
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrifluoromethylarylation of alkenes using anilines†
Carlos
Corral Suarez
 ab and
Ignacio
Colomer
*ab
ab and
Ignacio
Colomer
*ab
aInstituto de Química Orgánica General (IQOG), CSIC, Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: colomer@iqog.csic.es
bIMDEA Nanociencia, Faraday 9, 28049 Madrid, Spain
First published on 27th October 2023
Abstract
Nitrogen containing compounds, such as anilines, are some of the most widespread and useful chemical species, although their high and unselective reactivity has prevented their incorporation into many interesting transformations, such as the functionalization of alkenes. Herein we report a method that allows the trifluoromethylarylation of alkenes using anilines, for the first time, with no need for additives, transition metals, photocatalysts or an excess of reagents. An in-depth mechanistic study reveals the key role of hexafluoroisopropanol (HFIP) as a unique solvent, establishing a hydrogen bonding network with aniline and trifluoromethyl reagent, that is responsible for the altered reactivity and exquisite selectivity. This work uncovers a new mode of reactivity that involves the use of abundant anilines as a non-prefunctionalized aromatic source and the simultaneous activation of trifluoromethyl hypervalent iodine reagent.
Introduction
It is well recognised that alkenes are versatile substrates that offer great potential for diversification using a plethora of established reactions, including epoxidation, dihydroxylation, aminations, halogenations or borylations. In recent years chemists have sought to incorporate arylation and alkylation into the toolbox of available chemical reactions to functionalize alkenes. Furthermore, functionalizing both positions of an olefin in an intermolecular fashion, creating two new C–C bonds, by simultaneously introducing an aromatic and an aliphatic motif in a single step is certainly a powerful transformation, although underdeveloped.1 Given the relevance of the trifluoromethyl group,2 one outstanding example of such transformations is the trifluoromethylarylation of alkenes, for which there are a handful of methods, each one developed for a particular type of substrate (Fig. 1A). Seminal contribution revolved around a copper-catalyzed process using aryl boronic acids as a pre-functionalized aromatic source,3 later on expanded into an asymmetric version.4 Shortly after, anisoles and indoles were used in a photoredox approach,5 the latter recently engaging into an asymmetric transformation using chiral phosphoric acids under copper catalysis.6 In a beautiful and elegant approach the scope has been expanded to incorporate pyridines using pre-functionalized 4-cyanopyridines under reductive radical coupling.7 Alternatively, isoquinolines were recently reported to react with enol ethers via Minisci type reaction,8 while complementary intramolecular aryl migration has been reported, requiring engineered substrates.9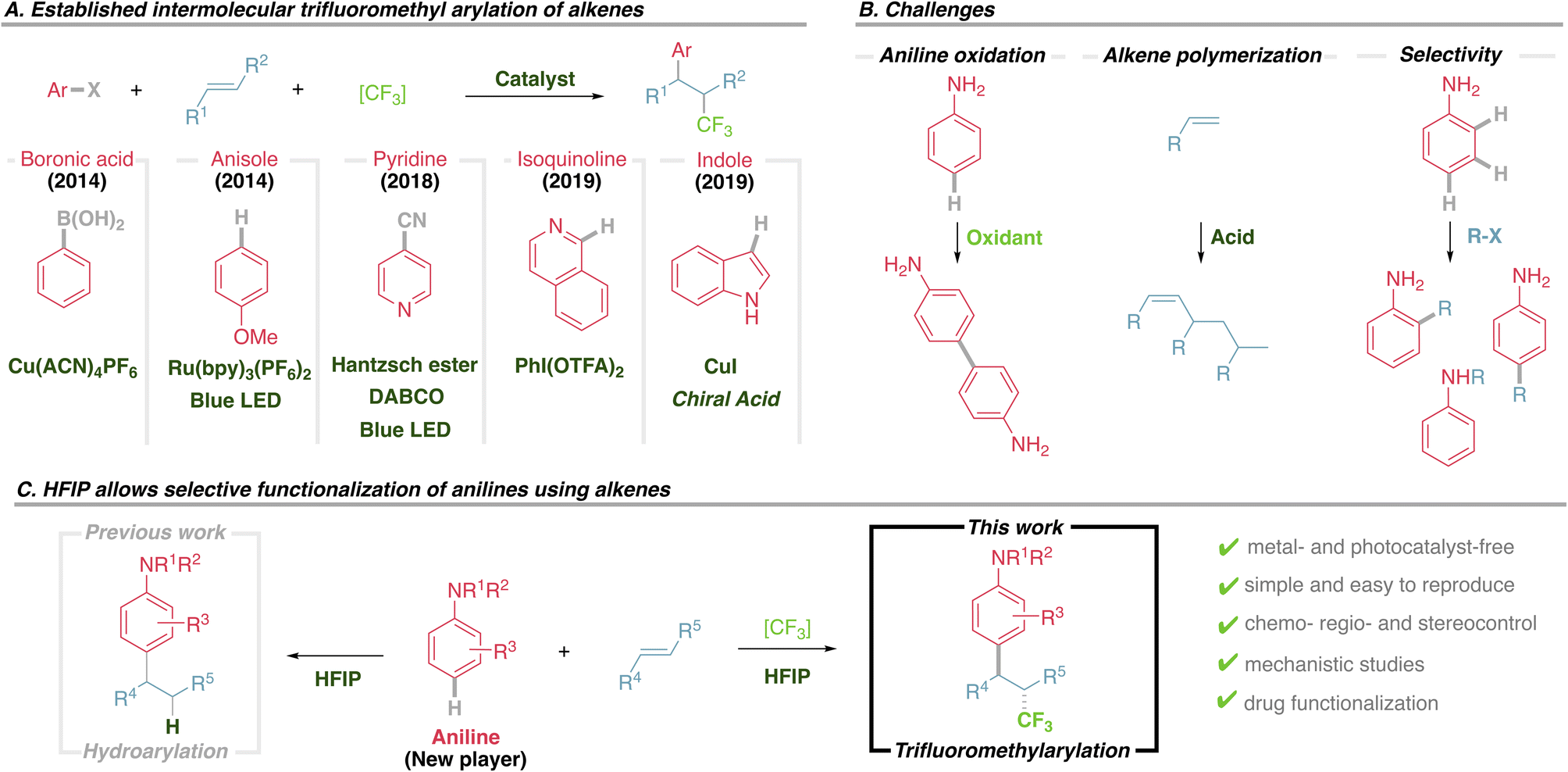 | ||
| Fig. 1 Trifluoromethylarylation of alkenes. (A) Established methods using different aromatic sources. (B) Using anilines entails challenges, such as oxidative side-reactions or non-regioselective functionalization. (C) HFIP is unique at promoting selective aniline functionalization. | ||
Thus, the scope of the aromatic partner is notably restricted, and incorporating other privileged motifs, such as anilines, is highly desirable. Although it could be in principle considered as a straightforward transformation, it is in fact a challenging reaction (Fig. 1B), among other reasons because: (1) anilines are easily oxidized to their radical cation affording a mixture of dimers and polymers,10 (2) alkenes are prone to polymerize under acidic conditions, especially electron rich ones,11 (3) multiple reactive positions of anilines tend to give unselective functionalization.12 These undesired effects have prevented anilines from being incorporated into this transformation,13 despite their prevalence in an uncountable number of bioactive molecules and their importance in agrochemical and medicinal industries. However, recent efforts are providing selective methods for ortho,14meta15 and para16 functionalization. Within this context we have recently reported the hydroarylation of alkenes using anilines, where the solvent hexafluoroisopropanol (HFIP) played a fundamental role, both transferring a proton to the alkene and also promoting the selective functionalization of anilines at the para position (Fig. 1C, previous work).17 As part of our programme to access new sustainable metal-free transformations we aim to exploit the amazing properties of HFIP not only to protonate an alkene, but also to promote the transfer of other interesting functional groups.18 HFIP has emerged as a powerful solvent that can be easily recycled, with very peculiar physical properties that promote a unique chemical reactivity, elongating the stability of polar and radical intermediates or enhancing the reactivity of chemical species.19
This work presents a new strategy for the trifluoromethylarylation of alkenes that allows the incorporation of anilines for the first time. This new approach is conceptually different from previous strategies and does not rely on transition metals or photocatalysts, but rather uses HFIP to promote this transformation, which could not be achieved otherwise (Fig. 1C, this work).
Results and discussion
Optimization and scope
Using model substrates N-benzyl aniline 1a and 4-methoxy styrene 2a, an extensive screening of conditions was performed, including trifluoromethyl reagents, solvent, temperature, stoichiometry and additives (ESI, Tables S1–S5†). Optimal conditions were found employing trifluoromethyl hypervalent iodine 3 and equimolecular amounts of reagents in HFIP at 40 °C, giving a 72% yield of the desired product 4a (Table 1, entry 1). It is important to emphasize that the hydroarylation background reaction is suppressed as it needs harsher conditions.17|
|
||
|---|---|---|
| Entry | Deviation from optimized conditions | Isolated yield, 4a (%) |
| 1 | None | 72% |
| 2 | HFIP (0.2 M) at 40 °C | 67% |
| 3 | HFIP (0.2 M) at rt | 58% |
| 4 | HFIP (0.2 M) at 80 °C | 53% |
| 5 |
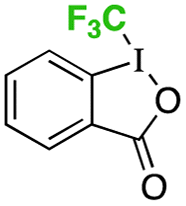 instead of 3 instead of 3 |
34% |
| 6 |
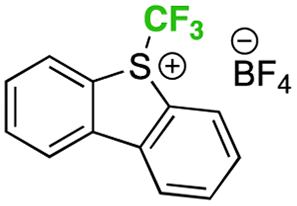 instead of 3 instead of 3 |
nr |
| 7 |
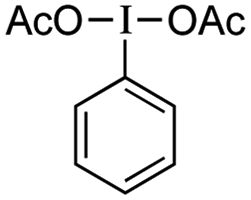 and CF3SO2Na instead of 3 and CF3SO2Na instead of 3 |
nr |
| 8 | CH2Cl2, CH3CN or AcOH (0.2 M) at rt | nr |
| 9 | TFE (0.2 M) at rt | 11% |
| 10 | 2.0 equiv. of 1a | 65% |
| 11 | 2.0 equiv. of 2a | 63% |
| 12 | 2.0 equiv. of 3 | 22% |
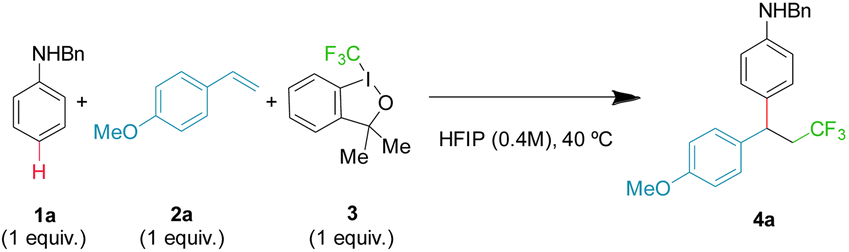
Most of the variables studied have little to moderate impact on the outcome. Neither changing the concentration (Table 1, entry 2) nor the temperature (Table 1, entries 3 and 4) of the reaction improves the isolated yields. However, both HFIP and reagent 3, with an ether backbone, are key for a productive transformation. Alternative trifluoromethyl reagents notably diminished the yield (Table 1, entries 5–7), while other common solvents gave no product (Table 1, entry 8), with analogous trifluoroethanol (TFE) providing very little product (Table 1, entry 9). Finally, using an excess of aniline 1a or alkene 2a slightly diminished the isolated yield (Table 1, entries 10 and 11), while using an excess of 3 has a very negative effect (Table 1, entry 12), probably as a result of competing non-desired oxidative pathways.
With these optimal conditions, the scope of the reaction was then explored. First, the aniline counterpart was evaluated (Fig. 2), where not only benzyl (4a), but also methyl (4b) or even the challenging free aniline (4c) are tolerated, as well as tertiary aniline (4d). The aromatic ring can be extensively modified in both ortho and meta positions. Concerning the ortho position, aliphatic (4e), benzylic (4f) and aromatic substituents (4g) are competent substrates. Electron donating heteroatoms can be incorporated with an exquisite para regiocontrol (4h), as well as electron-withdrawing halides, where both chloro and bromo derivatives are isolated with respectable yields (4i and 4j). The meta position was similarly investigated and both aliphatic (4k) and aromatic substituents (4l) can be accommodated. Moreover, functional groups, such as an alkyne (4m) or an ester (4n), that can serve as a handle for further modifications, are viable substrates. Electron deficient halides (fluoro, bromo and chloro) provide from excellent to good yields (4o–q). Substrates bearing more than one substituent with different patterns of substitution and electronic and steric properties complete the aniline scope (4r–t), while a fused bicycle gives a beautiful tetrahydronaphthalene derivative (4u).
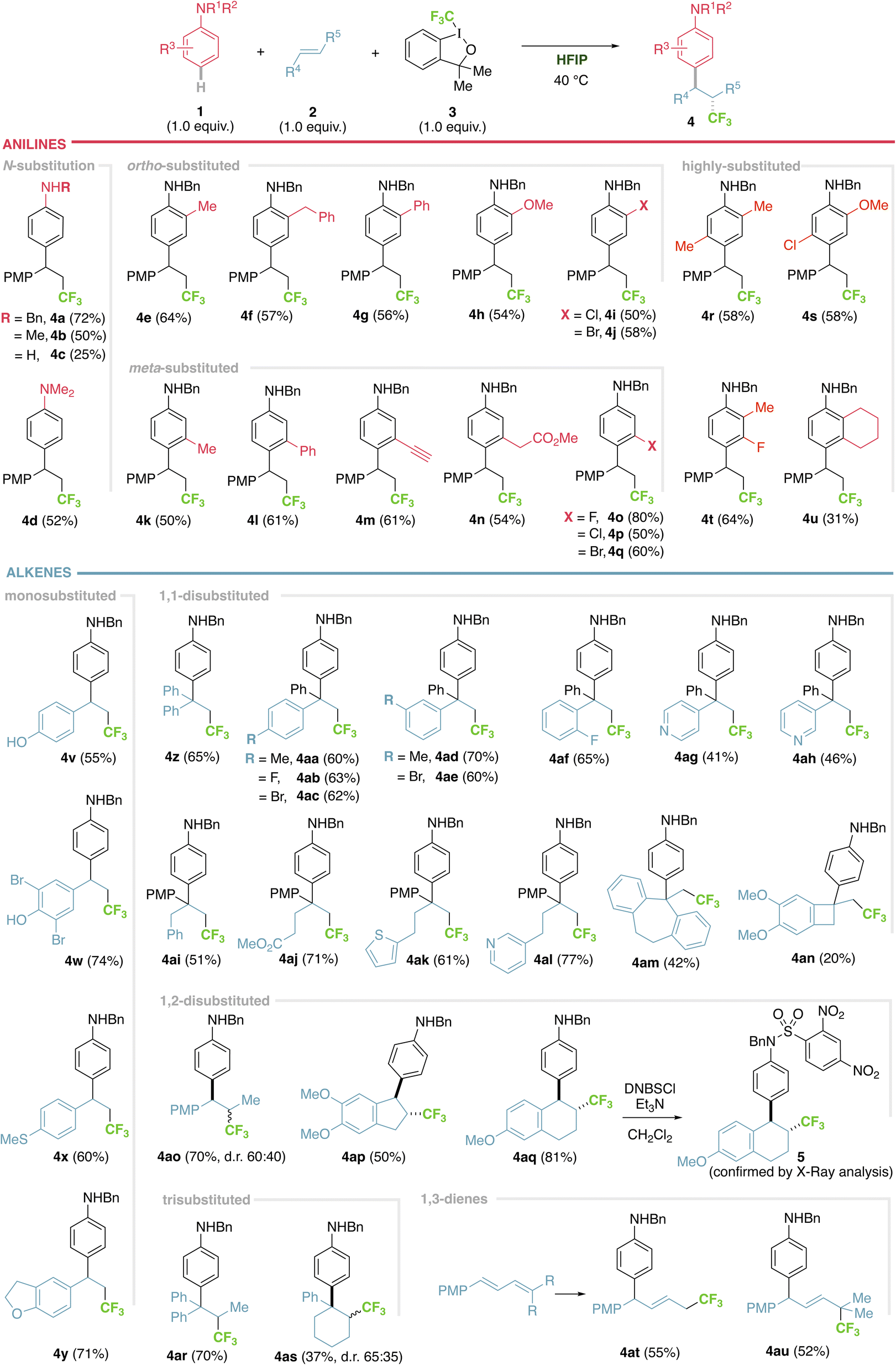 | ||
| Fig. 2 Scope of the trifluoromethylarylation of alkenes using anilines in HFIP. General conditions: 0.2 mmol of aniline 1, 0.2 mmol of alkene 2 and 0.2 mmol of 3 in 0.5 mL of HFIP. PMP: para-methoxyphenyl. DNBSCl: 2,4-dinitrobenzenesulfonyl chloride. | ||
We then moved to study the alkene component, exploring monosubstituted alkenes with aromatic substitution, which tolerated phenols (4v and 4w), thioether (4x) or a dihydrobenzofuran (4y) with very good results. Switching to 1,1-disubstituted alkenes bearing two aromatic groups, with no substitution (4z), aliphatic or halide substituents in para (4aa–ac), meta (4ad and 4ae) and ortho (4af) positions, or attaching heteroaromatic pyridyl motifs in variable locations (4ag and 4ah) resulted in moderate to good yields. Aliphatic substituents can be incorporated (4ai), bearing functional groups, such as an ester (4aj) or with a pendant valuable heteroaromatic thiophene (4ak) or pyridyl (4al) groups. Not only acyclic but also exocyclic terminal alkenes work, with different substitution patterns and ring sizes from cycloheptene (4am) to cyclobutene (4an). Moving on to 1,2-disubstituted alkenes, both acyclic trans-anethole (4ao) and cyclic indene or naphthalene derivatives (4ap–aq) performed extremely well, the latter producing single trans diastereoisomers, confirmed via the X-ray crystallographic structure of a sulfonamide derivative (5). Trisubstituted alkenes, both acyclic (4ar) and cyclic (4as), are also viable substrates, however a low diastereoselection is observed, probably due to a thermodynamically controlled process, where it is hard to discriminate Me vs. CF3 (for 4ao) or Ph vs. PhNHBn (for 4as). Finally, conjugated 1,3-dienes engage in the reaction, expanding the substrate scope with an excellent 1,4-addition outcome (4at), allowing access to the challenging CF3 all-carbon quaternary centre (4au).20 It is important to emphasize that an exquisite C- (vs. N-) and para (vs. ortho) regiocontrol was observed in all cases, isolating the products in moderate to good yields, within the same range for similar transformations.3–8 The ortho product is only observed when blocking the para position with a substituent (see ESI†), something that is in sharp contrast to the very few examples of amino trifluoromethylation of alkenes using anilines, that react via the amino group.21 We have also identified by-products, from side reactions, including trifluoromethyl olefins (that are not competent intermediates of the reaction, see ESI†) and aniline polymerization. Limitations in the scope include the use of electron rich alkenes (aliphatic alkenes such as 1-hexene or 3-hexene are not viable substrates, ESI†), while anilines appear to be unique substrates (other aromatic sources such as anisoles, phenol, durene or aminopyridines are not competent substrates).
Functionalization and applications
Further manipulation of the products includes debenzylation under reductive conditions (4c from 4a) or conversion of the PMP moiety into a valuable carboxylic acid under oxidative conditions,22 affording α-aryl β-trifluoromethyl carboxylic acid 6, otherwise challenging to obtain (Fig. 3A). Hydrogenation of alkene 4at entails aniline debenzylation and alkene reduction in one step to afford 7. Oxidation of PMP gives complementary α-aryl δ-trifluoromethyl carboxylic acid 8 (Fig. 3A). Alternatively, the method can be applied in the selective functionalization of bioactive molecules, such as the anti-inflammatory mefenamic acid,23 a challenging example bearing two aromatic rings susceptible to be functionalized, where a complete chemo control is observed (Fig. 3B, 4av–ax). The versatility of this method, combined with our previous hydroarylation reaction,17 was tested in a sequential transformation using dihydroquinoline 9, that first serves as the alkene component in a trifluoromethylarylation process, to render tetrahydroquinoline 10, that subsequently reacts as the aniline partner in a hydroarylation step, giving the highly functionalized tetrahydroquinoline 11 (Fig. 3C).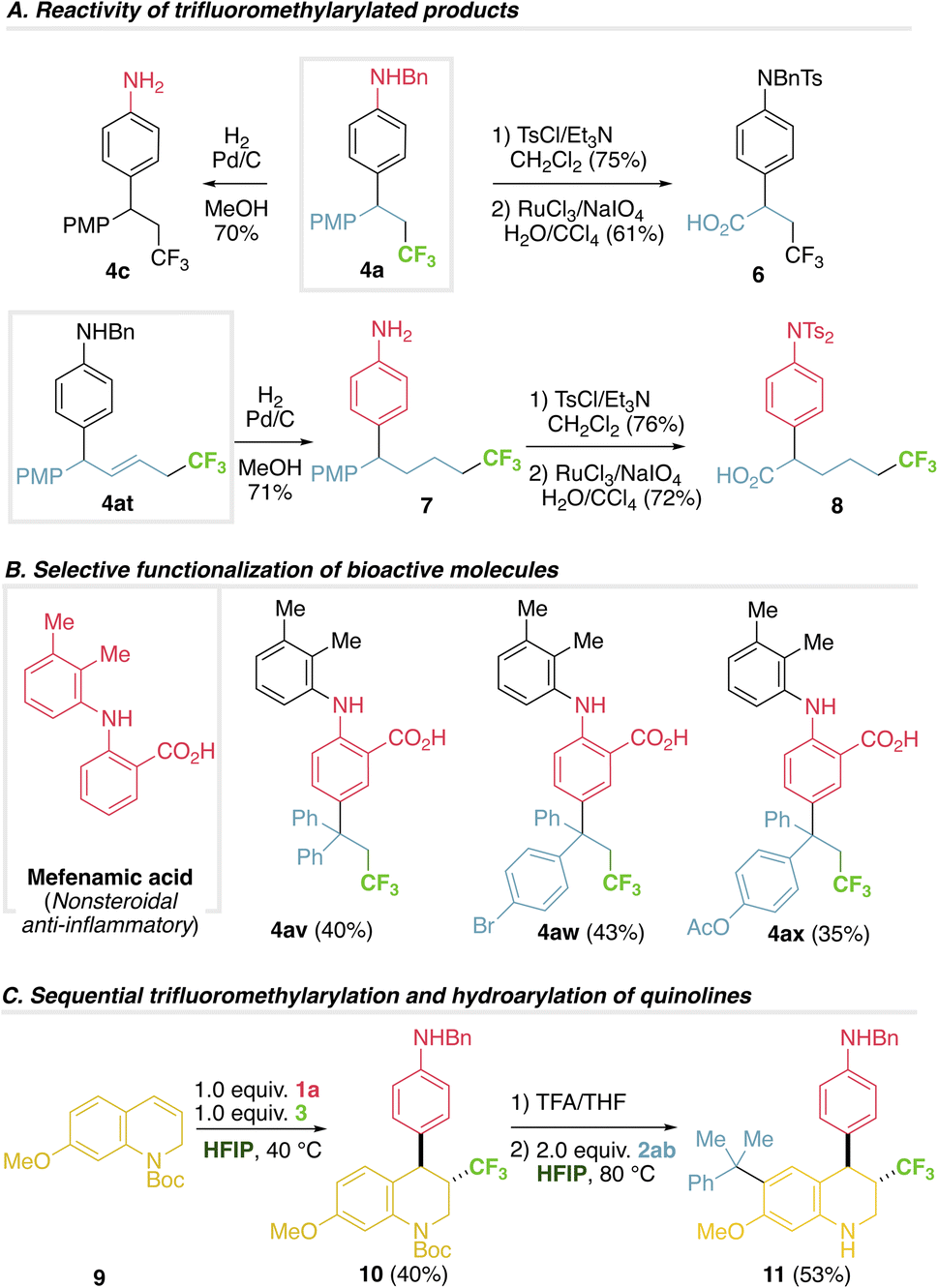 | ||
| Fig. 3 Derivatization and selective functionalization. | ||
Mechanistic experiments
As a first approach to elucidate the mechanism we first confirmed the exquisite preference of anilines to react under these conditions. In a competition experiment using aniline 1a and an excess of anisole, previously shown to be a competent substrate under photocatalysis,5 the product derived from aniline 4a was exclusively formed (Fig. 4A). Moreover, the anisole product was independently synthesized and submitted with aniline 1a and HFIP, recovering only starting materials, ruling out a fast retroarylation (see ESI†). Using radical scavengers BHT or TEMPO does not hamper the reaction (radical adducts CF3-BHT or CF3-TEMPO are not detected), refuting a radical mechanism with extra radical clock experiments (Fig. 4A and ESI†). This is in agreement with related computational studies supporting a cationic mechanism.24 Using pure E and Z acyclic alkene 2u affords the same stereochemical outcome, supporting a free rotating intermediate, such as a benzylic carbocation, and ruling out any concerted addition, such as an iodonium intermediate (Fig. 4A).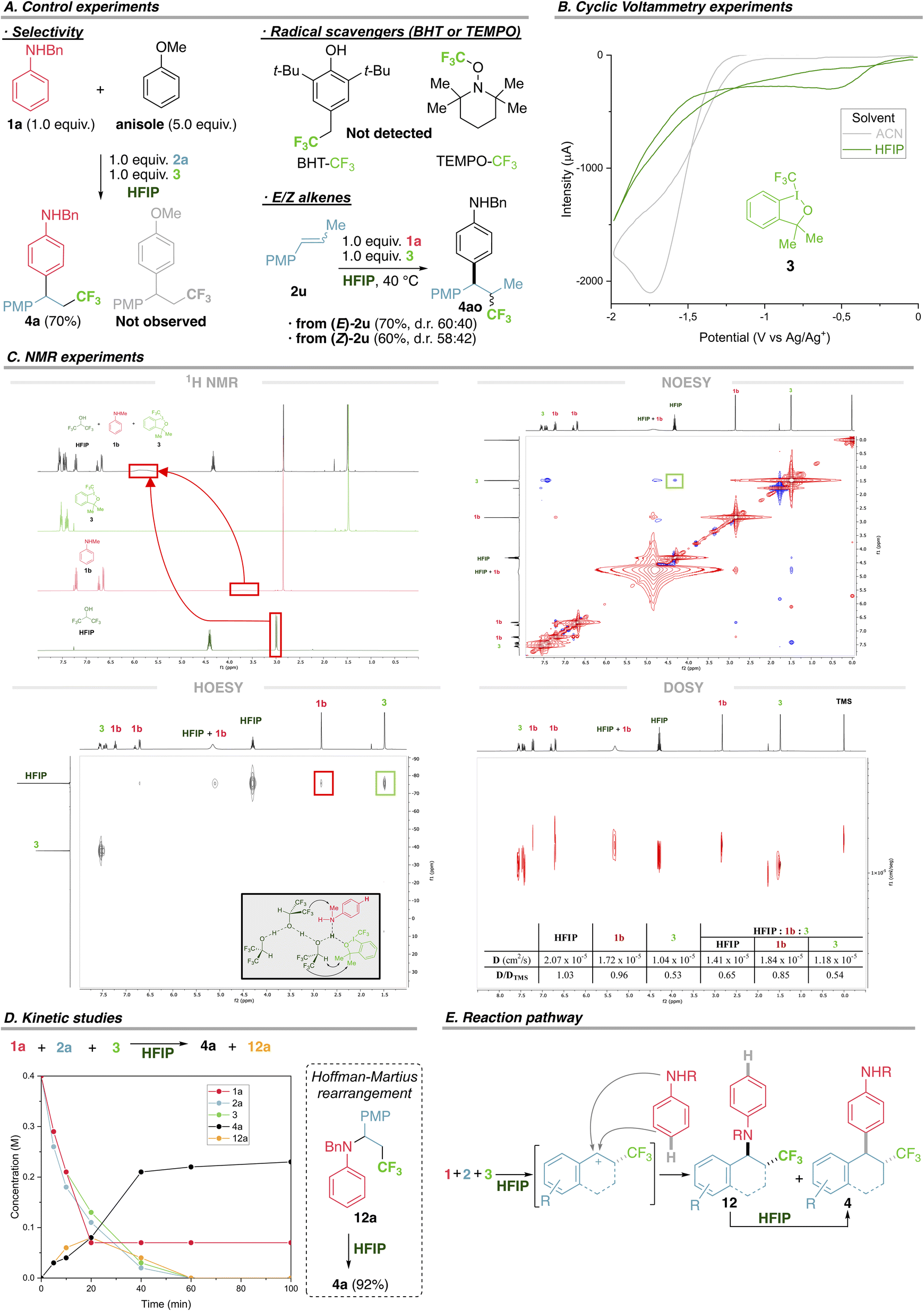 | ||
| Fig. 4 Experiments to elucidate the mechanism. | ||
We then directed our efforts to uncover the interactions that trigger the unique reactivity and selectivity displayed using HFIP. The unique behaviour of trifluoromethyl reagent 3 in HFIP prompted us to study its different electrophilic character both in a solvent where the reaction occurs (HFIP) and a solvent where the reaction does not proceed (acetonitrile, ACN). Cyclic voltammetry of 3 in ACN shows an irreversible reductive peak at −1.75 V, in agreement with a reported value,25 while in HFIP the analogous irreversible reductive peak is observed at −0.58 V (Fig. 4B). This 1.17 V decrease in the reductive potential of 3 using HFIP (not observed for non-competent trifluoromethyl reagents) points towards a more oxidative species, easier to be reduced, or in other words a more electrophilic trifluoromethyl hypervalent iodine reagent might be present in HFIP.
To elucidate the interactions between aniline 1, trifluoromethyl reagent 3 and HFIP that could be responsible of the altered reactivity, we performed an extensive and systematic NMR study including 1H NMR, 19F NMR, NOESY, HOESY and DOSY for both the individual species (HFIP, 1b and 3), binary mixtures of HFIP and 1b or 3, and the most representative and interesting ternary mixture (HFIP, 1b and 3), using TMS as a reference and CDCl3 as the supporting solvent due to its neutral behaviour with respect to interactions between species. Firstly, 1H NMR shows a notable downfield shift of the aniline NH and HFIP OH in the ternary mixture with respect to individual species (Fig. 4C – 1H NMR, red boxes), strong evidence for a H-bonded species. The same downfield shift was observed for the binary mixtures, reinforcing that HFIP establishes H-bonding with both anilines and trifluoromethyl reagent 3 (see ESI† for the complete study). Secondly, NOESY of the ternary mixture revealed, among other things, a strong spatial connection between CH of HFIP and Me signals of 3 (Fig. 4C – NOESY, green box). Thirdly, HOESY of the ternary mixture revealed a strong heteronuclear spatial correlation between CF3 of HFIP and both NMe of aniline 1b and Me signals of 3 (Fig. 4C – HOESY, see the red and green boxes respectively and ESI† for a complete study of binary mixtures). Finally, we designed a series of DOSY experiments to confirm the existence of such an intermolecular H-bonded species in solution. The diffusion rate of individual species was established using TMS as an internal standard, following the trend: HFIP (D/DTMS 1.03) > 1b (D/DTMS 0.96) > 3 (D/DTMS 0.53), with HFIP diffusing quicker and 3 the slowest. For the two binary mixtures (HFIP–1b and HFIP–3) both components of each mixture diffuse at the same rate, where HFIP lowers its diffusion rate (from 1.03 to 0.83 for HFIP–1b and from 1.03 to 0.51 for HFIP–3). These data perfectly correlate with the H-bonded species of a higher molecular weight and therefore diffuse more slowly, a variation that is stronger for the HFIP–3 mixture due to the heavy iodine atom (see ESI† for the complete study). A similar trend is observed for the ternary mixture where both HFIP and aniline 1b tend to diffuse more slowly (from 1.03 to 0.65 for HFIP and from 0.96 to 0.85 for 1b), while 3 remains at 0.54 (Fig. 4C – DOSY).
Finally, we performed kinetic studies to have a broader perspective. All starting materials (1a, 2a and 3) are initially consumed to form arylation product 4a and amination product 12a in equal amounts (8 mM), at a similar rate, during the first 20 min (Fig. 4D). Within the following 20 min, the rate of formation of 4a increases notably, probably due to a double contribution: direct synthesis of 4a from starting materials and conversion of intermediate 12a into 4a (Hoffman–Martius rearrangement,26 proved independently, Fig. 4D and ESI†).
Taken together, these results support the view that the hydrogen bonding between the HFIP network and both aniline and hypervalent iodine reagent remains tight in solution. Meanwhile, hypervalent iodine reagent is activated by HFIP,27 allowing the CF3 group to be transferred to the alkene, and both direct arylation and amination are possible. However, efficient Hoffman–Martius rearrangement of the latter contributes to the formation of 4a as the unique final product (Fig. 4E). This new mode of activating hypervalent iodine reagents via H-bonding to HFIP to transfer functional groups represents a good alternative to previous Brønsted and Lewis acid activation that generally used an excess of Zn, Re, Ti and other transition metals.28
Conclusions
In summary, a new protocol for the trifluoromethylarylation of alkenes using anilines has been developed for the first time that entails the use of HFIP as a unique solvent to promote the reaction. More than 50 examples validate this robust method that involves mild conditions, an atom-economical process that needs no additive or transition metal catalysts. Mechanistic experiments revealed that HFIP not only serves to activate hypervalent iodine, producing a more electrophilic species, but also directs the selective reactivity of anilines via the para position of the aromatic ring, establishing a hydrogen bonding network with both reagents, which is the physical origin of this efficient and unique transformation.Data availability
Data supporting the findings are provided within the article and in the ESI.† This includes experimental procedures, characterization data of all new compounds and NMR spectra. All data are also available from the authors upon request. Crystallographic data for compound 5 was deposited within the Cambridge Structural Database and is freely available via the Cambridge Crystallographic Data Centre under CCDC number 2219810.Author contributions
C. C. S. performed the experiments. I. C. conceived the work and guided the research. All authors contributed to designing, analysing, and discussing the experiments, writing, discussing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from MCIN (RyC2019-026674-I and PID2022-141911NB-I00) and CSIC (PIE 20218AT015 and PIE 202280I025). We thank Dr Josefina Perles (UAM) for performing X-ray structure analysis and Dr Zulay D. Pardo (IMDEA Nanociencia) for helping with DOSY experiments.Notes and references
- Representative examples using photochemical methods or transition metals, such as Ni, Cu, Co or Ru include: (a) Z.-F. Lu, Y.-M. Shen, J.-J. Yue, H.-W. Hu and J.-H. Xu, J. Org. Chem., 2008, 73, 8010–8015 CrossRef CAS PubMed; (b) S. KC, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, J. Am. Chem. Soc., 2018, 140, 9801–9805 CrossRef CAS PubMed; (c) X.-L. Lv, C. Wang, Q.-L. Wang and W. Shu, Org. Lett., 2019, 21, 56–59 CrossRef CAS PubMed; (d) S. Sakurai, A. Matsumoto, T. Kano and K. Maruoka, J. Am. Chem. Soc., 2020, 142, 19017–19022 CrossRef CAS PubMed; (e) X. Cheng, X. Liu, S. Wang, Y. Hu, B. Hu, A. Lei and J. Li, Nat. Commun., 2021, 12, 4366 CrossRef CAS PubMed; (f) Y.-Y. Luan, X.-Y. Gou, W.-Y. Shi, H.-C. Liu, X. Chen and Y.-M. Liang, Org. Lett., 2022, 24, 1136–1140 CrossRef CAS PubMed.
- Reviews covering trifluoromethyl functionalization of alkenes: (a) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (b) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294–8308 CrossRef CAS PubMed; (c) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed; (d) T. Koike and M. Akita, Chem, 2018, 4, 409–437 CrossRef CAS.
- F. Wang, D. Wang, X. Mu, P. Chen and G. Liu, J. Am. Chem. Soc., 2014, 136, 10202–10205 CrossRef CAS PubMed.
- L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904–2907 CrossRef CAS PubMed.
- (a) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Chem. Commun., 2014, 50, 14197–14200 RSC . For an alternative copper-catalyzed approach see:; (b) M.-Y. Min, R.-J. Song, X.-H. Ouyang and J.-H. Li, Chem. Commun., 2019, 55, 3646–3649 RSC.
- J.-S. Lin, T.-T. Li, J.-R. Liu, G.-Y. Jiao, Q.-S. Gu, J.-T. Cheng, Y.-L. Guo, X. Hong and X.-Y. Liu, J. Am. Chem. Soc., 2019, 141, 1074–1083 CrossRef CAS PubMed.
- (a) D. Chen, L. Xu, T. Long, S. Zhu, J. Yang and L. Chu, Chem. Sci., 2018, 9, 9012–9017 RSC; (b) J. Cao, G. Wang, L. Gao, H. Chen, X. Liu, X. Cheng and S. Li, Chem. Sci., 2019, 10, 2767–2772 RSC.
- J. M. Lear, J. Q. Buquoi, X. Gu, K. Pan, D. N. Mustafa and D. A. Nagib, Chem. Commun., 2019, 55, 8820–8823 RSC.
- Examples of semipinacol and boronate rearrangements: (a) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962–6966 CrossRef CAS PubMed; (b) R. J. Armstrong, C. Sandford, C. García-Ruiz and V. K. Aggarwal, Chem. Commun., 2017, 53, 4922–4925 RSC; (c) D. Wang, C. Mück-Lichtenfeld, C. G. Daniliuc and A. Studer, J. Am. Chem. Soc., 2021, 143, 9320–9326 CrossRef CAS PubMed.
- Anilines can be oxidized using transition metals, electrochemistry or metal-free oxidants, such as hypervalent iodine compounds: (a) K. Morimoto, D. Koseki, T. Dohi and Y. Kita, Synlett, 2017, 28, 2941–2945 CrossRef CAS; (b) S. Maddala, S. Mallick and P. Venkatakrishnan, J. Org. Chem., 2017, 82, 8958–8972 CrossRef CAS PubMed; (c) L. Bering, M. Vogt, F. M. Paulussen and A. P. Antonchick, Org. Lett., 2018, 20, 4077–4080 CrossRef CAS PubMed; (d) K. Matsumoto, K. Dougomori, S. Tachikawa, T. Ishii and M. Shindo, Org. Lett., 2014, 16, 4754–4757 CrossRef CAS PubMed; (e) K. Matsumoto, M. Yoshida and M. Shindo, Angew. Chem., Int. Ed., 2016, 55, 5272–5276 CrossRef CAS PubMed; (f) L. Schulz, M. Enders, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 4877–4881 CrossRef CAS PubMed.
- (a) The chemistry of cationic polymerization, ed. P. H. Plesch, 1963 Search PubMed; (b) R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905–6948 CrossRef CAS PubMed.
- The chemistry of anilines, ed. S. Patai and Z. Rappoport, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Few attempts have been made to use anilines in alkyl arylation of alkenes: (a) X. Yong, Y.-F. Han, Y. Li, R.-J. Song and J.-H. Li, Chem. Commun., 2018, 54, 12816–12819 RSC; (b) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed; (c) X.-L. Lv, C. Wang, Q.-L. Wang and W. Shu, Org. Lett., 2019, 21, 56–59 CrossRef CAS PubMed; (d) X. Wang, Y.-F. Han, X.-H. Ouyang, R.-J. Song and J.-H. Li, Chem. Commun., 2019, 55, 14637–14640 RSC; (e) F. J. R. Klauck, H. Yoon, M. J. James, M. Lautens and F. Glorius, ACS Catal., 2019, 9, 236–241 CrossRef CAS; (f) M. J. Cabrera-Afonso, A. Sookezian, S. O. Badir, M. El Khatib and G. A. Molander, Chem. Sci., 2021, 12, 9189–9195 RSC; (g) X. Cheng, X. Liu, S. Wang, Y. Hu, B. Hu, A. Lei and J. Li, Nat. Commun., 2021, 12, 4366 CrossRef CAS PubMed; (h) T. L. Buchanan, S. N. Gockel, A. M. Veatch, Y.-N. Wang and K. L. Hull, Org. Lett., 2021, 23, 4538–4542 CrossRef CAS PubMed; (i) P. Hu, L. Guo, L. Zhao, C. Yang and W. Xia, Org. Lett., 2022, 24, 7583–7588 CrossRef CAS PubMed.
- M. O. Tischler, M. B. Tóth and Z. Novák, Chem. Rec., 2017, 17, 184–199 CrossRef PubMed.
- (a) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed; (b) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed.
- (a) C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458–462 CrossRef CAS PubMed; (b) Y. Ma, B. Wang, L. Zhang and Z. Hou, J. Am. Chem. Soc., 2016, 138, 3663–3666 CrossRef CAS PubMed; (c) Q. Yin, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 3712–3717 CrossRef CAS PubMed; (d) J. A. Leitch, C. L. McMullin, A. J. Paterson, M. F. Mahon, Y. Bhonoah and C. G. Frost, Angew. Chem., Int. Ed., 2017, 56, 15131–15135 CrossRef CAS PubMed; (e) K. Naksomboon, J. Poater, F. M. Bickelhaupt and M. Á. Fernández-Ibáñez, J. Am. Chem. Soc., 2019, 141, 6719–6725 CrossRef CAS PubMed; (f) D. Lichte, N. Pirkl, G. Heinrich, S. Dutta, J. F. Goebel, D. Koley and L. J. Gooßen, Angew. Chem., Int. Ed., 2022, 61, e202210009 CrossRef CAS PubMed; (g) C. Haldar, R. Bisht, J. Chaturvedi, S. Guria, M. M. M. Hassan, B. Ram and B. Chattopadhyay, Org. Lett., 2022, 24, 8147–8152 CrossRef CAS PubMed; (h) N. Mali, J. G. Ibarra-Gutiérrez, L. I. Lugo Fuentes, R. Ortíz-Alvarado, L. Chacón-García, P. Navarro-Santos, J. O. C. Jiménez-Halla and C. R. Solorio-Alvarado, Eur. J. Org Chem., 2022, e202201067 CrossRef CAS.
- I. Colomer, ACS Catal., 2020, 10, 6023–6029 CrossRef CAS.
- (a) I. Colomer, R. C. Barcelos, K. E. Christensen and T. J. Donohoe, Org. Lett., 2016, 18, 5880–5883 CrossRef CAS PubMed; (b) C. Qi, G. Force, V. Gandon and D. Lebœuf, Angew. Chem., Int. Ed., 2021, 60, 946–953 CrossRef CAS PubMed; (c) Y. Li, J. Bao, Y. Zhang, X. Peng, W. Yu, T. Wang, D. Yang, Q. Liu, Q. Zhang and J. Fu, Chem, 2022, 8, 1147–1163 CrossRef CAS; (d) V. Pozhydaiev, M. Vayer, C. Fave, J. Moran and D. Lebœuf, Angew. Chem., Int. Ed., 2023, 62, e202215257 CrossRef CAS PubMed; (e) J. Binder, A. Biswas and T. Gulder, Chem. Sci., 2023, 14, 3907–3912 RSC; (f) G. Kirby, G. Prestat and F. Berhal, J. Org. Chem., 2023, 88, 4720–4729 CrossRef CAS PubMed.
- (a) I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem, 2017, 1, 0088 CrossRef CAS; (b) H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood and J. Aubé, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed.
- M. Vayer, R. J. Mayer, J. Moran and D. Lebœuf, ACS Catal., 2022, 12, 10995–11001 CrossRef CAS.
- (a) G. Dagousset, A. Carboni, E. Magnier and G. Masson, Org. Lett., 2014, 16, 4340–4343 CrossRef CAS PubMed; (b) H. Ge, B. Wu, Y. Liu, H. Wang and Q. Shen, ACS Catal., 2020, 10, 12414–12424 CrossRef CAS; (c) N. Chen and S.-L. Zhang, Adv. Synth. Catal., 2022, 364, 3941–3947 CrossRef CAS.
- P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936–3938 CrossRef CAS.
- J. K. Savjani, S. Mulamkattil, B. Variya and S. Patel, Eur. J. Pharmacol., 2017, 801, 28–34 CrossRef CAS PubMed.
- (a) L. Ling, K. Liu, X. Li and Y. Li, ACS Catal., 2015, 5, 2458–2468 CrossRef CAS; (b) M. Li, X.-S. Xue, J. Guo, Y. Wang and J.-P. Cheng, J. Org. Chem., 2016, 81, 3119–3126 CrossRef CAS PubMed.
- S. Mizuta, S. Verhoog, X. Wang, N. Shibata, V. Gouverneur and M. Médebielle, J. Fluorine Chem., 2013, 155, 124–131 CrossRef CAS.
- S. Wang, G. Force, R. Guillot, J.-F. Carpentier, Y. Sarazin, C. Bour, V. Gandon and D. Lebœuf, ACS Catal., 2020, 10, 10794–10802 CrossRef CAS.
- I. Colomer, C. Batchelor-McAuley, B. Odell, T. J. Donohoe and R. G. Compton, J. Am. Chem. Soc., 2016, 138, 8855–8861 CrossRef CAS PubMed.
- (a) R. Koller, K. Stanek, D. Stolz, R. Aardoom, K. Niedermann and A. Togni, Angew. Chem., Int. Ed., 2009, 48, 4332–4336 CrossRef CAS PubMed; (b) K. Niedermann, N. Früh, R. Senn, B. Czarniecki, R. Verel and A. Togni, Angew. Chem., Int. Ed., 2012, 51, 6511–6515 CrossRef CAS PubMed; (c) E. Mejía and A. Togni, ACS Catal., 2012, 2, 521–527 CrossRef; (d) S. Izquierdo, S. Essafi, I. del Rosal, P. Vidossich, R. Pleixats, A. Vallribera, G. Ujaque, A. Lledós and A. Shafir, J. Am. Chem. Soc., 2016, 138, 12747–12750 CrossRef CAS PubMed; (e) P. Liebing, J. Kalim, N. Arefyeva, F. Oehler, M. Wickleder and A. Togni, Angew. Chem., Int. Ed., 2019, 58, 8585–8588 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2219810. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03868h |
| This journal is © The Royal Society of Chemistry 2023 |