DOI:
10.1039/D3RA00209H
(Paper)
RSC Adv., 2023,
13, 9119-9127
Separation of flavonoids with significant biological activity from Acacia mearnsii leaves†
Received
11th January 2023
, Accepted 10th March 2023
First published on 20th March 2023
Abstract
Acacia mearnsii leaves, which are a rich source of flavonoids, were used to separate and purify myricitrin (W3) and myricetin-3-O-glucoside (W1). Further, the antioxidant and hypoglycemic activities of the two purified flavonoids were evaluated. The flavonoids were separated using solvent partition, macroporous adsorbent resin column, and Sephadex column chromatography, and purified using preparative reverse-phase high-performance liquid chromatography (HPLC). The purified flavonoids were characterized using HPLC, mass spectrometry, and nuclear magnetic resonance methods. A high yield (7.3 mg g−1 of crude extract) of W3 was obtained, with a high purity of 98.4%. Furthermore, the purity of W1 was over 95%. W1 and W3 showed strong antioxidant activity and significantly inhibited α-glucosidase. W3 also demonstrated substantial α-amylase inhibitory capacity. This study indicated that A. mearnsii leaves, which are discarded in significant amounts, can be used as a source of myricitrin, thus providing more adequate material for the production of antioxidants and type II diabetes inhibitors. Hence, A. mearnsii leaves have the potential to create great market economic value and environmental benefits.
1. Introduction
The Acacia mearnsii De Wild (common name: Black Wattle) is a perennial and spreading tree of the legume family. It is an easily transplanted, fast-growing, and highly adaptable native tree of southeast Australia1,2 and is now found in North and South America, Asia, Europe, and Africa.3 A. mearnsii is the only temperate species of Acacia that is grown commercially on a significant international scale.4 The bark of A. mearnsii contains high-quality and abundant tannins, and traditionally, the bark and trunk of A. mearnsii are used for tannin extraction, whereas the residue is applied to refine tar.5,6 Since A. mearnsii provides corrosion-resistant and high-density wood, it is used in producing furniture, charcoal, woodchip, and paper.4,7 Although A. mearnsii wood is used for producing several products, significant amounts of A. mearnsii leaves are typically discarded due to the lack of suitable processing methods, causing huge waste production and the proliferation of pests and diseases.8 Hence, developing strategies to utilize A. mearnsii leaves is important for economic as well as environmental benefits.
Previous studies have isolated the following seven flavonoids from A. mearnsii leaves: myricitrin, quercetin, catechin, gallocatechin, isoquercitrin,9,10 mearnsetin, and myricetin-4′-methyl ether-3-O-rhamnoside (mearnsitrin).11 In addition, electrospray ionization mass spectrometry (ESI-MS) analysis revealed the presence of a variety of flavonoids and proanthocyanidins in A. mearnsii leaves.12 Furthermore, A. mearnsii leaves were found to have anti-inflammatory,12 anti-bloat, antioxidant and antimicrobial properties.8
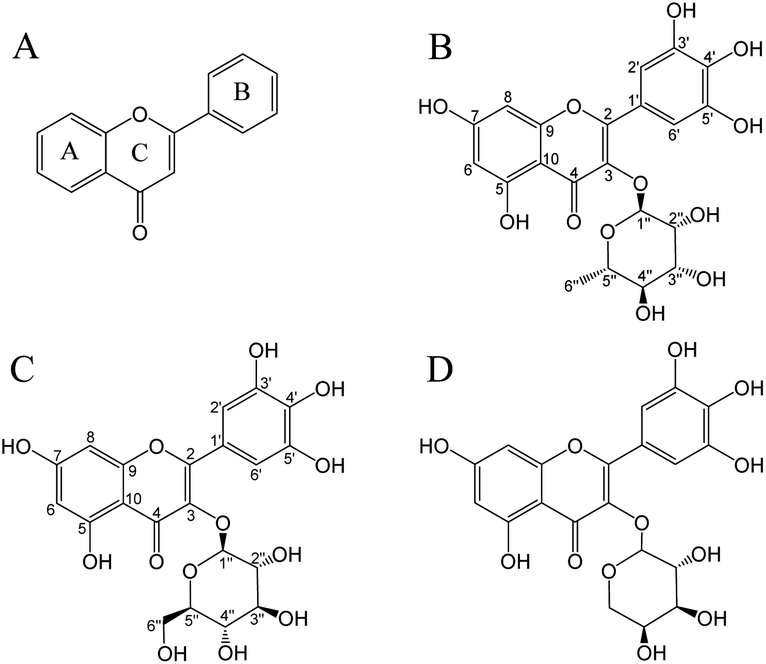
Flavonoids are important polyphenols consisting of two benzene rings linked by a heterocyclic six-membered pyrone ring (Fig. 1A).13 Flavonoids are a type of secondary metabolite naturally found in plants, vegetables, and fruits.14 Myricitrin (Fig. 1B) contains more phenolic hydroxyl groups and hence has a higher free radical scavenging capacity in comparison to other flavonoids such as rhamnosides or quercetin.15 The antioxidant activity of myricitrin was associated with a decreased risk of developing diabetes and fewer complications in patients with type II diabetes.16 Although diabetes complications mainly include nephropathy, retinopathy, and cardiovascular diseases, the most threatening complication is the development of malignant tumors and chronic diseases.17 It was demonstrated that flavonoids may show protective action against type II diabetes through mechanisms related to glucose tolerance and insulin sensitivity.18 In addition, flavonoids have anti-inflammatory,19 anti-cancer,20 anti-cardiovascular,21 anti-microbial,22 anti-SARS-CoV-2,23 and other biological activities.22
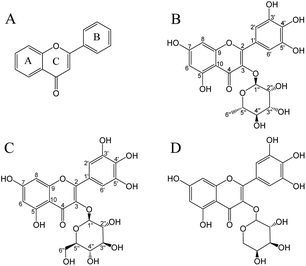 |
| | Fig. 1 Chemical structures of flavonoids (A), myricitrin (B), myricetin-3-O-glucoside (C), and myricetin-3-O-arabinoside (D). | |
This study aimed to establish a method for separating and purifying flavonoids (myricitrin and myricetin-3-O-glucoside; Fig. 1) from A. mearnsii leaves. We evaluated the yield, content, and purity of the isolated flavonoids. Furthermore, the antioxidant activity and hypoglycemic ability of isolated flavonoids were also investigated.
2. Materials and methods
2.1. Materials and chemicals
The analytical grade reagents used in this study include methanol, petroleum ether, dichloromethane, ethyl acetate, ethanol, acetone, formic acid, glacial acetic acid, hydrochloric acid, Tris–HCl, PBS, DMSO, and acetonitrile (HPLC grade). Rutin standard, vitamin C standard, acarbose standard, DPPH, ABTS, and TPTZ were purchased from Sigma-Aldrich (Shanghai, China). The other chemicals were analytical grade. AB-8 macroporous adsorbent resin was purchased from Zhengzhou Hecheng New Material Technology Co., Ltd (Zhengzhou, China). Dextran resin Sephadex LH-20 was purchased from Shanghai Yuanye Biotechnology Co., Ltd (Shanghai, China). The fresh raw A. mearnsii leaves were obtained from Guangxi State-owned Huangmian Forest Farm (Guangxi, China). A. mearnsii leaves were air-dried away from light and finely ground to particles of 40–80 mesh, with a moisture level of 9.17% for reserve.
2.2. Ultrasound-assisted extraction
The powdered leaf (1 kg) was mixed with 40 L of 80% (v/v) aqueous methanol solution and extracted twice in an ultrasonic cleaner (Nanjing Anxiu Instrument Equipment Co., Ltd, Nanjing, China). Each time, the ultrasonic-assisted conditions were as follows: frequency 40 kHz, power 150 W, temperature 60 °C, and time 75 minutes. The solution was filtered twice through qualitative filter paper to remove the residual powder, concentrated using a rotary evaporator at 50 °C, and then lyophilized as the crude extract.
2.3. Determination of total flavonoid content
The total flavonoid content of the samples was determined using the reported AlCl3 method.24 The sample solvent was methanol. In a 96-well microplate, the tested sample (100 μL) was added to 2% AlCl3 solution (100 μL) and placed at 20 °C for 1 h. The absorbance value at 415 nm was subsequently read using a microplate reader (Cytation 3, Vermont, USA). The regression equation of the rutin standard curve was obtained (Fig. S1†), and the concentration was expressed as mg rutin equivalent (mg RE) per g sample dw.
2.4. Solvent partition separation
The crude extract (20 g) was dissolved in deionized water (500 mL) and fractionated sequentially using petroleum ether, dichloromethane, ethyl acetate, and water. The petroleum ether layer (3 × 500 mL) was concentrated under reduced pressure and then lyophilized as L1. The manipulation of the dichloromethane layer (L2, 3 × 500 mL) and ethyl acetate layer (L3, 3 × 500 mL) was similar to the petroleum ether layer. The water layer was lyophilized as L4. All the layers were stored in desiccators.
2.5. Macroporous adsorbent resin column separation
The ethyl acetate fraction was separated using AB-8 macroporous adsorbent resin (0.3–1.25 mm particle size) combined with a rapid liquid preparation chromatography system. The column dimension was 4 cm × 20 cm and the sample mass was 8.0 g per loading. The elution flow rate was 15 mL min−1. The eluate was monitored using an ultraviolet-visible (UV-vis) diode array detector (DAD) at 255 nm and 357 nm. The eluate was collected for each 100 mL and detected by high-performance liquid chromatography (HPLC). Then seven fractions were obtained, including Fr1 (water elution), Fr2 (10% ethanol elution), Fr3 (20% ethanol elution), Fr4 (30% ethanol elution), Fr5 (40% ethanol elution), Fr6 (50% ethanol elution) and Fr7 (100% ethanol elution). The volume of each fraction was approximately 600 mL.
2.6. Sephadex column separation
Fractions Fr3 and Fr4 obtained from AB-8 macroporous adsorbent resin column were separated using Dextran resin Sephadex LH-20 (18–111 μm particle size) in the 4 cm × 20 cm column. After loading the column with 5.0 g of sample, the column was eluted with deionized water, 20% methanol, 40% methanol, 100% methanol, and 60% acetone as mobile phases. The elution flow rate was 8 mL min−1. The eluate was monitored using a UV-vis DAD at 255 nm and 357 nm. Earlier investigations served as the foundation for improving the purifying conditions.12 The effluent was collected for each 50 mL and detected by HPLC, and the same fractions were combined. Each fraction had roughly 350 mL of elution solvent.
2.7. Preparative reverse phase HPLC (RP-HPLC) purification
The fraction BFr3 was purified using a preparative RP-HPLC column: Spherical C18 (40–60 μm, 120 Å, L × I.D. 172.7 mm × 21.3 mm) (Changzhou Santai Technology Co., Ltd, Changzhou, China). Each sample mass for the column was 0.5 g. The temperature was set to 25 °C and the flow rate was 0.3 mL min−1. The detection wavelengths were 255 nm and 357 nm with UV-vis DAD. The mobile phases consisted of 0.1% formic acid in water (phase A) and acetonitrile (phase B). The elution gradient was as follows: 0–10 min, 5–15% B; 10–15 min, 15–20% B; 15–25 min, 20–20% B; 25–35 min, 20–50% B; 35–40 min, 50–5% B. Fractions were collected at 5 mL intervals. If the fractions are detected as identical by HPLC, the fractions are combined.
2.8. Characterization
2.8.1. High performance liquid chromatography (HPLC). The obtained fractions were used for RP-HPLC analysis (Agilent 1260 Series, California, USA), and the sample was categorized according to retention time and peak area. The samples in methanol were filtered and injected (5 μL) into a C18 column (250 mm × 4.6 mm, 5 μm, Agilent Eclipse XDB-C18). The mobile phases were 0.1% formic acid in water (phase A) and acetonitrile (phase B) at a flow rate of 1 mL min−1. The elution gradient was as follows: 0–15 min, 15–30% B; 15–25 min, 30–70% B; 25–30 min, 70–15% B. The detection wavelength of the UV detector was 255 nm and the column temperature was 30 °C.
2.8.2. Ultraviolet-visible absorption spectrometry (UV-vis). The UV absorption characteristics of the sample can be obtained by UV-vis spectrum. The UV-vis spectra of the isolated flavonoids were recorded by scanning in the range of 200–600 nm using a Shimadzu-2450 UV-vis spectrophotometer (Kyoto, Japan).
2.8.3. Mass spectrometry (MS). The determination of molecular masses and structural identification of the isolated flavonoids were carried out in a liquid chromatography-mass spectrometry (LC-MS), and the model is Agilent 1260 UPLC-DAD-6530 ESI-QTOF MS (Agilent, California, USA). MassHunter B0.05.0 workstation was utilized for assay development. The samples were separated using an Agilent Poroshell 120 EC-C18 column (100 mm × 3.0 mm, 2.7 μm). The flow rate was 0.4 mL min−1 and the detection wavelength was 255 nm. The column temperature was 35 °C with an injection volume of 5 μL. The mobile phase consisted of 0.1% formic acid in water (phase A) and acetonitrile (phase B). The elution conditions were as follows: 0–15 min, 15–25% B; 15–25 min, 25–100% B. MS analysis was performed in negative mode. Nebulizer pressure, 50 psi; N2 drying gas flow, 10 mL min−1; N2 drying gas temperature, 350 °C; capillary voltage, 3500 V; fragmentor voltage, 150 V; scanning range, 50–1500 m/z.
2.8.4. Nuclear magnetic resonance spectroscopy (NMR). The flavonoid products (15 mg in 0.55 mL, DMSO) were analyzed using a Bruker Avance III HD NMR spectrometer (Bruker biospin, AG, Switzerland) at 600 MHz. 1H-NMR, 13C-NMR, 1H–1H COSY, and HMBC spectra were recorded. The spectral width of 1H NMR was 0–15 ppm and that of 13C NMR was 0–200 ppm. The deuterated NMR solvent was dimethyl sulfoxide-d6 (DMSO-d6) containing 99.8% atom D, 0.03% (v/v) TMS (trimethylsilane).
2.9. Antioxidant activity
2.9.1. DPPH radical scavenging assay. The 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay was performed the previously described method with minor modifications.25 Fresh 0.2 mM DPPH ethanol solution was prepared, then 160 μL of DPPH ethanol solution and 80 μL of test sample were added to 96-well microplate. Vitamin C was a positive control. The mixture was left in darkness for 30 min, and the absorbance value was measured at 517 nm using a microplate reader. The DPPH radical scavenging rate was calculated by the following formula:| | |
Scavenging activity (%) = (A0 − As)/A0 × 100%
| (1) |
where A0 indicated the absorbance of the blank group, and As indicated the absorbance of the sample group.
2.9.2. ABTS radical scavenging assay. The ABTS stock solution was prepared by combining 7 mM ABTS with 2.45 mM potassium persulfate (K2S2O8) and incubated in dark conditions for 12–16 h.26 The working solution was obtained by diluting with deionized water so that the absorbance value at 734 nm was 0.70 ± 0.02. In a 96-well microplate, ABTS working solution (160 μL) and the tested sample (40 μL) were added. Vitamin C was a positive control. After the reaction for 10 min at room temperature, the absorbance at 734 nm was measured using a microplate reader. The ABTS radical scavenging rate was calculated by the following formula:| | |
Scavenging activity (%) = (A0 − As)/A0 × 100%
| (2) |
where A0 was the absorbance of the blank group, and As was the absorbance of the sample group.
2.9.3. Ferric-reducing antioxidant power (FRAP) assay. Working FRAP reagent was produced by mixing 300 mM sodium acetate buffer (pH 3.6), 10 mM TPTZ (dissolved in 40 mM hydrochloric acid solution) and 20 mM ferric chloride (FeCl3) with a volume ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.27 Vitamin C was a positive control. Working FRAP reagent (150 μL), acetate buffer (30 μL), and sample (20 μL) were added to a 96-well plate and reacted for 10 min at room temperature. Then absorbance value was measured at 593 nm using a microplate reader. Ferrous sulfate replacement sample was used to make the standard curve. The FRAP value was expressed as mmol ferrous sulfate equivalent (mmol FSE) g−1 sample dw.
:1:1.27 Vitamin C was a positive control. Working FRAP reagent (150 μL), acetate buffer (30 μL), and sample (20 μL) were added to a 96-well plate and reacted for 10 min at room temperature. Then absorbance value was measured at 593 nm using a microplate reader. Ferrous sulfate replacement sample was used to make the standard curve. The FRAP value was expressed as mmol ferrous sulfate equivalent (mmol FSE) g−1 sample dw.
2.10. Hypoglycemic capacity
2.10.1. α-Glucosidase inhibition assay. α-Glucosidase inhibition was determined using previous methods.28 α-Glucosidase from Saccharomyces cerevisiae (10.8 U mg−1) was added to 0.1 M sodium phosphate buffer (PBS, pH 6.9) and diluted to 1 U mL−1 of α-glucosidase working solution. Acarbose was a positive control. In a 96-well microplate, sample solvent (50 μL) was added to enzyme working solution (100 μL) and incubated for 10 min at 25 °C. Then, 50 μL of p-nitrophenyl-α-D-glucopyranoside (PNPG; 5 mM in PBS) was added to each well and incubated for 5 min at 25 °C. The absorption at 405 nm was measured using a microplate reader. The inhibitory activity was calculated as follows:| | |
Inhibitory activity (%) = [Ac − (As − A0)]/Ac × 100%
| (3) |
where Ac was the absorbance of the uninhibited enzyme, As was the absorbance of the enzyme treated with the sample, and A0 was the absorbance of the sample with the substrate (no enzyme).
2.10.2. α-Amylase inhibition assay. The methods for α-amylase inhibitory assay were adapted from Kellogg et al.29 The α-amylase from porcine pancreas was dissolved in Tris–HCl buffer (20 mM, pH 6.8) to a concentration of 0.1 mg mL−1. Each well of the 96-well plate was charged with sample (20 μL), soluble starch solution (20 μL, 0.1% w/v starch solution), and incubated for 5 min at 25 °C. 20 μL of the working enzyme solution was charged to each well, and the resulting solution was incubated for 10 min at 25 °C. Last, 100 μL of diluted Lugol's solution (1:1 dilution with deionized water) was added, and the absorbance was read at 595 nm using a microplate reader. As the α-amylase assay above, acarbose was a positive control. The inhibitory activity was calculated as follows:| | |
Inhibitory activity (%) = [(Ac1 − Ac2) − (As1 − As2)]/(Ac1 − Ac2) × 100%
| (4) |
where Ac1 was the absorbance of the blank solvent (no enzyme). Ac2 was the absorbance of the uninhibited enzyme, As1 was the absorbance of the sample with the substrate (no enzyme), and As2 was the absorbance of the enzyme treated with the sample.
3. Results and discussion
3.1. Ultrasound-assisted extraction
Ethyl acetate, ethanol, and methanol are the commonly used solvents for the extraction of flavonoids, among which methanol is considered suitable because it provides better solubility and higher extraction yield of flavonoids.30 To extract flavonoids, 1 kg of air-dried and ground A. mearnsii leaves were mixed with 40 L of an 80% aqueous methanol solution and extracted twice in an ultrasonic cleaner at a temperature of 60 °C for 75 min. The resulting solution was filtered and concentrated to obtain the crude extract (155.8 g; 15.58% of dried A. mearnsii leaves). The total flavonoid content was 68.7 ± 1.3 mg g−1 of crude extract when estimated using the aluminum chloride method.
3.2. Solvent partition separation
In this method, the crude methanolic extract was fractionated sequentially using petroleum ether, dichloromethane, ethyl acetate, and water. The yield of L3 (37.68%) and L4 (48.86%) was significantly higher than that of L1 (6.68%) and L2 (6.78%). Chlorophyll can be readily obtained by extracting plant substances with a wide polarity of organic solvents because the pigment is readily soluble in lipophilic solvents such as petroleum ether, alkanes, and chloroform.31 Hence, petroleum ether and dichloromethane extraction of the crude extract allowed the removal of less polar components, such as chlorophyll, from the crude extract. In contrast, polar compounds containing alcoholic and sugar groups were extracted in the water fraction.32 The flavonoid contents of L1, L2, L3, and L4 fractions were 103.1, 139.9, 94.2, and 34.4 mg g−1, respectively. Further, the ethyl acetate layer had the highest level of biological activity, and hence this layer was used for subsequent separation and purification.
3.3. Macroporous adsorbent resin column separation
Because of the high adsorption and desorption capacity, low cost, and straightforward equipment set-up, macroporous adsorbent resin chromatography is frequently used for the adsorption and enrichment of flavonoids.33 The ethyl acetate partition extract (58.71 g) was separated using an AB-8 macroporous adsorbent resin column (4 cm × 20 cm). Table 1 shows the weight, fraction yield, and flavonoid content of seven obtained fractions (Fr1, Fr2, Fr3, Fr4, Fr5, Fr6, and Fr7). The yields of Fr1, Fr2, Fr3, Fr4, Fr5, Fr6, and Fr7 were 0.66%, 5.08%, 8.06%, 40.84%, 16.11%, 13.13%, and 1.65%, respectively, while the flavonoid content was 7.4, 42.3, 144.3, 129.1, 64.1, 80.4, and 78.9 mg g−1, respectively. The Fr4 fraction had the highest flavonoid content, whereas the flavonoid content of the Fr3 and Fr4 fractions was higher than other fractions. Hence, for the subsequent step, Fr3 and Fr4 fractions were selected.
Table 1 Fraction weight, fraction yield, and flavonoid content after separated by macroporous adsorbent resin columna
| Fractions |
Weight (g) |
Yield (%) |
Flavonoid content (mg RE g−1 dw) |
| Values are means ± SD (n = 3). |
| Fr1 |
0.39 |
0.66 |
7.4 ± 0.6 |
| Fr2 |
2.98 |
5.08 |
42.3 ± 1.1 |
| Fr3 |
4.73 |
8.06 |
144.3 ± 3.6 |
| Fr4 |
23.98 |
40.84 |
129.1 ± 2.2 |
| Fr5 |
9.46 |
16.11 |
64.1 ± 1.3 |
| Fr6 |
7.71 |
13.13 |
80.4 ± 2.7 |
| Fr7 |
0.97 |
1.65 |
78.9 ± 2.4 |
3.4. Sephadex column separation
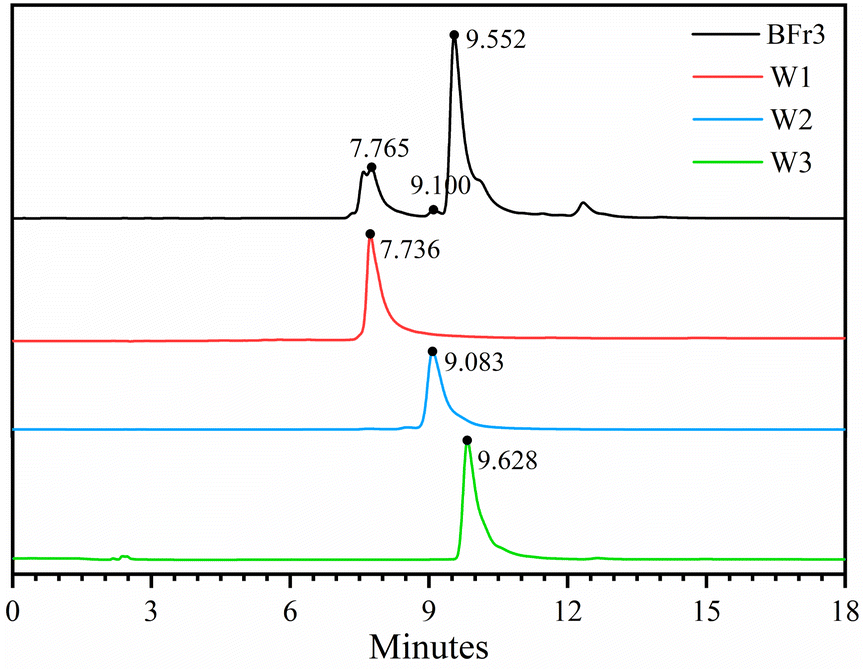
Sephadex LH-20 can separate substances with very similar structures because it combines methods of molecular filtration and adsorption chromatography.34 The Fr3 (4.73 g) and Fr4 (23.98 g) fractions from the previous step were separated using the Sephadex LH-20 column (4 cm × 20 cm). Table 2 shows the weight, yield, and flavonoid content of the five obtained fractions after the chromatography procedure. The yields of BFr1, BFr2, BFr3, BFr4, and BFr5 were 4.81%, 5.64%, 12.50%, 30.72%, and 38.63%, respectively, while the flavonoid content was 32.0, 65.3, 518.0, 75.3, and 57.2 mg g−1, respectively. The BFr3 fraction had a moderate yield, while the flavonoid content was the highest. Hence, the use of the Sephadex LH-20 column allowed for the effective enrichment of flavonoids. HPLC analysis (Fig. 2) of the BFr3 fraction showed that the fraction demonstrated only a few peaks with large areas, indicating a relatively simple composition with high content of each component.
Table 2 Fraction weight, fraction yield, and flavonoid content after separated by Sephadex columna
| Fractions |
Weight (g) |
Yield (%) |
Flavonoid content (mg RE g−1 dw) |
| Values are means ± SD (n = 3). |
| BFr1 |
1.38 |
4.81 |
32.0 ± 1.7 |
| BFr2 |
1.62 |
5.64 |
65.3 ± 2.1 |
| BFr3 |
3.59 |
12.50 |
518.0 ± 5.9 |
| BFr4 |
8.82 |
30.72 |
75.3 ± 1.9 |
| BFr5 |
11.09 |
38.63 |
57.2 ± 1.6 |
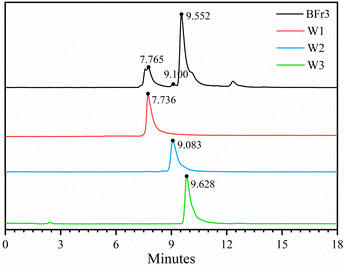 |
| | Fig. 2 HPLC chromatographic spectra of BFr3 fraction and isolated flavonoids: W1, W2, and W3. | |
3.5. Preparative reverse phase HPLC (RP-HPLC) purification
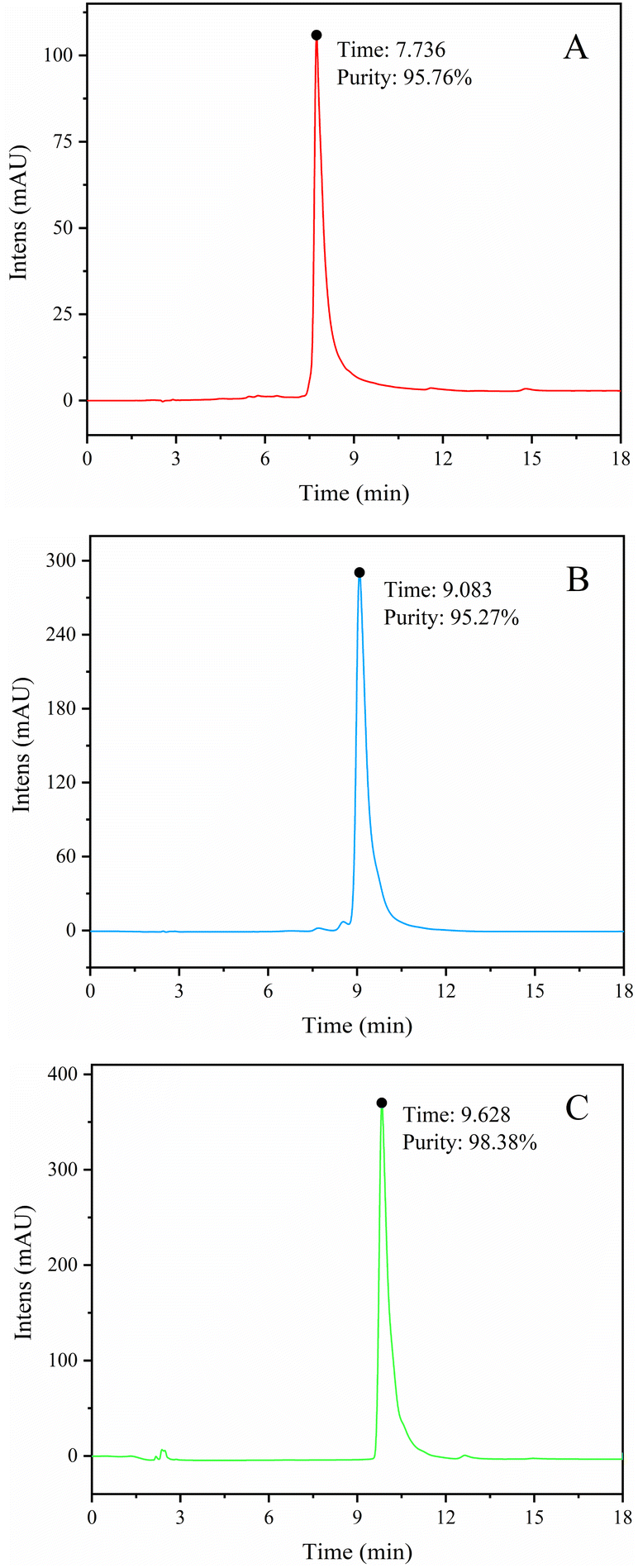
The BFr3 fraction (3.59 g) was further purified using a preparative RP-HPLC column (172.7 mm × 21.3 mm) with elution with a mixture of acetonitrile and deionized water. The flow rate was 5 mL min−1 and the wavelengths of detection were 255 and 357 nm. Peak fractions were collected and confirmed using HPLC, UV-vis, MS, and NMR. Fig. 3 shows the HPLC spectra of three flavonoid-containing fractions (W1, W2, and W3; Table 3). The yields of W1, W2, and W3 were 7.94%, 2.48%, and 31.69%, respectively, whereas the weight-based yields were 1.8, 0.6, and 7.3 mg g−1 of crude extract, respectively. So W3 was the main product. Finally, the purity of W1, W2, and W3 were 95.76%, 95.27%, and 98.38%, respectively.
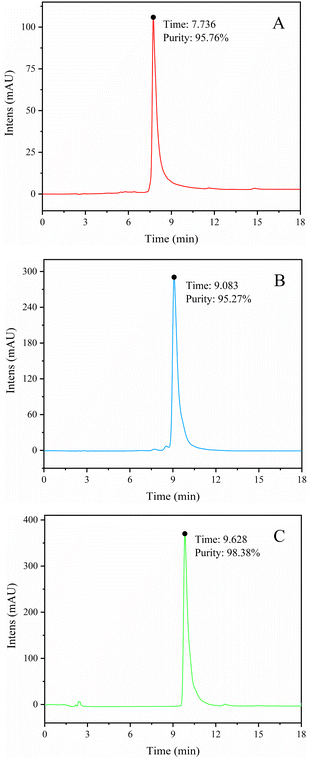 |
| | Fig. 3 HPLC spectra of W1 (myricetin-3-O-glucoside) (A), W2 (myricetin-3-O-arabinoside) (B), and W3 (myricitrin) (C). | |
Table 3 Yield of fractions separated by preparative RP-HPLC, purity, and flavonoid contenta
| Fractions |
Yield (%) |
Purity (%) |
Flavonoid content (mg g−1 crude extract) |
| Values are means ± SD (n = 3). |
| W1 |
7.94 |
95.76 |
1.8 ± 0.2 |
| W2 |
2.48 |
95.27 |
0.6 ± 0.1 |
| W3 |
31.69 |
98.38 |
7.3 ± 0.5 |
The retention of flavonoids in RP-HPLC is related to the polarity of the stationary and mobile phases, as well as the chemical structure of the flavonoids. The retention time is often favorably correlated with the chain length and the number of double bonds; the geometric isomerization of flavonoids also plays a significant role.32 The polarity difference between the three flavonoid-containing samples may be due to the presence of different glycosides at the C-3 position of flavonoids.
3.6. Structural identification
In the UV-vis spectra of flavonoids, B-ring cinnamyl group produces an absorption peak at 300–400 nm; A-ring benzoyl group produces an absorption peak at 220–280 nm. According to Fig. S2,† the UV-vis spectra of W1 (λmax 258.5, 353 nm), W2 (λmax 259, 352 nm), and W3 (λmax 256, 353 nm) were consistent with the UV absorption characteristics of flavonoids. The W3 purified from the BFr3 fraction was identified using ESI-MS and NMR. ESI-MS demonstrated a major parent molecular ion [M + H]− peak at m/z = 463.18 (Fig. S3†). Based on this, the molecular weight of W3 was determined to be 464, and the chemical formula was C21H20O12. The fragmentation peaks of the secondary mass spectrum (MS/MS) had m/z of 316.09 and 179.04; these values were consistent with those of myricitrin. For W1, the major fragment ion [M + H]− peak was obtained at m/z = 479.18 (Fig. S4†), and the molecular weight and chemical formula were 480 and C21H20O13, respectively. Further, MS/MS fragmentation of W1 yielded ions at m/z = 316.10, m/z = 271.09, and m/z = 179.05, representing myricetin-3-O-glucoside based on the database. For W2, the major parent molecular ion peak [M − H]− was at m/z = 449.16 (Fig. S5†), indicating a molecular weight of 450 and the chemical formula of C20H18O12. Fragmentation peaks showed m/z of 316.10, 271.09, and 179.05, revealing W2 as myricetin-3-O-arabinoside (Fig. 1D) based on the database. However, the information from the MS data should be validated by NMR analysis.
The 1H-NMR spectral results of the main product (W3, Fig. S6†) are illustrated in Table 4, revealing spectra (600 MHz in DMSO-d6) δ 6.89 (2H, s, 2′, 6′-H), 6.37 (1H, d, J = 2.1 Hz, 6-H), 6.20 (1H, d, J = 2.1 Hz, 8-H), 5.20 (1H, s, 1′′-H), 3.98 (1H, s, 5′′-H), 3.56–3.54 (1H, dd, J = 9.4, 2.9 Hz, 3′′-H), 3.37–3.34 (1H, m, 2′′-H), 3.17–3.14 (1H, t, J = 9.4 Hz, 4′′-H), 0.84 (3H, d, J = 6.2 Hz, 6′′-H), which were comparable with those reported for myricitrin standard,35 1H-NMR (600 MHz in DMSO-d6) δ 6.89 (2H, s, 2′, 6′-H), 6.37 (1H, d, J = 2.0 Hz, 6-H), 6.20 (1H, d, J = 2.0 Hz, 8-H), 5.20 (1H, s, 1′′-H), 3.78 (1H, s, 2′′-H), 3.99 (1H, s, 5′′-H), 3.55 (1H, dd, J = 9.4, 2.9 Hz, 3′′-H), 3.16 (1H, t, J = 9.4 Hz, 4′′-H), 0.84 (3H, d, J = 6.2 Hz, 6′′-H). Table 5 shows the 13C-NMR spectral results of W3 (Fig. S7†) in comparison to the spectra of the myricitrin standard. Notably, the reference and present studies were conducted at the same instrument MHz and both utilized deuterated NMR solvent (600 MHz, DMSO-d6). In addition, the 1H–1H COSY (Fig. S8†) and HMBC (Fig. S9†) spectra of W3 were compared with the literature,36,37 and the structure was confirmed as myricitrin (myricetin-3-O-α-L-rhamnoside). The 1H-NMR (Fig. S10†), 13C-NMR (Fig. S11†), 1H–1H COSY (Fig. S12†), and HMBC (Fig. S13†) spectra of W1 were compared with the literature,38,39 and W1 was identified as myricetin-3-O-β-D-glucoside. The results demonstrated the excellent purity of W3 and W1 obtained using preparative RP-HPLC.
Table 4 1H-NMR spectral data (1H-NMR) of obtained myricitrin (W3) in comparison to those of the previous report (1H-NMRref)35
| Position |
1H-NMR |
1H-NMRref |
| 6 |
6.37 (1H, d, J = 2.1 Hz) |
6.37 (1H, d, J = 2.0 Hz) |
| 8 |
6.20 (1H, d, J = 2.1 Hz) |
6.20 (1H, d, J = 2.0 Hz) |
| 2′, 6′ |
6.89 (2H, s) |
6.89 (2H, s) |
| 1′′ |
5.20 (1H, s) |
5.20 (1H, s) |
| 2′′ |
3.37–3.34 (1H, m) |
3.78 (1H, s) |
| 3′′ |
3.56–3.54 (1H, dd, J = 9.4, 2.9 Hz) |
3.55 (1H, dd, J = 9.4, 2.9 Hz) |
| 4′′ |
3.17–3.14 (1H, t, J = 9.4 Hz) |
3.16 (1H, t, J = 9.4 Hz) |
| 5′′ |
3.98 (1H, s) |
3.99 (1H, s) |
| 6′′ |
0.84 (3H, d, J = 6.2 Hz) |
0.84 (3H, d, J = 6.2 Hz) |
Table 5 Comparison of 13C-NMR chemical shift between the obtained myricitrin (W3) in this study and the myricitrin ref. 35
| Position |
13C-NMR |
13C-NMRref |
| 2 |
156.9 |
156.9 |
| 3 |
134.7 |
134.7 |
| 4 |
178.3 |
178.2 |
| 5 |
161.8 |
161.8 |
| 6 |
99.1 |
99.1 |
| 7 |
164.6 |
164.6 |
| 8 |
94.0 |
94.0 |
| 9 |
158.0 |
158.0 |
| 10 |
104.5 |
104.5 |
| 1′ |
120.1 |
121.1 |
| 2′ |
108.4 |
116.3 |
| 3′ |
146.2 |
146.2 |
| 4′ |
136.9 |
136.9 |
| 5′ |
146.2 |
146.5 |
| 6′ |
108.4 |
118.3 |
| 1′′ |
102.4 |
102.4 |
| 2′′ |
70.8 |
70.8 |
| 3′′ |
71.0 |
71.0 |
| 4′′ |
71.7 |
71.7 |
| 5′′ |
70.5 |
70.5 |
| 6′′ |
18.0 |
18.0 |
3.7. Antioxidant activity
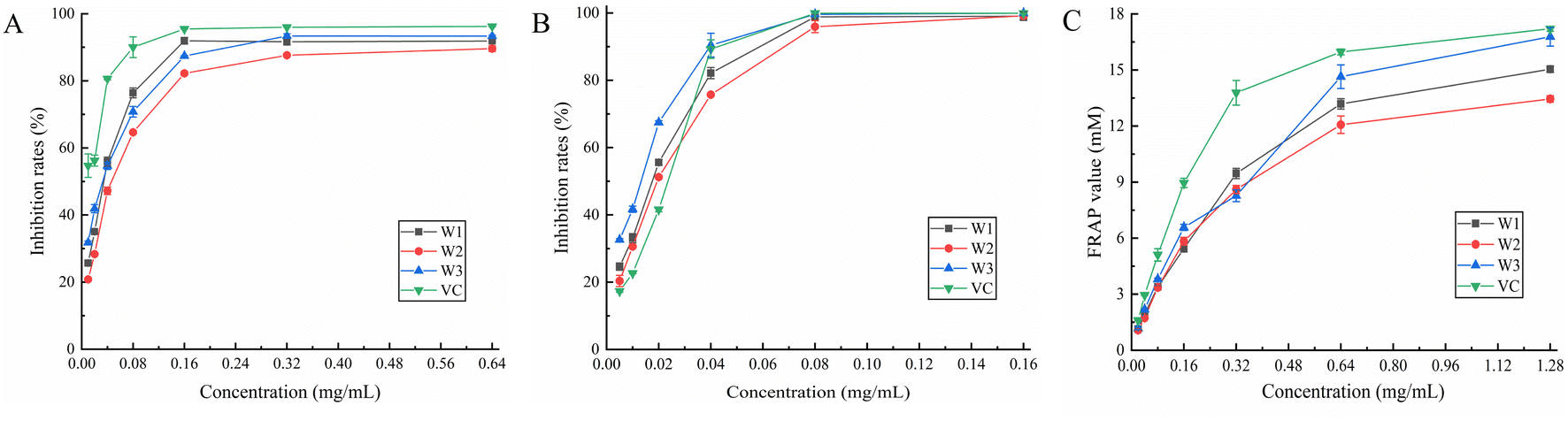
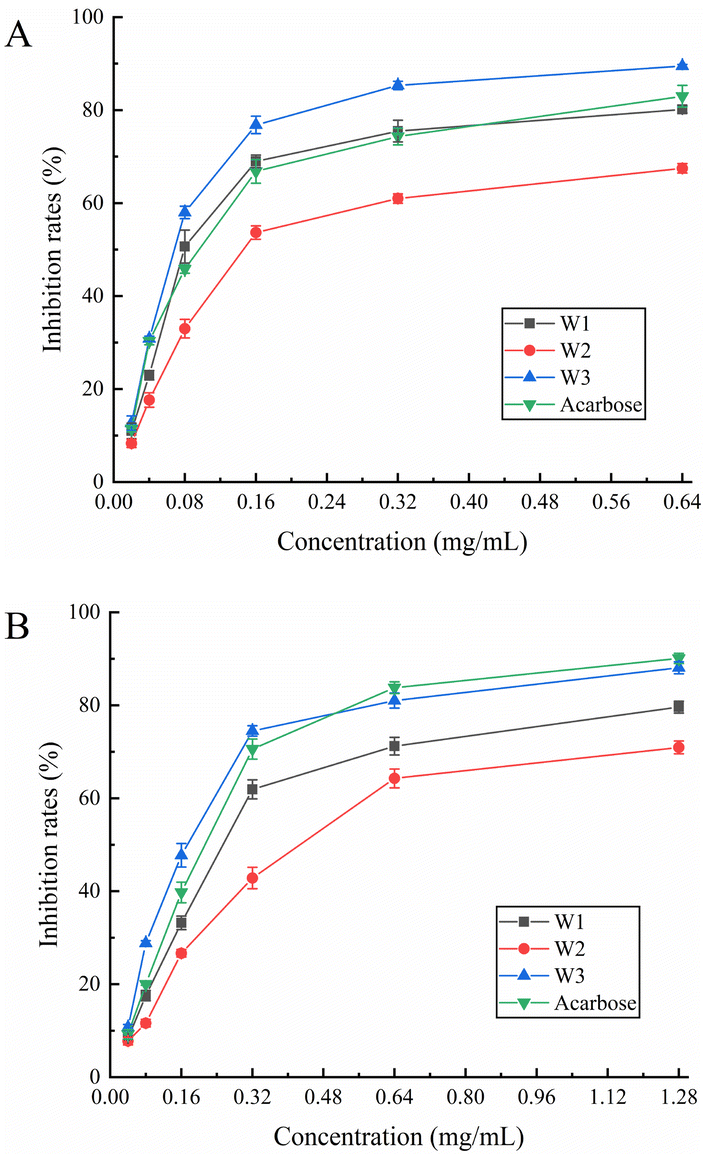
3.7.1. DPPH radical scavenging assay. It is important to assess the antioxidant properties of medicinal plants and plants that can be employed as food additives. It might be an effective technique to assess its overall quality and bioactive component content.40 DPPH is a consistent free radical and may be used to evaluate the antioxidant capability of compounds faster than other approaches.41 Fig. 4A shows the results of the scavenging ability of each compound against DPPH radicals. W1, W2, W3, and vitamin C demonstrated IC50 values of 0.030, 0.044, 0.027, and 0.010 mg mL−1, respectively, for DPPH radicals (Table 6). Furthermore, W1 and W3 showed the strongest scavenging abilities at 0.04–0.16 mg mL−1 and 0.32–0.64 mg mL−1, respectively. After the concentration of 0.16 mg mL−1, the scavenging ability of the three flavonoids against DPPH radicals increased slowly and was slightly weaker than that of the control (vitamin C). Among the three flavonoids, W3 showed the highest scavenging ability, reaching up to 93.34%. Overall, the three flavonoids are potential antioxidants due to their remarkable scavenging ability for DPPH radicals.
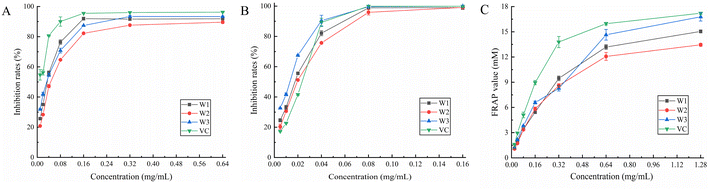 |
| | Fig. 4 Scavenging activity of W1, W2 and W3 on (A) DPPH radical, (B) ABTS radical, and their (C) FRAP value. | |
Table 6 The antioxidant activity, α-glucosidase and α-amylase inhibition of A. mearnsii leaves flavonoid compoundsa
| Compounds |
FRAP value (mmol FSE g−1 dw) |
DPPH radical (IC50, mg mL−1) |
ABTS radical (IC50, mg mL−1) |
α-Glucosidase (IC50, mg mL−1) |
α-Amylase (IC50, mg mL−1) |
| Values are means ± SD (n = 3). Different letters within a column indicate significant differences at p < 0.05. |
| Vitamin C |
17.21 ± 0.36a |
0.010 ± 0.003c |
0.019 ± 0.002a |
— |
— |
| Acarbose |
— |
— |
— |
0.102 ± 0.011b |
0.204 ± 0.017c |
| W1 |
15.04 ± 0.42b |
0.030 ± 0.005b |
0.018 ± 0.001a |
0.104 ± 0.016b |
0.286 ± 0.026b |
| W2 |
13.35 ± 0.46c |
0.044 ± 0.006a |
0.016 ± 0.001ab |
0.199 ± 0.023a |
0.441 ± 0.050a |
| W3 |
16.78 ± 1.31ab |
0.027 ± 0.003b |
0.013 ± 0.001b |
0.074 ± 0.007b |
0.176 ± 0.016c |
3.7.2. ABTS radical scavenging assay. In the presence of a strong oxidizing agent (potassium persulfate), ABTS reacts to create a persistent green radical. However, in the presence of an antioxidant, the formation of green radicals is suppressed. All three flavonoids (W1, W2, and W3) demonstrated dose-dependent ABTS radical scavenging ability (Fig. 4B), with IC50 values of 0.016, 0.018, and 0.013 mg mL−1, respectively (Table 6), which were smaller than that of the control (vitamin C; 0.019 mg mL−1). This indicated that the ABTS radical scavenging capacity was in the following order: W3 > W1 > W2 > vitamin C. Furthermore, the scavenging activity of W1, W2, and W3 for ABTS radicals was greater than that of the control at a dose of 0.005–0.02 mg mL−1. The scavenging capacity of W1, W2, and W3 for ABTS radicals was close to 100% at a concentration of 0.16 mg mL−1, demonstrating that the three flavonoids exhibited high ABTS radical scavenging ability in the experimental concentration range, and the difference between the three was not significant.
3.7.3. Ferric-reducing antioxidant power (FRAP) assay. The FRAP test determines overall reducing capacity by forming a blue-colored complex in the presence of antioxidants, and the change in absorbance at 593 nm directly represents the reducing capacity.42 At 0.32 mg mL−1, the reducing ability of W1 was the strongest among the three flavonoids, while W3 showed the greatest reducing ability in the range of 0.02–0.16 mg mL−1 (Fig. 4C), with a maximum FRAP value of 16.78 (Table 6). The highest FRAP value for W1, W2, and control (vitamin C) was 15.04, 13.35, and 17.21, respectively. In conclusion, all three flavonoids showed strong reducing ability, without any significant difference among them.The radical scavenging activity increases with the number of hydroxyl groups on the B-ring; in particular, C3′-OH is particularly important. The 5, 7 phenolic hydroxyl groups of the A-ring are also necessary for the efficient antioxidative action of flavonoids.43 Further, the antioxidant activity of flavonoids containing o-diphenolic hydroxyl groups is significantly better than that of flavonoids containing m-diphenolic hydroxyl groups in the B-ring.44 It might be because the three flavonoids in this study contained myricetin with differences only at the C-3 position, the antioxidant properties of these three flavonoids were relatively identical to each other.
3.8. Hypoglycemic capacity
3.8.1. α-Glucosidase inhibition assay. α-Glucosidase is a hydrolase that hydrolyzes a sugar's α-1,4-glycosidic bond and transforms it into glucose. In patients with diabetes, α-glucosidase inhibitors can efficiently postpone and alleviate postprandial glucose elevation.45 We found that all three flavonoids inhibited α-glucosidase in a dose-dependent manner (Fig. 5A). Throughout the measuring range, W3 demonstrated more inhibitory ability against α-glucosidase than acarbose control. W1 also demonstrated greater inhibitory efficacy than acarbose in the 0.08–0.32 mg mL−1 range. Further, W2 exhibited a moderate inhibitory effect. The highest inhibition rate of W3 was achieved at 1.28 mg mL−1 with 89.50%, whereas the highest inhibition rate of the acarbose control was 83.00%. The IC50 values of W1, W2, W3, and acarbose for α-glucosidase were 0.104, 0.199, 0.074, and 0.102 mg mL−1, respectively (Table 6). The results revealed that W3 and W1 significantly inhibited α-glucosidase activity and showed promising antidiabetic characteristics.
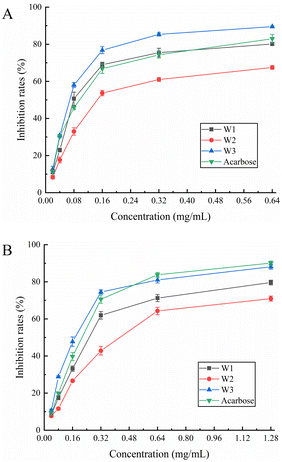 |
| | Fig. 5 Effect of W1, W2 and W3 of (A) α-glucosidase and (B) α-amylase activity. | |
3.8.2. α-Amylase inhibition assay. α-Amylase inhibitors reduce sugar intake by effectively inhibiting salivary and pancreatic amylase activity, thereby lowering blood glucose levels. Fig. 5B shows the α-amylase inhibitory ability of W1, W2, and W3. The α-amylase inhibitory activity of three flavonoids gradually increased at their increasing concentration in the solution. The inhibitory ability of W3 on α-amylase was comparable to that of acarbose control throughout the measurement range and was slightly stronger than that of acarbose in the range of 0.04–0.32 mg mL−1. At the highest dose (1.28 mg mL−1), the α-amylase inhibition rate of acarbose was 90.10%, and the W3 inhibition rate was 88.08%. The inhibition ability of W1 was slightly lower than that of acarbose whereas W2 demonstrated a moderate inhibition ability. Moreover, the IC50 values of W1, W2, W3, and acarbose for α-amylase were 0.286, 0.441, 0.176, and 0.204 mg mL−1, respectively (Table 6). Hence, W3 has a marked inhibitory effect on α-amylase, comparable to the acarbose control, and is a suitable α-amylase inhibitor.
4. Conclusions
A. mearnsii leaves are a rich source of flavonoids. In this study, a method was developed for the separation and purification of W3, the main product, and W1 from A. mearnsii leaves. From 1 kg of dried A. mearnsii leaves, 155.8 g of crude extract was produced, with a flavonoid content of 68.7 mg g−1 of crude extract. The ultimate yield of W3 and W1 was 7.3 and 1.8 mg g−1 of crude extract, with a purity of 98.4% and >95%, respectively. We found that W1 and W3 were potent antioxidants. W1 and W3 strongly inhibited α-glucosidase, whereas W3 had powerful α-amylase inhibitory capabilities. Hence, A. mearnsii leaves were identified as a novel source of myricitrin, while myricitrin and myricetin-3-O-glucoside were found to be excellent antioxidants and type II diabetes inhibitors. This research not only illustrates the significance of effective processing of A. mearnsii leaves but also provides a way to solve the problem of environmental impact caused by discarded A. mearnsii leaves.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We would like to acknowledge the financial support from the National Key Research and Development Plan of China (2022YFD2200805), the National Natural Science Foundation of China (21905138), the Top-Notch Academic Programs Project of Jiangsu Higher Education Institutions (PPZY2015C221).
References
- G. F. Silva, E. T. Souza and R. N. Almeida, et al., Molecules, 2022, 27, 970–989 CrossRef.
- O. O. Olajuyigbe and A. J. Afolayan, Int. J. Mol. Sci., 2012, 13, 4255–4267 CrossRef CAS.
- S. I. Pedro, T. Rosado and C. Barroca, et al., Plants, 2022, 11, 1442–1458 CrossRef CAS.
- G. R. Oliveira, F. Grasel and G. P. Pinho, et al., Ind. Crops Prod., 2020, 147, 112200 CrossRef CAS.
- C. E. Silva, D. M. Quevedo and V. D. Jahno, J. Clean. Prod., 2020, 264, 121632 CrossRef.
- F. S. Grasel, M. C. Marcelo and M. F. Ferrão, Anal. Methods, 2017, 3977–3982 RSC.
- F. A. Anjo, B. R. Saraiva and J. B. Silva, et al., Food Chem., 2021, 344, 128640 CrossRef CAS PubMed.
- T. Uushona, O. C. Chikwanha and T. Tayengwa, et al., J. Agric. Sci., 2022, 159, 743–756 CrossRef.
- H. M. Saayman and D. G. Roux, Biochem. J., 1965, 97, 794–801 CrossRef CAS PubMed.
- A. M. MacKenzie, Phytochemistry, 1969, 8, 1813–1815 CrossRef CAS.
- A. M. MacKenzie, Tetrahedron Lett., 1967, 8, 2519–2520 CrossRef.
- J. Xiong, M. H. Grace and D. Esposito, et al., Nat. Prod. Commun., 2016, 11, 649–653 CrossRef PubMed.
- J. Yang, X. Wang and C. Zhang, et al., Food Chem., 2021, 347, 129056 CrossRef CAS PubMed.
- L. Cui, Z. Ma and D. Wang, et al., Ultrason. Sonochem., 2022, 90, 106190 CrossRef CAS PubMed.
- B. Zhang, Q. Shen and Y. Chen, et al., Sci. Rep., 2017, 7, 44239 CrossRef CAS.
- A. Ahangarpour, A. A. Oroojan and L. Khorsandi, et al., Oxid. Med. Cell. Longev., 2018, 2018, 7496936 Search PubMed.
- S. S. Hawary, R. Mohammed and M. E. Din, et al., RSC Adv., 2021, 11, 16755–16767 RSC.
- M. D. Adu, C. P. Bondonno and B. H. Parmenter, et al., Food Funct., 2022, 13, 4459–4468 RSC.
- B. Silva, F. C. Biluca and L. V. Gonzaga, et al., Int. Food Res. J., 2020, 141, 110086 CrossRef PubMed.
- H. S. Chae, R. Xu and J. Y. Won, et al., Int. J. Mol. Sci., 2019, 20, 2420–2437 CrossRef PubMed.
- L. Yi, S. Ma and D. Ren, Phytochem. Rev., 2017, 16, 479–511 CrossRef CAS.
- D. K. Semwal, R. B. Semwal and S. Combrinck, et al., Nutrients, 2016, 8, 90–120 CrossRef PubMed.
- F. M. Mordy, M. M. Hamouly and M. T. Ibrahim, et al., RSC Adv., 2020, 10, 32148–32155 RSC.
- Y. T. Tung, J. H. Wu and C. Y. Hsieh, et al., Food Chem., 2009, 115, 1019–1024 CrossRef CAS.
- L. Wen, Y. Zhao and Y. Jiang, et al., Free Radic. Biol. Med., 2017, 110, 92–101 CrossRef CAS.
- R. C. Erhabor, M. A. Aderogba and J. O. Erhabor, et al., J. Ethnopharmacol., 2021, 273, 113981 CrossRef CAS.
- S. D. Maduwanthi and R. A. Marapana, Food Chem., 2021, 339, 127909 CrossRef CAS.
- S. M. Boue, K. W. Daigle and M. H. Chen, et al., J. Agric. Food Chem., 2016, 64, 5345–5353 CrossRef CAS PubMed.
- J. Kellogg, M. H. Grace and M. A. Lila, Mar. Drugs, 2014, 12, 5277–5294 CrossRef CAS PubMed.
- J. Zhou, L. Zhang and Q. Li, et al., Molecules, 2019, 24, 112–126 CrossRef PubMed.
- S. B. Kim, J. Bisson and J. B. Friesen, et al., J. Nat. Prod., 2020, 83, 1846–1858 CrossRef CAS PubMed.
- P. Yingyuen, S. Sukrong and M. Phisalaphong, Ind. Crops Prod., 2020, 149, 112307 CrossRef CAS.
- X. Chen, H. Wang and X. Huang, et al., Food Chem., 2021, 374, 131508 CrossRef PubMed.
- J. Mottaghipisheh and M. Iriti, Molecules, 2020, 18, 4146–4164 CrossRef PubMed.
- Y. Fu, Z. Li and J. Si, et al., J. Liq. Chromatogr. Relat. Technol., 2013, 36, 1503–1512 CrossRef CAS.
- A. A. Elegami, C. Bates and A. I. Gray, et al., Phytochemistry, 2003, 63, 727–731 CrossRef CAS PubMed.
- M. A. Syabana, N. D. Yuliana and I. Batubara, et al., J. Ethnopharmacol., 2022, 282, 114618 CrossRef CAS PubMed.
- J. C. Kuo, L. Zhang and H. Huang, et al., Molecules, 2020, 25, 4762–4778 CrossRef CAS.
- S. D. Ramalho, R. F. Sousa and C. M. Burger, et al., Nat. Prod. Res., 2015, 23, 2212–2214 CrossRef PubMed.
- M. B. Farhat, A. Landoulsi and R. Chaouch, et al., Ind. Crops Prod., 2013, 48, 904–914 CrossRef.
- P. S. Saravana, Y. Cho and H. Woo, et al., J. Clean. Prod., 2018, 198, 1474–1484 CrossRef CAS.
- D. Luo, T. Mu and H. Sun, Food Biosci., 2021, 39, 100801 CrossRef CAS.
- T. Miyake and T. Shibamoto, J. Agric. Food Chem., 1997, 45, 1819–1822 CrossRef CAS.
- M. N. Sarian, Q. U. Ahmed and S. Z. So'ad, et al., Biomed Res. Int., 2017, 2017, 106190 Search PubMed.
- X. Chen, J. Xiong and Q. He, et al., J. Chem., 2019, 2019, 4793047 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Lingxiao Heab,
Yu Zhang
ab,
Lingxiao Heab,
Yu Zhang