
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradable polyisoprene by radical ring-opening polymerization and application to polymer prodrug nanoparticles†
Maëlle
Lages
a,
Théo
Pesenti
 a,
Chen
Zhu
a,
Dao
Le
a,
Julie
Mougin
a,
Yohann
Guillaneuf
b and
Julien
Nicolas
*a
a,
Chen
Zhu
a,
Dao
Le
a,
Julie
Mougin
a,
Yohann
Guillaneuf
b and
Julien
Nicolas
*a
aUniversité Paris-Saclay, CNRS, Institut Galien Paris-Saclay, 17 Avenue des Sciences, 91400 Orsay, France. E-mail: julien.nicolas@universite-paris-saclay.fr; Web: https://twitter.com/julnicolas Tel: +33 1 80 00 60 81
bAix-Marseille-Univ., CNRS, Institut de Chimie Radicalaire, UMR 7273, F-13397 Marseille, France
First published on 6th March 2023
Abstract
Radical ring-opening polymerization (rROP) has received renewed attention to incorporate cleavable linkages into the backbones of vinyl polymers, especially from cyclic ketene acetals (CKAs). Among the monomers that hardly copolymerize with CKAs are (1,3)-dienes such as isoprene (I). This is unfortunate since synthetic polyisoprene (PI) and its derivatives are the materials of choice for many applications, in particular as elastomers in the automotive, sport, footwear, and medical industries, but also in nanomedicine. Thionolactones have been recently proposed as a new class of rROP-compatible monomers for insertion of thioester units in the main chain. Herein, we report the synthesis of degradable PI by rROP via the copolymerization of I and dibenzo[c,e]oxepane-5-thione (DOT). Free-radical polymerization as well as two reversible deactivation radical polymerization techniques were successfully used for the synthesis of (well-defined) P(I-co-DOT) copolymers with adjustable molecular weights and DOT contents (2.7–9.7 mol%). Reactivity ratios of rDOT = 4.29 and rI = 0.14 were determined, suggesting preferential incorporation of DOT in comparison to I. The resulting P(I-co-DOT) copolymers were successfully degraded (from −47% to −84% decrease in Mn) under basic conditions. As a proof of concept, the P(I-co-DOT) copolymers were formulated into stable and narrowly dispersed nanoparticles, showing similar cytocompatibility on J774.A1 and HUVEC cells compared to their PI counterparts. Furthermore, Gem-P(I-co-DOT) prodrug nanoparticles were synthesized by the “drug-initiated” method and exhibited significant cytotoxicity on A549 cancer cells. P(I-co-DOT) and Gem-P(I-co-DOT) nanoparticles were degraded under basic/oxidative conditions by bleach and under physiological conditions in the presence of cysteine or glutathione.
Introduction
Degradable vinyl polymers are receiving considerable attention due to their application in the biomedical field (e.g., safety of injected materials and facilitated excretion),1 but also to address environmental issues (e.g., environmental preservation and the circular plastic economy).2–4 In this context, there has been a renewed interest in radical ring-opening copolymerization (rROP)5 as a means of imparting degradability to vinyl polymers through the “cleavable comonomer” approach, particularly with cyclic ketene acetals (CKAs) which are precursors to ester groups in the polymer backbone.6 Yet, CKAs exhibit important limitations such as: (i) a low reactivity towards important classes of vinyl monomers (e.g., acrylates, styrenics, methacrylates, etc.), thus requiring at best a large excess of CKAs in the comonomer feed; (ii) an often significant proportion of ring-retained CKAs during polymerization, resulting in non-degradable acetal units in the copolymer backbone and (iii) a very poor stability in protic solvents or in the presence of traces of water, thus severely limiting their use for instance in polymerization in aqueous dispersed media.5,6Among the monomers that hardly copolymerize with CKAs are (1,3)-dienes such as isoprene (I). Synthetic polyisoprene (PI) and its derivatives are the materials of choice for many applications, in particular as elastomers in the automotive, sport, footwear and medical industries,7,8 with a global PI market size estimated at USD 2.15 billion in 2020.9 PI-based materials have also been extensively studied for application in colloids,10–12 materials science/nanotechnology13–16 and drug delivery owing to their biocompatibility and structural analogy with natural terpenoids.17–22 Moreover, the inclusion of double bonds in the polymer backbone provides an opportunity for further functionalization via silylation or thiol-ene chemistry.23,24 Even if PI can be degraded by thermal,25 chemical (e.g., ozone,16,26 chloranil,27 periodic acid,28 potassium persulfate,29 Grubbs catalysts,30,31 and photo-oxidation)32,33 and biological (enzymatic34–36 and bacterial)37 pathways (although they cannot occur under healthy physiological conditions), insertion of labile functionalities into PI backbones, allowing their degradation and potential recyclability, has never been a success. DFT calculations indeed recently predicted very unfavourable reactivity ratios for the free-radical polymerization (FRP) of I with main CKAs (e.g., rMDO = 0.02 and rI = 9.5; rBMDO = 0.006 and rI = 157 at 70 °C),38 suggesting very little incorporation of CKA units into the PI backbone. Experimental results confirmed the very low insertion of 2-methylene-1,3-dioxepane (MDO) while almost no open 5,6-benzo-2-methylene-1,3-dioxepane (BMDO) units were present. Therefore, an efficient pathway for PI degradation via rROP would certainly have major implications in many different areas such as sustainable materials and biomedical applications.
Thionolactones have been recently reported as a new class of cyclic monomers capable of polymerization by rROP and more specifically via a thiocarbonyl-addition-ring opening (TARO) mechanism, enabling insertion of labile thioester groups into the polymer backbone.5 In particular, it has been shown that dibenzo[c,e]oxepane-5-thione (DOT) exhibited superior reactivity towards several vinyl monomers (e.g., acrylates,39,40 acrylamides,41 acrylonitrile,39 styrene (S)42 and maleimide43), extensive stability in protic solvents and in aqueous media, and quantitative ring opening.39–41,43,44 Furthermore, it allows to diversify degradation pathways; from hydrolysis41,42,44,45 to aminolysis39–41 and thiolysis.43 Very recently, DOT was copolymerized with nBA or S by either conventional emulsion polymerization44 and polymerization-induced self-assembly,46 while DOT was also used to impart chemical recyclability to polystyrene.47 Moreover, it has been shown that DOT-containing copolymers can be degraded in the presence of physiologically relevant concentrations of cysteine or glutathione,48 which allows DOT-containing copolymers to be considered in nanomedicine.
Herein, we report the successful rROP of I and DOT by both FRP and two reversible deactivation radical polymerization techniques: nitroxide-mediated polymerization49 (NMP) and reversible addition–fragmentation chain-transfer50 (RAFT) polymerization (Fig. 1). Owing to favourable reactivity ratios, we demonstrated the formation of thioester-containing copolymers that can be readily degraded under basic conditions. To show the versatility and broad applicability of this copolymerization system, we also reported the formulation of the resulting copolymers into nanoparticles as well as the synthesis of degradable polymer prodrug nanoparticles for anticancer therapy.
 | ||
| Fig. 1 Synthesis of degradable vinyl polymer nanoparticles via radical ring opening copolymerization (rROP) of dibenzo[c,e]oxepane-5-thione (DOT) and isoprene (I) in solution followed by nanoprecipitation. | ||
Experimental part
Materials
Isoprene (I, 99%), dicumyl peroxide (DCP, 98%), potassium hydroxide (KOH, 90%), dioxane (anhydrous, 99.8%) and isopropylamine (≥99%) were purchased from Sigma-Aldrich and used as received. (N-tert-Butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy)-propionic acid alkoxyamine (BlocBuilder MA, BB) was kindly supplied by Arkema. 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, >98%) was purchased from TCI. Deuterated chloroform (CDCl3) and tetrahydrofuran (d8-THF) were purchased from Eurisotop. Tetrahydrofuran (THF, HPLC grade), methanol (MeOH, HPLC) and chloroform (CHCl3, HPLC grade) were obtained from VWR Chemicals. Hydrochloric acid (HCl, 37%) was supplied by Carlo-Erba. 2.5% Active chlorine bleach solution was purchased from Leroy Merlin (France). Dibenzo[c,e]oxepane-5-thione (DOT),39 gemcitabine-AMA-SG1 (Gem-AMA-SG1)19 and 2-ethylsulfanylthiocarbonylsulfanylpropionic acid ethyl ester (ETSPE)51 were prepared as reported elsewhere. Pressure tubes (Ace Glass 8648_164, 15 mL-capacity, fitted with a plunger valve and thermowell) were purchased from Sigma-Aldrich.Analytical methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 467 Hz, an acquisition time of 1 s and a relaxation delay of 34 s. The chemical shift scale was calibrated based on added diethyl phosphite at δ = 7.1 ppm.
000 g mol−1). Analyses were performed at 35 °C in chloroform (HPLC grade) at a flowrate of 1 mL min−1 using toluene as a flowrate marker. A conventional calibration curve was constructed based on polystyrene (PS) standards (peak molar masses: Mp = 575–126500 g mol−1) from Polymer Laboratories. A PI calibration curve was constructed by converting the PS standard peak molecular weights, MPS, to PI molecular weights, MPI, using MarkHouwink–Sakurada (MHS) constants determined for both polymers in CCl4 at 25 °C. For PI, the MHS constants used were KPI = 2.44 × 10−4 and αPI = 0.712. For PS, KPS = 7.1 × 10−4 and αPS = 0.54 (MW < 16700 g mol−1) or KPS = 1.44 × 10−4 and αPS = 0.713 (MW ≥ 16700 g mol−1).52,53 This technique allowed Mn (the number-average molar mass), Mw (the weight-average molar mass), and Mw/Mn (the dispersity, Đ) to be determined.
467 Hz, an acquisition time of 1 s and a relaxation delay of 34 s. The chemical shift scale was calibrated based on added diethyl phosphite at δ = 7.1 ppm.
000 g mol−1). Analyses were performed at 35 °C in chloroform (HPLC grade) at a flowrate of 1 mL min−1 using toluene as a flowrate marker. A conventional calibration curve was constructed based on polystyrene (PS) standards (peak molar masses: Mp = 575–126500 g mol−1) from Polymer Laboratories. A PI calibration curve was constructed by converting the PS standard peak molecular weights, MPS, to PI molecular weights, MPI, using MarkHouwink–Sakurada (MHS) constants determined for both polymers in CCl4 at 25 °C. For PI, the MHS constants used were KPI = 2.44 × 10−4 and αPI = 0.712. For PS, KPS = 7.1 × 10−4 and αPS = 0.54 (MW < 16700 g mol−1) or KPS = 1.44 × 10−4 and αPS = 0.713 (MW ≥ 16700 g mol−1).52,53 This technique allowed Mn (the number-average molar mass), Mw (the weight-average molar mass), and Mw/Mn (the dispersity, Đ) to be determined.
For purified and degraded samples, SEC was performed at 30 °C with two columns from Agilent (PL-gel MIXED-D; 300 × 7.5 mm; bead diameter, 5 μm; linear part, 400–400000 g mol−1) and a differential refractive index detector (Spectrasystem RI-150 from Thermo Electron Corp.), using CHCl3 as an eluent at a flowrate of 1 mL min−1 and toluene as a flowrate marker. A conventional calibration curve was constructed based on PS standards (peak molar masses: Mp = 575–126500 g mol−1) from Polymer Laboratories. A PI calibration curve was constructed in the same manner as described above. SEC of degraded copolymers was performed in the presence of 0.1% (v/v) of trifluoroacetic acid (TFA, 99%) in CHCl3 (in both the mobile phase and the sample) to avoid the formation of aggregates and/or interaction with the columns and sulfhydryle or hydroxyl chain ends.
Synthetic procedures
| Entry | f DOT,0 (mol%) | t (h) | DPn,th | Conv. Ia (wt%) | M n,th (g mol−1) | M n (g mol−1) | M w (g mol−1) | Đ | F DOT (mol%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by gravimetry. b Determined according to: Mn,th=Mn,BB + DPn,th × conv. MWI. c Determined by SEC on precipitated samples. d Determined by 1H NMR in d8-THF of precipitated samples by integrating the 8H (Ar) of open DOT at 6.7–8 ppm and protons from PI (1H for the (1,4) conformation at 5–5.5 ppm, 1H for the (1,2) conformation at 5.5–5.9 ppm and 4.4–5 ppm combining 2H of (1,2) and 2H of (3,4) conformations). For F1–F2 and R1–R2, the aromatic signal of DCP at 7.0–7.5 ppm was subtracted from the aromatic signal of DOT according to the DCP/PI ratio calculated on the F0 and R0 spectrum respectively. e The Gem-AMA-SG1 alkoxyamine was used instead of the BB alkoxyamine. | ||||||||||
| FRP | F0 | 0 | 30 | — | 37 | — | 7200 | 11000 |
1.53 | 0 |
| F1 | 1 | 30 | — | 38 | — | 10000 |
16400 |
1.64 | 2.7 | |
| F2 | 2 | 30 | — | 40 | — | 10400 |
17000 |
1.63 | 4.6 | |
| NMP | N0 | 0 | 48 | 100 | 33 | 2700 | 2400 | 2900 | 1.19 | 0 |
| N1 | 1 | 48 | 100 | 37 | 2900 | 3100 | 3900 | 1.25 | 2.7 | |
| N2 | 2 | 48 | 100 | 40 | 3100 | 3300 | 4400 | 1.32 | 4.4 | |
| N3 | 2 | 48 | 215 | 36 | 5700 | 5300 | 8200 | 1.54 | 5.3 | |
| N4 | 2 | 48 | 435 | 21 | 6600 | 4800 | 7100 | 1.48 | 9.7 | |
| N5 | 2 | 16 | 100 | 17 | 1800 | 1800 | 2200 | 1.29 | 6.6 | |
| N6 | 3 | 20 | 500 | 18 | 6500 | 7900 | 8400 | 1.07 | 6.1 | |
| N7 | 3 | 20 | 500 | 22 | 8100 | 9300 | 10700 |
1.15 | 6.9 | |
| RAFT | R0 | 0 | 24 | 500 | 11 | 3900 | 3400 | 3900 | 1.16 | 0 |
| R1 | 2 | 24 | 500 | 10 | 3800 | 4500 | 6100 | 1.36 | 5.5 | |
| R2 | 2 | 72 | 600 | 55 | 22900 |
11000 |
18200 |
1.67 | 3.9 | |
The same procedure was adapted by varying the initial feed ratio of DOT (fDOT,0) as follows: F0 [I (4 mL, 2.73 g, 40 mmol) and DCP (0.025 g, 0.093 mmol) in 5 mL of dioxane] and F2 [I (4 mL, 2.73 g, 40 mmol), DOT (0.185 g, 0.816 mmol, fDOT,0 = 0.02) and DCP (0.025 g, 0.093 mmol) in 5 mL of dioxane].
The determination of the reactivity ratios was performed according to the following protocol. In a typical procedure, I (0.08 mL, 54 mg, 0.79 mmol), DOT (179 mg, 0.79 mmol, fDOT,0 = 0.5), DCP (0.9 mg, 0.0036 mmol), anhydrous DMSO (61 mg, 0.79 mmol) used as the internal reference, and dioxane (7 mL) were introduced in a pressure tube fitted with a plunger valve and thermowell. The solution was subjected to three cycles of freeze-thaw degassing, and then backfilled with argon. The tube was placed in an oil bath at 115 °C for 1 h 30 and then cooled down at room temperature. The monomer conversions (kept at a low level; 34% maximum) were determined by 1H NMR in CDCl3 by integrating the signal at 6.4 ppm for I (dd, 1H), the signal at 8.2 ppm for DOT (d, 1H), and the signal of the internal reference at 2.6 ppm (d6-DMSO, s, 6H). The copolymer was then purified by two successive precipitations in MeOH to remove unreacted DOT and dried under vacuum at room temperature. The resulting copolymer was analysed by 1H NMR in d8-THF to determine FDOT. Different fDOT,0 values were considered (0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7 and 0.9) by keeping a total monomer concentration of 1.13 M (Table S1†). Evolution of FDOT with fDOT,0 was then plotted, and the reactivity ratios were determined using nonlinear least-squares (NLLS) analysis performed with the CONTOUR software.54
The same procedure was then adapted by varying fDOT,0 and the targeted number-average degree of polymerization of I (DPn,th) as follows: N0 [I (6 mL, 4.086 g, 60 mmol, 100 eq.), BB (0.229 g, 0.60 mmol, 1 eq.) and dioxane (6 mL)]; N1 [I (6 mL, 4.086 g, 60 mmol, 100 eq.), DOT (0.137 g, 0.61 mmol, 1.01 eq., fDOT,0 = 0.01), BB (0.229 g, 0.60 mmol, 1 eq.) and dioxane (6 mL)]; N3 [I (6 mL, 4.086 g, 60 mmol, 215 eq.), DOT (0.277 g, 1.22 mmol, 4.4 eq., fDOT,0 = 0.02), BB (0.106 g, 0.279 mmol, 1 eq.) and dioxane (6 mL)]; N4 [I (6 mL, 4.086 g, 60 mmol, 435 eq.), DOT (0.277 g, 1.22 mmol, 8.88 eq., fDOT,0 = 0.02), BB (0.053 g, 0.138 mmol, 1 eq.) and dioxane (6 mL)] and N6 [I (1.50 mL, 1.02 g, 15 mmol, 500 eq.), DOT (0.105 g, 0.46 mmol, 15.5 eq., fDOT,0 = 0.03), BB (0.0114 g, 0.03 mmol, 1 eq.) and dioxane (1.50 mL)]. For N3′ [I (6 mL, 4.086 g, 60 mmol, 215 eq.), DOT (0.277 g, 1.22 mmol, 4.4 eq., fDOT,0 = 0.02), BB (0.106 g, 0.279 mmol, 1 eq.), dioxane (6 mL) and anhydrous DMSO (0.056 mL, 0.123 g, 1.5 mmol)], anhydrous DMSO was added as an internal reference to determine the DOT conversion by 1H NMR in CDCl3.
The same procedure was then adapted by varying fDOT,0, DPn,th and reaction time as follows: R0 [I (2.86 g, 42 mmol, 500 eq.), ETSPE (0.02 g, 0.084 mmol, 1 eq.) and DCP (0.0045 g, 0.016 mmol, 0.2 eq.)] and R2 [I (1.47 g, 21.6 mmol, 600 eq.), DOT (0.099 g, 0.441 mmol, 12.25 eq., fDOT,0 = 0.02), ETSPE (0.008 g, 0.036 mmol, 1 eq.) and DCP (0.0019 g, 0.007 mmol, 0.2 eq.) for 72 h].
The same procedure was then adapted by targeting DPn,th = 500 as follows: N7 [I (1.30 mL, 0.885 g, 13 mmol, 500 eq.)], DOT (0.091 g, 0.40 mmol, 15 eq., fDOT,0 = 0.03), Gem-AMA-SG1 (0.016 g, 0.026 mmol, 1 eq.) and 1.30 mL of dioxane.
Nanoparticle preparation
Nanoparticles were prepared by the nanoprecipitation technique. Briefly, 10 mg of polymer N0–N5 (Table 1) were dissolved in 2 mL of THF and added dropwise to 3 mL MilliQ water under stirring. THF was removed under vacuum to reach a final concentration of 3.3 mg mL−1. For N6, 20 mg of polymer (Table 1) were dissolved in 8 mL of dioxane and added dropwise to 12 mL MilliQ water under stirring. Dioxane was removed under vacuum to reach a final concentration of 1.7 mg mL−1. For N7, volumes of dioxane and water were divided by two to reach a final concentration of 3.3 mg mL−1. Intensity-average diameter (Dz) and zeta potential measurements were carried out in triplicate.Degradation procedures
In vitro cytotoxicity
Results and discussion
Synthesis of DOT-containing polyisoprene
Copolymerization of DOT with I was first investigated by free-radical copolymerization in dioxane at 115 °C (Fig. 2). Initial molar fractions in DOT (fDOT,0) up to 2 mol% were considered due its low solubility in dioxane. After 30 h of copolymerization, experiments F1 (fDOT,0 = 0.01) and F2 (fDOT,0 = 0.02) resulted in ∼40% conversion of I and Mns of ∼10000 g mol−1 (Table 1).
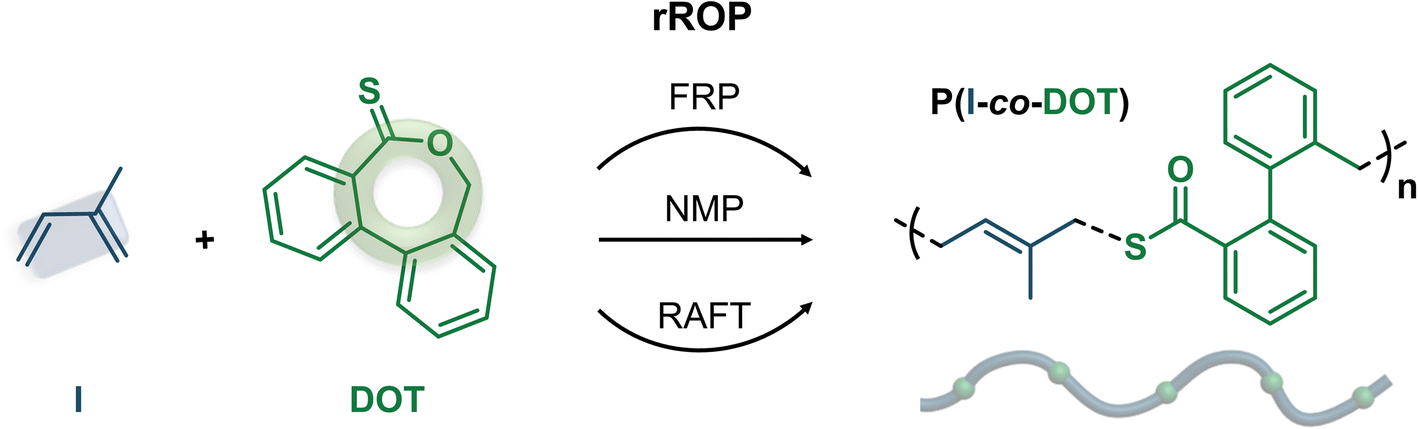 | ||
| Fig. 2 Synthesis of poly(isoprene-co-dibenzo[c,e]oxepane-5-thione) (P(I-co-DOT)) copolymers by radical ring-opening copolymerization (rROP) of DOT and I in dioxane at 115 °C by: (a) free-radical polymerization (FRP); (b) nitroxide-mediated polymerization (NMP) and (c) reversible addition–fragmentation chain-transfer (RAFT) polymerization. | ||
Homopolymerization of I under identical polymerization conditions gave similar results (F0, Table 1), suggesting no retardation effect of DOT with I, in contrast to other vinyl monomers.39,44 H1 NMR spectroscopy was then performed on the purified (co)polymers, showing successful insertion of DOT units to an extent of FDOT = 2.7 and 4.6 mol%, for experiments F1 and F2, respectively (Fig. 3a and Table 1). Importantly, CKAs required a much higher initial feed ratio of 75 mol% to only incorporate 7 mol% of MDO in the PI backbone.38 This was supported by theoretical DFT calculations that reported rMDO = 0.02 and rI = 9.5 (or rBMDO = 0.006 and rI = 157) at 70 °C for copolymerization between MDO (or BMDO) and I.38 This therefore suggested a superior reactivity of DOT towards I compared to CKAs. Reactivity ratios of this copolymerization system were estimated using a NLLS method performed with the CONTOUR software,54 giving rDOT = 4.29 and rI = 0.14 (the 95% joint confidence interval is given in Fig. S1†). This suggested a significant preferential incorporation of DOT over I and a high heterogeneity of the DOT fraction between chains if high conversion is reached (the latter expected from a FRP process). Similar trends, which can be explained by the structural similarity between DOT and RAFT agents, have been previously shown for the copolymerization of DOT with other vinyl monomers, such as N,N-dimethylacrylamide (rDOT = 1.89 and rDMAm = 0.34), N-2,3,4,5,6-pentafluorophenylmaleimide (rDOT = 0.198 and rPFPMI = 0.0078) and N-phenylmaleimide (rDOT = 0.348 and rPhMI = 0.0136).41
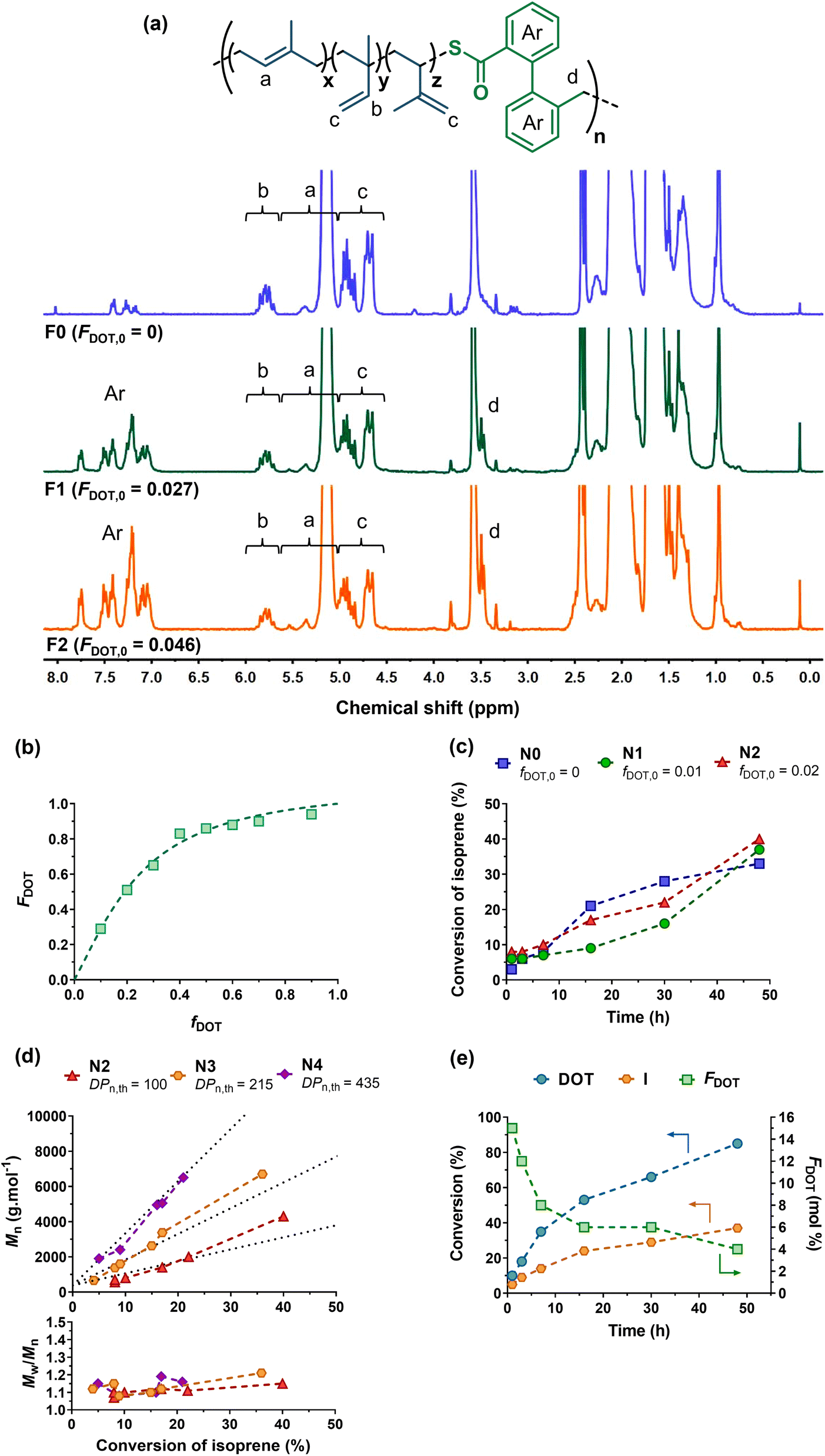 | ||
| Fig. 3 (a) 1H NMR spectrum (300 MHz, d8-THF) in the 0–8 ppm region of P(I-co-DOT) copolymers F0, F1 and F2 (Table 1). (b) Evolution of molar DOT content in copolymers with molar DOT in the monomer feed with nonlinear least-squares fitted curves with rDOT = 4.29 and rI = 0.14. (c) Evolution of I conversion vs. time as a function of fDOT,0 (N0, N1 and N2, Table 1). (d) Evolution of Mn and Đ vs. I conversion as a function of DPn,th (N2, N3 and N4, Table 1). (e) Evolution of I conversion, DOT conversion and average molar fraction of DOT (FDOT) in the copolymer vs. time (N3′). | ||
Copolymerizations via NMP and RAFT were then investigated to yield well-defined P(I-co-DOT) copolymers. NMP of I using BB as a SG1-based alkoxyamine was performed with fDOT,0 = 1 and 2 mol% in dioxane and with DPn,th = 100 (N0–N2, Table 1). 30–40% conversions of I were obtained after 48 h, with Mn values in the 2400–3300 g mol−1 range and low dispersities (Đ = 1.19–1.32), indicating a good control even in the presence of DOT. Interestingly, no noticeable retardation effect from DOT was obtained, despite the increasing initial molar fraction of DOT in the comonomer feed39,44 (Fig. 3c). At fDOT,0 = 2 mol%, different Mn,th were targeted by varying the DPn,th from 100 to 435 (N2–N4, Fig. 3c–e, Table 1). Interestingly, the copolymerizations exhibited all the characteristics of a controlled system, as confirmed by: (i) the linear evolutions of the Mn values with I conversion, in pretty good agreement with the theoretical ones (Fig. 3d) and (ii) the low dispersities (Đ < 1.2) throughout the reactions (Fig. 3d and S2†). The final copolymers were then purified and analysed by 1H NMR (Table 1, Fig. S3†), which successfully demonstrated insertion of open DOT units into the PI backbone at 4.4 and 5.3 mol% for N2 and N3, respectively. The final amount of DOT in the copolymer was correlated to: (i) the initial stoichiometry, as the higher fDOT,0, the higher the FDOT (see N1 and N2, Table 1) and (ii) the I conversion, as the lower the I conversion, the higher the FDOT, in agreement with the reactivity ratios and thus the expected composition drift of P(I-co-DOT) copolymers (see N2–N4, Table 1). In order to gain further insight into this copolymerization system, the evolutions of the individual conversions of DOT and I, as well as FDOT during the copolymerization were studied for N3. Note that N3 was repeated in the presence of DMSO as the internal reference and noted as N3′ (Fig. 3e and S4†). It appeared that DOT was indeed consumed faster than I, leading to 85% DOT conversion and 37% I conversion after 48 h. As expected, the evolution of FDOT progressively decreased from 15.5 to 4.3 mol% after 48 h in agreement with a gradient composition.
N3′ and N0 were also analysed by 31P NMR to perform in-depth chain-end characterization and give crucial insights into the nature of the copolymer terminal sequences and the living chain fractions.57 Interestingly, the phosphorus signals (24.4–24.6 ppm) from N3′ and N0 had the same shape and were characteristic of I-SG1 terminal sequences (Fig. S5†),53 thus ruling out accumulation of DOT as the last monomer unit and supporting its faster incorporation in the copolymer compared to I. By integrating these phosphorus signals and that of diethyl phosphite (7.1 ppm) used as an internal standard, the livingness of both (co)polymers was estimated to be ∼50%.
P(I-co-DOT) copolymers were also synthesized by RAFT polymerization using ETSPE as a RAFT agent56 and DCP as a source of radicals. After 24 h with fDOT,0 = 0.02, 10% of I conversion was achieved giving Mn = 4500 g mol−1 and Đ = 1.36 (R1, Table 1). The absence of DOT under the same conditions (R0, Table 1) led to very similar macromolecular characteristics, despite obtaining a lower dispersity (Đ = 1.16). To obtain higher molecular weights, the copolymerization reaction was then carried out for 72 h with DPn,th = 600 (R2, Table 1) instead of 500. This time, 55% conversion was obtained as well as Mn = 11000 g mol−1 with however a higher dispersity (Đ = 1.67). The DOT contents in the P(I-co-DOT) copolymers were determined to be 5.5 and 3.9 mol% for R1 and R2, respectively (Table 1 and Fig. S6†), which is also in line with the faster incorporation of DOT compared to I. Therefore, these results confirmed the ability of DOT to be inserted into the PI backbone by FRP, NMP and RAFT polymerization mechanisms.
Interestingly, the values of FDOTvs. conversion for F2, N2–N5, N3′ and R1–R2 are in fairly good agreement with the theoretical evolution of FDOTvs. conversion plotted using a PREDICI numerical simulation58 that predicts the compositional drift occurring during the copolymerization between vinyl and cyclic monomers, with rDOT, rI and fDOT,0 as the input parameters (Fig. S7†). This observation therefore validates the values of the reactivity ratios that we determined. It is also important to note that the copolymerization behaviour is dramatically different when the theoretical evolution of FMDOvs. conversion is plotted using the corresponding reactivity ratios38 and the same fMDO,0 value as for DOT. Indeed, FMDO values stay below 0.003 regardless of conversion, in contrast to FDOT values that range from 0.12 to 0.02 with conversion. This result highlights the strong added value of DOT over CKA in incorporating weak bonds into polyisoprene chains.
Degradation of P(I-co-DOT) copolymers
Degradation of the copolymers was then performed to confirm the successful insertion of thioester groups. Different degradation conditions were used (e.g., KOH, TBD and isopropylamine)39–44 to investigate hydrolysis and aminolysis as two distinct degradation modalities (in addition to physiological48 and oxidative59 degradations of thioester-containing copolymers). In general, whatever the polymerization methods used to generate the copolymers, significant degradation was observed after their treatment with either KOH or TBD for 16 h (Table 2, Fig. 4, S8 and S9†). The decrease in Mn and Mw ranged from −47 to −79% with KOH and from −55 to −84% with TBD (Table 2). Conversely, the Mn of PI homopolymers (F0, N0 and R0) after degradation under the same conditions stayed constant (Fig. S8–S10† and Table 2). The degradation was also in agreement with the DOT content as increasing FDOT,0 from 0.027 (F1, Table 2) up to 0.046 (F2, Table 2) led to a higher decrease in Mn and Mw in the presence of TBD or KOH compared to F1. A similar trend was observed for copolymers obtained by NMP (N1–N4, Table 2). In addition, Mn,deg values were in fairly good agreement with theoretical Mns values of the copolymers after degradation (Mn,deg.th, Table 2). However, no degradation was shown in the presence of isopropylamine, which could be correlated to the strong hydrophobicity of PI, similar to that observed with styrene.42,44| Entry | M n (g mol−1) | M w (g mol−1) | Đ | F DOT (mol%) | M n,deg.th (g mol−1) | Degradation by KOHd | Degradation by isopropylaminee | Degradation by TBDf | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M n,deg.KOH (g mol−1) (% Mn loss) | M w,deg.KOH (g mol−1) (% Mw loss) | Đ | M n,deg.isoprop (g mol−1) (% Mn loss) | M w,deg. isoprop (g mol−1) (% Mw loss) | Đ | M n,deg.TBD (g mol−1) (% Mn loss) | M w,deg.TBD (g mol−1) (% Mw loss) | Đ | |||||||
| a Determined by SEC on precipitated samples. b Determined by 1H NMR in d8-THF of precipitated samples by integrating the 8H (Ar) of open DOT at 6.7–8 ppm and protons from PI (1H for the (1,4) conformation at 5–5.5 ppm, 1H for the (1,2) conformation at 5.5–5.9 ppm and 4.4–5 ppm combining 2H of (1,2) and 2H of (3,4) conformations). For F1–F2 and R1–R2, the aromatic signal of DCP at 7.0–7.5 ppm was subtracted from the aromatic signal of DOT according to the DCP/PI ratio calculated on the F0 and R0 spectrum respectively. c Determined according to: Mn,deg.th = [((1 − FDOT)/FDOT) × MWI] + MWDOT. d Determined by SEC after degradation by KOH for 16 h. e Determined by SEC after degradation by isopropylamine for 16 h. f Determined by SEC after degradation by TBD for 16 h. % Mn loss is calculated according to: % Mn loss = [(Mn − Mn, deg)/Mn] × 100. g The Gem-AMA-SG1 alkoxyamine was used instead of the BB alkoxyamine. | |||||||||||||||
| FRP | F0 | 7200 | 11000 |
1.53 | 0 | 7200 | 7200 (0) | 11000 (0) |
1.52 | 7200 (0) | 10900 (−1) |
1.52 | 7200 (0) | 10900 (−1) |
1.51 |
| F1 | 10000 |
16400 |
1.64 | 2.7 | 2700 | 5300 (−47) | 10800 (−34) |
2.04 | 9600 (−4) | 14400 (−12) |
1.50 | 2700 (−73) | 4800 (−70) | 1.76 | |
| F2 | 10400 |
17000 |
1.63 | 4.6 | 1600 | 4100 (−61) | 8100 (−52) | 1.98 | 9600 (−8) | 15600 (−8) |
1.63 | 1700 (−84) | 3200 (−81) | 1.88 | |
| NMP | N0 | 2400 | 2900 | 1.19 | 0 | 2400 | 2400 (0) | 2900 (0) | 1.20 | 2400 (0) | 2900 (0) | 1.19 | 2400 (0) | 2900 (0) | 1.20 |
| N1 | 3100 | 3900 | 1.25 | 2.7 | 2700 | 1600 (−48) | 2700 (−30) | 1.69 | 3100 (0) | 3900 (0) | 1.26 | 1400 (−55) | 2400 (−38) | 1.71 | |
| N2 | 3300 | 4400 | 1.32 | 4.4 | 1700 | 1600 (−52) | 2800 (−36) | 1.73 | 3300 (0) | 4400 (0) | 1.35 | 1300 (−61) | 2200 (−50) | 1.66 | |
| N3 | 5300 | 8200 | 1.54 | 5.3 | 1400 | 1700 (−68) | 2900 (−64) | 1.73 | 5200 (−2) | 7600 (−7) | 1.47 | 1100 (−80) | 2300 (−72) | 2.10 | |
| N4 | 4800 | 7100 | 1.48 | 9.7 | 900 | 1000 (−79) | 1600 (−77) | 1.63 | 4700 (−3) | 6700 (−6) | 1.43 | 800 (−83) | 1700 (−76) | 2.10 | |
| N5 | 1800 | 2200 | 1.29 | 6.6 | 1200 | — | — | — | — | — | — | 700 (−61) | 1000 (−55) | 1.55 | |
| N6 | 7900 | 8400 | 1.07 | 6.1 | 1300 | 1700 (−78) | 2640 (−68) | 1.55 | — | — | — | — | — | ||
| N7 | 9300 | 10700 |
1.15 | 6.9 | 1150 | 2100 (−71) | 4650 (−56) | 2.22 | — | — | — | — | — | — | |
| RAFT | R0 | 3400 | 3900 | 1.16 | 0 | 3400 | 3400 (0) | 4300 (0) | 1.26 | — | — | — | 3400 (0) | 4400 (0) | 1.28 |
| R1 | 4500 | 6100 | 1.36 | 5.5 | 1400 | 1300 (−71) | 3500 (−43) | 2.63 | — | — | — | 900 (−80) | 1800 (−70) | 1.93 | |
| R2 | 11000 |
18200 |
1.67 | 3.9 | 1900 | 3300 (−70) | 7600 (−58) | 2.30 | — | — | — | 2300 (−79) | 4300 (−76) | 1.86 | |
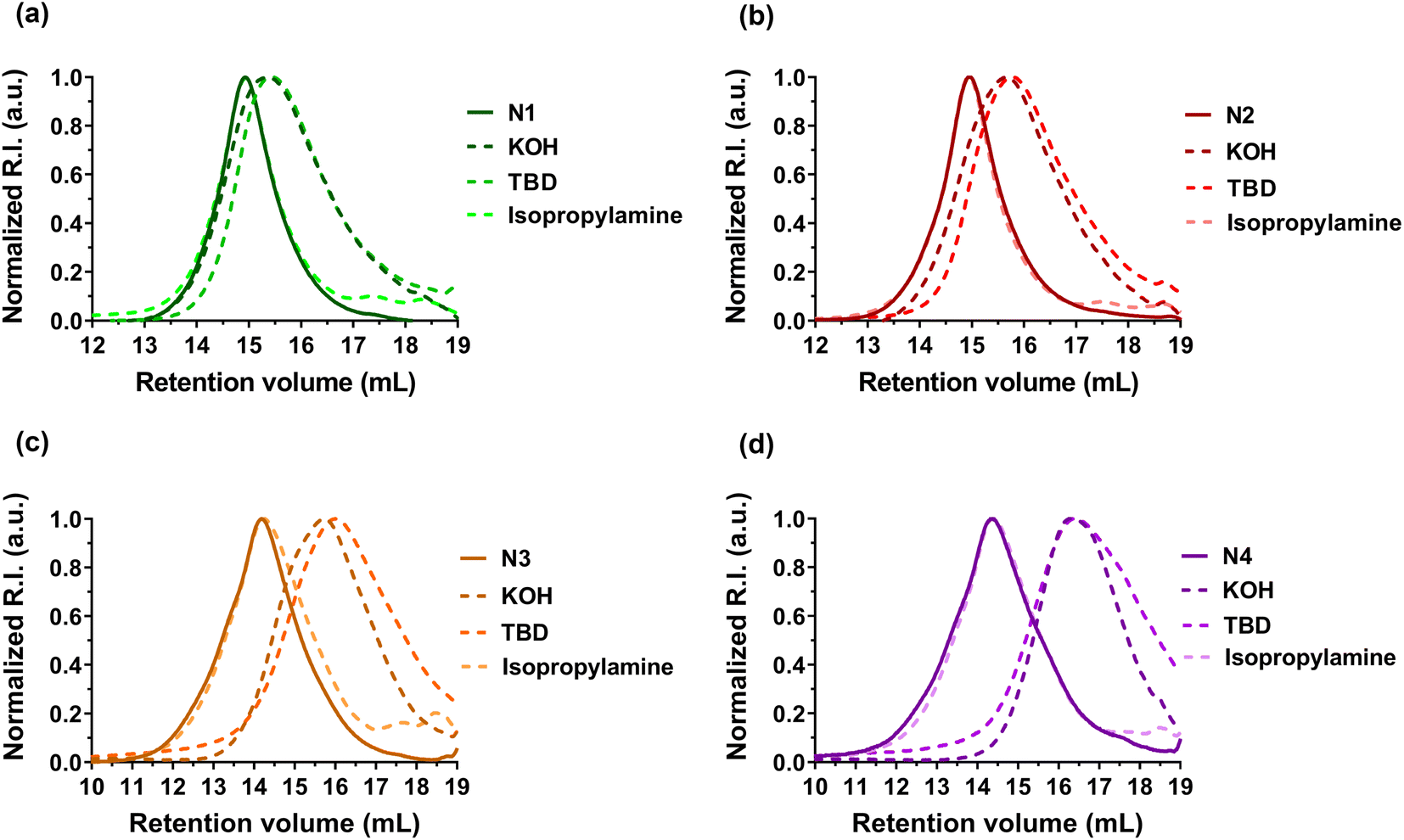 | ||
| Fig. 4 Evolution of the SEC chromatograms of P(I-co-DOT) copolymers: (a) N1, (b) N2, (c) N3, and (d) N4, after degradation under basic (KOH or TBD) or aminolytic (isopropylamine) conditions. | ||
Application to nanoparticles and polymer prodrug nanoparticles
Polymer nanoparticles for drug delivery purposes are usually obtained by formulation of preformed polymers such as aliphatic polyesters, poly(alkyl cyanoacrylate), etc.60–64 These polymers, which are few in number, are chosen because of their biocompatibility and biodegradability compared to non-degradable materials, which can accumulate in the body and induce prohibitive side effects. In order to diversify the range of polymers suitable for biomedical applications, the design of (bio)degradable vinyl polymers is currently of great interest.1We illustrated the potential of the I/DOT copolymerization system for drug delivery purposes by first investigating the formulation of P(I-co-DOT) copolymers into nanoparticles (N0–N4, Table 3). Narrowly dispersed nanoparticles were successfully formed by nanoprecipitation, exhibiting an average diameter between 105 and 150 nm (Table 3 and Fig. S11†). The nanoparticle diameters also tended to decrease with the increase of the copolymer chain length, in agreement with previous data on PI nanoparticles.19 This appeared to be attributed to an increase in hydrophobic interactions between PI chains as they lengthen, making the core of the nanoparticles more compact. All nanoparticles also exhibited strongly negative zeta potentials (Table 3), suggesting efficient electrostatic stabilisation, which was confirmed by their remarkable long-term colloidal stability up to one month (Table 3 and Fig. 5a, S12†). Cryo-TEM analysis also revealed the formation of nanoparticles with spherical morphologies (Fig. 5b). Overall, insertion of thioester units in the PI backbone did not impact the colloidal characteristics (size, stability and morphology) of the nanoparticles compared to their non-degradable PI counterparts.19
| Entry | D z (nm) | PDIa | ζ (mV) | % Mn loss, deg.TBDb | % Mw loss, deg.TBDb | Đ |
|---|---|---|---|---|---|---|
| a Determined by DLS. b Determined by SEC after degradation of the nanoparticles in water with TBD for 48 h. | ||||||
| N0 | 150 | 0.09 | −56 | 0 | −3 | 1.17 |
| N1 | 140 | 0.15 | −27 | −45 | −30 | 1.58 |
| N2 | 145 | 0.10 | −50 | −50 | −26 | 2.30 |
| N3 | 130 | 0.07 | −46 | −56 | −48 | 2.05 |
| N4 | 105 | 0.09 | −27 | −59 | −39 | 2.24 |
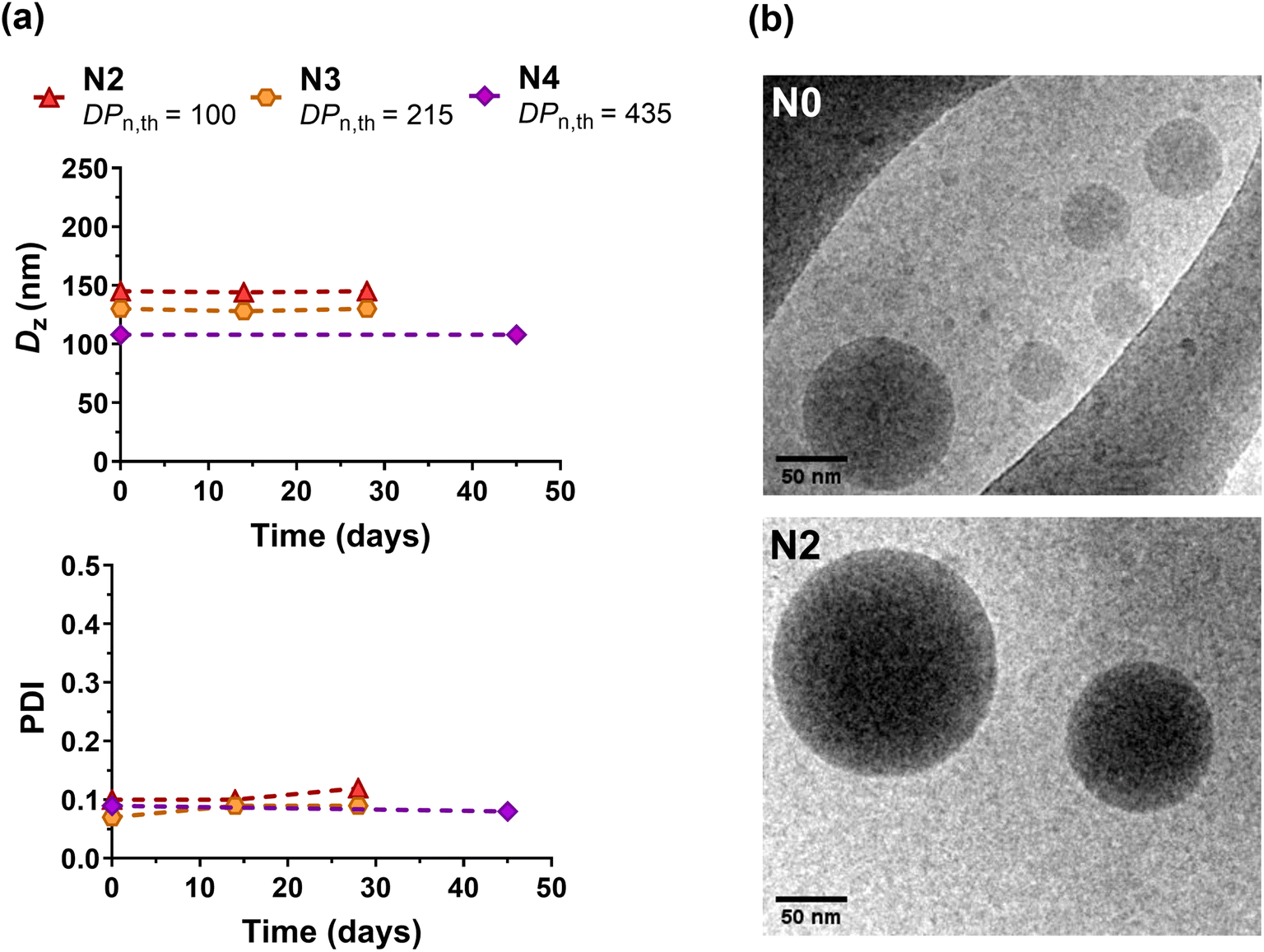 | ||
| Fig. 5 (a) Evolution of the intensity-average diameter (Dz) and polydispersity index (PDI) with time measured by DLS of P(I-co-DOT) nanoparticles (N2, N3 and N4, Table 3). (b) Representative cryo-TEM images of nanoparticles N0 and N2. | ||
Direct degradation of the nanoparticles was then attempted in the presence of TBD for 48 h at room temperature. Despite the strong hydrophobicity of the copolymer that may prevent efficient access of TBD to the hydrophobic core of nanoparticles, the decrease in Mn and Mw reached 40–59% and 26–48%, respectively (N1–N4, Table 3). The higher the FDOT, the greater the degradation, even if this trend was less pronounced than that for the copolymers, probably for reasons of accessibility to the thioester group in the nanoparticulate state. However, longer degradation times and/or higher concentrations of TBD may have certainly enhanced the degradation of the nanoparticles.
Cell viability experiments were then performed on two different healthy cell lines (J774.A1 and HUVEC), during which P(I-co-DOT) nanoparticles or the degradation products of the corresponding copolymers were evaluated (Fig. 6a and b). Half-maximal inhibitory concentrations (IC50) of cell proliferation of 7.9 μg mL−1 and 0.31 mg mL−1 were obtained for J774.A1 and HUVEC cells, respectively. These values are identical to the IC50 values obtained for PI nanoparticles (Fig. 6a and b), demonstrating the absence of the cytotoxicity effect from DOT units. Moreover, the degradation products exhibited similar IC50 values compared to the nanoparticles for HUVEC cells and much higher IC50 values on J774.A1 cells (0.11 mg mL−1). These results are very encouraging and let envision the use of P(I-co-DOT) as a carrier material for the design of degradable polymer nanoparticles for drug delivery.
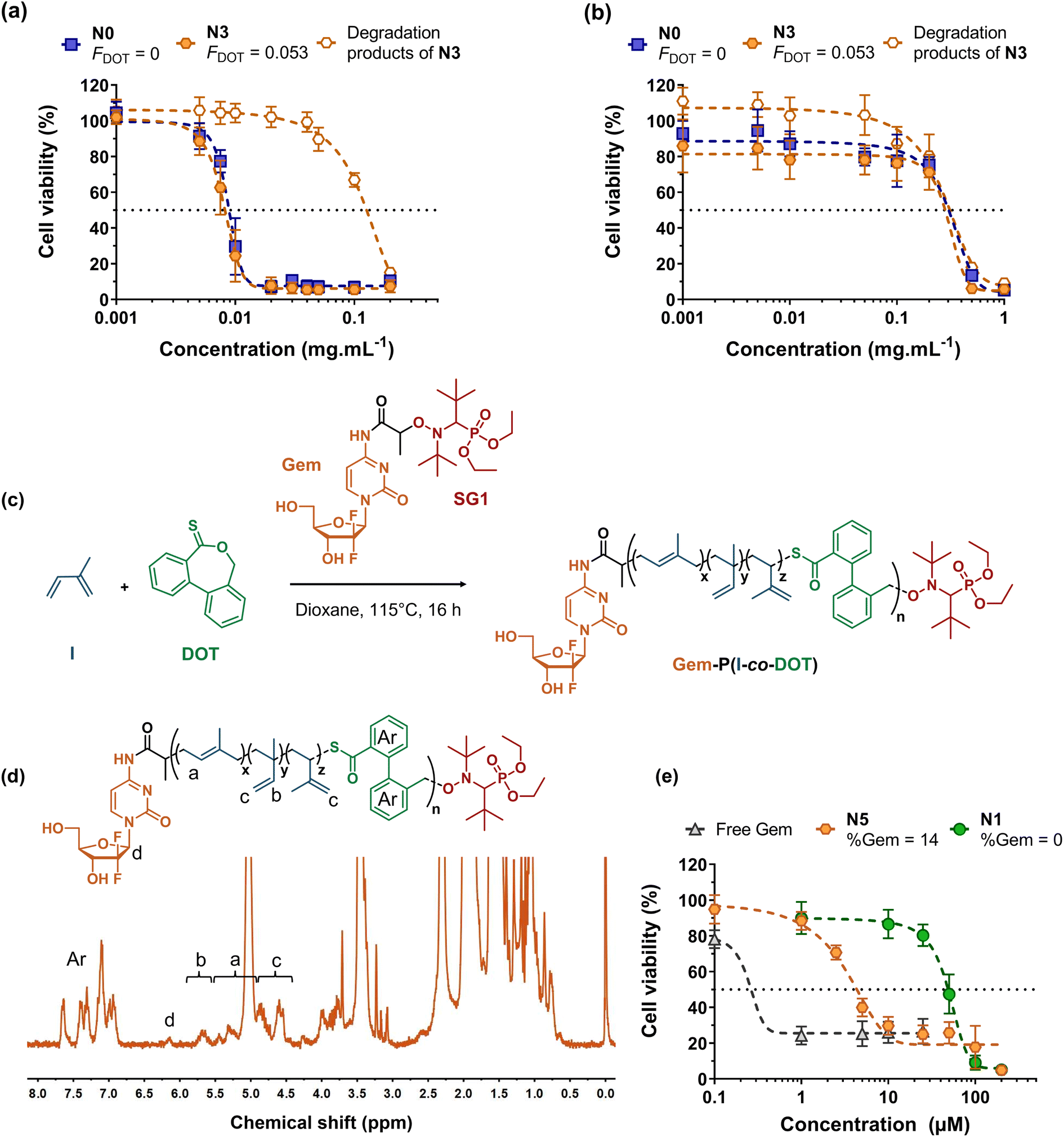 | ||
| Fig. 6 Cell viability of: (a) murine macrophage (J774.A1) and (b) human endothelial cell (HUVEC) lines as a function of the concentration of nanoparticles N3 and N0 (Table 3) and of N3 degradation products. (c) Synthesis of the Gem-P(I-co-DOT) polymer prodrug by radical ring-opening polymerization (rROP) of dibenzo[c,e]oxepane-5-thione (DOT) and isoprene (I) via nitroxide-mediated polymerization using a gemcitabine-functionalized macroalcoxyamine based on the nitroxide SG1 (Gem-AMA-SG1). (d) 1H NMR spectrum (300 MHz, d8-THF) in the 0–8 ppm region of the Gem-P(I-co-DOT) polymer prodrug N5 (Table 1). (e) Cell viability of the lung cancer cell line (A549) as a function of the concentration of Gem-P(I-co-DOT) nanoparticles (N5, Table 1), P(I-co-DOT) nanoparticles (N1, Table 1) and free gemcitabine. | ||
Of the many polymer-based drug delivery systems developed to date, polymer-drug nanocarriers, in which the drug is covalently bound to the polymer, are the most promising because they circumvent the limitations associated with physical encapsulation of drugs (e.g., burst release, poor drug loading, etc.).65–68 While grafting drugs to a preformed polymer is the most popular strategy to produce polymer prodrugs, the “drug-initiated” method,69 which consists in growing a polymer from a drug to obtain one drug molecule at the extremity of a well-defined polymer chain, has recently received increasing attention as an easy and scalable route to yield high drug loading, surfactant-free, vinyl polymer prodrug nanoparticles.17,19,21,22,70–72 For instance, this has been applied to the synthesis of the Gem-polyisoprene prodrug by NMP, whose nanoparticles exhibited in vitro and in vivo anticancer activities on different cancer cell lines. However, they were not degradable, which may severely limit their development because of potential accumulation in the body and harmful side effects. Herein, we propose to apply the I/DOT copolymerization system to the synthesis of degradable Gem-P(I-co-DOT) prodrug nanoparticles (Fig. 6c). To install a Gem moiety at the α-position of P(I-co-DOT) copolymer chains, the copolymerization of I and DOT (fDOT,0 = 0.02) was initiated by the Gem-AMA-SG1 alkoxyamine. Gem-AMA-SG1 was obtained by coupling unprotected Gem to the AMA-SG1 alkoxyamine, resulting in an amide bond that can be selectively cleaved by cathepsin B, whose overexpression is correlated with invasive and metastatic cancers.73,74 After 16 h (11% conv. in I), this yielded Gem-P(I-co-DOT) copolymers with Mn = 1800 g mol−1 and Đ = 1.29 (N5, Table 1). 1H NMR analysis of N5 confirmed the synthesis of the expected structure (Fig. 6d), particularly via the presence of Gem (peak d) and the characteristic proton signals of the PI backbone, and gave Mn,NMR = 2400 g mol−1 (by integrating peak a from PI and peak d from Gem, Fig. 6d) in rather good agreement with the Mn,SEC value (Table 1). Targeting such a low Mn allowed the prodrug to exhibit a Gem loading as high as 14 wt% with a FDOT value of 6.6 mol% (Fig. 6d and Table 1). TBD-assisted degradation of Gem-P(I-co-DOT) prodrug N5 led to a decrease in Mn and Mw of 61% and 55%, respectively (very close to the theoretical degradation), thus confirming the presence of thioester groups in the PI backbone and their successful cleavage (Table 2 and Fig. S13†). After nanoprecipitation, narrowly dispersed nanoparticles were obtained with Dz = 140 nm and a low PDI (0.11). They also showed a greater negative zeta potential value (−68 mV) than their drug-free counterparts (Table 3), suggesting the (partial) presence of Gem moieties at the surface nanoparticle surface, in agreement with molecular modelling experiments75 for polymer prodrugs obtained by the “drug-initiated” method.
Gem-P(I-co-DOT) prodrug nanoparticles were then tested for their in vitro cytotoxicity on human lung carcinoma (A549) cells. Whereas drug-free P(I-co-DOT) nanoparticles gave an IC50 of 49 μM, the prodrug nanoparticles led to a 35-fold-decrease in IC50 (1.4 μM) (Fig. 6e). This demonstrates the cytotoxic activity of Gem-P(I-co-DOT) prodrug nanoparticles. As expected, free Gem exhibited a much greater cytotoxicity (IC50 = 0.25 μM), since the free drug is immediately active whereas a prodrug must release the drug for it to be active. However, Gem is known to be quickly deaminated by deoxycytidine deaminase,76 thus preventing its direct administration in vivo and requiring the use of protecting nanocarriers/prodrugs.
After establishing the degradation of P(I-co-DOT) nanoparticles by TBD (Table 3), the final step of this work was to study their degradation under physiological conditions, which is a key issue when developing biodegradable drug delivery systems. To this end, two model copolymers were synthesized: P(I-co-DOT) N6 (Mn = 7900 g mol−1 and FDOT = 6.1 mol%) and Gem-P(I-co-DOT) N7 (Mn = 9300 g mol−1 and FDOT = 6.9 mol%). They were then formulated by nanoprecipitation into nanoparticles (Table 4) exhibiting high colloidal stability for up to at least 14 days (N6: Dz = 185 nm, PDI = 0.08 and N7: Dz = 130 nm, PDI = 0.08).
Spurred by the physiological thiolytic degradability of DOT-containing copolymers,48 the nanoparticles were subjected to degradation in the presence of: (i) chlorine bleach solution (2.5% sodium hypochlorite, 20 °C), which mimics both oxidative and base-catalysed hydrolysis due to the oxidative and alkaline nature of bleach, respectively59 and (ii) cysteine (10 mM, pH 7.4, PBS, 37 °C) or glutathione (10 mM, pH 7.2, PBS, 37 °C), which mimic reductive degradations under physiological conditions. Note that using degradation under alkaline conditions is a routine procedure in rROP, which is used to anticipate and predict the degradation of rROP-derived copolymers in the long run. For instance, this is usually performed with ester-containing copolymers obtained by rROP of CKA and vinyl monomers.77–80
Degradation of nanoparticles N6 and N7 under oxidative/alkaline conditions (Fig. 7a,b and Table 4) resulted in a significant decrease in Mn and Mw (∼60–80% after 12 days), which was similar to the degradation of the corresponding copolymers under alkaline conditions (KOH 5 wt%, see Table 2). More importantly, degradation of nanoparticles N6 and N7 under physiological reductive conditions successfully occurred (Table 4). As expected, the degradation kinetics were slower (∼10–20% decrease in Mn or Mw after 15 days) than under oxidative/alkaline conditions, but significantly faster than the hydrolytic degradation under physiological conditions of most CKA-containing copolymers, which typically degrade over several months and even a year.81–83 Overall, these results confirmed that P(I-co-DOT) and Gem-P(I-co-DOT) nanoparticles could be degraded under physiological conditions.
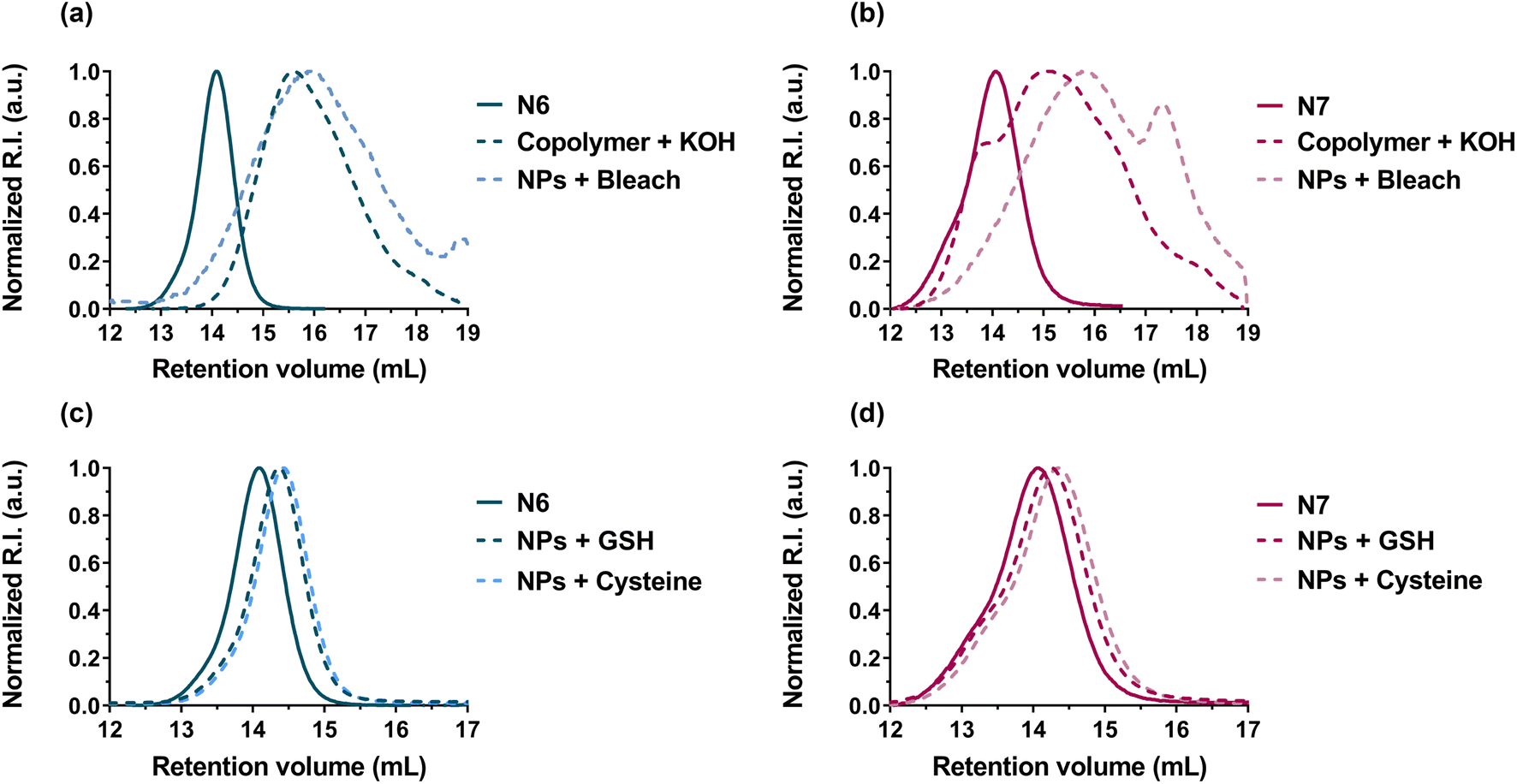 | ||
| Fig. 7 SEC chromatograms of: (a) P(I-co-DOT) polymer (N6, see Table 1) and (b) Gem-P(I-co-DOT) prodrug (N7, see Table 1) after degradation in the presence of KOH (5 wt%, 20 °C) on the polymer and chlorine bleach solution (2.5%, 20 °C) on the nanoparticles. SEC chromatograms of: (c) P(I-co-DOT) polymer (N6, see Table 1) and (d) Gem-P(I-co-DOT) prodrug (N7, see Table 1) after degradation in the presence of cysteine (pH 7.4, PBS, 37 °C) and glutathione (pH 7.2, PBS, 37 °C) on the nanoparticles. | ||
| Entry | D z (nm) | PDIa | ζ (mV) | Degradation by bleach solutionb | Degradation by cysteinec | Degradation by GSHd | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M n,deg.bleach (g mol−1) (% Mn loss) | M w,deg.bleach (g mol−1) (% Mw loss) | Đ | M n,deg.cysteine (g mol−1) (% Mn loss) | M w,deg.cysteine (g mol−1) (% Mw loss) | Đ | M n,deg.GSH (g mol−1) (% Mn loss) | M w,deg.GSH (g mol−1) (% Mw loss) | Đ | ||||
| a Determined by DLS. b Determined by SEC after degradation of the nanoparticles by 2.5% active chlorine bleach solution during 12 days at 20 °C. c Determined by SEC after degradation of the nanoparticles in the presence of cysteine in PBS pH 7.4 during 15 days at 37 °C. d Determined by SEC after degradation of the nanoparticles in the presence of GSH in PBS pH 7.2 during 15 days at 37 °C. e The Gem-AMA-SG1 alkoxyamine was used instead of BB alkoxyamine. | ||||||||||||
| N6 | 185 | 0.08 | −37 | 1800 (−77) | 2900 (−65) | 1.60 | 6300 (−20) | 6950 (−17) | 1.10 | 6500 (−18) | 7200 (−14) | 1.11 |
| N7 | 130 | 0.08 | −41 | 2600 (−65) | 3900 (−63) | 1.47 | 6600 (−19) | 8300 (−11) | 1.25 | 7100 (−12) | 8600 (−8) | 1.21 |
Conclusion
We successfully synthesized a small library of thionolactone-containing polyisoprene by radical ring-opening copolymerization of isoprene and DOT, under either FRP, NMP and RAFT polymerization conditions. The resulting P(I-co-DOT) copolymers exhibited predictable Mn, low dispersity and tuneable DOT contents (2.7–9.7 mol%) simply by adjusting the copolymerization conditions. The copolymers were significantly degraded under basic conditions, which confirmed the successful insertion of open DOT units in the PI chains. Cytocompatible, narrowly dispersed, surfactant-free nanoparticles were also prepared by nanoprecipitation of P(I-co-DOT) copolymers, with average diameters suitable for drug delivery purposes. As a proof of concept, well-defined Gem-P(I-co-DOT) polymer prodrug nanoparticles were produced, which exhibited significant cytotoxicity on A549 cancer cells, as well as degradation under oxidative/alkaline conditions and reductive physiological conditions. In conclusion, DOT represents a promising monomer that can easily introduce labile thioester groups into PI chains via rROP and advantageously broaden the range of degradable vinyl materials, not only in nanomedicine but also for other applications involving PI.Data availability
The authors declare that all data supporting the findings of this study are available within the article and ESI,† and raw data files are available from the corresponding author upon request.Author contributions
M. L.: investigation, writing – original draft, data curation, visualization. C. Z.: investigation, data curation. T. P.: investigation, data curation. D. L.: investigation, data curation. J. M.: investigation, data curation. Y. G.: formal analysis, software, writing – review & editing. J. N.: conceptualization, visualization, writing – review & editing, funding acquisition, supervision, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Agence Nationale de la Recherche (grant number ANR-18-CE06-0014 CKAPART). We thank Stéphanie Denis (IGPS, Université Paris-Saclay) for technical assistance in cell culture experiments and Dr Sylvain Trepout (Institut Curie, Orsay, France) and PICT-Ibisa for the experiments using a JEOL 2200FS TEM. We thank Pr. Alex Van Herk from Eindhoven University of Technology for sharing the CONTOUR software. This work has benefited from the facilities and expertise of the Platform for Transmission Electronic Microscopy of I2BC (Université Paris-Saclay).References
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- R. Wei, T. Tiso, J. Bertling, K. O'Connor, L. M. Blank and U. T. Bornscheuer, Nat. Catal., 2020, 3, 867–871 CrossRef CAS.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- The future of plastic, Nat. Commun., 2018, 9, 2157, https://doi.org/10.1038/s41467-018-04565-2 Search PubMed.
- T. Pesenti and J. Nicolas, ACS Macro Lett., 2020, 9, 1812–1835 CrossRef CAS PubMed.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- S. Nazhat, S. Parker, M. Patel and M. Braden, Biomaterials, 2001, 22, 2411–2416 CrossRef CAS PubMed.
- W. H. Kaminsky and B., Plast. Eng., 2005, 70, 333–380 CAS.
- A. Region Europe, And Segment Forecasts, 2021–2028 Search PubMed.
- S. Förster and E. Krämer, Macromolecules, 1999, 32, 2783–2785 CrossRef.
- J. K. Wegrzyn, T. Stephan, R. Lau and R. B. Grubbs, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2977–2984 CrossRef CAS.
- J. W. Bartels, S. I. Cauët, P. L. Billings, L. Y. Lin, J. Zhu, C. Fidge, D. J. Pochan and K. L. Wooley, Macromolecules, 2010, 43, 7128–7138 CrossRef CAS PubMed.
- S. C. Schmidt and M. A. Hillmyer, Macromolecules, 1999, 32, 4794–4801 CrossRef CAS.
- J. Wootthikanokkhan and B. Tongrubbai, J. Appl. Polym. Sci., 2003, 88, 921–927 CrossRef CAS.
- K. S. Murthy, Q. Ma, E. E. Remsen, T. Kowalewski and K. L. Wooley, J. Mater. Chem., 2003, 13, 2785–2795 RSC.
- C. Cheng, K. Qi, E. Khoshdel and K. L. Wooley, J. Am. Chem. Soc., 2006, 128, 6808–6809 CrossRef CAS PubMed.
- D. Vinciguerra, A. Degrassi, L. Mancini, S. Mura, J. Mougin, P. Couvreur and J. Nicolas, Biomacromolecules, 2019, 20, 2464–2476 CrossRef CAS PubMed.
- D. Vinciguerra, M. Jacobs, S. Denis, J. Mougin, Y. Guillaneuf, G. Lazzari, C. Zhu, S. Mura, P. Couvreur and J. Nicolas, J. Controlled Release, 2019, 295, 223–236 CrossRef CAS PubMed.
- S. Harrisson, J. Nicolas, A. Maksimenko, D. T. Bui, J. Mougin and P. Couvreur, Angew. Chem., Int. Ed., 2013, 52, 1678–1682 CrossRef CAS PubMed.
- Y. Bao, E. Guégain, V. Nicolas and J. Nicolas, Chem. Commun., 2017, 53, 4489–4492 RSC.
- Y. Bao, E. Guégain, J. Mougin and J. Nicolas, Polym. Chem., 2018, 9, 687–698 RSC.
- Y. Bao, T. Boissenot, E. Guégain, D. Desmaële, S. Mura, P. Couvreur and J. Nicolas, Chem. Mater., 2016, 28, 6266–6275 CrossRef CAS.
- S. Schlögl, M.-L. Trutschel, W. Chassé, I. Letofsky-Papst, R. Schaller, A. Holzner, G. Riess, W. Kern and K. Saalwächter, Polymer, 2014, 55, 5584–5595 CrossRef.
- W. M. Gramlich and M. A. Hillmyer, Polym. Chem., 2011, 2, 2062–2067 RSC.
- F. Chen and J. Qian, Fuel, 2002, 81, 2071–2077 CrossRef CAS.
- S.-Y. Chen, Y. Huang and R. C.-C. Tsiang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1964–1973 CrossRef CAS.
- E. I. Baptista, G. M. Campese, N. L. Zalloum, A. F. Rubira and E. C. Muniz, Polym. Degrad. Stab., 1998, 60, 309–315 CrossRef CAS.
- P. Berto, S. Grelier and F. Peruch, Polym. Degrad. Stab., 2018, 154, 295–303 CrossRef CAS.
- C. W. Phetphaisit and P. Phinyocheep, J. Appl. Polym. Sci., 2003, 90, 3546–3555 CrossRef CAS.
- S. S. Solanky, I. Campistron, A. Laguerre and J.-F. Pilard, Macromol. Chem. Phys., 2005, 206, 1057–1063 CrossRef CAS.
- S. Ouardad and F. Peruch, Polym. Degrad. Stab., 2014, 99, 249–253 CrossRef CAS.
- J. A. Blach, G. S. Watson, W. K. Busfield and S. Myhra, Polym. Int., 2002, 51, 12–20 CrossRef CAS.
- C. Adam, J. Lacoste and J. Lemaire, Polym. Degrad. Stab., 1991, 32, 51–69 CrossRef CAS.
- S. Sato, Y. Honda, M. Kuwahara and T. Watanabe, Biomacromolecules, 2003, 4, 321–329 CrossRef CAS PubMed.
- M. Enoki, Y. Doi and T. Iwata, Macromol. Biosci., 2003, 3, 668–674 CrossRef CAS.
- A.-L. Altenhoff, S. Thierbach and A. Steinbüchel, Biodegradation, 2021, 32, 113–125 CrossRef CAS PubMed.
- M. D. Chengalroyen and E. R. Dabbs, J. Polym. Environ., 2013, 21, 874–880 CrossRef CAS.
- A. Tardy, N. Gil, C. M. Plummer, C. Zhu, S. Harrisson, D. Siri, J. Nicolas, D. Gigmes, Y. Guillaneuf and C. Lefay, Polym. Chem., 2020, 11, 7159–7169 RSC.
- N. M. Bingham and P. J. Roth, Chem. Commun., 2019, 55, 55–58 RSC.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 1446–1451 CrossRef CAS PubMed.
- N. M. Bingham, Q. U. Nisa, S. H. L. Chua, L. Fontugne, M. P. Spick and P. J. Roth, ACS Appl. Polym. Mater., 2020, 2, 3440–3449 CrossRef CAS.
- N. Gil, B. Caron, D. Siri, J. Roche, S. Hadiouch, D. Khedaioui, S. Ranque, C. Cassagne, D. Montarnal and D. Gigmes, Macromolecules, 2022, 55, 6680–6694 CrossRef CAS.
- M. P. Spick, N. M. Bingham, Y. Li, J. de Jesus, C. Costa, M. J. Bailey and P. J. Roth, Macromolecules, 2020, 53, 539–547 CrossRef CAS.
- P. Galanopoulo, N. Gil, D. Gigmes, C. Lefay, Y. Guillaneuf, M. Lages, J. Nicolas, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2022, 61, e202117498 CrossRef CAS PubMed.
- N. Gil, C. Thomas, R. Mhanna, J. Mauriello, R. Maury, B. Leuschel, J.-P. Malval, J.-L. Clément, D. Gigmes and C. Lefay, Angew. Chem., Int. Ed., 2021, 134, e202117700 Search PubMed.
- M. Lages, N. Gil, P. Galanopoulo, J. Mougin, C. Lefay, Y. Guillaneuf, M. Lansalot, F. D'Agosto and J. Nicolas, Macromolecules, 2022, 55, 9790–9801 CrossRef CAS.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef CAS PubMed.
- N. Bingham, Q. Nisa, P. Gupta, N. Young, E. Velliou and P. Roth, Biomacromolecules, 2022, 23, 2031–2039 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- M. R. Wood, D. J. Duncalf, S. P. Rannard and S. Perrier, Org. Lett., 2006, 8, 553–556 CrossRef CAS PubMed.
- S. Allorio, S. Pispas, E. Siakali-Kioulafa and N. Hadjichristidis, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 2229–2234 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromolecules, 2011, 44, 9230–9238 CrossRef CAS.
- M. Van Den Brink, A. M. Van Herk and A. L. German, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3793–3803 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromol. Rapid Commun., 2012, 33, 805–810 CrossRef CAS PubMed.
- V. Jitchum and S. Perrier, Macromolecules, 2007, 40, 1408–1412 CrossRef CAS.
- V. Delplace, S. Harrisson, A. Tardy, D. Gigmes, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2014, 35, 484–491 CrossRef CAS PubMed.
- D. Gigmes, P. H. M. Van Steenberge, D. Siri, D. R. D'hooge, Y. Guillaneuf and C. Lefay, Macromol. Rapid Commun., 2018, 39, 1800193 CrossRef PubMed.
- O. Ivanchenko, S. Mazières, S. Harrisson and M. Destarac, Polym. Chem., 2022, 13, 5525–5529 RSC.
- C. Vauthier, C. Dubernet, E. Fattal, H. Pinto-Alphandary and P. Couvreur, Adv. Drug Delivery Rev., 2003, 55, 519–548 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- E. M. Pridgen, R. Langer and O. C. Farokhzad, Nanomedicine, 2007, 2, 669–680 CrossRef CAS PubMed.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS PubMed.
- M. Sumana, A. Thirumurugan, P. Muthukumaran and K. Anand, in Integrative Nanomedicine for New Therapies, Springer, 2020, pp. 231–246 Search PubMed.
- V. Delplace, P. Couvreur and J. Nicolas, Polym. Chem., 2014, 5, 1529–1544 RSC.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- M. L. Girase, P. G. Patil and P. P. Ige, Int. J. Polym. Mater. Polym. Biomater., 2020, 69, 990–1014 CrossRef CAS.
- P. Thakor, V. Bhavana, R. Sharma, S. Srivastava, S. B. Singh and N. K. Mehra, Drug Discovery Today, 2020, 25, 1718–1726 CrossRef CAS PubMed.
- J. Nicolas, Chem. Mater., 2016, 28, 1591–1606 CrossRef CAS PubMed.
- Y. Bao and J. Nicolas, Polym. Chem., 2017, 8, 5174–5184 RSC.
- E. Guégain, J. Tran, Q. Deguettes and J. Nicolas, Chem. Sci., 2018, 9, 8291–8306 RSC.
- D. Vinciguerra, S. Denis, J. Mougin, M. Jacobs, Y. Guillaneuf, S. Mura, P. Couvreur and J. Nicolas, J. Controlled Release, 2018, 286, 425–438 CrossRef CAS PubMed.
- C. S. Gondi and J. S. Rao, Expert Opin. Ther. Targets, 2013, 17, 281–291 CrossRef CAS PubMed.
- D. Dheer, J. Nicolas and R. Shankar, Adv. Drug Delivery Rev., 2019, 151–152, 130–151 CrossRef CAS PubMed.
- P. Gao, J. Nicolas and T. Ha-Duong, J. Am. Chem. Soc., 2021, 143, 17412–17423 CrossRef CAS PubMed.
- V. Heinemann, Y.-Z. Xu, S. Chubb, A. Sen, L. W. Hertel, G. B. Grindey and W. Plunkett, Cancer Res., 1992, 52, 533–539 CAS.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O'Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef CAS.
- G. G. Hedir, C. A. Bell, R. K. O'Reilly and A. P. Dove, Biomacromolecules, 2015, 16, 2049–2058 CrossRef CAS PubMed.
- C. Zhu, S. Denis and J. Nicolas, Chem. Mater., 2022, 34, 1875–1888 CrossRef CAS.
- V. Delplace, E. Guégain, S. Harrisson, D. Gigmes, Y. Guillaneuf and J. Nicolas, Chem. Commun., 2015, 51, 12847–12850 RSC.
- J. Undin, A. Finne-Wistrand and A.-C. Albertsson, Biomacromolecules, 2014, 15, 2800–2807 CrossRef CAS PubMed.
- E. Guégain, J.-P. Michel, T. Boissenot and J. Nicolas, Macromolecules, 2018, 51, 724–736 CrossRef.
- J. Tran, T. Pesenti, J. Cressonnier, C. Lefay, D. Gigmes, Y. Guillaneuf and J. Nicolas, Biomacromolecules, 2019, 20, 305–317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05316k |
| This journal is © The Royal Society of Chemistry 2023 |