
Revealing surface fine structure on PtAu catalysts by an in situ ATR-SEIRAS CO-probe method†
Guang
Li
a,
Zheng-Chao
An
a,
Jian
Yang
a,
Jin-Hong
Zheng
a,
Li-Fei
Ji
a,
Jun-Ming
Zhang
b,
Jin-Yu
Ye
*a,
Bin-Wei
Zhang
 *cd,
Yan-Xia
Jiang
*a and
Shi-Gang
Sun
ad
*cd,
Yan-Xia
Jiang
*a and
Shi-Gang
Sun
ad
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Engineering Research Center of Electrochemical Technologies of Ministry of Education, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: yxjiang@xmu.edu.cn
bSchool of Chemical and Material Science, Shanxi Normal University, Taiyuan 041001, China
cCenter of Advanced Energy Technology and Electrochemistry, Institute of Advanced Interdisciplinary Studies, Chongqing University, Chongqing 400044, China. E-mail: binwei@cqu.edu.cn
dSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China
First published on 7th June 2023
Abstract
The electrochemical performance of Pt-based catalysts depends on their surface structure. Nevertheless, it is still a challenge to investigate their intricate surface-active sites. Here, PtAu films were utilized as model catalysts to understand the surface fine structure and structure–activity relationship. Significantly, the in situ attenuated total reflectance-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) CO-probe method was established to reveal the surface-active sites of PtAu catalysts. By employing the second derivative spectrum (SDP) method and Gaussian fitting for the infrared spectra of the CO oxidation reaction (COR), three surface configurations, the PtisoAu, PtadjAu site and PtrichAu site, were unveiled. Additionally, the activity order and reactive species of the COR oxidation peaks were clarified by correlating COL intensities with the COR current curves. More importantly, this in situ ATR-SEIRAS CO-probe strategy can be extended from PtAu model films to PtAu nanoparticles.
 Bin-Wei Zhang | Dr Bin-Wei Zhang is an Associate Professor at Center of Advanced Energy Technology and Electrochemistry, Institute of Advanced Interdisciplinary Studies, and School of Chemistry and Chemical Engineering, Chongqing University. He received his bachelor’s and master’s degrees from Xiamen University in 2012 and 2015, respectively. In 2019, he obtained his PhD degree from Institute for Superconducting and Electronic Materials (ISEM), University of Wollongong (UOW). Then, he worked as postdoctoral fellow at the University of New South Wales and UOW. His current research interest is renewable energy storage and conversion, including electrocatalysis and sodium batteries. |
1. Introduction
Fuel cells, as energy conversion devices, have attracted significant attention due to their high efficiency and environmental friendliness.1–3 Pt-based catalysts usually show excellent performance in fuel cells; however, the high price and scarcity of Pt hinders their widespread application.4,5 To reduce the utilization of Pt and still maintain its outstanding activity, Pt alloys have been extensively studied.6–8 As electrochemical reactions usually take place on the surface of catalysts, it is important to study the surface Pt reactive sites of the Pt alloy.9–12 X-ray absorption spectroscopy (XAFS) and spherical-aberration-corrected transmission electron microscopy (SACTEM) have advanced in study of coordination environments and surface atomic structure, but it is still challenging to identify the practical active sites because of the lack of effective research tools to detect intermediate species.13–15 Therefore, it is highly demanded to develop an effective method to clarify the surface structure–activity relationship of Pt-based catalysts.16–19PtAu catalysts have been considered as promising candidates owing to their high activity and chemical stability towards electrocatalytic reduction reactions and various organic small-molecule electro-oxidation reactions, including HCOOH, methanol, and ethanol.20–23 For example, PtAu catalysts have been employed as efficient catalysts for oxygen reduction reactions.20,24 As these electrocatalytic reactions usually take place on the surface of the electrocatalysts, their surface structure plays an important role in the performance.10,25,26 Although great efforts have been made to improve their activity by modifying the electronic and geometric structure of the Pt-based catalysts, the intricate surface-active sites of these PtAu catalysts still needed to be studied further. Recently, we have developed CO as a probe molecule to characterize the d-band center of Pt-based catalysts.27,28 Although Au does not have a beneficial effect on anchoring either COad or OHad, it will modify the electronic and surface-site structure of the PtAu alloy, which influence the electrochemical performance towards CO electro-oxidation. Additionally, the CO adsorption and activity on Au sites are much weaker than on Pt sites, which is ideal for investigating the influence of the electronic and surface-site structure of the PtAu catalyst. Nevertheless, the surface fine structure and electronic structure of PtAu catalysts are unknown. Thus, it is very necessary to reveal the surface-active sites of PtAu catalysts, which can rationally guide the design of PtAu alloy.
In situ attenuated total reflectance-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) has recently been demonstrated to be a powerful tool to detect surface-adsorbed intermediate species and monitor reaction mechanism. Due to the limited penetration depth of the hidden wave, 100 nm in general, the in situ ATR-SEIRAS method was high surface sensitivity.29–32 Nevertheless, using the conventional ink drip method, the catalyst layer thickness of carbon nanomaterials is close to the micron level, which may lead to a worse infrared signal ratio due to exceeding the detection depth. Therefore, the development of an efficient strategy to study the surface fine structure of nanocatalysts is required. The electrodeposition and underpotential deposition methods are simple and useful ways to construct nanoscale surface structure by controlling quantity of electricity.33,34
In this work, the electrochemical in situ ATR-SEIRAS and CO probe molecule method were employed to investigate the surface fine structure on PtAu catalysts from model films to nanocatalysts. PtAu model films with various surface ratios were prepared using the electrodeposition strategy, which presented an enhanced infrared signal for CO intermediate species. Moreover, the second derivative spectrum (SDP) method and corresponding Gaussian fitting of in situ ATR-SEIRAS for CO oxidation were developed to locate the peak positions. Based on the structure characterization and surface content, there are three COL sites in the PtAu film catalysts: surface Pt atoms completely isolated by Au atoms (PtisoAu site), adjacent Pt active sites (PtadjAu site), and abundant continuous Pt atoms without Au atoms (PtrichAu site). Moreover, the activity order of the different sites was identified. Significantly, a consistent appearance and results were also verified in PtAu nanoparticles, which clarified the surface fine configurations on the nanocatalyst.
2. Experimental
2.1 Chemicals and reagents
H2PtCl6·H2O (wt% Pt: 37.5%), HAuCl4·4H2O (99.9%, trace metal basis), and HClO4 (70% aqueous solution, ACS) were purchased from Aladdin and used without purification. A silicon wafer (99.9%, 10 mm × 10 mm × 0.5 mm) was purchased from TEXCHEM. A ladder-type silicon prism was purchased from Jing Xaun Guang Dian (Shanghai, China). Ar (99.99%) and CO (99.999%) were purchased from Linde Industrial Gas. Deionized water used in electrochemical studies and device cleaning was from a Millipore Milli-Q system (resistivity 18.2 MΩ cm−1).2.2 Structure characterization
The surface morphologies of the electroless Au film and electrodeposited PtAu films on a silicon wafer were studied using field-emission scanning electron microscopy (SEM) (HITACHI-4800, HITACHI, Japan). The crystalline phases of the electrodeposited PtAu films were analyzed using powder X-ray diffraction (XRD) patterns using a Rigaku IV XRD with Cu Kα radiation (λ = 1.54 Å, step size: 0.02 for PtAu films on silicon wafer; current: 30 mA; and voltage: 40 kV). Surface electronic structures of the electrodeposited PtAu films were analyzed using an ESCALAB 250 XI X-ray photoelectron spectrometer (XPS, Thermo Fisher Scientific) with monochromatic Al Kα radiation (1486.6 eV; current: 12 mA; voltage: 12 kV; diameter beam spot: 500 μm), and the binding energies were calibrated with reference to the C 1s peak at 284.6 eV. The morphology of the PtAu nanomaterials was studied using transmission electron microscopy (TEM, TECNAI F20). The STEM imaging and EDS elemental mapping were conducted using a FEI Themis Z microscope equipped with a probe corrector operated at 300 kV.2.3 Preparation for electroless plated Au
The configurations and characteristics of the electrochemical IR cell were described in a previous work.35 The method of secondary electroless gold plating on the silicon prism/wafer was detailed in previous reported work: a silicon prism/wafer was ground with 1 μm, 0.3 μm and 0.05 μm Al2O3 and cleaned using ultrasonication with water and acetone. After treatment with piranha solution (H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30%H2O2 = 7:3) for 90 min, the plating bath was dropped on the silicon prism/wafer after being soaked in 40 wt% NH4F solution for 90 s, and electroless plating was conducted for 150 s at 60 °C. Aqua regia dissolved the unstable gold film and it was gilded again. The electrodeposited PtAu model films on the Au-coated silicon prisms were used for electrochemistry measurement and in situ ATR-SEIRAS measurements. The electrodeposited PtAu model films on the Au-coated silicon wafers were used for XRD and XPS measurement. The chemically synthesized PtAu nanocatalysts were used from our previous work.17 The cyclic voltammetry curve and structural characterization of the Au film on the Si prism are shown in Fig. S1.†
:30%H2O2 = 7:3) for 90 min, the plating bath was dropped on the silicon prism/wafer after being soaked in 40 wt% NH4F solution for 90 s, and electroless plating was conducted for 150 s at 60 °C. Aqua regia dissolved the unstable gold film and it was gilded again. The electrodeposited PtAu model films on the Au-coated silicon prisms were used for electrochemistry measurement and in situ ATR-SEIRAS measurements. The electrodeposited PtAu model films on the Au-coated silicon wafers were used for XRD and XPS measurement. The chemically synthesized PtAu nanocatalysts were used from our previous work.17 The cyclic voltammetry curve and structural characterization of the Au film on the Si prism are shown in Fig. S1.†
2.4 Electrochemistry measurements
The PtAu model films with different surface ratios were obtained by electrodepositing different concentration ratios of H2PtCl6 and HAuCl4 at 0.30 V (vs. RHE) under Ar saturation (molar ratios of electrodeposition solution: H2PtCl6:HAuCl4 = 9:1, 3:1, 1:1, 1:3, 1:9, total concentration: 2 mM, supporting electrolyte: 0.1 M HClO4). The electrodeposited PtAu films on Au-coated silicon wafer were used for analysis of structure and morphology. The electrodeposited PtAu on Au-coated silicon prism and PtAu nanocatalysts were used as the working electrodes for cyclic voltammetry, CO stripping measurement and in situ ATR-SEIRAS measurement in 0.1 M HClO4 solution by a potentiostat/galvanostat, 263A model, Princeton Applied Research, with a saturated calomel electrode (SCE) and a graphite rod as the reference and counter electrodes, respectively. The PtAu nanocatalyst was dispersed in 1 mL mixed solution (495 μL isopropanol + 495 μL water + 10 μL 5 wt% Nafion solution) and ultrasonically dispersed for 1 hour. 25 μL ink was dropped on the electroless Au@Si prism and dried in air for in situ ATR-SEIRAS measurement. In this study, all the indicated potentials were corrected to the reversible hydrogen electrode potential (RHE). After CO had been adsorbed for 8 min at 0.1 V and Ar-purged for 15 min, the electrochemistry and electrochemical in situ ATR-SEIRAS experiments of PtAu COR were conducted.
2.5 In situ ATR-SEIRAS measurements
Electrochemical in situ ATR-SEIRAS measurements were conducted using a Nicolet-8700 spectrometer equipped with a liquid-nitrogen-cooled MCT-A detector. A 263A model potentiostat was used to control the electrode potential. The electrodeposited PtAu on an Au-coated silicon prism was electrochemically cleaned until stable in Ar-saturated 0.1 M HClO4 solution. The multi-step FTIR method (MS-FTIR) was used to collect the spectrum from 0.1 V to 1.3 V at 0.05 V intervals. The spectra were recorded from 4000 cm−1 to 1000 cm−1 with a spectral resolution of 8 cm−1 and a scan number of 200. The spectra were calculated from the relative change in reflectivity (ΔR/R) with eqn (1) below. The R(ES) and R(ER) represent the single-beam spectrum recorded at the sample setting potential ES and the reference potential ER, respectively. In order to more intuitively display the changes in the spectra, the original infrared spectra were rotated and the direction of peaks were opposite.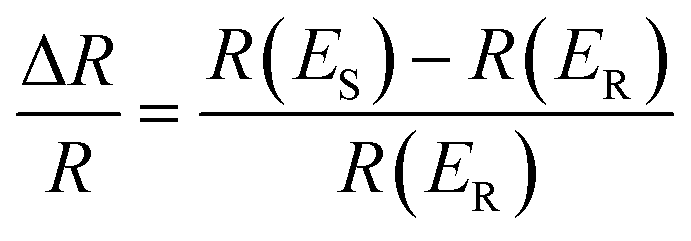 | (1) |
3. Results and discussion
3.1 Structural characterization of the PtAu films
The PtAu film model catalysts were prepared through the electrodeposition method at 0.3 V in solutions with different H2PtCl6:HAuCl4 ratios of 1.8:0.2, 1.5:0.5, 1:1, 0.5:1.5, and 0.2:1.8 with a total concentration of 2 mM. The total deposited charge was 0.10C to ensure the same thickness for all PtAu films (Fig. 1a). The obtained samples were labeled as Pt1.8Au0.2, Pt1.5Au0.5, Pt1Au1, Pt0.5Au1.5, and Pt0.2Au1.8, respectively. The cyclic voltammetry curves of the PtAu films with different ratios were shown in Fig. 1b. By integrating the hydrogen underpotential deposition region charge (210 μC cm−2) and Au redox peak charge (340 μC cm−2), the surface compositions of the PtAu films were calculated and were shown in Table S1.†17,36 The surface ratios of Pt for Pt1.8Au0.2, Pt1.5Au0.5, Pt1Au1, Pt0.5Au1.5, and Pt0.2Au1.8 were 0.92, 0.86, 0.68, 0.48 and 0.16, respectively.
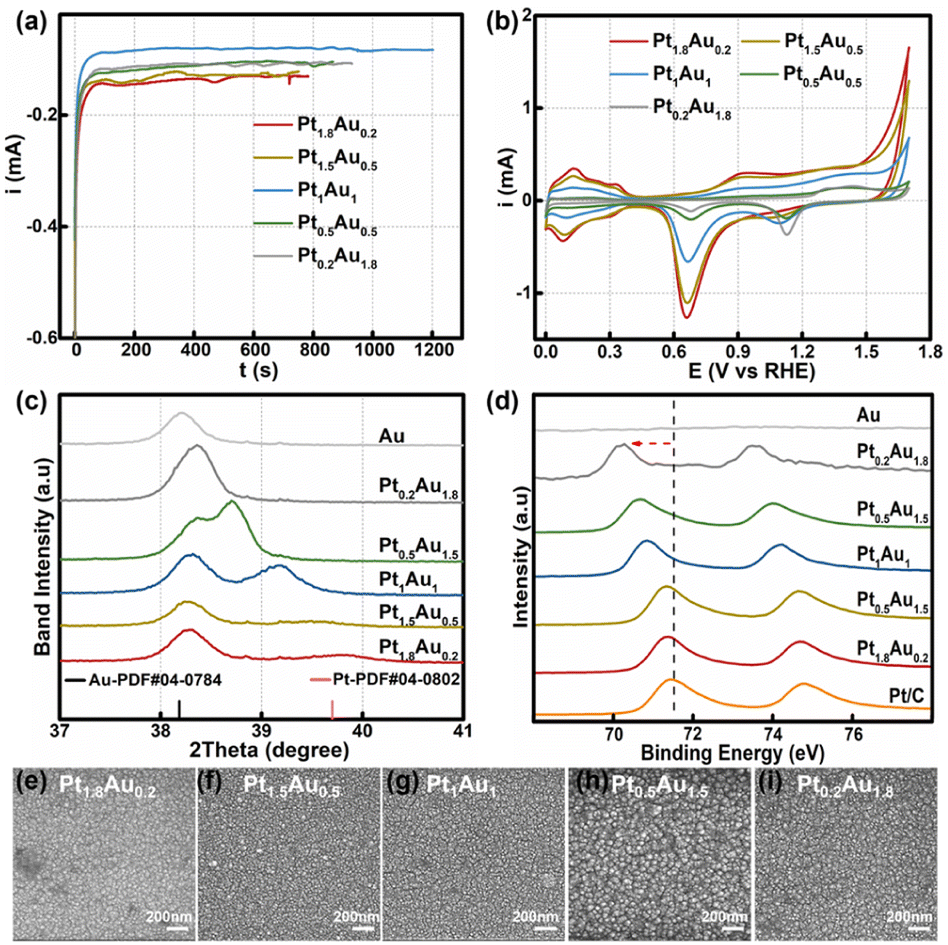 | ||
| Fig. 1 (a) Electrodeposition current–time curves for the various concentration ratios at 0.30 V. (b) Cyclic voltammetry curves of the PtAu film catalysts. (c) Magnification of the fine XRD patterns of the PtAu film model catalysts from 37° to 41°. (d) Pt XPS spectra of the PtAu films. (e)–(i) SEM images of the PtAu films. | ||
The powder X-ray diffraction (XRD) patterns of the PtAu films were shown in Fig. 1c and S2.† The magnified band details from 37.00° to 41.00° were shown in Fig. 1c to clarify the structure change. Compared with the Au(111) diffraction peak at 38.18° in the standard cards, the peak of the electroless-plated Au shifted positively to 38.20° due to lattice mismatch between Au and Si. Alloying with Pt, the diffraction peak positively shifted to 38.36° in the Pt0.2Au1.8 film. Moreover, new diffraction peaks appeared in the PtAu model films from Pt0.5Au1.5 to Pt1.8Au0.2. It gradually positive-shifted from 38.71° to 39.56° in the PtAu alloy phase with Pt ratio increasing. The Pt1.8Au0.2 film has the highest peak at 39.56°, which was close to the Pt(111) diffraction peak.17 This result indicated these PtAu model films have various surface structures. Moreover, we used Vegard's law to study the surface structures and the calculated lattice constants of the different diffraction peaks at different angles, as shown in Fig. S2.†37–39 It could clearly be seen that the lattice constants of the Pt0.2Au1.8 model film were well fitted by the linear relation of Vegard's law (red dotted line), indicating the formation of alloy phase. For the Pt0.5Au1.5 to Pt1.5Au0.5 model films, their high diffraction peaks were close to Vegard's law, also suggesting the formation of an alloy phase. Additionally, their lattice constants at the low-angle diffraction peaks showed mismatch with Vegard's law in Fig. S2d.† These results indicated that there were two alloy phases. Moreover, the lattice constants of the low-angle diffraction peak of the Pt0.5Au1.5 to Pt1.5Au0.5 model films were close to that of the Pt0.2Au1.8 model film, indicating that they may share the same PtAu phase, i.e., Au-rich PtAu alloy phase structure.
X-ray photoelectron spectroscopy (XPS) was used to analyze the surface ratios of the PtAu films, as shown in Fig. 1d and S3.† Their sub-subsurface component analyses were shown in Table S1,† and corresponded with the surface ratios calculated from the cyclic voltammetry results.27 Compared with Pt/C, the Pt binding energy peaks of PtAu film were negatively shifted, and with Pt ratio increasing, the Pt 4f binding energy of the PtAu film became close to that of Pt/C. This phenomenon also could be found in Au XPS results for the PtAu film catalysts in Fig. S3.† The shift in the XPS results is affected by the electronegativity and structure effect.40–42 As the electronegativity of Pt (2.1) is lower than that of Au (2.3), electrons should transfer from Pt to Au.43 In contrast, the atomic radius of Au is larger than Pt, and thus would generate tensile strain during the alloying of Au with Pt. This result corresponds with XRD results, i.e., the lattice constants gradually increase with Au content increasing. Significantly, the strain effect of PtAu is a long-range interaction, and would reduce the orbital overlap between the Pt atoms and increase the electron energy level of the d orbitals. Therefore, the binding energy of Pt would be negatively shifted and the d-band centre of Pt positively shifted, coinciding with the previous reported work.44,45 The morphologies of the PtAu films were studied using SEM as shown in Fig. 1e–i. It could clearly be seen that all PtAu surfaces consisted of uniform-size nanoparticles without any special shapes.
3.2 In situ ATR-SEIRAS of COR on PtAu film model catalysts
The COR curves of PtAu film model catalysts were measured in 0.1 M HClO4, as shown in Fig. 2a. There was one obvious CO oxidation peak at 0.71 V in the Pt electrode.46 However, in the Au electrode, there was no obvious oxidation peak, which may be attributed to CO being unable to stably adsorb on the Au electrode (Fig. S4†). For the PtAu model film catalysts, there were a pair of CO oxidation peaks in the COR curves, which was in agreement with previous works.47–49 These were usually assigned to the low/high potential oxidation peaks as the pre-oxidation and post-oxidation peak. A summary of the peak potentials of the pre-oxidation peaks and post-oxidation peaks were shown in Fig. 2b. Compared with those of the Pt electrode, the CO oxidation peaks of the PtAu model films gradually positively shifted. As the Pt ratio was reduced from Pt1.8Au0.2 to Pt0.5Au1.5, the pre-oxidation peak shifted positively from 0.73 V to 0.84 V; interestingly, the pre-oxidation COR peak disappeared in the Pt0.2Au1.8 film. In contrast, the post-oxidation peak was independent of the Pt content at 1.02 V. These results suggested that there were various reactive sites for CO oxidation on the PtAu model films. Additionally, these PtAu film model catalysts showed different activity towards COR: Pt1.8Au0.2 (0.95 mA@0.73 V) > Pt1.5Au0.5 (0.65 mA@0.82 V) > Pt1Au1 (0.15 mA@0.83 V) > Pt0.5Au1.5 (0.095 mA@0.83 V) > Pt0.2Au1.8 (0.10 mA@0.83 V). This result also indicated that their COR mechanisms were different. In general, the COad was oxidized by adjacent adsorbed OH* species (OH*) dissociated from H2O at free sites, i.e. the Langmuir–Hinshelwood mechanism (L–H mechanism).50,51 The emerging free sites originating from the oxidized CO sites could produce new OH* to accelerate COad oxidation. For the continuous Pt surface sites of the model film, the free Pt sites near the Pt–COad sites could provide OH* to oxidize other COad active sites. It was interesting that with Pt ratio increasing, the pre-oxidation peaks of the PtAu film catalysts were negatively shifted. Moreover, the post-oxidation peaks of these PtAu film catalysts were almost unchanged. This can be attributed to different OH generation potentials.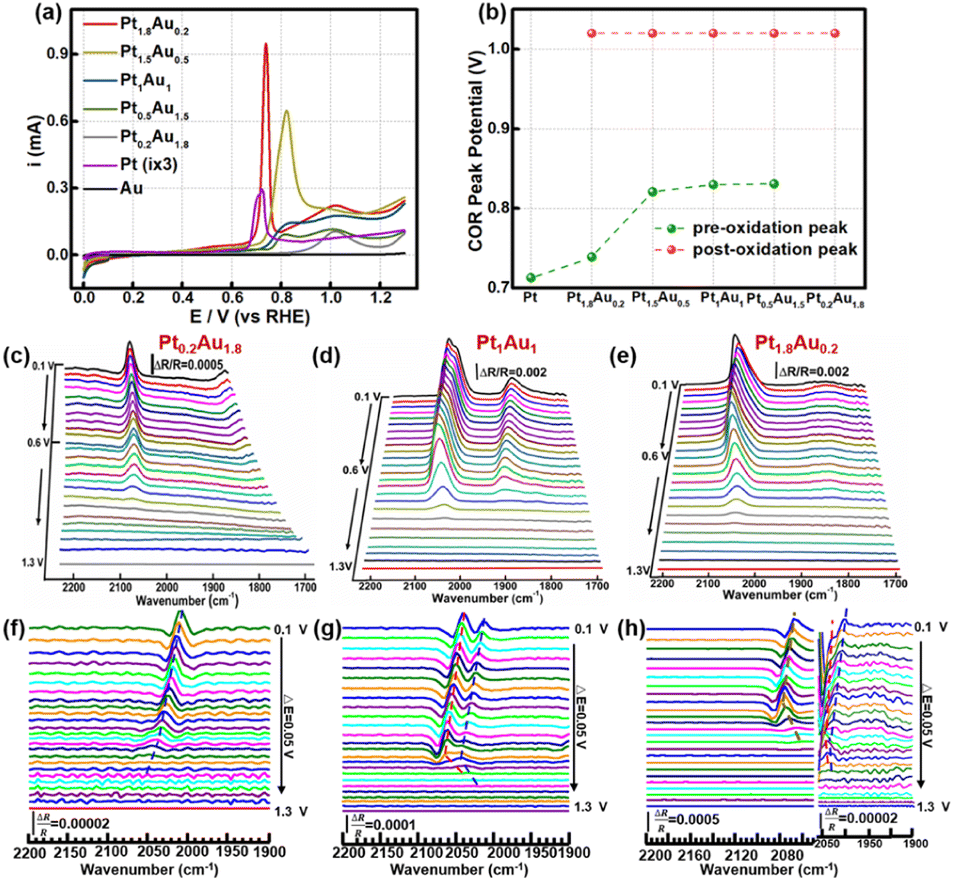 | ||
| Fig. 2 (a) CO oxidation reaction (COR) measurement of the PtAu film catalysts, Pt electrode and Au electrode in 0.1 M HClO4; the current of the Pt COR curve has been multiplied by 3 for clear display. (b) Oxidation potential of the pre-oxidation and post-oxidation peaks of (a). (c)–(e) CO oxidation infrared spectra of the PtAu model films: (c) Pt0.2Au1.8, (d) Pt1Au1, and (e) Pt1.8Au0.2. (f)–(h) Corresponding second-derivative spectra of (c)–(e) at 2200–1900 cm−1; the right part of (h) is the magnified spectra at 2050–1900 cm−1. | ||
Electrochemical in situ ATR-SEIRAS was employed to investigate the molecular behavior at the electrochemical interface and understand the structure–activity relationship of the PtAu catalyst. The infrared spectra of COR were recorded from 0.10 V to 1.3 V and the reference potential was set at 1.3 V. Pt0.2Au1.8, Pt1Au1, and Pt1.8Au0.2 were assigned as the research objects, and their spectra were shown in Fig. 2c–e. The bands at 2000–2100 cm−1 and 1900–1800 cm−1 were assigned as linear adsorbed CO (COL) and bridge-adsorbed CO (COB) on the PtAu films, respectively.52 No signal of CO2 generated by COR on the PtAu model films was detected in Fig. S5†.48,53 This was attributed to the generated CO2 rapidly diffusing to the solution phase due to the structural features of the electrolytic cell; the in situ ATR-SEIRAS method is insensitive to solution phase species.16,54,55 In order to investigate the CO oxidation on the Au catalysts, the spectra and peak intensity change of Au–CO also showed that CO was weakly adsorbed on the Au film at 2100 cm−1 and desorbed after Ar purging (Fig. S4†).32 This meant that CO was difficult to stable adsorption on Au site in the PtAu model films after Ar purged. It was interesting that with Au content increasing, the COL band red-shifted from 2063 cm−1 to 2008 cm−1. This could be ascribed to the enhancement of the electronic effect of Pt–CO by Au atoms.47 Au would affect the electronic structure of Pt, which would result in the positive shift of the d-band centre for Pt, and thus enhanced the d–π* feedback effect between Pt and CO.28 The result was conducive to weakening the bond energy of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O and coupling with OH* to oxidize CO to CO2.51 It was noteworthy that both the Pt0.2Au1.8 and Pt1.8Au0.2 catalysts presented one COL peak in their spectra; in contrast, the Pt1Au1 film showed two COL peaks. The different COL peaks indicated different CO adsorption sites on the surface of the PtAu films, which was confirmed by the COR curves and structure characterization. However, it was difficult to identify the COL peak sites of the CO bands. Therefore, it was necessary to clarify the CO peak sites and quantitative band intensity to study the surface fine configurations and activity of the PtAu catalysts.
O and coupling with OH* to oxidize CO to CO2.51 It was noteworthy that both the Pt0.2Au1.8 and Pt1.8Au0.2 catalysts presented one COL peak in their spectra; in contrast, the Pt1Au1 film showed two COL peaks. The different COL peaks indicated different CO adsorption sites on the surface of the PtAu films, which was confirmed by the COR curves and structure characterization. However, it was difficult to identify the COL peak sites of the CO bands. Therefore, it was necessary to clarify the CO peak sites and quantitative band intensity to study the surface fine configurations and activity of the PtAu catalysts.
The SDP method is convenient and effective for locating peak positions by mathematically calculating the extreme points. Hereby, SDP method was employed to identify the COL peaks on PtAu catalysts. The second-derivative spectra of these PtAu films were shown in Fig. 2f–h and S6.† In the second-derivative spectra at 0.10 V, Pt0.2Au1.8 showed one COL peak at 2005 cm−1 (Fig. 2f) and Pt1Au1 presented a pair of COL peaks at 2038 cm−1 and 2013 cm−1 (Fig. 2g), which were coincident with the original spectra. For both the Pt1.5Au0.5 and Pt0.5Au1.5 films, two COL peaks could be observed in the original spectra and second-derivative spectra (Fig. S4†). It was noteworthy that there were three peaks at 2063 cm−1, 2043 cm−1, and 2005 cm−1 for the Pt1.8Au0.2 film in the second-derivative spectrum (Fig. 2h). This suggested that there should be three COL peaks in the original unipolar spectra; however, the original spectra of Pt1.8Au0.2 only showed two peaks. This indicated that the COR mechanism should be different from the others, which suggested that the surface fine structures of these PtAu films were diverse.
To investigate the COR mechanism on these PtAu films, Gaussian fitting was employed on the ATR-SEIRAS spectra, as shown in Fig. 3 and S7.† The fitted COL peaks and a summary of the COL fitting curves of Pt0.2Au1.8, Pt1.5Au0.5, and Pt1.8Au0.2 at 0.10 V was shown in Fig. 3a–c. For the Pt0.2Au1.8 film catalyst, the surface composition was Au-rich (surface ratio of Pt: 16%), and there was only one COL peak at 2005 cm−1 (Fig. 3d) without COB. Therefore, there may be only one active site in Pt0.2Au1.8, i.e., its surface Pt atoms may be completely isolated by Au atoms (named as PtisoAu). The COad pattern on PtisoAu corresponding to its surface fine structure diagram was shown in Fig. 3d (left figure). For the Pt1Au1 film, there were two COL peaks at 2038 cm−1 and 2013 cm−1. This indicated that there were two surface configurations, in agreement with the XRD results. The low-wavenumber COL peak (2013 cm−1) was close to that of the PtisoAu site in Pt0.2Au1.8 (2005 cm−1). This result suggested that the Pt1Au1 film also possessed the PtisoAu sites. The high-wavenumber COL peak of 2038 cm−1 suggested that it may have another active site. Notably, Pt1Au1 showed a new COB peak at 1856 cm−1, which was usually attributed to the presence of continuous Pt atoms, that was, adjacent Pt active sites (named as PtadjAu site). Therefore, the Pt1Au1 film catalyst has PtisoAu sites and PtadjAu sites, and its surface fine structure corresponding with the COad diagram was shown in Fig. 3d (middle figure). Pt0.5Au1.5 and Pt1.5Au0.5 share similar active sites with Pt1Au1, as shown in Fig. S7.† For the Pt1.8Au0.2 film, three were three COL peaks at 2005 cm−1, 2043 cm−1 and 2063 cm−1. The peaks at 2005 cm−1 and 2043 cm−1 could be attributed to CO adsorbed on PtisoAu sites and PtadjAu sites, respectively. The highest-wavenumber COL peak (2063 cm−1) was close to that of CO adsorbed on pure Pt, which could be attributed to the high Pt surface ratio (92%).56,57 This indicated that it has a Pt-rich surface configuration, that was, abundant continuous Pt atoms without Au atoms (named as PtrichAu site). This corresponds with the results of the XRD pattern of the Pt1.8Au0.2 film. The COad diagram and surface fine structure diagram of Pt1.8Au0.2 were shown in Fig. 3d (right figure).
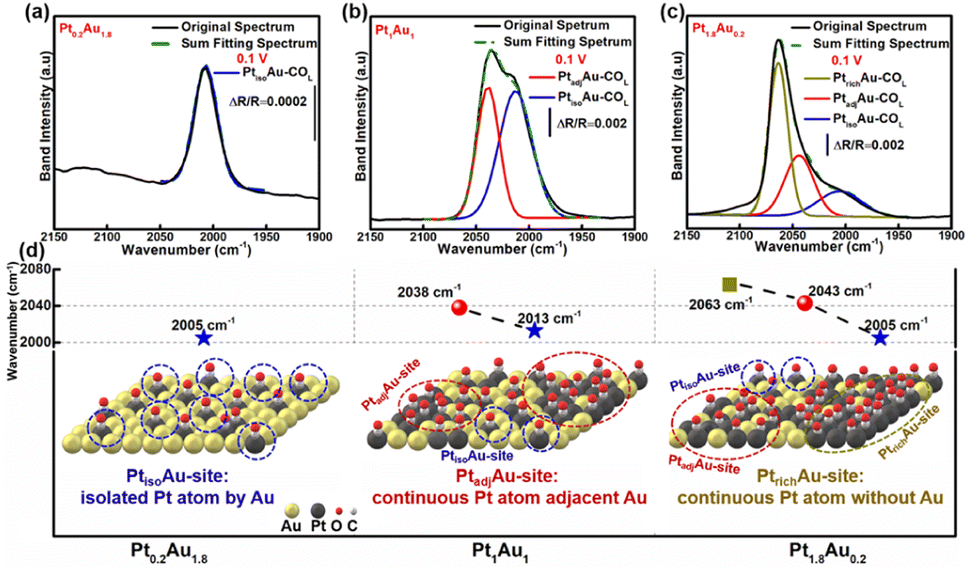 | ||
| Fig. 3 (a)–(c) Peak fitting of the COL bands of the PtAu model films at 0.1 V: (a) Pt0.2Au1.8, (b) Pt1Au1 and (c) Pt1.8Au0.2. (d) Wavenumbers of the fitting peak positions and COad diagrams of the surface fine structures of the PtAu model films: Pt0.2Au1.8 left figure, Pt1Au1 middle figure, Pt1.8Au0.2 right figure. | ||
To further verify the conformations of the PtAu sites of these PtAu films, the formic acid (HCOOH) electrooxidation reaction (FAOR) was carried out, as shown in Fig. S8.† As the FAOR is sensitive to the active site conformation, it is widely used to characterize the surface structure of Pt-based catalysts.58,59 HCOOH will go through the indirect pathway to produce CO via the dehydration reaction on contiguous Pt atom sites. For the isolated Pt sites, HCOOH is oxidized via the direct pathway without CO and its activity is higher than in the indirect pathway. There were two oxidation peaks at around 0.6 V (peak 1) and around 0.95 V (peak 2) in the forward scan curve in Fig. S8a.† This were attributed to the oxidation of HCOOH to CO/CO2 and oxidation of CO to CO2, corresponding to the direct and indirect oxidation pathways, respectively.60–62 Therefore, the values of peak 1 and peak 2 could be used to evaluate the surface structure of the PtAu films. The current densities of peak 1 and peak 2 are summarized in Fig. S8b.† It can be clearly seen that the Pt0.2Au1.8 model film only presents one peak at around 0.6 V, indicating that HCOOH goes through the direct pathway. This result demonstrated the PtisoAu site of Pt0.2Au1.8 model film. For the Pt1Au1 model films, the current density of peak 1 was 2 mA cm−2 and that of peak 2 was 2.3 mA cm−2, respectively. The increased activity of peak 2 indicated that it possessed the indirect pathway and direct pathway towards FAOR. This suggested continuous Pt sites, i.e., PtadjAu sites, for the Pt1Au1 model films. When the Pt content was increased to give the Pt1.8Au0.2 model films, the current density of peak 2 (1.5 mA cm−2) was higher than peak 1 (1.2 mA cm−2). This was beneficial to generate CO intermediate species via the indirect pathway due to the greater amount of continuous Pt atom sites, corresponding to the PtrichAu sites in the structure diagram of the Pt1.8Au0.2 model films.
3.3 Surface fine structure and CO oxidation activity
To further study the relationship between the surface-active sites of the PtAu films and the CO oxidation activity, the SDP method located peak position and Gaussian fittings of the various potential COL peaks were shown in Fig. 4 and S9–S11.† The fitted spectra at a potential interval of 0.10 V and the changes in the COL integrated intensities were shown in Fig. S9–S11.† As the onset potential of CO was at 0.1 V, the change in the CO integrated intensity and COR curves of Pt0.2Au1.8, Pt1Au1, and Pt1.8Au0.2 were summarized in Fig. 4a–c to reveal the relationship between the surface-active Pt sites and COR. For Fig. 4a, there were only PtisoAu-COL sites in the Pt0.2Au1.8 model film and the corresponding post-oxidation peak in the COR curve. The CO intensity of PtisoAu–COL decreased slightly at 0.20–0.85 V, which was in agreement with the previous work.29 As shown in Fig. 4d–f, the Stark tuning rate of PtisoAu–COL was near 55 cm−1 V−1 due to effect of Au in the film. It was more susceptible to potential variation than PtrichAu–CO. Based on the site structure, the PtisoAu–COL may induce conformational changes or convert intermediate species. In Fig. 4b, it was interesting that when the intensity of PtadjAu–COL decreased at 0.70 V, an oxidation current of the pre-oxidation peak appeared in the COR curve. At 0.85 V, the PtadjAu–COL was completely oxidized and the pre-oxidation disappeared. This could be attributed to PtadjAu–COL as the reactive species of the pre-oxidation peak. Furthermore, when the voltage exceeded 0.85 V, the intensity of PtisoAu–COL gradually decreased, corresponding to the oxidation of the post-oxidation peak. This indicated that PtisoAu–COL was the main reactive species of post-oxidation peak. The only CO peak and post-oxidation peak in Fig. 4a also confirmed the result. As depicted in Fig. 4c, similar to the Pt1Au1 film, PtisoAu–COL was also the main CO peak for the post-oxidation peak when the voltage surpassed 0.85 V. However, in the potential range of 0.65–0.80 V, the PtadjAu–COL and PtrichAu–COL were almost completely oxidized. This meant the PtrichAu–COL was also the reactive species of the post-oxidation peak. Due to the higher CO oxidation ability of the PtrichAu–COL, the pre-oxidation peak of Pt1.8Au0.2 shifted negatively 0.10 V to 0.73 V, compared with the other samples. The result also verified the site structure and mechanism speculation of the COR curves. The PtrichAu site was oxidized in the early potential range as free sites and dissociated H2O to OH*. The new OH* of the PtrichAu site contributed to oxidation of the adjacent COad of the PtadjAu sites. However, for the Pt atom isolated by Au of the PtisoAu–COL sites, the steric hindrance of Au may hinder OH* transfer to adjacent sites to oxidize PtisoAu–COL. The oxidation mechanism of PtisoAu–COL may be attributed to the Au product OH* at high potential to oxidize COad. Compared with the Pt1Au1 and Pt1.8Au0.2 model films in Fig. 4b–c, the intensity of PtisoAu–COL, PtadjAu–COL and PtrichAu–COL decreased to 0.65 V, 0.70 V and 0.85 V, respectively. This meant that the CO oxidation reactivity of the Pt surface could be expressed in the order PtrichAu–COL > PtadjAu–COL > PtisoAu–COL.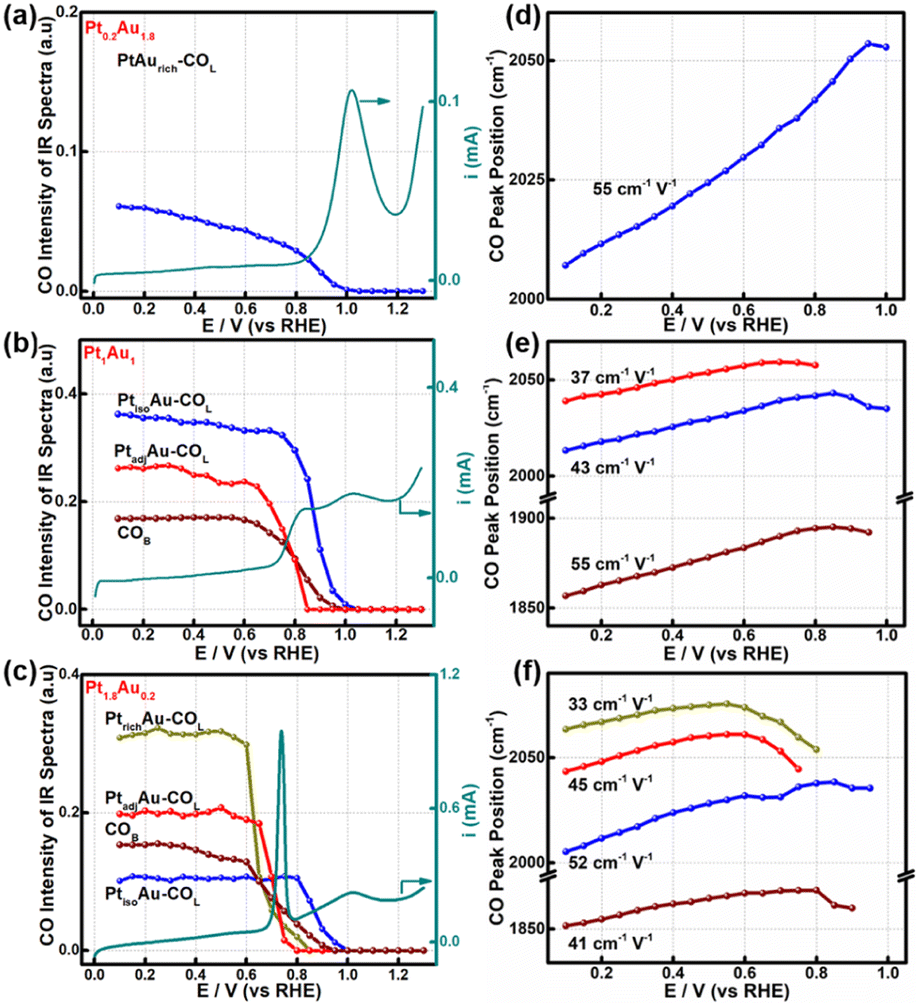 | ||
| Fig. 4 Integrated peak intensities and peak positions of the various CO species in the COR process plotted against electrode potential change. (a) and (d) Pt1.8Au0.2, (b) and (e) Pt1Au1, (c) and (f) Pt0.2Au1.8. PtrichAu–COL (dark yellow line), PtadjAu–COL (red line), PtisoAu–COL (blue line), COB (wine-colored line). The cyan solid lines are the COR curves of the PtAu samples in (a), (c) and (e). | ||
The Stark tuning rates of PtrichAu–COL, PtadjAu–COL and PtisoAu–COL in the different PtAu films were shown in Fig. 4d–f. In Fig. 4f, before CO oxidation, the Stark tuning rates of PtrichAu–COL, PtadjAu–COL and PtisoAu–COL gradually increased from 33 cm−1 V−1 to 45 cm−1 V−1 and 52 cm−1 V−1. The change in the Stark tuning rates could be attributed to the effect of Au. Compared to the PtrichAu sites, the Pt atoms were decorated with Au atoms in the PtadjAu sites and were isolated by Au atoms in the PtisoAu sites. The heightened electronic effect results in an increase in the Stark tuning rate. Additionally, the various COL were changeable in different surface-active and model films. This may be affected by the effect of COad–COad repulsions in the different active sites.29
3.4 In situ ATR-SEIRAS for studying the fine structure of the PtAu nanoparticles
To investigate the generalizability of the COR mechanism on the multiple active sites of the PtAu catalyst, we also prepared PtAu nanoparticles (NPs) using the NaBH4 reduction method reported in previous work.17 The structure characterization and electrochemistry tests of the PtAu NPs are shown in Fig. 5 and S12.† The XRD patterns of the PtAu NPs showed typical face-centered cubic (fcc) features in Fig. S12a,† without phase separation. The PtAu NPs had a uniform particle size and were homogeneously dispersed on the carbon substrate (Fig. S12b†). Atomic-resolution high-angle annular dark field scanning transmission electron microscopy (HAADF–STEM) images of the PtAu NPs (Fig. 5a) showed that the lattice spacing was 0.23 nm, which corresponded to PtAu(111). In addition, the elemental mapping in Fig. 5b confirmed that Pt and Au were homogeneously distributed in the PtAu NPs. As the PtAu model films present an island structure without special structure (Fig. 1), the uniform spherical structure of the PtAu nanoparticles ensures the reasonableness of extending the in situ ATR-SEIRAS CO-probe method.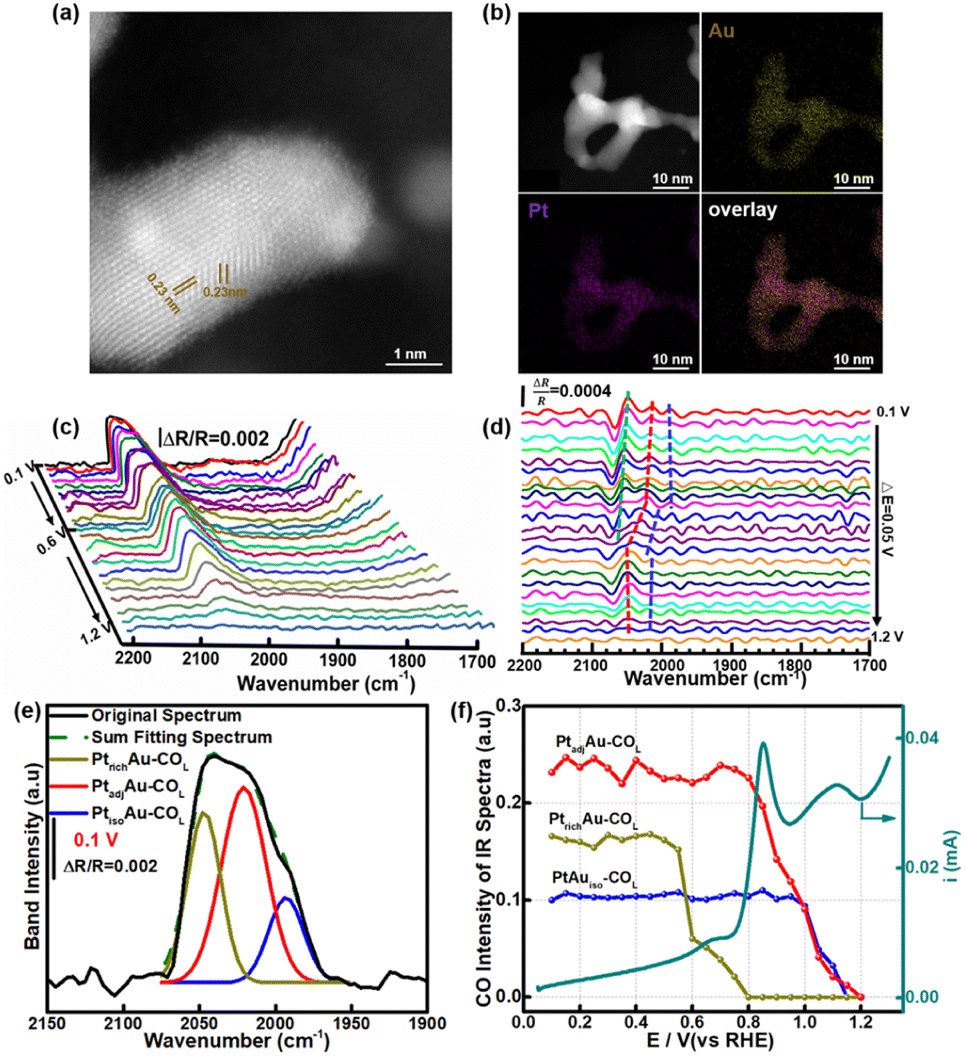 | ||
| Fig. 5 (a) HAADF-STEM image of the PtAu NPs. (b) Elemental mapping of PtAu NPs. (c) CO oxidation infrared spectra and the reference potential at 1.2 V. (d) Second-derivative spectra of the COR spectra; the dotted lines shows the peak position changes. (e) Fitting peaks of the COL band at 0.1 V. (f) Integrated peak intensities of the various CO species in the COR and COR curve. | ||
In the cyclic voltammetry tests (Fig. S12c†), by integrating the hydrogen underpotential deposition region charge and Au redox peak charge, the surface composition of the PtAu NPs was found to be Au rich (Pt:Au = 0.3:0.7). Surprisingly, there were also three obvious CO oxidation peaks in the PtAu NPs (Fig. S12d†). Their onset oxidation potentials were 0.60 V, 0.75 V, and 0.90 V, respectively. The result indicated that the PtAu alloy was heterogeneous on the surface, although there was not phase separation in the XRD pattern and TEM characterization. The similar appearance maybe exist in other binary and ternary Pt-based alloy nanocatalyst. In particular, the similar CO oxidation feature peaks in the COR curves in the PtAu model films and PtAu nanoparticles enabled development of this in situ ATR-SEIRAS CO-probe method. The CO probe molecule can not only characterize the d-band center of the surface, but also investigate the uniformity of surface for Pt-based catalysts. Using in situ ATR-SEIRAS, the surface fine structure of the PtAu NPs was analyzed in depth. The COR spectra of the PtAu NPs were recorded from 0.10 V to 1.2 V, and the reference potential was set at 1.2 V (Fig. 5c). The results present one COL band with multiple peaks around 2050 cm−1, without COB. The ATR-SEIRAS could detect detailed structural information due to the higher sensitivity for surface-adsorbed species, compared with only one COL peak in the in situ FTIR method. Moreover, second derivative spectra and Gaussian fitting were used to study the surface fine structure. In Fig. 5d, at 0.10 V, three obvious extreme points were observed at 2047 cm−1, 2021 cm−1 and 1993 cm−1. Similar to the PtAu model film, these could be attributed to three CO active sites, and their wavenumbers were close to those of PtrichAu–COL, PtadjAu–COL, and PtisoAu–COL, respectively.
The fitting spectra at 0.10 V and other potentials were shown in Fig. 5e and S13.† Comparing the integrated intensities of the three active sites, the CO onset oxidation potentials were 0.60 V, 0.75 V, and 0.95 V, respectively (Fig. 5f). The electrochemistry characteristics of the three active sites were also similar to PtrichAu–COL, PtadjAu–COL, and PtisoAu–COL. Therefore, based on the similar wavenumbers and CO oxidation ability, the three active sites of the PtAu NPs at 2047 cm−1, 2021 cm−1, and 1993 cm−1 could be assigned as PtrichAu–COL, PtadjAu–COL and PtisoAu–COL, respectively. Moreover, the fitting spectra at a potential interval of 0.10 V and the changes in the COL integrated intensity were shown in Fig. S11† and 5f. Comparing the COR curve, for the first oxidation peak at 0.60–0.80 V, the intensity of PtrichAu–COL gradually decreased. This meant that PtadjAu–COL was the main active species for the first oxidation peaks. When the voltage exceeded 0.80 V, PtadjAu–COL was gradually oxidized, corresponding to the second oxidation peak. At 1.2 V, PtadjAu–COL was completely oxidized. In addition, the intensity of PtisoAu–COL gradually decreased at 1.0 V, close to the third oxidation peak. The result indicated that the PtadjAu–COL was attributed as the reactive species for the second oxidation peak and the third oxidation peak, and PtisoAu–COL was the main active species for the third oxidation.
4. Conclusions
In summary, we successfully utilized the in situ electrochemical ATR-SEIRAS and CO-probe method to classify the surface fine structure of PtAu model films and nanocatalysts. The electrodeposition method was used to construct model PtAu films with PtisoAu sites, PtadjAu sites and PtrichAu sites. Additionally, the COR mechanism on these PtAu surface sites was revealed using in situ ATR-SEIRAS. By utilizing the SDP method located peak positions and Gaussian fitting to separate the complex CO band, three COL peaks in the COR spectra were disclosed. More importantly, correlating the changes in the integrated intensities of COL with the potentials, the CO oxidation reactivity could be placed in a sequence: PtrichAu–COL > PtadjAu–COL > PtisoAu–COL. The CO active sites could be assigned to the respective oxidation peaks in the COR curve. Finally, the surface fine configurations of the Au-rich PtAu nanoparticles were also determined.Author contributions
Guang Li: conceptualization, methodology, investigation, validation, data acquisition & analysis, visualization, software, writing – original draft. Zheng-Chao An: data curation, writing – original draft. Jian Yang: writing – review & editing. Jin-Hong Zheng: methodology, investigation, data acquisition & analysis. Li-Fei Ji: methodology, investigation. Jun-Ming Zhang: methodology, investigation. Jin-Yu Ye: methodology, investigation, writing – review & editing. Bin-Wei Zhang: methodology, investigation, writing – review & editing. Yan-Xia Jiang: conceptualization, methodology, funding acquisition, supervision, writing – review & editing. Shi-Gang Sun: conceptualization, methodology, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was granted by the National Key Research and Development Program of China (2017YFA0206500) and the National Natural Science Foundation of China (22172134, 22288102 and 22279011).Notes and references
- Ş. Neaţu, F. Neaţu, I. M. Chirica, I. Borbáth, E. Tálas, A. Tompos, S. Somacescu, P. Osiceanu, M. A. Folgado, A. M. Chaparro and M. Florea, J. Mater. Chem. A, 2021, 9, 17065–17128 RSC.
- X. Ren, Y. Wang, A. Liu, Z. Zhang, Q. Lv and B. Liu, J. Mater. Chem. A, 2020, 8, 24284–24306 RSC.
- S. Ponnada, M. S. Kiai, D. B. Gorle, A. Nowduri and R. K. Sharma, Energy Fuels, 2021, 35, 15265–15284 CrossRef CAS.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Chem. Rev., 2014, 114, 12397–12429 CrossRef CAS.
- J. Zhang, L. Shen, Y. Jiang and S. Sun, Nanoscale, 2020, 12, 19557–19581 RSC.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS.
- W. Xu, Z. Wu and S. Tao, J. Mater. Chem. A, 2016, 4, 16272–16287 RSC.
- C. Lyu, J. Cheng, Y. Yang, J. Wu, K. Wu, Y. Yang, W.-M. Lau, N. Wang, D. Pang and J. Zheng, J. Mater. Chem. A, 2023, 11, 4319–4333 RSC.
- A. R. Poerwoprajitno, L. Gloag, J. Watt, S. Cheong, X. Tan, H. Lei, H. A. Tahini, A. Henson, B. Subhash, N. M. Bedford, B. K. Miller, P. B. O'Mara, T. M. Benedetti, D. L. Huber, W. Zhang, S. C. Smith, J. J. Gooding, W. Schuhmann and R. D. Tilley, Nat. Catal., 2022, 5, 231–237 CrossRef CAS.
- P. N. Duchesne, Z. Y. Li, C. P. Deming, V. Fung, X. Zhao, J. Yuan, T. Regier, A. Aldalbahi, Z. Almarhoon, S. Chen, D.-e. Jiang, N. Zheng and P. Zhang, Nat. Mater., 2018, 17, 1033–1039 CrossRef CAS PubMed.
- Z. Wang, Y. Yang, X. Wang, Z. Lu, C. Guo, Y. Shi, H. Tan, L. Shen, S. Cao and C. Yan, J. Mater. Chem. A, 2022, 10, 12141–12149 RSC.
- Q. Wang, Q. Zhao, Y. Su, G. Zhang, G. Xu, Y. Li, B. Liu, D. Zheng and J. Zhang, J. Mater. Chem. A, 2016, 4, 12296–12307 RSC.
- T. Peng, T. Zhuang, Y. Yan, J. Qian, G. R. Dick, J. Behaghel de Bueren, S.-F. Hung, Y. Zhang, Z. Wang, J. Wicks, F. P. Garcia de Arquer, J. Abed, N. Wang, A. Sedighian Rasouli, G. Lee, M. Wang, D. He, Z. Wang, Z. Liang, L. Song, X. Wang, B. Chen, A. Ozden, Y. Lum, W. R. Leow, M. Luo, D. M. Meira, A. H. Ip, J. S. Luterbacher, W. Zhao and E. H. Sargent, J. Am. Chem. Soc., 2021, 143, 17226–17235 CrossRef CAS.
- I. I. Sadykov, M. Zabilskiy, A. H. Clark, F. Krumeich, V. Sushkevich, J. A. van Bokhoven, M. Nachtegaal and O. V. Safonova, ACS Catal., 2021, 11, 11793–11805 CrossRef CAS.
- S.-H. Yin, S.-L. Yang, G. Li, G. Li, B.-W. Zhang, C.-T. Wang, M.-S. Chen, H.-G. Liao, J. Yang, Y.-X. Jiang and S.-G. Sun, Energy Environ. Sci., 2022, 15, 3033–3040 RSC.
- J.-Y. Ye, Y.-X. Jiang, T. Sheng and S.-G. Sun, Nano Energy, 2016, 29, 414–427 CrossRef CAS.
- J.-H. Zheng, J. Zhang, G. Li, J.-M. Zhang, B.-W. Zhang, Y.-X. Jiang and S.-G. Sun, Mater. Today Energy, 2022, 27, 101028–101034 CrossRef CAS.
- S. Xie, W. Tan, C. Wang, H. Arandiyan, M. Garbrecht, L. Ma, S. N. Ehrlich, P. Xu, Y. Li, Y. Zhang, S. Collier, J. Deng and F. Liu, J. Catal., 2022, 405, 236–248 CrossRef CAS.
- Y.-C. Feng, X. Wang, Y.-Q. Wang, H.-J. Yan and D. Wang, J. Electrochem., 2022, 28, 2108531–2108543 Search PubMed.
- J. Li, H.-M. Yin, X.-B. Li, E. Okunishi, Y.-L. Shen, J. He, Z.-K. Tang, W.-X. Wang, E. Yücelen, C. Li, Y. Gong, L. Gu, S. Miao, L.-M. Liu, J. Luo and Y. Ding, Nat. Energy, 2017, 2, 17111–17119 CrossRef CAS.
- M. Gatalo, P. Jovanovič, G. Polymeros, J.-P. Grote, A. Pavlišič, F. Ruiz- Zepeda, V. S. Šelih, M. Šala, S. Hočevar, M. Bele, K. J. J. Mayrhofer, N. Hodnik and M. Gaberšček, ACS Catal., 2016, 6, 1630–1634 CrossRef CAS.
- A. Lu, Z.-P. Wu, B. Chen, D.-L. Peng, S. Yan, S. Shan, Z. Skeete, F. Chang, Y. Chen, H. Zheng, D. Zeng, L. Yang, A. Sharma, J. Luo, L. Wang, V. Petkov and C.-J. Zhong, J. Mater. Chem. A, 2018, 6, 5143–5155 RSC.
- J.-N. Zheng, S.-S. Li, X. Ma, F.-Y. Chen, A.-J. Wang, J.-R. Chen and J.-J. Feng, J. Mater. Chem. A, 2014, 2, 8386–8395 RSC.
- P. P. Lopes, D. Li, H. Lv, C. Wang, D. Tripkovic, Y. Zhu, R. Schimmenti, H. Daimon, Y. Kang, J. Snyder, N. Becknell, K. L. More, D. Strmcnik, N. M. Markovic, M. Mavrikakis and V. R. Stamenkovic, Nat. Mater., 2020, 19, 1207–1214 CrossRef CAS.
- C. Engelbrekt, N. Šešelj, R. Poreddy, A. Riisager, J. Ulstrup and J. Zhang, J. Mater. Chem. A, 2016, 4, 3278–3286 RSC.
- H.-X. Liu, N. Tian, M. P. Brandon, J. Pei, Z.-C. Huangfu, C. Zhan, Z.-Y. Zhou, C. Hardacre, W.-F. Lin and S.-G. Sun, Phys. Chem. Chem. Phys., 2012, 14, 16415–16423 RSC.
- J.-H. Zheng, G. Li, J.-M. Zhang, N. Cheng, L.-F. Ji, J. Yang, J. Zhang, B.-W. Zhang, Y.-X. Jiang and S.-G. Sun, Sci. China: Chem., 2023, 66, 279–288 CrossRef CAS.
- J. Zhang, X. Qu, Y. Han, L. Shen, S. Yin, G. Li, Y. Jiang and S. Sun, Appl. Catal., B, 2020, 263, 118345–118353 CrossRef CAS.
- D.-J. Chen, A. M. Hofstead-Duffy, I.-S. Park, D. O. Atienza, C. Susut, S.-G. Sun and Y. J. Tong, J. Phys. Chem. C, 2011, 115, 8735–8743 CrossRef CAS.
- D.-J. Chen and Y. J. Tong, Angew. Chem., Int. Ed., 2015, 54, 9394–9398 CrossRef CAS.
- E. Perez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. Koper, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef CAS PubMed.
- G. Cabello, R. A. Davoglio, J. F. Marco and A. Cuesta, J. Electroanal. Chem., 2020, 870, 114233–114237 CrossRef CAS.
- Y.-J. Zhang, Z.-F. Su, J.-F. Li and J. Lipkowski, J. Phys. Chem. C, 2020, 124, 13240–13248 CrossRef CAS.
- S. Zhu, X. Qin, Y. Yao and M. Shao, J. Am. Chem. Soc., 2020, 142, 8748–8754 CrossRef PubMed.
- S.-J. Huo, X.-K. Xue, Y.-G. Yan, Q.-X. Li, M. Ma, W.-B. Cai, Q.-J. Xu and M. Osawa, J. Phys. Chem. B, 2006, 110, 4162–4169 CrossRef CAS.
- J. Suntivich, Z. Xu, C. E. Carlton, J. Kim, B. Han, S. W. Lee, N. Bonnet, N. Marzari, L. F. Allard, H. A. Gasteiger, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef CAS PubMed.
- A. Habrioux, W. Vogel, M. Guinel, L. Guetaz, K. Servat, B. Kokoh and N. Alonso-Vante, Phys. Chem. Chem. Phys., 2009, 11, 3573–3579 RSC.
- K. Bhunia, M. Chandra, S. Khilari and D. Pradhan, ACS Appl. Mater. Interfaces, 2019, 11, 478–488 CrossRef CAS.
- W. Vogel, P. Britz, H. Bönnemann, J. Rothe and J. Hormes, J. Phys. Chem. B, 1997, 101, 11029–11036 CrossRef CAS.
- E. E. Benson, E. M. Miller, S. U. Nanayakkara, D. Svedruzic, S. Ferrere, N. R. Neale, J. van de Lagemaat and B. A. Gregg, Chem. Mater., 2017, 29, 2173–2179 CrossRef CAS.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2006, 110, 23489–23496 CrossRef CAS PubMed.
- S. C. Cook, J. D. Padmos and P. Zhang, J. Chem. Phys., 2008, 128, 154705–154715 CrossRef CAS.
- Z. J. Tan, V. Somjit, C. Toparli, B. Yildiz and N. Fang, Phys. Rev. Mater., 2022, 6, 105002–105013 CrossRef CAS.
- M. J. Prieto, E. A. Carbonio, S. Fatayer, R. Landers and A. d. Siervo, Phys. Chem. Chem. Phys., 2014, 16, 13329–13339 RSC.
- S. Yang, H. Noguchi and K. Uosaki, J. Phys. Chem. C, 2018, 122, 8191–8201 CrossRef CAS.
- G. Samjeské, K.-i. Komatsu and M. Osawa, J. Phys. Chem. C, 2009, 113, 10222–10228 CrossRef.
- W. Zhou, M. Li, L. Zhang and S. H. Chan, Electrochim. Acta, 2014, 123, 233–239 CrossRef CAS.
- D. Minudri, A. Y. Tesio, F. Fungo, R. E. Palacios, P. S. Cappellari, E. Pastor and G. A. Planes, J. Power Sources, 2021, 483, 229189–229198 CrossRef CAS.
- G. Cabello, R. A. Davoglio, F. W. Hartl, J. F. Marco, E. C. Pereira, S. R. Biaggio, H. Varela and A. Cuesta, Electrochim. Acta, 2017, 224, 56–63 CrossRef CAS.
- M. H. Mathew, A. Mattox, A. Johnson and S. Zou, J. Electrochem., 2012, 18, 521–536 Search PubMed.
- M. Su, J. C. Dong, J. B. Le, Y. Zhao, W. M. Yang, Z. L. Yang, G. Attard, G. K. Liu, J. Cheng, Y. M. Wei, Z. Q. Tian and J. F. Li, Angew. Chem., Int. Ed., 2020, 59, 23554–23558 CrossRef CAS.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS.
- G. A. Camara, J. F. Gomes, K. Bergamaski, E. Teixeira-Neto and F. C. Nart, J. Electroanal. Chem., 2008, 617, 171–178 CrossRef CAS.
- W. Chen, Y. Jiang and S. Sun, Chin. Sci. Bull., 2003, 48, 135–139 CrossRef CAS.
- Y.-G. Yan, Q.-X. Li, S.-J. Huo, M. Ma, W.-B. Cai and M. Osawa, J. Phys. Chem. B, 2005, 109, 7900–7906 CrossRef CAS PubMed.
- L. Tian, J.-T. Li, J.-Y. Ye, C.-H. Zhen and S.-G. Sun, J. Electroanal. Chem., 2011, 662, 137–142 CrossRef CAS.
- Y. X. Chen, S. Ye, M. Heinen, Z. Jusys, M. Osawa and R. J. Behm, J. Phys. Chem. B, 2006, 110, 9534–9544 CrossRef CAS PubMed.
- B.-W. Zhang, Z.-C. Zhang, H.-G. Liao, Y. Gong, L. Gu, X.-M. Qu, L.-X. You, S. Liu, L. Huang, X.-C. Tian, R. Huang, F.-C. Zhu, T. Liu, Y.-X. Jiang, Z.-Y. Zhou and S.-G. Sun, Nano Energy, 2016, 19, 198–209 CrossRef CAS.
- Y.-J. Deng, N. Tian, Z.-Y. Zhou, R. Huang, Z.-L. Liu, J. Xiao and S.-G. Sun, Chem. Sci., 2012, 3, 1157–1161 RSC.
- A. Ferre-Vilaplana, J. V. Perales-Rondón, C. Buso-Rogero, J. M. Feliu and E. Herrero, J. Mater. Chem. A, 2017, 5, 21773–21784 RSC.
- M. Tang, W. Chen, S. Luo, X. Wu, X. Fan, Y. Liao, X. Song, Y. Cheng, L. Li, L. Tan, Y. Liu and Z. Quan, J. Mater. Chem. A, 2021, 9, 9602–9608 RSC.
- F. Li, Y. Ding, X. Xiao, S. Yin, M. Hu, S. Li and Y. Chen, J. Mater. Chem. A, 2018, 6, 17164–17170 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The characterization and cyclic voltammetry of electroless Au films. Additional in situ ATR-SEIRAS spectra, second derivative spectra and Gaussian fitting peaks of COR on Pt1.5Au0.5 and Pt0.5Au1.5 surfaces. See DOI: https://doi.org/10.1039/d3ta01668d |
| This journal is © The Royal Society of Chemistry 2023 |