
Deciphering the shape selective conformational equilibrium of E- and Z-locked azobenzene–tetraethylammonium ion in regulating photo-switchable K+-ion channel blocking†
Rinsha
Cholasseri
 a and
Susmita
De
*b
a and
Susmita
De
*b
aTheoretical and Computational Chemistry Laboratory, Department of Chemistry, National Institute of Technology Calicut, Kozhikode, Kerala 673 601, India
bDepartment of Chemistry, University of Calicut, Calicut University P. O, Malappuram, Kerala 673 635, India. E-mail: susmita@uoc.ac.in
First published on 21st June 2024
Abstract
The search for photo-switchable optopharmacological agents that can block ion channels has been a prevalent area owing to its prime advantages of reversibility and specificity over the traditional blockers. However, the quest for a higher blocking ability shown by a less stable photo-isomer to perfectly suit the requirement of the optopharmacological agents is still ongoing. To date, only a marginal improvement in terms of blocking ability is observed by the less stable E-isomer of para-substituted locked azobenzene with TEA (LAB–TEA) for the K+-ion channel. Thus, rationalization of the limitation for achieving high activity by the E-isomer is rather essential to aid the improvement of the efficiency of photoswitchable blocker drugs. Herein, we report a molecular-level analysis on the mechanism of blocking by E- and Z-LAB–TEA with the bacterial KcsA K+-ion channel using Molecular Dynamics (MD) simulation and Quantum Mechanical (QM) calculations. The positively charged TEA fragment engages in stronger electrostatic interactions, while the neutral LAB fragment engages in weaker dispersive interactions. The binding free energy calculated by Molecular Mechanics Poisson–Boltzmann Surface Area (MMPBSA) for E-LAB–TEA (−22.3 kcal mol−1) shows less thermodynamic preference for binding with K+-ion channels than Z-LAB–TEA (−21.6 kcal mol−1) corroborating the experimental observation. The correlation between the structure and the binding ability of E- and Z-isomers of LAB–TEA indicates that the channel gate is narrow and acts as a bottleneck for the entry of the binder molecule inside the large cavity. Upon irradiation, the Z-isomer converts into a less stable but long and planar E-isomer (ΔE of photoisomerism = 7.0 kcal mol−1, at SA2-CASPT2(6,4)/6-31+G(d)//CASSCF(6,4)/6-31+G(d)), which is structurally more suitable to fit into the narrow channel gate rather than the curved and non-planar Z-LAB–TEA. Thus, a reduction in the ionic current is observed owing to the preferential entry and subsequent blocking by E-LAB–TEA. Discontinuing the irradiation leads to conversion to the Z-isomer, the curved nature of which hinders its spontaneous release outside the cavity, thereby contributing only a small increase in the ionic current.
Introduction
Azobenzene (AB) derivatives (Scheme 1) are the most common molecular photoswitches in photo-pharmacology,1–7 due to their notable difference in the size, geometry, and dipole moment between the thermodynamically stable linear trans (E) form and the less stable bent cis (Z) form.8–11 The photo-conversion can be achieved via ultraviolet (UV)/visible (VIS) light (Scheme 1) with low photochemical fatigue.4,12,13 While UV activated azobenzene derivatives are very popular as molecular photoswitches,14 their use is limited in biological systems,15–20 where exposure to UV light can cause severe damage15 to living cells. Furthermore, most reports2,4,6,21–27 indicate that the most stable linear trans (E) form is biologically active, whereas the less stable cis (Z) form is biologically inactive. Therefore, the direction of photoswitching activity (trans → cis) is counterintuitive from the pharmacological perspective. A possible solution has been suggested by introducing a –CH2–CH2– bridge between the ortho positions of the two aromatic rings12,28 (Locked AB or LAB, Scheme 1), which has not only changed the relative thermodynamic stability of the two isomers but also caused red-shift29–33 to the wavelength required for the photoswitching. | ||
| Scheme 1 Schematic representation of cis(Z)/trans(E) photoisomerization of (a) azobenzene (AB)34 and (b) locked azobenzene (LAB)13 and their relative energies with respect to the most stable isomer calculated at SA2-CASPT2(14,12)/6-31G(d)//CASSCF(14,12)/6-31G(d) for AB35 and at MS-SA2-CASPT2(6,4)/6-31G(d)//SA2-CASSCF(6,4)/6-31G(d) for LAB36 are given. | ||
Recently, there has been significant interest in photoswitchable channel blockers, exemplified by the azobenzene-containing quaternary ammonium compounds DENAQ (diethylamine–azobenzene–quaternary ammonium) and DMNAQ (dimethylamine–azobenzene–quaternary ammonium),37 which exhibit the ability to block or modulate the function of NMDA (N-methyl-D-aspartate) receptors in the brain (anti-NMDA activity). Importantly, the efficacy of these compounds and their responsiveness to illumination are strongly influenced by their structural characteristics. Even minor modifications in structure can lead to significant alterations in activity and sensitivity to light. Furthermore, the development of photochromic ligands, such as azobupivacaine 2 (AB2), has opened up new possibilities for light-mediated controlled modulation of cardiac electrophysiology.38 By reversibly blocking both voltage-gated Na+ and K+ channels, AB2 enables modulation of the ventricular effective refractory period and conduction velocity. Hence, the increasing demand for photoswitchable channel blockers emphasizes their importance in practical applications, driven by their ability to offer precise control over cellular function with spatiotemporal specificity. One of the vital drug targets for AB coupled quaternary ammonium ions (QAs) is potassium ion channels (Scheme 2), and they can be used as photo-switchable ion channel blocker drugs.39–41 The resulting para-substituted AB-QAs are reported to be excellent blockers, which reduces the prolonged blocking and adverse side effects42 of traditional QA blockers by improving the reversibility and specificity. However, AB-QA is not very effective in terms of its operating wavelength and thermodynamic stability (Scheme 1).15–20 In 2019, Trads et al.43 introduced the para-substituted locked AB-QAs as a significant improvement over para-substituted AB-QAs, where the operating wavelength was red-shifted and reversal of thermodynamic stability was attained. Their report highlighted the “pharmacological sign-inversion” concept with CAL (Cyclic Azobenzene version of Lidocaine, a locked AB-QA)43 and CLOGO (cyclic azobenzene light operated GIRK channel opener, a LAB derivative),43 which are a photoswitchable blocker and opener for voltage-gated potassium ion channels and G protein-coupled inwardly rectifying potassium (GIRK) channels, respectively. This expanded the plausible inclusion of diazocine (LAB) into photo-pharmacology to convert the dark-active molecules into dark-inactive molecules. The first study of the para-substituted LAB-tetraethylammonium ion (LAB–TEA) in living cells for controlling the signalling was reported by Thapaliya et al.13 It revealed the biological application of the LAB–TEA molecule as an intracellular blocker for the open potassium ion channel. However, measurement of potassium current (nA) through the channel depicted a similar blocking efficiency for the trans-(E) and cis-(Z) LAB–TEA counterpart, which is still far from the ideal goal of optopharmacological applications.
 | ||
| Scheme 2 Schematic representation of the open potassium ion channel, KcsA (PDB ID: 3F5W), showing only two diagonally opposite subunits. The cavity amino acids (in one subunit) pointing inward are labelled. The protein is represented in the new cartoon model and amino acid residues are shown in the ball and stick model. The horizontal distances are the distances from the similar residues located in diagonally opposite arms of KcsA and the vertical line, 15.5, represents the distance between the α-C of THR-75 at the top and THR-107 at the bottom of the same monomer chain in the CC. All the distances are given in Angstrom. | ||
Thus, rationalization of the limitation for achieving high activity by the E-isomer is rather essential to aid the improvement of the efficiency of photoswitchable blocker drugs. It is critical to comprehend why there is a marginal difference in the blocking efficiency in terms of ionic current between the two isomers (0.3 ± 0.1 nA) and why residual blockade persists in the absence of light. The improvement of the understanding of the blocking action at the molecular level is indeed a stepping stone to enable design of a better photoswitchable blocker. In this paper, we report a molecular-level study of the blocking action of E- and Z-isomers of para-substituted locked azobenzene–tetraethylammonium (TEA) with the bacterial KcsA K+-ion channel. Furthermore, the significant role of amino acid residues situated in the selectivity filter (SF), channel cavity (CC), and channel gate (CG) in the blocking action of both E- and Z-isomers has been discussed in detail using Molecular Dynamics (MD) simulation and Quantum Mechanical (QM) calculation. Our study provides insights into the structural modifications that can significantly enhance the difference in blocking efficiency between the isomers and in future could guide the complete reversibility of the blocking process. These compounds present promising opportunities for advancing research in neuroscience, pharmacology, and cardiology, with potential implications for therapeutic interventions across a range of diseases and disorders. However, further research is needed to optimize their properties and enhance their efficacy and safety for clinical applications.
Computational methodology
The optimized geometry of LAB–TEA at the B3LYP/6-31+G(d)44–47 level of theory using the Gaussian 09 program package48 is used as the starting geometry for generating parameters for the LAB–TEA molecule using the antechamber module of AMBER2049 with the generalized AMBER force field (GAFF)50,51 and RESP charge model with the Hartree–Fock (HF) method and the 6-31G(d)52 basis set to provide a better description of charge distribution. The undesirable torsional changes of the LAB moiety are restricted by modifying the torsional parameters for the same as that of standard azobenzene, as reported by Böckmann et al.53 The crystal structure of KcsA, with PDB ID: 3F5W,54 represents the open state. The amino acid residues viz. three ARG, one ASP, and one GLU residue per monomer of KcsA are protonated at pH = 4,55 and charge neutrality was achieved by adding eight chloride ions using the LEaP program in AMBER20.49 We have placed 30 water molecules inside the channel cavity at an intermolecular distance of van der Waals radius.56–58Modeling of starting structures for the simulations
The blocker molecules can enter into the KcsA-K+-ion channel when the channel is in its open state.54 Since LAB–TEA contains only two functional groups, one being TEA and the other LAB, there is a possibility that LAB–TEA may enter via the TEA part, as free TEA does, or through the LAB moiety. Hence, we considered two possible orientations as the starting structure for performing MD simulations for each isomer of LAB–TEA in the intracellular region with one K+-ion inside the channel (Scheme 3). Moreover, simulation with LAB–TEA inside the channel without K+-ion is also considered (Scheme S1, ESI†). These KcsA(-K+)-LAB–TEA systems were solvated by truncated octahedron TIP3PBOX at a 12 Å distance from the edge of the octahedron and HMassRepartition59 is used so that δt of 4 fs can be afforded during simulation. The solvated KcsA-(K+)-LAB–TEA systems were then relaxed by minimization.60 The non-bonded interactions were restricted to 9.0 Å, and long-range electrostatic interactions were treated using the particle mesh Ewald method.61–66 The systems were then gradually heated to 300 K with positional restraint on KcsA and LAB–TEA. Each orientation of Z- and E-isomers of the KcsA-K+-LAB–TEA systems is then subjected to eight different equilibrations and results in eight different starting structures. The equilibration started with a positional restraint of 6.0 kcal mol−1 Å−2 on the whole KcsA-K+-LAB–TEA system; then the restraint weight is gradually reduced such that at the end of equilibration only a restraint weight of 0.5 kcal mol−1 Å−2 is on the KcsA but zero on the LAB–TEA. However, one simulation of each Z- and E-isomer positional restraint similar to that on KcsA is kept on the K+-ion during the simulation to understand the effect of K+-ion on the binding of LAB–TEA inside the channel cavity. Similarly, for KcsA–LAB–TEA systems, five different equilibrations are performed following the same simulation conditions. The above equilibrations result in five starting structures for the following production run. Furthermore, simulations were performed by placing a K+-ion and LAB–TEA inside the channel cavity without any restraint weight, which revealed the existence of either LAB–TEA or a K+-ion inside the channel cavity.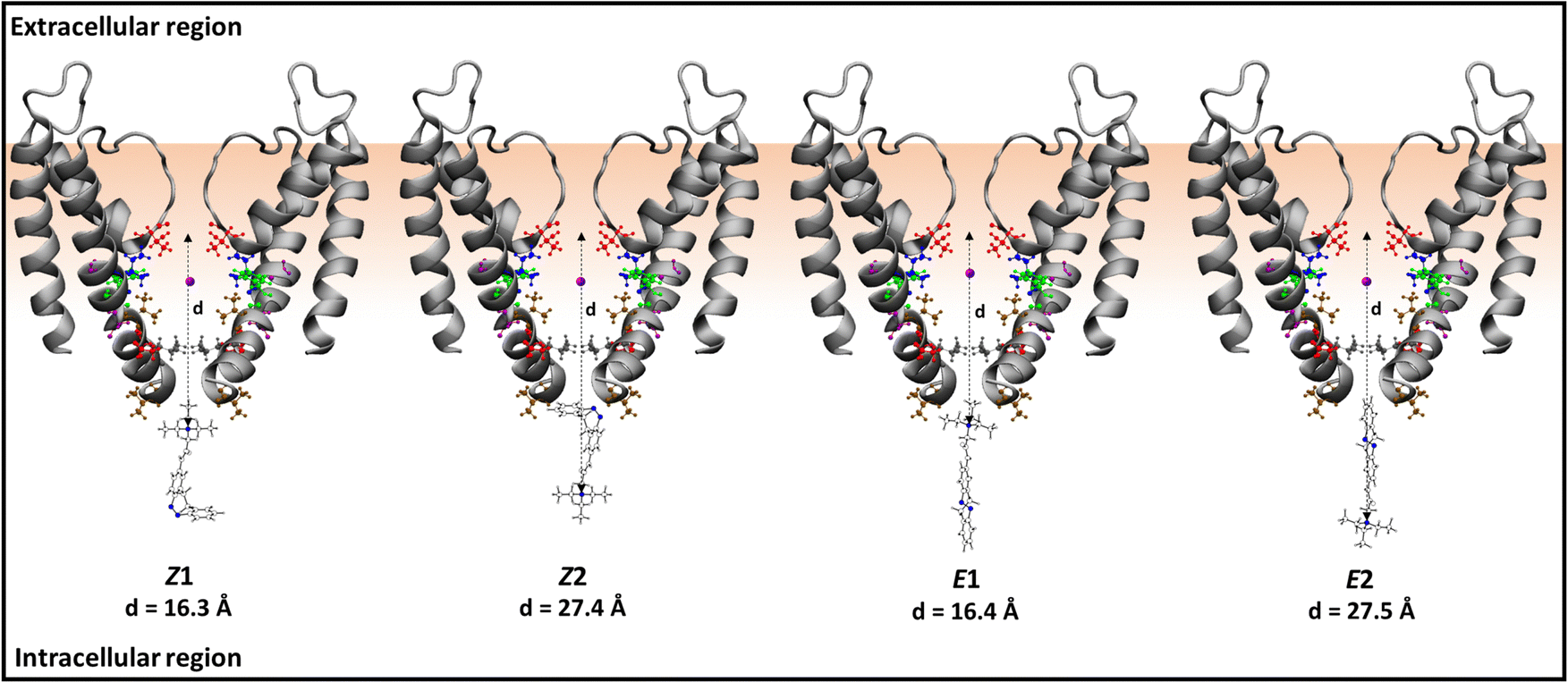 | ||
| Scheme 3 Schematic representation of KcsA–LAB–TEA starting conformations used for the simulation with K+-ions. The open KcsA (PDB ID: 3F5W) shows only two diagonally opposite subunits, and is represented as a new cartoon. The TEA and amino acids are represented by ball and stick model (red = threonine (THR-75 and THR-107), purple = glycine (GLY-99), blue = isoleucine (ILE-100), green = phenylalanine (PHE-103), ochre = valine (VAL-106 and VAL-115), grey = leucine (LEU-110)). The azo-nitrogens and TEA nitrogen are shown in blue colour. The purple ball represents the potassium ion. The d represents the distance between the center of α-C of THR-75 and the nitrogen of TEA. | ||
Molecular dynamics simulations
During all the production runs, a positional restraint of 0.5 kcal mol−1 Å−2 is kept on the α-C atom of KcsA to mimic the stabilizing effect of the lipid bilayer on the tetrameric ion channel. Since the binding of blocker molecules hardly affects the overall stability of the K+-ion channels and the interactions are barely affected by the membrane,67–70 the lipid bilayer is not incorporated to reduce the computational time. Also, previous simulation studies on the binding of blocker molecules with the K+-ion channels without a membrane have agreed well with the experimental data.71–75For the KcsA-K+-LAB–TEA system, simulation of a total 2.2 μs was performed on each isomer of LAB–TEA (Fig. S1, ESI†). On the other hand, for the KcsA–LAB–TEA system (Fig. S21, ESI†), 2.4 μs simulation was achieved for each isomer with the protein, KcsA. All the MD simulations59 were performed with the NPT76 ensemble in pmemd.cuda. The SHAKE59 algorithm and constant pressure periodic boundary conditions were applied in all directions. The resulting MD trajectories were examined visually using VMD software.77 The MD trajectories were analyzed using CPPTRAJ78 and the Qt version of xmgrace.79 The simulations are further clustered into different groups to represent the binding site in terms of the centroid. For that, the average-linkage hierarchical clustering algorithm80 provided in AMBER is used since it is reported to be an authentic approach for analyzing MD trajectories.81 The binding free energy calculations were carried out on the KcsA-(K+)-LAB–TEA systems using Molecular Mechanics Poisson Boltzmann Surface Area (MMPBSA).82,83 However, due to the heavy computational cost, changes in the conformational entropy are neglected as the relative binding free energies of the same ligand and protein sets are only calculated, where the large conformational changes are not expected.84 The nab program in AMBER20 was used to estimate the conformational entropy contributions to the binding free energy using the Kongsted Ryde entropy method85 on the selected trajectories. This calculation discloses the high entropy contribution for the binding conformations. Moreover, to highlight the critical residues involved in stabilizing the LAB–TEA, the native contacts for the residues within a 4 Å distance from the LAB–TEA during the simulation are analyzed.
To obtain the potential of mean force (PMF) of the LAB–TEA entry, the umbrella sampling (US) method86 was employed, under similar MD simulation conditions. In US simulations, the distance between the center of α-C of THR-75 and TEA was harmonically restrained with a force constant of 10.0 kcal mol−1 Å−2. PMF was calculated starting from the intracellular region into the channel cavity by the weighted histogram analysis method (WHAM) method87 using a histogram of 21 bins spanning from 10 to 30 Å. In this calculation, each window was run for 10.1 ns, with the first 0.1 ns considered as equilibration.
Quantitative analysis of KcsA–LAB–TEA interactions
Furthermore, the interaction energies between the LAB–TEA and amino acid residues in the selected conformations were quantified by the lower order symmetry adapted-perturbation theory calculation (SAPT0),88,89 using the quantum chemistry package PSI4,90 with jun-cc-pVDZ basis, density fitting and core electron freezing. The SAPT0 energy was reported to have good error cancellation with the jun-cc-pVDZ basis.91 Due to resource limitations, we were unable to incorporate all of the residues responsible for stabilization simultaneously. Instead, we performed SAPT0 on the LAB–TEA with one specific residue at a time.Results and discussion
The important geometrical parameters of Z- and E-LAB–TEA as well as the relative energy difference between them are given in Fig. 1. The Z → E isomerization energies for the LAB–TEA (7.0 kcal mol−1) and LAB (8.9 kcal mol−1)92 at the SA2-CASPT2(6,4)/6-31+G(d)//CASSCF(6,4)/6-31+G(d) level are similar; implying that the stability order of the isomer remains unaffected upon the para substitution. | ||
| Fig. 1 Optimized structures92 of Z- and E-isomers of LAB–TEA at the SA2-CASPT2(6,4)/6-31+G(d)//CASSCF(6,4)/6-31+G(d) level of theory. The key dihedral angles and the relative energies are given in degrees and kcal mol−1, respectively. | ||
By comparing the molecular electrostatic potential (MESP) plots of TEA,93 LAB, Z-LAB–TEA, and E-LAB–TEA qualitatively, we can understand that the electronic characteristics of the binder, TEA, hardly change upon coupling with LAB at the para position (Fig. 2). Thus, possible alkyl–alkyl and electrostatic interactions of these ethyl chains of LAB–TEA with the hydrophobic and hydrophilic amino acid residues of the ion channel can be expected similar to TEA.93 However, a significant shift in electrostatic potential is observed for the LAB moiety due to the substitution of positively charged TEA, resulting in an electron-withdrawing effect. After visual inspection of the MESP values of the LAB moiety of both Z- and E-LAB–TEA, we see that the minima are observed near the azo-nitrogens, followed by the phenyl rings, and maxima are observed near the hydrogens of the phenyl rings and ethyl bridges. These functional groups can thus direct the interactions that are responsible for the binding of LAB–TEA with the KcsA.
 | ||
| Fig. 2 Plots of the molecular electrostatic potential (MESP) on the van der Waals surface of the atoms in the TEA (QA), Z-LAB–TEA, and E-LAB–TEA at the B3LYP/6-31+G(d) level of theory. The MESP values are indicated in kcal mol−1. Blue indicates lower potential, and red indicates higher potential. | ||
A schematic representation of the ion channel, KcsA, in a state which allows the entry of incoming ions and molecules (open state)54 is shown in Scheme 2. The channel has three key regions viz., selectivity filter (SF), channel cavity (CC), and channel gate (CG). Internal channel blockers have been reported to enter the channel through the open channel gate (CG) and bind to the channel cavity (CC).71,93–97 As a result, these gate and cavity residues are involved in the passage and binding of internal blockers via significant interaction with them. The channel cavity consists of ten amino acid residues with the polar hydroxyl side chain of threonine at the top (THR-75) and the bottom (THR-107) of the central cavity. At the center, the non-polar side chains sec-butyl of isoleucine (ILE-100), benzyl of phenylalanine (PHE-103), isobutyl of leucine (LEU-105), and isopropyl of valine (VAL-106), as well as hydrogen of glycine (GLY-99 and GLY-104), are observed, which point towards the cavity. In addition to these non-polar side chains, polar side chain residues, viz., hydroxyl of threonine (THR-101) and hydroxymethyl of serine (SER-102) are observed to point away from the cavity. The channel gate is comprised of nine residues from THR-107 to VAL-115, incorporating polar hydroxyl of threonine (THR-107 and THR-112), non-polar methyl of alanine (ALA-108, ALA-109, and ALA-111), isobutyl of leucine (LEU-110), indole of tryptophan (TRP-113), benzyl of phenylalanine (PHE-114), and isopropyl of valine (VAL-115).
To rationalize the binding and internal blocking mechanism of Z- and E-isomers of LAB–TEA, we followed the progress of Z- and E-isomers from the intracellular region into CC in the presence of the K+-ion inside the channel. We have considered two possible orientations of the Z- and E-isomers of LAB–TEA with respect to the ion channel; one with the TEA pointing up (namely Z1 and E1) and one with the TEA pointing down (namely Z2 and E2) (Scheme 3). Each of them is then subjected to eight distinct equilibrations, resulting in eight starting structures (a–h) (Fig. S1, ESI†), which were then subjected to MD simulations giving a total of 1.1 μs simulations.
Visual inspection of the trajectories in almost all the simulations shows movement of LAB–TEA from the intracellular region to the channel cavity (CC) and movement of K+-ion from the CC to the selectivity filter (SF) (Fig. S4–S7, ESI†), which is according to the concentration gradient of the cell. In some simulations, it was visualized that Z-LAB–TEA approaches the CC through the positively charged TEA-end (Z1) rather than through the neutral LAB-end (Z2). However, the curved nature of the LAB restricts Z-LAB–TEA movement into the CC. Consequently, the TEA moiety binds to the CG residues and eventually detaches from the CG and moves to the intracellular region (Movie S1, ESI†).93 The above binding and unbinding process keeps repeating during the simulation. Only in one of the simulations it does move into the CC. However, a few scenarios have been observed, where the Z-isomer tries to enter through the neutral curved LAB-end (Z2). Among those, in one case, the Z-isomer moved to CC. The Z-isomer is observed inside the channel cavity for only 14.5% of the simulation time. The movements mentioned above are analyzed from the variation in the TEAN–CαTHR-75 distance along the trajectory (Scheme 3 and Fig. S8, S9, ESI†). These findings point out that the binding of Z-LAB–TEA inside the CC is less likely. Similar to the free TEA binding sites inside the cavity, the TEA moieties in Z1 and Z2 are either near the selectivity filter (SF) or the channel gate (CG), indicating the preference for TEA to selectively bind in these positions.93,94 Thus, one can interpret that the binding of Z-LAB–TEA is mainly governed by the binding preference of TEA.
The hierarchical clustering analyses (Fig. 3) indicate that for the Z-LAB–TEA, the major conformer that binds inside the channel cavity is Z1, with a binding energy of −21.6 kcal mol−1. In contrast, the binding energy outside the channel cavity is only −14.2 kcal mol−1, which is substantially low. On the other hand, no such preferential binding is seen for Z2 in terms of percentage population. It is found that for Z2, the binding energy inside (−17.0 kcal mol−1) the cavity is slightly higher than that in the outside (−13.0 kcal mol−1) of the channel cavity (Fig. 3). Thus, despite having higher binding energy for the Z-isomer inside the channel cavity, the experimental13 finding of lower blocking efficiency of the Z-isomer can be attributed to the curved structure of Z-LAB–TEA (Fig. 1), which might make the entry intrinsically difficult (Fig. 4 and Movie S1, ESI†). Additional simulations with LAB–TEA inside the cavity without the K+-ion also support the previous inference, in which most of the Z2 inside the cavity eventually converted to Z1 (Fig. S26 and Movie S2, ESI†). The binding energy of Z1 was calculated to be −22.9 kcal mol−1, which is close to the binding energy (−21.6 kcal mol−1) calculated from the simulations with K+-ions. The above observations indicate that the presence of K+-ions does not have a decisive role in guiding the orientation and the nature of binding of Z-LAB–TEA. Also, the contradicting factors such as adequate binding energy of the Z-isomer and the experimental finding of moderately low blocking efficiency of the Z-isomer indicate that the blocking ability is not thermodynamically guided; rather, it is more related to the intrinsic curved structure of the LAB moiety.
 | ||
| Fig. 3 Summary of hierarchical average-linkage clustering performed on trajectories of Z-LAB–TEA with one K+-ion showing binding of the Z-isomer in the channel cavity, when LAB–TEA placed in the cytoplasmic region. Each column represents different clusters. The centroid of each cluster is also shown, indicating the location of LAB–TEA relative to the ion channel. Z1 and Z2 are the conformations for Z-LAB–TEA inside the CC. The percentage population of clusters is given on the Y-axis. The corresponding binding energy (B.E.) is given in kcal mol−1 on the X-axis. Protein is represented as a ribbon with K+-ions (purple ball), and Z-LAB–TEA is shown in stick model (magenta colour). Enlarged versions of the figures are provided as separate images (Supplementary Data 1, ESI†). | ||
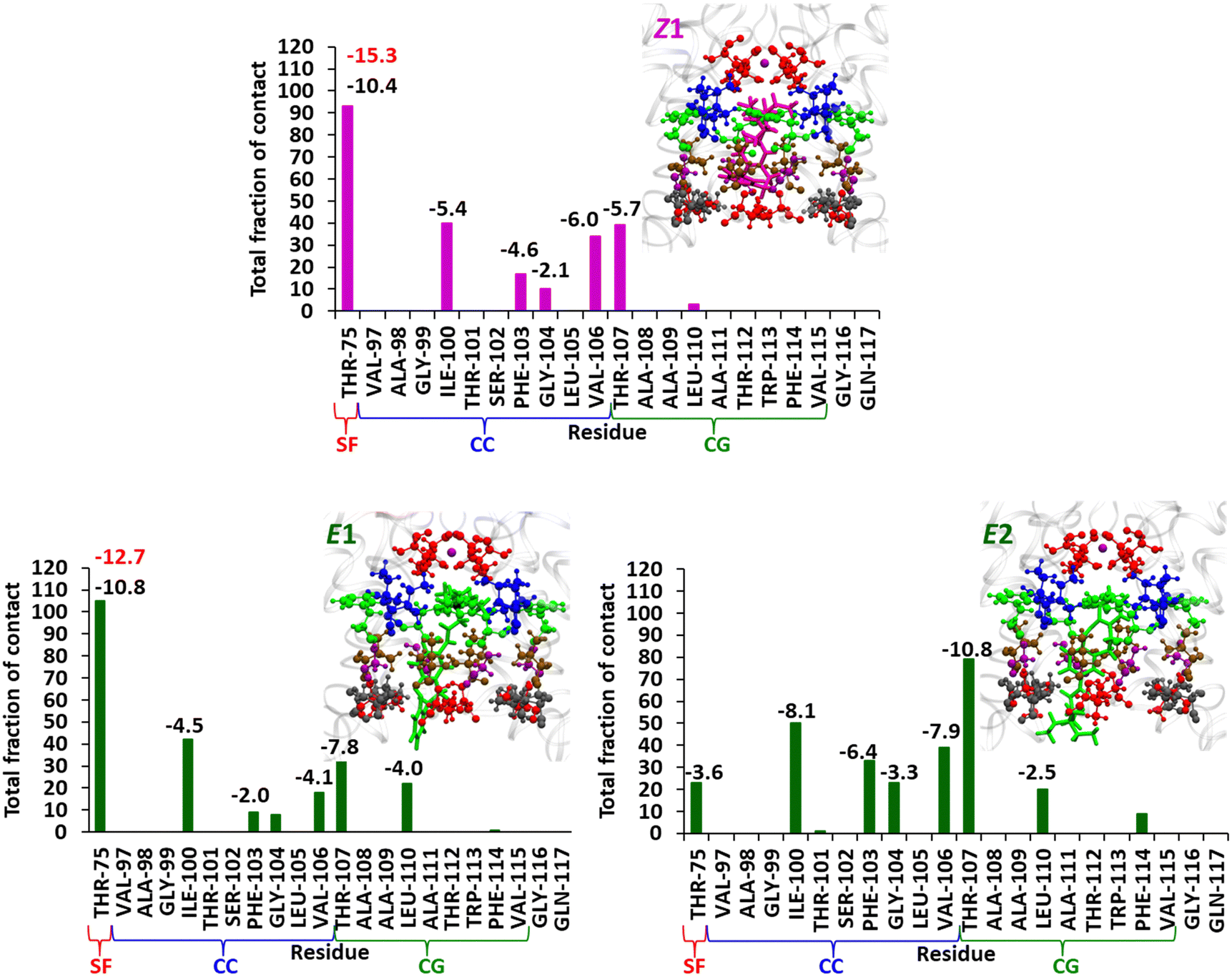 | ||
| Fig. 4 The contact plot for the Z1, E1, and E2 conformations in the KcsA-K+-ion channel and the interaction energy (electrostatic contribution (red) and dispersive contribution (black) in kcal mol−1) data from SAPT0 calculation (only energy values ≥2 kcal mol−1 are shown). The representative structures are shown. The protein and LAB–TEA are represented as the ribbon and stick (Z: magenta and E: green), respectively, and the interacting residues THR-75 (red), ILE-100 (blue), PHE-103 (green), GLY-104 (purple), VAL-106 (ochre), THR-107 (red), and LEU-110 (grey) from the extracellular to intracellular region are shown as a ball and stick model. The potassium ion is represented as a purple ball. The SF, CC, and CG represent the selectivity filter, channel cavity, and channel gate, respectively. Enlarged versions of the figures are provided as separate images (Supplementary Data 2, ESI†). | ||
The major contacts of Z1 were identified with THR-75, ILE-100, THR-107, VAL-106, and PHE-103 (Fig. 4 and 5). The nature of interaction and corresponding interaction energies of these amino acid residues were further evaluated with SAPT0 (Fig. 4). The TEA moiety of Z-LAB–TEA interacts electrostatically with THR-75 by non-classical hydrogen bonding between the hydroxyl oxygen of THR-75 and the ethyl hydrogen of TEA. The ethyl chain of TEA also shows dispersive interaction with the methyl group of THR-75 (Fig. 5). These two interactions with THR-75 are found to be the major contributor for stabilizing the Z1-isomer inside the channel cavity. In addition to that, TEA further interacts with ILE-100 and PHE-103 via alkyl–alkyl dispersive interactions. Alternatively, the LAB moiety in Z1 participates in the weak alkyl–alkyl dispersive interactions with the channel cavity and gate residues (Fig. 4 and 5). As the interaction energy of the TEA moiety with the cavity is higher than that of the LAB moiety of the blocker, we can say that the binding of Z inside the cavity is mainly directed by the positively charged TEA-end rather than the LAB-end. The binding nature of TEA in Z1 corroborates with the reported binding of free TEA in the cavity,98 highlighting the crucial role of the selectivity filter residue THR-75 in stabilizing free QAs.93
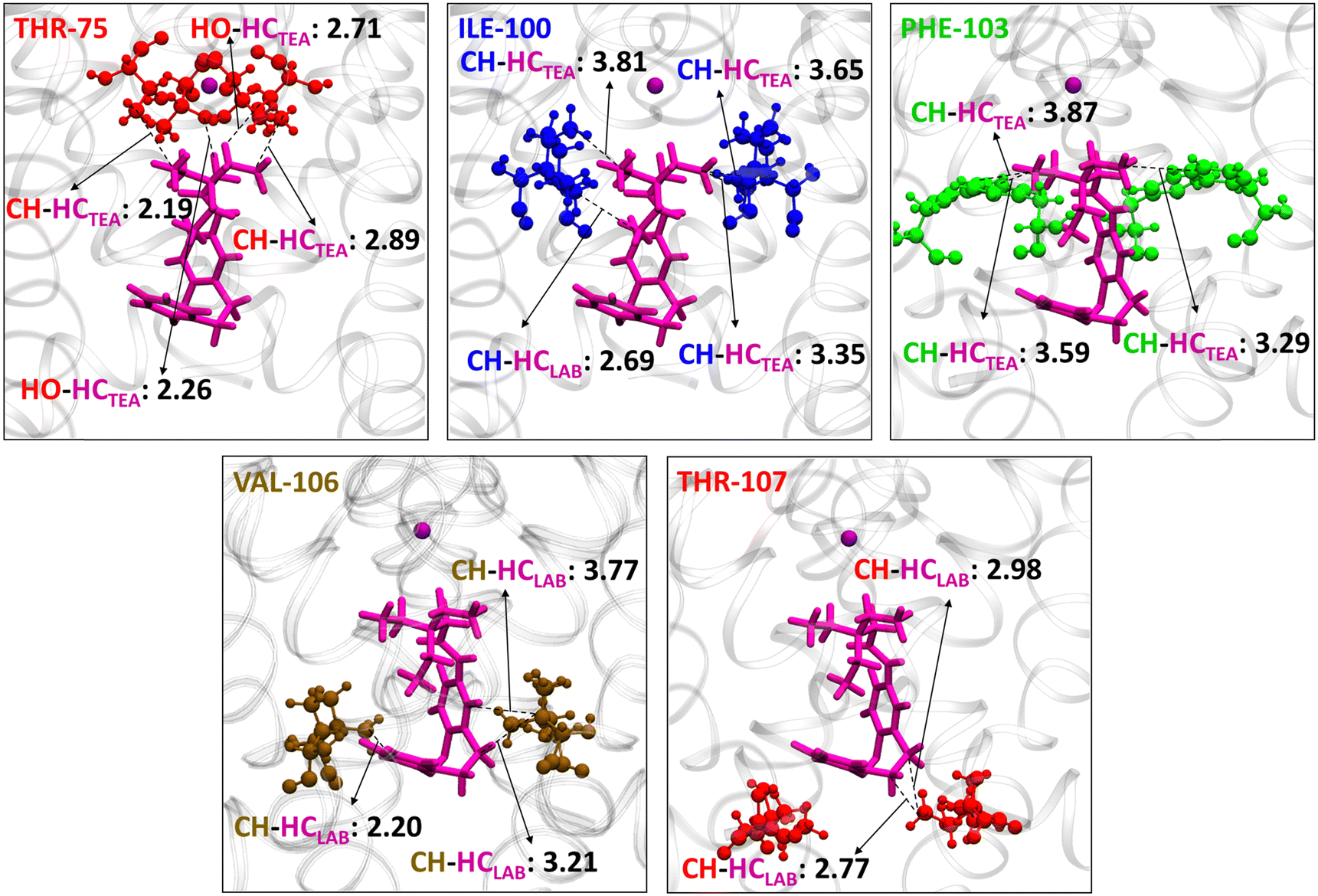 | ||
| Fig. 5 Interaction of the Z1 conformation with individual channel residues in the KcsA K+-ion channel cavity, when simulation was performed with one K+-ion (purple ball model) inside the channel and Z-LAB–TEA in the cytoplasmic region. The protein and Z1 are represented as ribbon and stick (magenta) respectively, and the interacting residues THR-75 (red), ILE-100 (blue), PHE-103 (green), VAL-106 (ochre), and THR-107 (red) from the extracellular to the intracellular region in the ball and stick model. The distances are given in Angstrom. Residue numbers are given in the upper left corner of each box. | ||
Contrary to the Z-isomer, the LAB moiety of the E-isomer is longer and planar, which is structurally suited to enter the channel cavity (CC) through the narrow channel gate (CG) (Scheme 3). The visual inspection of the trajectories reveals that the E-isomer enters CC through both the positively charged TEA-end (E1) and the LAB-end (E2). Moreover, the E-isomer was detected inside the channel cavity for 59.2% of the simulation time contrary to 14.5% for the Z-isomer. The movement is analyzed by calculating the change in the TEAN–CαTHR-75 distance during the simulation (Fig. S10 and S11, ESI†). These observations suggest that the E-LAB–TEA (E1 and E2) is more likely to enter the CC than the Z-isomer and thus might block the ion channel as well, which can be correlated to the reported greater blocking ability of the E-isomer.13 Inside the CC, the TEA moiety is close to SF and CG in the E1 and E2 conformations, respectively.
The analysis of E-LAB–TEA clustering data (Fig. 6) shows a higher percentage of cavity binding for the E-LAB–TEA with high binding energy (average binding energy for E1: −19.0 kcal mol−1 and E2: −18.0 kcal mol−1), as compared to the binding outside the channel cavity (average binding energy: −11.6 kcal mol−1). This indicates that both E1 and E2 have a similar thermodynamic preference for binding inside the channel cavity. Note that the E-isomer shows a higher likelihood of entering the channel cavity (13 out of 16 trajectories) as opposed to the Z-isomer (2 out of 16 trajectories) (Fig. 6). However, the highest binding energy that we have observed for both E- and Z-isomers are similar. Thus, it can be projected that the reported marginally higher blocking ability of the E-isomer over Z- is not thermodynamically driven, instead, the long and planar structure of the E-LAB–TEA might play a crucial role in the preferential entry to the channel cavity.13
 | ||
| Fig. 6 Summary of hierarchical average-linkage clustering performed on trajectories of E-LAB–TEA with one K+-ion (purple ball) showing binding of the E-isomer inside the channel cavity, when LAB–TEA is placed in the cytoplasmic region. Each column represents different clusters. The centroid of each cluster is also shown, indicating the location of LAB–TEA relative to the ion channel. E1 and E2 are the conformation of E-LAB–TEA inside the CC. The percentage population of clusters is given on the Y-axis. Corresponding binding energy (B.E.) is given in kcal mol−1 on the X-axis. Protein is represented as a ribbon with K+-ion (purple ball) and E-LAB–TEA is shown in stick model (green colour). Enlarged versions of the figures are provided as separate images (Supplementary Data 1, ESI†). | ||
Moreover, the comparable size of the E-isomer (15.9 Å, Fig. 1) and the channel cavity (15.5 Å, Scheme 2) restrict the free rotation of E-LAB–TEA inside the cavity throughout the simulation trajectories, as it may induce steric hindrance. However, once outside the cavity free rotation for the E-isomer is feasible. Hence simulations show entry through the LAB moiety (E2) to minimize the electrostatic repulsion with the K+-ion if present inside the cavity. Thus, it is possible for E1 to rotate outside the cavity and enter in the E2 conformation (Movie S3 and S4, ESI†).
Simulations were also performed with E-LAB–TEA inside the cavity without the K+-ion, where similar cavity binding probability for E1 and E2 with comparable binding energies (average: E1: −19.6 kcal mol−1 and E2: −18.5 kcal mol−1), is observed (Fig. S29 and S31, ESI†). This once again confirms that the K+-ion has no direct influence on the binding of E-LAB–TEA; however, it influences the direction of the TEA moiety pertaining to the entry of the E-isomer.
From Fig. 4 the residues that are involved in binding and stabilizing both E1 and E2 were identified; interestingly, they are the same viz., THR-75, ILE-100, PHE-103, GLY-104, VAL-106, THR-107, and LEU-110. However, the relative frequency of contact and the primary stabilization energy contributions coming from these residues, as calculated with SAPT0, are different (Fig. 4). For E1 the major contact is observed with THR-75, followed by ILE-100, THR-107, LEU-110, and VAL-106. Whereas, for E2, major contact is from THR-107, followed by ILE-100, VAL-106, PHE-103, GLY-104, and LEU-110. This variation can be correlated with the orientation of free TEA binding with the K+-ion channel as reported in our previous study.93 The ethyl hydrogens of positively charged TEA (Fig. 2) are involved in strong electrostatic interaction with the hydroxyl oxygen of THR-75 by non-classical hydrogen bonding as well as alkyl–alkyl dispersive interactions with the methyl side-chain of THR-75. These are the major contributors towards the stability of the E1 conformer. The TEA moiety is also involved in weak alkyl–alkyl interaction with the ILE-100 residue (Fig. 7). Thus, the interaction of TEA in E1 is similar to that of TEA in Z1 and free TEA.93 Conversely, the stabilizations from the remaining residues are identified as prominently weak dispersive in nature, resulting from their interaction with the LAB moiety. With THR-107, both alkyl–alkyl as well as alkyl–π interactions were observed followed by VAL-106, which shows alkyl–π interactions. Unlike the Z-isomer, alkyl–alkyl interaction with LEU-110 is possible for E1, which can be attributed to its planar and longer structure (Fig. 8). From this interaction analysis, we can confirm that the positively charged TEA fragment is responsible for the electrostatic interactions, while the neutral LAB fragment engages in dispersive interactions.
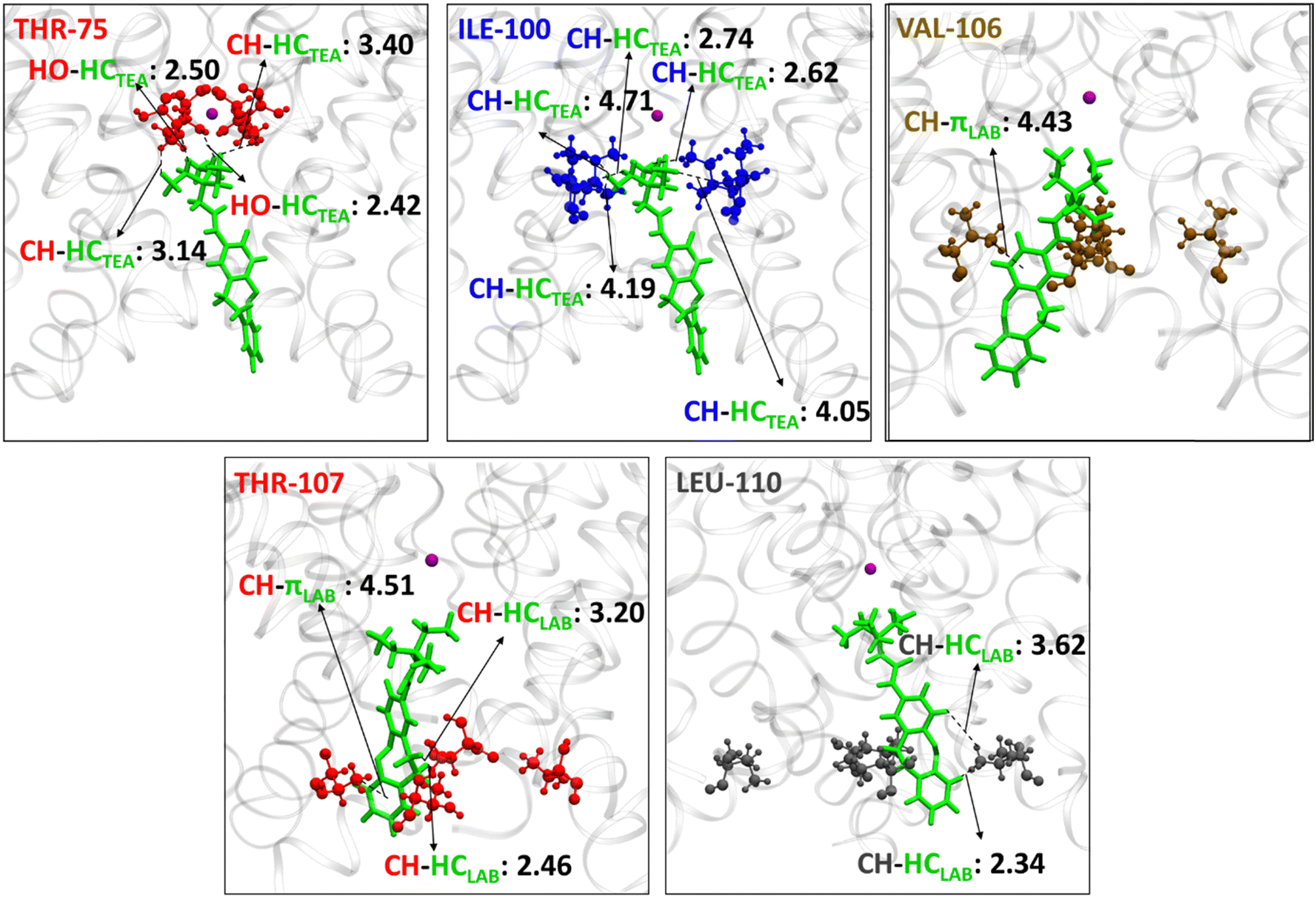 | ||
| Fig. 7 Interaction of the E1 conformation with individual channel residues in the KcsA K+-ion channel cavity, when simulation is performed with one K+-ion (purple ball model) inside the channel and E-LAB–TEA in the cytoplasmic region. The protein and E1 are represented as ribbon and stick (green) respectively, and the interacting residues THR-75 (red), ILE-100 (blue), VAL-106 (ochre), THR-107 (red), and LEU-110 (grey) from the extracellular to the intracellular region in ball and stick model. The distances are given in Angstroms. Residue numbers are given in the upper left corner of each box. | ||
 | ||
| Fig. 8 (a) Most probable binding conformations of LAB–TEA inside the KcsA channel cavity and (b) showing the benefits of the longer and planar structure of the E-isomer to have interactions with channel gate residue LEU-110. The protein is represented as a ribbon, LAB–TEA in licorice (Z: magenta and E: green), and channel gate residues THR-107 (red) and LEU-110 (grey) in the ball and stick model. | ||
On the other hand, E2 is expected to have stabilized by non-classical hydrogen bonding between the positively charged TEA moiety and THR-107 and attain stability by dispersive interaction with GLY-104, similar to the free TEA binding near the channel gate.93 However, the electrostatic contribution from the THR-107 was calculated to be very low (−2.8 kcal mol−1), and no interaction of GLY-104 with TEA is observed. This result can be attributed to the long E-isomer, which forces the TEA moiety in E2 to reside slightly below the THR-107 residue (Fig. 8). Consequently, TEA shows only dispersive interactions with THR-107 (Fig. 9). Thus, enforced by the longer and planar structure of LAB, the TEA binding in E2 differs from the previously reported free TEA binding.93 The THR-107 also participates in the alkyl–π interactions with the LAB moiety of E2; and contributes majorly to the E2 stabilization (−10.8 kcal mol−1). The rest of the stabilizing interactions for E2 result from the LAB moiety. Similar to THR-107, ILE-100 also shows alkyl–alkyl and alkyl–π interactions. The VAL-106 is involved in alkyl–alkyl interactions only. With PHE-103, CH–π (T-stacking) stabilization is observed. The GLY-104 has significant contact with E2; however, the energy contribution is less (Fig. 9). Thus, for E1, primary stabilization is from the TEA moiety, whereas for E2, the LAB moiety is the principal contributor towards stabilization.
 | ||
| Fig. 9 Interaction of the E2 conformation with individual channel residues in the KcsA K+-ion channel cavity, when simulation is performed with one K+-ion (purple ball model) inside the channel and E-LAB–TEA in the cytoplasmic region. The protein and E2 are represented as ribbon and stick (green), respectively, and the interacting residues ILE-100 (blue), PHE-103 (green), GLY-104 (purple), VAL-106 (ochre), and THR-107 (red) from the extracellular to the intracellular region in the ball and stick model. The distances are given in Angstrom. Residue numbers are given in the upper left corner of each box. | ||
In order to illustrate the mechanism for the entry of Z1, E1, and E2 from the intracellular region (left) into the channel cavity (right) the potential of mean force (PMF) was plotted (Fig. 10). The major changes are observed when the LAB–TEA comes in contact with the residues near and around the channel gate (from 24 to 14 Å). The channel gate residues THR-107 are known to be involved in the H-bonding interaction with the positively charged incoming group like TEA,72,93,99 which is the governing factor for the entry to the channel cavity. Thus, the Z1 and E1 moiety that enters through the TEA group facing the channel gate shows higher stabilization (23 to 22 Å) than E2, where the neutral hydrophobic LAB moiety shows an energy barrier of 5.2 kcal mol−1 (Fig. 10 and Fig. S34, ESI†). In the next step, the blocker molecules enter the channel cavity with a low overall energy barrier for Z1 (3.6 kcal mol−1), E1 (3.3 kcal mol−1), and E2 (4.7 kcal mol−1) (Fig. 10). Interestingly, Z1 demonstrates stabilizing interactions with the channel arms through its curved LAB moiety (Fig. S35, ESI†), which aligns perfectly with the channel arms, resulting in a ‘dip’ in the plot (18 Å). Conversely, such stabilization is not observed for E1 and E2, attributed to its linear and planar LAB moiety. Finally, when LAB–TEA binds inside the channel cavity, E1 exhibits higher stabilization as compared to Z1 and E2, as observed in the MMPBSA binding energy. Hence, the results reveal a difference, though small,13 in the binding energy and barrier height for the entry of the isomers into the cavity attributed to the shape of the molecules.
 | ||
| Fig. 10 Energy profile of Z1, E1, and E2 entry pathways: potential of mean force (PMF) from the intracellular region to the channel cavity obtained from umbrella sampling. The structures corresponding to the plateau of E1, the ‘dip’ in the plot for E2 and Z1, and the highest peaks for all isomers are given. The protein and LAB–TEA are represented as ribbon (grey) and stick, respectively. | ||
When administrated to the ion channel, LAB–TEA will exist in the thermodynamically most stable Z-isomeric inactive form in the dark. Owing to the disadvantage of having a short and curved structure, it is not fit to enter the narrow bottleneck of the channel gate as observed in the simulations in terms of less probability and slightly higher energy barrier. This result corroborates well with the fact that no significant reduction in ionic current is observed in the dark. Upon irradiation, with violet light (405 nm), the Z-isomer converts into the thermodynamically less stable E-isomer. Our theoretical study suggests that the planar and longer structure of E-LAB–TEA facilitates the entry, which is supported by lower energy barrier and, in turn, cavity binding, which is indicated by slightly higher stabilization energy, thereby reducing the ionic current. The longer and planar E-isomer can engage in CH⋯HC dispersive interaction with LEU-110, which is absent in the short and curved Z-isomer. Furthermore, photo-reversion using green light (530 nm) converts the E-isomer present inside the channel cavity back to the Z-isomer. However, due to the curved nature of the short Z-isomer, it probably remains inside the cavity. Thus, only a slight increase (0.3 ± 0.1 nA) in the ionic current is observed under green light. Therefore, a structural modification that can facilitate the release of the Z-isomer from the cavity under green light would be more effective in terms of practical use of a photoswitchable channel blocker.
Conclusions
The short and curved Z-LAB–TEA (E = 0.0 kcal mol−1, 12.4 Å) is thermodynamically more stable than the long and planar E-LAB–TEA (E = 7.0 kcal mol−1, 15.9 Å). The molecular-level analysis of the interaction of the LAB–TEA with the bacterial KcsA K+-ion channel using Molecular Dynamics (MD) simulation and Quantum Mechanical (QM) calculations identified preferable binding conformations for Z- (14.9%) and E-LAB–TEA (52.9%) inside the K+-ion channel. Simulations with a K+-ion inside the channel cavity and LAB–TEA in the cytoplasmic region show no direct influence of the K+-ion on the binding nature of the Z- and E-isomer; however, it guides the direction of the TEA moiety pertaining to the entry of the E-isomer to minimize the possible electrostatic repulsion between the positively charged TEA moiety and K+-ion inside the cavity. The binding energies for E-isomers (−22.3 kcal mol−1) are slightly higher than those of the Z-isomers (−21.6 kcal mol−1) inside the channel cavity. However, the structural difference in the isomers plays a crucial role in facilitating their entry/exit into and from the cavity. The E-isomer has a longer and planar LAB moiety, which facilitates the entry to the channel cavity through the narrow channel gate and in turn facilitates the blocking. On the contrary, the short and curved structure of LAB in the Z-isomer makes the entry a little challenging compared to the E-isomer. A local minimum is observed for the Z1 isomer near the channel gate residue, which is absent in E1, leading to a slightly higher energy barrier for the entry of Z1 as compared to E1. In both the E- and Z-isomers, the positively charged TEA moiety engages in strong electrostatic interactions by non-classical hydrogen bonding and dispersive interactions with the ion channel residues, while the neutral LAB moiety engages in weak dispersive hydrophobic interaction. Thus, the binding of LAB–TEA inside the cavity is mainly governed by the TEA interactions, which is in accordance with the preference observed in free TEA binding inside the cavity. In addition, the longer and planar E-isomer can engage in CH⋯HC dispersive interaction with LEU-110, which is absent in the short and curved Z-isomer. Thus, our results indicate that accelerated release of the inactive, Z-LAB–TEA from the cavity is essential for an efficient LAB–TEA-based photoswitchable potassium ion channel blocker. The release might be directed by introducing substituents on the LAB moiety that interact favourably with the hydrophobic residues at the channel gate. Hence, this work could aid future research to improve the rational design of photoswitchable blockers.Author contributions
Rinsha Cholasseri: methodology; data curation; writing – original draft. Susmita De: conceptualization; data curation; methodology; writing – review and editing; supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. D. thanks partial financial support from DST (IFA12-CH-76, CRG/2019/002891), SPG grant (SPG/2022/001350), SIRE grant (SIR/2022/000924) and partial support for the computational facility from computer centre of Calicut University. R. C. thanks UGC for providing the fellowship and also thanks the Centre for Computational Modelling and Simulation (CCMS) and Central Computer Center (CCC) at NITC.References
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- W. Szymanski, J. M. Beierle, H. A. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef CAS PubMed.
- N. Katsonis, M. Lubomska, M. M. Pollard, B. L. Feringa and P. Rudolf, Prog. Surf. Sci., 2007, 82, 407–434 CrossRef CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- A. S. Lubbe, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2017, 46, 1052–1079 RSC.
- P. Glock, J. Broichhagen, S. Kretschmer, P. Blumhardt, J. Mücksch, D. Trauner and P. Schwille, Angew. Chem., Int. Ed., 2018, 57, 2362–2366 CrossRef CAS PubMed.
- M. Reynders, B. S. Matsuura, M. Bérouti, D. Simoneschi, A. Marzio, M. Pagano and D. Trauner, Sci. Adv., 2020, 6, eaay5064 CrossRef CAS PubMed.
- H. Rau, Angew. Chem., Int. Ed. Engl., 1973, 12, 224–235 CrossRef.
- H. Fliegl, A. Kohn, C. Hattig and R. Ahlrichs, J. Am. Chem. Soc., 2003, 125, 9821–9827 CrossRef CAS PubMed.
- A. R. Dias, M. E. Minas da Piedade, J. A. Martinho Simões, J. A. Simoni, C. Teixeira, H. P. Diogo, Y. Meng-Yan and G. Pilcher, J. Chem. Thermodyn., 1992, 24, 439–447 CrossRef CAS.
- T. Tsuji, H. Takeuchi, T. Egawa and S. Konaka, J. Am. Chem. Soc., 2001, 123, 6381–6387 CrossRef CAS PubMed.
- R. Siewertsen, J. B. Schönborn, B. Hartke, F. Renth and F. Temps, Phys. Chem. Chem. Phys., 2011, 13, 1054–1063 RSC.
- E. R. Thapaliya, J. Zhao and G. C. Ellis-Davies, ACS Chem. Neurosci., 2019, 10, 2481–2488 CrossRef CAS PubMed.
- C. Renner and L. Moroder, ChemBioChem, 2006, 7, 868–878 CrossRef CAS PubMed.
- W. F. Cheong, S. A. Prahl and A. J. Welch, IEEE J. Quantum Electron., 1990, 26, 2166–2185 CrossRef.
- F. Klockmann, C. Fangmann, E. Zender, T. Schanz, C. Catapano and A. Terfort, ACS Omega, 2021, 6, 18434–18441 CrossRef CAS PubMed.
- M. A. Kienzler, A. Reiner, E. Trautman, S. Yoo, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2013, 135, 17683–17686 CrossRef CAS PubMed.
- D. Mulatihan, T. Guo and Y. Zhao, Photochem. Photobiol., 2020, 96, 1163–1168 CrossRef CAS PubMed.
- D. Hao, X. Tang, Y. An, L. Sun, J. Li, A. Dong, X. Shan and X. Lu, J. Phys. Chem. Lett., 2021, 12, 2011–2016 CrossRef CAS PubMed.
- L. Zou, C. J. Addonizio, B. Su, M. J. Sis, A. S. Braegelman, D. Liu and M. J. Webber, Biomacromolecules, 2021, 22, 171–182 CrossRef CAS PubMed.
- T. W. Colburn, A. C. Delmastro, M. Figueroa, F. Lopez and C. B. Cooper, J. Phys. Chem. C, 2022, 126, 15565–15572 CrossRef CAS.
- S. Jia and E. M. Sletten, ACS Chem. Biol., 2022, 17, 3255–3269 CrossRef CAS PubMed.
- D. C. Lee, K. N. Guye, R. K. Paranji, K. Lachowski, L. D. Pozzo, D. S. Ginger and S. H. Pun, Langmuir, 2021, 37, 10126–10134 CrossRef CAS PubMed.
- Z. Qiao, W. Fu, Q. Huang, Z. Li, C. Zhao and X. Shao, J. Agric. Food Chem., 2022, 70, 5541–5550 CrossRef CAS PubMed.
- W. Fu, Z. Shao, X. Sun, C. Zhou, Z. Xu, Y. Zhang, J. Cheng, Z. Li and X. Shao, J. Agric. Food Chem., 2022, 70, 4279–4290 CrossRef CAS PubMed.
- D. Kodura, L. L. Rodrigues, S. L. Walden, A. S. Goldmann, H. Frisch and C. Barner-Kowollik, J. Am. Chem. Soc., 2022, 144, 6343–6348 CrossRef CAS PubMed.
- S. Bhunia, A. Dolai, S. Bera and S. Samanta, J. Org. Chem., 2022, 87, 4449–4454 CrossRef CAS PubMed.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed.
- P. Lentes, E. Stadler, F. Röhricht, A. Brahms, J. Gröbner, F. D. Sönnichsen, G. Gescheidt and R. Herges, J. Am. Chem. Soc., 2019, 141, 13592–13600 CrossRef CAS PubMed.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- M. Hammerich, C. Schütt, C. Stähler, P. Lentes, F. Röhricht, R. Höppner and R. Herges, J. Am. Chem. Soc., 2016, 138, 13111–13114 CrossRef CAS PubMed.
- L. Liu, S. Yuan, W. H. Fang and Y. Zhang, J. Phys. Chem. A, 2011, 115, 10027–10034 CrossRef CAS PubMed.
- O. N. Oliveira, M. Raposo and A. Dhanabalan, Handbook of Surfaces and Interfaces of Materials 1st ed., Academic Press, San Diego, 2001 Search PubMed.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS PubMed.
- L. Liu, Y. Wang and Q. Fang, J. Chem. Phys., 2017, 146, 064308 CrossRef PubMed.
- M. V. Nikolaev, D. M. Strashkov, M. N. Ryazantsev and D. B. Tikhonov, Eur. J. Pharmacol., 2023, 938, 175448 CrossRef CAS PubMed.
- T. Fehrentz, E. Amin, N. Görldt, T. Strasdeit, S. E. Moussavi-Torshizi, P. Leippe, D. Trauner, C. Meyer, N. Frey, P. Sasse and N. Klöcker, BioRxiv, 2024 DOI:10.1101/2024.03.24.586505.
- A. Mourot, C. Herold, M. A. Kienzler and R. H. Kramer, J. Pharmacol., 2018, 175, 2296–2311 CAS.
- M. Banghart, K. Borges, E. Isacoff, D. Trauner and R. H. Kramer, Nat. Neurosci., 2004, 7, 1381–1386 CrossRef CAS PubMed.
- M. Schoenberger, A. Damijonaitis, Z. Zhang, D. Nagel and D. Trauner, ACS Chem. Neurosci., 2014, 5, 514–518 CrossRef CAS PubMed.
- Potassium-Channel Blockers (Class III Antiarrhythmics), https://www.cvpharmacology.com/antiarrhy/potassium-blockers, (accessed November 2022).
- J. B. Trads, K. Hüll, B. S. Matsuura, L. Laprell, T. Fehrentz, N. Görldt, K. A. Kozek, C. D. Weaver, N. Klöcker, D. M. Barber and D. Trauner, Angew. Chem., Int. Ed., 2019, 58, 15421–15428 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (version E.01), Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, AMBER 2020 (version 20), University of California, San Francisco, 2020 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- B. Wang and K. M. Merz Jr., J. Chem. Theory Comput., 2006, 2, 209–215 CrossRef CAS PubMed.
- A. Jakalian, B. L. Bush, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2000, 21, 132–146 CrossRef CAS.
- M. Böckmann, C. Peter, L. D. Site, N. L. Doltsinis, K. Kremer and D. Marx, J. Chem. Theory Comput., 2007, 3, 1789–1802 CrossRef PubMed.
- L. G. Cuello, V. Jogini, D. M. Cortes and M. Perozo, Nature, 2010, 466, 203–208 CrossRef CAS PubMed.
- A. Sumino, T. Sumikama, M. Iwamoto, T. Dewa and S. Oiki, Sci. Rep., 2013, 3, 1063 CrossRef PubMed.
- L. Guidonia, V. Torrea and P. Carlonia, FEBS Lett., 2000, 477, 37–42 CrossRef PubMed.
- M. Compoint, C. Boiteux, P. Huetz, C. Ramseyer and C. Girardet, Phys. Chem. Chem. Phys., 2005, 7, 4138–4145 RSC.
- H. Woo, A. R. Dinner and B. Roux, J. Chem. Phys., 2004, 121, 6392–6400 CrossRef CAS PubMed.
- J. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- M. A. Cauchy, C. R. Sci., 1847, 25, 536–538 Search PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- M. Crowley, T. Darden, T. Cheatham III and D. Deerfield II, J. Supercomput, 1997, 11, 255–278 CrossRef.
- C. Sagui and T. A. Darden, Simulation and Theory of Electrostatic Interactions in Solution, American Institute of Physics, Melville, NY, 1999 Search PubMed.
- A. Toukmaji, C. Sagui, J. Board and T. Darden, J. Chem. Phys., 2000, 113, 10913–10927 CrossRef CAS.
- C. Sagui, L. G. Pedersen and T. A. Darden, J. Chem. Phys., 2004, 120, 73–87 CrossRef CAS PubMed.
- B. Jouirou, S. Mouhat, N. Andreotti, M. de Waard and J. Sabatier, Toxicon, 2004, 43, 909–914 CrossRef CAS PubMed.
- K. M. Giangiacomo, Y. Ceralde and T. J. Mullmann, Toxicon, 2004, 43, 877–886 CrossRef CAS PubMed.
- J. Aiyar, J. M. Withka, J. P. Rizzi, D. H. Singleton, G. C. Andrews, W. Lin, J. Boyd, D. C. Hanson, M. Simon, B. Dethlefs, C. Lee, J. E. Hall, G. A. Gutman and K. G. Chandy, Neuron, 1995, 15, 1169–1181 CrossRef CAS PubMed.
- A. Gross and R. MacKinnon, Neuron, 1996, 16, 399–406 CrossRef CAS PubMed.
- J. D. Faraldo-Gómez, E. Kutluay, V. Jogini, Y. Zhao, L. Heginbotham and B. Roux, J. Mol. Biol., 2007, 365, 649–662 CrossRef PubMed.
- E. Kutluay, B. Roux and L. Heginbotham, Biophys. J., 2005, 88, 1018–1029 CrossRef CAS PubMed.
- Y. Wu, Z. Cao, H. Yi, D. Jiang, X. Mao, H. Liu and W. Li, Biophys. J., 2004, 87, 105–112 CrossRef CAS PubMed.
- M. Cui, J. Shen, J. M. Briggs, W. Fu, J. Wu, Y. Zhang, X. Luo, Z. Chi, R. Ji, H. Jiang and K. Chen, J. Mol. Biol., 2002, 318, 417–428 CrossRef CAS PubMed.
- M. A. L. Eriksson and B. Roux, Biophys. J., 2002, 83, 2595–2609 CrossRef CAS PubMed.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed , 27-8.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- P. J. Turner, XMGRACE (version 5.1.19), Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR, 2005 Search PubMed.
- P. N. Tan, M. Steinbach and V. Kumar, Introduction to Data Mining, Addison-Wesley, Boston, 2006, vol. 8, pp.487–568 Search PubMed.
- J. Shao, S. W. Tanner, N. Thompson and T. E. Cheatham, J. Chem. Theory Comput., 2007, 3, 2312–2334 CrossRef CAS PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham III, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- J. Srinivasan, T. E. Cheatham, P. Cieplak, P. A. Kollman and D. A. Case, J. Am. Chem. Soc., 1998, 120, 9401–9409 CrossRef CAS.
- H. Gohlke and D. A. Case, J. Comput. Chem., 2004, 25, 238–250 CrossRef CAS PubMed.
- J. Kongsted and U. Ryde, J. Comput.-Aided Mater. Des., 2009, 23, 63–71 CrossRef CAS PubMed.
- G. M. Torrie and J. P. Valleau, Chem. Phys. Lett., 1974, 28, 578–581 CrossRef CAS.
- A. Grossfield, The Weighted Histogram Analysis Method (version 2.0.9.1), https://membrane.urmc.rochester.edu/content/wham (accessed March 2019) Search PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno and C. D. Sherrill, J. Chem. Phys., 2014, 140, 094106 CrossRef PubMed.
- D. G. A. Smith, L. A. Burns, A. C. Simmonett, R. M. Parrish, M. C. Schieber, R. Galvelis, P. Kraus, H. Kruse, R. Remigio Di, A. Alenaizan, A. M. James, S. Lehtola, J. P. Misiewicz, M. Scheurer, R. A. Shaw, J. B. Schriber, Y. Xie, Z. L. Glick, D. A. Sirianni, J. S. O’Brien, J. M. Waldrop, A. Kumar, E. G. Hohenstein, B. P. Pritchard, B. R. Brooks, H. F. Schaefer III, A. Y. Sokolov, K. Patkowski, A. E. DePrince III, U. Bozkaya, R. A. King, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Phys., 2020, 152, 184108 CrossRef CAS PubMed.
- A. K. Wilson, T. V. Mourik and T. H. Dunning, J. Mol. Struct., 1996, 388, 339–349 CrossRef CAS.
- R. Cholasseri and P. Parameswaran, Int. J. Quantum Chem., 2023, 123, e27215 CrossRef CAS.
- R. Cholasseri and S. De, J. Phys. Chem. B, 2021, 125, 86–100 CrossRef CAS PubMed.
- S. De, C. H. Rinsha, A. H. Thamleena, A. Joseph, A. Ben and V. U. Krishnapriya, Phys. Chem. Chem. Phys., 2018, 20, 17517–17529 RSC.
- B. Hille, Ion Channels of Excitable Membranes, Oxford University Press, England, 2nd edn, 1992 Search PubMed.
- Y. Jiang, A. Lee, J. Chen, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2002, 417, 523–526 CrossRef CAS PubMed.
- M. Iwamoto, H. Shimizu, F. Inoue, T. Konno, Y. C. Sasaki and S. Oiki, J. Biol. Chem., 2006, 281, 28379–28386 CrossRef CAS PubMed.
- M. J. Lenaeus, D. Burdette, T. Wagner, P. J. Focia and A. Gross, Biochemistry, 2014, 53, 5365–5373 CrossRef CAS PubMed.
- V. B. Luzhkov and J. Åqvist, FEBS Lett., 2001, 495, 191–196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01604a |
| This journal is © the Owner Societies 2024 |