
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pillar[5]arene noncovalent assembly boosts a full-color lanthanide supramolecular light switch†
Wei-Lei
Zhou‡
ab,
Xian-Yin
Dai‡
ac,
Wenjing
Lin
a,
Yong
Chen
a and
Yu
Liu
 *a
*a
aCollege of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, China. E-mail: yuliu@nankai.edu.cn
bCollege of Chemistry and Material Science, Innovation Team of Optical Functional Molecular Devices, Inner Mongolia Minzu University, Tongliao 028000, P. R. China
cSchool of Chemistry and Pharmaceutical Engineering, Shandong First Medical University, Shandong Academy of Medical Sciences, Taian, 271016, China
First published on 30th May 2023
Abstract
A photo-responsive full-color lanthanide supramolecular switch was constructed from a synthetic 2,6-pyridine dicarboxylic acid (DPA)-modified pillar[5]arene (H) complexing with lanthanide ion (Ln3+ = Tb3+ and Eu3+) and dicationic diarylethene derivative (G1) through a noncovalent supramolecular assembly. Benefiting from the strong complexation between DPA and Ln3+ with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio, the supramolecular complex H/Ln3+ presented an emerging lanthanide emission in the aqueous and organic phase. Subsequently, a network supramolecular polymer was formed by H/Ln3+ further encapsulating dicationic G1via the hydrophobic cavity of pillar[5]arene, which greatly contributed to the increased emission intensity and lifetime, and also resulted in the formation of a lanthanide supramolecular light switch. Moreover, full-color luminescence, especially white light emission, was achieved in aqueous (CIE: 0.31, 0.32) and dichloromethane (CIE: 0.31, 0.33) solutions by the adjustment of different ratios of Tb3+ and Eu3+. Notably, the photo-reversible luminescence properties of the assembly were tuned via alternant UV/vis light irradiation due to the conformation-dependent photochromic energy transfer between the lanthanide and the open/closed-ring of diarylethene. Ultimately, the prepared lanthanide supramolecular switch was successfully applied to anti-counterfeiting through the use of intelligent multicolored writing inks, and presents new opportunities for the design of advanced stimuli-responsive on-demand color tuning with lanthanide luminescent materials.
:1 stoichiometric ratio, the supramolecular complex H/Ln3+ presented an emerging lanthanide emission in the aqueous and organic phase. Subsequently, a network supramolecular polymer was formed by H/Ln3+ further encapsulating dicationic G1via the hydrophobic cavity of pillar[5]arene, which greatly contributed to the increased emission intensity and lifetime, and also resulted in the formation of a lanthanide supramolecular light switch. Moreover, full-color luminescence, especially white light emission, was achieved in aqueous (CIE: 0.31, 0.32) and dichloromethane (CIE: 0.31, 0.33) solutions by the adjustment of different ratios of Tb3+ and Eu3+. Notably, the photo-reversible luminescence properties of the assembly were tuned via alternant UV/vis light irradiation due to the conformation-dependent photochromic energy transfer between the lanthanide and the open/closed-ring of diarylethene. Ultimately, the prepared lanthanide supramolecular switch was successfully applied to anti-counterfeiting through the use of intelligent multicolored writing inks, and presents new opportunities for the design of advanced stimuli-responsive on-demand color tuning with lanthanide luminescent materials.
Introduction
Intelligent supramolecular lanthanide luminescent materials with external modulation ability can be potentially applied in the fields of various artificial systems, such as bioimaging,1 optoelectronic devices,2 controllable catalysis,3 chemical sensing,4 and anti-counterfeit material5 due to their unique luminescence characteristics such as long-lived excited states, large Stokes shifts, visible-light emission, and narrow emission bandwidths.6 Recently, Gunnlaugsson et al.7 reported a supramolecular luminescence hydrogel composed of K+-stable bioconjugated dicarboxylic acid (DPA)–guanosine quadruplexes in which the chiral structure of the guanosine gel resulted in circular polarization luminescence (CPL) of lanthanide Eu3+, and the hydrogel exhibited satisfactory stability and self-healing properties.He's group prepared a multi-responsive (including pH, temperature, metal ions, sonication, and force) lanthanide luminescent hydrogel by copolymerizing a Eu-iminodiacetate complex with a coagulation polymer, and the gel was able to rapidly self-heal and exhibited tunable fluorescence color changes.8 Zhao et al. constructed a photoresponsive supramolecular coordination polyelectrolyte by electrostatic interaction between bis-DPA/Eu3+ coordination polymer and cationic diarylethene, which was regulated by the open-closed ring of diarylethene and can be used as anti-counterfeiting ink.9
Therefore, many studies have contributed to the construction of luminescent materials by integrating macrocyclic compounds (e.g., crown ethers, cyclodextrins, and cucurbiturils) and lanthanides, especially light-controlled lanthanide luminescence materials.10 Zhang's group harnessed the host–guest interaction of α-CD-modified down-conversion nanoparticles and azobenzene-modified up-conversion nanoparticles to construct supramolecular nanoparticles that were photocontrolled in an in vivo assembly and disassembly process to improve bioimaging.11 Li et al. presented a robust luminescent hybrid hydrogel based on a DPA–α-CD/Ln complex, guanidinium–azobenzene, and LAPONITE® by electrostatic interactions between guanidinium–azobenzene and LAPONITE® in which association or dissociation for photoisomerization of azobenzene and cyclodextrin governed the sol–gel transformation.12
Additionally, we reported a dual-modulated molecular switch constructed through host–guest competitive bonding and photo-response based on 24-crown-8-modified terpyridine/Eu3+ complexes with secondary ammonium salt diarylethene.13 The molecular switches based on a two-arm crown ether anthracene-modified-terpyridine/Ln assembly14 and γ-cyclodextrin anthracene-modified-DPA/Eu3+ supramolecular polymer were respectively constructed for lanthanide luminescence regulation through the oxidation and dimerization of anthracene.15
It is well known that pillararenes are a new class of macrocyclic compounds composed of hydroquinone units bridged by methylene groups at the para position.16 With their rigid backbones, rich π electrons, delocalized cavities, and the advantages of multiple facile functionalization possibilities, they can be employed as natural bridges to construct novel functional materials, among which the photo-responsive and luminescent materials are particularly attractive.17
For example, Ogoshi et al.18 developed a multilayer microporous membrane with azophenyl groups on the top surface via layer-by-layer assembly of cationic and anionic pillar[5]arene in which the isomerization of azobenzene was used as a light switch for controlling guest entry and exit. Sessler and Huang et al.19 prepared a novel supramolecular network invisible ink based on pillar[5]arene and spiropyran, in which time-dependent conversion of the colored mercyanine structure into the colorless spiropyran structure was achieved by adding unbound pillar[5]arene or nitrile guest competitors to adjust the cross-linking density.
A smart molecular chiral photoswitch based on azobenzene-fused bicyclic pillar[n]arene was successfully constructed where the conformational inversion of azobenzene into and out of the pillararene cavity enabled the planar chirality switching of molecular universal joints, while the inversion induced by the temperature change prevented the photoisomerization of azobenzene.20 A fluorescent supramolecular polymer with aggregation-induced emission properties was also reported based on thymine bilaterally modified-pillar[6]arene and tetraphenylethylene (TPE)-bridged bis(quaternary ammonium) through host–guest interaction that rapidly detected and removed Hg2+ through a change in luminescence in water.21
Recently, Tang et al.22 constructed a multicolor light-harvesting noncovalent polymer with excellent properties using TPE-modified pillar[5]arene and diketopyrrolopyrrole or anthracene derivatives with two cyano-triazole units via host–guest interaction. However, to the best of our knowledge, the enhanced luminescent properties and the regulated full-color luminescent behavior of supramolecular lanthanide by noncovalent assembly have not been reported.
Herein, we report a novel full-color lanthanide luminescent supramolecular switch based on a pillar[5]arene noncovalent assembly, which was composed of DPA-modified pillar[5]arene derivatives (H) that formed a complex with Tb3+/Eu3+ and dicationic diarylethene derivative (G1) (Scheme 1). The DPA moiety coordinated with Ln3+ in a 3:1 ratio, and the quaternary ammonium salt moiety of diarylethene assembled with pillar[5]arene through a 1:2 bonding ratio to form a lanthanide supramolecular assembly.17b,23
 | ||
| Scheme 1 Schematic illustration of the construction of a full-color lanthanide supramolecular light switch based on pillar[5]arene noncovalent assembly. | ||
Interestingly, when the host molecular complex H/Ln3+ is bonded to the photoresponsive guest molecule G1 by host–guest interaction, and both are in aqueous solution and in the organic phase, the luminescent properties of H/Ln3+ are greatly improved, accompanied by a 5-fold increase in emission intensity (lifetime from 0.870 ms to 1.03 ms) from the aqueous phase to a 100-fold increase in the organic phase (lifetime from 1.14 ms to 1.32 ms) for H/Eu3+. There was a corresponding 1.5-fold increase in H/Tb3+ emission intensity (lifetime from 0.82 ms to 0.84 ms) in water, and a 6.5-fold increase (lifetime from 0.84 ms to 1.09 ms) in CH2Cl2 solution.
The noncovalent assembly to build the network structure is different from previously reported linear assemblies,14 and it can effectively shield the coordination of water and the collision of oxygen, thereby greatly improving the lanthanide emission performance. More importantly, due to the blue fluorescence of the pillar[5]arene, panchromatic luminescence including white light (CIE: 0.31, 0.32 in water, CIE: 0.31, 0.33 in CH2Cl2 solution) can be obtained in aqueous and organic solutions by adjusting the different proportions of Eu3+ and Tb3+ within the H/Ln3+/G1 assembly. In addition, because the photo-induced conformational transition of diarylene regulates the energy transfer process between Ln3+ ions and diarylene units, it was utilized as a full-color light-controlled molecular switch.
This noncovalent assembly boosted the Ln3+ luminescent supramolecular assembly and exhibited the characteristic emission of Eu3+ or Tb3+ due to the poor overlap with the absorption of the ring-opened diarylethene. However, when the diarylethene unit was irradiated with UV light, its absorption band exactly overlapped with the emission band of Eu3+ or Tb3+, and then, after the diarylethene unit was irradiated with visible light, the emission of Eu3+ or Tb3+ was restored. This photo-responsive assembly can be used as a full-color lanthanide luminescent anti-counterfeiting invisible ink or hydrogel to erase and restore written characters by alternating UV and visible irradiation, which will provide a unique strategy for smart optical materials.
Experimental
Materials and methods
All chemicals were commercially available unless noted other-wise. Compound 1, compound 3, and G2 were prepared according to procedures from previous studies.23b,261H NMR spectra were recorded on a Bruker 400 instrument. UV-vis spectra were recorded on a Shimadzu UV-3600 spectrophotometer using a quartz cell (light path 10 mm) at 25 °C. Steady-state fluorescence emission spectra were recorded in a conventional quartz cell (10 × 10 × 45 mm) at 25 °C on a Varian Cary Eclipse equipped with a Varin Cary Single-Cell Peltier accessory to control temperature.Transmission electron microscopy (TEM) images were acquired by a high-resolution transmission electron microscope (FEI Tecnai 20 microscope) operating at an acceleration voltage of 200 keV. The samples were prepared by placing a drop of solution onto a carbon-coated copper grid with subsequent air drying. The zeta potential was recorded on a NanoBrook 173Plus (Brookhaven Company) at 25 °C.
Synthesis of H
Compound 1 (94.20 mg, 0.11 mmol), diethyl 4-hydroxypyridine-2,6-dicarboxylate (47.85 mg, 0.20 mmol), K2CO3 (69.10 mg, 0.50 mmol), and KI (3.65 mg, 0.02 mmol) were dissolved in a 100 mL round-bottom flask with 50 mL CH3CN. The mixture was refluxed with stirring for approximately 48 h, and was monitored by thin-layer chromatography (TLC). After removing the solvents under vacuum, CHCl3 (50 mL) was added and washed twice with H2O. The organic layer was collected and dried under reduced pressure to afford the crude product, which was separated by flash column chromatography using CH2Cl2/CH3OH. The product was combined and concentrated under vacuum to give white solid 2.Then, solid 2 was dissolved in 10 mL methanol, added to a solution of NaOH (16.05 mg, 0.40 mmol, 1 mL), and stirred for 4 h at room temperature. After removal of the methanol under vacuum, the pH of the solution was adjusted to 1 using 37% HCl solution. The yellow solid product was obtained through filtering, washing with water (3 × 30 mL), and drying under vacuum (87.50 mg, 94%). 1H NMR (400 MHz, DMSO-d6) δ 7.70 (s, 2H), 6.81–6.71 (m, 10H), 4.90 (t, 2H), 3.90 (t, 2H), 3.64 (m, 37H), 2.05–1.85 (m, 4H). HR-MS (ESI): m/z calcd for C55H59NO15+: 996.3783 [M + Na+]+, found: 996.3780.
Synthesis of G1
Compound 3 (250 mg, 0.45 mmol) was dissolved in 50 mL anhydrous acetone. Then, 1,4-dibromobutane (976 mg, 4.52 mmol) and potassium carbonate (624 mg, 4.52 mmol) were added to the above solution. The reaction mixture was stirred at 60 °C overnight. After the reaction was completed, insoluble substances were removed by filtration. The filtrate was concentrated and purified by column chromatography with eluents of petroleum ether/dichloromethane (20/1, v/v) to obtain the green residue of compound 4 (yield: 301 mg, 81%). 1H NMR (400 MHz, DMSO-d6) δ 7.53 (d, 2H), 7.34 (s, 1H), 6.99 (d, 2H), 4.03 (t, 2H), 3.61 (t, 2H), 1.96 (m, 5H), 1.84 (t, 2H). HR-MS (ESI): m/z calcd for C35H32Br2F6O2S2+: 821.0193 [M + H+]+, found: 821.0157.Compound 4 (100 mg, 0.12 mmol) and trimethylamine (1 mL, 30 wt% in ethanol) in acetonitrile (10 mL) were stirred for 24 h at 80 °C. After cooling to room temperature, excess solvent was removed by rotary evaporation. Then, the white solid powder was precipitated by the addition of 30 mL ether. The precipitate was filtered and washed with ether, and then dried under vacuum for 24 h to afford compound G1 as a light green solid (yield: 102 mg, 89%). 1H NMR (400 MHz, DMSO-d6) δ 7.56 (d, 2H), 7.37 (s, 1H), 6.99 (d, 2H), 4.06 (t, 2H), 3.37 (t, 2H), 3.07 (s, 9H), 1.95 (s, 3H), 1.86 (m, 2H), 1.75 (m, 2H). HR-MS (ESI): m/z calcd for C41H50F6N2O2S22+: 390.1604 [M − 2Br−]2+, found: 390.1605.
Synthesis of G2
Compound G2 (ref. 23b) was prepared according to protocols from previous studies. 1H NMR (400 MHz, D2O) δ 3.61 (t, 2H), 3.46 (s, 9H), 1.79–1.68 (m, 2H), 1.49–1.37 (m, 2H), 0.99 (t, 3H). HR-MS (ESI): m/z calcd for C7H18N [M − Br−]+: 116.1434, found: 116.1434.Results and discussion
The synthetic routes for host DPA-modified pillar[5]arene (H), guest dicationic diarylethene derivative (G1), and reference compound (G2) are shown in Scheme 2, Fig. S1 and S12.† Briefly, mono-bromine-modified pillar[5]arene (1) was synthesized by polymerization of 1,4-dimethoxybenzene and 1-(4-bromobutoxy)-4-methoxybenzene as raw materials, in the presence of paraformaldehyde and FeCl3. Then, DPA-modified pillar[5]arene H was obtained by a substitution and hydrolysis reaction between compound 1 and diethyl 4-hydroxypyridine-2,6-dicarboxylate. | ||
| Scheme 2 The synthetic route for host compound H, guest compound G1, and reference compound G2. | ||
G1 was prepared by the successive reaction of 2-methylthiophene such as Suzuki coupling, substitution, and protonation reaction. By utilizing Tb3+ as an example, the absorption of H at 295 nm gradually increased with the continuous addition of Tb3+ to the solution, which indicated the coordination process between Tb3+ and the DPA group of H. The characteristic emission peaks of Tb3+ at 490 nm (5D4 → 7F6), 545 nm (5D4 → 7F5), 583 nm (5D4 → 7F4), and 621 nm (5D4 → 7F3) emerged and gradually increased with green luminescence due to the energy transfer between DPA and Tb3+ (Fig. 1b).
 | ||
| Fig. 1 (a) Absorption spectra and (b) emission spectra of H (0.01 mM) upon addition of Tb3+ (from 0 to 2.0 eq.) in water at 298 K. (c) Changes in emission intensity at 545 nm vs. the Tb3+:H molar ratio (λex = 254 nm, 298 K). (d) Emission spectra of H upon addition of Eu3+ (from 0 to 1.0 eq.) in water. Inset: Photographic image of luminescence in aqueous solution under a UV light (λex = 254 nm, 298 K). | ||
The Tb3+:H coordination stoichiometry of 1:3 was obtained by analyzing the curve of ΔF (complex-induced fluorescence intensity change at 545 nm) versus the [Tb3+]/[H] molar ratio (Fig. 1c), which was in accordance with a previous report stating that 3 equivalents of DPA formed stable complexes with 1 equivalent of lanthanide ions (Eu3+ or Tb3+).9,12,15 Similarly, the characteristic emission peak of Eu3+ at 580 nm (5D0 → 7F1), 615 nm (5D0 → 7F2), and 680 nm (5D0 → 7F4) with red emission was given when Eu3+ was gradually added to the host molecule H (Fig. 1d), that is, when an energy transfer (ET) occurred from DPA to Eu3+.
Diarylethene functional groups exhibit reversible and bistable photochromic properties.24 Therefore, we designed and synthesized a quaternary ammonium salt-modified diarylethene guest molecule, G1, that could bond to the host molecule H. The optical properties were first investigated via the UV-vis absorption spectra (Fig. 2a and b), where a strong absorption peak at 295 nm was observed for the open-ring form of G1. After UV light (254 nm) irradiation, the guest molecule G1 changed from the open-ring state to the closed-ring state  and a new peak appeared at 600 nm. The absorption peak at 295 nm gradually decreased, with a blueshift of approximately 50 nm, and was accompanied by the appearance of an isoabsorptive point at 317 nm that resulted from the transition from open-ring to closed-ring forms.
and a new peak appeared at 600 nm. The absorption peak at 295 nm gradually decreased, with a blueshift of approximately 50 nm, and was accompanied by the appearance of an isoabsorptive point at 317 nm that resulted from the transition from open-ring to closed-ring forms.
 | ||
Fig. 2 (a) Structural formulae and solution color of the changes of G1 upon alternating with UV and visible light irradiation (298 K). (b) UV-vis absorption spectra changes of  at 600 nm ([G1] = 1.0 × 10−5 M, 298 K) upon irradiation with UV light (254 nm, 1.0 min). (c) Absorbance changes at 600 nm for G1 ([G1] = 1.0 × 10−5 M, 298 K) under alternant UV (254 nm, 1.0 min) and visible (>450 nm, 1.0 min) light irradiation. (d) Normalized partial absorption spectra (left axis) of the open form (G1, black line) and closed form ( at 600 nm ([G1] = 1.0 × 10−5 M, 298 K) upon irradiation with UV light (254 nm, 1.0 min). (c) Absorbance changes at 600 nm for G1 ([G1] = 1.0 × 10−5 M, 298 K) under alternant UV (254 nm, 1.0 min) and visible (>450 nm, 1.0 min) light irradiation. (d) Normalized partial absorption spectra (left axis) of the open form (G1, black line) and closed form ( blue line) after irradiation with 254 nm UV light for 60 s and luminescent emission of H/Eu3+ or H/Tb3+ (red line or green line, respectively, right axis, 298 K). blue line) after irradiation with 254 nm UV light for 60 s and luminescent emission of H/Eu3+ or H/Tb3+ (red line or green line, respectively, right axis, 298 K). | ||
After closed-ring  was irradiated with visible light (>450 nm), its solution not only changed from blue to the original colorless state, but the UV absorption also returned to the initial level, indicating that the
was irradiated with visible light (>450 nm), its solution not only changed from blue to the original colorless state, but the UV absorption also returned to the initial level, indicating that the 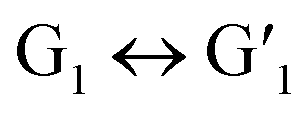 transformation is reversible. Importantly, this photochromic switch was alternately exposed to UV and visible light at least 5 times, and satisfactory reversibility and excellent fatigue resistance resulted (Fig. 2c).
transformation is reversible. Importantly, this photochromic switch was alternately exposed to UV and visible light at least 5 times, and satisfactory reversibility and excellent fatigue resistance resulted (Fig. 2c).
This reversible photocyclization/reversion process was also monitored by 1H NMR. As shown in Fig. S13a,† the protons on the G1 diarylethene skeleton underwent chemical shifts of different degrees with continuous UV irradiation, and exhibited little change until the photo-stable state was reached. Through the 1H NMR integration of the final product after UV irradiation, the ring-closing conversion rate of G1 to 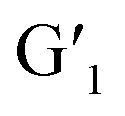 was found to be approximately 95%. More interestingly, when the transformed
was found to be approximately 95%. More interestingly, when the transformed  was irradiated with visible light (>450 nm), the 1H NMR spectrum completely recovered, indicating that it could be quantitatively transformed back to G1 (Fig. S13b†). In addition, the UV spectrum shows that the stability at a certain time and temperature for
was irradiated with visible light (>450 nm), the 1H NMR spectrum completely recovered, indicating that it could be quantitatively transformed back to G1 (Fig. S13b†). In addition, the UV spectrum shows that the stability at a certain time and temperature for  was satisfactory (Fig. S14†).
was satisfactory (Fig. S14†).
To study the host–guest bonding behavior of H and G1, a reference molecule quaternary ammonium salt G2 was synthesized. The host–guest binding behaviors of H with G2 were explored by UV and fluorescence titration experiments. According to the changes in the UV-vis spectra of H/G2 and H/H2O at the same concentration, Job's plots with the peak at 295 nm gave an inflection point at a molar ratio of 0.5, corresponding to a 1:1 H/G2 inclusion complex (Fig. S15†), which is consistent with previously reported results.17b,23
In Fig. S16,† the 0–0.5 equivalents of G2 were gradually titrated into an aqueous solution of H. Then, the UV-vis spectra of the peak at 225 nm gradually decreased, the peak at 293 nm gradually increased, and an isoabsorption point appeared at approximately 250 nm. With the continuous addition of G2, the fluorescence spectrum peak of the host molecule H at 360 nm gradually decreased, which indicated that the reference molecule G2 was bonded to the host molecule H. The complex formation constant (Ks) between G2 and H was calculated as Ks = 2.99 × 104 M−1 (298 K) by analyzing the sequential changes in the fluorescence spectrum (ΔF) of H at varying concentrations of G2 using a nonlinear least-squares curve-fitting method (Fig. S16c†). These experimental results indicated that the bilateral quaternary ammonium salt-modified diarylethene G1 can be bonded to H at a stoichiometric ratio of 1:2.
Importantly, the ring-closed form of this diarylethene exhibited a strong absorption peak at 600 nm, which overlaps very well with the luminescent emission of the constructed H/Ln3+ (H/Eu3+ or H/Tb3+) complex, while no absorption band of the open-ring form was observed at greater than 400 nm (Fig. 2d). The excellent anti-fatigue properties and the bonding behavior with pillar[5]arene can be used to construct a multicolor lanthanide supramolecular switch in water. After adding an equivalent amount of diarylethene derivative to the aqueous solution of the H/Ln3+ complex, the original luminescent emission of the H/Tb3+ complex increased by approximately 1.5 times. The quantum yield was enhanced from 0.42% to 1.63%, and the lifetime from 0.820 ms to 0.840 ms (Fig. 3a, b and S17†). The original luminescent emission of the H/Eu3+ complex increased approximately 5 times, along with the quantum yield increasing from 0.56% to 2.89% and the lifetime from 0.870 ms to 1.03 ms (Fig. 4a, b and S17†).
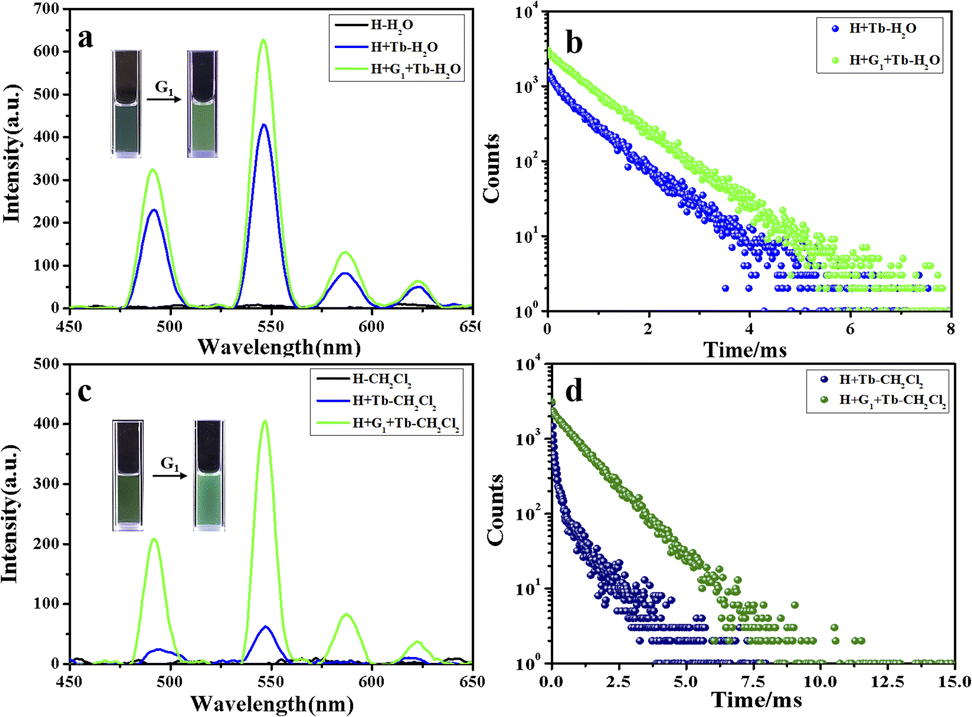 | ||
| Fig. 3 Luminescent emission spectra of (a) H (black line), H/Tb3+ (blue line), and H/Eu3+/G1 (green line), and time-resolved photoluminescence decay curves of (b) H/Tb3+ (blue line) and H/Tb3+/G1 (green line) in aqueous solution at 298 K. (c) Corresponding emission and (d) time-resolved photoluminescence decay curves in CH2Cl2 solution ([H] = 2.0 × 10−5 M) at 298 K. | ||
 | ||
| Fig. 4 Luminescent emission spectra of (a) H (black line), H/Eu3+ (blue line), and H/Eu3+/G1 (red line), and time-resolved photoluminescence decay curves of (b) H/Eu3+ (black line) and H/Eu3+/G1 (red line) in aqueous solution at 298 K. (c) Corresponding emission and (d) time-resolved photoluminescence decay curves in CH2Cl2 solution ([H] = 2.0 × 10−5 M) at 298 K. | ||
This may be due to the formation of noncovalent supramolecular network aggregates after the addition of two-armed quaternary ammonium salt-modified diarylethene guest molecules G1 to the H/Ln3+ complex, which assembled multiple luminescent centers together. The hydrophobic center of diarylethene and this aggregation state decreased the collision of water molecules, which increased the luminous efficiency. Subsequently, we considered the quaternary ammonium reference compound G2 without the diarylethene unit to be a guest molecule, and added it to an aqueous solution of the H/Eu3+ complex in an equivalent amount. No obvious luminescence enhancement phenomenon was observed, which further proved our hypothesis regarding the formation of the non-covalent supramolecular assembly H/Ln3+/G1 (Fig. S18a†).
It has been reported that pillar[5]arene can encapsulate quaternary ammonium salt not only in water,17b,23 but also in dichloromethane (CH2Cl2). Therefore, we further probed the optical properties of pillar[5]arene in organic phase CH2Cl2. Similarly, Ln3+ was added to a CH2Cl2 solution of H under excitation light of 254 nm, and the luminescence spectra at 490 nm (5D4 → 7F6), 545 nm (5D4 → 7F5), 583 nm (5D4 → 7F4), and 621 nm (5D4 → 7F3) exhibited clear characteristic emission peaks of Tb3+, and at 580 nm (5D0 → 7F1), 615 nm (5D0 → 7F2), and 693 nm (5D0 → 7F4) showed characteristic emission peaks of Eu3+ (Fig. S19†).
Interestingly, by adding equivalent G1 into CH2Cl2 solution of the H/Ln3+ complex, a 6.5-fold emission intensity increase occurred, with an increase in the quantum yield from 0.51% to 3.17%, and lifetime from 0.84 ms to 1.09 ms for H/Tb3+/G1 (Fig. 3c, d and S20†), as well as approximately a 100-fold emission intensity increase, with an increase in quantum yield from 0.66% to 6.41% and lifetime from 1.14 ms to 1.32 ms for the H/Eu3+/G1 assembly (Fig. 4c, d and S20†). However, reference molecule G2 was also added to the H/Eu3+ complex under the same conditions, and no obvious emission enhancement was observed (Fig. S18b†). Therefore, the luminescence enhancement of Ln3+ was apparent in the H/Ln3+ complex in the aqueous and organic phases, and occurred due to a noncovalent supramolecular assembly with diarylethene modified with two-arm quaternary ammonium salts.
The enhanced luminescent properties in this assembly were different from those in previously reported linear assemblies.14 The noncovalent assembly for building the network structure effectively suppressed molecular motion and shielded the coordination of water and the collision of oxygen, thus efficiently minimizing non-radiative decay and improving the luminescent performance. However, there was stronger luminescence in dichloromethane because the luminescent lanthanide centers were easily attacked by water molecules. A comparison of the optical transmittance changes in H and the H/Eu3+/G1 assembly showed that there was no significant change in the transmittance of the blank sample and the host H. Then, the optical transmittance at 500 nm sharply decreased after the formation of the assembly (Fig. S21†).
Furthermore, the morphology of H/Eu3+/G1 was observed by TEM (Fig. S22†). In TEM images, free H existed in a large number of 70 nm spherical forms that transformed into irregular lamellar morphology after forming H/Eu3+ complexes, and the H/Eu3+/G1 assembly existed in a larger area of lamellar form upon further addition of G1. The corresponding energy dispersive spectroscopy (EDS) data demonstrated the changes from no Eu, Eu, and then to Eu and F elements (Fig. S23†). The dynamic light scattering (DLS) test showed the hydrodynamic radius (Rh) of H as 107 nm and the larger particle Rh of H/Eu3+/G1 assembly as 464 nm, which is basically consistent with the TEM results (Fig. S24†). Moreover, the zeta potentials of H, H/Eu3+, H/Eu3+/G1, and  ranged from −22.50, −2.10, and 15.60 to 12.90 mV, indicating that the formation of complexes and the construction of assemblies increased the surface electronegativity of H (Fig. S25†). These further explained the formation of the noncovalent crosslinked network structure.
ranged from −22.50, −2.10, and 15.60 to 12.90 mV, indicating that the formation of complexes and the construction of assemblies increased the surface electronegativity of H (Fig. S25†). These further explained the formation of the noncovalent crosslinked network structure.
Benefiting from the photo-controlled properties of diarylethene derivatives, we subsequently investigated the photo-responsive behavior of this assembly. In the as-prepared noncovalent supramolecular polymers H/Ln3+/G1, no ET phenomenon was observed, and the characteristic spectra of Ln3+ (Eu3+ or Tb3+) appeared due to the absence of spectral overlap between the UV/vis absorption spectrum of G1 and the H/Eu3+ or H/Tb3+ emission spectrum (Fig. 2d). The H/Eu3+/G1 excitation spectrum shows a wide band centered at 254 nm due to absorption of the DPA moiety (Fig. S25†).
The characteristic emission peaks of Tb3+ (490 nm, 545 nm, 583 nm, and 621 nm) and Eu3+ (580 nm, 615 nm, and 692 nm) in the assembly are dominated by the 5D4 → 7F5 at 545 nm and 5D0 → 7F2 transition at 615 nm, respectively, where bright green and red light were emitted. Efficient ET processes from Ln3+ to  may occur in the
may occur in the  assembly because of the complete overlap between the emission spectra of the lanthanide complex H/Tb3+ or H/Eu3+ and the absorption spectra of the closed-ring state
assembly because of the complete overlap between the emission spectra of the lanthanide complex H/Tb3+ or H/Eu3+ and the absorption spectra of the closed-ring state  in the 500–700 nm range (Fig. 2d).
in the 500–700 nm range (Fig. 2d).
As expected, the luminescence of Tb3+ was quenched after UV irradiation (Fig. 5a), reaching a photo-stable state within 20 s, and the luminescence intensity was almost completely quenched. The original lifetime decreased from 0.840 ms to 0.274 ms in aqueous solution (Fig. S27a and S28a†). The same phenomenon was seen in the organic phase (Fig. 5b), where the lifetime decreased from 1.09 ms to 0.170 ms (Fig. S27b and S28b†). As shown in Fig. 5c and d, the same phenomenon was observed in the H/Eu3+/G1 assembly, with decreased lifetime from 1.03 ms to 0.109 ms in aqueous solution (Fig. S27a and S28c†) and from 1.32 ms to 0.127 ms in organic solutions (Fig. S27b and S28d†).
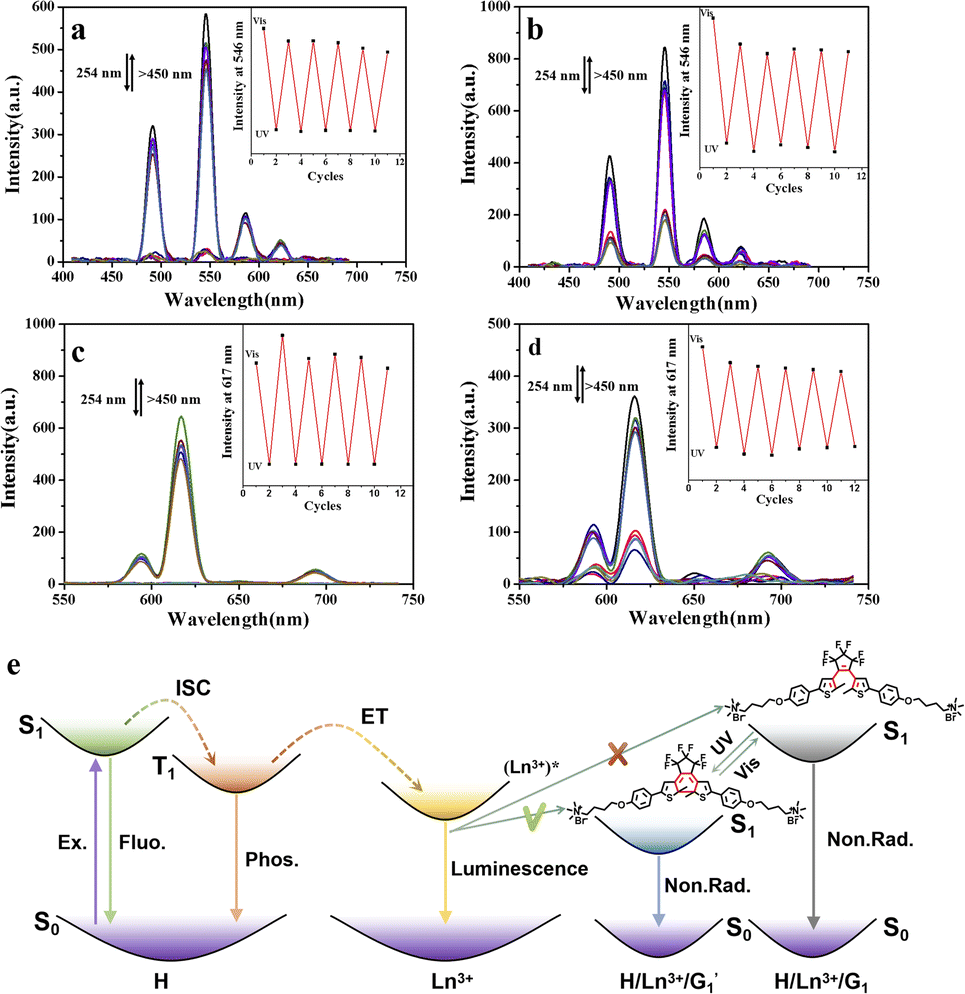 | ||
| Fig. 5 (a) Emission spectra for the H/Tb3+/G1 assembly upon alternating UV (254 nm, 5.5 min) and visible-light (>450 nm, 24 s) irradiation at 298 K in aqueous solution at 298 K (inset: intensity changes at 545 nm). (b) The corresponding data in CH2Cl2 solution at 298 K. (c) Emission spectra for the H/Eu3+/G1 assembly upon alternating UV (254 nm, 5.5 min) and visible-light (>450 nm, 24 s) irradiation at 298 K in aqueous solution (inset: intensity changes at 615 nm). (d) Corresponding data in CH2Cl2 solution. (e) Simplified Jablonski diagram to illustrate the possible mechanism for a reversible photo-controlled energy transfer process. | ||
These phenomena confirm the occurrence of ET processes from H/Eu3+ to the  closed forms. Subsequently, under visible light irradiation, the quenched
closed forms. Subsequently, under visible light irradiation, the quenched  red light was nearly restored to its original level due to the photo-cycloreversion reaction of the diarylene group (Fig. 5a–d). Importantly, this photo-controlled on/off luminescence is reversible, with no significant reduction in luminescence intensity after 5 consecutive cycles under alternating UV and visible light exposure (Fig. 5).
red light was nearly restored to its original level due to the photo-cycloreversion reaction of the diarylene group (Fig. 5a–d). Importantly, this photo-controlled on/off luminescence is reversible, with no significant reduction in luminescence intensity after 5 consecutive cycles under alternating UV and visible light exposure (Fig. 5).
The possible mechanisms for the luminescent transformation of the H/Ln3+/G1 assembly are displayed in Fig. 5e. After the complexation of the host compound H with Ln3+, the 2,6-pyridine dicarboxylic acid part of the host molecule can be used as a sensitizer or antenna to absorb the excitation light with a large absorption coefficient. Its energy is then transferred to the lanthanide ion through intersystem crossing so that Ln3+ is in the excited state. When the formed H/Ln3+ complex is further assembled with open-ring diarylethene derivative G1 to form a noncovalent supramolecular network polymer, this prevents the coordination of solvent molecules and increases the density of the luminescence centers, thus promoting the luminescence properties in solution.
In this process, energy transfer cannot occur due to spectral mismatch, resulting in lanthanide luminescent emission. Instead, when G1 is exposed to visible light and becomes the 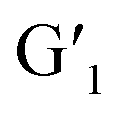 closed-ring form, its UV absorption changes. At this time, the Ln3+ emission of the assembly overlaps with it, leading to the ET process and inhibiting the process of energy transfer from DPA to Ln3+. This process can be governed by alternating UV/vis light irradiation of the diarylethene derivative. The noncovalent interaction between host and guest not only enhanced the luminescence properties of Ln3+, but it also played a crucial role in the ET process due to the decreased distance between the donor and acceptor chromophore.
closed-ring form, its UV absorption changes. At this time, the Ln3+ emission of the assembly overlaps with it, leading to the ET process and inhibiting the process of energy transfer from DPA to Ln3+. This process can be governed by alternating UV/vis light irradiation of the diarylethene derivative. The noncovalent interaction between host and guest not only enhanced the luminescence properties of Ln3+, but it also played a crucial role in the ET process due to the decreased distance between the donor and acceptor chromophore.
More interestingly, the assembly presents multicolor emission by adjusting the proportion of H emitting blue light, Eu3+ emitting red light, and Tb3+ emitting green light in the assembly. In the absence of Ln3+ ions, H/G1 emits blue fluorescence of the pillar[5]arene itself at 325 nm in aqueous solution or CH2Cl2 solution (Fig. 6a and d). As Tb3+ was added to the H/G1 assembly, the typical spectrum of Tb3+ appeared (Fig. 6a and d). The solution underwent a color change from blue to green under 254 nm excitation light (Fig. 6a and d), and the points on the CIE 1931 chromaticity diagram shifted from (0.20, 0.20) to (0.33, 0.55) in aqueous solution (Fig. 6c) and (0.19, 0.14) to (0.33, 0.54) in CH2Cl2 solution, respectively (Fig. 6f).
 | ||
| Fig. 6 Emission spectra of different colors (a) blue, green, yellow, red and (b) white doped with different proportions of Tb3+ and Eu3+ in the H/Ln3+/G1 assembly in aqueous solution at 298 K (inset: images of luminescence in solution). (c) CIE 1931 chromaticity diagram of the corresponding full-color luminescent coordinates. Emission spectra of different colors (d) blue, green, yellow, red and (e) white doped with different proportions of Tb3+ and Eu3+ in the H/Ln3+/G1 assembly in dichloride solution at 298 K (inset: images of luminescence in solution). (f) CIE 1931 chromaticity diagram of the corresponding full-color luminescent coordinates. (g) Under natural light, there is a color change in the letters ‘NKU’ written (left) by H/Ln3+/G1 hydrogel and (right) by H/Eu3+/G1 dichloride solution. (h) Under UV light, there is a change in the luminescent images of the letters ‘NKU’ written (left) by H/Ln3+/G1 hydrogel and (right) by H/Eu3+/G1 dichloride solution (the upper schematic image of changes) ([H] = 5.0 × 10−5 M). | ||
When Eu3+ was added to H/G1, the red emission of Eu3+ also appeared with the CIE coordinate (0.60, 0.32) and (0.63, 0.32) (Fig. 6a, c, d and f). In addition, when Eu3+ and Tb3+ were added to H/G1 in different proportions, the assembly showed multiple yellow light with CIE coordinate (0.43, 0.47) and (0.40, 0.49) (Fig. 6a, c, d and f). Importantly, a distinct white light was observed in the spectrum and the CIE 1931 chromaticity diagram (0.31, 0.32) with a 200:1 ratio for H/G1:Ln3+ in aqueous solution, and (0.31, 0.33) with a 150:1 ratio for H/G1:Ln3+ in dichloride solution (Fig. 6b, c, e and f).
The constructed H/Ln3+/G1 presented full-color emission including white light via the different ratios of blue-light H, green-light H/Tb3+/G1, and red-light H/Eu3+/G1, in water and in the organic phase. It also was endowed with reversible photo-control of lanthanide luminescence due to the ET process with the open/closed ring of diarylethene derivatives. Therefore, the full-color lanthanide light-emitting supramolecular optical switch boosted by a noncovalent assembly of pillar[5]arenes is suitable for a variety of anti-counterfeiting applications.
The developed assembly possesses important characteristics such as full-color, rapid response, outstanding fatigue resistance, and usage in multiphase solutions, which encourages us to further explore its performance in smart versatile anti-counterfeiting. The aqueous solution has the characteristics of environmental protection, while the organic solvent has the characteristics of volatility. Thus, we made use of their characteristics in the H/Ln3+/G1 solution to prepare a photo-controlled digital display for information encryption.
Hydrogels are an ideal substrate for biological applications because they can increase the information density per unit area, and can also be used as wearable or bio-anti-counterfeiting materials.25 Taking advantage of the photo-switchable properties of this assembly, a controllable luminescent hydrogel was easily obtained by simply dispersing agarose gelator in H/Ln3+/G1 aqueous solution. The colorless digital pattern ‘NKU’ under natural light was constructed by mixing a solution of H/Eu3+/G1, H/Eu3+/Tb3+/G1, and H/Tb3+/G1 with agarose, heating, and then cooling to room temperature (Fig. 6g, left and Fig. 6h, left), which displayed bright red, yellow, and green light, respectively, under UV light.
After being irradiated by 254 nm, due to the rapid conversion from H/Ln3+/G1 to  the digital pattern ‘NKU’ changed from colorless to blue under UV light, while the polychrome ‘NKU’ immediately disappeared upon the blue emission of H under UV light. Under visible light (>450 nm) irradiation, the pattern ‘NKU’ was restored to the initial state of colorful glowing for achieving information encryption and anti-counterfeiting. The hydrogel ‘NKU’ could be effectively concealed and then read by simply alternating between UV and visible light irradiation. Furthermore, invisible ink is a common anti-counterfeiting strategy to increase the security of paper information, and it can show or hide information through external stimulation.
the digital pattern ‘NKU’ changed from colorless to blue under UV light, while the polychrome ‘NKU’ immediately disappeared upon the blue emission of H under UV light. Under visible light (>450 nm) irradiation, the pattern ‘NKU’ was restored to the initial state of colorful glowing for achieving information encryption and anti-counterfeiting. The hydrogel ‘NKU’ could be effectively concealed and then read by simply alternating between UV and visible light irradiation. Furthermore, invisible ink is a common anti-counterfeiting strategy to increase the security of paper information, and it can show or hide information through external stimulation.
Because there is great volatility to the CH2Cl2 phase, the CH2Cl2 solution of H/Ln3+/G1 can be used as a writing ink for optical erasing. In a typical test (Fig. 6g, right and Fig. 6h, right), the letters ‘NKU’ were written on ordinary white paper using H/Ln3+/G1 dichloromethane solution as an ink. After rapidly drying in air, there was hardly any mark on the white paper under natural light. In contrast, the letters ‘NKU’ glowed with bright red, yellow, and green luminescence, respectively, under UV light. When irradiated with UV light (254 nm), the blue letters ‘NKU’ gradually appeared on white paper under natural light, whereas the multichrome written ‘NKU’ vanished under UV light.
This conversion was repeated at least three times without obvious attenuation (Fig. S29†). Similarly, the letters that emerged under natural light and disappeared under UV light were recovered via visible light (>450 nm) irradiation. Thus, the H/Ln3+/G1 assembly with satisfactory optical switching performance and noncovalent assembly promoting full-color luminescence can be used as a new multifunctional anti-counterfeiting material that can provide dual signals under natural light and ultraviolet light by simple alternation of UV and visible light irradiation. This could effectively increase the security level of anti-counterfeiting information, and could be used to write information by hydrogel or invisible ink to quickly and efficiently conceal or read. In a comparison, similar photoresponsive properties of commercial anti-counterfeit dyes composed of spiropyran derivatives were obtained for the noncovalent assembly (Fig. S30†).
Conclusions
We successfully constructed a full-color lanthanide supramolecular switch based on noncovalent assembly of a 2,6-pyridine dicarboxylic acid-modified pillar[5]arene/Ln3+ complex with a dicationic diarylethene derivative guest. The as-prepared lanthanide H/Ln3+ (Tb3+ or Eu3+) complexes exhibited significant assembly-induced luminescence enhancement in aqueous and dichloromethane solutions after interacting with the dicationic diarylethene derivative guest. Notably, there was a luminous shine from the enhanced lanthanide luminescent supramolecular assembly, H/Ln3+/G1, with prominent full-color emission ranging from blue, green, yellow to red light and even white light by the mixing of Tb3+/Eu3+ in appropriate ratios in aqueous solution or dichloromethane solution.Moreover, the energy transfer process between the lanthanide luminescence center and the diarylethene component was regulated via open-/closed-ring photoisomerization of the diarylethene group, resulting in the ability to reversibly switch the lanthanide luminescence on and off, which caused a visual change from colorless to blue under natural light due to the structural change of the diarylethene derivative. Through simple alternating UV/vis irradiation, this full-color enhanced lanthanide luminescent switch presented a double signal under natural light or UV light for effectively increasing the security level of information. This also applies in various solvents for producing hydrogels or invisible inks to broaden the application scope of anti-counterfeiting materials, and is a promising method for the construction of multifunctional controllable lanthanide anti-counterfeiting materials.
Data availability
The data that support the findings of this study are available in the ESI.†Author contributions
W.-L. Zhou and X.-Y. Dai contributed equally to this work. Y. Liu and Y. Chen conceived and designed the experiments. W.-L. Zhou and X.-Y. Dai synthesized and performed the chemical characterization. W. Lin performed anti-counterfeiting applications and drawings. W.-L. Zhou and W. Lin wrote the main manuscript. Y. Liu supervised the work and edited the manuscript. All authors analyzed and discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22101143, 22131008, and 21971127), the China Postdoctoral Science Foundation (2021M691661), the National Natural Science Foundation of Inner Mongolia (2021BS02014), the Young Scientific and Technological Talents of Inner Mongolia (NJYT22050), and the Inner Mongolia Minzu University Doctoral Research Fund (BS554).Notes and references
-
(a) A. Bagheri, H. Arandiyan, C. Boyer and M. Lim, Adv. Sci., 2016, 3, 1500437 CrossRef PubMed
; (b) U. Cho, D. P. Riordan, P. Ciepla, K. S. Kocherlakota, J. K. Chen and P. B. Harbury, Nat. Chem. Biol., 2018, 14, 15–21 CrossRef CAS PubMed
-
(a) W.-L. Chan, C. Xie, W.-S. Lo, J.-C. G. Bünzli, W.-K. Wong and K.-L. Wong, Chem. Soc. Rev., 2021, 50, 12189–12257 RSC
-
(a) N. D. Knöfel, H. Rothfuss, P. Tzvetkova, B. Kulendran, C. Barner-Kowollik and P. W. Roesky, Chem. Sci., 2020, 11, 10331–10336 RSC
-
(a) E. M. Surender, S. J. Bradberry, S. A. Bright, C. P. McCoy, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2017, 139, 381–388 CrossRef CAS PubMed
-
(a) F. Fueyo-González, E. Garcia-Fernandez, D. Martínez, L. Infantes, A. Orte, J. A. González-Vera and R. Herranz, Chem. Commun., 2020, 56, 5484–5487 RSC
-
(a) A. de Bettencourt-Dias, P. S. Barber and S. Bauer, J. Am. Chem. Soc., 2012, 134, 6987–6994 CrossRef CAS PubMed
- O. Kotova, C. O'Reilly, S. T. Barwich, L. E. Mackenzie, A. D. Lynes, A. J. Savyasachi, M. Ruether, R. P. M. E. Mobius and T. Gunnlaugsson, Chem, 2022, 8, 1395–1414 CAS
- X. Zhou, L. Wang, Z. Wei, G. Weng and J. He, Adv. Funct. Mater., 2019, 29, 1903543 CrossRef
- Z. Li, X. Liu, G. Wang, B. Li, H. Chen, H. Li and Y. Zhao, Nat. Commun., 2021, 12, 1363 CrossRef CAS PubMed
-
(a) T. Wang, S. Wang, Z. Liu, Z. He, P. Yu, M. Zhao, H. Zhang, L. Lu, Z. Wang, Z. Wang, W. Zhang, Y. Fan, C. Sun, D. Zhao, W. Liu, J.-C. G. Bünzli and F. Zhang, Nat. Mater., 2021, 20, 1571–1578 CrossRef CAS PubMed
- M. Zhao, B. Li, P. Wang, L. Lu, Z. Zhang, L. Liu, S. Wang, D. Li, R. Wang and F. Zhang, Adv. Mater., 2018, 30, 1804982 CrossRef PubMed
- Z. Li, G. Wang, Y. Wang and H. Li, Angew. Chem., Int. Ed., 2018, 57, 2194–2198 CrossRef CAS PubMed
- H.-B. Cheng, H.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2013, 135, 10190–10193 CrossRef CAS PubMed
- Y. Zhou, H.-Y. Zhang, Z.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2017, 139, 7168–7171 CrossRef CAS PubMed
- W. Zhou, Y. Chen, Q. Yu, P. Li, X. Chen and Y. Liu, Chem. Sci., 2019, 10, 3346–3352 RSC
-
(a) T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed
-
(a) M. A. H. Muhammed, L. K. Cruz, A.-H. Emwas, A. M. ElZohry, B. Moosa, O. F. Mohammed and N. M. Khashab, Angew. Chem., Int. Ed., 2019, 58, 15665–15670 CrossRef CAS PubMed
- T. Ogoshi, S. Takashima and T. Yamagishi, J. Am. Chem. Soc., 2018, 140, 1544–1548 CrossRef CAS PubMed
- H. Ju, C. N. Zhu, H. Wang, Z. A. Page, Z. L. Wu, J. L. Sessler and F. Huang, Adv. Mater., 2022, 34, 2108163 CrossRef CAS PubMed
- J. Yao, W. Wu, C. Xiao, D. Su, Z. Zhong, T. Mori and C. Yang, Nat. Commun., 2021, 12, 2600 CrossRef CAS PubMed
- D. Dai, Z. Li, J. Yang, C. Wang, J.-R. Wu, Y. Wang, D. Zhang and Y.-W. Yang, J. Am. Chem. Soc., 2019, 141, 4756–4763 CrossRef CAS PubMed
- L. Xu, Z. Wang, R. Wang, L. Wang, X. He, H. Jiang, H. Tang, D. Cao and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9908–9913 CrossRef CAS PubMed
-
(a) J.-F. Chen, Q. Lin, H. Yao, Y.-M. Zhang and T.-B. Wei, Mater. Chem. Front., 2018, 2, 999–1003 RSC
-
(a) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed
-
(a) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed
-
(a) Q. Zhao, G.-F. Gong, H.-L. Yang, Q.-P. Zhang, H. Yao, Y.-M. Zhang, Q. Lin, W.-J. Qu and T.-B. Wei, Polym. Chem., 2020, 11, 5455–5462 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01425h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |