
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCircularity in polymers: addressing performance and sustainability challenges using dynamic covalent chemistries
Tianwei
Yan
 ab,
Alex H.
Balzer
ab,
Katie M.
Herbert
*b,
Thomas H.
Epps
III
*abcd and
LaShanda T. J.
Korley
*abcd
ab,
Alex H.
Balzer
ab,
Katie M.
Herbert
*b,
Thomas H.
Epps
III
*abcd and
LaShanda T. J.
Korley
*abcd
aDepartment of Chemical & Biomolecular Engineering, University of Delaware, Newark 19716, Delaware, USA. E-mail: twyan@udel.edu; ahbalzer@udel.edu; lkorley@udel.edu; thepps@udel.edu
bCenter for Plastics Innovation (CPI), University of Delaware, Newark 19716, Delaware, USA. E-mail: herbertk@udel.edu
cDepartment of Materials Science and Engineering, University of Delaware, Newark 19716, Delaware, USA
dCenter for Research in Soft matter and Polymers (CRiSP), University of Delaware, Newark, 19716, Delaware, USA
First published on 5th May 2023
Abstract
The circularity of current and future polymeric materials is a major focus of fundamental and applied research, as undesirable end-of-life outcomes and waste accumulation are global problems that impact our society. The recycling or repurposing of thermoplastics and thermosets is an attractive solution to these issues, yet both options are encumbered by poor property retention upon reuse, along with heterogeneities in common waste streams that limit property optimization. Dynamic covalent chemistry, when applied to polymeric materials, enables the targeted design of reversible bonds that can be tailored to specific reprocessing conditions to help address conventional recycling challenges. In this review, we highlight the key features of several dynamic covalent chemistries that can promote closed-loop recyclability and we discuss recent synthetic progress towards incorporating these chemistries into new polymers and existing commodity plastics. Next, we outline how dynamic covalent bonds and polymer network structure influence thermomechanical properties related to application and recyclability, with a focus on predictive physical models that describe network rearrangement. Finally, we examine the potential economic and environmental impacts of dynamic covalent polymeric materials in closed-loop processing using elements derived from techno-economic analysis and life-cycle assessment, including minimum selling prices and greenhouse gas emissions. Throughout each section, we discuss interdisciplinary obstacles that hinder the widespread adoption of dynamic polymers and present opportunities and new directions toward the realization of circularity in polymeric materials.
 Tianwei Yan | Tianwei Yan received a BS in Chemistry from Peking University in 2016. He completed his PhD in 2021 at the University of Illinois Urbana-Champaign under the supervision of Prof. Damien Guironnet. His work primarily focused on the synthesis of polyolefin-based block copolymers. In the summer of 2021, he started a postdoctoral position under Prof. Thomas H. Epps, III and Prof. LaShanda T. J. Korley in the Center for Plastics Innovation (CPI) at the University of Delaware, where he works on valorization of plastics waste. |
 Alex H. Balzer | Alex H. Balzer received a BS in Chemical Engineering from North Carolina State University in 2015. He completed his PhD at Georgia Institute of Technology in 2022 under the supervision of Prof. Natalie Stingelin. His work focused on structure–property interrelations for polymers in photonics and electronics. Alex joined CPI at the University of Delaware in 2022 as a postdoctoral researcher working on valorizing plastics waste. |
 Katie M. Herbert | Katie M. Herbert received her BS (2011) and MS (2013) in Chemistry from Central Michigan University. She completed her PhD in Molecular Engineering at the University of Chicago (2019) and continued her studies at the University of Colorado Boulder as a Postdoctoral Associate (2019–2022). Her research focused on the design and synthesis of dynamic covalent and supramolecular polymer networks targeting multi-responsive materials for applications that included adhesives, robotics, and cell culture. In March 2022, she joined CPI at the University of Delaware as the Scientific Program Manager. |
 Thomas H. Epps, III | Thomas H. Epps, III is the Allan & Myra Ferguson Distinguished Professor of Chemical & Biomolecular Engineering at the University of Delaware and holds a joint appointment in Materials Science & Engineering. Prof. Epps is the Director of Center for Hybrid, Active, and Responsive Materials (CHARM) and the Center for Research in Soft matter & Polymers (CRiSP). He is also the Deputy Director of CPI. His research interests include renewable chemicals and polymers from biomass and polymer waste streams, ion-conducting nanostructured materials, polymer thin films, and self-assembling polymers for nucleic acid delivery. |
 LaShanda T. J. Korley | LaShanda T. J. Korley is a Distinguished Professor of Materials Science & Engineering at the University of Delaware and holds a joint appointment in the Department of Chemical and Biomolecular Engineering. Prof. Korley is the Director of CPI and the Co-Director of CHARM. She is also the Associate Director of CRiSP. Her research focuses on bio-inspired materials, film and fiber manufacturing, plastics recycling and upcycling, stimuli-responsive composites, fiber-reinforced hydrogels, and renewable materials derived from biomass. |
Introduction
Polymeric materials are indispensable in modern society due to their high strength-to-weight ratio and durability.1 Over the past ∼70 years, plastics use and production have outgrown those of most other man-made materials, such as cement and steel,2,3 and ∼390 million tons of plastics were produced globally in 2021.4 That amount is expected to grow exponentially over the next 30 years under various production scenarios.4,5Accompanying this rapid growth of plastics are the negative global impacts derived from the linear economy model, in which raw materials are collected, transformed into products, and eventually discarded as waste.6 Additionally, the majority of plastics production consumes non-renewable natural resources, and the current handling of post-consumer products results in further resource depletion,7,8 waste accumulation,9 and environmental (micro)plastics contamination.10–12 In response to these concerns, efforts have intensified to generate a more circular, closed-loop plastics consumption model.13 Within the closed-loop framework, one approach to minimize virgin resource input and waste accumulation is to adopt a reduce, reuse, remanufacture, and recycle system (Fig. 1).13–16
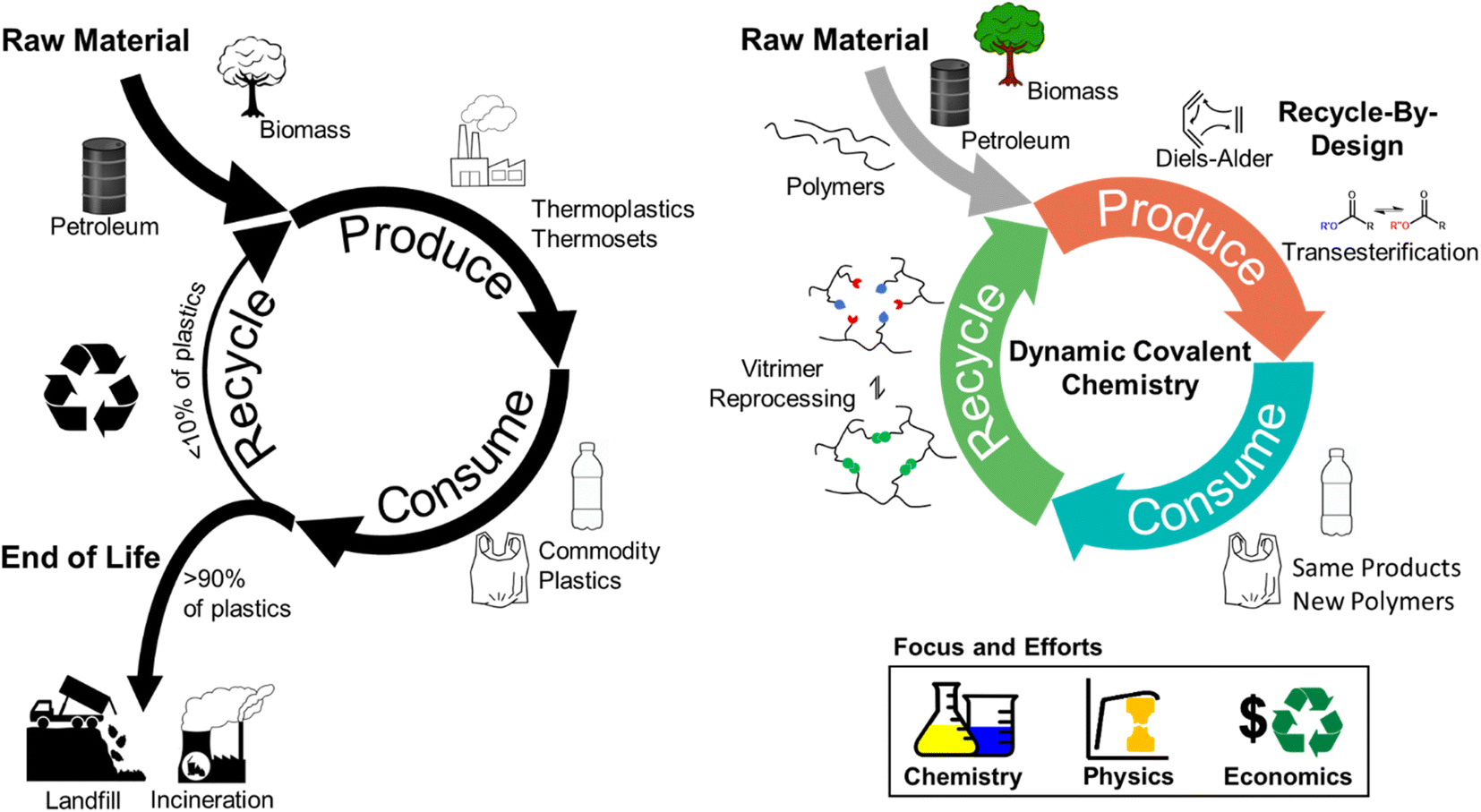 | ||
| Fig. 1 The circularity challenge. (Left) Conventional recycling efforts result in less than 10 wt% of plastics being recycled and over 90 wt% of plastics being landfilled or incinerated. Commodity plastics and plastic products are produced from petroleum- and bio-based feedstocks; however, challenges in conventional recycling processes limit full circularity. Recycling rates are illustrated by a thinner recycling arrow vs. end of life. (Right) Closed-loop circularity realized by dynamic covalent chemistries leads to increased retention of plastics in the production and consumption cycle. Polymeric materials are designed for specific recycling conditions through dynamic covalent chemistries, such as Diels–Alder and transesterification. Feedstocks can include petroleum, biomass, and existing commodity polymers. The same plastic products are produced using the new dynamic covalent chemistry-based polymers and reprocessed through reversible bond-breaking and reforming. Interdisciplinary efforts, including an expanded suite of available chemistries, structure–property relationships, and economic/environmental assessments, are still needed to realize the widespread implementation of dynamic covalent chemistry in closed-loop circularity. | ||
Although achievable in principle,17–21 several critical challenges prevent fully closed-loop circularity for polymer plastics waste.22–25 For example, thermosets (e.g., epoxy resin, vulcanized rubber) comprise ∼20 wt% of manufactured polymeric materials and are generally considered unrecyclable.26–28 The permanently crosslinked architecture of thermosets imbues these materials with solvent resistance and increased mechanical robustness; however, this structure also eliminates the ability to reprocess the networks via most mechanical or chemical means.27,29 Thus, thermoset-based polymers are typically landfilled.26,30 In contrast, thermoplastics (e.g., plastic bottles, films) are considered more recyclable using the aforementioned approaches.31 In thermoplastics, polymer chains associate mainly by intermolecular forces and can flow at elevated temperatures, yet polymer chains can irreversibly cleave or crosslink during mechanical recycling.22,32 The addition of virgin polymer is required to regenerate products with comparable properties to the original polymeric materials, which undermines full circularity.14,22
Another major obstacle to the circularity of polymeric materials is the unfavorable cost of recycling vs. virgin feedstock production.16 This high cost, present in both mechanical and chemical recycling, is the result of multiple factors, including waste collection, sorting, and total energy consumption.33 For example, sorting is necessary to separate different plastics and avoid significant property reduction of mechanically-recycled materials as a result of mixed waste.20,34 During chemical recycling, additives and impurities in real plastics waste also can adversely affect catalyst activity, decreasing process efficiency and increasing total energy demand.22,35–40 These recycling costs are rarely recovered in the value of recycled materials, leading to low recycling rates of plastics (e.g., 10 wt% for high-density polyethylene (HDPE), 5 wt% for low-density polyethylene (LDPE), less than 1 wt% for polystyrene (PS), polypropylene (PP), or poly(vinyl chloride) (PVC) in the U.S.),41,42 motivating the need for innovative recycling strategies.
Dynamic covalent chemistry (DCvC) in polymeric materials provides a synthetic pathway to address circularity challenges, including unrecyclable networks and irreversible chemical changes (Fig. 1).21,43,44 The characteristic high reversibility and stimuli-responsiveness of DCvC45–47 permit bond breaking and reforming during the recycling of corresponding modified polymeric networks (e.g., dynamically-crosslinked epoxy resins),48,49 unlike conventional thermosets. Furthermore, the irreversible structural changes and subsequent property deterioration from conventional recycling approaches can be mitigated or even circumvented by DCvCs. For example, industrially recycled poly(ethylene terephthalate) (PET) suffers from property deterioration (e.g., after 2 cycles, the melt viscosity decreased by 50%) due to thermal and shear degradation during the extrusion process,16,50 whereas a DCvC-modified PET with reconfigurable ester linkages displays reprocessability with minimal mechanical property loss over 3 cycles.51,52 Finally, DCvC can reduce recycling costs associated with reprocessing conditions, separations, and end-product valorization. The bond activity and tunable nature of DCvCs allow polymeric materials to be recycled at lower temperatures (e.g., sterically hindered urea bonds and alkoxyamine can lower the reprocessing temperatures of corresponding materials from over 140 °C to less than 40 °C), decreasing energy requirements.53–56 Beyond thermal reprocessing, DCvC provides opportunities to recycle plastics via selective dissolution and/or depolymerization under mild conditions. For example, a boroxine-crosslinked polymer is recoverable via depolymerization in an ethanol/N-methylpyrrolidone (NMP) solvent mixture at 30 °C, but remains suitable for many applications due to its mechanical strength, thermal stability, and hydrophobicity.57–59 The tunability of DCvC allows for potential implementation within current industrial manufacturing processes, leading to reduced investment in additional infrastructure.60,61 In some cases, DCvC incorporation has even been shown to improve select material properties (e.g., chemical sensing, shape memory effects),44,62 adding value to offset the recycling costs.63
Although the implementation of DCvC in polymeric materials is promising, there remain practical challenges (e.g., processing condition development, cost) before this strategy can be extended to the broad range of applications and recycling conditions inherent to polymer production. The synthetic design of DCvC for incorporation into polymeric materials must tailor properties to fit industry-level (re)processing requirements,44 such as fast stress relaxation64 and robustness, after repeated recycling.65 The balance between dimensional stability during application and material flow during reprocessing in dynamic polymeric materials is a challenge as the overall relaxation rates are dictated by DCvC bond reconfiguration and polymer chain motion.47 Thus, a fundamental understanding of structure–property relationships and physical models for DCvC-based polymers is necessary to predict performance and to develop appropriate (re)processing conditions to enable circularity.66 To align academic advances with wide-scale adoption, the generation of required data sets (e.g., reprocessing temperatures, performance after multiple cycles, relevant reagents) is pivotal but currently limited.22 Techno-economic analysis (TEA) and life-cycle assessment (LCA) will be critical to evaluate the costs and environmental impacts during the production/recycling processes of DCvC-containing polymers.
In this review, we describe the role and associated challenges of DCvCs in the development of closed-loop, recyclable polymer materials. Many reviews highlight synthetic advances in DCvCs and dynamic polymers;67–71 thus, that topic is not extensively covered herein. Instead, we focus on the application of these motifs towards circular polymers. First, we briefly summarize the key features of DCvCs and examine how each feature can be leveraged to promote the circularity of polymeric materials. Next, we outline the advantages and challenges in DCvC for both recycle-by-design polymers and existing commodity polymers. Then, we discuss the thermomechanical properties that impact dynamic covalent polymers from the perspective of their underlying polymer physics, which can guide the design and optimization of recycling processes. Finally, we highlight the practical limitations and opportunities toward the implementation of dynamic covalent chemistries in the circular economy using elements derived from economic and environmental studies (TEA and LCA), including minimum selling price and greenhouse gas emissions. By considering all facets of DCvC-containing polymer recycling, we provide a more comprehensive viewpoint on enhancing the circularity of polymeric materials.
Dynamic covalent chemistry for polymeric material design
The concept of dynamic covalent chemistry was described by Rowan et al. in 2002,45 as an extension of supramolecular chemistry to the covalent level. DCvC proceeds by the equilibration process of bond breaking and reforming, which leads to the efficient formation of products under thermodynamic control.45 At the molecular level, these bonds undergo reversible exchange without unwanted side reactions,45 making DCvC attractive in many arenas, including drug delivery,72,73 robotics,44,74 sensors,75,76 and recycle-by-design polymers.21,77,78In general, dynamic bonds are designed with three key properties in mind: thermodynamic reaction equilibrium, reaction rate, and stimuli-responsiveness (Fig. 2). The reaction equilibrium defines the reversibility of the bond, and the equilibrium constant, K, is exponentially proportional to the change in free energy upon product formation, or ΔG. Structural design strategies (e.g., altering steric hindrances,53 electronic effects79) can tune this equilibrium, providing pathways to develop additional DCvC reactions and modulate the dimensional stability of the resulting material in corresponding applications. One example is the use of a hindered urea bond to alter the equilibrium of C–N bonds in polyureas and poly(urethane-ureas).53 Although an unmodified urea bond is quite stable, the hindrance imposed on the nitrogen atom by bulky alkyl groups weakens the C–N bond by disturbing the coplanarity and conjugation effect of the carbonyl and amine groups, resulting in reversible, dynamic urea bonds80 and leading to reprocessable networks crosslinked by these bonds.81 In another illustration of the impact of equilibrium on material properties, imine-based dynamic covalent networks exhibited improved water-resistance due to the conjugative effect of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–X structures via the incorporation of electron-withdrawing functionalities,79,82,83 yet also maintained good recyclability via dynamic bond exchange in solution.84
N–X structures via the incorporation of electron-withdrawing functionalities,79,82,83 yet also maintained good recyclability via dynamic bond exchange in solution.84
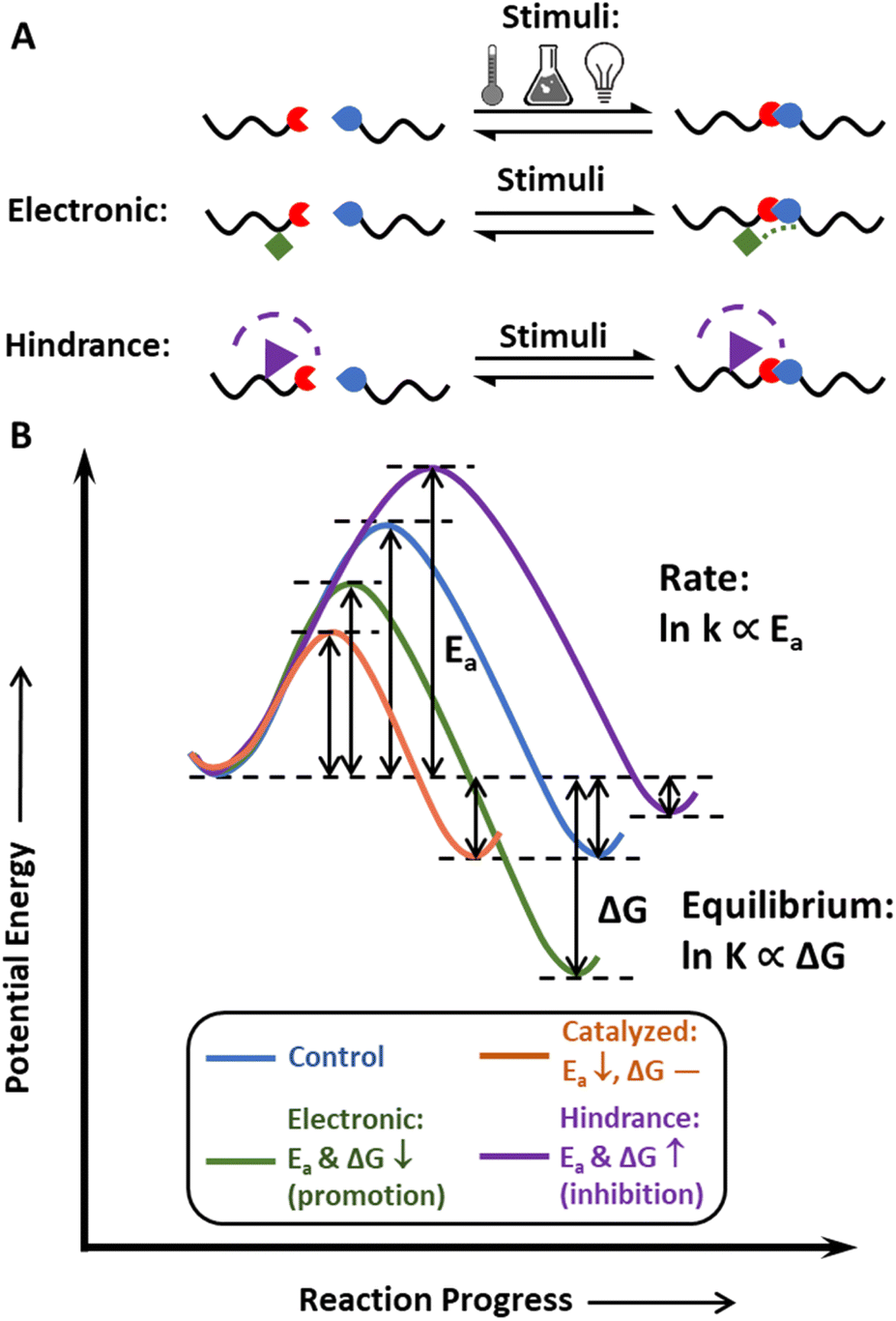 | ||
| Fig. 2 (A) Generic dynamic covalent chemistry scheme related to equilibrium control through electronic/steric effects. (B) Control of reaction equilibrium and rate (rate constant: k) via steric hindrance and electronic effects. Blue: control. Orange: catalyzed. Green: electronic effects (promote the reaction in this case). Purple: steric effects (inhibit the reaction in the case shown in (A)). | ||
Reaction equilibrium indicates the tendency of dynamic bonds to exchange, whereas reaction rate determines the time required for these bonds to rearrange. To display dynamic features on the macroscopic level, the reversible reaction must achieve equilibrium within a relevant timeframe (e.g., 10–103 s for common molding procedures) after exposure to an environmental stimulus.66,85 The reaction rate and reprocessing time of a dynamic polymer reflect the mechanism and kinetics of the specific DCvC, denoted by the activation energy, Ea (Fig. 2B).86,87 Some DCvCs are intrinsically fast with a relatively low Ea. For example, under mild temperatures (e.g., below 100 °C), the Diels–Alder (D–A) reaction (Ea ∼50 kJ mol−1)84 and boroxine exchange reactions (Ea ∼80 kJ mol−1)88,89 can lead to the complete reconfiguration of recyclable covalent networks within an hour without the aid of catalysts.86,87 Unlike the D–A and boroxine reactions, some of the DCvC networks exchange slowly with a high energy barrier, such that a catalyst is often necessary to lower the Ea and dramatically accelerate the reaction.90 Transesterification is an example of DCvC that is heavily dependent on both the catalyst type and the relative amount of catalyst present, as the relaxation time can be lowered to more desirable timescales for reprocessing (e.g., 10–103 s).90 In transesterification, the high reaction rate (and short relaxation time) achieved by optimizing the conditions and catalyst choice can facilitate reprocessing/recycling using methods, such as melt extrusion and injection molding.51,91 Therefore, the reaction rate dictates the framework for throughput, application conditions, available processing techniques,91 and related logistics associated with the dynamic polymer recycling processes.
Finally, tailored responsiveness with respect to specific stimuli, including heat, light, pH, and others, can lead to a range of responsive behavior in different environments. For example, heat is commonly used in dynamic covalent polymer recycling. As indicated by the Maxwell relations and Arrhenius equations,92 increasing the temperature accelerates both sides of a reversible reaction but often shifts the equilibrium towards dissociation, which promotes DCvC network reprocessing (e.g., D–A reaction).93 pH-responsive DCvCs can enable selective depolymerization under acidic or basic environments (e.g., hemiaminal,94 imine84) in mixed plastics,57–59 providing an alternative chemical recycling pathway to reduce sorting costs. Although light has not been widely used in recycling, some DCvCs facilitate rapid healing under UV irradiation (e.g., disulfide exchange),95 potentially supporting circularity by extending the product lifetime and reprocessing pathways that utilize this responsiveness.96 Conversely, in the absence of a triggering stimulus, the materials are highly stable and exhibit high creep resistance.92
Overall, reversibility, high reaction rates, and stimuli-responsiveness are essential starting points to achieve the circularity of polymeric materials via functionalization with dynamic covalent chemistries. Although macromolecular circularity requires long-term and iterative optimizations in both academic research and industrial practice, dynamic covalent chemistry, in principle, has the potential to facilitate the strategic, recycling-oriented design of polymeric systems. Therefore, it is imperative to develop a more comprehensive understanding of the chemistry (i.e., synthetic routes), physics (i.e., physical models), and industrial (i.e., economic and environmental impacts) perspectives through the lens of closed-loop recyclability.
Synthetic progress in DCvC-containing polymers
In general, recent synthetic advances related to DCvC in polymer recycling can be classified into two categories: polymer construction from functional monomers (i.e., recycle-by-design) and post-polymerization modification. Recycle-by-design focuses on inherently circular polymers at the point of invention whereas post-polymerization addresses challenges related to existing polymer waste. For example, epoxy resins and polyurethanes can be cured with crosslinkers that utilize DCvC (e.g., transesterification,97 Diels–Alder reaction98).99–102 Similarly, it is feasible to introduce DCvC into other common polymers, such as poly(meth)acrylate, polyolefins, polystyrene, and polydiene networks, via synthetic approaches from monomers and post-polymerization modification.48,81,103–106 Although both methods target a recyclable polymer product, they each have their own set of advantages, challenges, and opportunities.Recycle-by-design
Polymers synthesized with dynamic chemistries in mind comprise a significant portion of the research in the field. By modulating the molecular design of polymeric materials, chemists gain control over the dynamic properties of the system. This strategy enables the tuning of polymer architecture, bond equilibrium (i.e., reversibility), or stimuli response towards desired properties in a manner that offers more synthetic control than DCvC post-modification of existing polymers. Though this recycle-by-design approach results in a myriad of inherently recyclable polymers, it often is accompanied by a series of shortcomings (e.g., slow reaction rates, loss of crosslink density) that result in significant challenges related to scale up for specific applications.Numerous synthetic routes towards overcoming these challenges have been investigated since Leibler and collaborators proposed the concept of vitrimers in reprocessable epoxy resins.97 In their ground-breaking study of dynamic covalent networks,97 a diglycidyl ether of bisphenol A (DGEBA) was cured with carboxylic di- and tri-acids in the presence of two zinc catalysts to form the resultant network (Fig. 3). These vitrimers consisted of an associative network of ester linkages that broke and reformed concurrently, theoretically maintaining crosslink density (as opposed to dissociative reactions in which the linkage breaks first and a new linkage forms second). The networks exhibited reprocessability through compression molding at 240 °C within 3 min with comparable tensile strength and strain-at-break to the initial polymers.97 However, the reprocessed epoxy materials still displayed a slight decrease in Young's modulus,97 indicating a loss of crosslink density in the recycled networks. Additionally, estimations of the DCvC activation energy suggested that relaxation at room temperature would occur on the order of a few years, threatening the long-term dimensional stability of the material.97 In total, this initial investigation both established the opportunities in using DCvC in polymer networks and highlighted key limitations associated with these initial DCvC-based polymeric materials, (e.g., high reprocessing temperature requirements, property loss, creep) (Fig. 4).
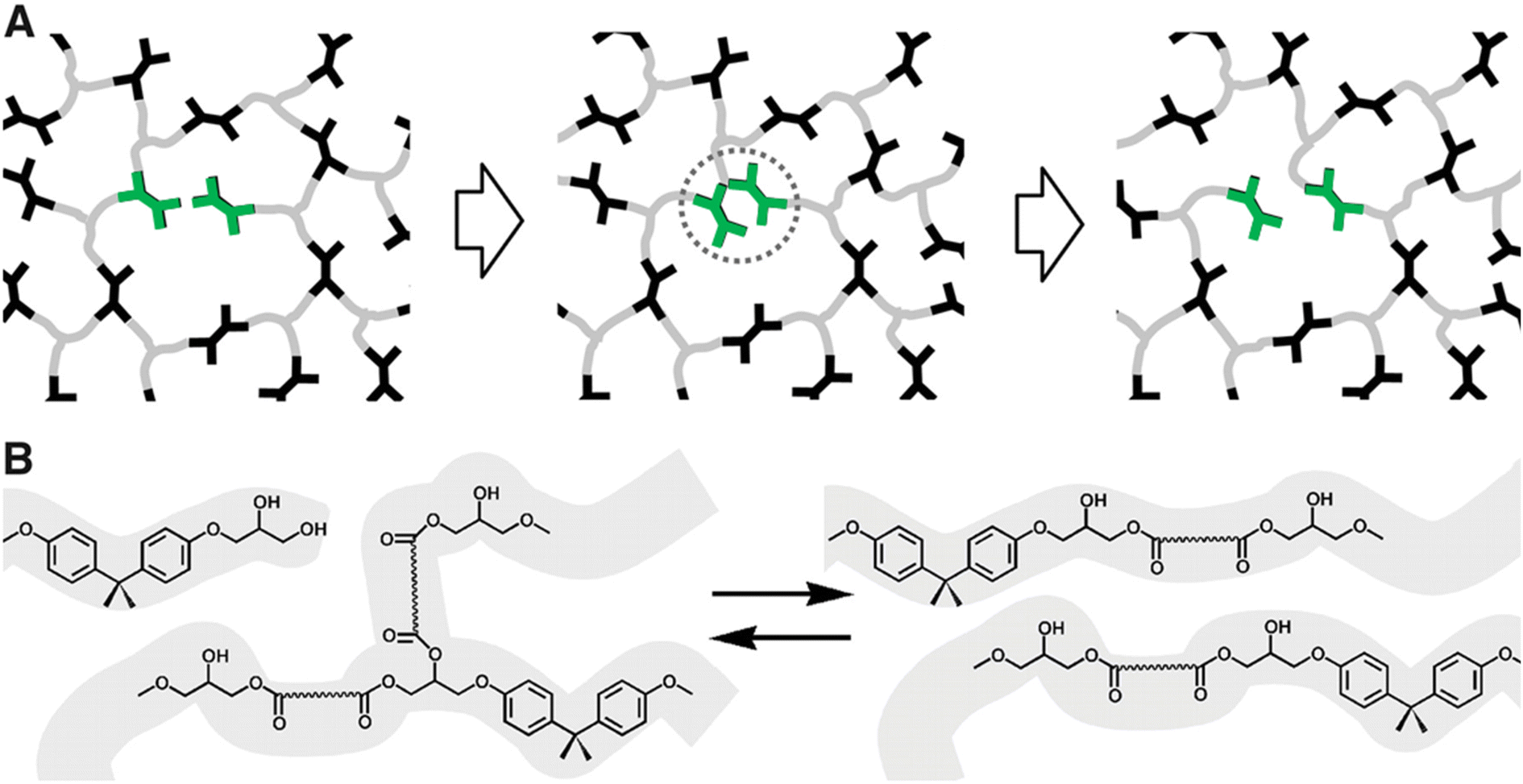 | ||
| Fig. 3 Rearrangement of reprocessable epoxy networks. (A) Schematic of a network with associative exchange processes (highlighted in green) that do not require depolymerization or loss of crosslink density. (B) Chemical illustration of a transesterification exchange process in hydroxy-ester networks. Adapted from ref. 97 with permission from the American Association for the Advancement of Science, copyright 2011. | ||
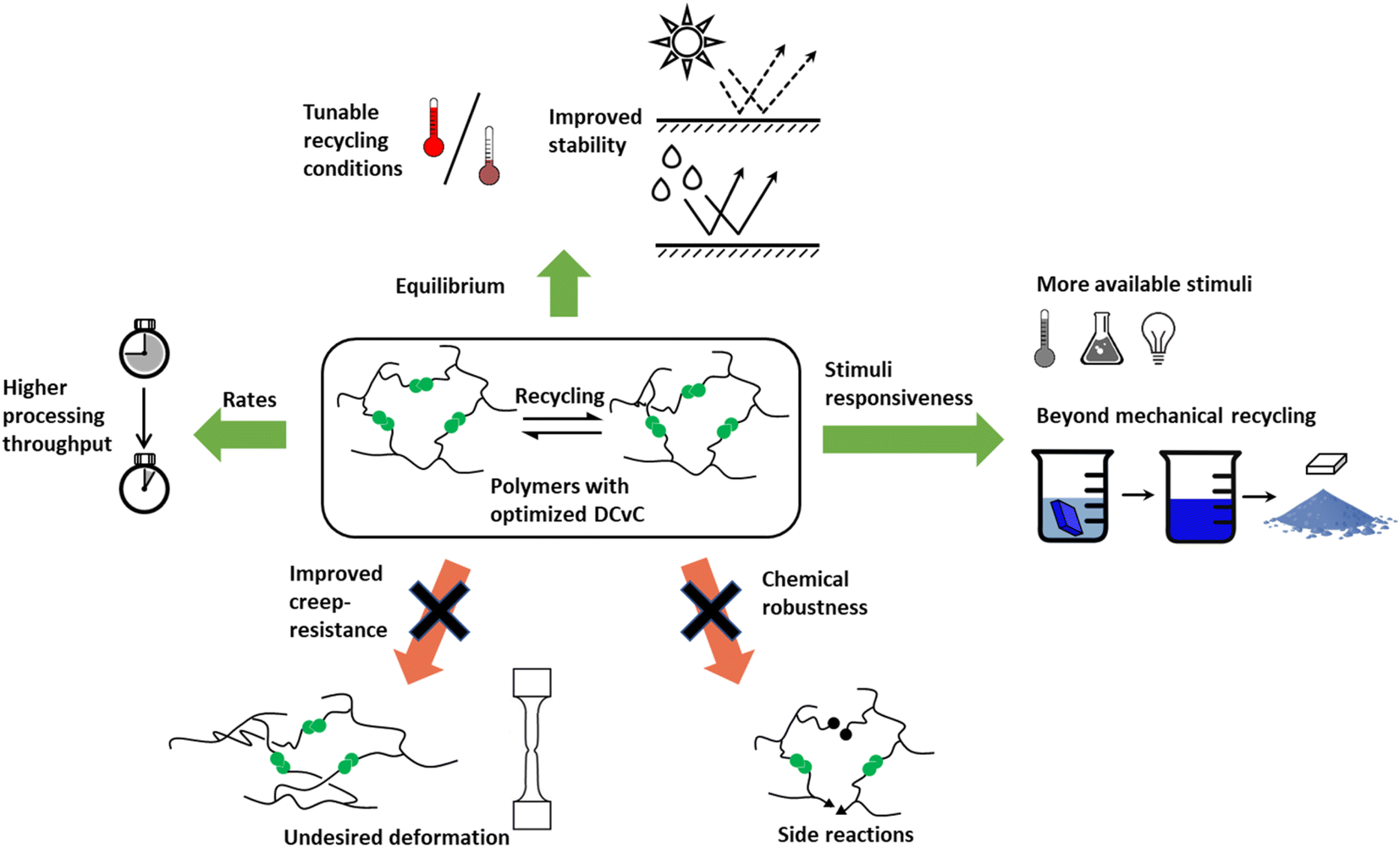 | ||
| Fig. 4 Achievable improvements in recyclable dynamic networks via recycling-by-design strategy. Optimized DCvC-containing polymeric materials can exhibit tunable recycling conditions, fast (re)processing, various recycling approaches, and improved stability against side reactions or undesirable deformation. | ||
Aside from catalyst design, catalyst loading also affects reprocessing conditions, although it does not influence the activation energy of the dynamic exchange. For instance, increasing the catalyst amount from 1 mol% to 5 mol% has been shown in some cases to decrease the stress relaxation time by approximately one order of magnitude, which subsequently decreases the reprocessing time.114 Albeit a promising route to tailor relaxation times, economic considerations also must be taken into account during catalyst selection to balance projected cost with returns on material property improvement. Using the previous example, it is important to evaluate whether the cost to increase catalyst loading from 1 mol% to 5 mol% (i.e., the cost of the catalyst itself) is offset by the expected value returned by a lower overall reprocessing time.
In addition to potential hurdles related to catalyst cost, concerns have been raised regarding catalyst compatibility, catalyst leaching, and environmental impacts.90 These issues have motivated researchers to design catalyst-free vitrimers by inserting catalyzing functional groups directly into the dynamic covalent polymer.115,116 When the inserted species are incorporated in close proximity to the reaction center (Fig. 5A), this effect is referred to as neighboring group participation (NGP).64,117–119 For example, catalyst-free dynamic ester exchange was achieved via the engagement of a neighboring acid group close to the transesterification center. This structural modification resulted in the formation of transient cyclic anhydrides and free alcohols, enabling a dissociative dynamic exchange.115 Expanding upon this phthalate monoester mechanism, Du Prez and collaborators introduced a tertiary amine on the beta position of the hydroxyls (Fig. 5B),64 further decreasing the relaxation times by a factor of 500 through a dual NGP mechanism (relaxation times, τ* ∼1 s below 160 °C).64 The resultant fast exchange and relaxation time enabled the reprocessing of chopped samples through industrially relevant processing techniques (e.g., extrusion), which is a promising improvement for physical recycling at scale.64,91 A recent publication reported an alternative NGP transesterification via beta-keto activated exchange instead of neighboring acid and amine participation, allowing 5 cycles of consecutive remolding at mild conditions (e.g., 70–100 °C).120 The concept of NGP also has been introduced to optimize other DCvCs to expand the reach of recycling conditions for related materials. For example, the incorporation of a neighboring tertiary amine accelerated the boronic ester exchange by five orders of magnitude.87 Yet another study integrated NGP-assisted silyl exchange into DCvC-modified polystyrene, increasing the reaction rate by three orders of magnitude and decreasing the corresponding reprocessing temperature by 30 °C.121 In general, these NGP approaches are a promising direction for significant rate enhancements in DCvCs while reducing the need for small-molecule catalyst additives.117
 | ||
| Fig. 5 (A) General NGP-assisted mechanism of DCvC (left). Non-NGP mechanism of DCvC (right). (B) Proposed NGP mechanism for rapid transesterification (left). Non-NGP transesterification (right). | ||
When incorporating the above strategies to accelerate DCvC reactions, the trade-off between exchange rates and material performance needs to be considered. Faster exchange rates in DCvC-based polymeric materials can facilitate reprocessing but also lower the upper limit of operating conditions under which the material will maintain mechanical integrity. The latter negatively impacts the application potential, particularly in high-performance thermoset materials expected to function in harsh environments (e.g., elevated temperatures, high mechanical stress).22,44,122 DCvC-based material designs that balance reprocessing conditions and target uses are crucial to enabling circularity without affecting performance.
Another pathway to suppress creep is to introduce an appropriate level of permanent crosslinking.47,125,126 In one study, crosslinked networks containing various fractions of dynamic boronic esters and irreversible linkages demonstrated that stress relaxation and creep can be moderated by the addition of permanent crosslinks that lend increased structural integrity.125 A hybrid network with ∼80 mol% permanent crosslinks suppressed the relaxation yet still permitted repeated healing.125 In another system, an epoxy resin crosslinked via dynamic transesterification incorporated 40 mol% of thiol-epoxy permanent crosslinks into the network. The relaxation time of the vitrimer almost doubled, and creep was reduced by 65–71%, yet the material maintained full reprocessability via hot pressing.47
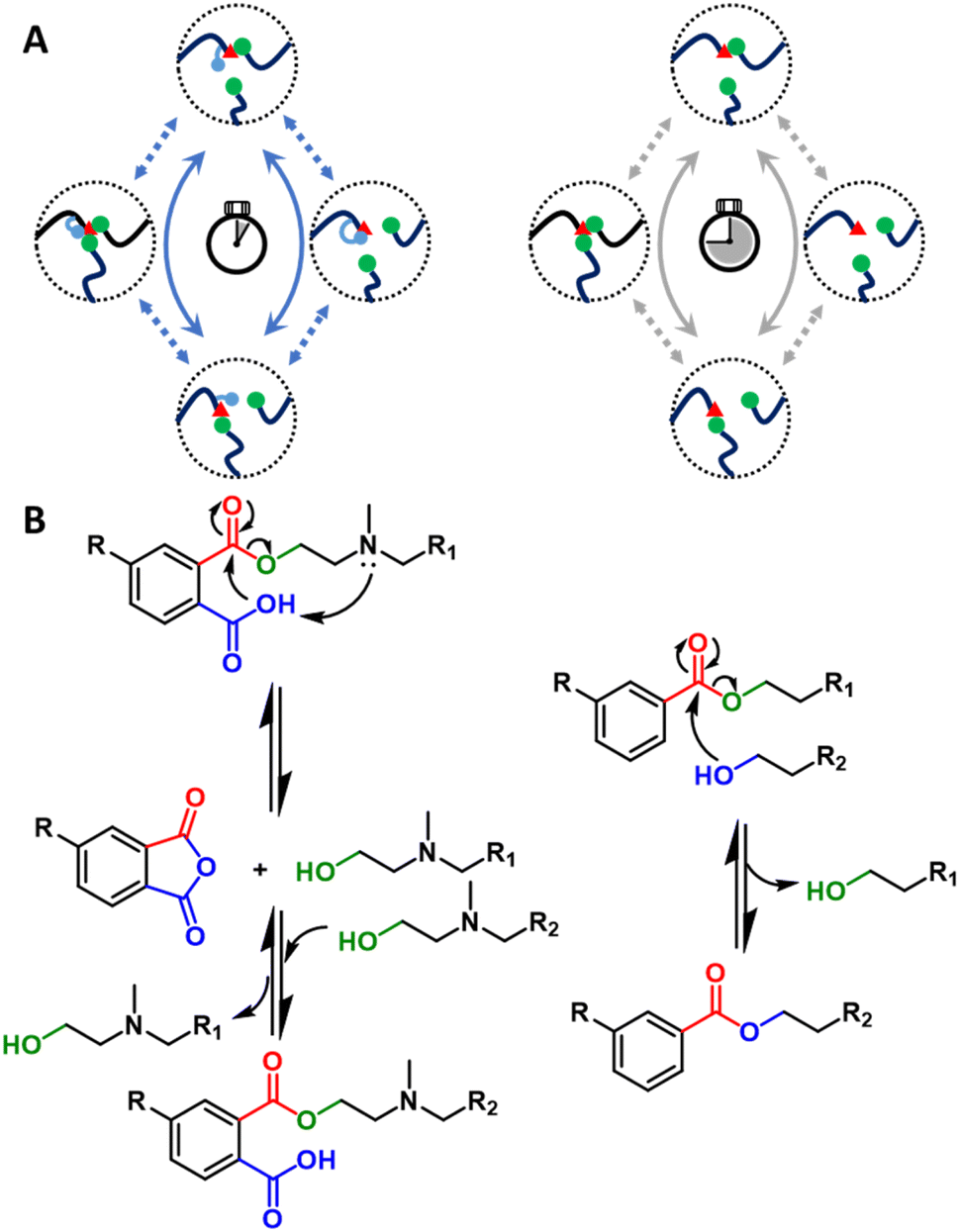
It is also possible to inhibit creep via combinational strategies of DCvC modification and non-covalent interactions. For example, a synergistic effect was achieved in a system of vitrimers featuring dynamic imine exchange with integrated metal-complexes.127 Adding 5 mol% of metal ions to coordinate with flexible imine bonds induced further crosslinking of the vitrimer via non-covalent bonding and simultaneously doubled the activation energy of dynamic imine exchange (107.3 kJ mol−1vs. 52.3 kJ mol−1). This combined approach enhanced the creep resistance by raising the initial creep temperature by 60 °C (from 60 °C to 120 °C, Fig. 6).127
 | ||
| Fig. 6 (Left) Polyimine–metal complex vitrimers synthesized from terephthaldehyde (TPA), diethylenetriamine (DETA), and tris(2-aminoethyl)amine (TREN). (Right) Strain recovery curves to show the creep response of the polyimine–metal complex vitrimers at (A) 60 °C, (B) 80 °C, (C) 100 °C, and (D) 120 °C. Adapted from ref. 127 with permission from the American Chemical Society, copyright 2020. | ||
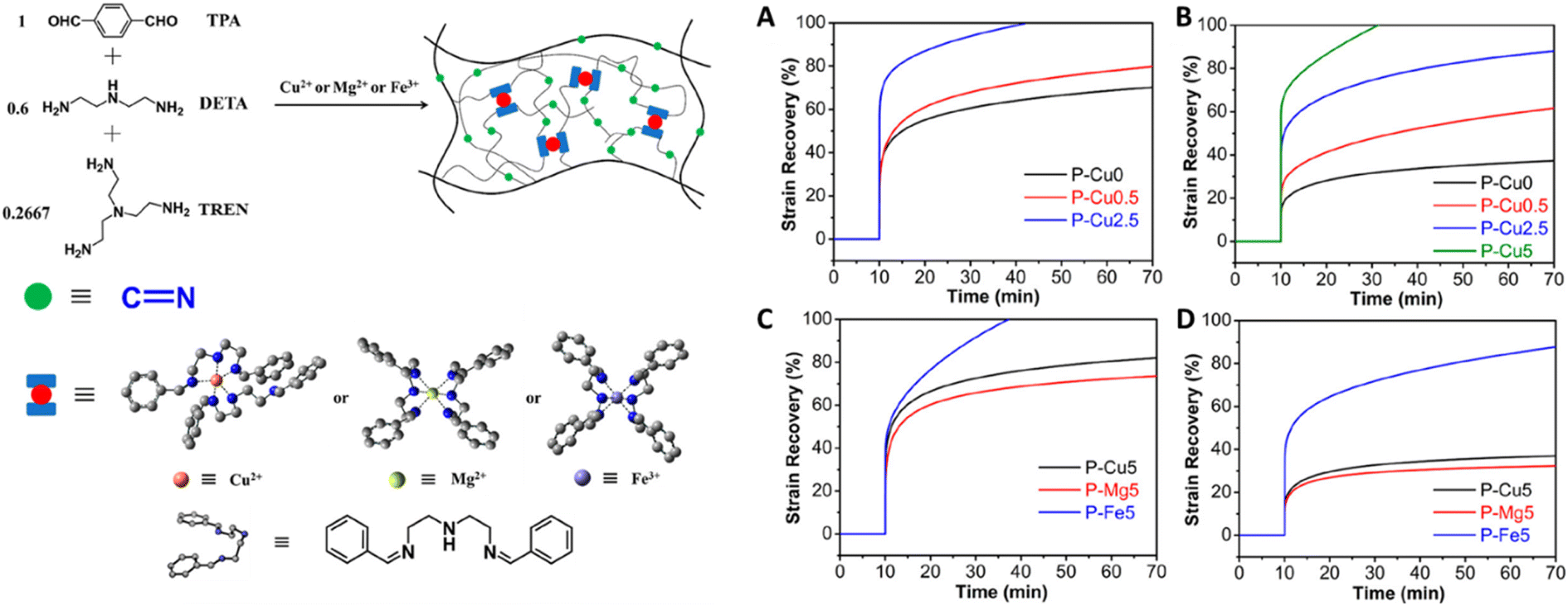
Tailoring the equilibrium also plays an important role in reprocessing conditions for DCvC-based polymers. For example, the homolysis temperature of alkoxyamine C–ON bonds can be adjusted from 140 °C124 to as low as 15 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 54,55 by leveraging the electron-withdrawing effect of neighboring substituents. With an appropriate molecular design to shift the equilibrium, it is even possible to convert a non-dynamic reaction into a DCvC that displays reprocessability under mild conditions.53 A well-known example is the hindered urea bond. A conventional urea bond is stable, but sterically hindering and twisting the C(O)–N bond facilitates dynamic dissociation and reconstruction.53 Inspired by the facile amidolysis of bulky amides, Cheng and collaborators measured the equilibrium constants and rate constants of urea bond dissociation with different bulky amines.53 By adjusting the bulkiness of the amines, the equilibrium constants varied by up to six orders of magnitude.53 Among the investigated amines, a secondary amine containing a tert-butyl urea group displayed a moderately reversible equilibrium constant (7.9 × 105 M−1) and a non-negligible dissociation rate (0.21 h−1 at 37 °C), indicating balanced bond stability and exchange rate.53 A crosslinked poly(urethane-urea) gel containing hindered urea bonds on the main chain was prepared with a diisocyanate, a bulky diamine, and polyol crosslinkers (Fig. 7).53 Self-healing (i.e., the failure strain recovered to 50% at 1 h, 87% at 12 h) was achieved at 37 °C. The results revealed the importance of manipulating equilibrium as a powerful tool in designing circular polymeric materials via facile adjustment of chemical structures.53
54,55 by leveraging the electron-withdrawing effect of neighboring substituents. With an appropriate molecular design to shift the equilibrium, it is even possible to convert a non-dynamic reaction into a DCvC that displays reprocessability under mild conditions.53 A well-known example is the hindered urea bond. A conventional urea bond is stable, but sterically hindering and twisting the C(O)–N bond facilitates dynamic dissociation and reconstruction.53 Inspired by the facile amidolysis of bulky amides, Cheng and collaborators measured the equilibrium constants and rate constants of urea bond dissociation with different bulky amines.53 By adjusting the bulkiness of the amines, the equilibrium constants varied by up to six orders of magnitude.53 Among the investigated amines, a secondary amine containing a tert-butyl urea group displayed a moderately reversible equilibrium constant (7.9 × 105 M−1) and a non-negligible dissociation rate (0.21 h−1 at 37 °C), indicating balanced bond stability and exchange rate.53 A crosslinked poly(urethane-urea) gel containing hindered urea bonds on the main chain was prepared with a diisocyanate, a bulky diamine, and polyol crosslinkers (Fig. 7).53 Self-healing (i.e., the failure strain recovered to 50% at 1 h, 87% at 12 h) was achieved at 37 °C. The results revealed the importance of manipulating equilibrium as a powerful tool in designing circular polymeric materials via facile adjustment of chemical structures.53
 | ||
| Fig. 7 Preparation and self-healing of a crosslinked poly(urethane-urea) gel with hindered urea bonds. (A) Illustration of the self-healing process via bond exchange. (B) Synthetic route to generate the polymer. (C) Selected snapshots during the self-healing experiment of poly(urethane-urea). The arrow indicates the cut position. Adapted from ref. 53 with permission from Springer Nature, copyright 2014. | ||
Cheng's work on self-healing poly(urethane-urea) gels inspired a series of recyclable polymeric networks that leverage the bulkiness of the amines to tune polymer backbone chemistry.81,129 A recent report highlighted the application of this hindered urea bond in a recyclable PMMA-based material.81 The PMMA-based network was crosslinked by a hindered urea-functionalized dimethacrylate and then reprocessed for three cycles to yield homogeneous films. The recycled material showed no decrease in gel content (>99%) and no significant change in tensile storage modulus, suggesting recovery of crosslink density.81
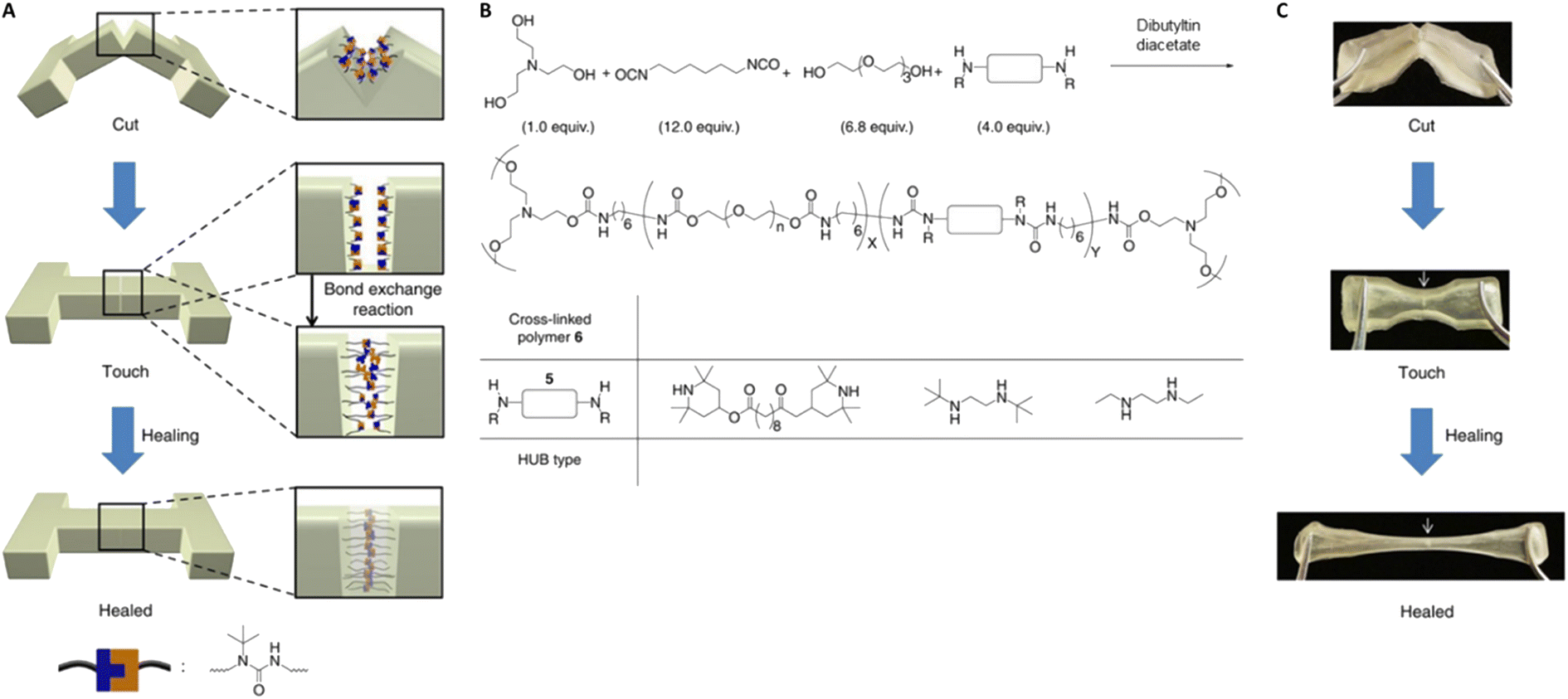
The loss of reversibility also can be caused by the gradual consumption of dynamic functional groups.122 Examples of these undesirable reactions include hydrolysis of esters and isocyanates,89,90,134 and dimerization/trimerization of isocyanates.135 The design of more chemically-robust DCvCs is a strategic focal point related to polymer circularity. Poly(siloxanes) represent one of the most well-known examples of robust, chemically-, and thermally-stable dynamic polymer networks.121 The equilibrium of siloxane exchange has been well-studied for almost 70 years136 but was just recently applied to dynamic polymer systems.137 The dynamic Si–O exchange can be initiated by silanolates and enables chain reconfiguration in crosslinked poly(dimethylsiloxane) (PDMS) and other siloxane-containing networks for self-healing and recycling purposes.137–139 As one example, a siloxane-crosslinked epoxy polymer was reprocessed twice at a high temperature (220 °C) without significant change in crosslink density, suggesting sufficient thermal stability during recycling.139 In addition to siloxanes, Si–O exchange also has played a critical role in polymers crosslinked by silyl ether linkages,65,121,140,141 which displayed reprocessability at elevated temperatures and thermal stability against side reactions. In a study of PE-based networks crosslinked by silyl ethers, these properties were exemplified by a high degradation temperature (>400 °C), as well as nearly constant tensile strength and failure strain after three cycles of reprocessing via compression molding (150 °C, 400 psi, 20 min).65
Another robust DCvC, the D–A reaction, occurs by a [4 + 2] cycloaddition mechanism to form a cyclohexene D–A adduct which can dissociate under elevated temperatures via a reversible pathway.93 The D–A reaction is a “click” reaction that exhibits several advantages for recyclable thermosets, including excellent efficiency, selectivity, and a catalyst- and byproduct-free nature. As one example, a series of small molecule-based thermosets were prepared via catalyst-free D–A curing142–144 from monomers composed of multiple furans/maleimides linked by various spacer groups.142–144 The reversibility of the D–A reaction was demonstrated through continuous reprocessing for five cycles,143 which was enabled by the D–A adducts that partially dissociate at elevated temperatures and fully recover at 80 °C during the post-curing.143 In another illustration, a poly(hydroxyaminoether) was formed utilizing an epoxy resin crosslinked with furfurylamine and bis(maleimide).98 This crosslinked network was recycled through compression molding, and by virtue of the dynamic reconfiguration, exhibited similar tensile strength (30.2 MPa vs. 30.2 MPa) and strain-at-break (159.0% vs. 142.7%) as compared to the original sample.98
For situations in which side reactions may be unavoidable, strategies to minimize their effect through DCvC mechanism regulation have been demonstrated with the aid of catalysts. For example, the recycling of polyurethanes (PUs) and non-isocyanate polyurethanes (NIPUs) has been achieved (e.g., 140 °C, 11 MPa for 2 h with a catalyst for reprocessing);145–147 however, selectivity and side reactions related to dissociative pathways hampered reprocessing without the catalyst.145,146 The reactions exhibited dual mechanisms involving both associative transcarbamoylation and dissociative reversible isocyanate/cyclic carbonate aminolysis.135,146,148 To suppress undesirable pathways, new and more efficient catalytic systems that lower reaction temperatures and balance these dynamic exchange processes have been developed to improve the circularity of PUs.135
DCvCs with similar mechanisms to urethane transcarbamoylation such as thiourethanes, also have been investigated as crosslinks with improved control against side reactions.99,149,150 For example, a recyclable (110 °C, 10 MPa for 15 min) poly(thiourethane-urethane) elastomer was prepared from PU prepolymers and a trifunctional isothiocyanate crosslinker.99 A small-molecule model study revealed that the dynamic thiourethane exchange displayed a similar dual mechanism as discussed above, such that both the dissociative reversible addition and the associative thiol exchange occurred.149 Incorporation of a small excess (10 mol%) of thiol groups promoted this duality to improve material circularity by suppressing deleterious side reactions (e.g., isocyanate dimerization/trimerization) that accompany the dissociative pathway.149
Aside from suppressing side reactions, expanding the toolbox of available stimuli beyond heat broadens potential applications and recycle/reuse scenarios. One of the most versatile DCvCs is disulfide exchange. Responding to various external stimuli, such as light,95 pH,158,159 redox potential,160 ultrasound,161 pressure,162 and mechanical force,163 the disulfide bond exhibits a responsive behavior that can be tailored to versatile reuse and recycle designs.95,103,164 This broad applicability is driven by the diversity of anionic or radical mechanisms in disulfide exchange or thiol-disulfide interchanges.158,159,165 The photo-responsiveness of disulfide bonds has been shown useful for polymer recycling via photo-degradation to recover monomers166,167 and healing166,168,169 through bond reconfiguration to repair scratches and cracks in the presence of UV light.95,170 For example, an epoxy resin utilizing a disulfide crosslinker exhibited healing after UV irradiation for 3 min,95 demonstrating the potential contribution of disulfide chemistry to circularity by promoting repair and reuse to extend product lifetime. Thermal treatment remains an option for reprocessing disulfide networks, similar to many other DCvCs.103,164,172 In one study, a homogeneous film with unaltered mechanical properties could be obtained by hot-pressing (150 °C, 30 bar, 20 min) a fragmented poly(urea-urethane) elastomer containing disulfide bridges.172
Disulfide networks also undergo decrosslinking in solution through reduction of the disulfide linkages into thiols in the presence of small molecule thiols or phosphines (e.g., 50 °C, 24 h in dimethylformamide),171 showcasing the potential for chemical recycling.95,166 The recycling of DCvC-crosslinked polymers via selective dissolution and decrosslinking under mild conditions provides a potential opportunity to simplify sorting procedures, as a complementary strategy to mechanical recycling.57–59 After solvent-based reprocessing, the refabricated polymers from recovered monomer exhibited no loss in properties, leading to an ideal, circular polymer economy.43 In fact, a large fraction of networks based on DCvCs, such as alkoxyamine,173 D–A reaction,98 transcarbamoylation,131 and thiourethane exchange, have exhibited recyclability via decrosslinking or depolymerization in solution with the assistance of the corresponding DCvC compounds.99 For example, PMMA networks crosslinked by alkoxyamine can be dissolved with an excess of small molecule alkoxyamines to yield linear polymers, and then re-crosslinked by adding more alkoxyamine-containing PMMA to shift the equilibrium back towards network formation (Scheme 1).173 However, the separation and purification of recovered polymer components, as well as the consumption of chemical reagents, can be limiting factors for the solution-based recycling of these dynamic covalent polymers.
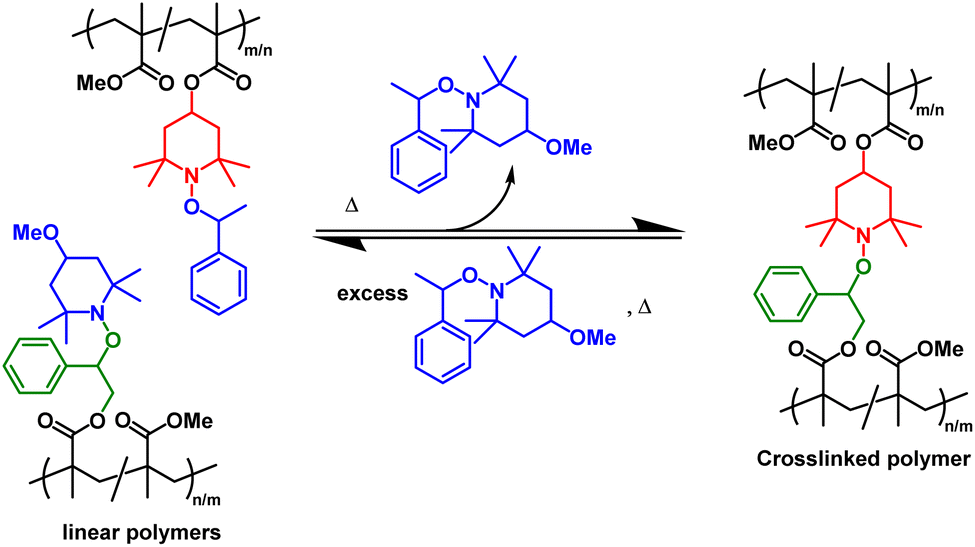 | ||
| Scheme 1 Reversible crosslinking/decrosslinking reactions between the complementary reactive alkoxyamine units (denoted in red and green) in linear polymethacrylates precursors. The reaction is tuned by the small-molecule alkoxyamine byproduct (denoted in blue) Adapted from ref. 173 with permission from Springer Nature, copyright 2013. | ||
To overcome the aforementioned obstacles, the use of pH and common solvents in solution-based recycling has attracted increasing attention. DCvCs, such as imines,174,175 boronic esters,176 and boroxines,58 are viable alternatives that don't require excess chemical consumption and related waste generation. In an example of solvolytic recyclability of imine-based dynamic covalent polymers, a DGEBA epoxy resin was cured using vanillin and m-xylylenediamine.174 Thermal remolding of these imines led only to partial recovery of the original material strength (∼40% loss). However, the imine hydrolysis under mildly acidic conditions (0.2 M HCl solution, VH2O/VTHF = 2/8, 25 °C, 8 h) and subsequent evaporation led to a defect-free recycled network with > 90% strength recovery.174,177 The improved properties of the chemically-recycled polymer in comparison to mechanically-recycled materials (e.g., almost unaltered tensile strength, failure strain, and Young's modulus vs. the original network)175 demonstrated that dissolution of DCvC-based polymeric materials is a promising and complementary pathway to circularity for common thermosetting materials, such as epoxy resins.174,175
Boronic esters and boroxines also are popular DCvCs in vitrimer design because of their reversible nature at ambient temperatures and facile decrosslinking with excess alcohols.176 For example, phenylboronic acid-terminated poly(aryl ether ketone) oligomers (PAEK) and (3-(3-carboxy-4-trifluoromethylphenoxy)-phenyl)boronic acid (CB) were used to design a recyclable PAEK-CB polymer.58 The PAEK-CB polymer was fabricated in solution via dynamic crosslinking of boroxine, displaying good mechanical performance and recyclability through dissolution and depolymerization in the presence of ethanol (30 °C, 1 h). Following filtration and evaporation, 90% and 93% of the chemically-intact PAEK and CB were recovered, respectively.58 The repolymerized PAEK-CB fabricated from recycled monomers showed nearly identical mechanical properties after five continuous cycles,58 indicating the recyclability via dissolution of this boroxine-crosslinked system.
A key feature in this solvent-responsive system is the ability to selectively recycle the PAEK-CB via depolymerization from mixed polymers, simulating a plastics waste stream (Fig. 8).58 This selectivity plays an important role in enabling recycling in complicated, heterogeneous, mixed plastics waste streams, reducing the effort required in separation and sorting, and ultimately, minimizing the property loss of recycled polymers caused by residual contaminants.33 Similar selective chemical recycling of DCvC networks via dissolution from mixed commodity polymers has been accomplished via diketoenamine hydrolysis and reversible nucleophilic aromatic substitution of cyanurate.57,59 The corresponding polymers were depolymerized and extracted in monomer form through hydrolysis (5 M H2SO4, 25 °C, 12 h) and base-catalyzed alcoholysis in refluxing ethanol (5 wt% K2CO3, 90 °C, 16 h), respectively.57,59 Although there is still room for improvement with respect to the time required for depolymerization, these recycling approaches are promising due to the dissolution and subsequent refabrication of the dynamic polymers from mixed waste streams. The selective responsiveness is attractive for simplifying sorting procedures and reducing costs in waste polymer recycling protocols without impacting the recycling process of other polymer components.
 | ||
| Fig. 8 Selective separation of the PAEK-CB polymer from mixed plastic waste streams. (A) PAEK-CB polymer (∼8 g) and five types of commodity plastics (∼42 g) were (B) combined in a beaker to generate a plastic mixture. (C) N-methylpyrrolidone (NMP)/ethanol was added to the plastic mixture and (D) stirred for 1 h at 30 °C. (E) The filtrate containing PAEK and CB monomers was separated from the remaining plastic solids and (F) PAEK and CB monomers were isolated from the filtrate. Adapted from ref. 58 with permission from the American Chemical Society, copyright 2022. | ||
There are considerably more reports on DCvC implementation in covalent networks than linear polymers (Table 1), yet a few dynamic thermoplastic polymers have been investigated in the context of solution-based recycling.150,178,179 For instance, Lehn and collaborators employed an imine-containing diol in a linear PU to introduce DCvC into this thermoplastic polymer.180 The polymer film was decomposed by immersion in water (50 °C for 14 h), whereby the number average molecular weight (Mn) decreased from 13 kDa to 7 kDa as a result of the dynamic hydrolysis of the imine linkages. After drying at 80 °C for 240 h, the imine linkages in the dynamic polymer (i.e., dynamer) reformed, and the polymer recovered back to the original Mn.180 A more recent study provided a general copolymerization approach towards chemically recyclable vinyl polymers through the introduction of a thionolactone cleavable comonomer.179 After deconstruction of the thioester-containing polymers with cysteamine, the thiol-terminated fragments could be reconfigured into new polymers via condensation of the disulfide groups with full recovery of elastic moduli at room temperature.179 In another example, an innovative three-component DCvC was developed to prepare linear dynamic polymers.178 This system involved the reaction of bis-salicylaldehyde, oligo(ethylene glycol) diamines, and 2-mercaptobenzaldehyde at room temperature to produce linear polymers (Mn ≤ 20.1 kDa).178 The polymer dynamically exchanged the monomer units with external 2-mercaptobenzaldehydes under mild heating (70 °C).178 Depolymerization was realized under mildly basic conditions (pH 8.0) to recover the constituent oligomers,178 facilitating thermomechanical reprocessing.
| Dynamic covalent chemistry | Advantages | Considerations |
|---|---|---|
| a This table presents a list of common attributes for the specified DCvC chemistry and is not intended to be comprehensive. | ||
| Transesterification | Tunable exchange rates | Metal salt catalysts: limited solubility, leaching, corrosion |
| Organic base catalysts: toxicity, high cost | ||
| Alkoxyamine | Tunable via electronic effect | Additional virgin feedstock required for decrosslinking/re-crosslinking |
| Imine | Chemically recyclable | Prone to hydrolysis |
| Hindered urea | Tunable via steric hindrance | Prone to hydrolysis |
| Siloxane; silyl ether | High thermal stability | Lewis acid catalysts: leaching, corrosion |
| Metal silanolate catalysts: catalyst deactivation, corrosion | ||
| Diels–Alder | Highly selective | Exchange rates predetermined by chemistry selection |
| (Thio)urethane | Accessibility | Prone to side reactions |
| Organotin catalysts: toxic | ||
| Rare earth metal catalysts: leaching, high cost | ||
| Organic base catalysts: prone to hydrolysis | ||
| Disulfide | Responsive to various stimuli (heat, light, redox, etc.) | Multiple, non-selective triggers for exchange |
| Susceptible to oxidation and side reactions | ||
| Boronic ester; boroxine | Mild reprocessing conditions | Prone to hydrolysis |
Commodity polymer valorization
While new dynamic covalent polymeric materials offer huge potential for reprocessability and recyclability under various conditions,49,106 it is equally important to translate these synthetic advances to existing plastics waste.2,21 Commodity plastics include LDPE, HDPE, PP, PVC, PS, and PET,181 and are the main contributors to the plastics accumulation problem due to an overall recycling rate below 10 wt%.41,42The utilization of DCvC to transform existing commodity polymers into dynamic, recyclable polymers is a promising pathway toward polymer circularity via post-polymerization modification.49,105 Although many of the commodity polymers are recyclable in principle, the downgrading of recycled materials and the unfavorable cost of reprocessing are general limitations to circularity.23 The introduction of reversible-bond exchange via DCvC addresses these issues,51 providing property enhancement and higher value to cover the cost of recycling.41,60,61 This industrial translation is critical to enabling circularity.105
DCvC post-polymerization modification on PE has been successful in minimizing undesirable chain alteration during recycling.61,105 One example involved the introduction of dioxaborolane into HDPE through reactive mixing.61,105 A maleimide with dioxaborolane functionality was grafted onto HDPE followed by dynamic crosslinking in the presence of a bis-dioxaborolane crosslinker via reactive extrusion.105 The successfully crosslinked HDPE was no longer soluble in xylene (gel content ∼31.7%) yet remained extrudable, and the tensile strength was maintained after three cycles of compression molding (150 °C, 10 min).105 These changes in solubility and reprocessability suggested the transformation of a commodity HDPE into a recyclable vitrimer. Decrosslinking and full dissolution were achieved on the reprocessed material in the presence of excess 1,2-diol,105 not only demonstrating the chemical recyclability of the network, but also highlighting the absence of permanent non-dynamic crosslinking after reprocessing, which is a common occurrence in PE mechanical recycling.16
In addition to the reprocessing of modified HDPE, the vitrimer crosslinked by dioxaborolane groups displayed efficient weldability with PMMA modified with the same DCvC, without the need for an extra compatibilizing tie layer.105 The chemical compatibility of the dioxaborolane-containing polymers improved general adhesion between PMMA/PE layers, whereas the dynamic bond exchange reinforced the welded seam through the formation of covalent bridges between the two vitrimers.105 The dynamic interfacial adhesion presents a promising route to recycling mixed, multi-layer plastics with reduced sorting and minimal contamination from additives (i.e., compatibilizers). The strategy is complementary to the selective dissolution approach above, but both methods can simplify the sorting process of mixed dynamic polymer waste streams and reduce the cost and labor of a closed-loop framework for the plastics circular economy.182
PP, another commodity plastic, also has been successfully valorized via a similar approach.183 PP was grafted with maleic anhydride in a melt compounder and subsequently crosslinked with a tetrafunctional thiol linker in the presence of an organic base catalyst during extrusion.183 The dynamic thioester crosslinking not only increased the tensile strength of PP, but also enabled versatile reprocessing via compression molding, injection molding, and fused filament fabrication 3D printing, with minimal impact on the mechanical properties (e.g., tensile strength, failure strain) after several cycles, if the PP vitrimer was cooled slowly to reconstruct the crosslinks.184
Dynamic crosslinking of polyolefins (e.g., PE, PP, ethylene/propylene copolymer rubbers) via grafting of maleic anhydride has become a popular approach. Followed by subsequent functionalization of the anhydride, other types of DCvC, such as the Diels–Alder reaction,185,186 transesterification (Fig. 9A),60,182 imine exchange,187 and silyl ether exchange,188 have been implemented in polyolefin vitrimers. Real post-consumer polyolefins also were utilized as feedstocks for transesterification-based vitrimers to highlight the compatibility of DCvCs with additives and contaminants, revealing the potential to upgrade real plastics waste.60,182 Overall, polyolefin vitrimers showed comparable, or even slightly improved, strength and Young's modulus, and enhanced creep resistance induced by crosslinking vs. their thermoplastic precursors (Fig. 9B and C).60
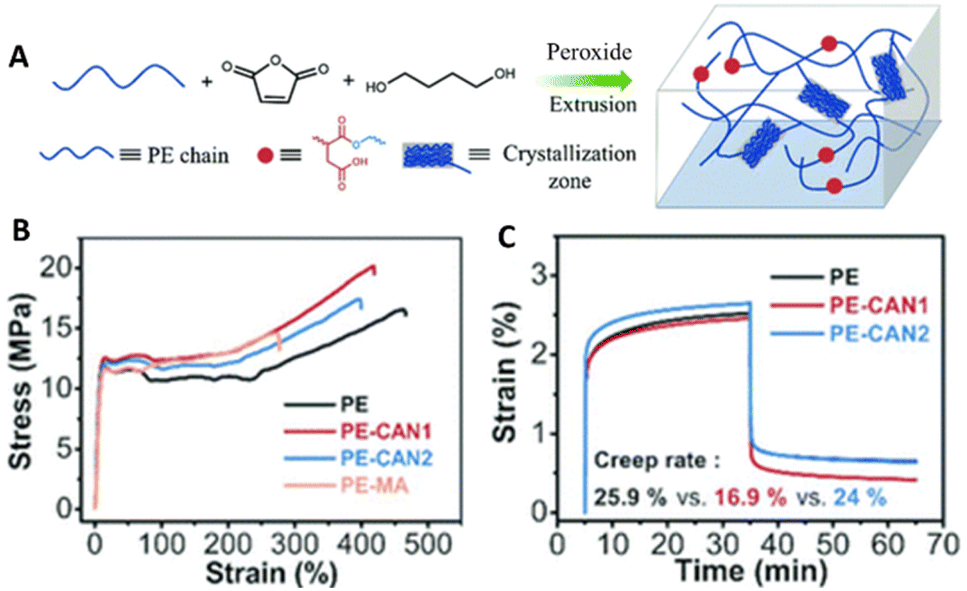 | ||
| Fig. 9 Preparation and properties of maleic anhydride grafted polyethylene (PE-MA) and polyethylene covalent adaptable networks (PE CANs). (A) Scheme of preparation and structure of PE CANs. (B) Tensile testing curves of PE, PE-MA, and PE CANs. (C) Creep rates and curves at 50 °C via dynamic mechanical analysis (DMA) before and after modification. Adapted from ref. 60 with permission from The Royal Society of Chemistry, copyright 2021. | ||
Polyolefins remain a key target for DCvC functionalization to improve recyclability; however, other commodity polymers also exhibit enhanced circularity with post-polymerization modifications. In a recent investigation, Saito and collaborators reported that the PS block in a styrene-ethylene-butylene-styrene (SEBS) block copolymer could be functionalized directly with boronic esters through iridium-catalyzed aromatic C–H borylation, followed by dynamic crosslinking with hydroxyls on the surface of silica nanoparticles to form a composite.189 The dynamic exchange in this crosslinked PS network led to reprocessability via compression molding in air; however, the tensile strength of the DCvC-containing PS gradually diminished during recycling (∼83% of original strength after three cycles) due to backbone oxidation, highlighting an area of improvement for future studies.189 In addition to the aromatic C–H activation, another study probed the use of azidoformate with dioxaborolane moieties to functionalize PS via an aliphatic C–H insertion mechanism of nitrenes.107 The functionalization was achieved via compression molding and reactive extrusion, suggesting the potential to implement this method at an industrial scale.107
As the world's third-most widely produced plastic,190 PVC suffers from the chemical instability of C–Cl bonds and a poor recycling rate as a consequence.41 In an effort to improve the recyclability of these polymers, Yagci and collaborators reacted commercial PVC with elemental sulfur, which effectively incorporated dynamic polysulfide linkages into the network.191 The crosslinked PVC exhibited reprocessability via thermal treatment up to three cycles, demonstrating the dynamic nature induced by the polysulfide linkages.191 However, deleterious side reactions (e.g., dehydrochlorination, irreversible crosslinking) on the PVC backbone progressively altered the polymer properties and eventually inhibited network reconfiguration after three cycles of reprocessing.191 Further research is needed in this area to develop DCvCs that also stabilize the PVC backbone or do not trigger main chain deterioration.
Apart from adapting commodity polymers for enhanced recyclability, incorporating DCvCs provides alternative recycling strategies for degradable condensation polymers, such as PET and PUs.51,147 For example, PET was reactively extruded via an autocatalyzed NGP transesterification in the presence of a polyol crosslinker containing an adjacent tertiary amine.51,52,192 The resultant PET vitrimer displayed enhanced creep resistance vs. the neat PET and was reprocessable to yield transparent and uniform products via compression molding, extrusion molding, and injection molding for 3 cycles with mechanical property (e.g., storage modulus) retention greater than 90%.51,52,192 In contrast, as mentioned earlier, PET recycled through conventional reprocessing suffered from property deterioration (e.g., brittleness, decreased melt viscosity) after two cycles of mechanical recycling and extrusion.51 Similar approaches have been investigated to enhance the properties of PUs. In a recent study, reactive extrusion reprocessing of a PU foam via dynamic transcarbamoylation in the presence of a tin catalyst was achieved which yielded extrudate with tensile strength (50.8 MPa vs. 49.7 MPa) and yield point (3.6% strain vs. 4.8% strain), similar to those of as-synthesized PU.147 Commodity PU foam with additives (e.g., surfactants, flame retardants, stabilizers) also can be reprocessed with this approach.147 The implementation of these continuous recycling approaches for PUs is of particular interest as they have the potential to reduce the demand for toxic precursors (e.g., phosgene, isocyanate) for new PU manufacturing by recreating materials from existing, post-consumer products.147 In comparison to conventional recycling, continuous recycling/upcycling of condensation polymers using a DCvC vitrimer framework has led to extended material lifetimes and a reduction in the (re)manufacturing costs.
In general, recycle-by-design polymers along with chemically modified commodity polymers that incorporate DCvC have been proven effective in improving the potential for circularity. Synthetic efforts have increased creep resistance, tuned equilibria, and expanded reprocessing conditions (e.g., temperature, time, stimulus). However, to further optimize conditions for reprocessing/recycling, synthetic knowledge must be coupled with a comprehensive physical understanding of dynamic covalent polymeric materials.
DCvC modeling, prediction, and physical behavior
The implementation of DCvCs into polymeric materials leads to challenges in the development of reliable physical models to predict the properties of dynamic polymers, particularly crosslinked structures and vitrimers. These physical models are necessary to guide the design of recyclable dynamic polymers suitable for industrial processing and various end uses.122 For example, engineering the characteristic relaxation time (τR) and tuning the Deborah number (De = τR/τobs, in which τobs is the time of observation or processing) of dynamic polymer systems are important aspects related to (re-)processing. For dimensional stability, the dynamic polymeric materials must exhibit slow network rearrangement (e.g., De ≫ 1, τR ∼103 s → ∞ at use temperatures),66 yet they must maintain reprocessability or recyclability at shorter time scales under appropriate stimuli for circularity. DCvC-containing polymers can possess several relaxation time- and length-scales that are integrally dependent on the specific chemistry, polymer architecture, and microphase structure of the material. Thus, the prediction of macroscopic properties from the macromolecular structures and dynamic bonding characteristics provides insight into the application and reprocessing of the polymer.Understanding the structure–property relationships of DCvC-based polymers involves the identification of a material's thermal transitions, and in the case of vitrimer networks, the topological freezing temperature (Tv), which classic thermoplastics and thermosets do not possess.97 The Tv is the temperature at which the dynamic covalent bond exchange becomes slower than the experimental time scale of relaxation, leading to a topologically frozen network.97,114 Generally, the Tv is the temperature at which the melt viscosity is equal to 1012 Pa s, which is also associated with the liquid-to-glass transition.97,193,194 This temperature is distinct from the glass transition temperature (Tg), which is the arresting of cooperative motion and decrease in molecular mobility. Although they are independent transitions, the Tg and Tv can be affected by similar phenomena, and both affect reprocessing conditions. For example, increasing the crosslinking density of a polylactide-based vitrimer increased both the Tg and Tv.195 However, when a higher loading of catalyst is added to improve transesterification rates, the Tg remained generally unaffected while the Tv was lowered by 50 °C (from 235 °C to 185 °C).114,195 The identification of the Tv in DCvC-based polymers influences the application and reprocessing of vitrimer networks, and the prediction of these transitions has gained significant interest.196–204
To understand the polymer relaxation modes and dynamics in dynamic covalent networks, the sticky reptation/Rouse theory provides a strong foundational basis.196–198 Originally designed for non-covalent systems (e.g., ionomers, biological macromolecules, supramolecular networks),196,199 these theories define ‘sticky points’ as the temporary crosslinks and describe the impact on motion in the entangled/unentangled linear chains. In comparison to the unmodified reptation/Rouse model, the chain mobility in the sticky reptation/Rouse model is constrained by the temporary crosslinks when the time scale is less than the average lifetime of the crosslink. On timescales equal to or longer than the lifetime of the crosslink, stress relaxation can occur through now-permitted chain diffusion.196 This relaxation time crossover is governed by, among other factors, the reaction potential of the dynamic covalent bond and bond strength thus highlighting the importance of an appropriately chosen chemistry for specific reprocessing conditions.197,205 Switching from non-covalent bonding to dynamic covalent bonding leads to significant differences in the lifetime and activation energy of the bond exchange, as well as the reaction pathway. The breaking and reforming of covalent bonds are energy intensive, and therefore, have longer lifetimes than their non-covalent counterparts.85,206 Vitrimers generally have a characteristic Arrhenius viscosity dependence with temperature,115,207 with larger activation energies leading to longer relaxation times. Activation energies of dynamic covalent bonds range from ∼10–200 kJ mol−1 depending on both the dynamic bond chemistry and the polymer structure;115 thus, several magnitudes of relaxation times are accessible. Other non-chemical factors include topological constraints, such as the number of crosslinks and chain entanglements, with entanglements between crosslinks increasing the power law dependence of viscosity on concentration in a good solvent.197,205
Numerous theoretical and computational studies about vitrimer networks have been conducted in recent years.200–203 To predict dynamic covalent bond energy, molecular dynamics studies introduced an optimal bond swap energy barrier (ΔEsw) with an Arrhenius temperature dependence to describe the bond exchange of the vitrimer network.200,201,204 The impact of ΔEsw was investigated and found to have an inverse relationship with extrusion rate.201 A theoretical and computational study using the inhomogeneous sticky Rouse (IHR) model revealed that,208–210 for crosslink exchange reactions, the viscous flow activation energy from rheology (Erha) is usually greater than the activation energy of small molecule analogues (Esma), which agreed well with the empirical observations.203 This difference highlights the reprocessing time dependence on the timescale of dynamic covalent crosslinks and polymer segmental relaxation.85,211 Neat polymers with lower Tgs will have faster relaxation times for the same dynamic covalent chemistry than a neat polymer with a higher Tg.203 Segmental relaxation of the polymer chain is generally disregarded at temperatures well above Tg. Nonetheless, as the temperature nears Tg, Williams–Landel–Ferry212 (WLF) and Vogel–Fulcher–Tammann213,214 (VFT) equations describe the non-Arrhenius temperature dependence. One study used a dynamically crosslinked PS copolymer with one fast-exchanging amino-modified group and one slow-exchanging unmodified silyl ether crosslinker, which changed the topological freezing temperature by 70 °C (from 117 °C to 47 °C).121 This design led to WLF-described dynamics at a temperature within 30 °C of the Tg, and eventually more Arrhenius-like dependence at higher temperatures.121 In addition to the exchange mechanism and temperature dependence, another notable difference among dynamic polymers is that many of these polymers contain dynamic bonds in the backbone (e.g., polyesters, imine-based dynamic polymer systems).122 Therefore, when the dynamic crosslinks change, the backbone relaxation timescale of the polymer chains also changes. The structural variation poses new challenges to models that attempt to predict activation energies and viscous flow behavior of DCvC-based polymers.
Dissociative chemistries also can experience structural variations, thus models with exclusively associative DCvCs at crosslinking points are inadequate to describe the behavior of dissociative networks. One major difference is that dissociative dynamic networks usually experience a decrease in crosslink density at elevated temperatures. The reduced crosslinking is determined by an enthalpy change (ΔH) of the dissociative reaction.78,215 Usually, the considerable ΔH value of the dynamic dissociative reactions, such as the Diels–Alder reaction (−157 kJ mol−1),88 supports the empirical conclusion of the temperature dependence of crosslink density. Yet, in anilinium-salt-based dynamic crosslinked networks, which undergo dissociative exchanges, the equilibrium constant (Keq) has minimal temperature dependence if the ΔH value is quite small (e.g., −2.8 kJ mol−1).215 As a result, the net decrosslinking is negligible over a wide temperature range, and the network exhibits the same Arrhenius viscosity dependence with temperature as vitrimers. The crosslink exchange mechanism does not necessarily influence the practical reprocessing behavior of dynamic covalent polymer networks, which suggests the design of recyclable dynamic polymers can be considered with fewer constraints. Thus, the focus can be shifted to parameters necessary for bond exchange (e.g., temperature, light) and recycling convenience.216 As a result of rational chemical design, 1,2,3-thiazolium-based poly(ionic liquid) networks displayed either an Arrhenius dependence with temperature like vitrimers or a significant decrease in crosslink density characteristic of dissociative networks, depending on the varied ΔH value of the dissociative reaction.217 The two different rheological responses influence recycling and refabrication processes, with the depolymerizable dissociative networks favoring applications requiring local or temporary flow.217
The physical explanation of the diverse rheological behavior and viscous flow activation energies in dynamic polymer networks inspires more subtle adjustments to the physical properties. Kalow et al.218 discovered from both theoretical prediction and experimental characterization that, in dynamically crosslinked polymer systems, acid-derived crosslinkers with a common exchange mechanism exhibited vastly different stress relaxation rates without impacting the stiffness or viscous flow activation energy. In other recent studies,195,219 supported by both theoretical and experimental investigations, researchers modified the polymer chains with pendant functional groups to adjust the viscous flow activation energy and relaxation behavior. The content of exchangeable groups had only a minimal effect on crosslink density.195,219 Additionally, this adjustment resulted in an improved recovery ratio of the strength and strain-at-break (>90%) after reprocessing. These insights provide innovative strategies to fine-tune specific aspects of the flow properties of dynamic polymer networks under recycling conditions.
In the discussion above, it is assumed that the DCvC groups are homogeneously dispersed throughout the polymer. However, the impact of phase separation between the dynamic crosslinks and polymer backbones should be considered when reprocessing real DCvC-containing polymers. The influence of phase behavior on dynamic exchange reactions is complicated and has various effects on polymer recycling. For example, the dioxaborolane-based HDPE vitrimer investigation discussed previously also showcased the impact of phase separation.105 When the HDPE was functionalized by 1,2-diol instead of dioxaborolane, phase separation induced by the diol prevented vitrimerization using bis-(dioxaborolane).105 Even in the subsequently crosslinked HDPE vitrimer, the gel fraction was lower in comparison to other HDPE vitrimers (∼32%).104,105,220 In a follow-up study,221 the authors revealed that the HDPE vitrimer experienced macroscopic phase separation into dioxaborolane maleimide-rich and -poor regions (Fig. 10) due to the polarity of DCvC functional groups. The phase separation, as indicated by the polymer's opaque appearance, prevented full crosslinking of the HDPE and resulted in a relatively high soluble fraction of the vitrimer.221 Interestingly, although a low gel fraction often is undesirable for polymer networks, the non-grafted phase-separated PE chains functioned as lubricants at the processing temperature and led to shorter stress relaxation times than anticipated, which could be exploited to adjust the recycling conditions of these vitrimers.221 This study exemplifies the complexity of modification of commodity polymers in comparison to small molecules.105
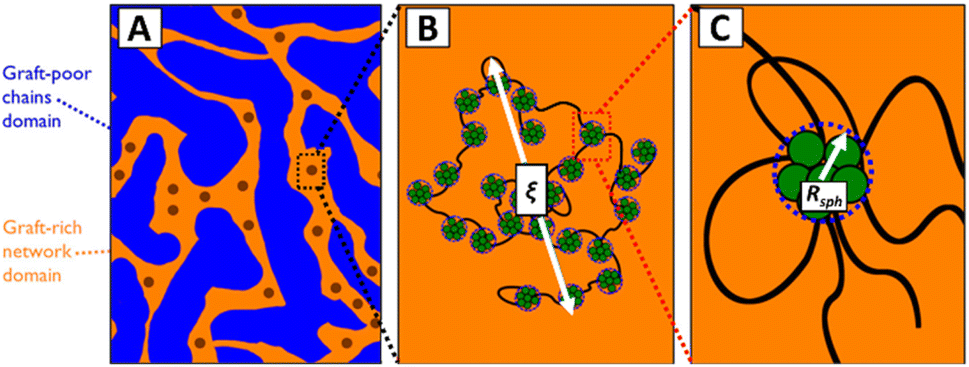 | ||
| Fig. 10 Hierarchical morphology of HDPE vitrimer with crosslinker/graft-rich and -poor phases. (A) Fractals are represented by dark circles. (B) ξ is the fractal length scale. The aggregates, PE chain bridges, and PE chain loops inside are illustrated. (C) A closer look at a single graft-rich aggregate is shown within the radius Rsph. Associated PE chain bridges, loops, and dangling ends also are displayed. Adapted from ref. 221 with permission from the American Chemical Society, copyright 2019. | ||
From another perspective, awareness of phase separation motivates approaches to enhance network homogeneity.104 For example, ethylene copolymerization with comonomers containing dioxaborolane groups led to a more uniform distribution of DCvC groups on the polymer backbone, increasing the gel content to 93–99% while maintaining the tensile strength after two reprocessing cycles.104 In addition to de novo copolymerization, implementing DCvC crosslinkers with good solubility and high reactivity into polyolefins could be an alternative approach to obtain homogeneous, recyclable dynamic covalent polymeric materials (e.g., carbene and nitrene crosslinkers with alkyl spacers to tune the polarity and solubility in the polymer phases).107,222
There are opportunities to leverage heterogeneities in DCvC-containing polymers to tune mechanical properties and recycling conditions. Common post-polymerization modifications to polyolefins, such as maleic anhydride grafting,55 can induce microphase separation. The high polarity of maleic anhydride leads to graft-rich clusters, but homogeneity is improved through the introduction of polar groups to the nonpolar polyolefin backbone.223,224 One study compared a nonpolar polyolefin (ethylene/propylene copolymer, EPM) and a polar polymer (ethylene/vinyl acetate copolymer, EVM)185 in which both EPM and EVM polymers were grafted with maleic anhydride, functionalized with a furan-containing imide, and then dynamically crosslinked with bis(maleimide) via the Diels–Alder reaction.185 Due to the polarity difference, microphase separation occurred in the EPM-based dynamic network, but not in the EVM-based dynamic network. Consequently, after melt reprocessing, the EPM-based network exhibited almost identical mechanical performance (97% retained hardness, 7% compression gain), whereas the EVM-based network displayed moderate property loss (83% retained hardness, 27% compression loss). The authors speculated that the microphase separation in the EPM-based network resulted in polar domains with enriched furans and maleimides remaining in close vicinity, which aided the recovery of the crosslink density after the reprocessing.185 Another study highlighted how the phase-separated clusters of associating bonds led to a high rubbery modulus and extended rubbery plateau region of the network.225 Although this investigation used a telechelic associating polymer, similar conclusions can be extrapolated to DCvC-containing polymers and how percolation of the phase-separated domains could lead to enhanced mechanical properties and recycling.225 Overall, aside from chemical explorations of DCvCs that have shown promise in research about enhanced circularity,22,44,122 addressing the structure–property relationships of DCvC-based polymers is important to realize the full potential of dynamic covalent chemistries for complete polymer circularity.
Economic and environmental assessments in DCvC-based polymer recycling
While progress in synthetic and physical understanding of dynamic polymers is promising, the potential economic and environmental impact of DCvC-containing polymeric materials remains relatively unknown. Quantitative value-chain benchmarks, such as minimum selling price (MSP) and life-cycle greenhouse gas (GHG) emissions, have become increasingly important in the scale up of specific processes, particularly when evaluating circular design vs. conventional, linear approaches.226 TEA and LCA are important tools for determining these quantitative values to provide additional insights about the overall effect of a new material or process on the economy or the environment. TEA typically incorporates technical and financial input parameters (e.g., costs, benefits, risks) to evaluate economic performance.227 An LCA study generally involves compiling energy and emissions information across material value chains (e.g., transportation, production, consumption) to assess environmental impacts.228 Over the last few years, several TEA and LCA studies have been published on the recycling of commercial polymers.33,229–236 In a recent example, the LCA of an exploratory, lab-scale synthetic process for the upcycling of poly(acrylic acid) revealed a 10-fold decrease in global warming potential (kg CO2 kg−1) and cumulative energy demand (MJ kg−1) when an acid-catalyzed hydrolysis approach was employed vs. a base-mediated route.237 The study revealed key method optimization parameters to minimize the impact on the environment at an industrial scale, which may have been overlooked in a lab-scale study that excluded LCA.In contrast to the cases of commercial polymers, TEA and LCA investigations of recyclable dynamic polymers are quite scarce due to the nascency of the field and the lack of industrial-scale application. For the rare instances in which TEA and LCA have been applied to recyclable dynamic polymer research,238 the results highlighted the potential of DCvC-based materials to reduce the cost and emissions during recycling.238 More importantly, TEA/LCA identifies the main contributors to elevated costs and emissions among the steps of the production/recycling process, motivating system optimization. For example, in practical application, only a fraction of the recycled polymer will be recovered from the waste stream and survive the recycling process. To maintain the material output of each cycle, recycled products must be supplemented with pristine polymer, increasing the expected cost and emissions of recycling dynamic polymers. Therefore, to lower the overall cost of the material, it is worthwhile to optimize the pristine material at a lab scale, using TEA/LCA to guide the synthetic pathways.
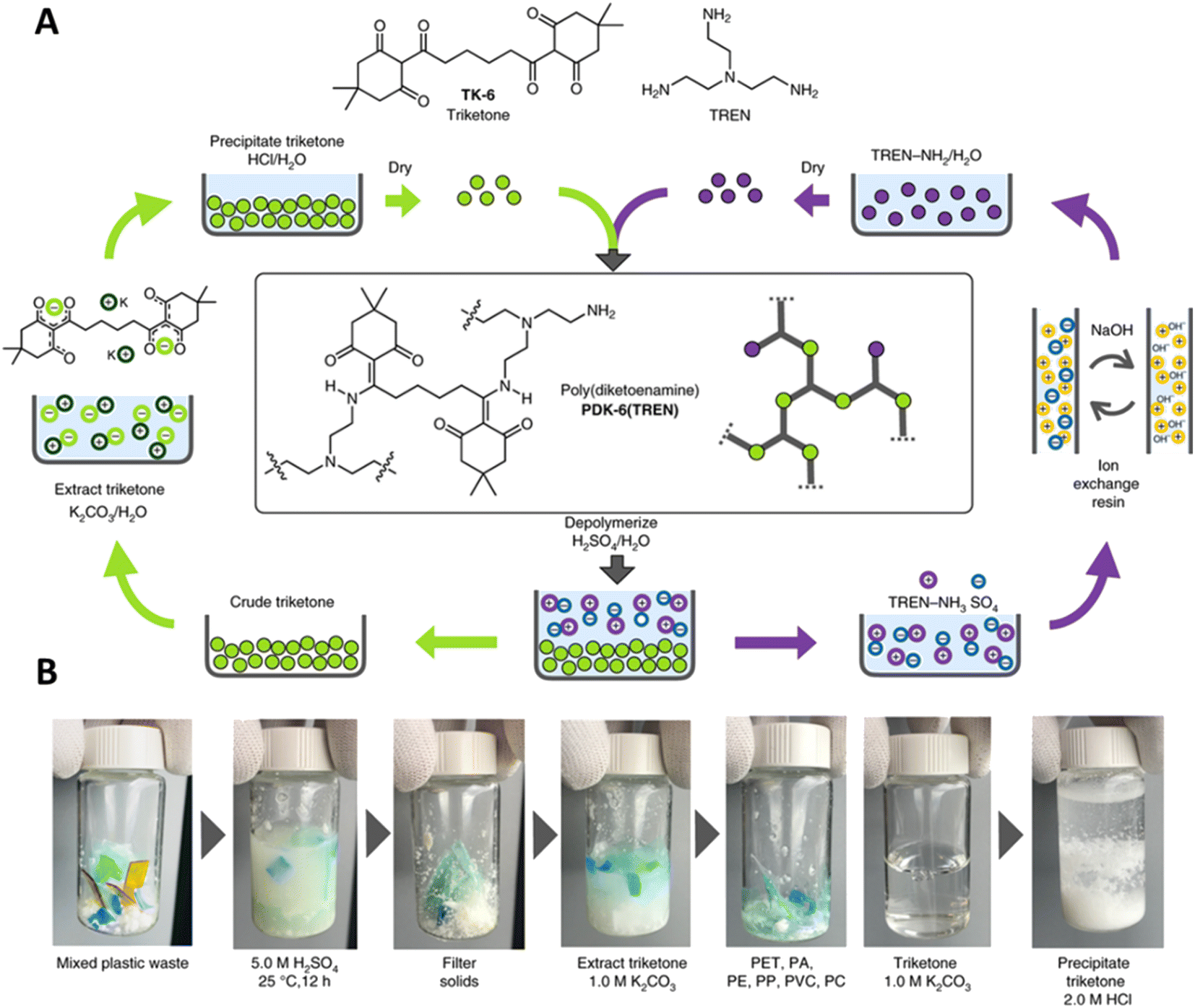
One of the few DCvC-containing materials with accompanying TEA and LCA is polydiketoenamine (PDK), which exhibits dynamic exchange of diketoenamines in the presence of an acid.238,239 The stiffness of PDK networks was readily tuned by adjusting the formulation with commercially-available soft and hard diamine monomers,238,240 indicating the versatility and applicability of PDKs for applications such as flexible packaging and vehicle/aircraft parts. Recycling was achieved using acid-triggered selective depolymerization and was compatible with additives and mixed plastics waste (Fig. 11),59 showcasing PDK as a promising DCvC-based polymer for circular design.
 | ||
| Fig. 11 The preparation and recycling of PDKs. (A) The circular design of network PDKs. The starting polyamines and ditopic triketones are recycled after hydrolyzing PDKs in a strong acid. (B) Photographs suggest the orthogonality of depolymerization and monomer recovery in the presence of PET, nylon-6,6 (PA), PE, PVC, and polycarbonate (PC). Adapted from ref. 59 with permission from Springer Nature, copyright 2019. | ||
According to TEA and LCA,238 ideal ‘circular’ PDK resins showed significant decreases in MSP ($1.5 kg−1vs. $45 kg−1) and GHG emissions (2 kg CO2e kg−1vs. 86 kg CO2e kg−1) per unit mass in comparison to the primary PDK resin.238 However, as discussed above, a fraction of the recyclate was lost during the waste recovery and recycling process.238 Even assuming 100% PDK waste recovery, the MSP was $9 kg−1 (as opposed to $1.5 kg−1 for the ideal circular resin) due to material losses during the chemical recycling process that needed to be supplemented with the more expensive primary PDK.238
In light of the TEA/LCA observations, subsequent work239 targeted optimization of the synthesis of primary PDK,238 such as limiting the use of N,N′-dicyclohexylcarbodiimide and 4-(dimethylamino) pyridine in favor of more cost-effective and environmental-friendly alternatives.239 By strategically modifying the synthetic route, the MSP and GHG emissions of primary PDK were reduced by 58% ($45 kg−1 to $19 kg−1) and 66% (86 kg CO2e kg−1 to 29 kg CO2e kg−1), respectively.239
In addition to PDKs, a dynamic polymer containing D–A adducts and hydrolyzable acetal functionalities was reported to be self-healable and closed-loop recyclable.241 This dynamic polymer was prepared from bis(maleimide), biomass-derived 5-hydroxymethyl furfural, and pentaerythritol. The dynamic D–A reaction led to self-healing under heat treatment (30 min, 130 °C), and chemical recycling was achieved via acid-catalyzed hydrolysis of cyclic acetal as well as retro-Diels–Alder reaction.241 A multi-solvent extraction method was employed to recover 95 wt% of the original polymer as monomer and D–A adducts by fractionation in different green solvents.241 TEA indicated that the MSP of the recycled polymer ($8.17 kg−1) was 19.5% lower than that of the virgin polymer, emphasizing a potential benefit of dynamic polymer usage and recycling.241
Advances in TEA and LCA of DCvC-containing polymers are critical to inform future studies that target polymer circularity. Open-source options exist for both TEA and LCA (e.g., Biorefinery Simulation and TEA Modules (BioSTEAM), openLCA, Materials Flows through Industry (MFI) tool) to aid in the preliminary design and calculation of economic and environmental impacts of newly synthesized DCvC-based polymers.227,242,243 The results will guide method optimization during lab-scale development and identify optimal application areas for DCvC-based polymers that utilize their unique features.239 Unfortunately, most dynamic polymers have yet to be evaluated in applied scenarios and are lacking the relevant data (e.g., GHG emissions, overall energy demands, costs) to make direct comparisons with incumbent materials. Moving forward, studies that focus on expanding the available economic and emissions data to include chemicals and processes relevant to DCvC-containing polymers will be crucial. These activities will include determining specific sources for comparison (e.g., solvents, feedstocks), identifying top process contributors (e.g., energy, GHG emissions), and considering the value chain as a whole to evaluate the impact of waste recovery vs. pristine polymer production.
Beyond the calculations of MSP and GHG emissions resulting from TEA/LCA studies, a number of other environmental impacts (e.g., biodegradability, microplastics formation) of DCvC-based polymers should be considered.244 As one example, a recent investigation showed a biobased vitrimer made with soybean oil and containing boronic ester dynamic bonds had superior seawater biodegradability vs. a corresponding thermoset with static crosslinks.244 The results suggested that an additional benefit of DCvC could be minimal waste accumulation,244 yet dynamic polymers are rarely studied for their environmental compatibility, which is anticipated to be strongly impacted by different functional groups and building blocks. Because a significant fraction of plastics enters the environment, it is imperative to investigate the potential effects of DCvC-based polymeric materials on the ecosystem.
Conclusion and outlook
The global plastics waste problem necessitates interdisciplinary innovations to promote circularity in plastics production and consumption. The path forward should include the design of new, sustainable, and recyclable materials, along with the ability to tackle existing plastics waste without compromising properties that make plastics desirable (e.g., high strength-to-weight ratio, durability, economic feasibility). As such, DCvC-containing polymeric materials are uniquely positioned to reduce plastics waste by enabling the recycling and reprocessing of post-consumer polymeric materials. Synthetic strategies guided by the principles of equilibrium, reaction kinetics, and stimuli-responsiveness for DCvC have produced numerous recycle-by-design strategies for new materials and post-polymerization modifications for existing commodity polymers. Physical models have been proposed to demonstrate the unique thermomechanical properties of dynamic covalent polymers that are crucial considerations in the optimization of processing and reprocessing techniques. Although currently limited by a lack of relevant data, TEA and LCA have in some cases quantified the economic and environmental impacts of recycling DCvC-based polymeric materials from a systematic perspective, guiding method optimizations towards more sustainable, cost-effective approaches.Yet, many of these current solutions have been obtained in the vacuum of a single field (synthesis, physics, engineering, data science, environmental science, economics, etc.) and approached only from the laboratory scale. The realization of circularity for plastics using dynamic polymeric materials will require tight integration of all fields and close collaborations between research institutes and industry. In particular, these interdisciplinary efforts should work towards establishing an integrated feedback loop between TEA/LCA and material design. Future studies should take advantage of TEA/LCA-informed experimental approaches, such as evaluating the role of cost in catalyst selection and estimating GHG emissions associated with changes to synthetic processes. Additionally, potential end-of-life impacts should be evaluated with the engagement of environmental scientists (e.g., microbiologists, ecologists). Prediction and exploration of physical properties and fine-tuning of chemical reaction behavior in DCvC-based polymers also can be enhanced through emerging artificial intelligence options, polymer-specific notations, and databases (e.g., BigSMILES, CRIPT).245–247 Aside from material-based concerns, social and behavioral challenges associated with waste plastics recovery also limit closed-loop recycling of polymer plastics waste.17–21 Long-lasting solutions toward circularity will require not only multidisciplinary scientific advances but policy reform from industry and government, and elimination of social barriers to recycling.17–21 With persistence on all fronts, dynamic covalent chemistries are poised to address several plastics' end-of-life challenges and accelerate the realization of closed-loop circularity in polymeric materials.
Author contributions
All authors contributed to the writing of this manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported as part of the Center for Plastics Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award DE-SC0021166.References
- S. Billiet and S. R. Trenor, ACS Macro Lett., 2020, 9, 1376–1390 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- Z. R. Hinton, M. R. Talley, P. A. Kots, A. V. Le, T. Zhang, M. E. Mackay, A. M. Kunjapur, P. Bai, D. G. Vlachos, M. P. Watson, M. C. Berg, T. H. Epps, III and L. T. J. Korley, Annu. Rev. Mater. Res., 2022, 52, 249–280 CrossRef.
- P. Europe, Plastics – the Facts, https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022, accessed December 15, 2022.
- L. Lebreton and A. Andrady, Palgrave Commun., 2019, 5, 6 CrossRef.
- T. Brydges, J. Cleaner Prod., 2021, 293, 126245 CrossRef.
- I. Capellán-Pérez, M. Mediavilla, C. de Castro, Ó. Carpintero and L. J. Miguel, Energy, 2014, 77, 641–666 CrossRef.
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181–189 CrossRef.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- Y. Zhou, G. He, X. Jiang, L. Yao, L. Ouyang, X. Liu, W. Liu and Y. Liu, J. Hazard. Mater., 2021, 411, 125178 CrossRef CAS PubMed.
- H. A. Leslie, M. J. M. van Velzen, S. H. Brandsma, A. D. Vethaak, J. J. Garcia-Vallejo and M. H. Lamoree, Environ. Int., 2022, 163, 107199 CrossRef CAS PubMed.
- E. S. Gruber, V. Stadlbauer, V. Pichler, K. Resch-Fauster, A. Todorovic, T. C. Meisel, S. Trawoeger, O. Holloczki, S. D. Turner, W. Wadsak, A. D. Vethaak and L. Kenner, Exposure Health, 2023, 15, 33–51 CrossRef CAS PubMed.
- M. Geissdoerfer, P. Savaget, N. M. P. Bocken and E. J. Hultink, J. Cleaner Prod., 2017, 143, 757–768 CrossRef.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- T. Hundertmark, M. Mayer, C. McNally, T. J. Simons and C. Witte, How plastics waste recycling could transform the chemical industry, https://www.mckinsey.com/industries/chemicals/our-insights/how-plastics-waste-recycling-could-transform-the-chemical-industry, accessed April 18, 2022.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- A. Cecchin, R. Salomone, P. Deutz, A. Raggi and L. Cutaia, Circ. Econ. Sustainability, 2021, 1, 83–97 CrossRef.
- P. Schröder, A. Lemille and P. Desmond, Resour., Conserv. Recycl., 2020, 156, 104686 CrossRef.
- Nature, 2021, 590, 363–364 Search PubMed.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- L. T. J. Korley, T. H. Epps, III, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- T. H. Epps, III, L. T. J. Korley, T. Yan, K. L. Beers and T. M. Burt, JACS Au, 2022, 2, 3–11 CrossRef PubMed.
- H. Mangold and B. von Vacano, Macromol. Chem. Phys., 2022, 223, 2100488 CrossRef CAS.
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić, J. M. Modak and H. S. Soo, Adv. Mater., 2022, 34, 2100843 CrossRef CAS PubMed.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- Y. L. Liu, Z. Yu, B. B. Wang, P. Y. Li, J. Zhu and S. Q. Ma, Green Chem., 2022, 24, 5691–5708 RSC.
- L. Shen, J. Haufe and M. K. Patel, Report for European polysaccharide network of excellence (EPNOE) and European bioplastics, 2009, p. 243 Search PubMed.
- W. Post, A. Susa, R. Blaauw, K. Molenveld and R. J. I. Knoop, Polym. Rev., 2020, 60, 359–388 CrossRef CAS.
- A. Durand-Silva and R. A. Smaldone, ACS Cent. Sci., 2020, 6, 836–838 CrossRef CAS PubMed.
- P. K. Mallick, in Materials, Design and Manufacturing for Lightweight Vehicles, ed. P. K. Mallick, Woodhead Publishing, 2010, pp. 174–207, DOI:10.1533/9781845697822.1.174.
- Y.-B. Zhao, X.-D. Lv and H.-G. Ni, Chemosphere, 2018, 209, 707–720 CrossRef CAS PubMed.
- B. D. Vogt, K. K. Stokes and S. K. Kumar, ACS Appl. Polym. Mater., 2021, 3, 4325–4346 CrossRef CAS.
- N. P. Balsara, L. J. Fetters, N. Hadjichristidis, D. J. Lohse, C. C. Han, W. W. Graessley and R. Krishnamoorti, Macromolecules, 1992, 25, 6137–6147 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis, E. A. Stach, W. Zheng and D. G. Vlachos, JACS Au, 2021, 1, 1422–1434 CrossRef CAS PubMed.
- H. E. van der Bij and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7406–7428 RSC.
- K. K. Ramasamy, M. A. Gerber, M. Flake, H. Zhang and Y. Wang, Green Chem., 2014, 16, 748–760 RSC.
- G. S. Foo, A. K. Rogers, M. M. Yung and C. Sievers, ACS Catal., 2016, 6, 1292–1307 CrossRef CAS.
- Z. R. Hinton, P. A. Kots, M. Soukaseum, B. C. Vance, D. G. Vlachos, T. H. Epps, III and L. T. J. Korley, Green Chem., 2022, 24, 7332–7339 RSC.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- A. Rahimi and J. M. Garcia, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- L. Yue, V. S. Bonab, D. Yuan, A. Patel, V. Karimkhani and I. Manas-Zloczower, Global chall., 2019, 3, 1800076 CrossRef PubMed.
- F. P. La Mantia and M. Vinci, Polym. Degrad. Stab., 1994, 45, 121–125 CrossRef CAS.
- J. Qiu, S. Ma, S. Wang, Z. Tang, Q. Li, A. Tian, X. Xu, B. Wang, N. Lu and J. Zhu, Macromolecules, 2021, 54, 703–712 CrossRef CAS.
- P. Li, B. Lan, X. Zhang, S. Lei, Q. Yang, P. Gong, C. B. Park and G. Li, Green Chem., 2022, 24, 5490–5501 RSC.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- Z. P. Zhang, M. Z. Rong, M. Q. Zhang and C. e. Yuan, Polym. Chem., 2013, 4, 4648–4654 RSC.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 3936–3943 CrossRef CAS.
- A. Erice, A. Ruiz de Luzuriaga, J. M. Matxain, F. Ruipérez, J. M. Asua, H.-J. Grande and A. Rekondo, Polymer, 2018, 145, 127–136 CrossRef CAS.
- Z. Lei, H. Chen, C. Luo, Y. Rong, Y. Hu, Y. Jin, R. Long, K. Yu and W. Zhang, Nat. Chem., 2022, 14, 1399–1404 CrossRef CAS PubMed.
- X. Lu, P. Xie, X. Xiang and J. Sun, Macromolecules, 2022, 55, 2557–2565 CrossRef CAS.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- S. Wang, S. Ma, J. Qiu, A. Tian, Q. Li, X. Xu, B. Wang, N. Lu, Y. Liu and J. Zhu, Green Chem., 2021, 23, 2931–2937 RSC.
- M. Maaz, A. Riba-Bremerch, C. Guibert, N. J. Van Zee and R. Nicolaÿ, Macromolecules, 2021, 54, 2213–2225 CrossRef CAS.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- S. Kim, M. A. Rahman, M. Arifuzzaman, D. B. Gilmer, B. Li, J. K. Wilt, E. Lara-Curzio and T. Saito, Sci. Adv., 2022, 8, eabn6006 CrossRef CAS PubMed.
- M. Delahaye, F. Tanini, J. O. Holloway, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5207–5215 RSC.
- C. A. Tretbar, J. A. Neal and Z. Guan, J. Am. Chem. Soc., 2019, 141, 16595–16599 CrossRef CAS PubMed.
- M. J. Webber and M. W. Tibbitt, Nat. Rev. Mater., 2022, 7, 541–556 CrossRef.
- B. Zhang, N. De Alwis Watuthanthrige, S. V. Wanasinghe, S. Averick and D. Konkolewicz, Adv. Funct. Mater., 2021, 32, 2108431 CrossRef.
- S. J. D. Lugger, S. J. A. Houben, Y. Foelen, M. G. Debije, A. P. H. J. Schenning and D. J. Mulder, Chem. Rev., 2022, 122, 4946–4975 CrossRef CAS PubMed.
- A. Campanella, D. Döhler and W. H. Binder, Macromol. Rapid Commun., 2018, 39, 1700739 CrossRef.
- F. Huang and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 5879–5880 RSC.
- T. Aida and E. W. Meijer, Isr. J. Chem., 2020, 60, 33–47 CrossRef CAS.
- Y. Zhang, Y. Qi, S. Ulrich, M. Barboiu and O. Ramström, Mater. Chem. Front., 2020, 4, 489–506 RSC.
- Y. Tao, S. Liu, Y. Zhang, Z. Chi and J. Xu, Polym. Chem., 2018, 9, 878–884 RSC.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Sci. Adv., 2018, 4, eaao3865 CrossRef PubMed.
- T.-P. Huynh and H. Haick, Adv. Mater., 2016, 28, 138–143 CrossRef CAS PubMed.
- J. Li, J. Liang, L. Li, F. Ren, W. Hu, J. Li, S. Qi and Q. Pei, ACS Nano, 2014, 8, 12874–12882 CrossRef CAS PubMed.
- B. Marco-Dufort, R. Iten and M. W. Tibbitt, J. Am. Chem. Soc., 2020, 142, 15371–15385 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- J. C. Stowell and S. J. Padegimas, J. Org. Chem., 1974, 39, 2448–2449 CrossRef CAS.
- M. A. Bin Rusayyis and J. M. Torkelson, ACS Macro Lett., 2022, 11, 568–574 CrossRef CAS PubMed.
- N. Roy, E. Buhler and J.-M. Lehn, Polym. Int., 2014, 63, 1400–1405 CrossRef CAS.
- N. Kuhl, S. Bode, R. K. Bose, J. Vitz, A. Seifert, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Adv. Funct. Mater., 2015, 25, 3295–3301 CrossRef CAS.
- G. Li, X. Zhang, S. Yang, T. Li, Y. Wang, M. Chen and W. Dong, ChemSusChem, 2021, 14, 4340–4348 CrossRef CAS PubMed.
- S. Samanta, S. Kim, T. Saito and A. P. Sokolov, J. Phys. Chem. B, 2021, 125, 9389–9401 CrossRef CAS PubMed.
- P. van den Tempel, F. Picchioni and R. K. Bose, Macromol. Rapid Commun., 2022, 43, 2200023 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- A. Buonerba, R. Lapenta, S. O. Sanchez, C. Capacchione, S. Milione and A. Grassi, Chemistryselect, 2017, 2, 1605–1612 CrossRef CAS.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef.
- T. Liu, B. Zhao and J. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- J. M. Garcia, G. O. Jones, K. Virwani, B. D. McCloskey, D. J. Boday, G. M. ter Huurne, H. W. Horn, D. J. Coady, A. M. Bintaleb, A. M. Alabdulrahman, F. Alsewailem, H. A. Almegren and J. L. Hedrick, Science, 2014, 344, 732–735 CrossRef CAS PubMed.
- Y. Li, Y. Zhang, O. Rios, J. K. Keum and M. R. Kessler, RSC Adv., 2017, 7, 37248–37254 RSC.
- F. Van Lijsebetten, T. Debsharma, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed. Engl., 2022, 61, e202210405 CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Fan, J. Liu, X. Li, J. Zhang and J. Cheng, Ind. Eng. Chem. Res., 2014, 53, 16156–16163 CrossRef CAS.
- C.-J. Fan, Z.-B. Wen, Z.-Y. Xu, Y. Xiao, D. Wu, K.-K. Yang and Y.-Z. Wang, Macromolecules, 2020, 53, 4284–4293 CrossRef CAS.
- J.-H. Chen, X.-P. An, Y.-D. Li, M. Wang and J.-B. Zeng, Chin. J. Polym. Sci., 2018, 36, 641–648 CrossRef CAS.
- X. Kuang, Y. Zhou, Q. Shi, T. Wang and H. J. Qi, ACS Sustainable Chem. Eng., 2018, 6, 9189–9197 CrossRef CAS.
- L. Lu, J. Pan and G. Li, J. Mater. Chem. A, 2017, 5, 21505–21513 RSC.
- M. A. Bin Rusayyis and J. M. Torkelson, Polym. Chem., 2021, 12, 2760–2771 RSC.
- L. Odenwald, F. P. Wimmer, N. K. Mast, M. G. Schußmann, M. Wilhelm and S. Mecking, J. Am. Chem. Soc., 2022, 144, 13226–13233 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- A. M. Wemyss, C. Bowen, C. Plesse, C. Vancaeyzeele, G. T. M. Nguyen, F. Vidal and C. Wan, Mater. Sci. Eng., R, 2020, 141, 100561 CrossRef.
- A. Breuillac, F. Caffy, T. Vialon and R. Nicolaÿ, Polym. Chem., 2020, 11, 6479–6491 RSC.
- W. Liu, D. F. Schmidt and E. Reynaud, Ind. Eng. Chem. Res., 2017, 56, 2667–2672 CrossRef CAS.
- Y. Liu, Z. Tang, Y. Chen, C. Zhang and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 2992–3001 CrossRef CAS PubMed.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, RSC Adv., 2016, 6, 88647–88655 RSC.
- S. Mu, Y. Zhang, J. Zhou, B. Wang and Z. Wang, ACS Sustainable Chem. Eng., 2020, 8, 5296–5304 CrossRef CAS.
- A. Demongeot, S. J. Mougnier, S. Okada, C. Soulié-Ziakovic and F. Tournilhac, Polym. Chem., 2016, 7, 4486–4493 RSC.
- D. Nakatake, Y. Yokote, Y. Matsushima, R. Yazaki and T. Ohshima, Green Chem., 2016, 18, 1524–1530 RSC.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- F. Cuminet, S. Caillol, É. Dantras, É. Leclerc and V. Ladmiral, Macromolecules, 2021, 54, 3927–3961 CrossRef CAS.
- F. Van Lijsebetten, J. O. Holloway, J. M. Winne and F. E. Du Prez, Chem. Soc. Rev., 2020, 49, 8425–8438 RSC.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS PubMed.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- H. Zhang, S. Majumdar, R. A. T. M. van Benthem, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2020, 9, 272–277 CrossRef CAS PubMed.
- S. Debnath, C. Upadhyay and U. Ojha, ACS Appl. Mater. Interfaces, 2022, 14, 9618–9631 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- L. Li, X. Chen, K. Jin, M. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2021, 54, 1452–1464 CrossRef CAS.
- J. J. Cash, T. Kubo, D. J. Dobbins and B. S. Sumerlin, Polym. Chem., 2018, 9, 2011–2020 RSC.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps, III and B. S. Sumerlin, J. Am. Chem. Soc., 2020, 142, 283–289 CrossRef CAS PubMed.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, K. Huang, Y. liu and J. Zhu, Macromolecules, 2020, 53, 2919–2931 CrossRef CAS.
- A. Liguori and M. Hakkarainen, Macromol. Rapid Commun., 2022, 43, e2100816 CrossRef PubMed.
- W. Sun, L. Zhang, S. Wang, J. Mao, J. Luo, Y. Chen and Y. Cheng, J. Mater. Chem. C, 2021, 9, 8579–8588 RSC.
- Z. P. Zhang, Y. Lu, M. Z. Rong and M. Q. Zhang, RSC Adv., 2016, 6, 6350–6357 RSC.
- W.-X. Liu, C. Zhang, H. Zhang, N. Zhao, Z.-X. Yu and J. Xu, J. Am. Chem. Soc., 2017, 139, 8678–8684 CrossRef CAS PubMed.
- J. Shi, T. Zheng, Y. Zhang, B. Guo and J. Xu, ACS Sustainable Chem. Eng., 2020, 8, 1207–1218 CrossRef CAS.
- P. Taynton, C. Zhu, S. Loob, R. Shoemaker, J. Pritchard, Y. Jin and W. Zhang, Polym. Chem., 2016, 7, 7052–7056 RSC.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- C. Bakkali-Hassani, D. Berne, V. Ladmiral and S. Caillol, Macromolecules, 2022, 55, 7974–7991 CrossRef CAS.
- R. C. Osthoff, A. M. Bueche and W. T. Grubb, J. Am. Chem. Soc., 1954, 76, 4659–4663 CrossRef CAS.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- W. Schmolke, N. Perner and S. Seiffert, Macromolecules, 2015, 48, 8781–8788 CrossRef CAS.
- T. Debsharma, V. Amfilochiou, A. A. Wróblewska, I. De Baere, W. Van Paepegem and F. E. Du Prez, J. Am. Chem. Soc., 2022, 144, 12280–12289 CrossRef CAS PubMed.
- S. Wu, Z. Yang, S. Fang, Z. Tang, F. Liu and B. Guo, J. Mater. Chem. A, 2019, 7, 1459–1467 RSC.
- Q. Liu, L. Jiang, Y. Zhao, Y. Wang and J. Lei, Macromol. Chem. Phys., 2019, 220, 1900149 CrossRef.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- J. D. Mayo and A. Adronov, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5056–5066 CrossRef CAS.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- S. Hu, X. Chen and J. M. Torkelson, ACS Sustainable Chem. Eng., 2019, 7, 10025–10034 CrossRef CAS.
- D. T. Sheppard, K. Jin, L. S. Hamachi, W. Dean, D. J. Fortman, C. J. Ellison and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 921–927 CrossRef CAS PubMed.
- J. S. Mahajan, R. M. O'Dea, J. B. Norris, L. T. J. Korley and T. H. Epps, III, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS.
- L. Li, X. Chen and J. M. Torkelson, Macromolecules, 2019, 52, 8207–8216 CrossRef CAS.
- Y. Yoshida, K. Ohnaka and T. Endo, Macromolecules, 2019, 52, 6080–6087 CrossRef CAS.
- L. C. Chambers, C. Barner-Kowollik, L. Barner, L. Michalek and H. Frisch, ACS Macro Lett., 2022, 11, 532–536 CrossRef CAS PubMed.
- S. R. Trenor, A. R. Shultz, B. J. Love and T. E. Long, Chem. Rev., 2004, 104, 3059–3078 CrossRef CAS PubMed.
- Y. Zheng, M. Micic, S. V. Mello, M. Mabrouki, F. M. Andreopoulos, V. Konka, S. M. Pham and R. M. Leblanc, Macromolecules, 2002, 35, 5228–5234 CrossRef CAS.
- C. M. Chung and M. Hasegawa, J. Am. Chem. Soc., 1991, 113, 7311–7316 CrossRef CAS.
- G. Kaur, P. Johnston and K. Saito, Polym. Chem., 2014, 5, 2171–2186 RSC.
- H. A. Houck, E. Blasco, F. E. Du Prez and C. Barner-Kowollik, J. Am. Chem. Soc., 2019, 141, 12329–12337 CrossRef CAS PubMed.
- V. X. Truong and C. Barner-Kowollik, Trends Chem., 2022, 4, 291–304 CrossRef CAS.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- F. García and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551–3577 CrossRef PubMed.
- Q. N. Bui, Y. Li, M.-S. Jang, D. P. Huynh, J. H. Lee and D. S. Lee, Macromolecules, 2015, 48, 4046–4054 CrossRef CAS.
- U. F. Fritze and M. von Delius, Chem. Commun., 2016, 52, 6363–6366 RSC.
- S. Sobczak, W. Drożdż, G. I. Lampronti, A. M. Belenguer, A. Katrusiak and A. R. Stefankiewicz, Chem.–Eur. J., 2018, 24, 8769–8773 CrossRef CAS PubMed.
- J. Kida, D. Aoki and H. Otsuka, ACS Macro Lett., 2021, 10, 558–563 CrossRef CAS PubMed.
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS PubMed.
- I. Azcune and I. Odriozola, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- C.-C. Chang and T. Emrick, Macromolecules, 2014, 47, 1344–1350 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS PubMed.
- M. Ochmann, A. Hussain, I. von Ahnen, A. A. Cordones, K. Hong, J. H. Lee, R. Ma, K. Adamczyk, T. K. Kim, R. W. Schoenlein, O. Vendrell and N. Huse, J. Am. Chem. Soc., 2018, 140, 6554–6561 CrossRef CAS PubMed.
- H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150–1152 RSC.
- C. Di Mauro, S. Malburet, A. Genua, A. Graillot and A. Mija, Biomacromolecules, 2020, 21, 3923–3935 CrossRef CAS PubMed.
- R. Martin, A. Rekondo, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, J. Mater. Chem. A, 2014, 2, 5710–5715 RSC.
- H. Otsuka, Polym. J., 2013, 45, 879–891 CrossRef CAS.
- X. Liu, L. Liang, M. Lu, X. Song, H. Liu and G. Chen, Polymer, 2020, 210, 123030 CrossRef CAS.
- H. Memon, H. Liu, M. A. Rashid, L. Chen, Q. Jiang, L. Zhang, Y. Wei, W. Liu and Y. Qiu, Macromolecules, 2020, 53, 621–630 CrossRef CAS.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- H. Memon, Y. Wei, L. Zhang, Q. Jiang and W. Liu, Compos. Sci. Technol., 2020, 199, 108314 CrossRef CAS.
- H. Liu, H.-H. Lu, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2021, 143, 20735–20746 CrossRef CAS PubMed.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef CAS PubMed.
- K. Fukuda, M. Shimoda, M. Sukegawa, T. Nobori and J.-M. Lehn, Green Chem., 2012, 14, 2907–2911 RSC.
- A. L. Andrady and M. A. Neal, Philos. Trans. R. Soc., B, 2009, 364, 1977–1984 CrossRef CAS PubMed.
- G. P. Kar, M. O. Saed and E. M. Terentjev, J. Mater. Chem. A, 2020, 8, 24137–24147 RSC.
- M. O. Saed, X. Lin and E. M. Terentjev, ACS Appl. Mater. Interfaces, 2021, 13, 42044–42051 CrossRef CAS PubMed.
- M. Podgórski, S. Mavila, S. Huang, N. Spurgin, J. Sinha and C. N. Bowman, Angew. Chem., Int. Ed., 2020, 59, 9345–9349 CrossRef PubMed.
- L. M. Polgar, E. Hagting, P. Raffa, M. Mauri, R. Simonutti, F. Picchioni and M. van Duin, Macromolecules, 2017, 50, 8955–8964 CrossRef CAS PubMed.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- Z. He, H. Niu, L. Liu, S. Xie, Z. Hua and Y. Li, Polymer, 2021, 229, 124015 CrossRef CAS.
- A. Zych, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Polymer, 2020, 199, 122567 CrossRef CAS.
- M. A. Rahman, C. Bowland, S. Ge, S. R. Acharya, S. Kim, V. R. Cooper, X. C. Chen, S. Irle, A. P. Sokolov, A. Savara and T. Saito, Sci. Adv., 2021, 7, eabk2451 CrossRef CAS PubMed.
- I. Fischer, W. F. Schmitt, H.-C. Porth, M. W. Allsopp and G. Vianello, in Ullmann's Encyclopedia of Industrial Chemistry, pp. 1–30, DOI:10.1002/14356007.a21_717.pub2.
- Z. Deliballi, R. Demir-Cakan, B. Kiskan and Y. Yagci, Macromol. Chem. Phys., 2023, 224, 2100423 CrossRef CAS.
- S.-S. Wu, Y.-D. Li, Z. Hu and J.-B. Zeng, ACS Sustainable Chem. Eng., 2023, 11, 1974–1984 CrossRef CAS.
- C. A. Angell, Science, 1995, 267, 1924–1935 CrossRef CAS PubMed.
- J. C. Dyre, Rev. Mod. Phys., 2006, 78, 953–972 CrossRef CAS.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS PubMed.
- L. Leibler, M. Rubinstein and R. H. Colby, Macromolecules, 1991, 24, 4701–4707 CrossRef CAS.
- M. Rubinstein and A. N. Semenov, Macromolecules, 2001, 34, 1058–1068 CrossRef CAS.
- M. Ahmadi, L. G. D. Hawke, H. Goldansaz and E. van Ruymbeke, Macromolecules, 2015, 48, 7300–7310 CrossRef CAS.
- L. L. De Lucca Freitas and R. Stadler, Macromolecules, 1987, 20, 2478–2485 CrossRef CAS.
- S. Ciarella, R. A. Biezemans and L. M. C. Janssen, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 25013–25022 CrossRef CAS PubMed.
- H. Zhao, X. Wei, Y. Fang, K. Gao, T. Yue, L. Zhang, V. Ganesan, F. Meng and J. Liu, Macromolecules, 2022, 55, 1091–1103 CrossRef CAS.
- L. Rovigatti, G. Nava, T. Bellini and F. Sciortino, Macromolecules, 2018, 51, 1232–1241 CrossRef CAS.
- R. G. Ricarte and S. Shanbhag, Macromolecules, 2021, 54, 3304–3320 CrossRef CAS.
- F. Sciortino, Eur. Phys. J. E: Soft Matter Biol. Phys., 2017, 40, 3 CrossRef PubMed.
- Z. Shen, H. Ye, Q. Wang, M. Kröger and Y. Li, Macromolecules, 2021, 54, 5053–5064 CrossRef CAS.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- B. Soman and C. M. Evans, Soft Matter, 2021, 17, 3569–3577 RSC.
- D. R. Hansen and M. Shen, Macromolecules, 1975, 8, 343–348 CrossRef CAS.
- W. H. Stockmayer and J. W. Kennedy, Macromolecules, 1975, 8, 351–355 CrossRef CAS.
- F. W. Wang and E. A. DiMarzio, Macromolecules, 1975, 8, 356–360 CrossRef CAS.
- Z. Zhang, Q. Chen and R. H. Colby, Soft Matter, 2018, 14, 2961–2977 RSC.
- M. L. Williams, R. F. Landel and J. D. Ferry, J. Am. Chem. Soc., 1955, 77, 3701–3707 CrossRef CAS.
- H. Vogel, Phys. Z., 1921, 22, 645–646 CAS.
- G. Tammann and W. Hesse, Z. fur Anorg. Allg., 1926, 156, 245–257 CrossRef CAS.
- P. Chakma, C. N. Morley, J. L. Sparks and D. Konkolewicz, Macromolecules, 2020, 53, 1233–1244 CrossRef CAS.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Polym. Chem., 2020, 11, 5339–5345 RSC.
- P. Miao, X. Leng, J. Liu, G. Song, M. He and Y. Li, Macromolecules, 2022, 55, 4956–4966 CrossRef CAS.
- F. Caffy and R. Nicolaÿ, Polym. Chem., 2019, 10, 3107–3115 RSC.
- R. G. Ricarte, F. Tournilhac and L. Leibler, Macromolecules, 2019, 52, 432–443 CrossRef CAS.
- S. Yang, S. Yi, J. Yun, N. Li, Y. Jiang, Z. Huang, C. Xu, C. He and X. Pan, Macromolecules, 2022, 55, 3423–3429 CrossRef CAS.
- M. E. L. Wouters, J. G. P. Goossens and F. L. Binsbergen, Macromolecules, 2002, 35, 208–216 CrossRef CAS.
- M. van Duin, Macromol. Symp., 2003, 202, 1–10 CrossRef CAS.
- S. Ge, S. Samanta, B. Li, G. P. Carden, P.-F. Cao and A. P. Sokolov, ACS Nano, 2022, 16, 4746–4755 CrossRef CAS PubMed.
- R. Mahmud, S. M. Moni, K. High and M. Carbajales-Dale, J. Cleaner Prod., 2021, 317, 128247 CrossRef.
- Y. Cortes-Peña, D. Kumar, V. Singh and J. S. Guest, ACS Sustainable Chem. Eng., 2020, 8, 3302–3310 CrossRef.
- E. N. Staff, LCA is a technique to assess the environmental aspects and potential impacts associated with a product, process, or service, by:/*Compiling an inventory of relevant energy and material inputs and environmental releases/*Evaluating the potential environmental impacts associated with identified inputs and releases/*Interpreting the results to help you make a more informed decision, https://web.archive.org/web/20120306122239/http://www.epa.gov/nrmrl/std/lca/lca.html, accessed May 20, 2022.
- A. Singh, N. A. Rorrer, S. R. Nicholson, E. Erickson, J. S. DesVeaux, A. F. T. Avelino, P. Lamers, A. Bhatt, Y. Zhang, G. Avery, L. Tao, A. R. Pickford, A. C. Carpenter, J. E. McGeehan and G. T. Beckham, Joule, 2021, 5, 2479–2503 CrossRef CAS.
- M. Larrain, S. Van Passel, G. Thomassen, B. Van Gorp, T. T. Nhu, S. Huysveld, K. M. Van Geem, S. De Meester and P. Billen, Resour., Conserv. Recycl., 2021, 170, 105607 CrossRef CAS.
- D. Shonnard, E. Tipaldo, V. Thompson, J. Pearce, G. Caneba and R. Handler, Procedia CIRP, 2019, 80, 602–606 CrossRef.
- P. Sarda, J. C. Hanan, J. G. Lawrence and M. Allahkarami, J. Polym. Sci., 2022, 60, 7–31 CrossRef CAS.
- X. Zhao and F. You, ACS Sustainable Chem. Eng., 2021, 9, 12167–12184 CrossRef CAS.
- R. R. Bora, R. Wang and F. You, ACS Sustainable Chem. Eng., 2020, 8, 16350–16363 CrossRef CAS.
- H. Almohamadi, M. Alamoudi, U. Ahmed, R. Shamsuddin and K. Smith, Korean J. Chem. Eng., 2021, 38, 2208–2216 CrossRef.
- T. Uekert, A. Singh, J. S. DesVeaux, T. Ghosh, A. Bhatt, G. Yadav, S. Afzal, J. Walzberg, K. M. Knauer, S. R. Nicholson, G. T. Beckham and A. C. Carpenter, ACS Sustainable Chem. Eng., 2023, 11, 965–978 CrossRef CAS.
- P. T. Chazovachii, M. J. Somers, M. T. Robo, D. I. Collias, M. I. James, E. N. G. Marsh, P. M. Zimmerman, J. F. Alfaro and A. J. McNeil, Nat. Commun., 2021, 12, 4524 CrossRef CAS PubMed.
- N. Vora, P. R. Christensen, J. Demarteau, N. R. Baral, J. D. Keasling, B. A. Helms and C. D. Scown, Sci. Adv., 2021, 7, eabf0187 CrossRef CAS PubMed.
- J. Demarteau, N. Vora, J. D. Keasling, B. A. Helms and C. D. Scown, ACS Sustainable Chem. Eng., 2022, 10, 2740–2749 CrossRef CAS.
- C. He, P. R. Christensen, T. J. Seguin, E. A. Dailing, B. M. Wood, R. K. Walde, K. A. Persson, T. P. Russell and B. A. Helms, Angew. Chem., Int. Ed., 2020, 59, 735–739 CrossRef CAS PubMed.
- H. Chang, M. S. Kim, G. W. Huber and J. A. Dumesic, Green Chem., 2021, 23, 9479–9488 RSC.
- P. Bakshi, A. Pappu, D. K. Bharti, B. Parmar, R. Patidar and A. K. Srivastava, ACS Sustainable Chem. Eng., 2022, 10, 13710–13721 CrossRef CAS.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- A. Zych, J. Tellers, L. Bertolacci, L. Ceseracciu, L. Marini, G. Mancini and A. Athanassiou, ACS Appl. Polym. Mater., 2021, 3, 1135–1144 CrossRef CAS.
- C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson and A. A. Lapkin, Chem. Rev., 2023, 123(6), 3089–3126 CrossRef CAS PubMed.
- D. J. Walsh, W. Zou, L. Schneider, R. Mello, M. E. Deagen, J. Mysona, T.-S. Lin, J. J. de Pablo, K. F. Jensen, D. J. Audus and B. D. Olsen, ACS Cent. Sci., 2023, 9(3), 330–338 CrossRef CAS PubMed.
- T.-S. Lin, C. W. Coley, H. Mochigase, H. K. Beech, W. Wang, Z. Wang, E. Woods, S. L. Craig, J. A. Johnson, J. A. Kalow, K. F. Jensen and B. D. Olsen, ACS Cent. Sci., 2019, 5, 1523–1531 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |