
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromaticity: Quo Vadis
Gabriel
Merino
 *a,
Miquel
Solà
*b,
Israel
Fernández
*c,
Cina
Foroutan-Nejad
*d,
Paolo
Lazzeretti
*e,
Gernot
Frenking
*f,
Harry L.
Anderson
g,
Dage
Sundholm
h,
Fernando P.
Cossío
i,
Marina A.
Petrukhina
j,
Jishan
Wu
k,
Judy I.
Wu
l and
Albeiro
Restrepo
m
*a,
Miquel
Solà
*b,
Israel
Fernández
*c,
Cina
Foroutan-Nejad
*d,
Paolo
Lazzeretti
*e,
Gernot
Frenking
*f,
Harry L.
Anderson
g,
Dage
Sundholm
h,
Fernando P.
Cossío
i,
Marina A.
Petrukhina
j,
Jishan
Wu
k,
Judy I.
Wu
l and
Albeiro
Restrepo
m
aDepartamento de Física Aplicada, Centro de Investigación y de Estudios Avanzados, Unidad Mérida, km 6 Antigua Carretera a Progreso, Apdo. Postal 73, Cordemex 97310, Mérida, Yucatán, Mexico. E-mail: gmerino@cinvestav.mx
bInstitut de Química Computacional i Catàlisi and Department de Química, Universitat de Girona, C/M. Aurèlia Capmany, 69, Girona 17003, Catalonia, Spain. E-mail: miquel.sola@udg.edu
cDepartamento de Química Orgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: israel@quim.ucm.es
dInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: cforoutan-nejad@icho.edu.pl
eDipartimento di Chimica e Biologia “A. Zambelli”, Università degli Studi di Salerno, via Giovanni Paolo II 132, Fisciano 84084, SA, Italy. E-mail: plazzeretti@unisa.it
fFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse 4, D-35043 Marburg, Germany. E-mail: frenking@chemie.uni-marburg.de
gDepartment of Chemistry, University of Oxford, Oxford, OX1 3TA, UK
hDepartment of Chemistry, Faculty of Science, University of Helsinki, P.O. Box 55, A. I. Virtasen aukio 1, FIN-00014 Helsinki, Finland
iDepartamento de Química Orgánica I, Instituto de Innovaciónen Química Avanzada (ORFEO-CINQA), University of the Basque Country (UPV/EHU), Paseo Manuel Lardizabal 3, 20018 Donostia/San Sebastián, Spain
jDepartment of Chemistry, University at Albany, State University of New York, Albany, New York 12222, USA
kDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore
lDepartment of Chemistry, University of Houston, Houston, Texas 77204, USA
mInstituto de Química, Universidad de Antioquia, Calle 70 No. 52-21, 050010, Medellín, Colombia
First published on 22nd February 2023
Abstract
Aromaticity is one of the most deeply rooted concepts in chemistry. But why, if two-thirds of existing compounds can be classified as aromatic, is there no consensus on what aromaticity is? σ−, π−, δ−, spherical, Möbius, or all-metal aromaticity… why are so many attributes needed to specify a property? Is aromaticity a dubious concept? This perspective aims to reflect where the aromaticity community is and where it is going.
Introduction
The concept of aromaticity has evolved to keep pace with the times.1–4 Although it was first used to describe compounds that resemble benzene, it is now employed to label any molecule with two- or three-dimensional circuits of highly delocalized electrons with presumably enhanced thermodynamic stability, bond-length equalization, particular reactivity, and distinct magnetic and spectroscopic properties. The so-called aromaticity criteria are derived from these characteristics, but none of them are free of ambiguities.In a field as broad as aromaticity, misunderstandings and disagreements are widespread regarding its definition and the aromatic nature of specific systems. Numerous manuscripts, reviews, and conferences are available, but the chemical community has not fully agreed on a general definition of aromaticity. Paul von Ragué Schleyer led one of the most important initiatives in 2001,1 compiling several review articles in Chemical Reviews that provided an overview of this concept. But considering recent events, it is necessary to have an updated broad perspective on this fundamental chemical idea. For that purpose, we gathered a range of viewpoints from experimental and theoretical experts on key questions we posed about the essence of aromaticity. Along the way, a few suggestions to the definition5 of aromaticity are suggested, and some factors that indicate a crisis in our community are listed.
Is there any misconception prevalent among the aromaticity community?
The chemical community interested in aromaticity has been relatively uniform in the past. However, a plethora of novel organometallic,6–13 all-metal and all-semimetal clusters,14,15 and inorganic compounds with attributes frequently found in “aromatic” compounds have been discovered, saturating the “aromatic character” in all branches of chemistry. Are they aromatic? Is aromaticity an exercise in chemical futility? Is it a suspicious or dubious concept?16 One thing is unquestionable, small cyclic and planar hydrocarbons are no longer the only species covered by the original conception.Chemistry is loaded with concepts, some of them deep-rooted, like aromaticity or chemical bonding, but in addition to them, there are also hybridization, atomic charge, conjugation, nucleophilicity, hardness and softness, donor–acceptor interactions, frontier orbitals, or acidity, to mention a few. Chemists are familiar with such notions and learn them during their professional training. Gernot Frenking calls them “unicorns in the world of chemistry” because everyone seems to know them even though no one has ever seen them.17 This is a crucial difference from physics, where the fundamental quantities are well-defined and generally valid.
Albeiro Restrepo argues that aromaticity is just one of the many ideas that make up the large conceptual pool that prevents chemistry from being reduced to physics (“the underlying physical laws necessary for the theory of the whole of chemistry are completely known”)18 or mathematics (“In so far as quantum mechanics is correct, chemical questions are problems in applied mathematics”).19 The lack of operators to unambiguously define useful concepts needed to rationalize experimental observations has not prevented chemists from devising rigorous calculation methods to quantify or determine whether a molecule is aromatic or not. Since these methods invoke different properties and strategies, it is not uncommon to find incomplete descriptions or inconsistent results. Diagnosing aromaticity from molecular wavefunctions brings challenges, which would require additional finesse even if we had the correct operator.
So, part of the confusion stems from the fact that many chemists believe that aromaticity is an observable property. While it is possible to measure specific energy values,20–22 structural data,23,24 magnetic response25–32 or other properties33–36 related to aromaticity, none of these can be stated unequivocally as a quantitative measure of it. In other words, we measure its consequences, but not aromaticity itself. This leads to other disruptions. For example, it is often believed that the most aromatic among a series of isomers is the most stable one. But other factors such as (hyper)conjugation, steric repulsions, or the occurrence of contacts such as hydrogen bonds or other non-covalent interactions may play a more critical role in determining the stability than aromaticity itself. As a result, it is easy to forget other stabilizing and destabilizing effects comparable to aromaticity. Judy I. Wu provides two clear examples: “The strain of the six σ C–C bonds in benzene is as destabilizing as aromaticity is stabilizing. Single C–C bonds are the strongest at an ideal distance of about 1.54 Å. Compressing them to a length of 1.40 Å incurs strain, and the σ-bond strain of benzene is near 30 kcal mol−1. Cyanuric acid tautomerizes to the nonaromatic keto form instead of the aromatic enol form due to enamine conjugation. There are many stabilizing or destabilizing effects, compared to widely accepted aromatic stabilization energy (ASE) of benzene of 30 kcal mol−1”.22 Another belief is that transient species cannot be aromatic, but transition states of allowed pericyclic reactions could be classified as aromatic,37 as could also the excited states (according to Baird's rule).38,39
Is the concept of aromaticity overused by chemists?
Harry L. Anderson mentions that aromaticity has the potential to contribute to further insights into molecular structure–property relationships in various areas of chemistry. “I am particularly interested in how aromaticity relates to molecular electronics. If a molecular ring is aromatic or antiaromatic then there must be a coherent wavefunction extending around the whole ring, which implies that charge transport can be controlled by quantum interference”.40–44 Marina A. Petrukhina has a similar idea: “The use of the concept and understanding of aromaticity opens the door into the discussion of properties at the molecular level, which then provides original insights into bulk material's properties and applications. This concept can also serve as a unifying ground for various molecular types, bringing different communities of chemists together for insightful discussions”.45–47 Jishan Wu comments that aromaticity “is applicable to (1) either π- or σ-electrons, (2) either closed-shell or open-shell systems, (3) either two- or three-dimensional conjugated molecules, (4) either through-bond or through-space interactions, and (5) either ground-state or transition/excited states. The molecules tend to adjust their geometry and spin states”.48–50 Fernando P. Cossío remarks that “Fig. 1 shows a monotonous growing bibliometric impact of aromaticity and antiaromaticity terms during this century. Since this evolution is supported by the peer review procedure and the scientific interest of researchers, it can be concluded that, despite possible overuse in some cases, this positive evolution reflects the usefulness of aromaticity as a central concept in chemistry. In contrast, the bibliometric impact of the antiaromaticity concept is much lower, perhaps because of the less appealing character of a negative concept”.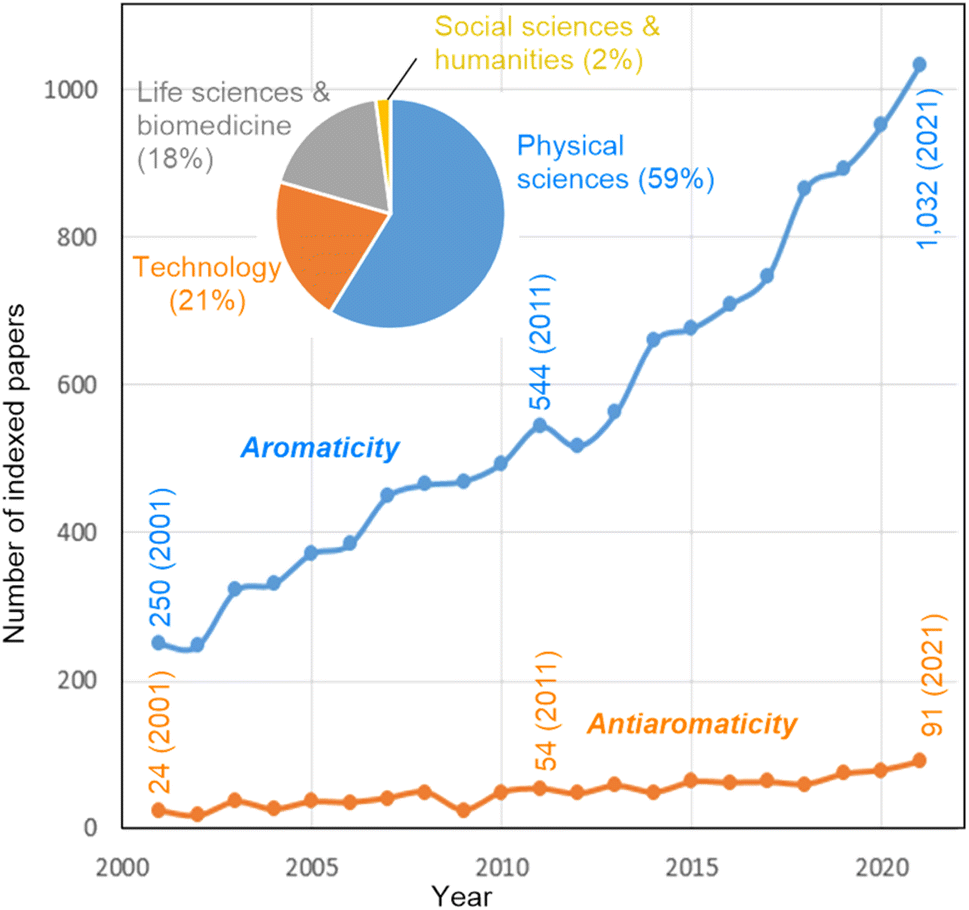 | ||
| Fig. 1 Evolution of the number of indexed papers including the words “aromaticity” and “antiaromaticity” in their title, abstract, and keywords along the first fifth part of the twenty-first century (Source: Web of Science. Data downloaded on October 28, 2022). | ||
According to Balaban and co-workers,51 about two-thirds of known molecules are aromatic or have aromatic rings. The majority of all known aromatic molecules are regular Hückel-aromatics. But not all these compounds behave in the same way, and maybe not all these behaviors are solely attributable to aromaticity. Nevertheless, there is an eagerness to announce the discovery of a new type of aromaticity whenever a newly synthesized molecule is described that exhibits properties that can be remotely associated with aromaticity. Dage Sundholm points out that “a unique aromatic character is often a motivation for publication in prestigious journals, but in some cases, the assessment of the aromaticity is not performed with the same expertise as the experimental part of the work. A more careful evaluation of the aromatic character often shows that the molecule in question does not have the expected aromatic nature”. This is consistent with a relatively recent provocative essay by Roald Hoffmann on “The many guises of aromaticity”, in which he states that “…but to me the labeling of molecules as aromatic seems to be motivated less by an intellectual desire to probe what aromatic means than by an attempt at distinction”.52 Miquel Solà adds that “if the concept of aromaticity is not used correctly, it gets devalued and becomes useless for chemical bonding analyses. It also introduces confusion in the field and, consequently, the aromaticity concept itself becomes suspicious”.
For instance, Gernot believes that “the extension to σ-aromaticity was an important addition to the concept of aromaticity, as was the discovery of homoaromatic compounds, but I view further proposals for “new” types of aromatic systems with skepticism”. Why are so many attributes needed to specify a substantive? Is this a warning of careless acceptance of a worn-out idea? Until 2017, 45 different types of aromaticity had been reported,53 which is a sign of sensationalism rather than an actual scientific discovery (vide infra).
An ad hoc way out to circumvent the aromaticity aporia has been proposed by some authors, putting forward the suggestive idea of aromaticity as a “multidimensional property”, which is a sort of conundrum including several different qualities challenging to assemble and reconcile within a comprehensive frame. Paolo Lazzeretti indicates that “In fact, such a locution [aromaticity as a “multidimensional property”] looks like a confession of incapacity to define aromaticity: it is equivalent to a capitulation”. Miquel thinks that, as pointed out by Bultinck,54 “multidimensionality cannot be used as an excuse to consider any indicator of aromaticity a good descriptor of this phenomenon”. Gernot considers that if one accepts the idea of multidimensionality, “this means that anything goes and all results and conclusions about the relative strength of aromaticity are valid”. Fernando agrees, saying, “In general, excessive multiparametric correlations should be avoided according to the Ockham's razor rule (Entia non sunt multiplicanda sine necessitate)”.
Are there any criteria for aromaticity that are superior (or more desirable) to the others?
Many chemists have attempted to grapple with universally acceptable notions of aromaticity, antiaromaticity, and nonaromaticity as molecular properties. The results so far are rather frustrating and indicate the difficulty, if not the impossibility, of ascribing a coherent set of standard distinguishing qualitative features to “aromatic” compounds. Despite that, chemists attempt to quantify it. Thus “aromaticity” would be something that can be measured via some proper yardstick. Several long-established “quantifiers” of different kinds are available – energetic, geometrical, magnetic, electronic, topological, etc. – sometimes incompatible with one another.Harry thinks that “criteria for aromaticity should be grounded in parameters that can be measured experimentally and calculated theoretically. Alternatively, if aromaticity becomes seen as primarily a theoretical quality, without experimental manifestations, it loses relevance”. So, what parameters should we choose? Paolo mentions that “at any rate, a few skeptical people are convinced that the concept of aromaticity could be disposed of without prejudice or detriment to chemistry unless it is related to something measurable”.
Starting from the original observation that led to the introduction of aromaticity as a chemical concept directly related to energetic stabilization by a specific number of conjugated electrons, the dilemma is that the stabilization energy is not unambiguously defined and depends on the choice of the reference system.21 Is this sufficient to invalidate this choice as a basis for defining aromaticity? Two additional elements must be considered. Aromaticity is a phenomenon related to electron delocalization in 2D or 3D rings (or closed circuits) that results in extra stabilization. Other descriptors not based on the stabilization energy, such as structural or magnetic phenomena, are secondary attributes, says Gernot, whose occurrence and strength are of interest and may sometimes show the same trend as the stabilization energy. However, as there is no absolute reference for ASE, aromaticity is often assessed indirectly by evaluating some physicochemical property in which aromaticity manifests itself. Because of the difficulties in obtaining reliable ASE, Miquel considers that “indicators based on the quantification of cyclic electron delocalization (one of the key properties of aromaticity) are the most reliable as showed in a series of tests of aromaticity that we proposed”.55,56 Unfortunately, electron delocalization is not an observable property that can be experimentally assessed. In this regard, Israel Fernández points out that while most methods to estimate ASE values involving isodesmic/homodesmotic reactions are indeed not reliable as they are typically contaminated by different flaws such as strain, hyperconjugation, “proto” branching, or syn-anti effects, there exist other modern approaches based on the Block Localized Wavefunction (BLW)22 or Energy Decomposition Analysis (EDA)20 methods which, although possess some limitations (for instance, EDA can be only applied to molecules having a mirror plane), allow us to accurately estimate reliable ASE values without recourse to external reference systems”.
Jishan states, “I prefer to use magnetic and structural criteria because they can be experimentally measured by NMR and X-ray crystallographic analysis, respectively”. The magnetic criteria are straightforward because if a ring sustains a diatropic (or paratropic) ring current when placed in an external magnetic field, it is classified as aromatic (or antiaromatic).26 The great advantage is that the presence of ring currents and the induced magnetic field can often be probed by NMR spectroscopy without the difficulty of identifying a suitable reference system. Regardless, there are situations where the magnetic criteria are difficult to apply and to conclude regarding the degree of aromaticity, for example, systems with very heavy metals or molecules in their excited states. Cina Foroutan-Nejad remarks that “a problem is prevalent among the proponents of the magnetic aromaticity criterion, where ring current is vastly interpreted as electron delocalization while the former is as mentioned a response property and the latter is a ground-state property that is measurable via any quantum topological approach and is related to the stability of a molecule”.57,58 One of the reviewers' comments that “The term ground state aromaticity is in my view not ideal as a reader may think that the opposite term is excited state aromaticity (which is not the case). The feature Cina refers to as ground state aromaticity is also found for the electronically excited state; the ground state is rather the electron configuration(s) of a particular state”. In this sense, a possibility is to use the term “intrinsic aromaticity” instead of “ground state aromaticity” as proposed by Ottosson et al.59 However, the choice of the aromaticity tool is up to the users. If we rely on magnetic properties as a probe of aromaticity, we might list some molecules as aromatic, which might not be considered as aromatic if we chose intrinsic criteria of aromaticity, i.e., energetic, structural, and electronic criteria. Dage remarks that “response properties are also ground-state properties. For instance, the dipole moment is the first-order response to an external electric field and the polarizability is next term in that series expansion. The dipole moment and the polarizability are ground-state properties, even though response theory is needed to determine polarizabilities”.
Nevertheless, Dage points out that “magnetically induced ring current density is the underlying property of all methods used to assess aromaticity by magnetic criteria”. The nucleus-independent chemical shift (NICS,60 one of the most popular aromaticity descriptors) and ring-current strengths are related via the Ampère–Maxwell integration law.61 Aromaticity can even be estimated by measuring 1H NMR chemical shifts, which can also be calculated from the Biot–Savart law by integrating the susceptibility of the magnetically induced current density multiplied by the derivative of the vector potential of the nuclear magnetic moment in the limit of evanescent perturbations.62,63 Nonetheless, while observable magnetic properties appear to provide reliable benchmarks of aromaticity, they are difficult to reconcile with thermodynamic stabilization criteria.
NICS deserves a special mention.64,65 Is NICS still the best indicator of aromaticity? This is a controversial question, but evidence of NICS's shortcomings has accumulated over the years.66–69 Judy states that “one of the reasons for the popularization of NICS is that this criterion originates from NMR experiments and shares the same language, one that all experimentalists are familiar with. Nearly all experimental groups working on “aromaticity” perform some form of NICS calculations”.70 NICS provides an average magnetic behavior in the molecular ring center (or in any selected point in the space). However, the first proposal of the NICS as a “measure of aromaticity” turned out to be a formidable weapon of mass distraction based on a misconception of the tensorial character of nuclear magnetic shielding. According to the benzenoid hydrocarbon ring current model, only the out-of-plane component of the magnetic tensors, i.e., the magnetizability, is biased by the diamagnetic flux induced in an electron cloud delocalized by a magnetic field perpendicular to the molecular plane. This analogous argument explains the downward NMR shift that decreases the out-of-plane shielding of peripheral hydrogen atoms in benzene and rationalizes the virtual shielding at points along the sixfold symmetry axis. Thus, the in-plane components' contributions to the magnetic tensors are spurious. Paolo states that “a crucial test yields an epistemological falsification of NICS as an indicator of “σ-aromaticity” in cyclopropane, where the in-plane components exceed the perpendicular by 18 ppm”.71 After a long and frank discussion at the Exeter workshop in 2003, in which the different opinions of Schleyer and Paolo were opposed, the proposal for a NICSzz was accepted. Still, using NICS as a black box leads to interpretation challenges.
The concept of “aromaticity” originated partly from the unique chemical reactivity of a family of benzene-containing molecules, which tend to undergo substitution reactions rather than electrophilic addition reactions. As Jishan mentions, “chemical reactivity depends on many factors, such as strain, alignment of the energy levels of the boundary molecular orbitals, oxidation states, etc., so it is difficult to clearly correlate the aromaticity of a molecule with its chemical reactivity”. Harry argues that “In my opinion, the reactivity criterion for aromaticity is obsolete; in every case where reactivity indicates aromaticity, the energetic, structural or magnetic criteria give the same conclusion more reliably”. But the concept of aromaticity has practical consequences beyond structure and reactivity. Judy recalls that Evans and Warhurst related the six π-electrons of benzene to the six π-electrons involved in the Diels–Alder transition state structure of butadiene and ethylene,72 which extended the concept of aromaticity to understand reaction barriers in pericyclic reactions. Similar relationships were generalized through the Woodward–Hoffmann rules and, later, the Dewar and Zimmerman rules for transition state structures with Hückel and Möbius topologies.73,74
On the other hand, no universal rule establishes when a compound can be classified as aromatic. Unlike conjugation, which describes a gradually increasing stabilization due to delocalization, aromaticity has an oscillating trend of stability which is appropriately described by the famous 4n + 2 rule (formulated as such by von Doering and Detert,75,76 although Hückel already realized that systems with 2, 6, 10, etc. π-electrons would exhibit greater stability)77 for annulenes. It was initially derived for benzene and related molecules exhibiting π-conjugation. In contrast, antiaromatic molecules have 4n electrons in the conjugated ring. Baird's rule for triplet-state aromaticity states the opposite, i.e., 4n electrons lead to aromaticity, and 4n + 2 electrons lead to antiaromaticity.38,39,78 The Hückel and Baird rules can be combined and generalized to states with higher spin multiplicity when the number of occupied conjugated orbitals is considered instead of the number of electrons. This combined rule then states that molecules with an even number of occupied conjugated orbitals in the ring are antiaromatic and aromatic rings have an odd number of occupied conjugated orbitals.79 The combined rule can assess the aromaticity of states with higher spin multiplicity. Another equivalent way to combine Hückel's and Baird's rule is the so-called Mandado's 2n + 1 rule for aromaticity of separate spins.80 The celebrated Clar, Hückel, Baird, or Wade–Mingos rules, as well as later generalizations by other authors, constitute milestones in the historical evolution of the notion and throw light on the vexata quaestio of “aromatic character”.16,81,82 But there are several examples where Baird aromaticity is used to explain certain phenomena unrelated to this type of aromaticity. So, when old rules fail, a search for new ones begins.
A reviewer mentions that “A general dilemma in the aromaticity research area is the cherry-picking of computational data to fit a working hypothesis concerning the (anti)aromatic character of a novel compound. Far too seldom are alternative rationalization models of the observations set up and tested. If the research on (anti)aromaticity should lead to an improved understanding of the phenomena, we need to strive for a better qualitative insight, not just numbers from various computational tools for (anti)aromaticity assessments (ultimately machine learning). In that regard, qualitative molecular orbitals and valence bond theoretical tools must be revived, learnt and used also by experimental chemists active in the area”.
Is it possible to find a universal definition of aromaticity?
The IUPAC definition. 5 The concept of spatial and electronic structure of cyclic molecular systems displaying the effects of cyclic electron delocalization which provide for their enhanced thermodynamic stability (relative to acyclic structural analogues) and tendency to retain the structural type in the course of chemical transformations. A quantitative assessment of the degree of aromaticity is given by the value of the resonance energy. It may also be evaluated by the energies of relevant isodesmic and homodesmotic reactions. Along with energetic criteria of aromaticity, important and complementary are also a structural criterion (the lesser the alternation of bond lengths in the rings, the greater is the aromaticity of the molecule) and a magnetic criterion (existence of the diamagnetic ring current induced in a conjugated cyclic molecule by an external magnetic field and manifested by an exaltation and anisotropy of magnetic susceptibility). Although originally introduced for characterization of peculiar properties of cyclic conjugated hydrocarbons and their ions, the concept of aromaticity has been extended to their homoderivatives (see homoaromaticity), conjugated heterocyclic compounds (heteroaromaticity), saturated cyclic compounds (σ-aromaticity) as well as to three-dimensional organic and organometallic compounds (three-dimensional aromaticity). A common feature of the electronic structure inherent in all aromatic molecules is the close nature of their valence electron shells, i.e., double electron occupation of all bonding molecular orbitals (MOs) with all antibonding and delocalized nonbonding MOs unfilled. The notion of aromaticity is applied also to transition states.This is not a simple question to answer, and in fact, opinions are diverse. A reviewer comments that “Why should we precisely define a concept that we do not know exactly what it is? IUPAC should certainly have a description (call it a “definition” if one wants) but it needs to be rather loose until we have gathered more knowledge. It could well be that the concept splits into several (connected) concepts… Still, it is vital for chemistry to discuss the aromaticity and antiaromaticity concepts, and I think the text adds to such a discussion. It should be important to identify means to curb severe overuse/misuse of the concepts, and I believe that it can be achieved through a revision of the IUPAC Gold Book description/definition”. Paolo states that “The IUPAC definition has a historical value. It provides timely documentation of cultural heritage, giving a snapshot of chemical theory in the second decade of the third millennium. As such, it should not be changed: it would possibly be abandoned soon, owing to our incapacity to give adequate definitions… In a memorable paper, Gerhard Binsch seems to postulate that a universal definition of aromaticity is impractical or non-compatible with the general laws constituting chemical theory.83 He wrote that “Attempts to sharpen our definitions must and will continue, but it appears advisable not to permit such attempts to become an obsession; undue emphasis on conceptual rigor might otherwise well turn into rigor mortis”. I subscribe to this point of view and confess that I feel uncomfortable when I must teach my students something I do not understand”. Gernot is in line with this view and points out that “the current definition by the IUPAC correctly describes the concept of aromaticity based on energy criteria with the note that other criteria (structural and magnetic phenomena) are important and complementary but not elementary. I have no problem with the further statements, and I see no reason to make changes. Aromaticity remains a fuzzy concept in the zoo of chemical models, and all attempts to define it more clearly create more confusion than clarity”.
In contrast, Miquel thinks, “In the last decades, the aromaticity concept has enormously widened. For this reason, the current definition by the IUPAC of 1999 must be updated. In particular, the current definition does not refer to excited state aromaticity, metalloaromaticity, spherical aromaticity, multiple aromaticity, or conflicting aromaticity. Moreover, it gives too much relevance to the magnetic criteria to measure aromaticity and does not consider using electron delocalization measures to quantify it. Finally, it overemphasizes the resonance energies, which in many cases are very difficult to assess”. Marina agrees with this view: “In my view, the definition of aromaticity requires updating (and that must be done regularly). It is hard, if not impossible, to offer an all-inclusive general definition of this complex fundamental concept”. Jishan supports this position as well: “Recent theoretical and experimental works have extended the concept from 2D to 3D π-/σ-conjugated systems, from closed-shell to open-shell molecules, and from ground state to excited states and transition states. Some descriptions could be modified: A quantitative assessment of the degree of aromaticity is given by the value of the resonance energy. It may also be evaluated by the energies of relevant isodesmic and homodesmotic reactions. It is practically difficult to measure resonance energy, and calculations on complicated systems may not be reliable”.
Cina adds that “A simple solution to define aromaticity as a universal phenomenon is to remain fully loyal to the IUPAC definition of aromaticity. That means we can call a molecule aromatic if it does satisfy all criteria of aromaticity, i.e. structural, energetic, electronic, and magnetic, at the same time. This involves that if a molecule satisfies the magnetic criterion of aromaticity but fails in energetic test, we should not call the system aromatic and vice versa. This puts all intrinsic and response-based criteria of aromaticity at an equal position. Following this advice will eliminate a great number of controversial species out of the league of aromaticity, among troublesome transition metal, lanthanide, and actinide molecules”.84
Fernando recounts other attempts to define this concept. “Aromaticity is a manifestation of electron delocalization in closed circuits, either in two or in three dimensions. This results in energy lowering, often quite substantial, and a variety of unusual chemical and physical properties. These include a tendency toward bond length equalization, unusual reactivity, and characteristic spectroscopic features”. This definition was provided in 2005 by Chen et al. “It is very general and, in principle, includes polycyclic systems, excited states, and chemical reactivity, although these aspects are not explicitly mentioned”.64
Harry is more precise: “It is not possible to find a universal definition of aromaticity or a single quantitative measure for aromaticity. This is not necessarily a problem, and some other fundamental chemistry concepts are similarly difficult to define (e.g., atomic radius and oxidation state). It is easier to precisely define an aromatic ring current and quantify ring current susceptibility (or ‘ring-current strength’). Rather than asking whether a compound is aromatic, it is often more constructive to ask: does it exhibit an aromatic (diatropic) ring current, and if so, is the ring current predominantly local or global?”.
Harry and Dage also suggest the following changes to the current IUPAC definition of aromaticity:
(a) Remove the reactivity criterion, i.e., delete the phrase “and tendency to retain the structural type in the course of chemical transformations”. This reactivity effect is a consequence of ASE, and it is not helpful to view reactivity as a separate criterion.
(b) Remove the statement that “A quantitative assessment of the degree of aromaticity is given by the value of the resonance energy”. Why base the quantitative assessment of aromaticity on ASE rather than ring current susceptibility? Aromaticity is not an on/off property. The strength of the ring-current susceptibility tells how strong the aromaticity/antiaromaticity is. Nonaromatic molecules sustain very weak net ring currents.
(c) Even though the ring-current criterion has been used for many years, it is mentioned in the IUPAC definition within parenthesis. The fact that the 1H NMR chemical shifts are used in experimental studies to assess aromatic properties is not mentioned.
(d) Remove the sentence “A common feature of the electronic structure inherent in all aromatic molecules is the close nature of their valence electron shells, i.e., double electron occupation of all bonding MOs with all antibonding and delocalized nonbonding MOs unfilled”. This statement is generally true, but a closed-shell electronic configuration is not particularly unique to aromatic molecules, and aromaticity can arise in open-shell systems, such as triplet excited states.
(e) Mention excited state aromaticity, perhaps by changing the final sentence to: “The notion of aromaticity is applied also to transition states and electronic excited states”.
(f) Harry considers that “the definition should mention the Hückel 4n + 2 and 4n rules. If a molecular framework is studied in a range of oxidation states and becomes more delocalized/stable/diatropic when it has a circuit of 4n + 2 electrons and more localized/unstable/paratropic when it has a circuit of 4n electrons, this is a clear sign of aromaticity and/or antiaromaticity”. However, Dage partially agrees with that statement: “The electronic structure of antiaromatic molecules is delocalized because they sustain a paratropic ring current”.
Change of definition or a new paradigm
According to Gabriel Merino, the sum of all the reactions clearly shows that we are in a crisis, as defined by Thomas S. Kuhn.85 We have reached the point where specific newly found systems may be categorized as anomalies or systems that somehow defy the assumptions made possible by the aromaticity paradigm. When an anomaly is more than just another enigma, the transition to crisis and non-ordinary science begins; that is when the anomaly becomes perceptible. The perception of these anomalies is a prerequisite for a change of paradigm, as in the aromaticity community. Indeed, as a community, we have initiated an exploration of the area encompassing these anomalies to isolate them and give them a structure, which has led to frequent and in-depth discussions on methodologies, problems, and solutions, even though these discussions have served more to form schools than to produce agreements. Even though the rules are no longer entirely correct, we have applied them more forcefully to delimit the impact of the crisis. Paraphrasing Kuhn, the crisis weakens the rules of enigma solving and allows the proliferation of paradigm versions. Every day, we see new interpretations of aromaticity, but interpretation can only articulate a paradigm, not correct it. Paradigms cannot be corrected by normal science, and normal science leads only to recognizing anomalies and crises. Moreover, these are ended not by deliberation or interpretation but by a relatively sudden and unstructured event.There are other apparent symptoms of crisis, for example, the proliferation of versions of the concept. Scientists adopt a different attitude towards existing paradigms when confronted with anomalies or crises. Consequently, the nature of their research changes. Even though they may begin to lose faith and consider other options, they do not give up the paradigm that has led to the crisis. As Paolo put it, they will invent numerous ad hoc modifications of their theory to eliminate any apparent conflict. This is when there is a proliferation of competing definitions, the readiness to try everything, the expression of explicit discontent, the recourse to philosophy and the debate on foundations, which are symptoms of a transition from normal to non-ordinary research. We might be tempted to wait for the crisis to pass, especially if there are many other interesting problems. Nevertheless, the symptoms are apparent, there is a crisis, and all crises conclude with the emergence of a new paradigm candidate and the subsequent struggle to cement it. Scientific revolutions occur only when there is no other option. So, a crisis is a prelude to the emergence of new theories, of a scientific revolution!
Historically, the researchers who make such paradigm changes have been very young, as they are not committed to the traditional rules; they could see that the existing rules no longer provide a coherent explanation to the problems posed by the community and conceive another set that can replace them. Do we need a change of the definition? Perhaps that is not the right question. Maybe the right question is, when will the paradigm change that defines the activities of our “aromaticity” community take place? Not all authors believe that the definition should be changed. It is not a matter of being theoretical or experimental. Several theoreticians oppose changing the IUPAC definition, and one of the experimental authors proposes even more modifications. Everything is relative; nothing is black and white, and numerous shades of grey exist. But it becomes clear that a change is required, and it will come when there are enough anomalies challenging our currently accepted view of aromaticity.
Data availability
This perspective gathers a series of thoughts and as such it did not generate any data.Author contributions
GM, MS, IF, and CFN conceived the idea. All authors contributed to the text by writing a few manuscript pages. The corresponding authors have combined the contributions to a single text that all authors have polished.Conflicts of interest
There are no conflicts to declare.References
- P. v. R. Schleyer, Chem. Rev., 2001, 101, 1115–1118 CrossRef CAS PubMed.
- N. Martín and L. T. Scott, Chem. Soc. Rev., 2015, 44, 6397–6400 RSC.
- G. Merino and M. Solà, Phys. Chem. Chem. Phys., 2016, 18, 11587–11588 RSC.
- M. Solà, A. I. Boldyrev, M. K. Cyranski, T. M. Krygowski and G. Merino, Aromaticity and Antiaromaticity: Basics and Applications, John Wiley & Sons, Inc., Hoboken, p. 2023 Search PubMed.
- V. I. Minkin, Pure Appl. Chem., 1999, 71, 1919–1981 CrossRef CAS.
- I. Fernández and G. Frenking, Chem. - Eur. J., 2007, 13, 5873–5884 CrossRef PubMed.
- J. R. Bleeke, Chem. Rev., 2001, 101, 1205–1228 CrossRef CAS PubMed.
- I. Fernández, G. Frenking and G. Merino, Chem. Soc. Rev., 2015, 44, 6452–6463 RSC.
- D. Chen, Q. Xie and J. Zhu, Acc. Chem. Res., 2019, 52, 1449–1460 CrossRef CAS PubMed.
- D. Chen, Y. Hua and H. Xia, Chem. Rev., 2020, 120, 12994–13086 CrossRef CAS PubMed.
- Y. Zhang, C. Yu, Z. Huang, W.-X. Zhang, S. Ye, J. Wei and Z. Xi, Acc. Chem. Res., 2021, 54, 2323–2333 CrossRef CAS PubMed.
- F. Feixas, E. Matito, J. Poater and M. Solà, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 105–122 CAS.
- V. Y. Lee and A. Sekiguchi, Angew. Chem., Int. Ed., 2007, 46, 6596–6620 CrossRef CAS PubMed.
- X. Li, A. E. Kuznetsov, H.-F. Zhang, A. I. Boldyrev and L.-S. Wang, Science, 2001, 291, 859–861 CrossRef CAS PubMed.
- A. I. Boldyrev and L.-S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- M. Solà, Front. Chem., 2017, 5, 22 Search PubMed.
- G. Frenking and A. Krapp, J. Comput. Chem., 2007, 28, 15–24 CrossRef CAS PubMed.
- P. A. M. Dirac, Proc. R. Soc. London, 1929, 123, 714–733 CAS.
- H. Eyring, J. Walter and G. E. Kimball, Quantum Chemistry, Wiley, New York, 14th edn, 1967 Search PubMed.
- I. Fernández and G. Frenking, Faraday Discuss., 2007, 135, 403–421 RSC.
- M. K. Cyrański, Chem. Rev., 2005, 105, 3773–3811 CrossRef PubMed.
- Y. Mo and P. v. R. Schleyer, Chem. – Eur. J., 2006, 12, 2009–2020 CrossRef CAS PubMed.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385–1420 CrossRef CAS PubMed.
- T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS PubMed.
- J. A. N. F. Gomes and R. B. Mallion, Chem. Rev., 2001, 101, 1349–1384 CrossRef CAS PubMed.
- P. Lazzeretti, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 1–88 CrossRef CAS.
- G. Merino, T. Heine and G. Seifert, Chem. – Eur. J., 2004, 10, 4367–4371 CrossRef CAS PubMed.
- P. Lazzeretti, Phys. Chem. Chem. Phys., 2004, 6, 217–223 RSC.
- J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed.
- R. Islas, T. Heine and G. Merino, Acc. Chem. Res., 2012, 45, 215–228 CrossRef CAS PubMed.
- D. Sundholm, H. Fliegl and R. J. F. Berger, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- F. De Proft and P. Geerlings, Chem. Rev., 2001, 101, 1451–1464 CrossRef CAS PubMed.
- J. Poater, M. Duran, M. Solà and B. Silvi, Chem. Rev., 2005, 105, 3911–3947 CrossRef CAS PubMed.
- F. Feixas, E. Matito, J. Poater and M. Solà, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- G. Merino, A. Vela and T. Heine, Chem. Rev., 2005, 105, 3812–3841 CrossRef CAS PubMed.
- P. v. R. Schleyer, J. I. Wu, F. P. Cossío and I. Fernández, Chem. Soc. Rev., 2014, 43, 4909–4921 RSC.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- H. Ottosson, Nat. Chem., 2012, 4, 969–971 CrossRef CAS PubMed.
- M. Jirásek, H. L. Anderson and M. D. Peeks, Acc. Chem. Res., 2021, 54, 3241–3251 CrossRef PubMed.
- M. D. Peeks, T. D. W. Claridge and H. L. Anderson, Nature, 2017, 541, 200–203 CrossRef CAS PubMed.
- M. D. Peeks, J. Q. Gong, K. McLoughlin, T. Kobatake, R. Haver, L. M. Herz and H. L. Anderson, J. Phys. Chem. Lett., 2019, 10, 2017–2022 CrossRef CAS PubMed.
- M. Rickhaus, M. Jirasek, L. Tejerina, H. Gotfredsen, M. D. Peeks, R. Haver, H.-W. Jiang, T. D. W. Claridge and H. L. Anderson, Nat. Chem., 2020, 12, 236–241 CrossRef CAS PubMed.
- T. Stuyver, M. Perrin, P. Geerlings, F. De Proft and M. Alonso, J. Am. Chem. Soc., 2018, 140, 1313–1326 CrossRef CAS PubMed.
- B. D. Steinberg, E. A. Jackson, A. S. Filatov, A. Wakamiya, M. A. Petrukhina and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 10537–10545 CrossRef CAS PubMed.
- A. V. Zabula, A. S. Filatov, S. N. Spisak, A. Y. Rogachev and M. A. Petrukhina, Science, 2011, 333, 1008–1011 CrossRef CAS PubMed.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Y. Rogachev and M. A. Petrukhina, Acc. Chem. Res., 2018, 51, 1541–1549 CrossRef CAS PubMed.
- C. Liu, Y. Ni, X. Lu, G. Li and J. Wu, Acc. Chem. Res., 2019, 52, 2309–2321 CrossRef CAS PubMed.
- Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857 RSC.
- C. Liu, Y. Ni, X. Lu, G. Li and J. Wu, Acc. Chem. Res., 2019, 52, 2309–2321 CrossRef CAS PubMed.
- A. T. Balaban, D. C. Oniciu and A. R. Katritzky, Chem. Rev., 2004, 104, 2777–2812 CrossRef CAS PubMed.
- R. Hoffmann, Am. Sci., 2015, 103, 18 CrossRef.
- J. Grunenberg, Int. J. Quantum Chem., 2017, 117, e25359 CrossRef.
- P. Bultinck, Faraday Discuss., 2007, 135, 347–365 RSC.
- F. Feixas, E. Matito, J. Poater and M. Solà, J. Comput. Chem., 2008, 29, 1543–1554 CrossRef CAS PubMed.
- F. Feixas, J. O. C. Jiménez-Halla, E. Matito, J. Poater and M. Solà, J. Chem. Theory Comput., 2010, 6, 1118–1130 CrossRef CAS.
- Z. Badri and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2016, 18, 11693–11699 RSC.
- T. Janda and C. Foroutan-Nejad, ChemPhysChem, 2018, 19, 2357–2363 CrossRef CAS PubMed.
- P. Preethalayam, N. Proos Vedin, S. Radenković and H. Ottosson, J. Phys. Org. Chem., 2023, 36, e4455 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- R. J. F. Berger, M. Dimitrova, R. T. Nasibullin, R. R. Valiev and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 624–628 RSC.
- R. M. Stevens, R. M. Pitzer and W. N. Lipscomb, J. Chem. Phys., 1963, 38, 550–560 CrossRef CAS.
- C. J. Jameson and A. D. Buckingham, J. Phys. Chem., 1979, 83, 3366–3371 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- R. Gershoni-Poranne and A. Stanger, Chem. Soc. Rev., 2015, 44, 6597–6615 RSC.
- J. Poater, M. Solà, R. G. Viglione and R. Zanasi, J. Org. Chem., 2004, 69, 7537–7542 CrossRef CAS PubMed.
- R. Islas, G. Martínez-Guajardo, J. O. C. Jiménez-Halla, M. Solà and G. Merino, J. Chem. Theory Comput., 2010, 6, 1131–1135 CrossRef CAS.
- L. Zhao, R. Grande-Aztatzi, C. Foroutan-Nejad, J. M. Ugalde and G. Frenking, ChemistrySelect, 2017, 2, 863–870 CrossRef CAS.
- M. Mauksch and S. B. Tsogoeva, Chem. – Eur. J., 2021, 27, 14660–14671 CrossRef CAS PubMed.
- R. H. Mitchell, Chem. Rev., 2001, 101, 1301–1316 CrossRef CAS PubMed.
- S. Pelloni, P. Lazzeretti and R. Zanasi, J. Phys. Chem. A, 2007, 111, 8163–8169 CrossRef CAS PubMed.
- M. G. Evans and E. Warhurst, Trans. Faraday Soc., 1938, 34, 614 RSC.
- M. J. S. Dewar, Tetrahedron, 1966, 22, 75–92 CrossRef.
- H. E. Zimmerman, J. Am. Chem. Soc., 1966, 88, 1564–1565 CrossRef CAS.
- W. v. E. Doering and F. L. Detert, J. Am. Chem. Soc., 1951, 73, 876–877 CrossRef CAS.
- G. Frenking, Theor. Chem. Acc., 2000, 103, 187–189 Search PubMed.
- E. Hückel, Z. Phys., 1931, 70, 204–286 Search PubMed.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- R. R. Valiev, T. Kurten, L. I. Valiulina, S. Y. Ketkov, V. N. Cherepanov, M. Dimitrova and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 1666–1674 RSC.
- M. Mandado, A. M. Graña and I. Pérez-Juste, J. Chem. Phys., 2008, 129, 164114 CrossRef PubMed.
- M. Solà, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1404 Search PubMed.
- M. Solà, Nat. Chem., 2022, 14, 585–590 CrossRef PubMed.
- G. Binsch, Naturwissenschaften, 1973, 60, 369–374 CrossRef CAS.
- B. J. R. Cuyacot, Z. Badri, A. Ghosh and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2022, 24, 27957–27963 RSC.
- T. S. Kuhn, The Structure of Scientific Revolutions, University of Chicago Press, Chicago, 2nd edn, 1970 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |