
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in supramolecular fullerene chemistry†
Xingmao
Chang
 ab,
Youzhi
Xu
*a and
Max
von Delius
*b
ab,
Youzhi
Xu
*a and
Max
von Delius
*b
aCollege of Chemistry and Molecular Sciences, Henan University, Kaifeng 475004, China. E-mail: youzhixu@henu.edu.cn
bInstitute of Organic Chemistry, Ulm University, Ulm 89081, Germany. E-mail: max.vondelius@uni-ulm.de
First published on 18th October 2023
Abstract
Fullerene chemistry has come a long way since 1990, when the first bulk production of C60 was reported. In the past decade, progress in supramolecular chemistry has opened some remarkable and previously unexpected opportunities regarding the selective (multiple) functionalization of fullerenes and their (self)assembly into larger structures and frameworks. The purpose of this review article is to provide a comprehensive overview of these recent developments. We describe how macrocycles and cages that bind strongly to C60 can be used to block undesired addition patterns and thus allow the selective preparation of single-isomer addition products. We also discuss how the emergence of highly shape-persistent macrocycles has opened opportunities for the study of photoactive fullerene dyads and triads as well as the preparation of mechanically interlocked compounds. The preparation of two- or three-dimensional fullerene materials is another research area that has seen remarkable progress over the past few years. Due to the rapidly decreasing price of C60 and C70, we believe that these achievements will translate into all fields where fullerenes have traditionally (third-generation solar cells) and more recently been applied (catalysis, spintronics).
 Xingmao Chang | Xingmao Chang studied chemistry at Shanxi Normal University and obtained the BS in 2012. He completed his PhD in 2019 at Shaanxi Normal University (China, Prof. Yu Fang, 2012–2019), and was a joint-trained PhD student and post-doctor at the University of Utah (USA, Prof. Peter J Stang, 2017–2018 and 2019–2021, respectively), as well as a research assistant at Shaanxi Normal University (China, Prof. Yu Fang, 2021–2022). He moved to Germany in April 2022 and joined Prof. Max von Delius group at Ulm University as a postdoctoral researcher. He was awarded a research fellowship by the Alexander von Humboldt Foundation in November 2021. His current research is focused on the supramolecular chemistry of complex π-systems. |
 Youzhi Xu | Youzhi Xu is a professor of organic chemistry at Henan University in China. He earned his MSc in organic chemistry from Zhengzhou University under the guidance of Prof. Zheng Duan from 2012 to 2015. Subsequently, in 2019, He completed his PhD at Ulm University in Germany, under the supervision of Prof. Max von Delius. From 2020 to 2022, He held the position of postdoctoral fellow at the University of Hong Kong, working with Prof. Vivian W.-W. Yam. He was awarded a Chinese government award for outstanding self-financed students abroad in 2019 and Henan youth talent program in 2023. His research focus is on the synthesis of new curved π-conjugated functional materials, fullerene and supramolecular chemistry. |
 Max von Delius | Max von Delius is Professor of Organic Chemistry at Ulm University (Germany). He obtained his PhD at the University of Edinburgh and was a Postdoctoral Fellow at the University of Toronto, before establishing his independent research group at FAU Erlangen-Nürnberg (Germany) in 2013. His research interests include supramolecular chemistry, systems chemistry and the synthesis of functional organic materials. He has been awarded an ERC Starting Grant (“SUPRANET”) and has received the Cram-Lehn-Pedersen Prize (2022) and the Prix Forcheurs Jean-Marie Lehn (2023). |
1. Introduction
Carbon allotropes come in many different forms and flavours (Fig. 1), all of which have distinct properties that are exploited in diverse functional organic materials.1 Diamond and graphite occur naturally and are both polymeric, but exhibit very different mechanical and electronic properties mainly as a result of sp3vs. sp2 hybridization of carbon. One of the unique aspects of graphene is that it can be made by exfoliation from natural graphite2 and by chemical bottom-up synthesis (e.g. by chemical vapour deposition).3 Synthetic carbon allotropes have seen important new arrivals such as γ-graphyne, which was prepared in bulk thanks to dynamic covalent synthesis,4 and a discrete, sp-hybridized member of the cyclocarbon family (cyclo[18]carbon, C18),5 which so far is only accessible via on-surface synthesis.6 Fullerenes7 and carbon nanotubes8 can be prepared in bulk and comprise sp2-hybridized carbon frameworks as well as a significant degree of curvature that influences reactivity, optoelectronic properties and non-covalent interactions. While C60 is the most abundant member of the fullerene family, higher fullerenes,9 endohedral metallofullerenes (EMFs)10 and heterofullerenes11 have unique properties due to their lower symmetry, anisotropic curvature and the incorporation of elements other than carbon (Fig. 1, grey box). | ||
| Fig. 1 Selected examples for carbon allotropes (top row): diamond, graphite, γ-graphyne,4 carbon nanotube and cyclo[18]carbon.6 Selected examples for fullerenes Cn.12,13 Grey box: the most abundant endohedral metallofullerene Sc3N⊂Ih-C80 and heterofullerene (C59N)2 as examples for relevant compounds beyond carbon allotropes.10,14 | ||
Fullerenes are engaged in a long-standing relationship with supramolecular chemistry. Concerning higher fullerenes and EMFs, this relationship is built on necessity, because their isolation from complex soot mixtures is far from trivial and requires specialized HPLC columns or innovative uses of host–guest chemistry. What makes C60 special as a supramolecular guest is its high symmetry (Ih), relatively large size, complete rigidity, well-defined curvature and lack of heteroatoms. While its high symmetry and large size implies that sometimes C60 is used as a “guest of last resort” (e.g. in a relatively large self-assembled cage), we found that the majority of recent studies goes beyond proof-of-principle science. In this article, we therefore aim to put emphasis on advances showing exceptional originality either from the perspective of fullerene or supramolecular chemistry (or ideally: both). Because we focus mainly on advances from the past decade and we do not discuss all aspects of the field in equal detail, we wish to direct the reader to relevant work here. A book entitled “Supramolecular Chemistry of Fullerenes and Carbon Nanotubes” (edited by Martin and Nierengarten) comprises the state-of-the-art until 2012.15 Older review articles include overviews on metalloporphyrin hosts (2007),16 curved fullerene receptors (2008),17 open-cage fullerenes (2010),18 fullerene assemblies (2010)19 and endohedral fullerenes (2013).10 Among the more recent review articles, several rather narrow articles focus on methods for fullerene binding/release (2016),20 fullerene purification (2017, 2020)21,22 and selective functionalization (2020, 2021).22,23
It is our hope that this review article will lend further momentum to the promising research directions described herein. For the first time since 1990, two dreams of fullerene chemists have come within reach: (i) the purification of fullerene mixtures (incl. EMFs) without use of chromatography and (ii) the selective synthesis of single-isomer addition products thanks to the use of supramolecular templates. Due to the rapidly decreasing price of C60 (currently as low as 20 USD per gram) and C70, further progress towards these aims could make a real difference in key 21st century technologies, including photovoltaics, photocatalysis and quantum information processing.24,25
2. Selective fullerene functionalization
Exploiting the properties of the most abundant fullerenes C60 and C70 in solution-processed devices typically requires their covalent functionalization. Attaching one or two substituents to the fullerene core increases solubility, lowers the LUMO level, which is important for any application relying on electron transport, and modulates the morphology of solid-state materials. In this section, we will provide a brief introduction into unique challenges associated with the selective (multiple) functionalization of fullerenes and discuss supramolecular approaches towards meeting these challenges.Phenyl-C61-butyric acid methyl ester (PC61BM) and phenyl-C71-butyric acid methyl ester (PC71BM) are typical examples of fullerene mono-adducts acting as electron acceptors or electron transport materials in bulk heterojunction or perovskite solar cells, respectively.26,27Fig. 2 gives an overview on the most common methods used for the functionalization of C60, such as the Bingel(–Hirsch) reaction, the Prato reaction, the Diels–Alder reaction and trifluoromethylation.28 C70 has a lower symmetry than C60 and therefore has four different types of reactive sites (α-, β-, γ-, and δ-site, see Fig. 4). This implies that even mono-adducts of C70 (such as the above mentioned PC71BM) are obtained as mixture of (regio)isomers that are hard to separate. With spherical C60, complex isomer mixtures are obtained whenever 2–5 groups are added to the fullerene core.
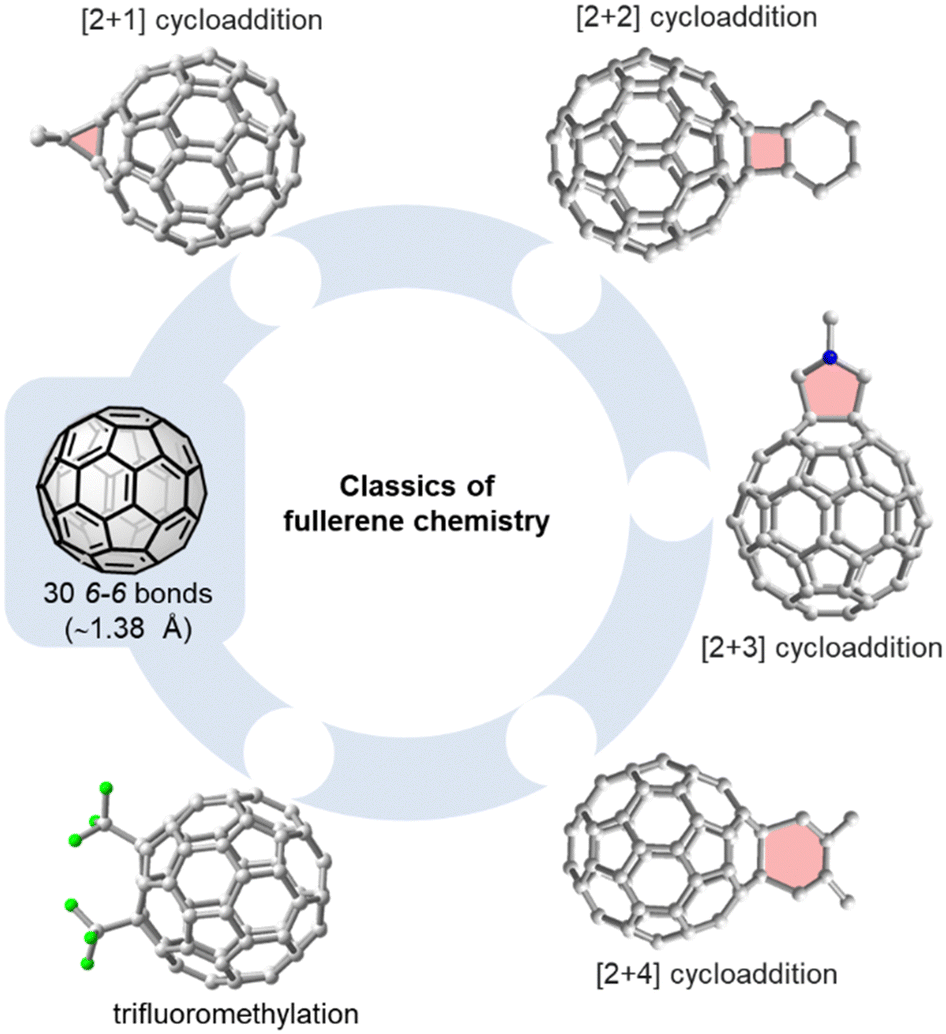 | ||
| Fig. 2 Selected methods for the functionalization of C60: [2+1] cycloaddition (e.g. Bingel–Hirsch reaction29,30), [2+2] cycloaddition,31,32 [2+3] cycoaddition (e.g. Prato reaction33), or trifluoromethylation.34 | ||
Recent work by diverse groups in materials science established that significantly higher device efficiencies can be obtained when using isomerically pure fullerenes rather than mixtures of regioisomers, diastereomers or racemates.23,35–37 The classic approach to address the regioisomer problem in C60 bis-addition is the use of a tether. Pioneered by Diederich, this method has the disadvantage that the tether remains attached to the fullerene, thus restricting the scope of products, unless degradable linkers are used, such as the dialkoxysilanes recently employed by Nierengarten.38–40 Tethers can also be stimuli-responsive,41 and their regioisomer-directing effect can be enhanced by a non-covalent interaction, as in the example by Hirsch shown in Fig. 3a.42
 | ||
| Fig. 3 Selected methods for regioselective twofold addition reactions to C60. (a) π–π interaction assistance. (b) Supramolecular nanohoop template. (c) Supramolecular cage template, 3 equiv. N-methylglycine was used. (d) Three-shell supramolecular mask strategy. | ||
While Kräutler's highly selective synthesis of a trans-1 bisadduct (96%) in the solid-state can be regarded as a supramolecular strategy,43 it took until 2016 for researchers to strategically employ non-covalent interactions for addressing the regioisomer problem. Torres and coworkers relied on strong intermolecular π–π interactions between two porphyrins to achieve a high regioselectivity for cis-substituted C60 bis-adducts in a Prato reaction.44 In 2018, our group utilized [10]CPP as a supramolecular template to synthesize [2]rotaxanes comprising a central fullerene bis-adduct as binding site for the CPP ring. The regioselectivity was not perfect, with the trans-1, trans-2 and trans-3 bis-adducts formed in 4%, 43% and 52% relative yield, respectively (Fig. 3b).45 Beuerle and Ribas independently developed the “supramolecular shadow mask” strategy that is particularly powerful, wherever a multi-addition product is desired that matches the symmetry and number of “windows” in the cage (e.g. tris-addition and trans-3 relationship between substituents in the case of the Beuerle cage shown in Fig. 3c). However, when a bis-adduct is desired, supramolecular shadow masks either give a suboptimal reaction outcome (Fig. 3c) or the reaction progress has to be stopped at precisely the right time.22 By combining our [10]CPP strategy and Ribas’ nanocapsule approach, we were able to achieve exclusive trans-3 regioselectivity for the Bingel bis-addition reaction to C60.46 This three-shell supramolecular mask strategy (Fig. 3d) required the design of an extended Pd-based cage to allow the encapsulation of the [10]CPP⊃C60 complex. Interestingly, the trans-3 C60 bis-adduct is symmetry-mismatched with the outer shell (three-fold vs. four-fold symmetry), and scope studies revealed that Rebek's 55% rule47 can be used to rationalize and predict the limitations of the approach.
Because C70 has the shape of an American football, multi-adducts to this second most abundant fullerene present a particular challenge.48–50 Due to the decreased symmetry of the molecule eight distinct types of C–C double bonds (6,6 and 5,6), such that even mono-addition reactions give rise to isomer mixtures. While supramolecular approaches for the selective mono-functionalization of C70 are still elusive, Echegoyen and coworkers achieved an impressive yield of 68% for the C70 bis-addition 1 and obtained only one regioisomer using Kräutler's solvent-free Diels–Alder reaction process (Fig. 4). The researchers found that the C70 bis-adducts can be converted into the “α”-mono-adduct 2 at a temperature of 190 °C, which represents an indirect solution to the mono-addition challenge. Moreover, both mono- and bis-adducts can be reverted to pristine C70 at 250 °C. This anthracene addition strategy may prove beneficial as a way of protecting groups to guide multiple fullerene additions.51
 | ||
| Fig. 4 Four distinct types of double bonds in C70 and dynamic multi-additions of C70 resulting from Diels–Alder reactions in molten anthracene. | ||
In fullerene tris- or higher addition reactions it is notoriously difficult to achieve itero- and regioselectivity. So far, only a few successful strategies have been developed based on privileged addition patterns of C60 encapsulated inside supramolecular masks. For instance, Beuerle and coworkers synthesized a trigonal bipyramidal covalent organic cage and observed strong affinity for C60/70 (Ka > 105 M−1) in CHCl3. Thanks to the threefold symmetry of the cage (Fig. 3c), to the authors were able to obtain the trans-3–trans-3–trans-3 C60 Prato tris-adduct in an impressive relative yield of 25%.52 In a Pd-based nanocapsule shadow mask, Ribas and coworkers reported the first synthesis of an e,e,e,e-tetrakis-C60 Bingel adduct in quantitative yield (Fig. 5).53 The nanocapsule's four perpendicular “windows” led to the observed regioselectivity, and the authors were also able to achieve the selective and quantitative formation of exclusively tetrakis-adducts using a biphasic protocol and catalytic amounts of the nanocapsule. More recently, Torres, Torre and coworkers realized the efficient Diels–Alder reaction between C60 and anthracene in water by using a metallo–organic Pd(II)-subphthalocyanine (SubPc) capsule as the catalytic host.54 In summary, supramolecular masks represent a promising new approach to achieve selective fullerene addition reactions, but atom economy is an obvious problem of this approach, because the masks typically have a higher molecular weight than the encapsulated fullerene and/or contain precious metals. It is therefore noteworthy that the masks can be recovered, and an important next goal is to develop methods that only require catalytic quantities in homogeneous solution.
 | ||
| Fig. 5 Biphasic catalytic protocol for the synthesis of e,e,e,e-tetrakis-diethylmalonate-C60 adduct 4 by using a sophisticated Pd-based nanocapsule 3 as the mask. | ||
Because most fullerene multi-adducts are chiral, achieving enantioselectivity is a formidable challenge whose importance has been underscored by recent work by Fuchter demonstrating that homochiral fullerenes outperform the corresponding racemates as electron transport layers in perovskite solar cells.37 While this challenge has been approached previously using chiral reagents/catalysts,55 one recent report by Nitschke and coworkers demonstrated the first supramolecular approach enabling the enantioselective functionalization of fullerenes (Fig. 6).56 The authors utilized an enantiopure metal–organic cage (5) that was self-assembled from Fe(NTf2)2, chiral 2-formylpyridine, and 1,5-anthracene-based dianiline. The cage was shown to react with encapsulated C60 to produce a highly diastereoselective e,e,e-tris adduct (6) via a chemo-, regio-, and enantio-selective Diels–Alder cycloaddition with the anthracene component of the cage. Encouraged by the successful diastereoselective reaction observed with C60, the researchers investigated the reaction with PC61BM. They found that PC61BM reacted with only one of the six anthracene components on the opposite hemisphere to the original substituent, resulting in an adduct (7) that exhibited excellent diastereoselectivity for trans-3 addition. The chiral adducts produced by this method were released from the cage by adding excess tris(2-aminoethyl)amine, which led to disintegration of the cage. The chiroptical properties of these adducts were also investigated, which make them promising materials for chiral organic electronics.57 While these findings represent a significant advance in the field of enantioselective fullerene functionalization, a method that facilitates enantioselective addition reactions with external reagents (rather than with components of the cage) would be the next logical step.
 | ||
| Fig. 6 Enantioselective synthesis of an e,e,e-tris adduct-C606 and trans-3-bisadduct-PC61BM 7 through stereochemical information transfer from a chiral self-assembled cage 5. | ||
3. Supramolecular fullerene dyads and triads
Electrochemical experiments show that C60 exhibits six equidistant reduction waves, with the lowest reduction potential at ca. −0.44 V (vs. SCE). The unique spherical and rigid framework of sp2 carbon atoms in C60 offers exceptional properties as an electron acceptor.58,59 It has been established that C60 derivatives possess relatively small reorganization energies during an electron transfer process, which makes them ideal electron acceptors for energy conversion and storage applications.60 In this section, we will review recent work on the electron-accepting properties of fullerenes in supramolecular dyads and triads with electron donors.To mimic the multicomponent photosynthesis process, various covalently/non-covalently bridged C60-based donor–acceptor (D–A) dyads have been developed since the first pioneering work revealed that charge-recombination is considerably slower than charge-separation in porphyrin–fullerene dyads.32,61 The design of efficient photoinduced electron transfer systems depends on several factors, including aromaticity, planarity, and energetics of the electron donor, the distance and orientation between the donor and acceptor, and the nature of the bridging unit. Numerous C60-based D–A systems, containing donors such as porphyrin, ferrocene, platinum complex, tetrathiafulvalene (TTF), boron dipyrromethene (BODIPY), oligothiophene, phenothiazine or phthalocyanine have been studied to investigate the photoinduced charge separation and solar cell characteristics of these dyads.62–65
Although covalently bridged D–A systems based on fullerenes have shown promising results, non-covalently bridged D–A systems resemble more closely natural photosynthetic systems, particularly in respect to the bridge between donor and acceptor. A wide range of non-covalent interactions (e.g. π–π, electrostatic, metal–ligand, H bonds) have been employed for the construction of D–A assemblies.66 [10]Cycloparaphenylene ([10]CPP) exhibits a strong supramolecular association with fullerenes due to concave–convex π–π interactions.67 Our group has used [10]CPP as a supramolecular junction to create modular dyads between zinc porphyrin and five representative fullerenes (Fig. 7a).68 Fluorescence titrations revealed that all fullerene derivatives have a remarkably high association constant with 8 (Ka > 105 M−1). Time-resolved transient absorption studies showed efficient charge separation and recombination across the non-covalent [10]CPP junction. This efficient supramolecular connection allowed studying the electron-accepting properties of the rather unstable dimer (C60)2, and interesting stoichiometry and concentration dependent effects on charge recombination were observed. For example, desymmetrization of the C60 moieties resulted in two distinct charge recombination processes in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex. While only one electron-recombination process was observed in the more symmetric 2:1 complex, a lifetime of up to 542 ns was observed, which is among the longest-lived charge-separated states in porphyrin–fullerene containing D–A systems found to date.
:1 complex. While only one electron-recombination process was observed in the more symmetric 2:1 complex, a lifetime of up to 542 ns was observed, which is among the longest-lived charge-separated states in porphyrin–fullerene containing D–A systems found to date.
 | ||
| Fig. 7 (a) Efficient electron transfer in modular 8⊃fullerene complexes. (b) Selected examples of hydrogen bond bridged C60-based D–A dyads (9 and 10) and corresponding charge transfer process. (c) Supramolecular complexation between ditopic porphyrin receptor 11 and fullerene derivative 12. | ||
Hydrogen bonding-based noncovalent interactions have been widely employed to control the electronic coupling between D–A dyads. For instance, Hirsch and coworkers have reported the assembly of a C60-based Hamilton receptor and metalloporphyrin-based cyanuric acid motifs.69 Sessler and coworkers have reported zinc porphyrin-appended cytidine and fullerene-appended guanosine (9).70 In these supramolecular wires, the photoinduced charge transfer processes have occurred exclusively through the hydrogen bonding bridges. The attenuation factor (β) is a key parameter of a molecular bridge that determines the magnitude of the electronic coupling between the redox sites and the energy of the charge transfer states localized at the two ends. Martín and colleagues have reported a series of noncovalent C60-based hybrids (10), which combine a zinc porphyrin with p-(2-fulleropyrrolidinyl)benzoates of different lengths (Fig. 7b).71 The authors achieved an exceptionally small β value of ca. 0.07 Å−1, benefitting from the strong supramolecular interactions in the carboxylate/amidinium salt bridge. Nierengarten and coworkers have investigated the supramolecular complexation of a C60 derivative 12 (Fig. 7c) with a porphyrin dimer and a porphyrin tape (11) endowed with two crown ether rings.72,73 Both ditopic porphyrin systems formed complexes with 1:1 and 1:2 stoichiometry and exhibit negative cooperativity, indicating a reduced binding constant for the complex of the second fullerene unit. The formation of these complexes is driven by the complementary π–π interactions and ammonium-crown ether hydrogen bonding interactions between the porphyrin tape and the C60 moieties.
The use of fullerenes and carbon nanotubes to mimic natural photosynthesis in supramolecular multicomponent D–A assemblies via metal–ligand coordination is another promising field of research.74,75 Porphyrins and phthalocyanines have been extensively employed as electron donors in the construction of fullerene-based dyads/triads due to their exceptional photophysical and photochemical properties, as well as their capacity to form metal–ligand coordination bonds.76–79 For instance, Lengo and coworkers effectively fabricated a three-component multichromophoric assembly 13, which consists of a fullerene monoadduct, an aluminium(III)-monopyridylporphyrin, and a ruthenium(II)-tetraphenylporphyrin (Fig. 8a).80 The photophysical properties of this triad have been examined on the femtosecond–nanosecond timescale using pump–probe spectroscopy. Upon excitation of the aluminium(III)-monopyridylporphyrin, the strong emission characteristic of this moiety was quenched. The transient absorption experiments provided evidence for the occurrence of stepwise photoinduced electron and hole transfer processes, resulting in the formation of a charge-separated state between the fullerene acceptor and the ruthenium-porphyrin donor (see Fig. 8a for the Jablonski diagram). Architectures of higher complexity such as tetrads, pentads, and hexads have also shown promise for photoinduced electron-transfer processes.81–84 The non-covalent assembly between multiple fullerenes or chromophores is another promising approach for preparing organic photosensitizers.85–87
 | ||
| Fig. 8 (a) Self-assembled ruthenium(II)porphyrin–aluminium(III)porphyrin-fullerene triad 13. Insert: Photophysical processes occurring in the triad (in CH2Cl2). (b) Schematic illustration of host–guest complexation facilitated triplet–triplet annihilation upconversion (TTA-UC) of 15⊃14. Adapted from ref. 88 with permission from American Chemical Society, copyright 2016. | ||
The process of triplet–triplet annihilation upconversion (TTA-UC) is of interest for efficiently harvesting diffuse visible/near-IR light and achieving high upconversion quantum yields.89 To improve TTA-UC efficiency, Yang and coworkers developed a new strategy using host–guest complexation between an alkyl nitrile chain functionalized C60-BODIPY sensitizer (14) and a tetraperylene-based pillar[5]arene emitter (15) (Fig. 8b).88 This supramolecular complexation facilitated triplet–triplet energy transfer (TTET) and TTA processes between the sensitizer and emitter, resulting in a significant increase in TTA-UC intensity and a high upconversion quantum yield (ΦUC) of up to 3.2% even at a very low emitter concentration of 6 × 10−5 M. This innovative supramolecular approach for bringing several components into spatial proximity improves TTA-UC efficiency without altering the intrinsic photophysical properties of the sensitizers and emitters.
Fullerene-based materials are being increasingly recognized for their electrophilic nature and their ability to stabilize radicals, which makes suitable C60 derivatives promising in the field of (photo)catalysis.66,90 For instance, Heredia and coworkers have synthesized a novel boron pyrrol hydrazine-C60 (BOPHY-C60) dyad that not only produces singlet oxygen (1O2) and superoxide radical anion (O2˙−) under irradiation with visible light (470 nm), but also demonstrates the ability to photoinactivate microorganisms.91 Similarly, Martín and coworkers have employed metallo-fulleropyrrolidines as homogeneous/heterogeneous catalysts for hydrogen transfer reactions (Fig. 9a), which resulted in a quantitative yield of ketone reduction and alcohol N-alkylation with only 0.5 mol% and 0.125 mol% iridiumfulleropyrrolidine catalyst 16 loading, respectively.92 The catalyst is easily separable from the reaction mixture and has also been successfully utilized for the alkylation of aniline with aliphatic alcohols in the presence of MgSO4. For example, benzylamine underwent quantitative alkylation with cyclohexanol using a 1.25% iridiumfulleropyrrolidine catalyst.
 | ||
| Fig. 9 (a) Transfer hydrogenation by metallo-fulleropyrrolidine 16. (b) Anionic Diels–Alder reaction of 17 and 18; insert: proposed anionic transition state for exo products on C60 surfaces. | ||
Anion–π catalysis involves stabilizing anionic transition states and intermediates through anion–π interactions on aromatic surfaces.93,94 Fullerenes, which offer a highly symmetric π system devoid of heteroatoms, provide an excellent platform to investigate the importance of polarizability in anion–π catalysis, without the complications of substituents, positive quadrupole moments or in-plane dipoles.95 Matile and coworkers discovered that a fullerene mono-adduct catalyst 19 exhibited good selectivity for enolate addition and improved the exo/endo diastereoselectivity of the Diels–Alder reaction.96 Using the anionic [4+2] cycloaddition of dienophile 18 and 3-hydroxy-2-pyrones 17 as an example, strong endo selectivity was observed. Fullerenes with flexible tethers failed to alter this intrinsic selectivity. The best exo-20/endo-20 ratio of 0.56 with 55% ee was achieved using fullerene 19 with conformationally constrained tethers as catalysts. The experimental data suggests that anion–π stabilization persists during the subsequent charge delocalization in the intrinsically disfavored exo transition state of the [4+2] cycloaddition.
4. Fullerene host–guest chemistry
4.1. Fullerenes as hosts
Open-cage fullerenes with their extremely rigid, all-carbon backbones exhibit unique cavities for the binding of neutral or charged guests.10,97–100 Although endohedral metallofullerenes (EMFs) do not meet the definition of a supramolecular complex but rather of a carceplex,101 we will also discuss this compound class, because non-covalent interactions in open-cage intermediates are relevant during statistical or rational EMF syntheses.Since the first report of a metal-encapsulating fullerene in 1985,102 endohedral fullerenes have been studied extensively.97–99,103 Endohedral fullerenes can be classified according to their “imprisoned” guests as (i) endohedral metallofullerenes (EMFs) and (ii) nonmetal endohedral fullerenes.10 The first category includes mono-, di-, and trimetallofullerenes,104–109 as well as cluster metallofullerenes110 (e.g. oxide clusters,103 nitride clusters,111–113 cyano clusters,114 sulfide clusters, and carbide clusters),10,97–99,115–120 while the second category includes H2, noble gases,121 water and other non-metal guests.10,100 (Fig. 10) The synthesis of endohedral fullerenes is a challenging task, for which several strategies have been developed, including the vaporization of graphite in the presence of additives, the implantation of atoms through the walls of the pre-existing carbon framework, as well as multi-step chemical synthesis involving the opening of orifices in the fullerene scaffold.
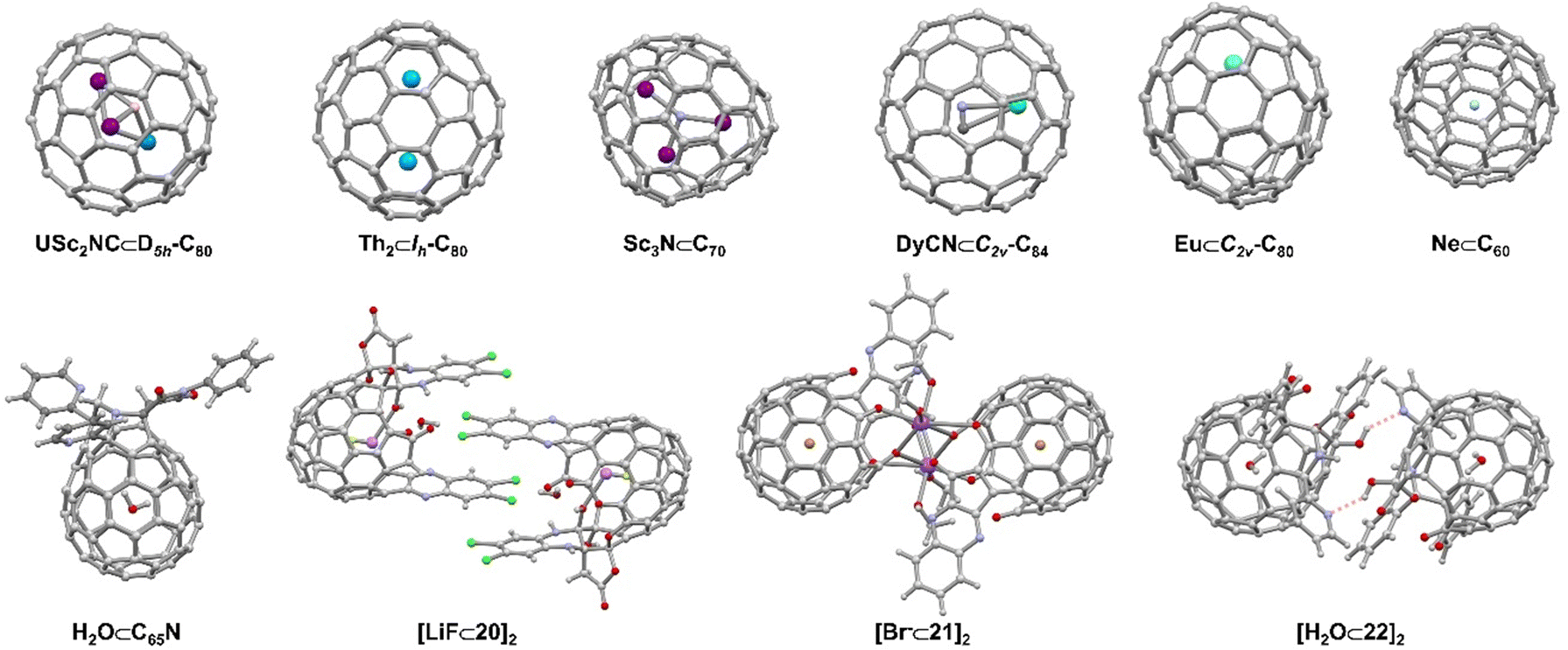 | ||
| Fig. 10 Selected endohedral fullerenes and open-cage fullerenes acting as hosts104,109,112,114,119,121 and supramolecular open cage fullerene-based dimers.122–125 | ||
The first open-cage fullerene was synthesized by using singlet oxygen to oxidize an azafullerene.126 Pioneering work by a number of research groups18,127,128 led to a wide scope of open-cage fullerenes, which offer the possibility to encapsulation and entrap guests within their cavity. The encapsulation of H2, O2, CO2, NH3, CH4, HF, HCN, HCCH, CH3OH, and H2O was achieved in this way and progress in this field has been reviewed recently by Gan.127,129 In addition to the neutral molecules listed above, halide anions, LiF ([LiF⊂20]2, Fig. 10), and cationic [BeF]+ have also been encapsulated in the fullerene cage by Gan and coworkers.123,124 The authors demonstrated that a 19-membered open-cage fullerene with four carbonyl groups, an ether oxygen and a quinoxaline moiety on the rim of the orifice could act as the container for F−, Cl−, Br− ([Br⊂21]2, Fig. 10) and I−. Fullerenes containing halide anions exhibit higher polarity compared to the respective empty fullerenes. The functional groups attached to open-cage fullerenes, such as amines, alcohols, and aromatic moieties, not only enable the selective recognition of guests, but also provide possibilities for further assembling higher-order structures or enabling the study of fundamental guest properties. For example, hydrogen-bonded fullerene dimers were obtained utilizing amide-125 ([H2O⊂22]2, Fig. 10) and bis(hemiketal)-130 containing open-cage[60]fullerenes. Dimers were also found in other work due to coordination between metal ions (such as Na, Ag, and Pt) and donor atoms as well as intermolecular π–π interactions.123,124,131–133 Murata and coworkers constructed a supramolecular complex by encapsulating a 3O2 molecule into an open-cage C60 derivative, of which the EPR spectra exhibited triplet state character as well as the anisotropy of the 3O2.134
Endohedral fullerenes, while technically not host–guest complexes, nevertheless offer a unique opportunity to study metal–fullerene interactions,135,136 unusual metal–metal interactions,107,109,116,137 and other special bonds within fullerenes. These effects can give rise to single-molecule magnetism.138–141 For example, lanthanide dimetallofullerenes featuring a single-electron Ln–Ln bond behave like single-molecule magnets and are therefore potential qubits for molecule-based quantum computing.99 Recently, Popov and coworkers discovered that anionic metallofullerenes can react with the Umemoto reagent II, resulting in the addition of CF3 groups to fullerenes, indicating electrophilic trifluoromethylation might be a useful method to derivate fullerenes.142 By employing this approach, M2⊂C80(CF3) (M = Tb, Y) monoadducts were synthesized, of which Tb2⊂C80(CF3) exhibits robust and remarkable magnetic properties with magnetic hysteresis up to 27 K. Apart from encapsulation of metals by larger fullerenes, Shinohara successfully obtained crystalline C60-based metallofullerenes Gd⊂C60(CF3)5 and La⊂C60(CF3)5 from their CS2 solution by vapor diffusion, in which CF3 groups highly improved the stability of these metallofullerenes.143 The electronic properties of endohedral fullerenes are tunable by the insertion of different metals or metal clusters.144,145 For instance, Lu3N⊂C80, a typical structure within the trimetallic nitride template (TNT) family, has a lower oxidation potential than C60 and exhibits high stability compared to other EMFs. This property makes it an ideal electron donor, and in 2019, Martin and coworkers used Lu3N⊂C80 in combination with C60 as an acceptor to construct an “all-fullerene” donor–acceptor system.146
4.2. Molecular tweezers as fullerene hosts
Molecular tweezers are molecular hosts with an open cavity and two identical binding sites for capturing guests (Fig. 11). By controlling the balance between rigidity and flexibility in the tweezers, distinct advantages can be provided for molecular recognition.147 Generally, the two binding sites can be bridged by a pH-responsive (e.g. pyridyl), ion-responsive (e.g. dipyridyl and crown ether), or photo-responsive (e.g. dithienylethene and azobenzene) linker, allowing the molecular tweezers to capture and release guests.148–150 This controllable recognition property has made molecular tweezers attractive tools in sensing, drug delivery and mixture separation. In the field of fullerene chemistry, tweezers have been studied for selectively extracting C60/C70 or other higher fullerenes from carbon soot.151–164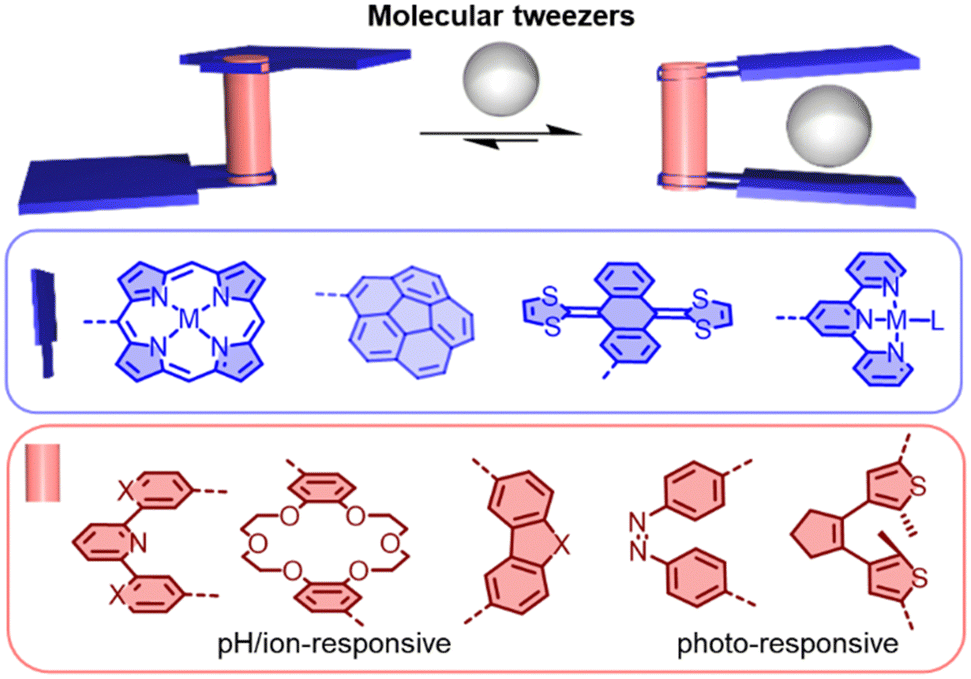 | ||
| Fig. 11 Schematic representation of switchable molecular tweezers (based on various linkers and binding motifs). | ||
Porphyrins have a strong affinity for fullerenes, thanks to their large π surface and the capability to undergo charge transfer interactions. For instance, Boyd and colleagues devised molecular tweezers for binding C60 and C70, using porphyrin units connected via a metal coordination bond.165,166 Álvarez and colleagues synthesized double-tweezers with a single porphyrin core containing eight pyrene units, capable of binding with C60 (and C70) and forming a 1:2 complex. Interestingly, the double-tweezers can transform into single-tweezers by coordinating the porphyrin core with Zn2+.167 Meanwhile, π-extended tetrathiafulvalene (exTTF), with its concave geometry and high electron-donating capacity, has been extensively utilized for binding fullerenes and carbon nanotubes.168,169 Notably, Martín and colleagues have shown that exTTF-based molecular tweezers can be covalently linked to carbon nanotubes. By binding C60 relatively strongly (logKa ≈ 3.0–3.1), interesting non-covalent C60/CNT hybrid materials can be obtained.170
Corannulene is a bowl-shaped polycyclic aromatic hydrocarbon (PAH), which is predisposed for the binding fullerenes, because it represents a fragment of C60. In recent years, several π-extended and N-embedded “buckybowls” have been synthesized.151 These buckybowls exhibit high affinity towards fullerenes through concave–convex π–π interactions when incorporated in tweezers architectures.171,172 Sygula and coworkers provided the first strong evidence of supramolecular binding between a corannulene-based molecular tweezers and C60 in solution and in the solid state. The authors found that this double concave host strongly binds to C60 to form a stable complex (Ka = 8600 M−1 in tolulene-d8).173 Stuparu and coworkers designed a family of corannulene-based amphiphilic polymers that can yield fullerene-rich water-soluble materials.174,175 Shinokubo and coworkers reported two types of azabuckybowl-based molecular tweezers that exhibited different affinities towards C60 and C70.151 The carbazole-linked tweezers preferentially binds to C70 over C60, while the phenanthrene-linked tweezers associated with C60 more strongly than with C70. Most host–guest systems can recognize guests, but releasing the guest without destroying the host structure is often a significant challenge. Alvarez and coworkers designed 2,2′-bipyridine-bridged molecular tweezers 23 that can capture and release fullerenes through in situ Cu(I) complexation and decomplexation. In the presence of Cu(I), the tweezers exhibited good binding affinities with C60 and C70 (Ka (C60) = 2 × 103 M−1, Ka (C70) = 5 × 103 M−1 (Fig. 12a), and the reversibility of the fullerene capture and release was demonstrated.176
 | ||
| Fig. 12 (a) Chemical structure of 2,2′-bipyridine-bridged molecular tweezers 23 and its in situ coordination/decoordination of C70 as monitored using 1H-NMR in CD2Cl2 at 298 K. Adapted from ref. 176 with permission from Royal Society of Chemistry, Copyright 2021. (b) Schematic representation of the formation of supramolecular polymers based on biscalix[5]arenes tweezers 24 and dumbbell fullerenes 25. Adapted from ref. 177 with permission from American Chemical Society, copyright 2021. | ||
In addition to PAH motifs, macrocycles can also act as binding sites in molecular tweezers. Haino and coworkers showed that biscalix[5]arenes covalently tethered together possess an ideal cavity for accommodating fullerenes. The Ka between the biscalix[5]arenes tweezers and C60 was found to be approximately 104 M−1 in toluene, significantly higher than the Ka between a single calix[5]arene and C60.178 When a molecule is equipped with two or more biscalix[5]arene tweezers, as demonstrated by molecule 24, supramolecular polymers and networks can be formed simply by addition of the dumbbell fullerene 25 (Fig. 12b). These hierarchically assembled supramolecular architectures are expected to pave the way for the development of stimuli-responsive fullerene-containing polymeric materials.177,179
4.3. Macrocycles as fullerene hosts
Pedersen, Lehn, and Cram's pioneering work on macrocyclic compounds, such as crown ethers and cryptands, established the foundations of supramolecular chemistry. Since then, there have been significant advances in the design and synthesis of new macrocyclic molecules, which have in many cases led to applications in drug delivery, sensing, and chemical separation technologies. Examples include cyclodextrins,180 calixarenes, cucurbiturils, pillar[n]arenes, pagoda[n]arene and cyclobis(paraquat-p-phenylene).181–190 Additionally, fully conjugated macrocycles like [n]CPPs, cyclo-porphyrins, and [n]cyclo-2,8-chrysenylenes with well-defined diameters and unique radial conjugation exhibit fascinating electronic and optical properties.191–204 These shape-persistent macrocycles offer significant advantages in constructing 1D nanotubes, 2D networks, and 3D complexes via self-assembly.205This review article primarily focusses on covalent macrocycles and metallomacrocycles, which have demonstrated their effectiveness as hosts for fullerene recognition (Fig. 13).162,206–243 These include highly strained and π-conjugated compounds such as [10]CPP and its derivatives,244–251 [4]cyclo-2,8-chrysenylene (26),252–254 porphyrinylene nanohoop (31),255 and [n]cyclodibenzopentalenes (33),256,257 which all exhibit exceptionally high affinity towards fullerenes (Ka > 105 M−1). Among these macrocycles, [4]cyclo-2,8-chrysenylene (26) and porphyrinylene nanohoop (31) exhibit outstanding fullerene affinity (Ka > 108 M−1) due to an extended π surface that maximizes concave–convex π–π interactions and further decreases the degrees of freedom in the host. One powerful strategy for constructing macrocyclic structures is coordination-driven self-assembly.258,259 For instance, Peris and coworkers synthesized a palladium-cornered metallomacrocycle 42 with four pyrene-bis(imidazolylidene) bridging ligands, which can encapsulate both C60 and C70 to form 42⊃C60 (Ka = 5.4 × 103 M−1 in CD3CN) and 42⊃C70 (Ka = 7.1 × 104 M−1 in CD3CN) complexes.260 By introducing additional homo-/hetero-macrocyclic structures to the backbone of macrocycles, e.g. in bismacrocycles, compounds with the capability to bind to multiple, distinct guests can be obtained.261–266 For example, Cong and coworkers reported a conjugated figure-of-eight oligoparaphenylene nanohoop (40) with adaptive cavities that can form 1:2 host–guest complexes with C60 and C70.267 More recently, Xu, Yam and von Delius reported the synthesis of two [n]cycloparaphenylene-pillar[5]arene ([n]CPP-P[5]A, n = 8 and 10) bismacrocycles by integrating P[5]A into the [n]CPP backbone. [n]CPP-P[5]A exhibits multiple guest recognition and promising properties of circularly polarized luminescence (glum ≈ 0.02), with [10]CPP-P[5]A showing potential for use in supramolecular polymer preparation.268
 | ||
| Fig. 13 Selected fullerene hosts: covalent macrocycles and metallomacrocycles. Side-chains are omitted for clarity. | ||
Nano-Saturn complexes consist of a fullerene molecule as a planetary body surrounded by a macrocycle featruing a perpendicular π-system. Achieiving such complexes is not trivial, because the perpendicular arrangement of π-system limits the strength of the van der Waals (vdW) dispersion interaction. Toyota and coworkers have nevertheless demonstrated that a cyclo-2,7-anthrylene hexamer can form a disk-type nano-Saturn complex with C60 through multiple CH–π interactions (Ka = 2.3 × 103 M−1 in toluene).269 Another promising candidate for preparing nano-Saturn complexes with fullerene is metal coordination macrocycles. However, constructing a stable and size-compatible disk-type metallomacrocycle is challenging because of the inherent lability of coordination interactions and the effects of steric crowding in the planar metallomacrocycle. Zhan and coworkers have recently reported the selective synthesis of [Cu10(2-methylimidazolate)10] 46 using C60 as a template.270 Remarkably, the ten methyl groups of the metallomacrocycle provide almost thirty CH–π interactions with the C60 or C70 molecule, stabilizing the disk-type metallomacrocycle (Fig. 14a). The interaction energy of their nano-Saturn complex is calculated to be much larger than that of most reported disk-type nano-Saturn complexes.
 | ||
| Fig. 14 (a) Selective synthesis of [Cu10(2-methylimidazolate)10] 46 utilizing C60 as the template, and the solid-state structure of nano-Saturn complex 46⊃C60. (b) Molecular models of megalo-cavitands 47 and corresponding C60 and C70 complexes. Adapted from ref. 271 with permission from Wiley-VCH, copyright 2022. (c) Synthesis of a supramolecular double-decker cage 48 and its selectively recognition with C70. (d) Formation of linear array of fullerenes in self-assembled porphyrin nanotube. Adapted from ref. 272 with permission from American Chemical Society, copyright 2014. | ||
In addition to their direct use as fullerene hosts, macrocyclic molecules can also serve as backbones to prepare molecular cavitands, cages and nanotubes that enable fullerene encapsulation in the third dimension. For example, Tiefenbacher and coworkers have recently reported a megalo-cavitand 47 with volumes of up to 814 Å3 by using an acridane[4]arene and four triptycenes as building blocks (Fig. 14b).271 They found that 47 exhibits a higher affinity for C70 (Ka = 1.2 × 106 M−1 in toluene) than C60 (Ka = 6.5 × 104 M−1 in toluene). Moreover, the authors discovered that cavitand 47 can selectively bind to C70 in the presence of C60, which may be useful for fullerene purification. Tanaka and coworkers have described the synthesis of a supramolecular double-decker cage 48 composed of two shape-persistent imine-bridged tetranuclear ZnII-macrocycles and four 1,4-diazabicyclo-[2.2.2]octane molecules for the specific recognition of ellipsoidal fullerenes (Fig. 14c).273 Additionally, Tani and coworkers have designed a family of phenothiazine/alkynyl-bridged cyclic porphyrin dimers bearing self-assembling 4-pyridyl groups (Fig. 14d). The phenothiazine-bridged dimers exhibit a higher affinity with both C60 (Ka ≈ 106 M−1 in toluene) and C70 (Ka ≈ 107 M−1 in toluene), and these dimers can self-assemble into a nanotube through π–π interactions of the pyridyl groups and C–H⋯N hydrogen bonds between porphyrin β-CH groups and pyridyl nitrogen donors.272,274 Thus, macrocycle-based cavitands, macrocyclic double-deckers and self-assembled nanotubes offer opportunities for the specific recognition of fullerene derivatives.275
The importance of fullerene radicals in energy conversion and storage applications has been highlighted in photoinduced charge transfer processes in fullerene-based D–A systems. However, these radicals are typically short-lived and labile under air. To address this challenge, Tagmatarchis and coworkers developed a supramolecular approach to stabilize fullerene radicals.276 By continuously illuminating a mixture of [10]CPP⊃(C59N)2⊂[10]CPP in 1-chloronaphthalene, the highly reactive azafullerene radical (C59N˙) was generated and immediately shielded by formation of the stable [10]CPP⊃C59N˙ complex. This shielding effect leads to exceptionally long-lived C59N˙ radicals, which are of interest for quantum information processing technologies,277 because dimerization is prevented (Fig. 15a). Additionally, Tao, Du and coworkers introduced [9]CPP to the active layer of fullerene organic solar cells (OSCs). This not only promotes charge transfer between poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene-co-3-fluorothieno[3,4-b]-thiophene-2-carboxylate] (PTB7-Th) and PC71BM, but also enhances charge transport between the PC71BM molecules by adjusting intermolecular π–π stacking. As a result, the ternary OSCs made from PTB7-Th, [9]CPP, and PC71BM achieved a high power conversion efficiency (PCE) of around 11%, almost one fifth higher than the PTB7-Th and PC71BM binary OSC devices.278
 | ||
| Fig. 15 (a) Structure and time dependence of the X-band EPR signal in 1-chloronaphthalene after the illumination at 532 nm has been switched off of [10]CPP⊃C59N˙. Adapted from ref. 276 with permission from Wiley-VCH, copyright 2019. (b) Conformation transformation of “Figure-eight” macrocycle 49 by adding guest 7,7,8,8-tetracyanoquinodimethane and C60, the side-chains are omitting for clarity. | ||
Conjugated macrocycles typically have rigid, inflexible cavities that restrict their ability to recognize a wide range of guest molecules and sometimes limit them to bind only a single type of guest. However, in 2021, Liu and coworkers reported a remarkable “Figure-eight” macrocycle 49, that possesses the ability to flexibly adjust its conformation in response to changes in the external environment, allowing it to accommodate a variety of guest molecules (Fig. 15b).279 This unique property of macrocycle 49 was demonstrated through its ability to assemble with planar, electron-deficient guest molecules, such as 7,7,8,8-tetracyanoquinodimethane, by adopting a boat-shaped conformation. The Ka of this complex was determined to be 1.0 × 103 M−1 in toluene at 323 K. Furthermore, the macrocycle demonstrated an even higher affinity towards C60, with a Ka of 1.1 × 104 M−1 in toluene at 323 K, and single crystal X-ray diffraction (SCXRD) confirmed that the macrocycle changed into a belt-shaped conformation in the presence of C60. These results suggest that macrocycle 49 is a promising candidate for the development of versatile host molecules capable of accommodating a diverse range of guest molecules.
Compared to covalent macrocycles, metallomacrocycles have demonstrated greater synthetic efficiency and diversity due error correction during their formation from small subcomponents. Metal coordination-driven self-assembly is a highly efficient strategy for constructing supramolecular architectures.280,281 Thanks to their kinetic reversibility, metallomacrocycles have wide applications in host–guest chemistry, particularly in the encapsulation and release of fullerenes.282–285 In 2020, Oppel reported a new torus-shaped metallomacrocycle 50 with an outer diameter of 31.7 Å. Zn(II) ions are octahedrally coordinated between two ligands of alternating orientation which in solution, bind fullerenes C60 and C70 in their spherical cavities (Fig. 16a).208 The fullerene encapsulations were characterized by SCXRD and NMR spectroscopy, and theoretical calculations were conducted to gain a deep understanding of host–guest interactions in these metallomacrocycles.
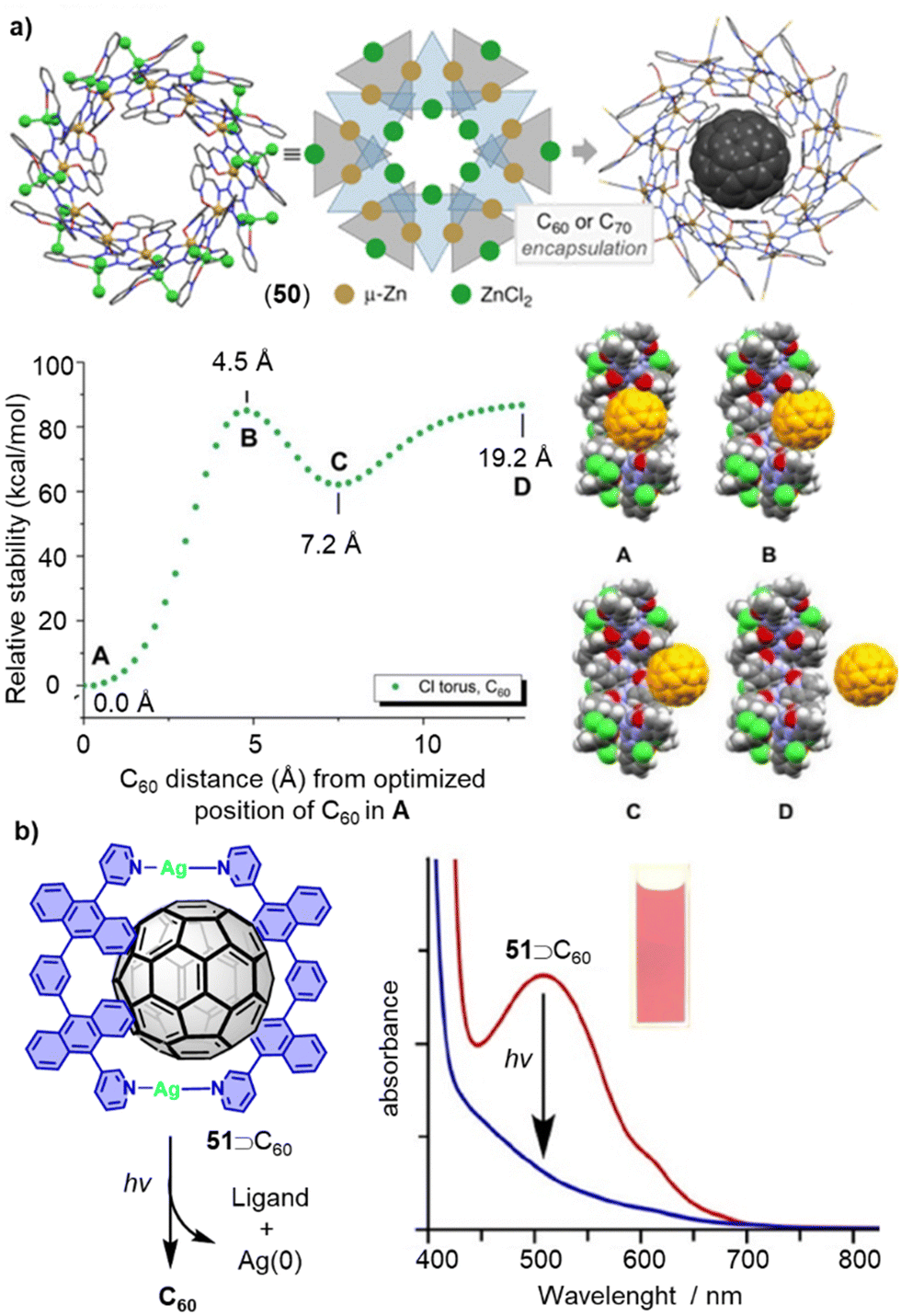 | ||
| Fig. 16 (a) Supramolecular metallocycle 50, schematic drawing and inclusion complex 51⊃C60 as well as energy diagram showing the relative stability as a function of the C60 distance from its position at the BP86/6-31G level of theory (A). Adapted from ref. 208 with permission from Wiley-VCH, copyright 2020. (b) Fullerene release from 51⊃C60 complex by photoirradiation. Adapted from ref. 214 with permission from American Chemical Society, copyright 2013. | ||
For certain applications of fullerene hosts, the release of encapsulated guest molecules from the host molecule is necessary. Although many macrocycles exhibit excellent fullerene recognition ability, the subsequent release of fullerenes remains challenging, especially in cases where the binding constant is very high. Typically, guest release from host–guest complexes can be triggered by chemical, electrochemical, heat stimuli, or the addition of secondary guests in a guest–exchange process. Yoshizawa and coworkers developed a method for releasing captured C60 upon UV-vis irradiation (Fig. 16b).214 Metallomacrocycle 51 was formed only in the presence of guest C60. A pale-yellow solution with a powdery suspension of solids was observed after exposing 51⊃C60 in CH3CN to UV-vis irradiation produced by a 36 W incandescent light bulb at room temperature for 1.5 h. The broad UV-vis absorption band of 52⊃C60 (400–700 nm) disappeared, indicating the release of C60 from the metallomacrocycle. A recovery yield of 68% was determined for C60, and the complex can be regenerated in ca. 60% yield from the UV-vis irradiated sample by adding AgNO3 at room temperature.
4.4. Cages as fullerene hosts
Cages are a unique class of supramolecular hosts due to their three-dimensional and rigid molecular structure, typically exhibiting a well-defined cavity. In contrast to covalent or metal–organic frameworks, cages are discrete and can be characterized with solution-based techniques. Syntheses are typically achieved by self-assembly, which is why cages often feature dynamic covalent bonds,286 labile metal–ligand bonds280,287 or weak non-covalent interactions.288 In recent years, the encapsulation of fullerenes in cages has been pursued by a large number of groups.16,20,289–292 This research is typically motivated by the desire to explore new host–guest chemistry and to endow the encapsulated fullerene with unusual properties. For instance, encapsulated fullerenes can exhibit improved solubility, (radical) stability, catalytic activity or modulated redox potentials.293–295 As discussed in section 2, encapsulated fullerenes can also undergo itero-, regio- and stereoselective addition reactions.52,296,297 The selective encapsulation of fullerenes is of interest for obtaining pure fullerenes from complex mixtures (e.g. soot).298–300PAH moieties are often employed as the core building blocks incorporated into cages due to their strong π–π or charge transfer interactions with fullerenes. Examples of such moieties include porphyrin,16,301,302 pyrene,303 anthracene,304 tetrathiafulvalene,305 carbon bowls, as well as some electron-deficient groups like naphthalene diimide (NDI), and perylene bisimide (PBI), etc.306 Coordination-driven self-assembled cages, with diverse shapes and sizes, also known as metallocages or metal–organic cages, have been constructed and widely used for the encapsulation of fullerenes.20,22,307,308 The dynamic nature of metallocages, owing to labile coordination bonds (e.g. Pd–N) allows large fullerene guests to enter cages with small windows and transformations between different complexes,207,302,309–312 Some exceptionally large metallocages were shown to bind multiple fullerenes,313 offering an opportunity to study cluster of fullerenes.302,314–316 For example, a large tetrahedron metallocage 52 containing nickel(II) porphyrins and Zn metals assembled by Nitschke and coworkers was used to encapsulate 1–4 equiv. of C60.316 Interestingly, co-encapsulation within the metallocage made it easier to reduce C60 to the C60˙− radical anion, in which according to theory is due to vdW interactions between multiple fullerenes (Fig. 17a). In related work, Yoshizawa and coworkers used a peanut-shaped polyaromatic metallocage 53,282,317 assembled from “W” ligands and Pd(II) metals, which allowed the encapsulation of two fullerenes separated by a distance of 6.4 Å (Fig. 17b). This discrete, non-contacted fullerene dimer undergoes sequential reduction in the cavity of the metallocage to generate (C60˙−)2, C60˙−·C602˙−, and (C602˙−)2. Furthermore, the stepwise encapsulation of two C60 molecules was achieved in a temperature-controlled fashion.
 | ||
| Fig. 17 Selected redox properties of fullerenes encapsulated within the cages. (a) Cyclic voltammograms porphyrin cage 52 encapsulating 0–4 equiv. C60. Adapted from ref. 316 with permission from American Chemical Society, copyright 2017. (b) Assembly of peanut-shaped polyaromatic metallocage-C60 complex [crystal structure of (C60)2⊂53] and schematic representation of the sequential reduction processes of (C60)2⊂53. Adapted from ref. 282 with permission from Wiley-VCH, copyright 2020. | ||
Purely organic cages have been synthesized using the well-established toolbox of dynamic covalent chemistry (DCvC).286,318,319 DCvC cages are typically more robust than metallocages and the products of self-assembly can be kinetically inert. The encapsulation of fullerenes into organic cages can therefore depend on the size of the cage windows, unless the cage is still dynamic, as in Beuerle's boronic ester cage.52 As an example for an organic cage with large windows, Zhang and coworkers synthesized a porphyrin and carbazole moieties-contained rectangular prismatic organic cage by utilizing alkyne metathesis.320 This cage exhibits highly selective encapsulation of C70 over C60 (KC70/KC60 > 1000), which was successfully used to isolate C70 from a mixture with C60. Of note, the encapsulated C70 was released by addition of excess trifluoroacetic acid and encapsulation of the fullerene was possible upon addition of triethylamine, highlighting the robustness of organic cages, which in this case enables the controlled a repeated guest encapsulation and release.
Trigonal prism-shaped cages also offer the opportunity to encapsulate fullerenes, as long as their size is in a suitable range. Recently, a trigonal prismatic nanobarrel constituted by three pyrene panels and two triangular windows with a diameter of 12.7 Å was synthesized by employing dynamic imine bond.321 This pyrene cage allows the encapsulation of C60 in poor solvents for the fullerene, which is a useful method for dissolving C60 in non-aromatic solvents (such as dichloromethane, chloroform, and 1,1,2,2-tetrachloroethane). In order to provide a better prediction for rational design of DCvC cages for C60, Jelfs and coworkers used an evolutionary algorithm to identify potential hosts for C60. The study showed that promising imine-based cages for encapsulation of C60 need to have suitable size, planar tri-topic aldehyde units with a low number of rotating single bonds, di-topic amine building blocks with functionality on adjacent carbon atoms and overall a highly symmetrical structure.322 Besides self-assembly based on reversible interactions, organic cages can also be synthesized by high-yielding irreversible reactions.323 Wu and coworkers reported a three-dimensional π-conjugated polyradicaloid prism-like cage consisting two benzene-1,3,5-triyl and three Chichibabin's hydrocarbon motifs as linker, which was prepared by Ni(COD)2-mediated Yamamoto homocoupling.324 The large cavity in the conjugated cage allowed selective encapsulation C70 over C60.
A new class of organic cationic viologen/porphyrin cages was developed by Stoddart and coworkers in recent years.325 A tetragonal organic porphyrin cage 54 with 8 positive charges was utilized to encapsulate C60 or C70 due to the suitable cavity within cage and the favorable D–A interaction between the porphyrins and fullerene guests (Fig. 18a).326 More recently, Lipke and coworkers reported the gram-scale synthesis of a different cationic porphyrin cage by the formation of pyridinium linkages between two complementary porphyrin “bowls” in the last step. This new cage 55 was found to bind fullerenes very strongly (Ka > 108 M−1 in MeCN) (Fig. 18b).327 Interestingly, although the cage binds C60 or C70 with strong affinities, the redox properties of fullerenes are not affected due to encapsulation, which is in contrast to other porphyrin metallocages. As both cages 54 and 55 exhibit stronger binding ability towards C70 over C60, the selective extraction of C70 from C60/C70 mixtures has also been achieved.
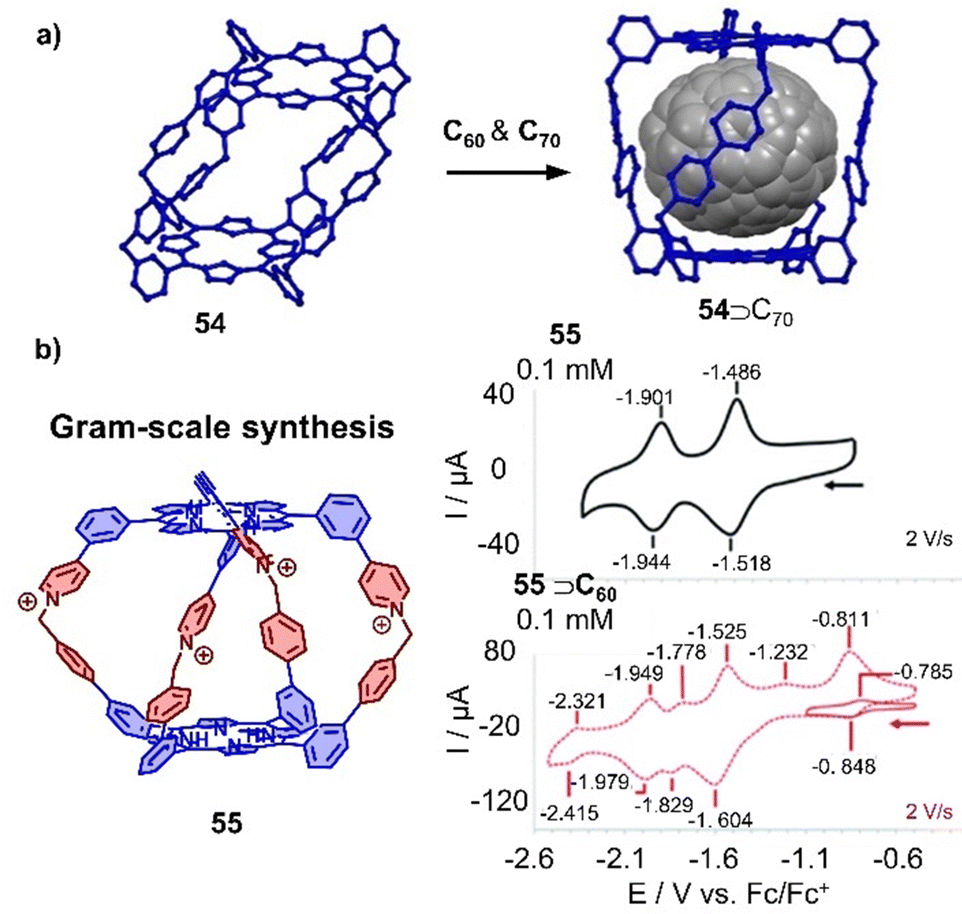 | ||
| Fig. 18 Organic cationic cages encapsulating fullerenes. (a) Selective extraction of C70 by a tetragonal prismatic porphyrin cage 54 (crystal structures). (b) Tetracationic cage 55 and cyclic voltammograms of 55 in comparison with C60⊂55. Adapted from ref. 327 with permission from Royal Society of Chemistry, copyright 2020. | ||
Encapsulation of fullerenes within cages has not only improved the solubility in common solvents,321 but also may tune the electronic properties of fullerenes due to strong π–π interactions with cage, which provide a reaction platform to modify fullerenes. Kim and coworkers constructed a series of well-defined 3D porphyrin boxes by employing dynamic covalent chemistry approach,328 of which the large cavity and suitable windows allow to encapsulate fullerenes and even occur those harsh condition reactions (Fig. 19a). Recently, they successfully conducted the inverse-electron-demand Diels–Alder reaction between fullerenes and 1,2,4,5-tetrazine within a Zn–porphyrin box 56, a reaction that normally requires very harsh reaction condition and longer reaction time with low yield. Interestingly, C60-tetrazine adduct is transferred to a bent-shaped C60-pyrazoline adduct through a hydration reaction and then released from the box with the addition of excess axle. The confined microenvironment within a cage also allows for photoredox chemical transformations of encapsulated fullerenes. Recently, a self-assembled subphthalocyanine capsule 57 was used as a reactor to accelerate the additions of (diaryl) methylamine and trifuoroethyl radicals to C60 under green light irradiation, which are typically photoredox transformations that occur within a cage (Fig. 19b).329,330
 | ||
| Fig. 19 Tuning the reactivity of fullerenes within cages. (a) Representation of insertion of tetrazine-based linear axle inside a porphyrin box 56 and functionalization of C60/C70 inside box. Adapted from ref. 328 with permission from Wiley-VCH, copyright 2022. (b) Assembly of a subphthalocyanine capsule-based molecular reactor (57) and the photoredox transformations of fullerenes. | ||
Traditionally, electron-rich moieties are adopted as the main components to build hosts for electron-deficient fullerenes, which enhances host–guest interactions through charge transfer. However, electron-deficient moieties like PBI,309,314 NDI,331 and triptycene carboxylic dianhydride294,332 can also be employed to construct cages, resulting in different host–guest interactions with fullerenes. For example, Würthner and coworkers found that a Fe4(PBI)6 tetrahedron cage assembled from octahedral Fe(II) ions and linear 2,2′-bipyridine modified PBI at the imide positions was capable of encapsulating two equivalents of C60.314 Encapsulation of C60 exhibits almost no effect on the absorption of cage, indicating weak interaction between PBI and C60 at the ground state, possibly because bulky groups at bay positions prevent the contact. Stang, Fang and coworkers also observed encapsulation of fullerenes in a PBI-based trigonal prism,309 assembled from Pt(II) and tetrapyridyl connected PBI at ortho positions. Similar as in Würthner's study, C60 or C70 encapsulation had a negligible effect on the both absorption and fluorescence spectra of the trigonal prism, indicating that the interaction is mainly based on dispersion. The encapsulation of fullerenes in cages comprising electron deficient subcomponents offers the opportunity to stabilize fullerene or cage once either of the two is reduced to the radical anion due to the favourable interaction between acceptor and the newly formed donor (radical anion). For example, Nitschke and colleagues constructed an NDI-based ZnII4L6 cage 58,331 where the NDIs could be reduced to the radical anion and used as the catalyst for the oxidative coupling of different tetraaryl borates to give biaryls (Fig. 20a). Furthermore, the catalytic reactivity of the radical cage was further enhanced by the presence of the C60 guest that plays an important role in stabilizing the NDI radical. C60˙− could act as a carrier for efficient charge harvesting and can be generated by obtaining single electrons from suitable electron donors. Generally, the lifetime of C60˙− is rather short (<1 s) in solution. Recently, Clever and coworkers found that a self-assembled electron-deficient cage 59 consisting of four triptycene carboxylic dianhydride ligands and two Pd(II) cations294 stabilized the C60˙− radical anion, which was attributed to the strong interaction between C60˙− and electron-deficient cage 59 (Fig. 20b). The chemical reduction of encapsulated C60 was achieved by adding 1-benzyl-1,4-dihydronicotinamide as a reducing regent with the 2 min irradiation of a white LED, and encapsulation which extended the half-life of C60˙− to 14 min in air and 893 min under inert conditions.
 | ||
| Fig. 20 (a) C–C bond formation catalyzed by C60⊂58. (b) Solid state structure of C60⊂59 and the UV-Vis-NIR spectra of C60˙−⊂59 measured under N2 atmosphere over time. Adapted from ref. 294 with permission from American Chemical Society, copyright 2021. | ||
The interaction between cages and fullerenes not only affects their electronic properties, but also their spin state properties. For example, Lützen and coworkers reported a metallocage with spin crossover behavior, assembled by using 5,10,15,20-tetrakis(4-aminophenyl)porphyrin or its zinc(II) complex,333 1H-4-imidazolecarbaldehyde, and iron(II) salts. The iron(II) centers in this metallocage 60 exhibit the high-spin state at room temperature and low-spin state in solution at low temperature. More interestingly, a “high-spin-stabilizing effect” through encapsulation of C70 was observed (Fig. 21).
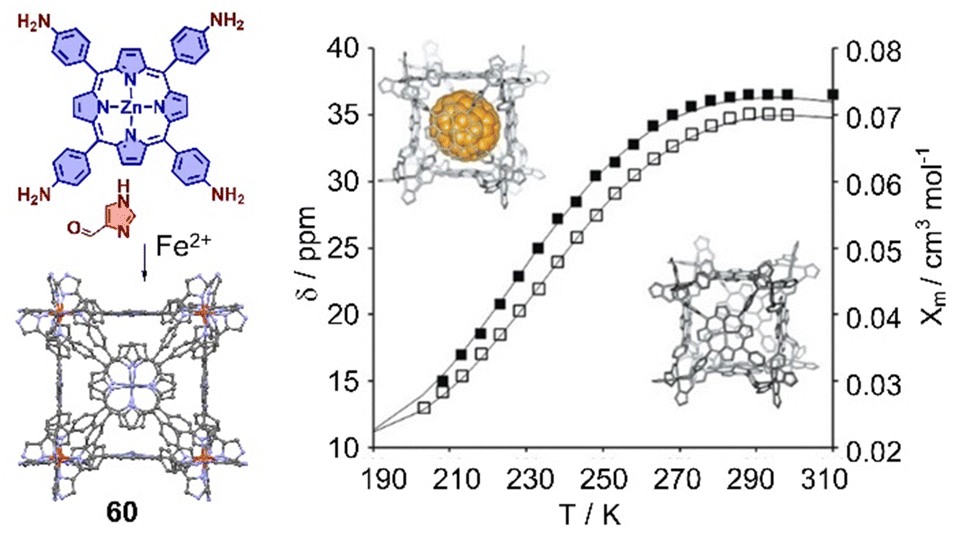 | ||
| Fig. 21 Self-assembly of an octanuclear metallocage 60 and chemical shift of selected protons in temperature-dependent 1H NMR experiments in CD3OD. Black lines represent the calculated molar susceptibility χm based on the ideal solution model. Adapted from ref. 333 with permission from Wiley-VCH, copyright 2017. | ||
4.5. Other fullerene hosts
Hosts for fullerene binding are not just limited to tweezers, macrocycles and cages. The recent use of other types of hosts will be summarized in this section, including curved or bowl-shaped molecules,17 proteins,334–336 polymers337,338 and carbon nanotubes.339,340Curved or bowl-shaped π-conjugated molecules are inherently more shape-complementary to fullerenes than planar compounds and possess other unique properties.17,341–354 For instance, many curved molecules are chiral and are more soluble than comparable planar analogues. Corannulene, which has already been discussed in the section on tweezers, is a prototypical curved π-conjugated molecule. As a fragment of C60, it has been a building block of choice for the binding of fullerenes.355,356 Dibenzo-[a,g]corannulene,357 a simple corannulene derivative, formed a co-crystal with C60 and C70 in 1:1 and 2:1 stoichiometry, respectively. The solid state structures indicated a strong interaction between corannulene and the fullerenes, with the shortest distance being only 3.14 Å. Shinokubo and coworkers used a nitrogen-embedded buckybowl as host for C60, and the 1:1 association of was supported by X-ray diffraction and UV-vis absorption and fluorescence titration experiments in 1,2-dichlorobenzene.348 A flexible decapyrrylcorannulene (DPC) host,358 developed by Zhang, Yang, Xie and coworkers, possesses ten pyrrole groups which resemble flexible ‘fingers’ on the periphery of the corannulene core. This host could bind 15 different fullerenes, including almost all commonly known types of fullerenes, such as pristine (C60, C70, C90), exohedral (six methanofullerene derivatives, three fullerene hydride derivatives, and one fulleroid derivative), endohedral (Sc3N@C80), dimeric and heteroderivatized [(C59N)2] structures, fulleroid and pentagon-fused fullerenes. Crystal structures of fullerenes with DPC reveal the high adaptivity that results from the presence of ten electron-rich ‘fingers’ at the outer rim of corannulene. Corannulene was also incorporated into organometallic complexes with the purpose of binding fullerenes. Peris and coworkers used a corannulene-functionalized di-N-heterocyclic carbene-AuI complex as receptor for C60.345 The binding was found to be on the order of 103 M−1 (toluene) and a host–guest ratio of up to 3:1 was confirmed by NMR spectroscopy and ITC titrations. Apart from corannulene derivatives,359–361 Nabeshima and colleagues developed a series of chiral concave π-systems comprising phosphorus atoms,343,347 which bind C60 due to the concave–convex interactions. In addition to buckybowls,362–365 a bowl-shaped nanobelt reported by Wu and coworkers, which could selectively capture C70 with a large binding constant (log(Ka) ≈ 5; in toluene) from a mixture of C60/C70 due to size and shape complementarity.366 Isobe and coworkers synthesized a nanometer-sized geodesic phenylene bowl comprising 20 phenylene units, which forms a 1:1 ball-in-bowl complex with C60.367 The association constant in chloroform was found on the order of 104 M−1 by 1H NMR spectroscopy and the complex structure was confirmed by SCXRD. Osuka, Kim and coworkers reported a series of porphyrin trimers, bearing additional carbonyl groups or methylene groups inserted between one of the β–β linkages of the porphyrin tapes. Among these hosts for C60, the methylene-linked syn-Ni(II) porphyrin trimer exhibited the strongest association constant of ca. 107 M-1 in toluene at 25 °C.368,369
π-Extended nanographenes with suitably sized cavities have also been utilized as hosts for fullerenes.370–372 For example, Wang and coworkers reported a Janusarene 61,342 possessing nineteen phenyl rings (seven constituting the hexaphenylbenzene core and the other twelve forming a “fence” around the two faces of the core), which was synthesized via an efficient cobalt-catalyzed cyclotrimerization of alkyne precursors. Host–guest complexation of 61 and C60 was supported by SCXRD, which revealed a ratio of 1:1 in the solid state. The crystal packing showed a 1D alternating supramolecular polymer (Fig. 22a),373 in which fullerene nestles inside the cavity formed by two janusarene molecules, indicating concave–convex interactions between 61 and C60. Wei and coworkers also reported a ‘Janus’ hexabenzocoronene derivative consisting of three triptycene units fused onto the periphery of coronene.350 The synthesis featured the condensation of syn-triveratrylbenzene and 2-formyltriptycene in one pot with high yield, and the product was found to host C60 (Ka = 4.9 × 103 M−1) and C70 (Ka = 6.9 × 103 M−1) in toluene. Wust and coworkers designed a series of D4-symmetric tetraoxa[8]circulenes,374 which possess an extended planar π-conjugation due to the introduction of oxygen atoms. One [8]circulene 62 was fused with four triptyceno subunits and could therefore serve as host for C60, which was demonstrated by SCXRD (Fig. 22b). Recently, Würthner and coworkers observed the co-assembly of C60 and a negatively curved polycyclic aromatic hydrocarbon 63, forming a supramolecular complex with an unusual 1:4 stoichiometry, which was characterized and analyzed by SCXRD and theoretical calculations.375 Interestingly, the complex topologically resembles of the fascinating carbon allotrope Schwarzite (Fig. 22c).376
 | ||
| Fig. 22 Co-crystals of Janusarene 61 (a), tetraoxa[8]circulene 62 (b) and negatively curved polycyclic aromatic hydrocarbon 63 (c) with C60. | ||
In addition to using large π-conjugated molecules, some small molecules also form host–guest complexes with fullerenes due to hydrogen bonding.377–383 For instance, Stefankiewicz and coworkers reported a hydrogen-bonded capsule, self-assembled by eight amino acid functionalized molecules 64 through 48 hydrogen bonds between the carboxyl and amine moieties of adjacent monomers (Fig. 23a), which was confirmed by SCXRD and multiple NMR methods in chlorinated solvents.384 The crystallographic analysis revealed that the monomer 64 formed an octameric supramolecular nanocapsule with the cavity volume up to 1719 Å3. The large inner cavity of capsule has been used to selectively encapsulate C70 from a mixture of C60/C70 in tetrachloroethane. Orentas, Wärnmark and coworkers developed a series of ureidopyrmidinone (UPy)-based hydrogen bonding tubes that exhibit intriguing dynamic behavior, such as self-sorting and solvent/guest-induced rearrangement.380,381,385,386 By adding C60, an octameric tube (65)4 comprising multiple hydrogen bonds was rearranged to capsular complex C60⊂(66)4 due to encapsulation of C60 (Fig. 23b), which was investigated by multiple NMR measurements.386 Recently, Maeda and coworkers reported a self-associating curved π-system,387 dipyrrolylbenzodiazepines (67), which forms a 1D supramolecular polymer in the solid state driven by the hydrogen bonds between a pyrrole NH as H bond donor and the diazepine nitrogen atom as H bond acceptor (Fig. 23c). The crystal structures revealed that this supramolecular polymer was successfully transformed into a large supramolecular macrocycle by the addition of C60, forming a supramolecular complex assembled by six compound 67 and one C60. The arrangement of monomer units and the strong concave–convex D–A interactions between 67 and C60 result in ultrafast electron transfer from 67 to C60.
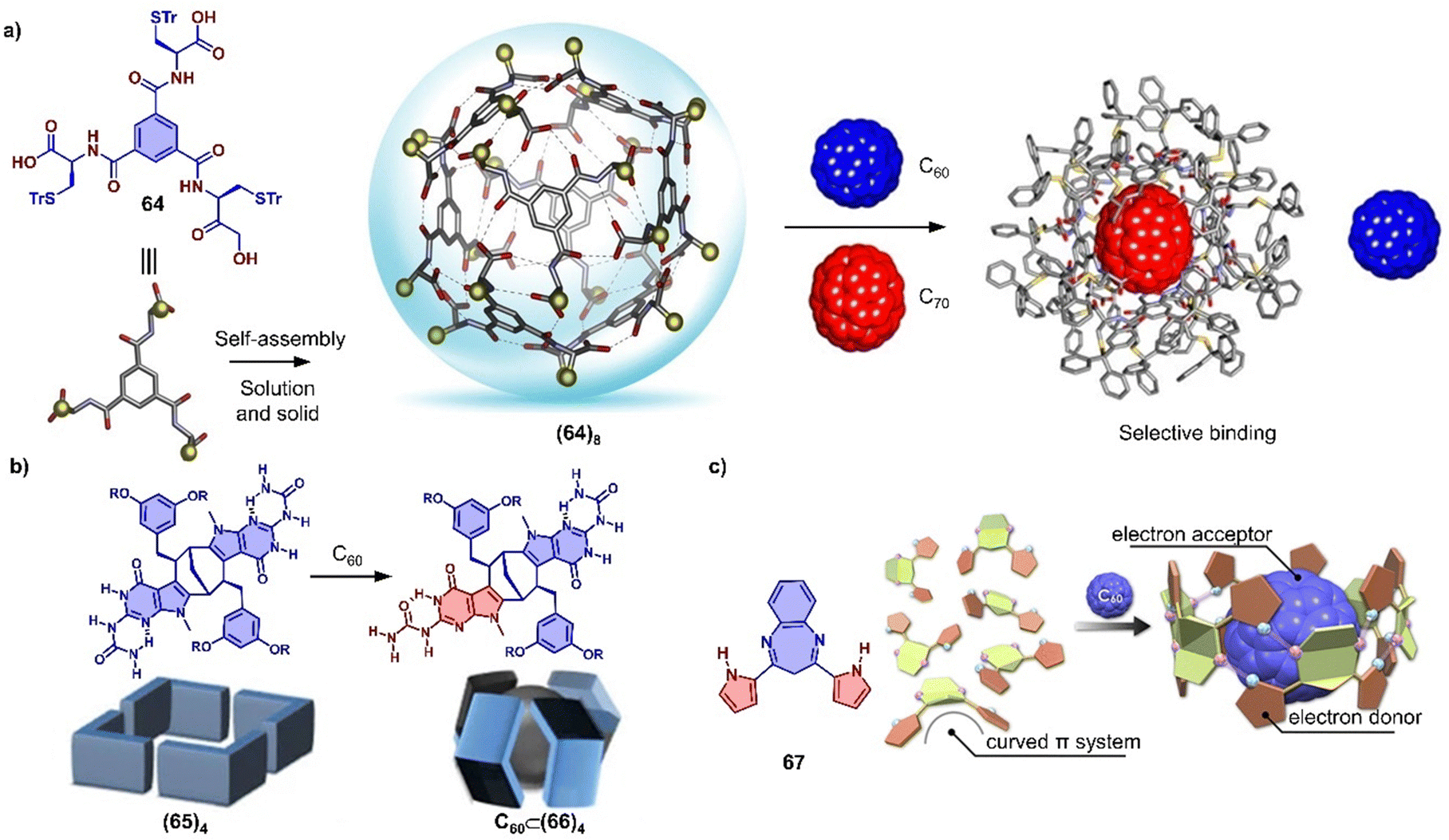 | ||
| Fig. 23 Hydrogen bond associated, adaptive systems. (a) Self-assembly of supramolecular nanocapsule (64)8 from compound 64 and selective encapsulation of C70 from a mixture of C60/C70. Adapted from ref. 384 with permission from Springer Nature, copyright 2017. (b) The symmetry breaking of monomer by rearrangement and schematic representation of starting tetramer (65)4 and capsular complex C60⊂(66)4 Adapted from ref. 386 with permission from Springer Nature, copyright 2017. (c) Structure of DPB 67 and schematic representation of supramolecular polymer to macrocycle transformation with addition of C60. Adapted from ref. 387 with permission from American Chemical Society, copyright 2020. | ||
Fullerenes are typically hosted by molecules with a rigid backbone, large π system and a suitably sized cavity. However, encapsulation of fullerenes by proteins, driven by multiple weak interactions, was also reported.388,389 The interactions between proteins with fullerenes and their derivatives include a very broad range, such as π–π, dispersion, hydrophobic, surfactant-like, cation–π, anion–π, hydrogen bonding, as well as electrostatic interactions, and are presumably acting synergistically,334 involving multiple functional groups. For instance, aliphatic moieties from amino acids like methionine, proline, alanine, leucine, valine, and isoleucine interact with the surface of C60,390,391 while amino acids with aromatic rings (e.g. phenylalanine) establish strong π–π stacking interactions with the fullerene.392 In addition, functional groups possessing positive and/or negative charges (e.g. lysine, aspartate and others) undergo surfactant-like interactions.391,393 Understanding the interaction between fullerene and protein by multiple methods (such as SCXRD, NMR, UV-vis, and fluorescence) is a complex challenge that promises insights into protein structure and dynamics.334
Utilizing multiple weak interactions to host fullerenes also represents a new method to construct capsules.394,395 Remarkably, Yoshizawa and coworkers achieved the encapsulation of fullerene in water by using a water-soluble adamantane capsule,394 where the driving force was the hydrophobic effect, dispersion and CH–π interactions. This approach demonstrates the potential of using weak interactions to create highly selective and efficient encapsulation systems.
5. Mechanically interlocked molecules
The mechanical bond396 is of interest for 21st century material science because it offers access to unique structural and dynamic properties as well as the ability to respond to external stimuli. Among the various types of mechanically interlocked molecules (MIMs), fullerenes are evidently good candidates as stoppers in rotaxanes, because the large and rigid carbon scaffold can prevent the dissociation of a macrocycle from a thread.397–399 Furthermore, fullerene-based D–A systems have been found to exhibit rapid charge separation and low reorganization energy values during photoinduced processes (see above).400 This unique property allows for the investigation of molecular motion and control of intramolecular photoinduced charge separation processes, which are essential steps in natural photosynthesis. As a result, the development of molecular fullerene-based MIM photosynthetic systems has gained considerable attention in recent years.398,400 The first fullerene-based mechanically interlocked molecule was reported in 1995 by Sauvage and colleagues,401 and since then, several methodologies have been developed to effectively incorporate fullerenes into pseudorotaxanes, rotaxanes, and catenanes. Metal–ligand exchange, hydrogen bonding and π–π interactionshave been instrumental for achieving these MIM syntheses.402–404Porphyrin–fullerene MIMs are the most frequently employed in photosynthetic model systems because they reliably provide photoinduced charge separation. On many occasions, both fullerenes and porphyrins have been utilized as stoppers in rotaxanes. In 2019, Weiss and coworkers reported a novel rotaxane 68 containing a strapped porphyrin as the shuttle and a fullerene stopper at the end of a mobile dumbbell (Fig. 24a).405 The position of the porphyrin-based shuttle was fixed via metal–ligand coordination and could be switched by the addition of coordinating ligands such as 10% pyridine-d5 in benzene-d6. Pyridine binds to the Zn-center opposite to the triazole, relocating the triazole outside of the phenanthroline pocket. The motion of the shuttle was clearly supported by comparing the chemical shifts of the alkyl protons. Moreover, transient absorption measurements revealed that solvent polarity influenced molecular motion of the rotaxanes upon charge separation.
 | ||
| Fig. 24 (a) Comparison of the chemical shifts and position of the dumbbell of 68 in benzene-d6 upon addition of 10% pyridine-d5. (b) Schematic depiction of a switchable D–A four-station C60-based [3]rotaxane 69via anion-induced molecular motion. | ||
Beer and coworkers have developed a multicomponent [3]rotaxane 69 as part of their efforts to create photoactive devices with multiple switching capabilities. The rotaxane includes a four-station bis-NDI axle, a centrally positioned C60 bis-triazolium component, and two macrocycles containing ferrocenyl-isophthalamide. The rotaxane was prepared using a chloride anion template methodology (Fig. 24b).406 In the chloride form, two ferrocenyl-functionalized macrocycles reside at the center of the axle, which triggers the activation of the NDI fluorophore and formation of a C60 fullerene-based charge-separated state. Conversely, when the chloride anions were replaced with hexafluorophosphate anions, the ferrocenyl-functionalized macrocycles shuttle to the peripheral NDI axle stations, leading to the formation of an NDI-containing charge-separated state and quenching of the NDI emission.
Macrocyclic arenes have garnered significant attention in supramolecular chemistry due to their easy synthesis and ability to selectively recognize guests.407 The small, electron-rich cavity of macrocyclic arenes allows them to capture electron-deficient molecules such as alkyl nitriles, pyridinium salts, and urea derivatives, making them ideal for developing stimuli-responsive materials and constructing MIMs.408,409 Nierengarten and coworkers functionalized C60 with pillar[5]arene (P[5]A) to create a fulleropillar[5]arene derivative 70, where each fullerene moiety interacts with three P[5]A subunits through intermolecular π–π and electrostatic interactions in the solid state.410,411 By encapsulating a diacyl chloride in P[5]A and carrying out a stoppering reaction, the authors were able to synthesize a photoactive [2]rotaxane 71 that comprises a central fullerene moiety and two terminal BODIPY stoppers (Fig. 25a). Even though the C60 moiety and the BODIPY stoppers are not covalently connected, efficient through-space excited state interactions have been observed in this architecture. In 2020, Wu and coworkers reported a “fulleropillar[4]arene”: a pillararene in which one arene moiety has been replaced by a trans-4 fullerene bisadduct. Interestingly, this unusual macrocycle exhibits significantly higher association (Ka ≈ 6600 M−1 in DMSO) with viologen derivatives than P[5]A,412 which allowed the authors to prepare a new type of pseudorotaxane.
 | ||
| Fig. 25 (a) Solid state packing of fulleropillar[5]arene (70) and corresponding photoactive [2]rotaxane 71. (b) Optimized structure of [10]cycloparaphenylene–fullerene [2]rotaxane 72. | ||
Constructing MIMs with large size and shape-persistent cavities, such as those found in prototypical carbon nanohoops or nanobelts, presents significant challenges, not least because these componds typically lack heteroatoms. In 2018, the von Delius group overcame this difficulty by reporting the successful synthesis of two discrete [2]rotaxanes (72) that feature a strained carbon nanoring [10]CPP, which exhibited intriguing photoinduced charge transfer properties (Fig. 25b).45 The strong concave–convex π–π interactions between [10]CPP and C60 monoadduct (Ka = 1.8 × 106 M−1 in toluene) enabled the highly efficient and regioselective synthesis of these two rotaxanes (trans-2: 10% and trans-3: 26%). Transient absorption studies revealed snapshots of the macrocycle's “bound” and “unbound” states along the trajectory of the thread. The exceptionally slow dissociation of the CPP ring from the fullerene binding site, along with very fast translation along the thread was later confirmed in high-level meta dynamics simulations413 and could pave the way towards MIMs in which the ring is transporting charge, which would have relevance for photovoltaics and photocatalysis.
Recently, the Ribas and von Delius groups used their Matryoshka approach (see Fig. 3) for the trans-3 selective bis-functionalization of C60 to achieve the synthesis of a new type of [2]catenane 74.414 These catenanes comprise a [10]CPP ring that is mechanically interlocked with a larger macrocycle that is formed in the final step of the synthetic route by Bingel bis-addition to C60. Because the malonate esters (73) are unsymmetric, this reaction could lead to an astonishingly complex reaction mixture with up to 22 spectroscopically distinguishable isomeric products. The combination of nanocapusle shadowmask (degraded in a controlled fashion after the reaction), [10]CPP template (ring incorporated into the product) and tether effect (use of bismalonates connected through a long spacer) was thus necessary to obtain >97% selectivity for trans-3 isomers along with ca. 30% yield (Fig. 26a). The authors demonstrate that for the optimal tether length (C14) an isomer fraction of up to 87% was obtained for one out of the three distinct trans-3 diastereomers (in,out-trans-3). The dynamic properties of this type of catenane were explored in silico by the Pavan group, who reported extremely fast translation of the [10]CPP along the non-fullerene parts of the larger macrocycle as well as a very long average residence time of 30 s on the fullerene binding site (Fig. 26b and C).
 | ||
| Fig. 26 (a) The synthetic strategy of C60/[10]CPP-catenanes (74). (b) Atomistic molecular model of the C14-C60/[10]CPP-catenane. (c) Free-energy diagram as a function of the angular position of [10]CPP along the larger ring of the catenane. | ||
6. 2D and 3D assemblies
Fullerenes and their derivatives have emerged as valuable building blocks to construct functional carbon materials, which are widely used in different fields, such as sensors, liquid crystals, optoelectronics, catalysts, and energy conversion.19,171,415,416 To obtain highly ordered fullerene 2D or 3D fullerene materials, numerous approaches have been developed.417,418 While covalent cross-linking establishes robust 2D frameworks, the principles of supramolecular chemistry are vital for establishing order during synthesis and can be harnessed to generate 3D vdW (hetero)structures.6.1. Assembly of ordered fullerene materials
2D carbon materials have attracted increasing attention due to their unique fundamental properties that promise applications in electronic and optical devices, as well as in charge storage, heterojunctions, hydrogenation and photovoltaic applications.417,418 Within the fullerene family, C60 has been widely used as a building block to construct 2D structures owing to its spherical shape and commercial availability. While the band gap of crystalline C60 is around 2.3 eV,419 theoretical studies predict that the band gap of both polymeric and vdW-based C60 monolayers ranges from 0.6 to 1.0 eV,420 which is significantly lower than that of any experimentally isolated monolayer of an organic semiconductor. Furthermore, the band gap of C60-based 2D materials highly depends on the symmetry of the structure and the distance between individual C60 moieties.420,421 Recently, some important advances have been made in preparations of highly ordered 2D fullerene-based materials, representing a new approach to synthesize 2D materials with novel (optoelectronic) properties.422–424 For example, Zheng and coworkers successfully synthesized covalently bonded C60 networks by using an organic cation slicing strategy.422 C60 molecules formed radical anions upon heating with Mg, and new C–C bonds formed between adjacent C60 moieties through [2+2] cycloaddition reactions. X–ray diffraction studies revealed that the bulk crystals exhibit a layered structure of alternative polymeric C60 layers and Mg atomic layers. The Mg layers act as linkers between the adjacent C60 layers by Mg–C bonds with the length of 2.22–2.37 Å, which made the C60 layers cannot be exfoliated by simple mechanical exfoliation.Thus, the authors used an organic cation slicing strategy treating the bulk crystals with tetrabutylammonium salicylate (TBAS). This led to cleavage of the Mg–C bonds and the formation of magnesium salicylate and tetrabutylammonium cations (NBu4+) that replaced the Mg cations. (Fig. 27a) The NBu4+-intercalated C60 crystals were subsequently exfoliated by gentle manual shaking furnishing a few-layer quasi-tetragonal phase (qTP) and a monolayer quasi-hexagonal phase (qHP). Both two 2D materials exhibit great crystallinity and unique topological structures. Compared with graphene and individual fullerene molecules, the monolayer polymeric C60 possesses a moderate band gap of 1.6 eV and good thermodynamic stability. The asymmetric lattice structure of the monolayer induces significant in-plane anisotropic properties, such as anisotropic phonon modes and conductivity, which can be attributed to the unique structure of the monolayer. Later in the same year, Peng rationalized the experimental band gap of these polymeric C60 materials in silico. The calculations also suggested that these monolayer C60 materials have suitable band gaps, charge carrier mobilities and band edges for photocatalytic water splitting.425
 | ||
| Fig. 27 (a) Schematic of organic cation slicing exfoliation and crystal structures of the bulk single crystal of qHP-C60 and qTP-C60 as well as their monomer C60. Adapted from ref. 422 with permission from Springer Nature, copyright 2022. (b) Schematic of the CVT technique used for the growth of (Mg4C60)∞ single crystals, iFFT and AFM image of a few-layer “graphullerene” flake, the log of the conductance (σ) versus temperature (T) for a 70 nm-thick (Mg4C60)∞-based device (a fit to a thermally activated (Arrhenius) model (dashed green line), inset is a typical device and corresponding four-terminal measurement scheme). Adapted from ref. 423 with permission from Springer Nature, copyright 2023. | ||
Recently, Roy, Nuckolls and coworkers reported on “graphullerene”,423 a graphene-like hexagonal layer of polymeric C60 linked by covalent bonds (Fig. 27b), synthesized using the chemical vapour transport (CVT) method to grow single crystals of a (Mg4C60)n network, followed by removal of magnesium by dilute acid. This material exhibits a much higher thermal conductivity compared to molecular C60, likely due to the in-plane covalent bonding of layered polymeric C60. In addition, Moiré-type superlattices were found by high-resolution transmission electron microscopy (HR-TEM) and near-field nano-photoluminescence spectroscopy. However, the methods used for small-scale preparation of carbon structures with covalently cross-linked C60 limit detailed characterization and represent a hurdle for applications. In this respect, an important advance was made in 2023 by Zhu, Ruoff and coworkers who reported the gram-scale synthesis of long-range ordered porous carbon (LOPC),424 a new type of carbon. The synthesis started from C60 and was catalyzed by α-Li3N at 550 °C and ambient pressure (Fig. 28). LOPC consists of broken C60 cages mostly bonded via sp2 carbon atoms, as evidenced by X-ray diffraction, Raman spectroscopy, magic-angle spinning solid-state NMR spectroscopy, aberration-corrected transmission electron microscopy and neutron scattering. LOPC possesses remarkable conductivity, with a value of 1.17 × 10−2 S cm−1 at room temperature and the conduction is close to the result from a combination of metallic-like transport over short distances punctuated by carrier hopping at a temperature lower than 30 K. Simulations of LOPC reveal that graphullerene is a metastable structure that exists as intermediate during the transformation from a fullerene-type to a graphene-type carbon allotrope and represents a transition from semiconducting to metallic property with increasing temperature. Recently, Du and coworkers explored the photocatalytic water splitting properties of a few-layer C60 network, which furnished production rates of H2 and H2O2 of 91 and 116 μmol g−1 h−1, respectively.426 Collectively, these recent breakthroughs offer new opportunities for exploring the fundamental properties, supramolecular properties and potential applications of covalently bonded 2D carbon materials.
 | ||
| Fig. 28 Atomic structure of LOPC, TEM images of LOPC particles, Cu Kα (λ = 0.15418 nm) X-ray diffraction patterns with simulation for LOPC, and direct current voltage–current curves of three membranes made by mixing each carbon material with 5wt%; polytetrafluoroethylene. Adapted from ref. 424 with permission from Springer Nature, copyright 2023. | ||
2D or 3D fullerene crystals are different from these covalently cross-linked 2D materials, because they rely only on non-covalent forces such as π- and vdW interactions.19,417,418,427,428 A large amount of 2D fullerene crystals with great diversity of shapes, such as 2D hexagons, 2D nanorhombus or hexagonal 2D nanosheets have been obtained by using the liquid–liquid interfacial participation (LLIP) method.429–431 For example, Ariga and coworkers obtained a new class of C60 crystals with bimodal pore architectures by employing LLIP with the use of solvents isopropyl alcohol (IPA), benzene and carbon tetrachloride (CCl4). By changing the ratio of benzene and CCl4, the shapes of 2D crystals were tuned. Interestingly, the bimodal pore crystals exhibited 2D hexagonal plate-like morphology and offered enhanced electrochemically active surface areas compared to pristine C60.432 Sathish and coworkers reported 2D hexagonal C60 nanosheets using the LLIP method at the CCl4/alcohol interface. The size of 2D C60 crystals could be tuned by changing the alcohol (anti)solvent and the diameters of hexagonal nanosheets varied from ∼7.5 μm, ∼2.5 μm, and ∼500 nm (IPA, ethanol, and methanol, respectively).433
The synthesis of 3D fullerene crystals, which are mostly based on C60 or C70 and their derivatives, is highly dependent on the shapes of fullerenes.434 C70 3D crystals were first prepared by Choi and coworkers in 2010 by using the precipitation approach.435 Since then, several other methods have been developed to obtain fullerene 3D crystals, including static liquid–liquid precipitation, re-precipitation, solvent vapor annealing, and drop-drying.417 When using different methods, polymorphic structures of 3D fullerene crystals can be obtained that exhibit distinct properties such as enhanced fluorescence emission, hydrophobicity and photocurrent response, high surface-to-volume ratio beneficial for electrocatalytic hydrogen evolution reaction and sensing.417,418 Recently, Yang and coworkers developed a universal approach based on the LLIP method associated with ultrasonication to prepare the endohedral fullerene-based 3D crystals with tunable crystal shape.434 Three metal nitrile “clusterfullerene” (M3N⊂Ih-C80, M = Tb, Er, and Sc) 3D crystals were successfully obtained, and the shape of crystals could be easily switched by changing the solvent ratio of solvent to antisolvent. The crystal shape-dependent emission of the three clusterfullerenes was studied, revealing that crystals with dice shape emit stronger light than the cube-shaped crystals (Fig. 29a). Such an enhancement of photoluminescence in highly crystalline C70 and higher fullerene C78 relative to their powder states was also found by others, and was attributed to the increased crystallinity of fullerenes together with the decreased intermolecular interactions.435–437 The formation of fullerene-based 3D materials not only provides an ordered arrangement of carbon cages, but also produces a regular network of pores that are typically larger in the case of less symmetric C70. By changing the crystal preparation method, the porous structure can also be tuned. Shrestha, Ariga and coworkers prepared highly crystalline C70 cubes with the average edge lengths of ca. ∼3.4 ± 0.4 μm that possess holes extending 1–1.5 μm deep from the surfaces via dynamic LLIP.438 Interestingly, the holes on the surfaces of the C70 cube could be closed by regrowing a thin layer of fullerene, and subsequently be reopened by local irradiation via electron beam.
 | ||
| Fig. 29 (a) SEM images of cube- and dice-shaped Tb3N⊂C80 microcrystals and fluorescence spectra of Tb3N⊂C80 dice, cube, and powder. Adapted from ref. 434 with permission from Wiley-VCH, copyright 2019. (b) Dynamic behavior of a hexakis[60]fullerene 75. Adapted from ref. 439 with permission from Royal Society of Chemistry, copyright 2021. | ||
In addition to the formation of 3D crystalline materials by using pristine fullerenes, fullerene derivatives have also been utilized as building blocks to construct 3D frameworks or porous materials.440–445 These frameworks are assembled by non-covalent interactions, and their structures can be tuned by manipulating the dynamic interactions within frameworks. Beuerle and coworkers reported a series of hexakis-substituted C60 adducts bearing twelve carboxylic acid groups, and their incorporation as the polytopic organic linker into the hydrogen-bonding frameworks in solid state446,447 and MOFs interlinked by metal ions (such as Zn2+, Ca2+, Cu2+, and Cd2+).448,449 The interfullerene distances within the frameworks could be controlled by either alkyl space length between C60 and carboxylic acid group or the type of cross-linking.446–449 Recently, Martin, Costa and coworkers created dynamic molecular crystalline frameworks by using weak “sticky fingers” vdW interactions439,450 based on a hexakis-substituted C60 adduct 75 synthesized using the Bingel–Hirsch reaction (Fig. 29b). The hexakis-substituted C60 adduct 75 forms two different crystals from ethanol with and without Fe(BF4)2. Transformation between two polymorphs could be achieved by heating with and without ethanol. These materials are highly dynamic, such that exposure to hydrazine vapor induced the selective hydrogenation of crystalline materials and a structural change. The molecular movements in the lattice and the selective reaction can be observed directly by single-crystal to single-crystal diffraction (Fig. 29b).
Self-assembled monolayers (SAMs) have been widely used for surface modification and act as the crucial interlayers and electronically active layers in organic electronic devices (such as organic light emitting diodes, organic photovoltaics, and organic thin film transistors). Not surprisingly, C60-functionalized SAMs exhibit interesting properties.451–456 For example, Peukert and coworkers tuned the molecular order of C60 functionalized phosphonic acid monolayers by changing the ratio of alkyl phosphonic acids (PA) and C60-functionalized octadecyl phosphonic acids (C60-PA) on alumina substrates.455 A pronounced maximum in sum-frequency intensity of the C60 band is observed for SAMs with ∼75% C60-PA and ∼25% PA. By using the same method, Clark, Halik and coworkers further confirmed that a mixture of C60-functionalized and nonfunctionalized spacer molecules can lead to a morphology with improved charge transport in self-assembled monolayer field-effect transistors.453 By changing the ratio of C60-PA and PA from 100:0, 70:30, 50:50, and 30:70, the maximum drain currents of these monolayers were successfully tuned and quantum mechanical calculations revealed conduction pathways within the fullerene monolayers.
A series of fullerene amphiphiles have been synthesized,457–459 different from neutral derivatives, whose relevant interfacial chemistry has been summarized in 2019.460 Recently, Nakamura and coworkers described a pseudo-C5 symmetric fullerene amphiphile 76 attached with five 4-benzoic acid groups. The toluene/1-butanol/water solution of this compound spontaneously forms a 3 nm thick, free-standing 2D film as 1-butanol and toluene are evaporating gradually during a few hours at the water/air interface. (Fig. 30) The film was stabilized by hydrogen bonding between two fullerene layers. The size of this large-area film was up to several tens of cm2 and the photoconductivity of this film (transferred to a gold comb electrode) was determined as 1.4 × 10−4 S cm−1. Furthermore, the film was laminated into a multilayer film either by using large amount of 76 solution or repeating the preparation procedure several times.461
 | ||
| Fig. 30 Structure of pseudo-C5 symmetric fullerene amphiphile 76, assembly of 2D film, the 2D film on 160.6 ± 0.1 nm thick SiO2/Si wafer (φ = 10.2 cm) (the thickness of six points were calculated by interference fringe shifts from visible-light reflection spectra), and sequential lamination of film on 288.8 nm thick SiO2/Si wafer to form single, double, and triple films. Adapted from ref. 461 with permission from Wiley-VCH, copyright 2022. | ||
Fullerene liquid crystals (LCs) have attracted considerable attention, because they offer a combination of the excellent optoelectronic properties of fullerenes and the unique properties of liquid crystals. There are two approaches to construct fullerene LCs: the “molecular LC approach” is based on the covalent linking of fullerenes with large liquid crystal mesogens. The “supramolecular LC” method is based on supramolecular self-assembly.462 Using the molecular LC approach, nematic, cholesteric, smectic, and columnar phases could be achieved by introducing cholesterol and other functional groups to C60via flexible spacers or via a rigid “shuttlecock” geometry.463 However, the molecular LC approach is limited by the low content of fullerenes, which “dilutes” the optical and photophysical properties of fullerenes. The supramolecular LC approach was first developed by Nakamura and coworkers in 2002464 and further refined by other groups.465 This method relies on fullerene derivatives consisting of two parts: fullerenes and/or other PAHs provide π–π interactions between aromatic moieties, whereas and soft parts such as long alkyl chains provide vdW interactions. Both parts facilitate the assembly of highly ordered supramolecular structures with high aspect ratios, which exhibit liquid crystalline behaviour. The high fullerene content endows the fullerene-based LC with optoelectronic properties that are difficult to achieve with conventional molecular LCs.466 Combining fullerene LCs with 2D crystals or superlattices takes these materials properties even further.467 Recently, Tu, Li and coworkers employed a series of tetrablock-mimic azobenzene-containing C60 dyads 77 (n = 4, 7, 8, 9, 12) to construct supramolecular LCs.468 This approach allows the manipulation of smectic supramolecular LC phases by changing the alkyl tail length of dyads (Fig. 31a). These materials exhibit excellent electron mobility of ca. 1.5 × 10−3 cm2 V−1 s−1 due to the favourable combination of LC properties and 2D crystals. In a more recent study, well-defined superlattices were observed in supramolecular fullerene LCs by utilizing hierarchical self-assembly with a sphere-cone molecule as the building block 78. The formation of the superlattice was shown to improve the transient electron conductivity of the material (Fig. 31b).463
 | ||
| Fig. 31 (a) Molecular structure of tetrablock-mimicazobenzene-containing C60 dyads 77 (n = 4, 7, 8, 9, 12), molecular packing model for dyads in the ST phase and SQ phase, with face-centered tetragonal fullerene packing in the 2D crystals viewed along the [001] zone and the [110] zone (blue dots indicate voids between the packed molecules and orange rectangles alkyl-tail-substituted azobenzene moieties). Adapted from ref. 468 with permission from Wiley-VCH, copyright 2018. (b) The sphere-cone molecule 78, representation of molecular packing, and TEM image of layered superlattice structure. Adapted from ref. 463 with permission from American Chemical Society, copyright 2020. | ||
Self-assembled fullerene materials can act as sensors, catalysts, photoconductors or semiconductors in field-effect transistors.417,418 For instance, Li and coworkers prepared long-range M3N⊂C80 (M = Sc, Lu) single-crystal microwires by utilizing a pillar-structured template, which exhibited highly sensitive photoconductivity with a response time as low as 0.1 s, and also were employed in field-effect transistors.469 Molecular gels have been used as spatially confined templates for the growth of C60 crystals to obtain super-long crystalline C60 fibers and large-area 2D crystals, which were utilized as photodetectors with high performance by Liu, Fang, and coworkers.470,471 The dice-shaped Sc3N⊂C80 3D crystals have been utilized as a support for the Pt-catalyzed methanol oxidation by Yang and coworkers. Catalyst performance was shown to be improved due to the larger surface area when compared with the cube shape crystals.472 Shrestha, Ariga and coworkers prepared hierarchically structured C70 cubes and corn-husk-shaped fullerene C60 crystals, which have been successfully applied as sensors for volatile aromatic solvents and acid vapors, respectively.473,474
6.2. Co-assembly of fullerenes and organic molecules
Fullerenes and their derivatives have been studied as building blocks to construct supramolecular (co)polymers in solution. Beside the work by Haino on helically organized fullerene arrays (see Section 4.2 or Fig. 12b),177 many other groups have made efforts to construct fullerene-based supramolecular polymers. For example, Langa and coworkers synthesized a fullerene-bis-Zn–porphyrin e-bisadduct as the monomer,475 which assembled into large donut structures driven by charge transfer interactions between porphyrin-bearing arms and fullerenes. This self-assembled D–A polymer possesses long-lived charge separated states upon light irradiation, specially, the lifetime of final charge-separated state is upon to 40 μs, which is relevant for single-component light harvesting devices. Sessler and coworkers employed a thiopropyl-functionalized tetrathiafulvalene-annulated calix[4]pyrrole (TTP-C[4]P) and phenyl C61 butyric acid (PCBA) as monomers to assemble an alternating supramolecular polymer in a 7:3 mixture of CHCl3 and CS2. The self-assembly is driven by the charge transfer interaction between electron rich pocket of TTF-C[4]P and fullerene and the hydrogen bonding between TTF-C[4]P and carbolic acid moiety.476 The supramolecular polymer could be disaggregated by addition of organic acid or electrolysis. Later, Sessler and coworkers reported an extended tetrathiafuvalene-porphyrin macrocycle, which acts as a ball-and-socket receptor for C60 and C70. This macrocycle and fullerenes assemble into 3D supramolecular organic frameworks (SOFs) in the solid state. The C70-based SOF exhibits remarkable electrical conductivity (σ = 1.3 × 10−8 S cm−1 at 298 K).211 These supramolecular polymers offer unique advantages over traditional covalent polymers due to the reversible nature of noncovalent interactions, such as response to the redox, pH, heating, light, or small molecules.
Pristine fullerenes have been successfully co-assembled with molecules to fabricate novel functional materials. For example, Jeong, Jang, and coworkers prepared well-defined hierarchical nanostructures, consisting of a host–guest complex between pyrene-based tweezers 79 and C60. Due to its layered structure that comprises a 2D array of C60 moieties, the material exhibited high electron mobility of 1.7 × 10−2 cm2 V−1 s−1 (Fig. 32a).477 Although several porphyrin/fullerene supramolecular co-assemblies/crystals have been reported,478–481 photoinduced charge transfer between these two organic semiconductors was rarely studied, and these co-assemblies/crystals exhibited short lifetimes of charger transfer states and low charge mobility. In a co-crystal of a zinc-metalated porphyrin box 80 with C60/C70,482 Kim and coworkers observed a tightly packed square-planar core of four C60 or C70 surrounded by six porphyrin boxes. This tight packing pattern provides high charge mobility and allows for forming long-lived charge-separated states. Relative to crystalline box 80, a significant enhancement of photoconductivity was observed in architectures, 10-fold and 3-fold enhancement in φΣμ for 80/4C60 and 80/4C70, respectively. The photoconductivity of C70-based material is lower than C60-based material, which was ascribed to different electronic couplings between porphyrins and C60/C70. (Fig. 32b).
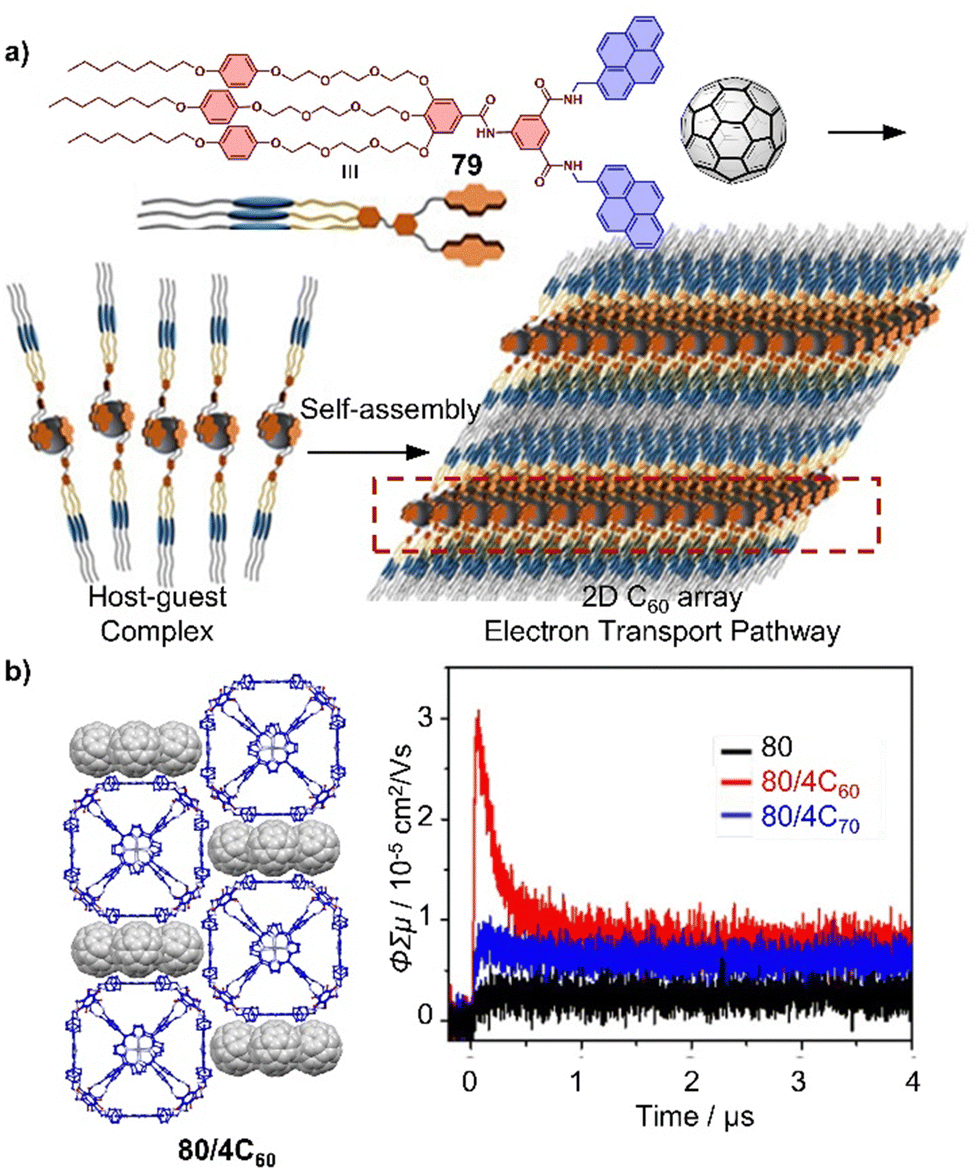 | ||
| Fig. 32 (a) Hierarchical assembly of bispyrene tweezers 79 and fullerene. Adapted from ref. 477 with permission from American Chemical Society, copyright 2019. (b) Co-assembly of porphyrin box 80 with four equivalents C60 and flash-photolysis time-resolved microwave conductivity of 80, 80/4C60, and 80/4C70. Adapted from ref. 482 with permission from American Chemical Society, copyright 2020. | ||
Metal–organic frameworks (MOFs) and covalent-organic frameworks (COFs) have emerged as promising materials with potential applications in a wide range of research fields.483 Because most MOFs or COFs have relatively large pores, the encapsulation of guests has been widely explored and fullerenes are an evident choice due to their relatively large size and optoelectronic properties.484–494 Berna and coworkers employed a flexible benzylic amide macrocycle attached with two carboxylic acid groups as the ligand to prepare copper(II)- and zinc(II)-based MOFs, which could selectively encapsulate C60 from a mixture of C60 and C70.484 The encapsulation of fullerenes was used to modulate the optoelectronic properties of MOFs due to the electronic interaction with hosts, leading to the unique properties and applications. For instance, Zhu and colleagues employed a zirconium-based MOF 81 as a host,489 composed of 1,3,6,8-tetrakis(4-benzoate) pyrene (TBAPy) linkers and Zr-oxo nodes, to encapsulate C60 (Fig. 33a). The uneven charge distribution in C60⊃81 provides a robust built-in electric field, which is 10.7 times higher than that in 81. Using this material, photocatalytic hydrogen evolution was found to be significantly enhanced thanks to the encapsulation of C60. The interplay between MOF hosts and fullerenes was also demonstrated to boost photoelectric conductivity.486,488 Heinke and coworkers constructed a crystalline porphyrin-based MOF (82) incorporating C60,487 providing rapid charge separation. Thanks to the efficient formation of holes and electrons, good photoconductivity was observed with an on–off photocurrent ratio of two orders of magnitude (Fig. 33b). The chirality of complex fullerene derivatives (e.g. certain fullerene bis-adducts) is a fascinating topic. However, imparting chirality to pristine C60 without installation of substituents is far from trivial. To this end, Uemura and coworkers employed a chiral MOF (83) hosting the highly symmetric C60 using a self-assembly strategy (Fig. 33c), of which C60 could be incorporated into the chiral channels of MOF.485 This approach can therefore endow highly symmetric, achiral compounds with chirality.
 | ||
| Fig. 33 Selected encapsulations of fullerenes into MOFs to achieve: (a) improved photocatalytic hydrogen evolution, (b) improved photoconduction, (c) chirality transfer from MOF to C60. Adapted from ref. 489, 487 and 485 with permission from Wiley-VCH, copyright 2023, 2019, 2021 for (a), (b) and (c), respectively. | ||
Surface co-assembly is a powerful method for creating well-organized 2D arrays that exhibit excellent performance in photovoltaic devices, sensors, and catalysts.427 For example, the Rabe group used hydrogen bonding to self-assemble a trimesic acid monolayer as the template for mono- or bilayers of complexes of fullerenes and oligothiophene macrocycles.206 Recently, Tanaka and coworkers reported the formation of a periodic monolayer of spatially separated C60 moieties on an Au(111) surface using carbazole-salphens or Ni-salphens containing macrocycles as fullerenes hosts. The pattern of discrete C60 units on the surface was thermally stable up to 200 °C under ambient pressure.212 In related work, Zeng and coworkers created ordered 2D C60 patterns on a highly oriented pyrolytic graphite (HOPG) surface using host–guest complexes of aggregation-induced emissive macrocycles and fullerenes.495 Rosei and coworkers employed a 2D covalent organic framework as surface-confined template to host the C70 moieties, imposing anchoring effects on the LC growth of C70 molecules, forming several fullerene-based LC mesophases, which cannot be observed under other conditions.496 These findings highlight the potential of fullerene-based surface co-assemblies for developing advanced materials and functional devices.
7. Summary and outlook
Thanks to the rapidly decreasing price of C60 (as low as 20 USD per gram) and the relevance of fullerenes as organic electronic materials, fullerene-based supramolecular chemistry has witnessed remarkable advances in the past decade. For instance, selective fullerene functionalization methods were developed by encapsulating fullerenes in supramolecular hosts and thus shutting down undesired reaction pathways or enhancing reactivity. With the help of such selectively modified fullerenes, supramolecular dyads and mechanically interlocked architectures have been prepared that exhibit unique optoelectronic systems. Endohedral fullerenes and open-cage fullerenes offer not only an opportunity to tune fundamental properties of fullerenes, but also a unique “playground” for the creation of functional supramolecular architectures.Over the past decade, a large number of effective hosts for fullerenes have been synthesized, and the relevant host–guest complexes have shown significant potential in selective functionalization reactions, catalysis, the stabilization of short -lived compounds and the tuning of spin properties. Fullerenes have also been utilized as building blocks to construct covalent or non-covalent 2D/3D carbon materials, which show great potential for electron transport, conductivity, liquid crystallinity and catalysis. We expect that the recent trend towards the preparation of ordered, high-performance fullerene materials will continue and that increasing effort will be devoted towards harnessing the unique spin properties of non-conventional fullerenes, which will require further advances in both synthesis and supramolecular chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. X. acknowledges funding from the National Science Foundation of China (22201064). X. C. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. M. v. D. acknowledges funding by the ERC (802428, SUPRANET), the German Research Foundation (DFG, projects DE1830/2-1, DE1830/5-1, SFB953 “Synthetic Carbon Allotropes”, TRR234 “CataLight”, EXC 2154 “POLiS: Post Lithium Ion Storage”) and the Federal Ministry of Education and Research (BMBF, project QuE-MRT).References
- A. Hirsch, Nat. Mater., 2010, 9, 868–871 CrossRef CAS PubMed.
- K. S. Novoselov, K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- Y. Hu, C. Wu, Q. Pan, Y. Jin, R. Lyu, V. Martinez, S. Huang, J. Wu, L. J. Wayment, N. A. Clark, M. B. Raschke, Y. Zhao and W. Zhang, Nat. Synth., 2022, 1, 449–454 CrossRef.
- H. L. Anderson, C. W. Patrick, L. M. Scriven and S. L. Woltering, Bull. Chem. Soc. Jpn., 2021, 94, 798–811 CrossRef CAS.
- K. Kaiser, L. M. Scriven, F. Schulz, P. Gawel, L. Gross and H. L. Anderson, Science, 2019, 365, 1299–1301 CrossRef CAS PubMed.
- H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- F. Diederich, R. Ettl, Y. Rubin, R. L. Whetten, R. Beck, M. Alvarez, S. Anz, D. Sensharma, F. Wudl, K. C. Khemani and A. Koch, Science, 1991, 252, 548–551 CrossRef CAS.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS.
- O. Vostrowsky and A. Hirsch, Chem. Rev., 2006, 106, 5191–5207 CrossRef CAS PubMed.
- H. W. Kroto, Nature, 1987, 329, 529–531 CrossRef CAS.
- F. Diederich and R. L. Whetten, Acc. Chem. Res., 1992, 25, 119–126 CrossRef CAS.
- O. Vostrowsky and A. Hirsch, Chem. Rev., 2006, 106, 5191–5207 CrossRef CAS PubMed.
- N. Martin and J.-F. Nierengarten, Supramolecular Chemistry of Fullerenes and Carbon Nanotubes, Wiley-VCH, 2012 Search PubMed.
- K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189–197 RSC.
- E. M. Perez and N. Martin, Chem. Soc. Rev., 2008, 37, 1512–1519 RSC.
- G. C. Vougioukalakis, M. M. Roubelakis and M. Orfanopoulos, Chem. Soc. Rev., 2010, 39, 817–844 RSC.
- S. S. Babu, H. Mohwald and T. Nakanishi, Chem. Soc. Rev., 2010, 39, 4021–4035 RSC.
- C. Garcia-Simon, M. Costas and X. Ribas, Chem. Soc. Rev., 2016, 45, 40–62 RSC.
- H. Yi, G. Zeng, C. Lai, D. Huang, L. Tang, J. Gong, M. Chen, P. Xu, H. Wang, M. Cheng, C. Zhang and W. Xiong, Chem. Eng. J., 2017, 330, 134–145 CrossRef CAS.
- C. Fuertes-Espinosa, M. Pujals and X. Ribas, Chem, 2020, 6, 3219–3262 CAS.
- S. B. Beil and M. von Delius, Org. Mater., 2021, 3, 146–154 CrossRef CAS.
- J. Zheng, L. Huang, C.-H. Cui, Z.-C. Chen, X.-F. Liu, X. Duan, X.-Y. Cao, T.-Z. Yang, H. Zhu, K. Shi, P. Du, S.-W. Ying, C.-F. Zhu, Y.-G. Yao, G.-C. Guo, Y. Yuan, S.-Y. Xie and L.-S. Zheng, Science, 2022, 376, 288–292 CrossRef CAS PubMed.
- L. Jia, M. Chen and S. Yang, Mater. Chem. Front., 2020, 4, 2256–2282 RSC.
- A. T. Barrows, A. J. Pearson, C. K. Kwak, A. D. F. Dunbar, A. R. Buckley and D. G. Lidzey, Energy Environ. Sci., 2014, 7, 2944–2950 RSC.
- W. Xu, L. Zheng, T. Zhu, L. Liu and X. Gong, ACS Appl. Mater. Interfaces, 2019, 11, 34020–34029 CrossRef PubMed.
- A. Montellano López, A. Mateo-Alonso and M. Prato, J. Mater. Chem., 2011, 21, 1305–1318 RSC.
- C. Bingel, Chem. Ber., 1993, 126, 1957–1959 CrossRef CAS.
- A. Hirsch, I. Lamparth and H. R. Karfunkel, Angew. Chem., Int. Ed. Engl., 1994, 33, 437–438 CrossRef.
- S. H. Hoke, J. Molstad, D. Dilettato, M. J. Jay, D. Carlson, B. Kahr and R. G. Cooks, J. Org. Chem., 1992, 57, 5069–5071 CrossRef CAS.
- P. A. Liddell, J. P. Sumida, A. N. Macpherson, L. Noss, G. R. Seely, K. N. Clark, A. L. Moore, T. A. Moore and D. Gust, Photochem. Photobiol., 1994, 60, 537–541 CrossRef CAS.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- O. V. Boltalina, A. A. Popov, I. V. Kuvychko, N. B. Shustova and S. H. Strauss, Chem. Rev., 2015, 115, 1051–1105 CrossRef CAS PubMed.
- T. Cao, N. Chen, G. Liu, Y. Wan, J. D. Perea, Y. Xia, Z. Wang, B. Song, N. Li, X. Li, Y. Zhou, C. J. Brabec and Y. Li, J. Mater. Chem. A, 2017, 5, 10206–10219 RSC.
- F. Zhang, W. Shi, J. Luo, N. Pellet, C. Yi, X. Li, X. Zhao, T. J. S. Dennis, X. Li, S. Wang, Y. Xiao, S. M. Zakeeruddin, D. Bi and M. Gratzel, Adv. Mater., 2017, 29, 1606806 CrossRef.
- W. Shi, Q. Zhuang, R. Zhou, X. Hou, X. Zhao, J. Kong and M. J. Fuchter, Adv. Energy Mater., 2023, 13, 2300054 CrossRef CAS.
- T. M. N. Trinh, F. Schillinger, S. Guerra, E. Meichsner, I. Nierengarten, U. Hahn, M. Holler and J. F. Nierengarten, Eur. J. Org. Chem., 2021, 3770–3786 CrossRef CAS.
- M. J. van Eis, P. Seiler, F. Diederich, R. J. Alvarado and L. Echegoyen, Chem. Commun., 2000, 1859–1860 RSC.
- M. J. van Eis, P. Seiler, L. A. Muslinkina, M. Badertscher, E. Pretsch, F. Diederich, R. J. Alvarado, L. Echegoyen and I. Pérez Núñez, Helv. Chim. Acta, 2002, 85, 2009–2055 CrossRef CAS.
- L. Ethordevic, L. Casimiro, N. Demitri, M. Baroncini, S. Silvi, F. Arcudi, A. Credi and M. Prato, Angew. Chem., Int. Ed., 2021, 60, 313–320 CrossRef.
- J. Dannhauser, W. Donaubauer, F. Hampel, M. Reiher, B. Le Guennic, B. Corzilius, K. P. Dinse and A. Hirsch, Angew. Chem., Int. Ed., 2006, 45, 3368–3372 CrossRef.
- B. Kräutler, T. Müller, J. Maynollo, K. Gruber, C. Kratky, P. Ochsenbein, D. Schwarzenbach and H.-B. Bürgi, Angew. Chem., Int. Ed. Engl., 1996, 35, 1204–1206 CrossRef.
- G. Bottari, O. Trukhina, A. Kahnt, M. Frunzi, Y. Murata, A. Rodriguez-Fortea, J. M. Poblet, D. M. Guldi and T. Torres, Angew. Chem., Int. Ed., 2016, 55, 11020–11025 CrossRef CAS PubMed.
- Y. Xu, R. Kaur, B. Wang, M. B. Minameyer, S. Gsanger, B. Meyer, T. Drewello, D. M. Guldi and M. von Delius, J. Am. Chem. Soc., 2018, 140, 13413–13420 CrossRef CAS PubMed.
- E. Ubasart, O. Borodin, C. Fuertes-Espinosa, Y. Xu, C. Garcia-Simon, L. Gomez, J. Juanhuix, F. Gandara, I. Imaz, D. Maspoch, M. von Delius and X. Ribas, Nat. Chem., 2021, 13, 420–427 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek, Jr., Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- M. R. Ceron, M. Izquierdo, A. Aghabali, J. A. Valdez, K. B. Ghiassi, M. M. Olmstead, A. L. Balch, F. Wudl and L. Echegoyen, J. Am. Chem. Soc., 2015, 137, 7502–7508 CrossRef CAS PubMed.
- M. Chen, Y. Zeng, G. Chen and Y. Qiu, Nanomaterials, 2022, 12, 2355 CrossRef CAS PubMed.
- M. Nambo, Y. Segawa, A. Wakamiya and K. Itami, Chem. – Asian J., 2011, 6, 590–598 CrossRef CAS PubMed.
- V. S. Neti, M. R. Ceron, A. Duarte-Ruiz, M. M. Olmstead, A. L. Balch and L. Echegoyen, Chem. Commun., 2014, 50, 10584–10587 RSC.
- V. Leonhardt, S. Fimmel, A. M. Krause and F. Beuerle, Chem. Sci., 2020, 11, 8409–8415 RSC.
- C. Fuertes-Espinosa, C. García-Simón, M. Pujals, M. Garcia-Borràs, L. Gómez, T. Parella, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem, 2020, 6, 169–186 CAS.
- A. Salazar, M. Moreno-Simoni, S. Kumar, J. Labella, T. Torres and G. de la Torre, Angew. Chem., Int. Ed., 2023, 62, e202311255 CrossRef CAS PubMed.
- E. E. Maroto, S. Filippone, M. Suarez, R. Martinez-Alvarez, A. de Cozar, F. P. Cossio and N. Martin, J. Am. Chem. Soc., 2014, 136, 705–712 CrossRef CAS PubMed.
- Z. Lu, T. K. Ronson, A. W. Heard, S. Feldmann, N. Vanthuyne, A. Martinez and J. R. Nitschke, Nat. Chem., 2023, 15, 405–412 CrossRef CAS PubMed.
- Y. Hashikawa, S. Okamoto, S. Sadai and Y. Murata, J. Am. Chem. Soc., 2022, 144, 18829–18833 CrossRef CAS PubMed.
- M. A. Lebedeva, T. W. Chamberlain and A. N. Khlobystov, Chem. Rev., 2015, 115, 11301–11351 CrossRef CAS PubMed.
- D. M. Guldi, Chem. Commun., 2000, 321–327 RSC.
- H. Imahori, K. Hagiwara, T. Akiyama, M. Aoki, S. Taniguchi, T. Okada, M. Shirakawa and Y. Sakata, Chem. Phys. Lett., 1996, 263, 545–550 CrossRef CAS.
- K. G. Thomas, M. V. George and P. V. Kamat, Helv. Chim. Acta, 2005, 88, 1291–1308 CrossRef CAS.
- G. N. Lim, C. O. Obondi and F. D'Souza, Angew. Chem., Int. Ed., 2016, 55, 11517–11521 CrossRef CAS PubMed.
- S. H. Lee, C. T. Chan, K. M. Wong, W. H. Lam, W. M. Kwok and V. W. Yam, J. Am. Chem. Soc., 2014, 136, 10041–10052 CrossRef CAS PubMed.
- W. Wang, R. Sun, J. Guo, J. Guo and J. Min, Angew. Chem., Int. Ed., 2019, 58, 14556–14561 CrossRef CAS.
- M. Daboczi, J. Kim, J. Lee, H. Kang, I. Hamilton, C. T. Lin, S. D. Dimitrov, M. A. McLachlan, K. Lee, J. R. Durrant and J. S. Kim, Adv. Funct. Mater., 2020, 30, 2001482 CrossRef CAS.
- F. D'Souza and O. Ito, Chem. Commun., 2009, 4913–4928 RSC.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Angew. Chem., Int. Ed., 2011, 50, 8342–8344 CrossRef CAS PubMed.
- Y. Xu, B. Wang, R. Kaur, M. B. Minameyer, M. Bothe, T. Drewello, D. M. Guldi and M. von Delius, Angew. Chem., Int. Ed., 2018, 57, 11549–11553 CrossRef CAS PubMed.
- F. Wessendorf, J. F. Gnichwitz, G. H. Sarova, K. Hager, U. Hartnagel, D. M. Guldi and A. Hirsch, J. Am. Chem. Soc., 2007, 129, 16057–16071 CrossRef CAS PubMed.
- J. L. Sessler, J. Jayawickramarajah, A. Gouloumis, T. Torres, D. M. Guldi, S. Maldonado and K. J. Stevenson, Chem. Commun., 2005, 1892–1894 RSC.
- S. Vela, S. Bauroth, C. Atienza, A. Molina-Ontoria, D. M. Guldi and N. Martin, Angew. Chem., Int. Ed., 2016, 55, 15076–15080 CrossRef CAS PubMed.
- L. Moreira, J. Calbo, J. Arago, B. M. Illescas, I. Nierengarten, B. Delavaux-Nicot, E. Orti, N. Martin and J. F. Nierengarten, J. Am. Chem. Soc., 2016, 138, 15359–15367 CrossRef CAS PubMed.
- L. Moreira, J. Calbo, B. M. Illescas, J. Arago, I. Nierengarten, B. Delavaux-Nicot, E. Orti, N. Martin and J. F. Nierengarten, Angew. Chem., Int. Ed., 2015, 54, 1255–1260 CrossRef CAS.
- C. B. Kc and F. D'Souza, Coord. Chem. Rev., 2016, 322, 104–141 CrossRef CAS.
- O. Trukhina, M. Rudolf, G. Bottari, T. Akasaka, L. Echegoyen, T. Torres and D. M. Guldi, J. Am. Chem. Soc., 2015, 137, 12914–12922 CrossRef CAS PubMed.
- N. Zarrabi, C. O. Obondi, G. N. Lim, S. Seetharaman, B. G. Boe, F. D'Souza and P. K. Poddutoori, Nanoscale, 2018, 10, 20723–20739 RSC.
- A. Bagaki, H. B. Gobeze, G. Charalambidis, A. Charisiadis, C. Stangel, V. Nikolaou, A. Stergiou, N. Tagmatarchis, F. D'Souza and A. G. Coutsolelos, Inorg. Chem., 2017, 56, 10268–10280 CrossRef CAS PubMed.
- C. B. Kc, K. Ohkubo, P. A. Karr, S. Fukuzumi and F. D'Souza, Chem. Commun., 2013, 49, 7614–7616 RSC.
- N. Zarrabi, C. Agatemor, G. N. Lim, A. J. Matula, B. J. Bayard, V. S. Batista, F. D’Souza and P. K. Poddutoori, J. Phys. Chem. C, 2018, 123, 131–143 CrossRef.
- A. Amati, P. Cavigli, A. Kahnt, M. T. Indelli and E. Iengo, J. Phys. Chem. A, 2017, 121, 4242–4252 CrossRef CAS PubMed.
- R. F. Enes, J. J. Cid, A. Hausmann, O. Trukhina, A. Gouloumis, P. Vazquez, J. A. Cavaleiro, A. C. Tome, D. M. Guldi and T. Torres, Chem. – Eur. J., 2012, 18, 1727–1736 CrossRef CAS PubMed.
- B. Fu, Y. Che, X. Yuan, L. Sun, H. Xu, J. Zhao and L. Liu, Dyes Pigm., 2021, 196, 109754 CrossRef CAS.
- M. A. Collini, M. B. Thomas, V. Bandi, P. A. Karr and F. D'Souza, Chem. – Eur. J., 2017, 23, 4450–4461 CrossRef CAS PubMed.
- Y. Che, X. Yuan, L. Sun, H. Xu, X. Zhao, F. Cai, L. Liu and J. Zhao, J. Mater. Chem. C, 2020, 8, 15839–15851 RSC.
- M. E. El-Khouly, D. K. Ju, K. Y. Kay, F. D'Souza and S. Fukuzumi, Chem. – Eur. J., 2010, 16, 6193–6202 CrossRef CAS PubMed.
- D. Badgurjar, S. Seetharaman, F. D'Souza and R. Chitta, Chem. – Eur. J., 2021, 27, 2184–2195 CrossRef CAS PubMed.
- C. B. Kc, G. N. Lim, P. A. Karr and F. D'Souza, Chem. – Eur. J., 2014, 20, 7725–7735 CrossRef CAS PubMed.
- C. Fan, W. Wu, J. J. Chruma, J. Zhao and C. Yang, J. Am. Chem. Soc., 2016, 138, 15405–15412 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- E. E. Maroto, M. Izquierdo, S. Reboredo, J. Marco-Martinez, S. Filippone and N. Martin, Acc. Chem. Res., 2014, 47, 2660–2670 CrossRef CAS PubMed.
- E. J. Gonzalez Lopez, A. M. Sarotti, S. R. Martinez, L. P. Macor, J. E. Durantini, M. Renfige, M. A. Gervaldo, L. A. Otero, A. M. Durantini, E. N. Durantini and D. A. Heredia, Chem. – Eur. J., 2022, 28, e202103884 CrossRef CAS PubMed.
- S. Vidal, J. Marco-Martinez, S. Filippone and N. Martin, Chem. Commun., 2017, 53, 4842–4844 RSC.
- Y. Zhao, Y. Cotelle, L. Liu, J. Lopez-Andarias, A. B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS PubMed.
- J. Lopez-Andarias, A. Bauza, N. Sakai, A. Frontera and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 10883–10887 CrossRef CAS.
- M. Yamada, K. Sahara, M. Koizumi, Y. Maeda and M. Suzuki, Chem. – Eur. J., 2023, 29, e202300877 CrossRef CAS PubMed.
- J. Lopez-Andarias, A. Frontera and S. Matile, J. Am. Chem. Soc., 2017, 139, 13296–13299 CrossRef CAS PubMed.
- L. Bao, P. Peng and X. Lu, Acc. Chem. Res., 2018, 51, 810–815 CrossRef CAS PubMed.
- W. Cai, C. H. Chen, N. Chen and L. Echegoyen, Acc. Chem. Res., 2019, 52, 1824–1833 CrossRef CAS PubMed.
- F. Liu, L. Spree, D. S. Krylov, G. Velkos, S. M. Avdoshenko and A. A. Popov, Acc. Chem. Res., 2019, 52, 2981–2993 CrossRef CAS PubMed.
- S. Jalife, J. Arcudia, S. Pan and G. Merino, Chem. Sci., 2020, 11, 6642–6652 RSC.
- D. J. Cram and J. M. Cram, Container Molecules and Their Guests,The, Royal Society of Chemistry, 1997 Search PubMed.
- J. R. Heath, S. C. O'Brien, Q. Zhang, Y. Liu, R. F. Curl, F. K. Tittel and R. E. Smalley, J. Am. Chem. Soc., 1985, 107, 7779–7780 CrossRef CAS.
- L. Feng, Y. Hao, A. Liu and Z. Slanina, Acc. Chem. Res., 2019, 52, 1802–1811 CrossRef CAS PubMed.
- L. Bao, Y. Li, P. Yu, W. Shen, P. Jin and X. Lu, Angew. Chem., Int. Ed., 2020, 59, 5259–5262 CrossRef CAS PubMed.
- W. Cai, J. Alvarado, A. Metta-Magana, N. Chen and L. Echegoyen, J. Am. Chem. Soc., 2020, 142, 13112–13119 CrossRef CAS PubMed.
- Q. Deng and A. A. Popov, J. Am. Chem. Soc., 2014, 136, 4257–4264 CrossRef CAS PubMed.
- A. Moreno-Vicente, Y. Rosello, N. Chen, L. Echegoyen, P. W. Dunk, A. Rodriguez-Fortea, C. de Graaf and J. M. Poblet, J. Am. Chem. Soc., 2023, 145, 6710–6718 CrossRef.
- W. Xiang, X. Jiang, Y. R. Yao, J. Xin, H. Jin, R. Guan, Q. Zhang, M. Chen, S. Y. Xie, A. A. Popov and S. Yang, J. Am. Chem. Soc., 2022, 144, 21587–21595 CrossRef CAS.
- J. Zhuang, R. Morales-Martinez, J. Zhang, Y. Wang, Y. R. Yao, C. Pei, A. Rodriguez-Fortea, S. Wang, L. Echegoyen, C. de Graaf, J. M. Poblet and N. Chen, Nat. Commun., 2021, 12, 2372 CrossRef CAS PubMed.
- Q. Meng, L. Abella, W. Yang, Y. R. Yao, X. Liu, J. Zhuang, X. Li, L. Echegoyen, J. Autschbach and N. Chen, J. Am. Chem. Soc., 2021, 143, 16226–16234 CrossRef CAS PubMed.
- M. Chen, R. Guan, B. Li, L. Yang, C. Niu, P. Jin, G. W. Wang and S. Yang, Angew. Chem., Int. Ed., 2021, 60, 7880–7886 CrossRef CAS PubMed.
- Y. Hao, Y. Wang, V. Dubrovin, S. M. Avdoshenko, A. A. Popov and F. Liu, J. Am. Chem. Soc., 2021, 143, 612–616 CrossRef CAS PubMed.
- Q. Meng, L. Abella, Y. R. Yao, D. C. Sergentu, W. Yang, X. Liu, J. Zhuang, L. Echegoyen, J. Autschbach and N. Chen, Nat. Commun., 2022, 13, 7192 CrossRef CAS.
- R. Guan, M. Chen, J. Xin, X. M. Xie, F. Jin, Q. Zhang, S. Y. Xie and S. Yang, J. Am. Chem. Soc., 2021, 143, 8078–8085 CrossRef CAS PubMed.
- J. Roukala, M. Straka, S. Taubert, J. Vaara and P. Lantto, Chem. Commun., 2017, 53, 8992–8995 RSC.
- F. Jin, J. Xin, R. Guan, X. M. Xie, M. Chen, Q. Zhang, A. A. Popov, S. Y. Xie and S. Yang, Chem. Sci., 2021, 12, 6890–6895 RSC.
- Y. Shen, X. Yu, Q. Meng, Y. R. Yao, J. Autschbach and N. Chen, Chem. Sci., 2022, 13, 12980–12986 RSC.
- P. Yu, W. Shen, L. Bao, C. Pan, Z. Slanina and X. Lu, Chem. Sci., 2019, 10, 10925–10930 RSC.
- H. Jiang, X. Yu, M. Guo, Y. R. Yao, Q. Meng, L. Echegoyen, J. Autschbach and N. Chen, J. Am. Chem. Soc., 2023, 145, 5645–5654 CrossRef CAS PubMed.
- R. Guan, Z. C. Chen, J. Huang, H. R. Tian, J. Xin, S. W. Ying, M. Chen, Q. Zhang, Q. Li, S. Y. Xie, L. S. Zheng and S. Yang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202563119 CrossRef CAS PubMed.
- G. Hoffman, M. C. Walkey, J. Grasvik, G. R. Bacanu, S. Alom, S. Bloodworth, M. E. Light, M. H. Levitt and R. J. Whitby, Angew. Chem., Int. Ed., 2021, 60, 8960–8966 CrossRef CAS PubMed.
- S. Zhang, Y. Hashikawa and Y. Murata, J. Am. Chem. Soc., 2021, 143, 12450–12454 CrossRef CAS PubMed.
- R. Gao, Z. Liu, Z. Liu, T. Liang, J. Su and L. Gan, Angew. Chem., Int. Ed., 2023, 62, e202300151 CrossRef CAS PubMed.
- S. Sun, Z. Liu, F. Colombo, R. Gao, Y. Yu, Y. Qiu, J. Su and L. Gan, Angew. Chem., Int. Ed., 2022, 61, e202212090 CrossRef CAS PubMed.
- Y. Li, N. Lou, D. Xu, C. Pan, X. Lu and L. Gan, Angew. Chem., Int. Ed., 2018, 57, 14144–14148 CrossRef CAS PubMed.
- J. C. Hummelen, M. Prato and F. Wudl, J. Am. Chem. Soc., 1995, 117, 7003–7004 CrossRef CAS.
- L. Gan, Acc. Chem. Res., 2019, 52, 1793–1801 CrossRef CAS.
- M. Murata, Y. Murata and K. Komatsu, Chem. Commun., 2008, 6083–6094 RSC.
- L. Shi and L. Gan, J. Phys. Org. Chem., 2013, 26, 766–772 CrossRef CAS.
- Y. Hashikawa, S. Sadai and Y. Murata, ChemPluschem, 2023, 88, e202300136 CrossRef CAS.
- R. Gao, Z. Liu, Z. Liu, J. Su and L. Gan, J. Am. Chem. Soc., 2023, 145, 18022–18028 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, A. F. Shestakov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, J. Am. Chem. Soc., 2016, 138, 16592–16595 CrossRef CAS PubMed.
- A. Aghabali, S. Jun, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2016, 138, 16459–16465 CrossRef CAS PubMed.
- T. Futagoishi, T. Aharen, T. Kato, A. Kato, T. Ihara, T. Tada, M. Murata, A. Wakamiya, H. Kageyama, Y. Kanemitsu and Y. Murata, Angew. Chem., Int. Ed., 2017, 56, 4261–4265 CrossRef CAS PubMed.
- Y. R. Yao, Y. Rosello, L. Ma, A. R. Puente Santiago, A. Metta-Magana, N. Chen, A. Rodriguez-Fortea, J. M. Poblet and L. Echegoyen, J. Am. Chem. Soc., 2021, 143, 15309–15318 CrossRef CAS PubMed.
- Y. Z. Tan, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Chem., 2009, 1, 450–460 CrossRef CAS PubMed.
- Z. Zhu and J. Tang, Chem. Soc. Rev., 2022, 51, 9469–9481 RSC.
- Y. C. Chen and M. L. Tong, Chem. Sci., 2022, 13, 8716–8726 RSC.
- M. Nie, J. Liang, C. Zhao, Y. Lu, J. Zhang, W. Li, C. Wang and T. Wang, ACS Nano., 2021, 15, 19080–19088 CrossRef CAS PubMed.
- Y. Li, R. Biswas, W. P. Kopcha, T. Dubroca, L. Abella, Y. Sun, R. A. Crichton, C. Rathnam, L. Yang, Y. W. Yeh, K. Kundu, A. Rodriguez-Fortea, J. M. Poblet, K. B. Lee, S. Hill and J. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202211704 CrossRef CAS PubMed.
- H. Nie, C. Zhao, Z. Shi, C. Jia and X. Guo, ACS Mater. Lett., 2022, 4, 1037–1052 CrossRef CAS.
- Y. Wang, G. Velkos, N. J. Israel, M. Rosenkranz, B. Buchner, F. Liu and A. A. Popov, J. Am. Chem. Soc., 2021, 143, 18139–18149 CrossRef CAS PubMed.
- A. Nakagawa, M. Nishino, H. Niwa, K. Ishino, Z. Wang, H. Omachi, K. Furukawa, T. Yamaguchi, T. Kato, S. Bandow, J. Rio, C. Ewels, S. Aoyagi and H. Shinohara, Nat. Commun., 2018, 9, 3073 CrossRef PubMed.
- M. Zalibera, F. Ziegs, S. Schiemenz, V. Dubrovin, W. Lubitz, A. Savitsky, S. H. M. Deng, X. B. Wang, S. M. Avdoshenko and A. A. Popov, Chem. Sci., 2021, 12, 7818–7838 RSC.
- Y. Chai, L. Liu, Y. Xu, X. Liu, C. Wang, Y. Bo, Y. Zhang, Z. Wang, Y. Weng, D. M. Guldi, B. Wu and C. Wang, J. Am. Chem. Soc., 2023, 145, 14190–14195 CrossRef CAS PubMed.
- M. Izquierdo, B. Platzer, A. J. Stasyuk, O. A. Stasyuk, A. A. Voityuk, S. Cuesta, M. Sola, D. M. Guldi and N. Martin, Angew. Chem., Int. Ed., 2019, 58, 6932–6937 CrossRef CAS PubMed.
- A. K. Chan and V. W.-W. Yam, Acc. Chem. Res., 2018, 51, 3041–3051 CrossRef CAS PubMed.
- C. M. Davis, J. M. Lim, K. R. Larsen, D. S. Kim, Y. M. Sung, D. M. Lyons, V. M. Lynch, K. A. Nielsen, J. O. Jeppesen, D. Kim, J. S. Park and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 10410–10417 CrossRef CAS.
- K. Luan, Q. F. Lin, F. F. Xie, Y. Wang, Y. F. Li, L. Wang, L. L. Deng, S. Y. Xie and L. S. Zheng, ACS Omega, 2022, 7, 31442–31447 CrossRef CAS PubMed.
- S. J. Wezenberg, Chem. Commun., 2022, 58, 11045–11058 RSC.
- M. Takeda, S. Hiroto, H. Yokoi, S. Lee, D. Kim and H. Shinokubo, J. Am. Chem. Soc., 2018, 140, 6336–6342 CrossRef CAS.
- V. H. Le, M. Yanney, M. McGuire, A. Sygula and E. A. Lewis, J. Phys. Chem. B, 2014, 118, 11956–11964 CrossRef CAS PubMed.
- C. M. Alvarez, G. Aullon, H. Barbero, L. A. Garcia-Escudero, C. Martinez-Perez, J. M. Martin-Alvarez and D. Miguel, Org. Lett., 2015, 17, 2578–2581 CrossRef CAS.
- P. L. Abeyratne Kuragama, F. R. Fronczek and A. Sygula, Org. Lett., 2015, 17, 5292–5295 CrossRef CAS.
- H. Gotfredsen, T. Holmstrom, A. V. Munoz, F. E. Storm, C. G. Tortzen, A. Kadziola, K. V. Mikkelsen, O. Hammerich and M. B. Nielsen, Org. Lett., 2018, 20, 5821–5825 CrossRef CAS PubMed.
- K. Mulla, H. Shaik, D. W. Thompson and Y. Zhao, Org. Lett., 2013, 15, 4532–4535 CrossRef CAS PubMed.
- M. Yanney, F. R. Fronczek and A. Sygula, Angew. Chem., Int. Ed., 2015, 54, 11153–11156 CrossRef CAS PubMed.
- S. Ferrero, H. Barbero, D. Miguel, R. García-Rodríguez and C. M. Álvarez, J. Org. Chem., 2020, 85, 4918–4926 CrossRef CAS PubMed.
- C. M. Alvarez, H. Barbero, S. Ferrero and D. Miguel, J. Org. Chem., 2016, 81, 6081–6086 CrossRef CAS PubMed.
- R. M. Calderon, J. Valero, B. Grimm, J. de Mendoza and D. M. Guldi, J. Am. Chem. Soc., 2014, 136, 11436–11443 CrossRef CAS PubMed.
- G. Zango, M. Krug, S. Krishna, V. Marinas, T. Clark, M. V. Martinez-Diaz, D. M. Guldi and T. Torres, Chem. Sci., 2020, 11, 3448–3459 RSC.
- T. Maulbetsch, P. Frech, M. Scheele, K. W. Tornroos and D. Kunz, Chem. – Eur. J., 2023, e202302104 CrossRef CAS PubMed.
- D. C. Yang, M. Li and C. F. Chen, Chem. Commun., 2017, 53, 9336–9339 RSC.
- M. F. Abdollahi and Y. Zhao, J. Org. Chem., 2021, 86, 14855–14865 CrossRef CAS PubMed.
- D. Y. Sun, F. S. Tham, C. A. Reed, L. Chaker, M. Burgess and P. D. W. Boyd, J. Am. Chem. Soc., 2000, 122, 10704–10705 CrossRef CAS.
- D. Sun, F. S. Tham, C. A. Reed and P. D. W. Boyd, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5088–5092 CrossRef CAS PubMed.
- S. Ferrero, H. Barbero, D. Miguel, R. Garcia-Rodriguez and C. M. Alvarez, J. Org. Chem., 2019, 84, 6183–6190 CrossRef CAS PubMed.
- A. Lopez-Moreno, J. Villalva and E. M. Perez, Chem. Soc. Rev., 2022, 51, 9433–9444 RSC.
- B. Balakrishna, A. Menon, K. Cao, S. Gsanger, S. B. Beil, J. Villalva, O. Shyshov, O. Martin, A. Hirsch, B. Meyer, U. Kaiser, D. M. Guldi and M. von Delius, Angew. Chem., Int. Ed., 2020, 59, 18774–18785 CrossRef CAS PubMed.
- J. Mateos-Gil, J. Calbo, L. Rodriguez-Perez, M. Angeles Herranz, E. Orti and N. Martin, ChemPlusChem, 2019, 84, 730–739 CrossRef CAS PubMed.
- G. A. Leith and N. B. Shustova, Chem. Commun., 2021, 57, 10125–10138 RSC.
- C. M. Alvarez, L. A. Garcia-Escudero, R. Garcia-Rodriguez, J. M. Martin-Alvarez, D. Miguel and V. M. Rayon, Dalton Trans., 2014, 43, 15693–15696 RSC.
- V. G. Jimenez, A. H. G. David, J. M. Cuerva, V. Blanco and A. G. Campana, Angew. Chem., Int. Ed., 2020, 59, 15124–15128 CrossRef CAS.
- M. C. Stuparu, Angew. Chem., Int. Ed., 2013, 52, 7786–7790 CrossRef CAS.
- T. Eom, V. Barat, A. Khan and M. C. Stuparu, Chem. Sci., 2021, 12, 4949–4957 RSC.
- A. Sacristan-Martin, H. Barbero, S. Ferrero, D. Miguel, R. Garcia-Rodriguez and C. M. Alvarez, Chem. Commun., 2021, 57, 11013–11016 RSC.
- T. Hirao, Y. Iwabe, N. Fujii and T. Haino, J. Am. Chem. Soc., 2021, 143, 4339–4345 CrossRef CAS PubMed.
- T. Hirao, Y. Iwabe, N. Hisano and T. Haino, Chem. Commun., 2020, 56, 6672–6675 RSC.
- T. Hirao and T. Haino, Chem. – Asian J., 2022, 17, e202200344 CrossRef CAS PubMed.
- A. Ikeda, M. Ishikawa, R. Aono, J. Kikuchi, M. Akiyama and W. Shinoda, J. Org. Chem., 2013, 78, 2534–2541 CrossRef CAS.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS PubMed.
- X. N. Han, Y. Han and C. F. Chen, Nat. Commun., 2021, 12, 6378 CrossRef CAS PubMed.
- X. N. Han, Y. Han and C. F. Chen, J. Am. Chem. Soc., 2020, 142, 8262–8269 CrossRef CAS PubMed.
- D. X. Wang and M. X. Wang, Acc. Chem. Res., 2020, 53, 1364–1380 CrossRef CAS PubMed.
- Q. Shi, X. Wang, B. Liu, P. Qiao, J. Li and L. Wang, Chem. Commun., 2021, 57, 12379–12405 RSC.
- H. Zheng, L. Fu, R. Wang, J. Jiao, Y. Song, C. Shi, Y. Chen, J. Jiang, C. Lin, J. Ma and L. Wang, Nat. Commun., 2023, 14, 590 CrossRef CAS PubMed.
- J. Q. Wang, Y. Han and C. F. Chen, Chem. Commun., 2021, 57, 3987–3990 RSC.
- F. Jia, D. H. Li, T. L. Yang, L. P. Yang, L. Dang and W. Jiang, Chem. Commun., 2016, 53, 336–339 RSC.
- W. Y. Cha, A. Ahn, T. Kim, J. Oh, R. Ali, J. S. Park and D. Kim, Chem. Commun., 2019, 55, 8301–8304 RSC.
- M. Hermann, D. Wassy and B. Esser, Angew. Chem., Int. Ed., 2021, 60, 15743–15766 CrossRef CAS PubMed.
- Y. Fan, J. He, L. Liu, G. Liu, S. Guo, Z. Lian, X. Li, W. Guo, X. Chen, Y. Wang and H. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202304623 CrossRef CAS PubMed.
- C. Zhao, H. Meng, M. Nie, X. Wang, Z. Cai, T. Chen, D. Wang, C. Wang and T. Wang, J. Phys. Chem. C, 2019, 123, 12514–12520 CrossRef CAS.
- W. Li, F. Qu, L. Liu, Z. Zhang, J. Liang, Y. Lu, J. Zhang, L. Wang, C. Wang and T. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116854 CrossRef CAS PubMed.
- J. Xia, J. W. Bacon and R. Jasti, Chem. Sci., 2012, 3, 3018–3021 RSC.
- D. Lu, Q. Huang, S. Wang, J. Wang, P. Huang and P. Du, Front. Chem., 2019, 7, 668 CrossRef CAS PubMed.
- S. Cui, Q. Huang, J. Wang, H. Jia, P. Huang, S. Wang and P. Du, Org. Lett., 2019, 21, 5917–5921 CrossRef CAS PubMed.
- S. Wang, X. Li, X. Zhang, P. Huang, P. Fang, J. Wang, S. Yang, K. Wu and P. Du, Chem. Sci., 2021, 12, 10506–10513 RSC.
- Y. Zhou, G. Zhuang and P. Du, Chin. Chem. Lett., 2023, 34, DOI:10.1016/j.cclet.2023.108593.
- M. B. Minameyer, Y. Xu, S. Fruhwald, A. Gorling, M. von Delius and T. Drewello, Chem. – Eur. J., 2020, 26, 8729–8741 CrossRef CAS PubMed.
- E. J. Leonhardt, J. M. Van Raden, D. J. Miller, L. N. Zakharov, B. J. Alemán and R. Jasti, Nano Lett., 2018, 18, 7991–7997 CrossRef CAS PubMed.
- F. Schwer, S. Zank, M. Freiberger, R. Kaur, S. Frühwald, C. C. Robertson, A. Görling, T. Drewello, D. M. Guldi and M. von Delius, Org. Mater., 2022, 4, 7–17 CrossRef CAS.
- N. Grabicki, S. Fisher and O. Dumele, Angew. Chem., Int. Ed., 2023, 62, e202217917 CrossRef CAS PubMed.
- M. Freiberger, M. B. Minameyer, I. Solymosi, S. Frühwald, M. Krug, Y. Xu, A. Hirsch, T. Clark, D. M. Guldi, M. von Delius, K. Amsharov, A. Görling, M. E. Pérez-Ojeda and T. Drewello, Chem. – Eur. J., 2023, 29, e202203734 CrossRef CAS PubMed.
- Y. Xu and M. von Delius, Angew. Chem., Int. Ed., 2020, 59, 559–573 CrossRef CAS PubMed.
- J. D. Cojal Gonzalez, M. Iyoda and J. P. Rabe, Nat. Commun., 2017, 8, 14717 CrossRef PubMed.
- L. Chai, Y. Y. Ju, J. F. Xing, X. H. Ma, X. J. Zhao and Y. Z. Tan, Angew. Chem., Int. Ed., 2022, 61, e202210268 CrossRef CAS PubMed.
- C. R. Gob, A. Ehnbom, L. Sturm, Y. Tobe and I. M. Oppel, Chem. – Eur. J., 2020, 26, 3609–3613 CrossRef PubMed.
- S. Goeb, S. Bivaud, P. I. Dron, J. Y. Balandier, M. Chas and M. Salle, Chem. Commun., 2012, 48, 3106–3108 RSC.
- Q. Huang, G. Zhuang, H. Jia, M. Qian, S. Cui, S. Yang and P. Du, Angew. Chem., Int. Ed., 2019, 58, 6244–6249 CrossRef CAS PubMed.
- R. Kaur, S. Sen, M. C. Larsen, L. Tavares, J. Kjelstrup-Hansen, M. Ishida, A. Zieleniewska, V. M. Lynch, S. Bahring, D. M. Guldi, J. L. Sessler and A. Jana, J. Am. Chem. Soc., 2020, 142, 11497–11505 CrossRef CAS PubMed.
- S. I. Kawano, M. Nakaya, M. Saitow, A. Ishiguro, T. Yanai, J. Onoe and K. Tanaka, J. Am. Chem. Soc., 2022, 144, 6749–6758 CrossRef CAS PubMed.
- X. S. Ke, T. Kim, J. T. Brewster, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2017, 139, 4627–4630 CrossRef CAS PubMed.
- N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H. F. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976–12979 CrossRef CAS PubMed.
- S. Lee, E. Chenard, D. L. Gray and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 13814–13817 CrossRef CAS PubMed.
- D. Lu, G. Zhuang, H. Wu, S. Wang, S. Yang and P. Du, Angew. Chem., Int. Ed., 2017, 56, 158–162 CrossRef CAS PubMed.
- Y. Lu, Z. D. Fu, Q. H. Guo and M. X. Wang, Org. Lett., 2017, 19, 1590–1593 CrossRef CAS PubMed.
- L. Mao, M. Zhou, Y.-F. Niu, X.-L. Zhao and X. Shi, Org. Chem. Front., 2021, 8, 4678–4684 RSC.
- P. Mondal and S. P. Rath, Chem. – Asian J., 2017, 12, 1824–1835 CrossRef CAS PubMed.
- T. Ogoshi, N. Ueshima, F. Sakakibara, T. A. Yamagishi and T. Haino, Org. Lett., 2014, 16, 2896–2899 CrossRef CAS PubMed.
- S. Sen, F. Ishiwari, R. Kaur, M. Ishida, D. Ray, K. Kikuchi, T. Mori, S. Bahring, V. M. Lynch, A. Saeki, D. M. Guldi, J. L. Sessler and A. Jana, J. Am. Chem. Soc., 2023, 145, 1031–1039 CrossRef CAS PubMed.
- Q. Wang, C. Zhang, B. C. Noll, H. Long, Y. Jin and W. Zhang, Angew. Chem., Int. Ed., 2014, 53, 10663–10667 CrossRef CAS PubMed.
- J. Xie, X. Li, Z. Du, Y. Liu and K. Zhu, CCS Chem., 2023, 5, 958–970 CrossRef CAS.
- J. Xie, X. Li, S. Wang, A. Li, L. Jiang and K. Zhu, Nat. Commun., 2020, 11, 3348 CrossRef CAS PubMed.
- S. Xue, D. Kuzuhara, N. Aratani and H. Yamada, Org. Lett., 2019, 21, 2069–2072 CrossRef CAS PubMed.
- C. Yu, H. Long, Y. Jin and W. Zhang, Org. Lett., 2016, 18, 2946–2949 CrossRef CAS PubMed.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef PubMed.
- L.-J. Feng, H. Li, Q. Chen and B.-H. Han, RSC Adv., 2013, 3, 6985–6990 RSC.
- H. Chen, Z. Xia and Q. Miao, Chem. Sci., 2022, 13, 2280–2285 RSC.
- M. F. Abdollahi and Y. Zhao, J. Org. Chem., 2023, 88, 3451–3465 CrossRef CAS PubMed.
- H. Danjo, K. Iwaso, M. Kawahata, K. Ohara, T. Miyazawa and K. Yamaguchi, Org. Lett., 2013, 15, 2164–2167 CrossRef CAS PubMed.
- T. Nakamura, S. Tsukuda and T. Nabeshima, J. Am. Chem. Soc., 2019, 141, 6462–6467 CrossRef CAS PubMed.
- N. Zhang, L. Yang, W. Li, J. Zhu, K. Chi, D. Chang, Y. Qiao, T. Wang, Y. Zhao, X. Lu and Y. Liu, J. Am. Chem. Soc., 2022, 144, 21521–21529 CrossRef CAS PubMed.
- W. S. Ren, L. Zhao and M. X. Wang, Org. Lett., 2016, 18, 3126–3129 CrossRef CAS PubMed.
- H. Shimizu, J. D. Cojal Gonzalez, M. Hasegawa, T. Nishinaga, T. Haque, M. Takase, H. Otani, J. P. Rabe and M. Iyoda, J. Am. Chem. Soc., 2015, 137, 3877–3885 CrossRef CAS PubMed.
- S. X. Fa, L. X. Wang, D. X. Wang, L. Zhao and M. X. Wang, J. Org. Chem., 2014, 79, 3559–3571 CrossRef CAS PubMed.
- A. Ikeda, T. Hida, J.-I. Kikuchi, K. Nobusawa and T. Matsuo, Org. Lett., 2013, 15, 6194–6197 CrossRef CAS PubMed.
- M. Yanagisawa, K. Tashiro, M. Yamasaki and T. Aida, J. Am. Chem. Soc., 2007, 129, 11912–11913 CrossRef CAS PubMed.
- Y. D. Yang and H. Y. Gong, Chem. Commun., 2019, 55, 3701–3704 RSC.
- X. Li, L. Jia, W. Wang, Y. Wang, D. Sun and H. Jiang, J. Mater. Chem. C, 2023, 11, 1429–1434 RSC.
- B. Kang, R. K. Totten, M. H. Weston, J. T. Hupp and S. T. Nguyen, Dalton Trans., 2012, 41, 12156–12162 RSC.
- J. Song, N. Aratani, H. Shinokubo and A. Osuka, J. Am. Chem. Soc., 2010, 132, 16356–16357 CrossRef CAS PubMed.
- T. Iwamoto, Y. Watanabe, H. Takaya, T. Haino, N. Yasuda and S. Yamago, Chem. – Eur. J., 2013, 19, 14061–14068 CrossRef CAS PubMed.
- J. Volkmann, D. Kohrs and H. A. Wegner, Chem. – Eur. J., 2023, 29, e202300268 CrossRef CAS PubMed.
- D. Kohrs, J. Volkmann and H. A. Wegner, Eur. J. Org. Chem., 2023, e202300575 CrossRef CAS.
- J. Jaksic, I. Solymosi, A. Hirsch, M. E. Perez-Ojeda, A. Mitrovic and V. Maslak, Chem. – Eur. J., 2023, 29, e202301061 CrossRef CAS PubMed.
- T. A. Schaub, A. Zieleniewska, R. Kaur, M. Minameyer, W. Yang, C. M. Schusslbauer, L. Zhang, M. Freiberger, L. N. Zakharov, T. Drewello, P. O. Dral, D. M. Guldi and R. Jasti, Chem. – Eur. J., 2023, 29, e202300668 CrossRef CAS PubMed.
- T. Iwamoto, Z. Slanina, N. Mizorogi, J. Guo, T. Akasaka, S. Nagase, H. Takaya, N. Yasuda, T. Kato and S. Yamago, Chem. – Eur. J., 2014, 20, 14403–14409 CrossRef CAS PubMed.
- F. Bernt and H. A. Wegner, Chem. – Eur. J., 2023, 29, e202301001 CrossRef CAS PubMed.
- J. P. Mora-Fuentes, M. D. Codesal, M. Reale, C. M. Cruz, V. G. Jimenez, A. Sciortino, M. Cannas, F. Messina, V. Blanco and A. G. Campana, Angew. Chem., Int. Ed., 2023, 62, e202301356 CrossRef CAS PubMed.
- K. Wei, J. Li, W. Zhang, B. Yuan, M.-D. Li and P. Du, Chin. Chem. Lett., 2023, 34 DOI:10.1016/j.cclet.2023.109055.
- H. Isobe, S. Hitosugi, T. Yamasaki and R. Iizuka, Chem. Sci., 2013, 4, 1293 RSC.
- T. Matsuno, S. Sato, R. Iizuka and H. Isobe, Chem. Sci., 2015, 6, 909–916 RSC.
- S. Sato, T. Yamasaki and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8374–8379 CrossRef CAS PubMed.
- Y. Xu, S. Gsanger, M. B. Minameyer, I. Imaz, D. Maspoch, O. Shyshov, F. Schwer, X. Ribas, T. Drewello, B. Meyer and M. von Delius, J. Am. Chem. Soc., 2019, 141, 18500–18507 CrossRef CAS PubMed.
- J. S. Wossner, D. Wassy, A. Weber, M. Bovenkerk, M. Hermann, M. Schmidt and B. Esser, J. Am. Chem. Soc., 2021, 143, 12244–12252 CrossRef PubMed.
- M. Hermann, D. Wassy, J. Kohn, P. Seitz, M. U. Betschart, S. Grimme and B. Esser, Angew. Chem., Int. Ed., 2021, 60, 10680–10689 CrossRef CAS PubMed.
- J. H. Tang, Y. Li, Q. Wu, Z. Wang, S. Hou, K. Tang, Y. Sun, H. Wang, H. Wang, C. Lu, X. Wang, X. Li, D. Wang, J. Yao, C. J. Lambert, N. Tao, Y. W. Zhong and P. J. Stang, Nat. Commun., 2019, 10, 4599 CrossRef PubMed.
- Z. L. Sinclair, N. L. Bell, J. R. Bame, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2023, 62, e202214203 CrossRef CAS PubMed.
- V. Martinez-Agramunt, T. Eder, H. Darmandeh, G. Guisado-Barrios and E. Peris, Angew. Chem., Int. Ed., 2019, 58, 5682–5686 CrossRef CAS PubMed.
- X. Zhang, H. Shi, G. Zhuang, S. Wang, J. Wang, S. Yang, X. Shao and P. Du, Angew. Chem., Int. Ed., 2021, 60, 17368–17372 CrossRef CAS PubMed.
- K. Li, Z. Xu, H. Deng, Z. Zhou, Y. Dang and Z. Sun, Angew. Chem., Int. Ed., 2021, 60, 7649–7653 CrossRef CAS PubMed.
- K. Miki, T. Matsushita, Y. Inoue, Y. Senda, T. Kowada and K. Ohe, Chem. Commun., 2013, 49, 9092–9094 RSC.
- Y. Yang, S. Huangfu, S. Sato and M. Juríček, Org. Lett., 2021, 23, 7943–7948 CrossRef CAS PubMed.
- J. He, M. H. Yu, Z. Lian, Y. Q. Fan, S. Z. Guo, X. N. Li, Y. Wang, W. G. Wang, Z. Y. Cheng and H. Jiang, Chem. Sci., 2023, 14, 4426–4433 RSC.
- W. Xu, X. D. Yang, X. B. Fan, X. Wang, C. H. Tung, L. Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2019, 58, 3943–3947 CrossRef CAS PubMed.
- L. Zhan, C. Dai, G. Zhang, J. Zhu, S. Zhang, H. Wang, Y. Zeng, C. H. Tung, L. Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2022, 61, e202113334 CrossRef CAS PubMed.
- Y. Xu, F. Steudel, M. Y. Leung, B. Xia, M. von Delius and V. W. Yam, Angew. Chem., Int. Ed., 2023, 62, e202302978 CrossRef CAS PubMed.
- Y. Yamamoto, E. Tsurumaki, K. Wakamatsu and S. Toyota, Angew. Chem., Int. Ed., 2018, 57, 8199–8202 CrossRef CAS PubMed.
- S. Z. Zhan, J. H. Li, G. H. Zhang, M. D. Li, S. Sun, J. Zheng, G. H. Ning, M. Li, D. B. Kuang, X. D. Wang and D. Li, Chem. Commun., 2020, 56, 3325–3328 RSC.
- J. Pfeuffer-Rooschuz, S. Heim, A. Prescimone and K. Tiefenbacher, Angew. Chem., Int. Ed., 2022, 61, e202209885 CrossRef PubMed.
- K. Sakaguchi, T. Kamimura, H. Uno, S. Mori, S. Ozako, H. Nobukuni, M. Ishida and F. Tani, J. Org. Chem., 2014, 79, 2980–2992 CrossRef CAS PubMed.
- S. I. Kawano, T. Fukushima and K. Tanaka, Angew. Chem., Int. Ed., 2018, 57, 14827–14831 CrossRef CAS PubMed.
- Y. Ooyama, K. Uenaka, T. Kamimura, S. Ozako, M. Kanda, T. Koide and F. Tani, RSC Adv., 2016, 6, 16150–16158 RSC.
- R. Alvarez-Yebra, A. Sors-Vendrell and A. Lledo, Chem. Commun., 2023, 59, 11556–11559 RSC.
- A. Stergiou, J. Rio, J. H. Griwatz, D. Arcon, H. A. Wegner, C. P. Ewels and N. Tagmatarchis, Angew. Chem., Int. Ed., 2019, 58, 17745–17750 CrossRef CAS PubMed.
- Y. Tanuma, A. Stergiou, A. Buzan Bobnar, M. Gaboardi, J. Rio, J. Volkmann, H. A. Wegner, N. Tagmatarchis, C. P. Ewels and D. Arcon, Nanoscale, 2021, 13, 19946–19955 RSC.
- Y. Tang, J. Li, P. Du, H. Zhang, C. Zheng, H. Lin, X. Du and S. Tao, Org. Electron., 2020, 83, 105747 CrossRef CAS.
- J. Wang, Y. Y. Ju, K. H. Low, Y. Z. Tan and J. Liu, Angew. Chem., Int. Ed., 2021, 60, 11814–11818 CrossRef CAS PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- H. Y. Lin, Y. T. Wang, X. Shi, H. B. Yang and L. Xu, Chem. Soc. Rev., 2023, 52, 1129–1154 RSC.
- K. Matsumoto, S. Kusaba, Y. Tanaka, Y. Sei, M. Akita, K. Aritani, M. A. Haga and M. Yoshizawa, Angew. Chem., Int. Ed., 2019, 58, 8463–8467 CrossRef CAS PubMed.
- J. Yuan, W. Lv, A. Li and K. Zhu, Chem. Commun., 2021, 57, 12848–12851 RSC.
- A. Saura-Sanmartin, A. Martinez-Cuezva, M. Marin-Luna, D. Bautista and J. Berna, Angew. Chem., Int. Ed., 2021, 60, 10814–10819 CrossRef CAS PubMed.
- S. Z. Zhan, Y. L. Liu, H. Cai, M. D. Li, Q. Huang, X. D. Wang, M. Li, L. Dang, S. W. Ng, W. Lu and D. Li, Angew. Chem., Int. Ed., 2023, 62, e202312698 CrossRef CAS PubMed.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889–3919 RSC.
- L. Liang, W. Zhao, X. J. Yang and B. Wu, Acc. Chem. Res., 2022, 55, 3218–3229 CrossRef CAS PubMed.
- W. Liu and J. F. Stoddart, Chem, 2021, 7, 919–947 CAS.
- D. Canevet, E. M. Perez and N. Martin, Angew. Chem., Int. Ed., 2011, 50, 9248–9259 CrossRef CAS PubMed.
- R. Banerjee, D. Chakraborty and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 7692–7711 CrossRef CAS PubMed.
- X. Q. Guo, L. P. Zhou, S. J. Hu, L. X. Cai, P. M. Cheng and Q. F. Sun, J. Am. Chem. Soc., 2021, 143, 6202–6210 CrossRef CAS PubMed.
- B. Huang, L. Mao, X. Shi and H. B. Yang, Chem. Sci., 2021, 12, 13648–13663 RSC.
- S. Hasegawa, S. L. Meichsner, J. J. Holstein, A. Baksi, M. Kasanmascheff and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 9718–9723 CrossRef CAS PubMed.
- T. H. Wong, J. C. Chang, C. C. Lai, Y. H. Liu, S. M. Peng and S. H. Chiu, J. Org. Chem., 2014, 79, 3581–3586 CrossRef CAS PubMed.
- M. Pujals, T. Pèlachs, C. Fuertes-Espinosa, T. Parella, M. Garcia-Borràs and X. Ribas, Cell Rep. Phys. Sci., 2022, 3, 100992 CrossRef CAS.
- B. Chen, J. J. Holstein, S. Horiuchi, W. G. Hiller and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 8907–8913 CrossRef CAS PubMed.
- W. Brenner, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 75–78 CrossRef CAS PubMed.
- C. Garcia-Simon, M. Garcia-Borras, L. Gomez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef CAS PubMed.
- W. Sun, Y. Wang, L. Ma, L. Zheng, W. Fang, X. Chen and H. Jiang, J. Org. Chem., 2018, 83, 14667–14675 CrossRef CAS PubMed.
- C. S. Wood, C. Browne, D. M. Wood and J. R. Nitschke, ACS Cent. Sci., 2015, 1, 504–509 CrossRef CAS PubMed.
- F. J. Rizzuto and J. R. Nitschke, Nat. Chem., 2017, 9, 903–908 CrossRef CAS PubMed.
- T. K. Ronson, A. B. League, L. Gagliardi, C. J. Cramer and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 15615–15624 CrossRef CAS PubMed.
- T. K. Ronson, B. S. Pilgrim and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 10417–10420 CrossRef CAS PubMed.
- S. Goeb and M. Salle, Acc. Chem. Res., 2021, 54, 1043–1055 CrossRef CAS PubMed.
- Q.-H. Ling, J.-L. Zhu, Y. Qin and L. Xu, Mater. Chem. Front., 2020, 4, 3176–3189 RSC.
- C. Garcia-Simon, C. Colomban, Y. A. Cetin, A. Gimeno, M. Pujals, E. Ubasart, C. Fuertes-Espinosa, K. Asad, N. Chronakis, M. Costas, J. Jimenez-Barbero, F. Feixas and X. Ribas, J. Am. Chem. Soc., 2020, 142, 16051–16063 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed.
- X. Chang, S. Lin, G. Wang, C. Shang, Z. Wang, K. Liu, Y. Fang and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 15950–15960 CrossRef CAS PubMed.
- N. Kishi, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2014, 53, 3604–3607 CrossRef CAS PubMed.
- P. C. Purba, M. Maity, S. Bhattacharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2021, 60, 14109–14116 CrossRef CAS PubMed.
- R. Banerjee, D. Chakraborty, W. T. Jhang, Y. T. Chan and P. S. Mukherjee, Angew. Chem., Int. Ed., 2023, 62, e202305338 CrossRef CAS PubMed.
- E. O. Bobylev, D. A. Poole, 3rd, B. de Bruin and J. N. H. Reek, J. Am. Chem. Soc., 2022, 144, 15633–15642 CrossRef CAS PubMed.
- K. Mahata, P. D. Frischmann and F. Wurthner, J. Am. Chem. Soc., 2013, 135, 15656–15661 CrossRef CAS PubMed.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- F. J. Rizzuto, D. M. Wood, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 11008–11011 CrossRef CAS PubMed.
- K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914 CrossRef CAS PubMed.
- R.-C. Brachvogel and M. von Delius, Eur. J. Org. Chem., 2016, 3662–3670 CrossRef CAS.
- F. B. L. Cougnon and J. K. Sanders, Acc. Chem. Res., 2012, 45, 2211–2221 CrossRef CAS PubMed.
- C. Zhang, Q. Wang, H. Long and W. Zhang, J. Am. Chem. Soc., 2011, 133, 20995–21001 CrossRef CAS PubMed.
- S. Bera, S. Das, M. Melle-Franco and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2023, 62, e202216540 CrossRef CAS PubMed.
- M. Miklitz, L. Turcani, R. L. Greenaway and K. E. Jelfs, Commun. Chem., 2020, 3, 10 CrossRef CAS PubMed.
- S. Cui, G. Zhuang, D. Lu, Q. Huang, H. Jia, Y. Wang, S. Yang and P. Du, Angew. Chem., Int. Ed., 2018, 57, 9330–9335 CrossRef CAS PubMed.
- Y. Ni, F. Gordillo-Gamez, M. Pena Alvarez, Z. Nan, Z. Li, S. Wu, Y. Han, J. Casado and J. Wu, J. Am. Chem. Soc., 2020, 142, 12730–12742 CrossRef CAS PubMed.
- E. J. Dale, N. A. Vermeulen, M. Juricek, J. C. Barnes, R. M. Young, M. R. Wasielewski and J. F. Stoddart, Acc. Chem. Res., 2016, 49, 262–273 CrossRef CAS PubMed.
- Y. Shi, K. Cai, H. Xiao, Z. Liu, J. Zhou, D. Shen, Y. Qiu, Q. H. Guo, C. Stern, M. R. Wasielewski, F. Diederich, W. A. Goddard, 3rd and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13835–13842 CrossRef CAS PubMed.
- D. A. Rothschild, W. P. Kopcha, A. Tran, J. Zhang and M. C. Lipke, Chem. Sci., 2022, 13, 5325–5332 RSC.
- A. Dhamija, A. Gunnam, X. Yu, H. Lee, I. C. Hwang, Y. Ho Ko and K. Kim, Angew. Chem., Int. Ed., 2022, 61, e202209326 CrossRef CAS PubMed.
- M. Moreno-Simoni, T. Torres and G. de la Torre, Chem. Sci., 2022, 13, 9249–9255 RSC.
- I. Sanchez-Molina, B. Grimm, R. M. Krick Calderon, C. G. Claessens, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2013, 135, 10503–10511 CrossRef CAS PubMed.
- Z. Lu, R. Lavendomme, O. Burghaus and J. R. Nitschke, Angew. Chem., Int. Ed., 2019, 58, 9073–9077 CrossRef CAS PubMed.
- S. Hasegawa, A. Baksi, B. Chen and G. H. Clever, Org. Mater., 2022, 4, 222–227 CrossRef CAS.
- N. Struch, C. Bannwarth, T. K. Ronson, Y. Lorenz, B. Mienert, N. Wagner, M. Engeser, E. Bill, R. Puttreddy, K. Rissanen, J. Beck, S. Grimme, J. R. Nitschke and A. Lutzen, Angew. Chem., Int. Ed., 2017, 56, 4930–4935 CrossRef CAS PubMed.
- M. Di Giosia, F. Zerbetto and M. Calvaresi, Acc. Mater. Res., 2021, 2, 594–605 CrossRef CAS.
- X. Zhou, W. Xi, Y. Luo, S. Cao and G. Wei, J. Phys. Chem. B, 2014, 118, 6733–6741 CrossRef CAS PubMed.
- Y. Sun, A. Kakinen, C. Zhang, Y. Yang, A. Faridi, T. P. Davis, W. Cao, P. C. Ke and F. Ding, Nanoscale, 2019, 11, 11933–11945 RSC.
- Y. Chen, D. Zhao and Y. Liu, Chem. Commun., 2015, 51, 12266–12269 RSC.
- F. Vidal, L. Falivene, L. Caporaso, L. Cavallo and E. Y. Chen, J. Am. Chem. Soc., 2016, 138, 9533–9547 CrossRef CAS PubMed.
- A. J. Stasyuk, O. A. Stasyuk, M. Solà and A. A. Voityuk, Chem. Commun., 2020, 56, 12624–12627 RSC.
- Z. Sun, T. Mio, K. Ikemoto, S. Sato and H. Isobe, J. Org. Chem., 2019, 84, 3500–3507 CrossRef CAS PubMed.
- B. D. Gliemann, V. Strauss, J. F. Hitzenberger, P. O. Dral, F. Hampel, J. P. Gisselbrecht, T. Drewello, W. Thiel, D. M. Guldi and M. Kivala, Chem. – Eur. J., 2017, 23, 12353–12362 CrossRef CAS PubMed.
- T. Li, L. Fan, H. Gong, Z. Xia, Y. Zhu, N. Jiang, L. Jiang, G. Liu, Y. Li and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 9473–9477 CrossRef CAS PubMed.
- M. Yamamura, D. Hongo and T. Nabeshima, Chem. Sci., 2015, 6, 6373–6378 RSC.
- Z. Liu, W. Song, S. Yang, C. Yuan, Z. Liu, H. L. Zhang and X. Shao, Chem. – Eur. J., 2022, 28, e202200306 CrossRef CAS PubMed.
- C. Mejuto, L. Escobar, G. Guisado-Barrios, P. Ballester, D. Gusev and E. Peris, Chem. – Eur. J., 2017, 23, 10644–10651 CrossRef CAS PubMed.
- G. Gao, M. Chen, J. Roberts, M. Feng, C. Xiao, G. Zhang, S. Parkin, C. Risko and L. Zhang, J. Am. Chem. Soc., 2020, 142, 2460–2470 CrossRef CAS PubMed.
- M. Yamamura, T. Saito and T. Nabeshima, J. Am. Chem. Soc., 2014, 136, 14299–14306 CrossRef CAS PubMed.
- H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed.
- V. Garcia-Calvo, J. V. Cuevas, H. Barbero, S. Ferrero, C. M. Alvarez, J. A. Gonzalez, B. Diaz de Grenu, J. Garcia-Calvo and T. Torroba, Org. Lett., 2019, 21, 5803–5807 CrossRef CAS PubMed.
- Y. Yang, K. Cheng, Y. Lu, D. Ma, D. Shi, Y. Sun, M. Yang, J. Li and J. Wei, Org. Lett., 2018, 20, 2138–2142 CrossRef CAS PubMed.
- Y. M. Liu, D. Xia, B. W. Li, Q. Y. Zhang, T. Sakurai, Y. Z. Tan, S. Seki, S. Y. Xie and L. S. Zheng, Angew. Chem., Int. Ed., 2016, 55, 13047–13051 CrossRef CAS PubMed.
- A. Heskia, T. Maris, P. M. Aguiar and J. D. Wuest, J. Am. Chem. Soc., 2019, 141, 18740–18753 CrossRef CAS PubMed.
- K. Yoshida and A. Osuka, Chem. – Eur. J., 2016, 22, 9396–9403 CrossRef CAS PubMed.
- X. Lu, T. Y. Gopalakrishna, Y. Han, Y. Ni, Y. Zou and J. Wu, J. Am. Chem. Soc., 2019, 141, 5934–5941 CrossRef CAS PubMed.
- Y.-T. Wu and J. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- M. C. Stuparu, Acc. Chem. Res., 2021, 54, 2858–2870 CrossRef CAS PubMed.
- A. S. Filatov, M. V. Ferguson, S. N. Spisak, B. Li, C. F. Campana and M. A. Petrukhina, Cryst. Growth Des., 2013, 14, 756–762 CrossRef.
- Y. Y. Xu, H. R. Tian, S. H. Li, Z. C. Chen, Y. R. Yao, S. S. Wang, X. Zhang, Z. Z. Zhu, S. L. Deng, Q. Zhang, S. Yang, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Commun., 2019, 10, 485 CrossRef CAS PubMed.
- M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635–661 RSC.
- M. C. Stuparu, Chem. Mater., 2023, 35, 1836–1848 CrossRef CAS.
- S. Ferrero, H. Barbero, D. Miguel, R. Garcia-Rodriguez and C. M. Alvarez, J. Org. Chem., 2020, 85, 4918–4926 CrossRef CAS PubMed.
- H. Yokoi, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Chem. Sci., 2018, 9, 819–824 RSC.
- I. Sánchez-Molina, C. G. Claessens, B. Grimm, D. M. Guldi and T. Torres, Chem. Sci., 2013, 4, 1338–1344 RSC.
- M. R. Ivancevic, M. L. Ball, V. Bhat, J. A. Wisch, S. R. Parkin, C. Risko, B. P. Rand and Y.-L. Loo, Chem. Mater., 2023, 35, 5524–5531 CrossRef CAS.
- Y. Sun, X. Wang, B. Yang, M. Chen, Z. Guo, Y. Wang, J. Li, M. Xu, Y. Zhang, H. Sun, J. Dang, J. Fan, J. Li and J. Wei, Nat. Commun., 2023, 14, 3446 CrossRef CAS PubMed.
- X. Lu, T. Y. Gopalakrishna, Y. Han, Y. Ni, Y. Zou and J. Wu, J. Am. Chem. Soc., 2019, 141, 5934–5941 CrossRef CAS PubMed.
- K. Ikemoto, R. Kobayashi, S. Sato and H. Isobe, Org. Lett., 2017, 19, 2362–2365 CrossRef CAS PubMed.
- N. Fukui, T. Kim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2017, 139, 9075–9088 CrossRef CAS PubMed.
- K. Wang, Y. Rao, L. Xu, M. Zhou, N. Aratani, A. Osuka and J. Song, Chem. – Eur. J., 2023, e202301955, DOI:10.1002/chem.202301955.
- P. Haines, R. Kaur, M. M. Martin, M. B. Minameyer, S. Frühwald, S. Bönisch, D. Lungerich, F. Hampel, A. Görling, T. Drewello, N. Jux and D. M. Guldi, Adv. Energy Mater., 2021, 11, 2100158 CrossRef CAS.
- S. Zank, J. M. Fernandez-Garcia, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Sola, D. M. Guldi and N. Martin, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS PubMed.
- Q. Chen, A. L. Thompson, K. E. Christensen, P. N. Horton, S. J. Coles and H. L. Anderson, J. Am. Chem. Soc., 2023, 145, 11859–11865 CrossRef CAS PubMed.
- N. B. Shustova, I. V. Kuvychko, A. A. Popov, M. von Delius, L. Dunsch, O. P. Anderson, A. Hirsch, S. H. Strauss and O. V. Boltalina, Angew. Chem., Int. Ed., 2011, 50, 5537–5540 CrossRef CAS PubMed.
- N. G. Petrov, P. Chartier, T. Maris and J. D. Wuest, J. Am. Chem. Soc., 2021, 144, 556–572 CrossRef PubMed.
- C. Zhu, K. Shoyama, M. A. Niyas and F. Wurthner, J. Am. Chem. Soc., 2022, 144, 16282–16286 CrossRef CAS PubMed.
- M.-W. Wang, Z. Li, Y. Liu, W. Jiang and Z. Wang, Org. Chem. Front., 2023, 10, 2808–2812 RSC.
- A. R. Stefankiewicz, E. Tamanini, G. D. Pantos and J. K. Sanders, Angew. Chem., Int. Ed., 2011, 50, 5725–5728 CrossRef CAS PubMed.
- N. Ponnuswamy, G. D. Pantos, M. M. Smulders and J. K. Sanders, J. Am. Chem. Soc., 2012, 134, 566–573 CrossRef CAS PubMed.
- M. Grajda, M. J. Lewinska and A. Szumna, Org. Biomol. Chem., 2017, 15, 8513–8517 RSC.
- D. Rackauskaite, R. Gegevicius, Y. Matsuo, K. Warnmark and E. Orentas, Angew. Chem., Int. Ed., 2016, 55, 208–212 CrossRef CAS PubMed.
- A. Jozeliu Naite, A. Neniskis, A. Bertran, A. M. Bowen, M. Di Valentin, S. Raisys, P. Baronas, K. Kazlauskas, L. Vilciauskas and E. Orentas, J. Am. Chem. Soc., 2023, 145, 455–464 CrossRef CAS PubMed.
- K. Eichstaedt, K. Szpotkowski, M. Grajda, M. Gilski, S. Wosicki, M. Jaskolski and A. Szumna, Chem. – Eur. J., 2019, 25, 3091–3097 CrossRef CAS PubMed.
- S. Langis-Barsetti, T. Maris and J. D. Wuest, J. Org. Chem., 2017, 82, 5034–5045 CrossRef CAS PubMed.
- G. Markiewicz, A. Jenczak, M. Kolodziejski, J. J. Holstein, J. K. M. Sanders and A. R. Stefankiewicz, Nat. Commun., 2017, 8, 15109 CrossRef PubMed.
- Q. Shi, K. E. Bergquist, R. Huo, J. Li, M. Lund, R. Vacha, A. Sundin, E. Butkus, E. Orentas and K. Warnmark, J. Am. Chem. Soc., 2013, 135, 15263–15268 CrossRef CAS PubMed.
- Q. Shi, T. Javorskis, K. E. Bergquist, A. Ulcinas, G. Niaura, I. Matulaitiene, E. Orentas and K. Warnmark, Nat. Commun., 2017, 8, 14943 CrossRef PubMed.
- Y. Haketa, M. Miyasue, Y. Kobayashi, R. Sato, Y. Shigeta, N. Yasuda, N. Tamai and H. Maeda, J. Am. Chem. Soc., 2020, 142, 16420–16428 CrossRef CAS PubMed.
- M. Di Giosia, P. H. H. Bomans, A. Bottoni, A. Cantelli, G. Falini, P. Franchi, G. Guarracino, H. Friedrich, M. Lucarini, F. Paolucci, S. Rapino, N. Sommerdijk, A. Solda, F. Valle, F. Zerbetto and M. Calvaresi, Nanoscale, 2018, 10, 9908–9916 RSC.
- M. Liutkus, A. Lopez-Andarias, S. H. Mejias, J. Lopez-Andarias, D. Gil-Carton, F. Feixas, S. Osuna, W. Matsuda, T. Sakurai, S. Seki, C. Atienza, N. Martin and A. L. Cortajarena, Nanoscale, 2020, 12, 3614–3622 RSC.
- K. H. Kim, D. K. Ko, Y. T. Kim, N. H. Kim, J. Paul, S. Q. Zhang, C. B. Murray, R. Acharya, W. F. DeGrado, Y. H. Kim and G. Grigoryan, Nat. Commun., 2016, 7, 11429 CrossRef CAS PubMed.
- M. Calvaresi, A. Bottoni and F. Zerbetto, J. Phys. Chem. C, 2015, 119, 28077–28082 CrossRef CAS.
- M. Di Giosia, F. Nicolini, L. Ferrazzano, A. Solda, F. Valle, A. Cantelli, T. D. Marforio, A. Bottoni, F. Zerbetto, M. Montalti, S. Rapino, A. Tolomelli and M. Calvaresi, Bioconjugate Chem., 2019, 30, 808–814 CrossRef CAS PubMed.
- A. Giełdoń, M. M. Witt, A. Gajewicz and T. Puzyn, Struct. Chem., 2017, 28, 1775–1788 CrossRef.
- Y. Katagiri, Y. Tsuchida, Y. Matsuo and M. Yoshizawa, J. Am. Chem. Soc., 2021, 143, 21492–21496 CrossRef CAS PubMed.
- Y. Satoh, L. Catti, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2019, 141, 12268–12273 CrossRef CAS PubMed.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond, John Wiley & Sons, 2016 Search PubMed.
- A. Mateo-Alonso, D. M. Guldi, F. Paolucci and M. Prato, Angew. Chem., Int. Ed., 2007, 46, 8120–8126 CrossRef CAS PubMed.
- J. D. Megiatto, D. M. Guldi and D. I. Schuster, Chem. Soc. Rev., 2020, 49, 8–20 RSC.
- A. Mateo-alonso, Supramolecular Chemistry of Fullerenes and Carbon Nanotubes, Wiley-VCH Verlag GmbH & Co. KGaA., 2012, pp. 107–126 Search PubMed.
- D. I. Schuster, K. Li and D. M. Guldi, C. R. Chim., 2006, 9, 892–908 CrossRef CAS.
- F. Diederich, C. Dietrich-Buchecker, J.-F. Nierengarten and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1995, 781–782 RSC.
- R. Z. Pavlović, M. S. Bjelaković and D. R. Milić, RSC Adv., 2016, 6, 37246–37253 RSC.
- T. Da Ros, D. M. Guldi, A. F. Morales, D. A. Leigh, M. Prato and R. Turco, Org. Lett., 2003, 5, 689–691 CrossRef CAS PubMed.
- N. Watanabe, N. Kihara, Y. Furusho, T. Takata, Y. Araki and O. Ito, Angew. Chem., Int. Ed., 2003, 42, 681–683 CrossRef CAS PubMed.
- M. Wolf, A. Ogawa, M. Bechtold, M. Vonesch, J. A. Wytko, K. Oohora, S. Campidelli, T. Hayashi, D. M. Guldi and J. Weiss, Chem. Sci., 2019, 10, 3846–3853 RSC.
- T. A. Barendt, I. Rasovic, M. A. Lebedeva, G. A. Farrow, A. Auty, D. Chekulaev, I. V. Sazanovich, J. A. Weinstein, K. Porfyrakis and P. D. Beer, J. Am. Chem. Soc., 2018, 140, 1924–1936 CrossRef CAS PubMed.
- X. N. Han, Y. Han and C. F. Chen, Chem. Soc. Rev., 2023, 52, 3265–3298 RSC.
- X. Q. Wang, W. J. Li, W. Wang and H. B. Yang, Acc. Chem. Res., 2021, 54, 4091–4106 CrossRef CAS PubMed.
- G. Wu, J.-R. Wu, Y. Wang and Y.-W. Yang, Chem, 2023, 9, 1–13 Search PubMed.
- E. Meichsner, I. Nierengarten, M. Holler, M. Chessé and J.-F. Nierengarten, Helv. Chim. Acta, 2018, 101, e1800059 CrossRef.
- M. Remy, I. Nierengarten, B. Park, M. Holler, U. Hahn and J. F. Nierengarten, Chem. – Eur. J., 2021, 27, 8492–8499 CrossRef CAS PubMed.
- Y. Mi, J. Yao, J. Ma, L. Dai, C. Xiao, W. Wu and C. Yang, Org. Lett., 2020, 22, 2118–2123 CrossRef CAS PubMed.
- L. Leanza, C. Perego, L. Pesce, M. Salvalaglio, M. von Delius and G. M. Pavan, Chem. Sci., 2023, 14, 6716–6729 RSC.
- F. M. Steudel, E. Ubasart, L. Leanza, M. Pujals, T. Parella, G. M. Pavan, X. Ribas and M. von Delius, Angew. Chem., Int. Ed., 2023, 62, e202309393 CrossRef CAS PubMed.
- A. M. Rice, E. A. Dolgopolova and N. B. Shustova, Chem. Mater., 2017, 29, 7054–7061 CrossRef CAS.
- Y. Yang and M. Juricek, ChemPlusChem, 2021, 87, e202100468 CrossRef PubMed.
- L. Bao, T. Xu, K. Guo, W. Huang and X. Lu, Adv. Mater., 2022, 34, e2200189 CrossRef PubMed.
- A. V. Baskar, M. R. Benzigar, S. N. Talapaneni, G. Singh, A. S. Karakoti, J. Yi, A. A. H. Al-Muhtaseb, K. Ariga, P. M. Ajayan and A. Vinu, Adv. Funct. Mater., 2021, 32, 2106924 CrossRef.
- R. W. Lof, M. A. van Veenendaal, B. Koopmans, H. T. Jonkman and G. A. Sawatzky, Phys. Rev. Lett., 1992, 68, 3924–3927 CrossRef CAS PubMed.
- K. Lee, B. Choi, I. J. Plante, M. V. Paley, X. Zhong, A. C. Crowther, J. S. Owen, X. Zhu and X. Roy, Angew. Chem., Int. Ed., 2018, 57, 6125–6129 CrossRef CAS PubMed.
- C. H. Xu and G. E. Scuseria, Phys. Rev. Lett., 1995, 74, 274–277 CrossRef CAS PubMed.
- L. Hou, X. Cui, B. Guan, S. Wang, R. Li, Y. Liu, D. Zhu and J. Zheng, Nature, 2022, 606, 507–510 CrossRef CAS PubMed.
- E. Meirzadeh, A. M. Evans, M. Rezaee, M. Milich, C. J. Dionne, T. P. Darlington, S. T. Bao, A. K. Bartholomew, T. Handa, D. J. Rizzo, R. A. Wiscons, M. Reza, A. Zangiabadi, N. Fardian-Melamed, A. C. Crowther, P. J. Schuck, D. N. Basov, X. Zhu, A. Giri, P. E. Hopkins, P. Kim, M. L. Steigerwald, J. Yang, C. Nuckolls and X. Roy, Nature, 2023, 613, 71–76 CrossRef CAS PubMed.
- F. Pan, K. Ni, T. Xu, H. Chen, Y. Wang, K. Gong, C. Liu, X. Li, M. L. Lin, S. Li, X. Wang, W. Yan, W. Yin, P. H. Tan, L. Sun, D. Yu, R. S. Ruoff and Y. Zhu, Nature, 2023, 614, 95–101 CrossRef CAS PubMed.
- B. Peng, J. Am. Chem. Soc., 2022, 144, 19921–19931 CrossRef CAS PubMed.
- T. Wang, L. Zhang, J. Wu, M. Chen, S. Yang, Y. Lu and P. Du, Angew. Chem., Int. Ed., 2023, 62, e202311352 CrossRef CAS PubMed.
- D. Bonifazi, O. Enger and F. Diederich, Chem. Soc. Rev., 2007, 36, 390–414 RSC.
- K. Vimalanathan, Z. Zhang, J. Zou and C. L. Raston, Chem. Commun., 2023, 59, 9698–9701 RSC.
- M. Chen, R. Guan and S. Yang, Adv. Sci., 2019, 6, 1800941 CrossRef PubMed.
- L. K. Shrestha, Q. Ji, T. Mori, K. Miyazawa, Y. Yamauchi, J. P. Hill and K. Ariga, Chem. – Asian J., 2013, 8, 1662–1679 CrossRef CAS PubMed.
- K. Ariga and L. K. Shrestha, Mater. Adv., 2021, 2, 582–597 RSC.
- L. K. Shrestha, Y. Yamauchi, J. P. Hill, K. Miyazawa and K. Ariga, J. Am. Chem. Soc., 2013, 135, 586–589 CrossRef CAS PubMed.
- M. Sathish and K. Miyazawa, J. Am. Chem. Soc., 2007, 129, 13816–13817 CrossRef CAS PubMed.
- J. Wu, X. Zhu, Y. Guan, Y. Wang, F. Jin, R. Guan, F. Liu, M. Chen, Y. Tian and S. Yang, Angew. Chem., Int. Ed., 2019, 58, 11350–11354 CrossRef CAS PubMed.
- C. Park, E. Yoon, M. Kawano, T. Joo and H. C. Choi, Angew. Chem., Int. Ed., 2010, 49, 9670–9675 CrossRef CAS PubMed.
- N. Jannatun, N. Chen, P. Yu, W. Shen and X. Lu, Chem. – Eur. J., 2021, 27, 348–353 CrossRef CAS PubMed.
- P. Bairi, T. Tsuruoka, S. Acharya, Q. Ji, J. P. Hill, K. Ariga, Y. Yamauchi and L. K. Shrestha, Mater. Horiz., 2018, 5, 285–290 RSC.
- P. Bairi, K. Minami, J. P. Hill, K. Ariga and L. K. Shrestha, ACS Nano, 2017, 11, 7790–7796 CrossRef CAS PubMed.
- E. Fernandez-Bartolome, A. Gamonal, J. Santos, S. Khodabakhshi, E. Rodriguez-Sanchez, E. C. Sanudo, N. Martin and J. Sanchez Costa, Chem. Sci., 2021, 12, 8682–8688 RSC.
- Y. L. Liu, S. Z. Zhan, J. X. Sun, H. Cai, Z. L. Yuan, H. F. Zhang, M. Li, L. Dang, S. F. Ni, S. Weng Ng, W. Lu and D. Li, Angew. Chem., Int. Ed., 2023, 62, e202306748 CrossRef CAS PubMed.
- D. E. Williams, E. A. Dolgopolova, D. C. Godfrey, E. D. Ermolaeva, P. J. Pellechia, A. B. Greytak, M. D. Smith, S. M. Avdoshenko, A. A. Popov and N. B. Shustova, Angew. Chem., Int. Ed., 2016, 55, 9070–9074 CrossRef CAS PubMed.
- J. F. Nierengarten, Chem. Commun., 2017, 53, 11855–11868 RSC.
- S. Uchikawa, A. Kawasaki, N. Hoshino, T. Takeda, S.-I. Noro, K. Takahashi, T. Nakamura, N. Sato, K. Kokubo, H. Sakurai and T. Akutagawa, J. Phys. Chem. C, 2019, 123, 23545–23553 CrossRef CAS.
- M. A. Mezour, R. M. Choueiri, O. Lukoyanova, R. B. Lennox and D. F. Perepichka, Nanoscale, 2016, 8, 16955–16962 RSC.
- R. Sekine, P. Ravat, H. Yanagisawa, C. Liu, M. Kikkawa, K. Harano and E. Nakamura, J. Am. Chem. Soc., 2021, 143, 2822–2828 CrossRef CAS PubMed.
- A. Kraft, M. Gsänger and F. Beuerle, Eur. J. Org. Chem., 2014, 523–528 CrossRef CAS.
- A. Kraft, J. Stangl, A. M. Krause, K. Muller-Buschbaum and F. Beuerle, Beilstein J. Org. Chem., 2017, 13, 1–9 CrossRef CAS PubMed.
- A. Kraft, P. Roth, D. Schmidt, J. Stangl, K. Muller-Buschbaum and F. Beuerle, Chem. – Eur. J., 2016, 22, 5982–5987 CrossRef CAS PubMed.
- A. Kraft, C. Roger, D. Schmidt, J. Stangl, K. Muller-Buschbaum and F. Beuerle, Chem. – Eur. J., 2017, 23, 15864–15868 CrossRef CAS PubMed.
- E. Fernandez-Bartolome, J. Santos, A. Gamonal, S. Khodabakhshi, L. J. McCormick, S. J. Teat, E. C. Sanudo, J. S. Costa and N. Martin, Angew. Chem., Int. Ed., 2019, 58, 2310–2315 CrossRef CAS PubMed.
- A. Khassanov, H. G. Steinruck, T. Schmaltz, A. Magerl and M. Halik, Acc. Chem. Res., 2015, 48, 1901–1908 CrossRef CAS PubMed.
- H. Chen, W. Zhang, M. Li, G. He and X. Guo, Chem. Rev., 2020, 120, 2879–2949 CrossRef CAS PubMed.
- C. M. Jager, T. Schmaltz, M. Novak, A. Khassanov, A. Vorobiev, M. Hennemann, A. Krause, H. Dietrich, D. Zahn, A. Hirsch, M. Halik and T. Clark, J. Am. Chem. Soc., 2013, 135, 4893–4900 CrossRef PubMed.
- F. K. Leung, F. Ishiwari, T. Kajitani, Y. Shoji, T. Hikima, M. Takata, A. Saeki, S. Seki, Y. M. Yamada and T. Fukushima, J. Am. Chem. Soc., 2016, 138, 11727–11733 CrossRef CAS PubMed.
- A. Rumpel, M. Novak, J. Walter, B. Braunschweig, M. Halik and W. Peukert, Langmuir, 2011, 27, 15016–15023 CrossRef CAS PubMed.
- H. K. Yang, M. Khadem, O. V. Penkov and D. E. Kim, Nanoscale, 2019, 11, 2863–2870 RSC.
- O. A. Kraevaya, A. V. Novikov, A. F. Shestakov, E. S. Ershova, E. A. Savinova, L. V. Kameneva, N. N. Veiko, D. Schols, J. Balzarini, S. V. Kostyuk and P. A. Troshin, Chem. Commun., 2020, 56, 10203–10206 RSC.
- L. Wasserthal, B. Schade, K. Ludwig, C. Bottcher and A. Hirsch, Chem. – Eur. J., 2014, 20, 5961–5966 CrossRef CAS PubMed.
- M. Kunkel, S. Sutter and S. Polarz, Angew. Chem., Int. Ed., 2019, 58, 15620–15625 CrossRef CAS PubMed.
- K. Harano and E. Nakamura, Acc. Chem. Res., 2019, 52, 2090–2100 CrossRef CAS PubMed.
- P. Ravat, H. Uchida, R. Sekine, K. Kamei, A. Yamamoto, O. Konovalov, M. Tanaka, T. Yamada, K. Harano and E. Nakamura, Adv. Mater., 2022, 34, e2106465 CrossRef PubMed.
- L. Zhu and C. Y. Li, Liquid Crystalline Polymers, Springer Nature Switzerland AG, 2020 Search PubMed.
- H. Lu, X. Zhang, T. Sakurai, X. Li, Y. Tu, J. Guo, S. Seki, C. Y. Li, G. Ungar and S. Z. D. Cheng, Nano Lett., 2020, 20, 8647–8653 CrossRef CAS PubMed.
- S. Barland, J. R. Tredicce, M. Brambilla, L. A. Lugiato, S. Balle, M. Giudici, T. Maggipinto, L. Spinelli, G. Tissoni, T. Knodl, M. Miller and R. Jager, Nature, 2002, 419, 699–702 CrossRef CAS PubMed.
- F. Lu, E. A. Neal and T. Nakanishi, Acc. Chem. Res., 2019, 52, 1834–1843 CrossRef CAS PubMed.
- T. Zhu, X. Zhang, Z. Li, C. H. Hsu, W. Chen, T. Miyoshi, X. Li, X. Yang, Y. Tu and C. Y. Li, Chem. Commun., 2017, 53, 8336–8339 RSC.
- M. Lehmann, M. Dechant, M. Holzapfel, A. Schmiedel and C. Lambert, Angew. Chem., Int. Ed., 2019, 58, 3610–3615 CrossRef CAS PubMed.
- Y. Hu, K. Y. Wu, T. Zhu, P. Shen, Y. Zhou, X. Li, C. L. Wang, Y. Tu and C. Y. Li, Angew. Chem., Int. Ed., 2018, 57, 13454–13458 CrossRef CAS PubMed.
- M. M. Su, Y. J. Hu, S. T. Yang, A. Yu, P. Peng, L. Yang, P. Jin, B. Su and F. F. Li, Adv. Electron. Mater., 2022, 8, 2100753 CrossRef CAS.
- K. Liu, S. Gao, Z. Zheng, X. Deng, S. Mukherjee, S. Wang, H. Xu, J. Wang, J. Liu, T. Zhai and Y. Fang, Adv. Mater., 2019, 31, e1808254 CrossRef PubMed.
- C. Shen, P. Han, Z. Zheng, W. Jiang, S. Gao, C. Hua, C. L. Chen, F. Xia, T. Zhai, K. Liu and Y. Fang, Adv. Sci., 2022, 9, e2203662 CrossRef PubMed.
- Y. Xu, X. Chen, F. P. Liu, X. Chen, J. H. Guo and S. F. Yang, Mater. Horiz., 2014, 1, 411–418 RSC.
- P. Bairi, K. Minami, W. Nakanishi, J. P. Hill, K. Ariga and L. K. Shrestha, ACS Nano, 2016, 10, 6631–6637 CrossRef CAS PubMed.
- Z. Wei, J. Song, R. Ma, K. Ariga and L. K. Shrestha, Chemosensors, 2022, 10, 16 CrossRef CAS.
- R. Caballero, M. Barrejon, J. Cerda, J. Arago, S. Seetharaman, P. de la Cruz, E. Orti, F. D'Souza and F. Langa, J. Am. Chem. Soc., 2021, 143, 11199–11208 CrossRef CAS PubMed.
- D. S. Kim, J. Chang, S. Leem, J. S. Park, P. Thordarson and J. L. Sessler, J. Am. Chem. Soc., 2015, 137, 16038–16042 CrossRef CAS PubMed.
- M. Park, K. I. Hong, M. Kang, T. W. Kim, H. Lee, W. D. Jang and K. U. Jeong, ACS Nano, 2019, 13, 6101–6112 CrossRef CAS PubMed.
- P. D. W. Boyd, Acc. Chem. Res., 2005, 38, 235–242 CrossRef CAS PubMed.
- L. Bao, B. Wang, P. Yu, C. Huang, C. Pan, H. Fang, T. Akasaka, D. M. Guldi and X. Lu, Chem. Commun., 2019, 55, 6018–6021 RSC.
- Goudappagouda, M. Gedda, G. U. Kulkarni and S. S. Babu, Chem. – Eur. J., 2018, 24, 7695–7701 CrossRef CAS PubMed.
- B. Wang, S. Zheng, A. Saha, L. Bao, X. Lu and D. M. Guldi, J. Am. Chem. Soc., 2017, 139, 10578–10584 CrossRef CAS PubMed.
- X. Yu, B. Wang, Y. Kim, J. Park, S. Ghosh, B. Dhara, R. D. Mukhopadhyay, J. Koo, I. Kim, S. Kim, I. C. Hwang, S. Seki, D. M. Guldi, M. H. Baik and K. Kim, J. Am. Chem. Soc., 2020, 142, 12596–12601 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. Saura-Sanmartin, A. Martinez-Cuezva, M. Marin-Luna, D. Bautista and J. Berna, Angew. Chem., Int. Ed., 2021, 60, 10814–10819 CrossRef CAS PubMed.
- S. W. Lo, T. Kitao, Y. Nada, K. Murata, K. Ishii and T. Uemura, Angew. Chem., Int. Ed., 2021, 60, 17947–17951 CrossRef CAS PubMed.
- D. Ray, S. Goswami, J. Duan, J. T. Hupp, C. J. Cramer and L. Gagliardi, Chem. Mater., 2021, 33, 1182–1189 CrossRef CAS.
- X. Liu, M. Kozlowska, T. Okkali, D. Wagner, T. Higashino, G. Brenner-Weiss, S. M. Marschner, Z. Fu, Q. Zhang, H. Imahori, S. Brase, W. Wenzel, C. Woll and L. Heinke, Angew. Chem., Int. Ed., 2019, 58, 9590–9595 CrossRef CAS PubMed.
- S. M. Pratik, L. Gagliardi and C. J. Cramer, J. Phys. Chem. C, 2019, 124, 1878–1887 CrossRef.
- L. Liu, H. Meng, Y. Chai, X. Chen, J. Xu, X. Liu, W. Liu, D. M. Guldi and Y. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202217897 CrossRef CAS PubMed.
- L. Vujević, B. Karadeniz, N. Cindro, A. Krajnc, G. Mali, M. Mazaj, S. M. Avdoshenko, A. A. Popov, D. Žilić, K. Užarević and M. Kveder, Chem. Sci., 2023, 14, 9389–9399 RSC.
- Y. Li, X. Su, W. Zheng, J. J. Zheng, L. Guo, M. Bonn, X. Gao, H. I. Wang and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202216795 CrossRef CAS PubMed.
- T. Ohmura, A. Usuki, Y. Mukae, H. Motegi, S. Kajiya, M. Yamamoto, S. Senda, T. Matsumoto and K. Tatsumi, Chem. – Asian J., 2016, 11, 700–704 CrossRef CAS PubMed.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed.
- J. Han, H. Wu, H. Fan, L. Ding, G. Hai, J. Caro and H. Wang, J. Am. Chem. Soc., 2023, 145, 14793–14801 CrossRef CAS PubMed.
- C. Chen, Y. Feng, P. Wang, S. Zhang, B. Tu, Y. Liu, W. Duan and Q. Zeng, Langmuir, 2021, 37, 7486–7491 CrossRef CAS PubMed.
- D. Cui, J. M. MacLeod and F. Rosei, Small, 2019, 15, e1903294 CrossRef PubMed.
Footnote |
| † Dedicated to the 100th anniversary of chemistry at Henan University. |
| This journal is © The Royal Society of Chemistry 2024 |