 | 0.05 M H2SO4 | <2 h@1 mA cm−2 | 113 | ||
| Cu1.5Mn1.5O4 | 0.5 M H2SO4 | 310 mV@10 mA cm−2 | 24 h@1.55 V vs. RHE | 114 | |
| Mn7.5O10Br3 | 0.5 M H2SO4 | ∼295 mV@10 mA cm−2 | 300 h@100 mA cm−2 | 115 | |
| γ-MnO2 | 1.0 M H2SO4 + 0.5 M Na2SO4 | ∼489 mV@10 mA cm−2 | 2.0 V@127 mA cm−2@25 °C | 350 h@10 mA cm−2 | 116 |
| Co3O4/CeO2 | 0.5 M H2SO4 | 327 mV@10 mA cm−2 | 117 | ||
| 1T-MoS2 | 0.5 M H2SO4 | 322 mV@10 mA cm−2 | 1 h@10 mA cm−2 | 118 | |
| 1T-TaS2 | 0.5 M H2SO4 | 282 mV@10 mA cm−2 | 1 h@10 mA cm−2 | 118 | |
| Nanoporous IrO2 | 0.5 M H2SO4 | 282 mV@10 mA cm−2 | 1.649 V@1 A cm−2@80 °C | 3000 cycles (1.06–1.66 V in PEM electrolyzer) | 136 |
| Ir/WxTi1−xO2 | 0.1 M HClO4 | 1.75 V@2 A cm−2@80 °C | 1200 h@1.5 A cm−2 | 141 |
4.2 Design strategy for viable anode catalysts towards PEMWE
As discussed above, the performance of the OER catalyst in MEA is affected by several factors, and the design of the catalyst needs comprehensive consideration. Three strategies for obtaining desirable MEA anode catalysts are listed below.Firstly, designing the catalysts with high intrinsic activity. A key factor affecting the catalytic activity is the electronic structure of the electrocatalyst,132,133 and it can be modulated by doping heteroatoms, crystal phase engineering, and introducing lattice stress. For example, the introduction of some transition metal heteroatoms into Ir-based catalysts not only reduces the content of noble metals but also adjusts the Ir d orbital and optimizes the adsorption of oxygen intermediates to improve its OER activity. By introducing Ta and Tm atoms into the IrO2 lattice, Hao et al. prepared torsion-strain Ta0.1Tm0.1Ir0.8O2−δ OER electrocatalyst with a large number of grain boundaries.134 The ligand effect of heteroatoms and Ir–O bond with torsional strain both regulate the adsorption energy of oxygen intermediates, leading to improved catalytic performance. The MEA based on Ta0.1Tm0.1Ir0.8O2−δ anode exhibited much lower initial potential and better catalytic performance than IrO2, benefiting from an ultra-low oxygen evolution overpotential, and could operate for 500 hours at 1.5 A cm−2 current density with a low catalysis load of 0.2 mg cm−2.
Secondly, nanostructure engineering and morphology control of the catalyst. Nanomaterials with special structures are widely used in the catalytic field because of their outstanding performance. Their large specific surface area leads to more exposure to active sites and improved utilization of PGM.135 Li et al. reported a micro/mesoporous IrO2 catalyst with a surface area of more than 300 m2 g−1. Porous nano IrO2 was synthesized by the ammonia induction pore formation method without the use of templates.136 The PEM electrolyzer based on this OER catalyst achieved 1 A cm−2 at the low voltage of 1.649 V as well as its outstanding stability was proved by the accelerated durability tests. In addition, it has been reported that the pore structure of the catalyst layer is conducive to oxygen transport. However, micropores are too small for the ionomer to penetrate, resulting in the formation of “dead” sites, while too many macropores are not conducive to the construction of proton transport networks.29 To achieve the best mass and load transfer, the optimum pore size distribution is a large number of mesoporous structures between 5–20 nm.137 3M corporation invented a nanostructured thin film (NSTF) MEA using a rod-like array on the polymer membrane.138 The performance of MEA based on the NSTF catalyst was greatly improved due to the increased activity and the enhanced electron transfer between CL and membrane.
Thirdly, the construction of a supported catalyst. It is a general strategy to select appropriate catalyst support to reduce the content of PGM as well as increase the electrocatalytic activity and stability.139 Carbon is the most wildly used electrocatalytic support substrate because of its good conductivity and low cost. Unfortunately, the high potential in the anode of the PEM electrolyzer makes C tend to be oxidized to CO2, accompanied by the shedding of catalytic species.140 As described above, the optimal supports for anode catalysts require a large surface area, high conductivity, and strong acid and oxidation resistance. It should be noted that if the carrier material with poor conductivity is selected, more load of noble metal is required to obtain a higher overall conductivity. Zhao et al. dispersed Ir nanoparticles (Ir NPs) on tungsten-doped TiO2 support by the polyol reduction method.141 The large specific surface area of Ir NPs formed a chain-like conductive network and showed high-quality activity. The catalyst durability was evaluated by MEA, with only 20 mV voltage degradation after 1200 h operation at 1.5 A cm−2.
4.3 Intermediate technologies for performance evaluation
Considering the significant difference between three-electrode cells and the PEM electrolyzer, some intermediate test technologies that cover both the operating conditions close to MEA and the simple structure similar to that of the RDE have been developed. For example, a half-cell gas diffusion electrode (GDE) test system has similar components to a real cell and has been used as an intermediate characterization technique in the PEM fuel cell (PEMFC) and PEMWE. A half-cell GDE setup is separated by a polymer membrane into two parts, one side connecting with the GDL and flow field-like in a real PEM electrolyzer, and the other side directly facing towards the aqueous acidic electrolyte (Fig. 13). The catalyst can be coated on the membrane or GDL with the mg cm−2 to μg cm−2 scale catalyst covering.29 Moreover, the half-cell GDE technology can operate at a high current density of more than 1000 mA cm−2, which is close to the actual operating conditions. Inaba et al. tested commercial Pt/C catalysts in half-cell GDE and found that the ECSA measured was almost the same as that in MEA.142 Recently, Schröder et al. demonstrated that the half-cell GDE test system is more suitable than RDE for evaluating the mass activity, activation energy, and current density of IrO2 with the catalyst load at the PEMWE level, and can also be used for short-term stability testing.125 In a word, half-cell GDE is a prospective intermediate test technology for PEMWE. | ||
| Fig. 13 Schematic illustration of half-cell GDE configuration. | ||
In recent years, with the expansion of PEMWE technology in the industry and related developments in chemistry and materials science, miniaturized commercial water electrolyzers suitable for laboratory applications are becoming cheaper and more readily available. The small active area (usually several to tens of cm2) reduces catalyst consumption to an acceptable level. Although they are different from the industrial stacks in scale, the same operating conditions are sufficient to evaluate the reliability of catalysts in MEA systems. In conclusion, we suggest that (i) pre-screen high-throughput catalysts quickly by the RDE test; (ii) develop and improve the intermediate test technology from RDE to MEA (e.g., the half-cell GDE test system); (iii) Consider the performance of catalysts in actual PEM electrolyzers as the evaluation standard for their prospective application.
5 Summary and outlook
The growth of the PEMWE hydrogen production industry relies on the innovation and development of anode catalysts. In this review, we introduce the mechanism in PEMWE, as well as summarize the advances and status of anode catalysts towards PEMWE hydrogen production technology. Then, we elucidate the intrinsic reasons why the superior RDE-level activity and stability of anode catalysts are rarely translated into high MEA-level performance in the PEM electrolyzer. Strategies for bridging this gap are also provided. To promote the scale up of the PEMWE hydrogen production technology, the further development of anode catalysts needs efforts in the following areas (Fig. 14): | ||
| Fig. 14 Future directions of anode catalysts towards PEMWE. | ||
(i). Rationally designing and synthesizing anode catalysts for PEMWE. Reasonable design and synthesis of the anode catalysts is the prerequisite for the synergic optimization of cost, activity, and stability. In the respect of design principle, density functional theory (DFT) calculation is a helpful tool to simulate and predict promising anode catalysts. For example, the adsorption strength of oxygen intermediates, and the position of the O 2p band center are strongly related to the catalytic activity of materials. The thermodynamic stabilities of the potential catalysts and supports can be identified by calculating their Pourbaix diagrams. Thus, DFT calculation guides us to efficiently design the catalyst candidate. In addition, effective synthesis methods can make the catalyst design strategy into reality. It is essential to exploit multiple synthesis methods to prepare the catalysts with a specific element distribution, crystal orientation, crystallinity, or morphology structure, as well as the catalyst supports with high surface area, conductivity, and corrosion resistance. Besides, in order to meet the needs of industrial applications of PEMWE, it is important to develop macroscopic-scale preparation methods to produce highly uniform anode catalysts on a kilogram scale.
(ii). Developing in situ characterization techniques. At present, the structural evolution, actual active phase, and OER course of anode catalysts are ambiguous. Thus, in order to reveal the catalytic/degradation mechanism, researchers have employed in situ characterization techniques to monitor the structure evolutions and key reaction intermediates of catalysts. For example, in situ X-ray diffraction (XRD) can be employed to observe the transformation in the crystal phase during OER catalysis. To characterize the electronic structure in real-time and geometric coordination environment of each element, near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS), in situ XAS, and in situ Raman spectroscopy are powerful tools. The source of the O atom in the O2 product can be determined by 18O isotope labelling coupled with in situ differential electrochemical mass spectrometry (DEMS). By combining the results obtained by in situ characterizations, we are getting closer to the clarified catalytic/degradation mechanism. However, the reality must be acknowledged that technical bottleneck remains for in situ characterization. On the one hand, it is urgent to develop in situ characterization techniques with higher resolution, stronger signal sources, and more accurate selected areas to obtain more accurate structural information. On the other hand, the high criteria for reactors restrict the availability of in situ characterization techniques. Taking the PEMWE anode catalysts as an example, it is difficult but necessary to design the matched in situ reactor to simulate the PEMWE operating conditions.
(iii). Taking the long-term stability under high current density into account. The stability of the catalytic material fundamentally affects the lifetime of PEMWE, thereby determining whether the anode catalyst possesses industrial practicability. However, the importance of stability is ignored in the pursuit of low-cost and active anode catalysts. When assessing the durability in the laboratory, researchers measure the short-term stability at a low current density (10 mA cm−2) by chronopotentiometry or chronoamperometry. However, the operating current density of PEMWE is more than 1 A cm−2, and the lifetime is measured in years. Obviously, current testing methods in the laboratory are not competent for identifying the applicable catalyst used in PEMWE. Therefore, the long-term stability of the anode catalyst at a higher current density is crucial to be tested in the laboratory. The conventional glassy carbon electrode may no longer be suitable because it is prone to be passivated at high voltages. The anode catalysts can be drop-casted on the substrate with high corrosion and oxidation resistant, such as carbon paper, carbon cloth, and titanium felt. Furthermore, and more directly, the durability of MEA constructed using anode catalysts can be tested under practical operating conditions to identify the applicability of anode catalysts for PEMWE.
(iv). Adding MEA-level tests into the performance evaluation protocol. Although the three-electrode cell test system is capable of rapidly identifying promising catalysts, these catalysts are most likely not applicable to PEMWE. This is due to discrepancies in testing/operating conditions (e.g., pH, temperature, pressure, voltage). For example, the test voltage of the RDE test system hardly exceeds 1.5 V vs. RHE due to the limitations of the measurement equipment and artifacts. As a result, it is impossible to assess the activity and stability of the catalyst at the high-voltage operating conditions (∼2 V) for PEMWE. In addition, the structural stabilities of the anode catalysts will face severe tests under the high-temperature conditions for MEA, during which the degradation behaviors (e.g., Ir leaching, irreversible oxidation, nanoparticle agglomeration, structure collapse) will be aggravated. The instability of the anode catalyst at high temperatures is fatal to MEA, unfortunately, it is difficult to be predicted accurately in the RDE test system. It can be seen that the MEA test is necessary to identify whether the anode catalysts can actually be applied in PEMWE, no matter how long it takes or how much it costs. Therefore, we can preliminarily screen the active and stable catalysts by the RDE test system, then, further, measure the performance and lifetime of candidates in MEA to identify the anode catalysts that can meet the demands of industrial applications.
Author contributions
Qiannan Wu, Yuannan Wang, Kexin Zhang, Zhoubing Xie, Ke Sun and Wei An: writing-original draft; Xiao Liang and Xiaoxin Zou: writing-review & editing; all authors had approved the final version.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
X. Z. thanks for the financial support from the National Key R & D Program of China (2021YFB4000200), the National Natural Science Foundation of China (NSFC) (grant no. 21922507 and 22179046). X. L. acknowledges the NSFC (no. 22205072), the financial support from China Postdoctoral Science Foundation (no. 2021M701377). We also thank the NSFC (no. 21621001) and the 111 Project (no. B17020) for additional financial support.References
- J. Rogelj, M. den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Paris Agreement climate proposals need a boost to keep warming well below 2 °C, Nature, 2016, 534, 631–639 CrossRef CAS.
- M. Hulme, 1.5 °C and climate research after the Paris Agreement, Nat. Clim. Change, 2016, 6, 222–224 CrossRef.
- J. Chi and H. Yu, Water electrolysis based on renewable energy for hydrogen production, Chinese, J. Catal., 2018, 39, 390–394 CrossRef CAS.
- G. He, D. S. Mallapragada, A. Bose, C. F. Heuberger-Austin and E. Gençer, Sector coupling via hydrogen to lower the cost of energy system decarbonization, Energy Environ. Sci., 2021, 14, 4635–4646 RSC.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, An overview of hydrogen production technologies, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- X. Zou and Y. Zhang, Noble metal-free hydrogen evolution catalysts for water splitting, Catal. Today, 2015, 44, 5148–5180 CAS.
- Z. Yan, L. Hitt Jeremy, A. Turner John and E. Mallouk Thomas, Renewable electricity storage using electrolysis, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12558–12563 CrossRef CAS.
- M. Z. Jacobson and M. A. Delucchi, Providing all global energy with wind, water, and solar power, Part I: Technologies, energy resources, quantities and areas of infrastructure, and materials, Energy Policy, 2011, 39, 1154–1169 CrossRef CAS.
- A. Velazquez Abad and P. E. Dodds, Green hydrogen characterisation initiatives: Definitions, standards, guarantees of origin, and challenges, Energy Policy, 2020, 138, 111300 CrossRef.
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy, H. Chen and X. Zou, Status and perspectives of key materials for PEM electrolyzer, Nano Res, Energy, 2022, 1, e9120032 Search PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, A comprehensive review on PEM water electrolysis, Int. J. Hydrog. Energy, 2013, 38, 4901–4934 CrossRef CAS.
- S. Zaman, M. Wang, H. Liu, F. Sun, Y. Yu, J. Shui, M. Chen and H. Wang, Carbon-based catalyst supports for oxygen reduction in proton-exchange membrane fuel cells, Trends Chem., 2022, 4, 886–906 CrossRef CAS.
- J. H. Scott, The Development of Fuel Cell Technology for Electric Power Generation: From NASA's Manned Space Program to the “Hydrogen Economy”, Proc. IEEE, 2006, 94, 1815–1825 CAS.
- C. V. Pham, D. Escalera-López, K. Mayrhofer, S. Cherevko and S. Thiele, Essentials of High Performance Water Electrolyzers – From Catalyst Layer Materials to Electrode Engineering, Adv. Energy Mater., 2021, 11, 2101998 CrossRef CAS.
- O. Omoniyi, T. Bacquart, N. Moore, S. Bartlett, K. Williams, S. Goddard, B. Lipscombe, A. Murugan and D. Jones, Hydrogen Gas Quality for Gas Network Injection: State of the Art of Three Hydrogen Production Methods, Processes, 2021, 9, 1056 CrossRef CAS.
- Y. Liu, X. Liang, H. Chen, R. Gao, L. Shi, L. Yang and X. Zou, Iridium-containing water-oxidation catalysts in acidic electrolyte, Chinese, J. Catal., 2021, 42, 1054–1077 CrossRef CAS.
- A. Ursua, L. M. Gandia and P. Sanchis, Hydrogen Production From Water Electrolysis: Current Status and Future Trends, Proc. IEEE, 2012, 100, 410–426 CAS.
- G. Glenk and S. Reichelstein, Economics of converting renewable power to hydrogen, Nat. Energy, 2019, 4, 216–222 CrossRef CAS.
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Perspectives on Low-Temperature Electrolysis and Potential for Renewable Hydrogen at Scale, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 219–239 CrossRef CAS.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C.-H. Yan, Recent Development of Oxygen Evolution Electrocatalysts in Acidic Environment, Adv. Mater., 2021, 33, 2006328 CrossRef CAS PubMed.
- M. Bernt, A. Hartig-Weiß, M. F. Tovini, H. A. El-Sayed, C. Schramm, J. Schröter, C. Gebauer and H. A. Gasteiger, Current Challenges in Catalyst Development for PEM Water Electrolyzers, Chem. Ing. Tech., 2020, 92, 31–39 CrossRef CAS.
- H. J. Song, H. Yoon, B. Ju and D.-W. Kim, Highly Efficient Perovskite-Based Electrocatalysts for Water Oxidation in Acidic Environments: A Mini Review, Adv. Energy Mater., 2021, 11, 2002428 CrossRef CAS.
- Y. Wei, D. Zhao, J. Wan and D. Wang, Electron–orbital–lattice interactions in hollow multishelled structures, Trends Chem., 2022, 4, 1021–1033 CrossRef CAS.
- C. Lei, W. Li, G. Wang, L. Zhuang, J. Lu and L. Xiao, Improving the Catalytic Efficiency of NiFe-LDH/ATO by Air Plasma Treatment for Oxygen Evolution Reaction, Chem. Res. Chin. Univ., 2021, 37, 293–297 CrossRef CAS.
- E. Antolini, Iridium As Catalyst and Cocatalyst for Oxygen Evolution/Reduction in Acidic Polymer Electrolyte Membrane Electrolyzers and Fuel Cells, ACS Catal., 2014, 4, 1426–1440 CrossRef CAS.
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, The Stability Challenges of Oxygen Evolving Catalysts: Towards a Common Fundamental Understanding and Mitigation of Catalyst Degradation, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef.
- L. She, G. Zhao, T. Ma, J. Chen, W. Sun and H. Pan, On the Durability of Iridium-Based Electrocatalysts toward the Oxygen Evolution Reaction under Acid Environment, Adv. Funct. Mater., 2022, 32, 2108465 CrossRef CAS.
- H. Wang, J. Li, F. Li, D. Guan, X. Wang, W. Su and J. Xu, Strategies with Functional Materials in Tackling Instability Challenges of Non-aqueous Lithium-Oxygen Batteries, Chem. Res. Chin. Univ., 2021, 37, 232–245 CrossRef CAS.
- J. Fan, M. Chen, Z. Zhao, Z. Zhang, S. Ye, S. Xu, H. Wang and H. Li, Bridging the gap between highly active oxygen reduction reaction catalysts and effective catalyst layers for proton exchange membrane fuel cells, Nat. Energy, 2021, 6, 475–486 CrossRef CAS.
- R. Abbasi, B. P. Setzler, S. Lin, J. Wang, Y. Zhao, H. Xu, B. Pivovar, B. Tian, X. Chen and G. Wu, A Roadmap to Low-Cost Hydrogen with Hydroxide Exchange Membrane Electrolyzers, Adv. Mater., 2019, 31, 1805876 CrossRef.
- S. Shiva Kumar and V. Himabindu, Hydrogen production by PEM water electrolysis – A review, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- G.-B. Jung, S.-H. Chan, C.-J. Lai, C.-C. Yeh and J.-W. Yu, Innovative Membrane Electrode Assembly (MEA) Fabrication for Proton Exchange Membrane Water Electrolysis, Energies, 2019, 12, 4218 CrossRef CAS.
- Z. Kang, J. Mo, G. Yang, S. T. Retterer, D. A. Cullen, T. J. Toops, J. B. Green Jr, M. M. Mench and F.-Y. Zhang, Investigation of thin/well-tunable liquid/gas diffusion layers exhibiting superior multifunctional performance in low-temperature electrolytic water splitting, Energy Environ. Sci., 2017, 10, 166–175 RSC.
- Q. Zhang, H. Chen, L. Yang, X. Liang, L. Shi, Q. Feng, Y. Zou, G.-D. Li and X. Zou, Non-catalytic, instant iridium (Ir) leaching: A non-negligible aspect in identifying Ir-based perovskite oxygen-evolving electrocatalysts, Chin. J. Catal., 2022, 43, 885–893 CrossRef CAS.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, The stability number as a metric for electrocatalyst stability benchmarking, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
- R. L. Doyle and M. E. G. Lyons, in Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, ed. S. Giménez and J. Bisquert, Springer International Publishing, Cham, 2016, pp. 41–104 DOI:10.1007/978-3-319-29641-8_2.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Electrolysis of water on (oxidized) metal surfaces, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS.
- A. Vojvodic and J. K. Nørskov, New design paradigm for heterogeneous catalysts, Natl. Sci. Rev., 2015, 2, 140–143 CrossRef CAS.
- X. Rong, J. Parolin and A. M. Kolpak, A Fundamental Relationship between Reaction Mechanism and Stability in Metal Oxide Catalysts for Oxygen Evolution, ACS Catal., 2016, 6, 1153–1158 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- A. Grimaud, A. Demortière, M. Saubanère, W. Dachraoui, M. Duchamp, M.-L. Doublet and J.-M. Tarascon, Activation of surface oxygen sites on an iridium-based model catalyst for the oxygen evolution reaction, Nat. Energy, 2016, 2, 16189 CrossRef.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- M. T. M. Koper, Theory of multiple proton–electron transfer reactions and its implications for electrocatalysis, Chem. Sci., 2013, 4, 2710–2723 RSC.
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. C. Fisher, F. Cheng, X. Wang, H. Zhang and Z. J. Xu, Enlarged Co–O Covalency in Octahedral Sites Leading to Highly Efficient Spinel Oxides for Oxygen Evolution Reaction, Adv. Mater., 2018, 30, 1802912 CrossRef.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- O. Kasian, S. Geiger, T. Li, J.-P. Grote, K. Schweinar, S. Zhang, C. Scheu, D. Raabe, S. Cherevko, B. Gault and K. J. J. Mayrhofer, Degradation of iridium oxides via oxygen evolution from the lattice: correlating atomic scale structure with reaction mechanisms, Energy Environ. Sci., 2019, 12, 3548–3555 RSC.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Anionic redox processes for electrochemical devices, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- A. Lončar, D. Escalera-López, S. Cherevko and N. Hodnik, Inter-relationships between Oxygen Evolution and Iridium Dissolution Mechanisms, Angew. Chem., Int. Ed., 2022, 61, e202114437 CrossRef PubMed.
- H. Su, M. A. Soldatov, V. Roldugin and Q. Liu, Platinum single-atom catalyst with self-adjustable valence state for large-current-density acidic water oxidation, eScience, 2022, 2, 102–109 CrossRef.
- G. N. Martelli, R. Ornelas and G. Faita, Deactivation mechanisms of oxygen evolving anodes at high current densities, Electrochim. Acta, 1994, 39, 1551–1558 CrossRef CAS.
- C. Wang, L. Jin, H. Shang, H. Xu, Y. Shiraishi and Y. Du, Advances in engineering RuO2 electrocatalysts towards oxygen evolution reaction, Chin. Chem. Lett., 2021, 32, 2108–2116 CrossRef CAS.
- O. Kasian, J.-P. Grote, S. Geiger, S. Cherevko and K. J. J. Mayrhofer, The Common Intermediates of Oxygen Evolution and Dissolution Reactions during Water Electrolysis on Iridium, Angew. Chem., Int. Ed., 2018, 57, 2488–2491 CrossRef CAS PubMed.
- R. E. Fuentes, J. Farell and J. W. Weidner, Multimetallic Electrocatalysts of Pt, Ru, and Ir Supported on Anatase and Rutile TiO2 for Oxygen Evolution in an Acid Environment, Electrochem. Solid-State Lett., 2011, 14, E5–E7 CrossRef CAS.
- T. Ioroi, N. Kitazawa, K. Yasuda, Y. Yamamoto and H. Takenaka, Iridium Oxide/Platinum Electrocatalysts for Unitized Regenerative Polymer Electrolyte Fuel Cells, J. Electrochem. Soc., 2000, 147, 2018 CrossRef CAS.
- W. H. Carothers and R. Adams, Platinum oxide as a catalyst in the reduction of organic compounds. V. The preparation of primary alcohols by the catalytic hydrogenation of aldehydes1, J. Am. Chem. Soc., 1924, 46, 1675–1683 CrossRef CAS.
- W. H. Lee and H. Kim, Oxidized iridium nanodendrites as catalysts for oxygen evolution reactions, Catal. Commun., 2011, 12, 408–411 CrossRef CAS.
- L. Fu, F. Yang, G. Cheng and W. Luo, Ultrathin Ir nanowires as high-performance electrocatalysts for efficient water splitting in acidic media, Nanoscale, 2018, 10, 1892–1897 RSC.
- L. Fu, X. Zeng, C. Huang, P. Cai, G. Cheng and W. Luo, Ultrasmall Ir nanoparticles for efficient acidic electrochemical water splitting, Inorg. Chem. Front., 2018, 5, 1121–1125 RSC.
- Y. Pi, N. Zhang, S. Guo, J. Guo and X. Huang, Ultrathin Laminar Ir Superstructure as Highly Efficient Oxygen Evolution Electrocatalyst in Broad pH Range, Nano Lett., 2016, 16, 4424–4430 CrossRef CAS PubMed.
- J. Lim, D. Park, S. S. Jeon, C.-W. Roh, J. Choi, D. Yoon, M. Park, H. Jung and H. Lee, Ultrathin IrO2 Nanoneedles for Electrochemical Water Oxidation, Adv. Funct. Mater., 2018, 28, 1704796 CrossRef.
- H. Su, V. Linkov and B. J. Bladergroen, Membrane electrode assemblies with low noble metal loadings for hydrogen production from solid polymer electrolyte water electrolysis, Int. J. Hydrog. Energy, 2013, 38, 9601–9608 CrossRef CAS.
- B. Jiang, J. Kim, Y. Guo, K. C. W. Wu, S. M. Alshehri, T. Ahamad, N. Alhokbany, J. Henzie and Y. Yamachi, Efficient oxygen evolution on mesoporous IrOx nanosheets, Catal. Sci. Technol., 2019, 9, 3697–3702 RSC.
- H. Yu, N. Danilovic, Y. Wang, W. Willis, A. Poozhikunnath, L. Bonville, C. Capuano, K. Ayers and R. Maric, Nano-size IrOx catalyst of high activity and stability in PEM water electrolyzer with ultra-low iridium loading, Appl. Catal., B, 2018, 239, 133–146 CrossRef CAS.
- S. Chen, T. Luo, K. Chen, Y. Lin, J. Fu, K. Liu, C. Cai, Q. Wang, H. Li, X. Li, J. Hu, H. Li, M. Zhu and M. Liu, Chemical Identification of Catalytically Active Sites on Oxygen-doped Carbon Nanosheet to Decipher the High Activity for Electro-synthesis Hydrogen Peroxide, Angew. Chem., Int. Ed., 2021, 60, 16607–16614 CrossRef CAS PubMed.
- S. Siracusano, N. Van Dijk, E. Payne-Johnson, V. Baglio and A. S. Aricò, Nanosized IrOx and IrRuOx electrocatalysts for the O2 evolution reaction in PEM water electrolysers, Appl. Catal., B, 2015, 164, 488–495 CrossRef CAS.
- T. Audichon, E. Mayousse, S. Morisset, C. Morais, C. Comminges, T. W. Napporn and K. B. Kokoh, Electroactivity of RuO2–IrO2 mixed nanocatalysts toward the oxygen evolution reaction in a water electrolyzer supplied by a solar profile, Int. J. Hydrog. Energy, 2014, 39, 16785–16796 CrossRef CAS.
- H. Lv, S. Wang, J. Li, C. Shao, W. Zhou, X. Shen, M. Xue and C. Zhang, Self-assembled RuO2@IrOx core-shell nanocomposite as high efficient anode catalyst for PEM water electrolyzer, Appl. Surf. Sci., 2020, 514, 145943 CrossRef CAS.
- S. Du, R. Chen, W. Chen, H. Gao, J. Jia, Z. Xiao, C. Xie, H. Li, L. Tao, J. Huo, Y. Wang and S. Wang, Activation of iridium site by anchoring ruthenium atoms on defects for efficient anodic catalyst in polymer electrolyte membrane water electrolyzers, J. Energy Chem., 2022, 75, 260–266 CrossRef CAS.
- C. Spöri, L. J. Falling, M. Kroschel, C. Brand, A. Bonakdarpour, S. Kühl, D. Berger, M. Gliech, T. E. Jones, D. P. Wilkinson and P. Strasser, Molecular Analysis of the Unusual Stability of an IrNbOx Catalyst for the Electrochemical Water Oxidation to Molecular Oxygen (OER), ACS Appl. Mater. Interfaces, 2021, 13, 3748–3761 CrossRef.
- M. Elmaalouf, M. Odziomek, S. Duran, M. Gayrard, M. Bahri, C. Tard, A. Zitolo, B. Lassalle-Kaiser, J. Y. Piquemal, O. Ersen, C. Boissiere, C. Sanchez, M. Giraud, M. Faustini and J. Peron, The origin of the high electrochemical activity of pseudo-amorphous iridium oxides, Nat. Commun., 2021, 12, 3935 CrossRef CAS PubMed.
- S. D. Ghadge, O. I. Velikokhatnyi, M. K. Datta, P. M. Shanthi, S. Tan, K. Damodaran and P. N. Kumta, Experimental and Theoretical Validation of High Efficiency and Robust Electrocatalytic Response of One-Dimensional (1D) (Mn,Ir)O2:10F Nanorods for the Oxygen Evolution Reaction in PEM-Based Water Electrolysis, ACS Catal., 2019, 9, 2134–2157 CrossRef CAS.
- G. Jiang, H. Yu, J. Hao, J. Chi, Z. Fan, D. Yao, B. Qin and Z. Shao, An effective oxygen electrode based on Ir0.6Sn0.4O2 for PEM water electrolyzers, J. Energy Chem., 2019, 39, 23–28 CrossRef.
- K. Kadakia, M. K. Datta, O. I. Velikokhatnyi, P. Jampani, S. K. Park, P. Saha, J. A. Poston, A. Manivannan and P. N. Kumta, Novel (Ir,Sn,Nb)O2 anode electrocatalysts with reduced noble metal content for PEM based water electrolysis, Int. J. Hydrog. Energy, 2012, 37, 3001–3013 CrossRef CAS.
- K. Kadakia, M. K. Datta, O. I. Velikokhatnyi, P. H. Jampani and P. N. Kumta, Fluorine doped (Ir,Sn,Nb)O2 anode electro-catalyst for oxygen evolution via PEM based water electrolysis, Int. J. Hydrog. Energy, 2014, 39, 664–674 CrossRef CAS.
- J. He, G. Fu, J. Zhang, P. Xu and J. Sun, Multistage Electron Distribution Engineering of Iridium Oxide by Codoping W and Sn for Enhanced Acidic Water Oxidation Electrocatalysis, Small, 2022, 18, e2203365 CrossRef PubMed.
- S. Hao, H. Sheng, M. Liu, J. Huang, G. Zheng, F. Zhang, X. Liu, Z. Su, J. Hu, Y. Qian, L. Zhou, Y. He, B. Song, L. Lei, X. Zhang and S. Jin, Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers, Nat. Nanotechnol., 2021, 16, 1371–1377 CrossRef CAS PubMed.
- S. R. Chemler and M. T. Bovino, Catalytic Aminohalogenation of Alkenes and Alkynes, ACS Catal., 2013, 3, 1076–1091 CrossRef CAS PubMed.
- R. D. Cowling and H. E. Hintermann, The Corrosion of Titanium Carbide, J. Electrochem. Soc., 1970, 117, 1447 CrossRef CAS.
- M. Bernt and H. A. Gasteiger, Influence of Ionomer Content in IrO2/TiO2Electrodes on PEM Water Electrolyzer Performance, J. Electrochem. Soc., 2016, 163, F3179–F3189 CrossRef CAS.
- X. Min, Y. Shi, Z. Lu, L. Shen, T. O. Ogundipe, P. Gupta, C. Wang, C. Guo, Z. Wang, H. Tan, S. Mukerjee and C. Yan, High performance and cost-effective supported IrOx catalyst for proton exchange membrane water electrolysis, Electrochim. Acta, 2021, 385, 138391 CrossRef CAS.
- E.-J. Kim, J. Shin, J. Bak, S. J. Lee, K. H. Kim, D. Song, J. Roh, Y. Lee, H. Kim, K.-S. Lee and E. Cho, Stabilizing role of Mo in TiO2-MoOx supported Ir catalyst toward oxygen evolution reaction, Appl. Catal., B, 2021, 280, 119433 CrossRef CAS.
- C. Yang, G. Rousse, K. Louise Svane, P. E. Pearce, A. M. Abakumov, M. Deschamps, G. Cibin, A. V. Chadwick, D. A. Dalla Corte, H. Anton Hansen, T. Vegge, J. M. Tarascon and A. Grimaud, Cation insertion to break the activity/stability relationship for highly active oxygen evolution reaction catalyst, Nat. Commun., 2020, 11, 1378 CrossRef CAS PubMed.
- X. Shi, H.-J. Peng, T. J. P. Hersbach, Y. Jiang, Y. Zeng, J. Baek, K. T. Winther, D. Sokaras, X. Zheng and M. Bajdich, Efficient and Stable Acidic Water Oxidation Enabled by Low-Concentration, High-Valence Iridium Sites, ACS Energy Lett., 2022, 7, 2228–2235 CrossRef CAS.
- Z. Shi, J. Li, J. Jiang, Y. Wang, X. Wang, Y. Li, L. Yang, Y. Chu, J. Bai, J. Yang, J. Ni, Y. Wang, L. Zhang, Z. Jiang, C. Liu, J. Ge and W. Xing, Enhanced Acidic Water Oxidation by Dynamic Migration of Oxygen Species at the Ir/Nb(2)O(5-x) Catalyst/Support Interfaces, Angew. Chem., Int. Ed., 2022, 61, e202212341 CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- C. Shang, C. Cao, D. Yu, Y. Yan, Y. Lin, H. Li, T. Zheng, X. Yan, W. Yu, S. Zhou and J. Zeng, Electron Correlations Engineer Catalytic Activity of Pyrochlore Iridates for Acidic Water Oxidation, Adv. Mater., 2019, 31, 1805104 CrossRef.
- K.-X. Zhang, Y.-C. Shen, K. Sun, L. Shi, H. Chen, K.-Y. Zheng, X.-X. Zou and X. Liang, Perovskite-Type Water Oxidation Electrocatalysts, J. Electrochem., 2022, 28, 2214004 Search PubMed.
- O. Diaz-Morales, S. Raaijman, R. Kortlever, P. J. Kooyman, T. Wezendonk, J. Gascon, W. T. Fu and M. T. M. Koper, Iridium-based double perovskites for efficient water oxidation in acid media, Nat. Commun., 2016, 7, 12363 CrossRef CAS.
- L. Yang, G. Yu, X. Ai, W. Yan, H. Duan, W. Chen, X. Li, T. Wang, C. Zhang, X. Huang, J.-S. Chen and X. Zou, Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing IrO6 octahedral dimers, Nat. Commun., 2018, 9, 5236 CrossRef CAS PubMed.
- L. Yang, K. Zhang, H. Chen, L. Shi, X. Liang, X. Wang, Y. Liu, Q. Feng, M. Liu and X. Zou, An ultrathin two-dimensional iridium-based perovskite oxide electrocatalyst with highly efficient {001} facets for acidic water oxidation, J. Energy Chem., 2022, 66, 619–627 CrossRef CAS.
- X. Liang, L. Shi, Y. Liu, H. Chen, R. Si, W. Yan, Q. Zhang, G.-D. Li, L. Yang and X. Zou, Activating Inert, Nonprecious Perovskites with Iridium Dopants for Efficient Oxygen Evolution Reaction under Acidic Conditions, Angew. Chem., Int. Ed., 2019, 58, 7631–7635 CrossRef CAS PubMed.
- X. Liang, L. Shi, R. Cao, G. Wan, W. Yan, H. Chen, Y. Liu and X. Zou, Perovskite-Type Solid Solution Nano-Electrocatalysts Enable Simultaneously Enhanced Activity and Stability for Oxygen Evolution, Adv. Mater., 2020, 32, 2001430 CrossRef CAS PubMed.
- H. Chen, L. Shi, X. Liang, L. Wang, T. Asefa and X. Zou, Optimization of Active Sites via Crystal Phase, Composition, and Morphology for Efficient Low-Iridium Oxygen Evolution Catalysts, Angew. Chem., Int. Ed., 2020, 59, 19654–19658 CrossRef CAS PubMed.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, Recent Advances and Prospective in Ruthenium-Based Materials for Electrochemical Water Splitting, ACS Catal., 2019, 9, 9973–10011 CrossRef CAS.
- R. Kötz, S. Stucki, D. Scherson and D. M. Kolb, In-situ identification of RuO4 as the corrosion product during oxygen evolution on ruthenium in acid media, J. Electroanal. Chem. Interfacial Electrochem., 1984, 172, 211–219 CrossRef.
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy, H. Chen and X. Zou, Status and perspectives of key materials for PEM electrolyzer, Nano Research, Energy, 2022, 1, e9120032 Search PubMed.
- F. Zhou, L. Zhang, J. Li, Q. Wang, Y. Chen, H. Chen, G. Lu, G. Chen, H. Jin, S. Wang and J. Wang, Novel engineering of ruthenium-based electrocatalysts for acidic water oxidation: A mini review, Eng. Rep., 2021, 3, e12437 CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Recent Progress in Advanced Electrocatalyst Design for Acidic Oxygen Evolution Reaction, Adv. Mater., 2021, 33, 2004243 CrossRef CAS PubMed.
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Dopants fixation of Ruthenium for boosting acidic oxygen evolution stability and activity, Nat. Commun., 2020, 11, 5368 CrossRef CAS PubMed.
- M. A. Hubert, A. M. Patel, A. Gallo, Y. Liu, E. Valle, M. Ben-Naim, J. Sanchez, D. Sokaras, R. Sinclair, J. K. Nørskov, L. A. King, M. Bajdich and T. F. Jaramillo, Acidic Oxygen Evolution Reaction Activity–Stability Relationships in Ru-Based Pyrochlores, ACS Catal., 2020, 10, 12182–12196 CrossRef CAS.
- S. Hirai, T. Ohno, R. Uemura, T. Maruyama, M. Furunaka, R. Fukunaga, W.-T. Chen, H. Suzuki, T. Matsuda and S. Yagi, Ca1−xSrxRuO3 perovskite at the metal–insulator boundary as a highly active oxygen evolution catalyst, J. Mater. Chem. A, 2019, 7, 15387–15394 RSC.
- M. Retuerto, L. Pascual, F. Calle-Vallejo, P. Ferrer, D. Gianolio, A. G. Pereira, Á. García, J. Torrero, M. T. Fernández-Díaz, P. Bencok, M. A. Peña, J. L. G. Fierro and S. Rojas, Na-doped ruthenium perovskite electrocatalysts with improved oxygen evolution activity and durability in acidic media, Nat. Commun., 2019, 10, 2041 CrossRef PubMed.
- J. Kim, P.-C. Shih, K.-C. Tsao, Y.-T. Pan, X. Yin, C.-J. Sun and H. Yang, High-Performance Pyrochlore-Type Yttrium Ruthenate Electrocatalyst for Oxygen Evolution Reaction in Acidic Media, J. Am. Chem. Soc., 2017, 139, 12076–12083 CrossRef CAS PubMed.
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen, T.-W. Song, Z. Yin, H.-W. Liang, S. Zhang, G. Wang and H. Wang, Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis, Nat. Mater., 2023, 22, 100–108 CrossRef CAS PubMed.
- J. Gao, H. Tao and B. Liu, Progress of Nonprecious-Metal-Based Electrocatalysts for Oxygen Evolution in Acidic Media, Adv. Mater., 2021, 33, 2003786 CrossRef CAS PubMed.
- Z. Wang, Y.-R. Zheng, I. Chorkendorff and J. K. Nørskov, Acid-Stable Oxides for Oxygen Electrocatalysis, ACS Energy Lett., 2020, 5, 2905–2908 CrossRef CAS.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Design of template-stabilized active and earth-abundant oxygen evolution catalysts in acid, Chem. Sci., 2017, 8, 4779–4794 RSC.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Toward an Active and Stable Catalyst for Oxygen Evolution in Acidic Media: Ti-Stabilized MnO2, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- P. P. Patel, M. K. Datta, O. I. Velikokhatnyi, R. Kuruba, K. Damodaran, P. Jampani, B. Gattu, P. M. Shanthi, S. S. Damle and P. N. Kumta, Noble metal-free bifunctional oxygen evolution and oxygen reduction acidic media electro-catalysts, Sci. Rep., 2016, 6, 28367 CrossRef CAS PubMed.
- S. Pan, H. Li, D. Liu, R. Huang, X. Pan, D. Ren, J. Li, M. Shakouri, Q. Zhang, M. Wang, C. Wei, L. Mai, B. Zhang, Y. Zhao, Z. Wang, M. Graetzel and X. Zhang, Efficient and stable noble-metal-free catalyst for acidic water oxidation, Nat. Commun., 2022, 13, 2294 CrossRef CAS PubMed.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Stable Potential Windows for Long-Term Electrocatalysis by Manganese Oxides Under Acidic Conditions, Angew. Chem., Int. Ed., 2019, 58, 5054–5058 CrossRef CAS PubMed.
- J. Huang, H. Sheng, R. D. Ross, J. Han, X. Wang, B. Song and S. Jin, Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid, Nat. Commun., 2021, 12, 3036 CrossRef CAS PubMed.
- J. Wu, M. Liu, K. Chatterjee, K. P. Hackenberg, J. Shen, X. Zou, Y. Yan, J. Gu, Y. Yang, J. Lou and P. M. Ajayan, Exfoliated 2D Transition Metal Disulfides for Enhanced Electrocatalysis of Oxygen Evolution Reaction in Acidic Medium, Adv. Mater. Interfaces, 2016, 3, 1500669 CrossRef.
- Y. Yang, H. Yao, Z. Yu, S. M. Islam, H. He, M. Yuan, Y. Yue, K. Xu, W. Hao, G. Sun, H. Li, S. Ma, P. Zapol and M. G. Kanatzidis, Hierarchical Nanoassembly of MoS2/Co9S8/Ni3S2/Ni as a Highly Efficient Electrocatalyst for Overall Water Splitting in a Wide pH Range, J. Am. Chem. Soc., 2019, 141, 10417–10430 CrossRef CAS PubMed.
- J. S. Mondschein, K. Kumar, C. F. Holder, K. Seth, H. Kim and R. E. Schaak, Intermetallic Ni2Ta Electrocatalyst for the Oxygen Evolution Reaction in Highly Acidic Electrolytes, Inorg. Chem., 2018, 57, 6010–6015 CrossRef CAS PubMed.
- A. Buttler and H. Spliethoff, Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- Z. Shi, J. Li, J. Jiang, Y. Wang, X. Wang, Y. Li, L. Yang, Y. Chu, J. Bai, J. Yang, J. Ni, Y. Wang, L. Zhang, Z. Jiang, C. Liu, J. Ge and W. Xing, Enhanced Acidic Water Oxidation by Dynamic Migration of Oxygen Species at the Ir/Nb2O5−x Catalyst/Support Interfaces, Angew. Chem., Int. Ed., 2022, 61, e202212341 CAS.
- J. Xu, Z. Lian, B. Wei, Y. Li, O. Bondarchuk, N. Zhang, Z. Yu, A. Araujo, I. Amorim, Z. Wang, B. Li and L. Liu, Strong Electronic Coupling between Ultrafine Iridium–Ruthenium Nanoclusters and Conductive, Acid-Stable Tellurium Nanoparticle Support for Efficient and Durable Oxygen Evolution in Acidic and Neutral Media, ACS Catal., 2020, 10, 3571–3579 CrossRef CAS.
- Z. Fan, Y. Ji, Q. Shao, S. Geng, W. Zhu, Y. Liu, F. Liao, Z. Hu, Y.-C. Chang, C.-W. Pao, Y. Li, Z. Kang and M. Shao, Extraordinary acidic oxygen evolution on new phase 3R-iridium oxide, Joule, 2021, 5, 3221–3234 CrossRef CAS.
- J. Schröder, V. A. Mints, A. Bornet, E. Berner, M. Fathi Tovini, J. Quinson, G. K. Wiberg, F. Bizzotto, H. A. El-Sayed and M. Arenz, The gas diffusion electrode setup as straightforward testing device for proton exchange membrane water electrolyzer catalysts, JACS Au, 2021, 1, 247–251 CrossRef PubMed.
- M. P. Browne, J. Dodwell, F. Novotny, S. Jaśkaniec, P. R. Shearing, V. Nicolosi, D. J. Brett and M. Pumera, Oxygen evolution catalysts under proton exchange membrane conditions in a conventional three electrode cell vs. electrolyser device: a comparison study and a 3D-printed electrolyser for academic labs, J. Mater. Chem. A, 2021, 9, 9113–9123 RSC.
- M. Bernt, A. Hartig-Weiß, M. F. Tovini, H. A. El-Sayed, C. Schramm, J. Schröter, C. Gebauer and H. A. Gasteiger, Current Challenges in Catalyst Development for PEM Water Electrolyzers, Chem. Ing. Tech., 2020, 92, 31–39 CrossRef CAS.
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy and H. Chen, Status and perspectives of key materials for PEM electrolyzer, Nano Res, Energy, 2022, 1, e9120032 Search PubMed.
- H. A. El-Sayed, A. Weiß, L. F. Olbrich, G. P. Putro and H. A. Gasteiger, OER catalyst stability investigation using RDE technique: a stability measure or an artifact?, J. Electrochem. Soc., 2019, 166, F458 CrossRef CAS.
- J. Knöppel, M. Möckl, D. Escalera-López, K. Stojanovski, M. Bierling, T. Böhm, S. Thiele, M. Rzepka and S. Cherevko, On the limitations in assessing stability of oxygen evolution catalysts using aqueous model electrochemical cells, Nat. Commun., 2021, 12, 2231 CrossRef PubMed.
- K. Ehelebe, D. Escalera-López and S. Cherevko, Limitations of aqueous model systems in the stability assessment of electrocatalysts for oxygen reactions in fuel cell and electrolyzers, Curr. Opin. Electrochem., 2021, 29, 100832 CrossRef.
- C. Wang, W. Chen, D. Yuan, S. Qian, D. Cai, J. Jiang and S. Zhang, Tailoring the nanostructure and electronic configuration of metal phosphides for efficient electrocatalytic oxygen evolution reactions, Nano Energy, 2020, 69, 104453 CrossRef CAS.
- Y. Wang, M. Qiao, Y. Li and S. Wang, Tuning surface electronic configuration of NiFe LDHs nanosheets by introducing cation vacancies (Fe or Ni) as highly efficient electrocatalysts for oxygen evolution reaction, Small, 2018, 14, 1800136 CrossRef PubMed.
- S. Hao, H. Sheng, M. Liu, J. Huang, G. Zheng, F. Zhang, X. Liu, Z. Su, J. Hu and Y. Qian, Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers, Nat. Nanotechnol., 2021, 16, 1371–1377 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan and P. Strasser, Electrocatalytic oxygen evolution reaction (OER) on Ru, Ir, and Pt catalysts: a comparative study of nanoparticles and bulk materials, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- G. Li, S. Li, M. Xiao, J. Ge, C. Liu and W. Xing, Nanoporous IrO2 catalyst with enhanced activity and durability for water oxidation owing to its micro/mesoporous structure, Nanoscale, 2017, 9, 9291–9298 RSC.
- H. Wendt, Preparation, morphology and effective electrocatalytic activity of gas evolving and gas consuming electrodes, Electrochim. Acta, 1994, 39, 1749–1756 CrossRef CAS.
- K. A. Lewinski, D. van der Vliet and S. M. Luopa, NSTF Advances for PEM Electrolysis - the Effect of Alloying on Activity of NSTF Electrolyzer Catalysts and Performance of NSTF Based PEM Electrolyzers, ECS Trans., 2015, 69, 893 CrossRef CAS.
- H. Lv, G. Zhang, C. Hao, C. Mi, W. Zhou, D. Yang, B. Li and C. Zhang, Activity of IrO2 supported on tantalum-doped TiO2 electrocatalyst for solid polymer electrolyte water electrolyzer, RSC Adv., 2017, 7, 40427–40436 RSC.
- C. Daiane Ferreira da Silva, F. Claudel, V. Martin, R. Chattot, S. Abbou, K. Kumar, I. Jiménez-Morales, S. Cavaliere, D. Jones and J. Rozière, Oxygen evolution reaction activity and stability benchmarks for supported and unsupported IrOx electrocatalysts, ACS Catal., 2021, 11, 4107–4116 CrossRef CAS.
- S. Zhao, A. Stocks, B. Rasimick, K. More and H. Xu, Highly active, durable dispersed iridium nanocatalysts for PEM water electrolyzers, J. Electrochem. Soc., 2018, 165, F82 CrossRef CAS.
- M. Inaba, A. W. Jensen, G. W. Sievers, M. Escudero-Escribano, A. Zana and M. Arenz, Benchmarking high surface area electrocatalysts in a gas diffusion electrode: measurement of oxygen reduction activities under realistic conditions, Energy Environ. Sci., 2018, 11, 988–994 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2023 |