
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirecting group assisted para-selective C–H alkynylation of unbiased arenes enabled by rhodium catalysis†
Uttam
Dutta‡
a,
Gaurav
Prakash‡
 a,
Kirti
Devi
a,
Kongkona
Borah
a,
Xinglong
Zhang
*b and
Debabrata
Maiti
*a
a,
Kirti
Devi
a,
Kongkona
Borah
a,
Xinglong
Zhang
*b and
Debabrata
Maiti
*a
aIIT Bombay, Department of Chemistry, Powai, Mumbai 400076, India. E-mail: dmaiti@iitb.ac.in
bInstitute of High Performance Computing (IHPC), Agency for Science, Technology and Research (A*STAR), Singapore, Singapore. E-mail: zhang_xinglong@ihpc.a-star.edu.sg
First published on 20th September 2023
Abstract
Regioselective C–H alkynylation of arenes via C–H activation is challenging yet a highly desirable transformation. In this regard, directing group assisted C(sp2)–H alkynylation of arenes offers a unique opportunity to ensure precise regioselectivity. While the existing methods are mainly centered around ortho-C–H alkynylation and a few for meta-C–H alkynylation, the DG-assisted para-selective C–H alkynylation is yet to be reported. Herein we disclose the first report on Rh-catalyzed para-C–H alkynylation of sterically and electronically unbiased arenes. The para-selectivity is achieved with the assistance of a cyano-based directing template and the selectivity remained unaltered irrespective of the steric and electronic influence of the substituents. The post-synthetic modification of synthesized para-alkynylated arenes is also demonstrated. The mechanistic intricacies of the developed protocol are elucidated through experimental and computational studies.
The acetylene motif is prevalent in various natural products, agrochemicals, pharmaceuticals, and in materials.1 It not only provides a unique linear and rigid backbone in molecular arrangement but it also renders a platform to harness the benefit of the extended π-conjugated system in organo-electronic materials.2 The easily transformable nature of the alkyne is an additional advantage as it offers a unique opportunity to diversify the drug molecules and biologically active molecules via triple bond functionalization.3 Therefore, alkynylation of arenes is considered as one of the most desirable transformations. In this regard, the palladium catalyzed Sonogashira coupling reaction is the most versatile and generalized method to synthesize aryl alkynes using aryl (pseudo)halides and terminal alkynes.4 However, utilization of prefunctionalized aryl(pseudo)halides is the major drawback of this method and it inhibits wide application in synthetic chemistry.
In search of alternative approaches that preclude the usage of prefunctionalized arenes, a number of methods were developed in which arene-C(sp2)–H alkynylation was achieved using terminal alkynes or activated alkynes, e.g. ethynylbenziodoxolone (EBX) reagents5 or haloalkynes.6 Notably, the success of these methods requires electronically activated arenes7 or arenes bearing a chelating directing group (DG).8 While the former strategy delivers an alkynylated product based on the electronic effect of its substituents, the latter ensures the regioselective alkynylation of the targeted arene depending on the nature of the directing group. The applicability of the electronically controlled C(sp2)–H alkynylation method is limited to a certain class of substrates bearing electron releasing functional groups. Contrary to that, the DG-assisted C–H activation strategy offers a broad scope and practical method to ensure regioselective C–H alkynylation. However, the majority of the available methods are limited to ortho-C–H alkynylation, which proceed via a thermodynamically favourable five to six membered metallacyclic intermediate. Examples pertaining to the DG-assisted distal meta- or para-C–H alkynylation, which proceeded via a large macrocyclic cyclophane-type intermediate,9 are scarce in the literature. However, the state-of-the art methodologies that are available to perform para-selective C–H functionalization of arenes, either rely on the electronic control of the arene or the prudent design of the templates. In this regard, para-selective borylation was achieved by the group of Itami with the help of an elegantly designed bulky ligand.10 The group of Nakao reported the para-selective alkylation and borylation under nickel11 and iridium12 catalyzed conditions, respectively, by exploiting the steric governance of Al-based Lewis acids. A specialized L-shaped template was developed by Chattopadhyay and co-workers to perform para-selective borylation of aromatic esters.13Para-selective alkylation of aniline derivatives was demonstrated by Frost under Ru-catalyzed conditions in 2017.14 Subsequently, a similar catalytic platform was used by Zhao to perform para-selective difluoromethylation of anilides15 and ketoximes.16 In 2020, they also reported para-selective difluoromethylation using iron(tetraporphyriinato) chloride [Fe(TPP)Cl].17 Evidently, these protocols are extremely limited to a certain class of substrates as well as functionalizations. In order to establish a robust protocol to access selective distal C–H alkynylation of sterically and electronically unbiased arenes, we rely on the directing group assisted C–H activation technique. The group of Yu, in this regard, has demonstrated a meta-C–H alkynylation protocol using an ortho-directing group under palladium-norbornene (Pd-NBE) cooperative catalysis.18 Our group has also developed meta-selective alkynylation protocols employing previously developed meta-directing groups under both palladium19a and rhodium19b catalyzed conditions. However, the notable development in the realm of selective para-C–H alkynylation is yet to be reported.
A suitable directing group to ensure the selective para-C–H activation requires to maintain a precise geometric orientation between the desired C–H bond and the directing group, so that the transition metal catalyst can be accommodated at the proximity of the desired para-C–H bond.9c,20 In this regard, a cyano-based D-shaped template was contemplated by our group in 2015 to execute the para-selective C–H functionalization.20 With this D-shaped template, thus far, para-selective C–H olefination,20,21a acetoxylation,20,21b silylation,21c ketonization,21d cyanation,21e and arylation21f have been achieved. In 2021, Li and coworkers developed a novel pyridine-based para-directing template to harness para selective olefination under Pd-catalyzed conditions.22 Despite the seminal progress in this realm, the widespread application of the template assisted para-selective C–H activation is yet to be achieved. We, therefore, became interested in harnessing para-selective C–H alkynylation of arenes by employing the D-shaped template. Nevertheless, para-selective alkynylation of aniline derivatives was achieved by Waser7c and Fernández-Ibáñez7d using Au and Pd catalysts, respectively. These methods were found to be effective with electron rich aniline derivatives. van Gemmeren and co-workers also reported sterically controlled C–H alkynylation of arenes under Pd-catalyzed conditions at the distal meta-position.23
Our investigation in para-selective C–H functionalization relying on template assistance is mainly centered around the palladium catalyzed conditions in combination with the super stoichiometric silver oxidant.20,21 In sharp contrast, broader catalytic potential of other transition metals is yet to be explored. To the best of our knowledge, para-selective C–H olefination is the only instance, reported to date under Rh-catalyzed conditions (Scheme 1a).24 As our concerted focus is devoted to diversifying the template assisted regioselective distal C–H functionalization method, herein we disclosed the first example of DG-assisted para-C–H alkynylation of arenes via an inverse Sonogashira coupling reaction utilizing a Rh-catalyst (Scheme 1b). More importantly, the desired transformation is unattainable under Pd-catalyzed conditions. Therefore, the present method provides a complementary platform to Pd-catalysis in achieving selective distal C–H functionalization of arenes.
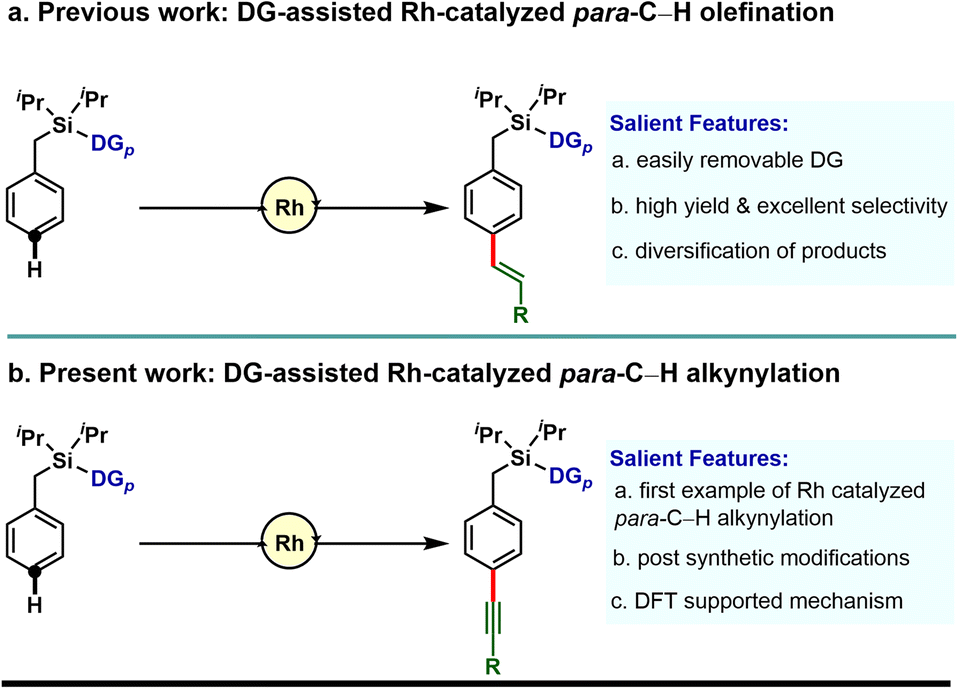 | ||
| Scheme 1 Directing group assisted Rh catalyzed para-C–H functionalization of arenes. | ||
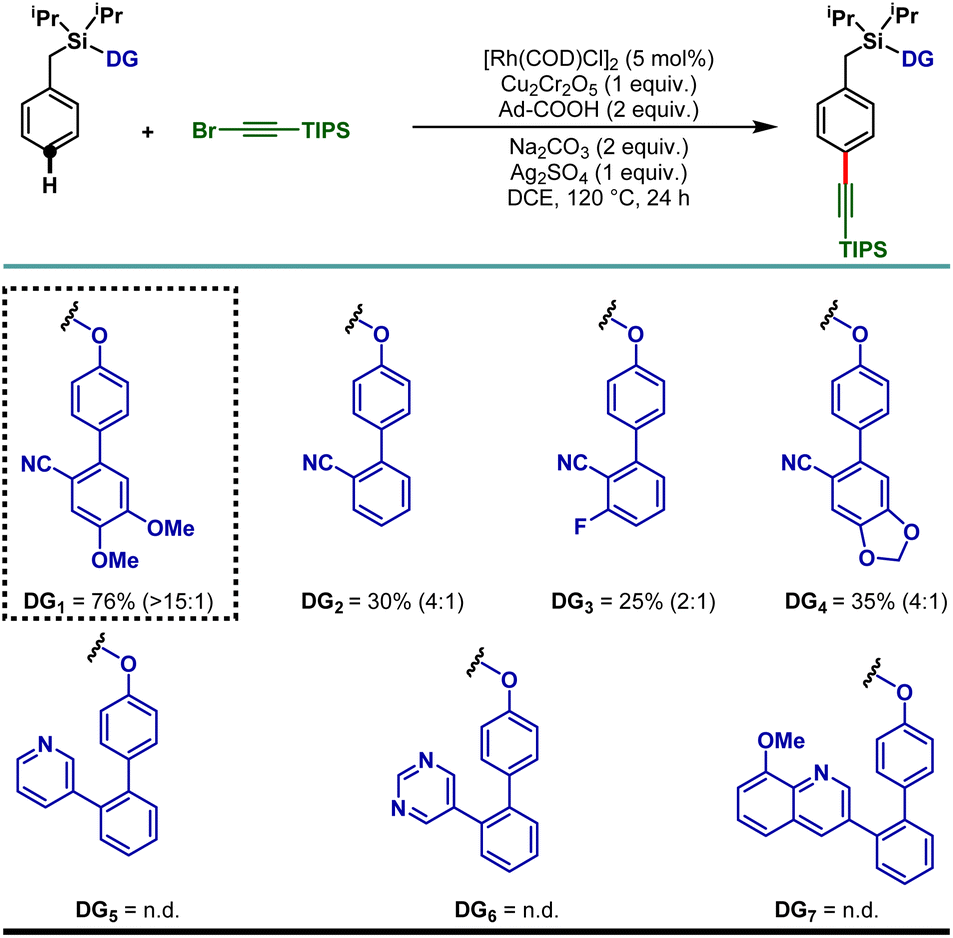
To demonstrate the feasibility of the para-selective C–H alkynylation reaction, we chose a model toluene scaffold, equipped with the electron rich, dimethoxy substituted cyano-based D-shaped template (DG1) and (bromoethynyl)triisopropylsilane as an alkynylating coupling partner. Initial attempt of para-C–H alkynylation using [RhCp*Cl2]2 as catalyst, CuCl2 as oxidant and Ag2CO3 as co-oxidant in the presence of trifluoroacetic acid (TFA) in dichloroethane (DCE) solvent gave the desired product in 20% yield. While optimizing the Rh-catalysts, a slight improvement in yield was observed with [Rh(COD)Cl]2. Rh2(OAc)4 and Rh(PPh3)3Cl were found to be ineffective. Further optimization of the reaction parameters revealed that the combination of dual additives consisting of Cu2Cr2O5 and Ag2SO4 furnished the desired compound in 51% yield. Finally, synthetically useful yield (76% isolated) and selectivity (para![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :others >15:1) were achieved when a combination of 1-adamantanecarboxylic acid and Na2CO3 was used as an acid and base additive, respectively. We also tried using oxygen as a green oxidant along with a catalytic amount of Cu2Cr2O5 but the reaction outcome was not good compared to Cu2Cr2O5 as the stoichiometric oxidant.25 Notably, electronically modified cyano-based weakly coordinating directing groups (DG2, DG3, and DG4) were also examined and all of them were found to be less efficient in comparison to DG1 (Table 1). Additionally, relatively strongly coordinating pyridine (DG5), pyrimidine (DG6) and quinoline (DG7) based directing groups failed to produce the alkynylated compounds under the developed reaction conditions.
:others >15:1) were achieved when a combination of 1-adamantanecarboxylic acid and Na2CO3 was used as an acid and base additive, respectively. We also tried using oxygen as a green oxidant along with a catalytic amount of Cu2Cr2O5 but the reaction outcome was not good compared to Cu2Cr2O5 as the stoichiometric oxidant.25 Notably, electronically modified cyano-based weakly coordinating directing groups (DG2, DG3, and DG4) were also examined and all of them were found to be less efficient in comparison to DG1 (Table 1). Additionally, relatively strongly coordinating pyridine (DG5), pyrimidine (DG6) and quinoline (DG7) based directing groups failed to produce the alkynylated compounds under the developed reaction conditions.
|
With the optimized reaction conditions, the mono substituted toluene-based scaffolds tethered with DG1 were examined to demonstrate the generality of the developed method (Scheme 2). Both the electron donating as well as electron withdrawing substituents delivered the desired para-alkynylated compounds with excellent yield and acceptable selectivity.
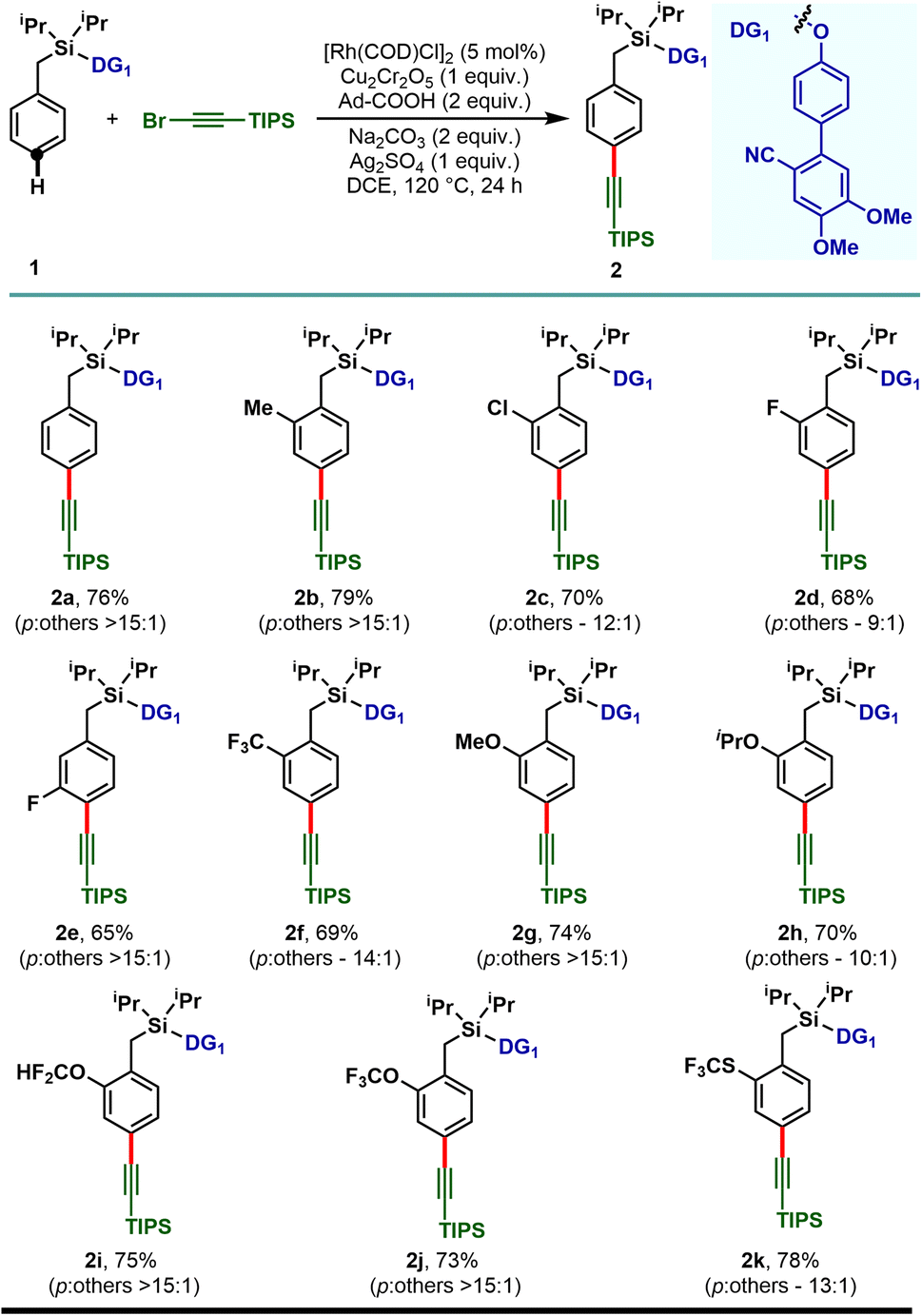 | ||
| Scheme 2 Rh-catalyzed para-C–H alkynylation of mono-substituted arenes. | ||
The electron releasing methyl substituent provided the desired product (2b) in 79% yield with greater than 15:1 selectivity and the electron deficient haloarenes (2c–2e) and ortho-trifluoromethyl arene also gave the para-alkynylated compounds (2f) in good yield and selectivity. Additionally, arenes bearing ethers (1g–1j) and thioether (1k) were tolerated under the reaction conditions and the desired products were obtained without compromising the yield and selectivity. The versatility of the developed protocol was further demonstrated with disubstituted arenes. para-C–H alkynylation with both the 2,6- (1l–1p) and 2,5-disubstituted arenes (1q–1s) proceeded smoothly and the desired products were obtained in synthetically useful yield and selectivity (Scheme 3). Notably, while the meta-F substrate provided the para-alkynylated compound in 65% yield with >15:1 selectivity, relatively bulky meta-substituents such as meta-CH3, meta-Cl, and meta-CF3 scaffolds delivered mainly the meta-alkynylated compounds. We reasoned that the bulkiness of these substituents prevents the accessibility of the para-C–H bond and therefore the sterically less hindered meta-C–H bond was activated. Further, the applicability of the protocol was extended to α-substituted toluene derivatives (1t and 1u). Notably, the α-phenyl toluene derivative mainly afforded the mono-alkynylated product (2t, mono:di – 10:1). It is evident from the scope of the reaction that the electronic nature of the arenes and steric influence of the substituents did not alter the reaction outcome in terms of yield and para-selectivity. Thus, the protocol offers an opportunity to expand the scope of regioselective distal para-C–H alkynylation to accommodate a wide range of steric and electronic demands of substrates. However, the developed protocol failed to accommodate other alkyne coupling partners such as 1-bromo-2-phenylacetylene, 1-bromohept-1-yne, ethyl 3-bromopropiolate and (bromoethynyl)trimethylsilane. The phenomenon could be justified by the coordinating propensity of π bonds to the Rh center, which was also highlighted in the previous literature reports.26
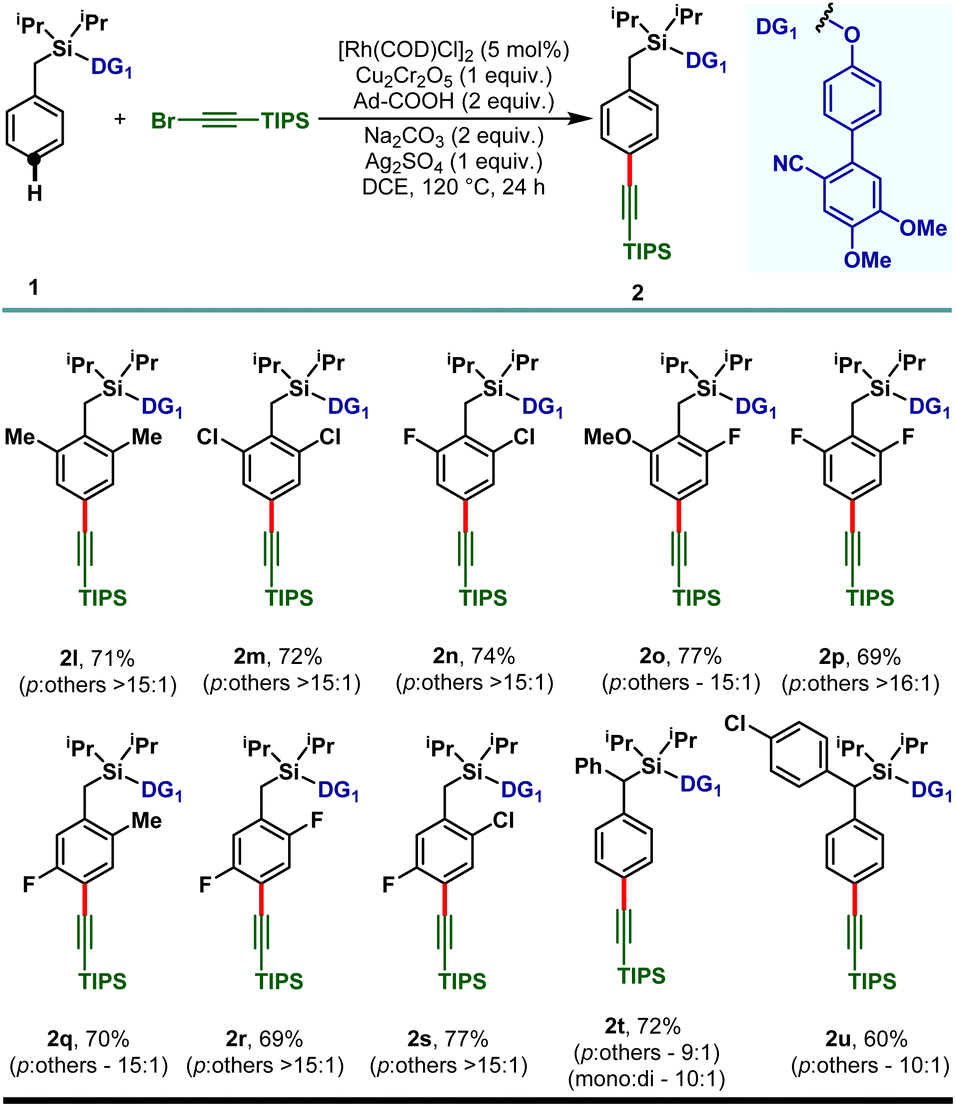 | ||
| Scheme 3 Rh-catalyzed para-C–H alkynylation of di-substituted arenes. | ||
The synthetic utility of the developed para-selective C–H alkynylation protocol was further demonstrated through the removal of the appended directing group as well as through functional group interconversion (Scheme 4). Treatment of 2a with tetra-butylammonium fluoride (TBAF) furnished p-tolylacetylene (3) along with the directing group (DG1) in quantitative yield. It is worth noting that the 4′-hydroxy-4,5-dimethoxy-[1,1′-biphenyl]-2-carbonitrile (DG1) could be attached further to prepare our starting materials, highlighting the reusability of the directing group. Compound 2a treated with p-toluenesulfonic acid in methanol resulted in a silanol derivative, 4. Taking advantage of ortho-directing capability of the silanol motif, 4 was subjected to Pd catalyzed ortho-C–H olefination reaction conditions and an ortho-olefinated and para-alkynylated compound, 5 was obtained in 68% yield. Considering the easy transformability of alkynes, the alkynylated product (3) was reduced to corresponding alkane (6) and alkene (7), and oxidized to benzonitrile (8), benzoic acid (9), and phenyl acetic acid (10) derivatives. Additionally, oxynitration (11), oxydibromination (12), and cyclopropenation (13) reactions were also achieved with good to excellent yields.
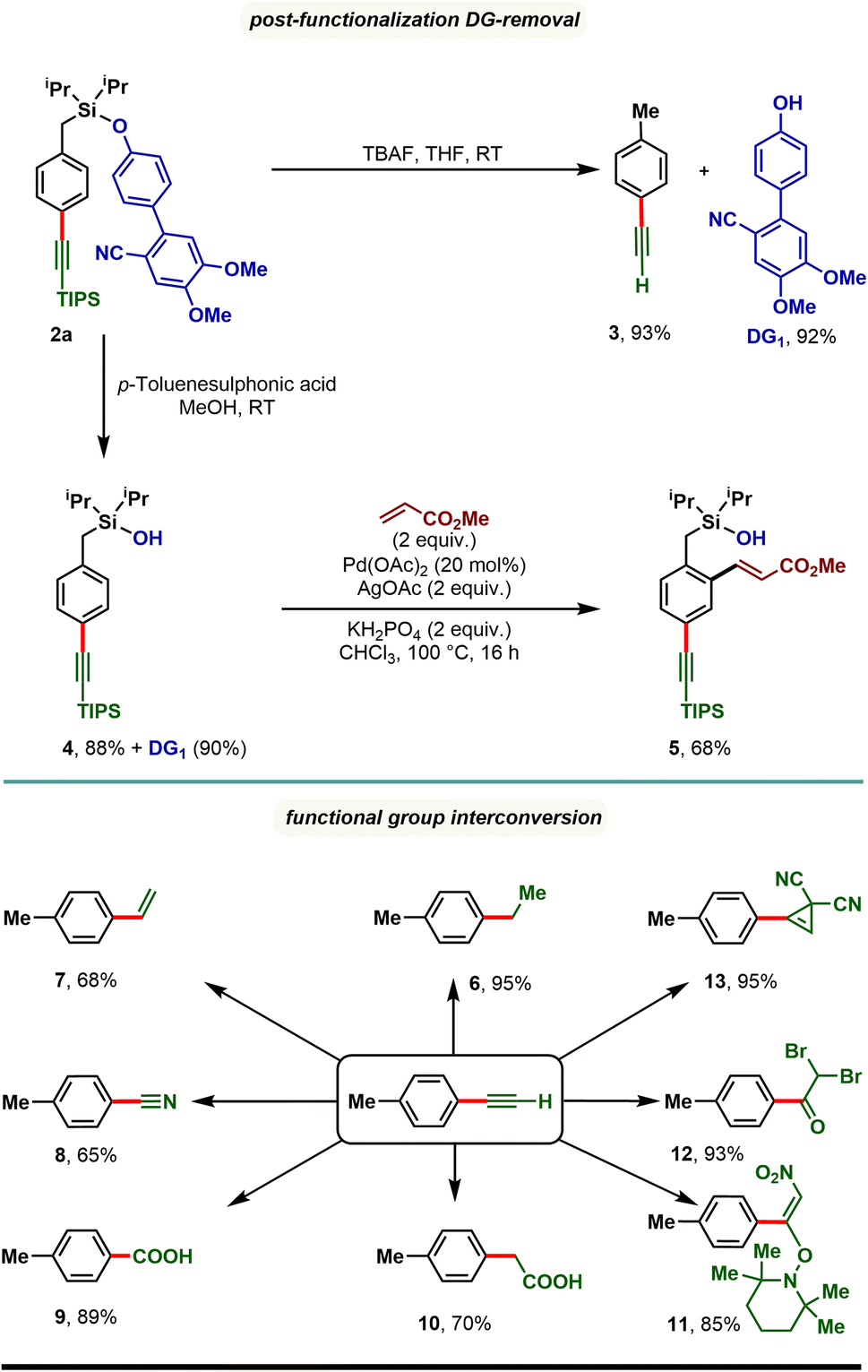 | ||
| Scheme 4 Directing group (DG) removal and post-synthetic modification of para-alkynylated arenes. | ||
Isotope labeling experiments were performed involving an intermolecular competition using the substrate and its deuterated analogue d7 and a PH/PD value of 1.04 and kH/kD value of 1.03 were obtained (Scheme 5). This implies that the C–H activation step is not the overall rate-limiting step of this reaction. Furthermore, order determination studies with respect to the substrate revealed that the reaction was first order with respect to the substrate and first order with respect to the alkyne coupling partner, indicating that both the substrate and the alkyne coupling partner are involved in the rate-limiting step.
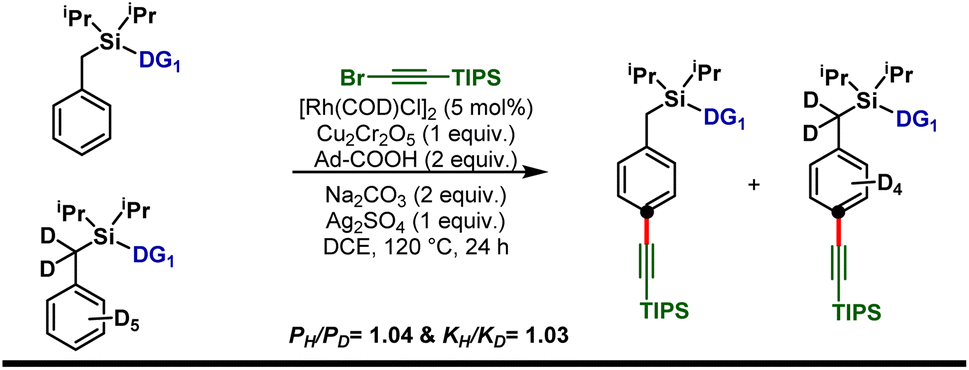 | ||
| Scheme 5 Kinetic isotopic experiments with the deuterium-labeled substrate. | ||
Computational studies
For computational investigation of the mechanism of present Rh-catalyzed para-selective C(sp2)–H alkynylation of arenes, density functional theory (DFT) was employed. Simplified arene, SM1, where the isopropyl groups on the Si atom were replaced by methyl groups, and bromoethynyltriisopropyllsilane (hereafter bromoalkyne) were used for modelling. The use of iPr groups on Si benefits from the favorable Thorpe–Ingold effect making the formation of rhodacycle easier than Me groups, but this simplification should not affect the reaction mechanism – any favorable barriers calculated with this simplified model are expected to be favorable for the iPr analogue. The adamantane-1-carboxylate is simplified to acetate in the DFT calculations to save computational cost. The adamantyl group provides steric hinderance to the molecules and in computational modelling we avoid conformations that would give rise to clashes if adamantane-1-carboxylate were used instead of acetate. We expect the carboxylate group of the simplified acetate to provide similar metal–ligand interactions with the Rh center as the adamantane-1-carboxylate. Gibbs energy profiles were computed at the SMD (dichloroethane)-MN15 (ref. 27)/def2-QZVP//MN15/GENECP (def2-TZVPD for Br,28 Rh29 and Ag29 + def2-SVP30,31 for others) level of theory where a mixed basis set was used for geometry optimization (See ESI Section 6† for full details).32The overall Gibbs free energy profile is shown in Fig. 1. First, the Cu2Cr2O5 additive may play a role in oxidising the Rh(I) precatalyst to Rh(III) which actively participates in the catalytic cycle. Computed C–H activation barriers indicate a kinetic preference for the activation of the para-C–H bond (see ESI, Section 6.3†) by a factor of 16:1 over the meta position and ∼1.5 milion:1 over the ortho position. Detailed investigation of the steric and electronic influences of these TSs suggests that ring strains in the meta-C–H activation transition structures (meta-TS1-site1 and meta-TS1-site2, Fig. S5†) are the lowest (Table S9†) while para-activation has the most favorable orbital overlap between the d-orbital on Rh-metal and the π-orbital on the arene (Fig. S6†). Taken together, the activation of the para-C–H bond (TS1) is lower than the activation of the meta-C–H bond (by 2.2 kcal mol−1 for meta-TS1-site1 and by 3.9 kcal mol−1 for meta-TS1-site2) and the activation of the ortho-C–H bond (by 11.1 kcal mol−1 for ortho-TS1-site1 and by 13.9 kcal mol−1 for ortho-TS1-site2). Additionally, the C–H activation step was found not to be the turnover frequency-determining transition state (TDTS) of the reaction as the formation of the rhodacycle after C–H activation is highly exergonic and irreversible, with the subsequent barrier for 1,2-migratory insertion (19.7 kcal mol−1 from the C–H activated complex INT2 to TS2) lower than the barrier from INT2 back to INT1 (23.0 kcal mol−1viaTS1). In the absence of silver ion participation, the β-bromide elimination step (TS3) has a barrier of 17.9 kcal mol−1. In the presence of silver ions, however, the β-bromide elimination step has a much lower barrier (TS3′, barrier of 1.5 kcal mol−1); this is potentially favored by the exergonic formation of AgBr salt.19a In either case, the 1,2-migratory insertion of bromoalkyne, TS2, will be the overall TDTS and the C–H activated rhodacycle, INT2, will be the TDI. Since the bromoalkyne enters after the TDI and participates in the TDTS, this predicts a first order dependence of the reaction rate on the concentration of the bromoalkyne substrate. In addition, as the C–H activated substrate also participates in the TDTS, a first order dependence on the substrate is similarly expected from the computed energy profile. Both of these predictions are consistent with our experimental order determination measurements (first order with respect to the substrate and bromoalkyne). Finally, the alkynylation product dissociates from the catalyst–product complex INT5′, which releases the product and enters the next catalytic cycle by regenerating INT1. Combined experimental and computational mechanistic investigations suggest that the present Rh-catalyzed C(sp2)–H alkynylation proceeds via non-rate-determining, irreversible regioselective C–H activation at the para-position, followed by turnover-frequency determining 1,2-migratory insertion of the bromoalkyne substrate. This is then followed by the β-bromide elimination step which can be greatly facilitated in the presence of silver ion participation. Our computed energy profile is in agreement with experimental kinetic isotope effects showing that C–H activation is not rate-determining and the measured rate law with first order dependence on both the substrate and the alkyne coupling partner.
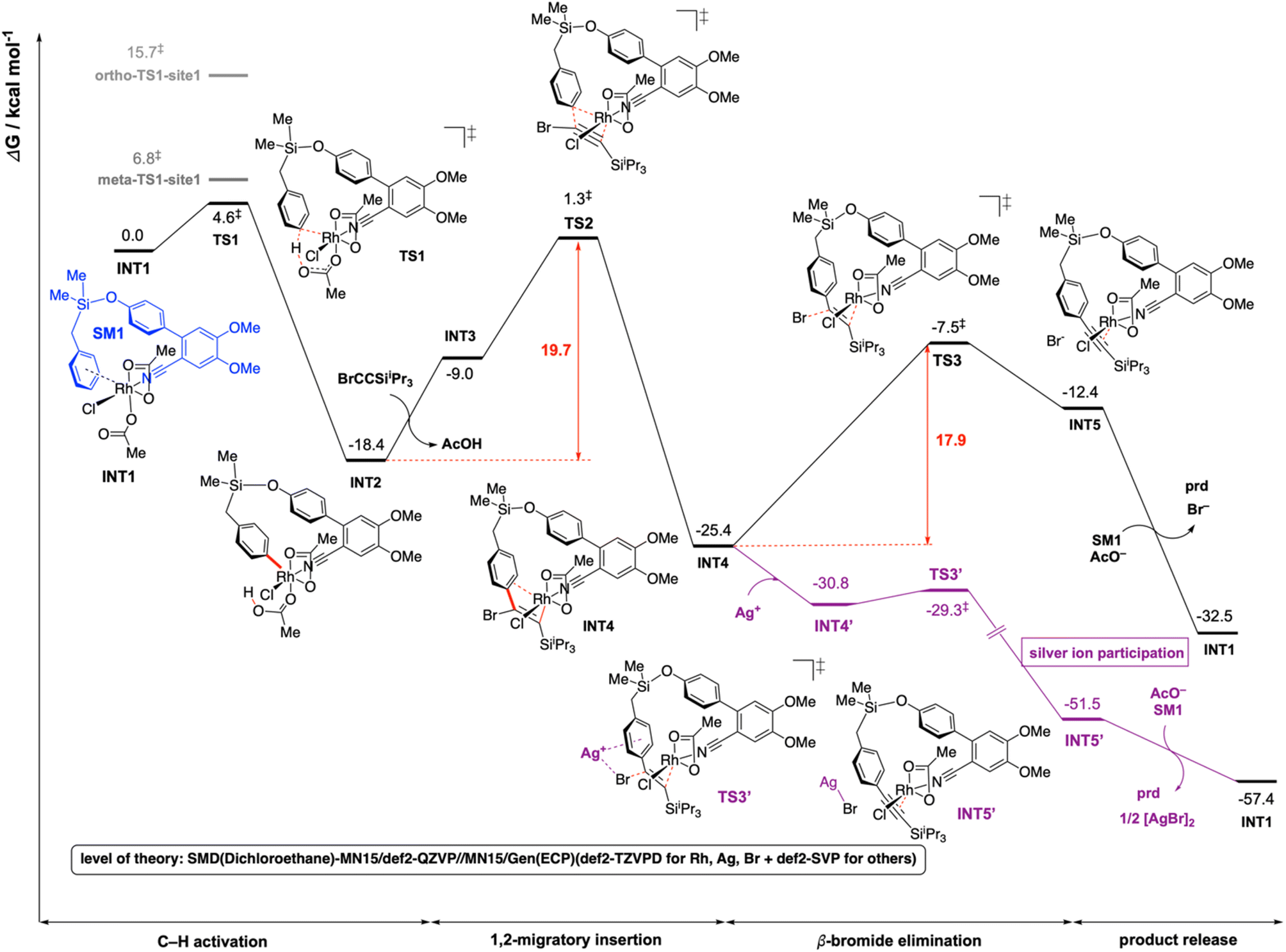 | ||
| Fig. 1 Gibbs energy profile for the Rh-catalyzed C(sp2)–H alkynylation of arenes. DFT optimized key transition structures are shown in Fig. 2. | ||
 | ||
| Fig. 2 DFT optimized TS structures for the C–H activation at different arene sites (TS1s), migratory insertion (TS2) and β-bromide elimination without (TS3) and with (TS3′) silver ion participation. All Gibbs energies are taken with INT1 as zero reference. See Fig. S5† for C–H activation TSs of all possible sites. | ||
Conclusions
In summary, we have developed an unprecedented route for reusable template-assisted Rh catalyzed para-C–H alkynylation. The protocol was well tolerated with different kinds of substituents on the arene ring. Further late-stage modification of the product and functional group inter-conversion were performed. The mechanistic studies were supported by computational studies proving that although regio-determining, the C–H activation is not the overall rate limiting step. Instead, migratory insertion of the alkyne coupling partner is overall rate-determining. We anticipate that this methodology will greatly broaden the scope of functionalization in the realm of remote para-C–H activation.Data availability
Data supporting the manuscript are provided in the ESI,† including the experimental and computational methods and protocols for this study. All DFT-optimized structures have been deposited and uploaded to https://zenodo.org/record/7585280 (DOI: 10.5281/zenodo.7585280). Additional relevant data are available from the corresponding authors upon reasonable request.Author contributions
U. D., G. P. and D. M. conceived the project. U. D., G. P., K. D. and K. B. completed the experimental work. X. Z. designed and performed the computational studies and analysed the results. X. Z. and D. M. wrote the manuscript and supervised the work. All authors contributed to writing the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This activity was funded by SERB-India (TTR/2021/000108). Financial support received as fellowship from CSIR-India (to G. P.) is gratefully acknowledged. X. Z. acknowledges the support from the Agency for Science, Technology and Research (A*STAR) under its Career Development Fund (CDF Project Number C210812008) and Manufacturing, Trade and Connectivity (MTC) Young Individual Research Grants (YIRG grant number M22K3c0091) for this work. X. Z. acknowledges the partial use of supercomputers in the A*STAR Computational Resource Center (A*CRC) for computations performed in this work.Notes and references
- (a) Acetylene Chemistry, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed; (b) Triple Bonded Functional Groups, ed. G. V. Boyd and S. Patai, Wiley, Hoboken, NJ, 1994, ch. 5 Search PubMed; (c) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (d) A. Fürstner and P. W. Davies, Chem. Commun., 2005, 2307 RSC; (e) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC; (f) M. G. Finn and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1231 RSC; (g) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC; (h) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902–912 CrossRef CAS PubMed; (i) L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953–965 CrossRef CAS PubMed; (j) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (k) D. Hashmi and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC; (l) R. Talpur, K. Cox and M. Duvic, Efficacy and safety of topical tazarotene: a review, Expert Opin. Drug Metab. Toxicol., 2009, 5, 195 CrossRef CAS PubMed; (m) Z.-Y. Yang, J.-Q. Yuan, M.-Y. Di, D.-Y. Zheng, J.-Z. Chen, H. Ding, X.-Y. Wu, Y.-F. Huang, C. Mao and J.-L. Tang, Gemcitabine Plus Erlotinib for Advanced Pancreatic Cancer: A Systematic Review with Meta-Analysis, PLoS One, 2013, 8, e57528 CrossRef CAS PubMed.
- (a) T. M. Swager, Semiconducting Poly(arylene ethylene)s, in Acetylene Chemistry: Chemistry, Biology and Material Science, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) F. Diederich and Y. Rubin, Angew. Chem., Int. Ed., 1992, 31, 1101 CrossRef; (c) U. H. F. Bunz, Y. Rubin and Y. Tobe, Chem. Soc. Rev., 1999, 28, 107 RSC.
- (a) L. Hintermann and A. Labonne, Synthesis, 2007, 8, 1121–1150 CrossRef; (b) R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407–1420 RSC; (c) R. Chinchilla and C. Najera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed; (d) J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed; (e) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (f) A. Fürstner, Handbook of Metathesis, Wiley-VCH Verlag GmbH & Co. KGaA: 2015, p. 445 Search PubMed; (g) J. Li and D. Lee, Handbook of Metathesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, p. 381 Search PubMed.
- Selected examples of the Sonogashira coupling reaction: (a) K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS; (b) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2018 CrossRef CAS PubMed; (c) A. O. King and N. Yasuda, Top. Organomet. Chem., 2004, 6, 205–245 CrossRef CAS; (d) H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed; (e) H. Plenio, Angew. Chem., Int. Ed., 2008, 47, 6954–6956 CrossRef CAS PubMed; (f) R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC; (g) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed; (h) D. Wang and S. Gao, Org. Chem. Front., 2014, 1, 556–566 RSC.
- For selected reviews on use of EBX see: (a) V. V. Zhdankin and P. J. Stang, Tetrahedron, 1998, 54, 10927–10966 CrossRef CAS; (b) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed.
- For use of haloalkynes see: (a) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096–2098 CrossRef CAS PubMed; (b) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984–12986 CrossRef CAS PubMed; (c) Y. Ano, M. Tobisu and N. Chatani, Synlett, 2012, 23, 2763–2767 CrossRef CAS; (d) J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387–3390 CrossRef CAS PubMed; (e) K. Kobayashi, M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 8528–8529 CrossRef CAS PubMed; (f) I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742–7743 CrossRef CAS PubMed.
- Electronic controlled alkynylation: (a) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512–1513 CrossRef CAS PubMed; (b) Y. Wei, H. Zhao, J. Kan, W. Su and M. Hong, J. Am. Chem. Soc., 2010, 132, 2522–2523 CrossRef CAS PubMed; (c) J. P. Brand and J. Waser, Org. Lett., 2012, 14, 744–747 CrossRef CAS PubMed; (d) K.-Z. Deng, W.-L. Jia and M. A. Fernández-Ibáñez, Chem. - Eur. J., 2021, e202104107 Search PubMed.
- Selected examples on DG-assisted C(sp2)–H alkynylation with alkynyl halides: (a) M. Tobisu, Y. Ano and N. Chatani, Org. Lett., 2009, 11, 3250–3252 CrossRef CAS PubMed; (b) Y. Ano, M. Tobisu and N. Chatani, Org. Lett., 2012, 14, 354–357 CrossRef CAS PubMed; (c) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590–11593 CrossRef CAS PubMed; (d) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780–4787 CrossRef CAS PubMed; (e) H. M.-F. Viart, A. Bachmann, W. Kayitare and R. Sarpong, J. Am. Chem. Soc., 2017, 139, 1325–1329 CrossRef CAS PubMed; (f) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 2722–2726 ( Angew. Chem. , 2014 , 126 , 2760–2764 ) CrossRef CAS PubMed; (g) Y.-H. Liu, Y.-J. Liu, S.-Y. Yan and B.-F. Shi, Chem. Commun., 2015, 51, 11650–11653 RSC; (h) N. Sauermann, M. J. Gonzalez and L. Ackermann, Org. Lett., 2015, 17, 5316–5319 CrossRef CAS PubMed; (i) Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094–4097 CrossRef CAS PubMed; (j) Y.-J. Liu, Y.-H. Liu, X.-S. Yin, W.-J. Gu and B.-F. Shi, Chem. - Eur. J., 2015, 21, 205–209 CrossRef CAS PubMed; (k) P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P.-X. Shen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092–14099 CrossRef CAS PubMed; (l) R. Boobalan, P. Gandeepan and C.-H. Chieng, Org. Lett., 2016, 18, 3314–3317 CrossRef CAS PubMed; (m) V. G. Landge, G. Jaiswal and E. Balaraman, Org. Lett., 2016, 18, 812–815 CrossRef CAS PubMed; (n) Z. Ruan, S. Lackner and L. Ackermann, ACS Catal., 2016, 6, 4690–4693 CrossRef CAS; (o) Z. Ruan, N. Sauermann, E. Manoni and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 3172–3176 CrossRef CAS PubMed; (p) E. Tan, O. Quinonero, M. Elena de Orbe and A. M. Echavarren, ACS Catal., 2018, 8, 2166–2172 CrossRef CAS PubMed and the references therein..
- (a) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS PubMed; (b) S. Sasmal, U. Dutta, G. K. Lahiri and D. Maiti, Chem. Lett., 2020, 49, 1406–1420 CrossRef CAS; (c) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS PubMed; (d) U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, eabd5992 CrossRef CAS PubMed; (e) W. Ali and G. Prakash, Chem. Sci., 2021, 12, 2735–2759 RSC; (f) J. Grover, G. Prakash, N. Goswami and D. Maiti, Nat. Commun., 2022, 13, 1085 CrossRef CAS PubMed.
- Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193–5198 CrossRef CAS PubMed.
- (a) S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed; (b) S. Okumura and Y. Nakao, Org. Lett., 2017, 19, 584–587 CrossRef CAS PubMed.
- L. Yang, K. Semba and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 4853–4857 ( Angew. Chem. , 2017 , 129 , 4931–4935 ) CrossRef CAS PubMed.
- M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745–7748 CrossRef CAS PubMed.
- J. A. Leitch, C. L. McMullin, A. J. Paterson, M. F. Mahon, Y. Bhonoah and C. G. Frost, Angew. Chem., Int. Ed., 2017, 56, 15131–15135 ( Angew. Chem. , 2017 , 129 , 15327–15331 ) CrossRef CAS PubMed.
- C. Yuan, L. Zhu, C. Chen, X. Chen, Y. Yang, Y. Lan and Y. Zhao, Nat. Commun., 2018, 9, 1189–1198 CrossRef PubMed.
- C. Yuan, L. Zhu, R. Zeng, Y. Lan and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1277–1281 ( Angew. Chem. , 2018 , 130 , 1291–1295 ) CrossRef CAS PubMed.
- W.-T. Fan, Y. Li, D. Wang, S.-J. Ji and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 20524–20530 CrossRef CAS PubMed.
- P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P.-X. Shen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092–14099 CrossRef CAS PubMed.
- (a) S. Porey, X. Zhang, S. Bhowmick, V. Singh, S. Guin, R. S. Paton and D. Maiti, J. Am. Chem. Soc., 2020, 142, 3762–3774 CrossRef CAS PubMed; (b) S. Sasmal, G. Prakash, U. Dutta, R. Laskar, G. K. Lahiri and D. Maiti, Chem. Sci., 2022, 13, 5616–5621 RSC.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef CAS PubMed.
- (a) T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS PubMed; (b) M. Li, M. Shang, H. Xu, X. Wang, H.-X. Dai and J.-Q. Yu, Org. Lett., 2019, 21, 540–544 CrossRef CAS PubMed; (c) A. Maji, S. Guin, S. Feng, A. Dahiya, V. K. Singh, P. Liu and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 14903–14907 CrossRef CAS PubMed; (d) A. Maji, A. Dahiya, G. Lu, T. Bhattacharya, M. Brochetta, G. Zanoni, P. Liu and D. Maiti, Nat. Commun., 2018, 9, 3582 CrossRef PubMed; (e) S. Pimparkar, T. Bhattacharya, A. Maji, A. Saha, R. Jayarajan, U. Dutta, G. Lu, D. W. Lupton and D. Maiti, Chem. - Eur. J., 2020, 26, 11558 CrossRef CAS PubMed; (f) S. Maiti, Y. Li, S. Sasmal, S. Guin, T. Bhattacharya, G. K. Lahiri, R. S. Paton and D. Maiti, Nat. Commun., 2022, 13, 3963 CrossRef CAS PubMed.
- X. Chen, S. Fan, M. Zhang, Y. Gao, S. Lia and G. Li, Chem. Sci., 2021, 12, 4126 RSC.
- A. Mondal, H. Chen, L. Flamig, P. Wedi and M. van Gemmeren, J. Am. Chem. Soc., 2019, 141, 18662–18667 CrossRef CAS PubMed.
- U. Dutta, S. Maiti, S. Pimparkar, S. Maiti, L. R. Gahan, E. H. Krenske, D. W. Lupton and D. Maiti, Chem. Sci., 2019, 10, 7426–7432 RSC.
- S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443–10450 CrossRef PubMed.
- J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387–3390 CrossRef CAS PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105–134116 CrossRef PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al.Gaussian 16, Revision B.01. 2016 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03528j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |