
Electronic, optical and charge transport properties of Zn–porphyrin–C60 MOFs: a combined periodic and cluster modeling†
Kevin
Granados-Tavera
 ab and
Gloria
Cárdenas-Jirón
*a
ab and
Gloria
Cárdenas-Jirón
*a
aLaboratory of Theoretical Chemistry, Faculty of Chemistry and Biology, University of Santiago de Chile (USACH), Santiago, Chile. E-mail: gloria.cardenas@usach.cl
bFacultad de Ciencias Básicas, Universidad de la Amazonia, Florencia, Colombia
First published on 19th August 2024
Abstract
Density functional theory (DFT) calculations were performed on the 5,15 meso-positions of nine porphyrin-containing MOFs; Zn2(TCPB)–(NMe2–ZnP); (H4TCPB = 1,2,4,5-tetrakis(4-carboxyphenyl)benzene), (NMe2–ZnP = [5,15-bis[(4-pyridyl)-ethynyl]-10,20-bis-(dimethylamine) porphinato]zinc(II)) functionalized with nitrogen-, oxygen-, and sulfur-containing groups to study their effects on the electronic, optical and transport properties of the materials. The properties of these materials have also been investigated by encapsulating fullerene (C60) in their pores (C60@MOFs). The results indicate that the guest C60 in the MOF generates high photoconductivity through efficient porphyrin/fullerene donor–acceptor (D–A) interactions, which are facilitated by oxygen and sulfur functionalities. DFT calculations show that C60 interacts favorably in MOFs due to negative Eint values. Encapsulated C60 molecules modify the electronic band structure, affecting the conduction band and unoccupied states of MOFs corresponding to C60 p orbitals. TD-DFT calculations show that incorporating C60 promotes D–A interactions in MOFs, leading to charge transfer in the near-infrared and visible photoinduced electron transfer (PET) from porphyrins to C60. Nonequilibrium Green's function-based calculations for MOFs with sulfur group, with and without C60, performed using molecular junctions with Au(111)-based electrodes show increased charge transport for the doped MOF. These insights into tuning electronic/optical properties and controlling charge transfer can aid in the design of new visible/near-infrared MOF-based optoelectronic devices.
1. Introduction
Metal–organic frameworks (MOFs) are a class of crystalline polymers characterized by a porous structure composed of metallic nodes and organic linkers. MOFs offer periodic porosity, tunable functionality, and diverse framework structures, making them promising materials for a wide range of applications, including sensors, gas adsorption and separation, biomedicine, energy conversion, and proton/ion conductivity.1–9 One area of particular interest is the development of conductive MOFs, which have attracted considerable attention for catalyzing various chemical reactions such as oxygen evolution reaction (OER) and oxygen reduction reaction (ORR), as well as in the field of fuel cells, solar cells, energy storage, supercapacitors, flexible electronics, and devices electronics.10–15 However, many existing MOFs suffer from limited electrical conductivity, which poses a challenge for their use in these applications.16,17 To address this issue, a promising approach is to design conductive MOFs with enhanced electrical conductivity as a primary goal. To overcome the limitations associated with bulk conductive MOFs, such as limited surface accessibility and exposure of metallic nodes to electrolytes, several innovative strategies have emerged. These strategies include downsizing MOFs to the nanometer or atomic scale,18 manipulating defects within MOFs,19 creating internal voids or cavities within the MOF structure,20 incorporating different metallic nodes into MOFs,21 and anchoring MOFs to conductive substrates.22 These approaches aim to improve the performance and stability of conductive MOFs, making them even more promising materials for various conductive applications.12,23–25Promising conductive MOFs are based on intra-layer π-electron delocalization enabled by conjugated planar ligands and trigonal geometries, and tight energetic alignment between metal and ligand interfaces.15 Optimization of these factors, often by direct synthesis or modification of MOF constituents,26–28 enhances charge transport, resulting in remarkable electrical conductivity. When selecting ligands for constructing MOFs, porphyrin and its derivative ligands have garnered significant attention due to their exceptional photosensitivity. They are frequently employed in photocatalytic reactions, solar cells, catalysis, optoelectronics, and photodynamic therapy (PDT).29–38 These ligands can be obtained by substituting several functional groups at the meso- and β-positions of porphin (C20H14N4).39 When exposed to visible light, the extensive π-conjugated porphyrin structure can generate electron–hole pairs,40,41 leading to a substantial charge flow within the MOF. On the other hand, there have been several intriguing examples demonstrating that stacking porphyrin units in an eclipsed mode can provide a pathway for conduction, facilitating efficient carrier transport. Jiang et al.42 discovered that the eclipsed stacking orientation of porphyrin molecules promotes the rapid transport of charge carriers within porphyrin columns. In addition, the introduction of metals into the porphyrin core further enhances the electron transport capabilities. Chen et al.29 also observed that the AA stacking structures of porphyrin molecules enhance carrier dynamics. In this scenario, photogenerated electron transport relies on the metal pathway at the center of the porphyrin, while the macrocycle facilitates the transfer of photogenerated holes, leading to effective carrier separation. Moreover, incorporating porphyrins as ligands into the crystal lattices of MOFs can increase their versatility and offer promising properties that build on their inherent properties.43 These include exceptional solution stability, energy transport, exciton migration and enhanced optoelectronic properties.43–45
After decades of progress, the semiconductor industry has indeed reached a pivotal point in its history. Electronic devices are moving towards miniaturization, intelligence, and greater energy efficiency. Consequently, it is imperative to discover conductive solid structures that can facilitate these goals, and porphyrin-based MOFs are emerging as a compelling choice due to their unique properties. One approach to enhance the carrier mobility of porphyrin-based MOFs is to incorporate charge-accepting molecules into the porous framework.46,47 In this context, the establishment of donor–acceptor chemical interactions within photoconductive MOF systems could be a highly effective strategy for creating novel conductive MOFs. We are particularly interested in the MOF previously reported in our recent research,48 designated as 5: Zn2(TCPB)–(NMe2–ZnP); (H4TCPB = 1,2,4,5-tetrakis(4-carboxyphenyl)benzene), (NMe2–ZnP = [5,15-bis[(4-pyridyl)-ethynyl]-10,20-bis-(dimethylamine)porphinato]zinc(II)). The inception of this MOF can be traced back to the experimental work conducted by Son et al.,49 followed by subsequent theoretical investigations by Pratik et al.47
Our aim in this work is to evaluate the conductive properties of systems based on the 5@C60 MOF, referred to as C60@Rpy in this paper, where the fullerene acts as an effective electron acceptor, while the porphyrin ligand acts as an efficient electron donor, generating acceptor–donor chemical interactions. For this purpose, we modified the porphyrin ligand in their 5,15 meso-positions with different functional groups (Scheme 1), which differ in electron-donating ability. Fullerene-containing MOFs are designated as C60@Rpyr, C60@Rthiopyr, C60@Ramine, C60@Rphen, C60@Rsulf, C60@Ramine,biph, C60@Roxy,biph, C60@Rsulf,biph. We performed various atomic-scale modeling approaches for the MOFs with and without C60. First, we used periodic modeling to study the structure and electronic properties, then we performed molecular configuration modeling to study the optical properties through electronic absorption spectra. Finally, we used molecular junction configuration to estimate the charge transport properties of Rsulf and C60@Rsulf clusters model.
 | ||
| Scheme 1 Molecular structure of the MOFs investigated. | ||
2. Computational methods
Unit cell optimization of the MOFs (Fig. 1a and b and S1†) was performed using density functional theory (DFT) methods.50 We used the Linear Combination of Atomic Orbitals (LCAO), Perdew–Burke–Ernzerhof (PBEsol) exchange–correlation functional for solids,51 double-ζ-polarized (DZP) basis set, a 2 × 2 × 2 k-point sampling grid and PseudoDojo norm-conserving pseudopotentials52 to represent all atoms. Each SCF calculation was energetically converged to 1 × 10−4 eV, with density mesh cutoff of 250 Rydberg, and the maximum Hellmann–Feynman force of each atomic was converged to be less than 0.05 eV Å−1. Once the optimized crystal geometries were obtained, single point calculations for the electronic properties (band structures, interaction energy (Eint), density of states and electrical conductivity (σ)) were performed using the Perdew–Burke–Ernzerhof (PBE) exchange correlation functional53 and the empirical DFT-D3 method of Grimme54 to better account for dispersion interactions. In our previous work, we reported that PBE-D3 is the most appropriate method for calculating the electronic properties of these materials.48 One molecule of C60 was incorporated per unit cell (Fig. 1b and S1†), and the interaction energy was calculated as Eint = E(C60@MOF) − E(MOF) − E(C60). The energy values for MOF and C60 were determined in their respective optimized geometries within the C60@MOF. To account for any potential basis set superposition error (BSSE),55 we applied the counterpoise method to correct these energy values.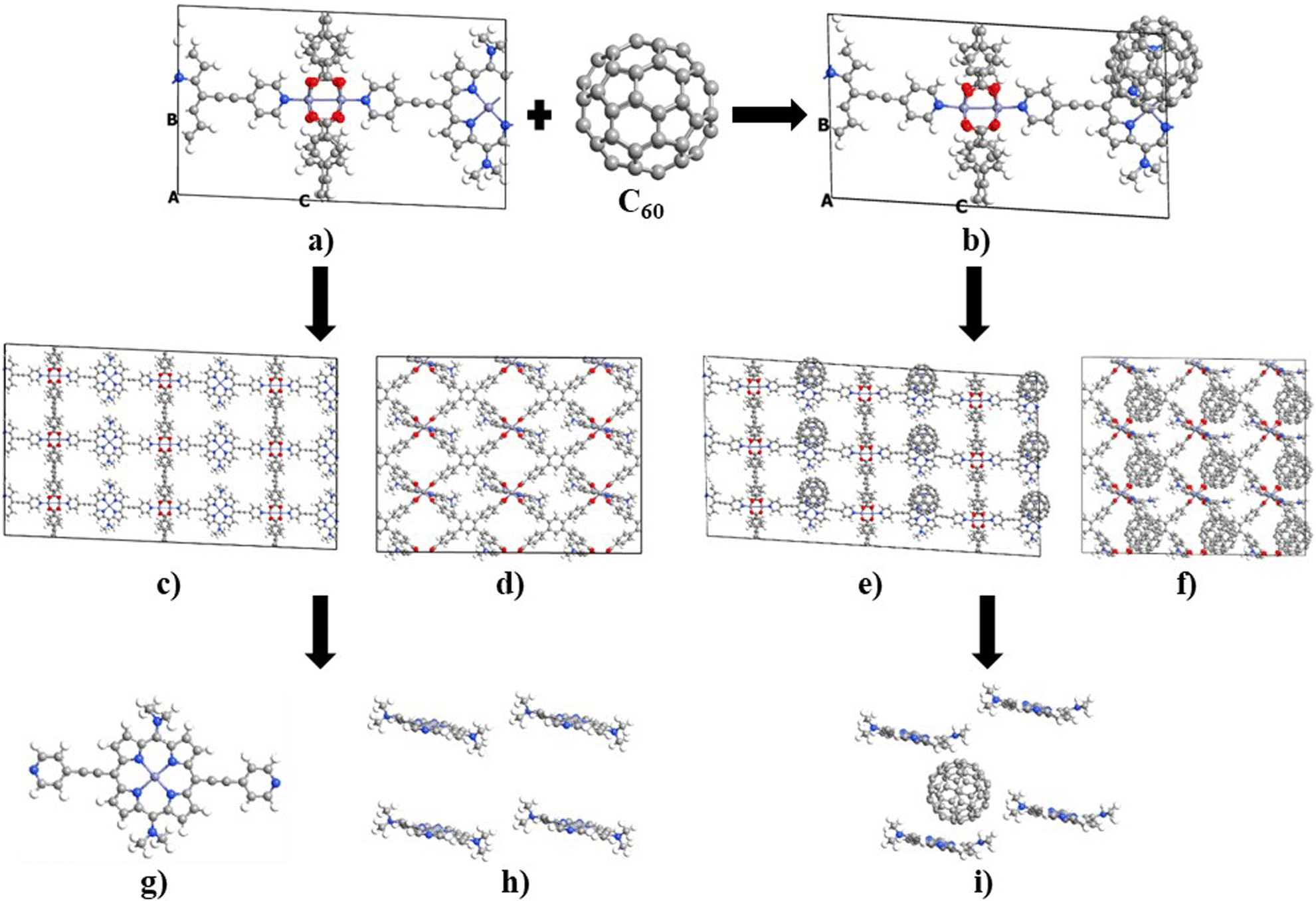 | ||
| Fig. 1 Molecular structure of the Rpy and C60@Rpy configurations; (a) unit cell optimized of Rpy. (b) Unit cell optimized of C60@Rpy. (c) Rpy bulk (front view). (d) Rpy bulk (side view). (e) C60@Rpy bulk (front view). (f) C60@Rpy bulk (side view). (g) Rpy porphyrin unit. (h) Rpy cluster. (i) C60@Rpy cluster. Color code: H (white), C (gray), N(blue), O (red), and Zn (purple). | ||
Density functional theory calculations can predict the electrical conductivity (σ) for semiconductor MOFs consisting of electron-donating and electron-accepting molecules. This latter property is the product of the number of charge carriers (n) with the electron's charge (e) and the carriers’ mobility (μ) given by the expression σ = n e μ.56 In a semiconductor, the intrinsic density of the charge carriers is directly proportional to 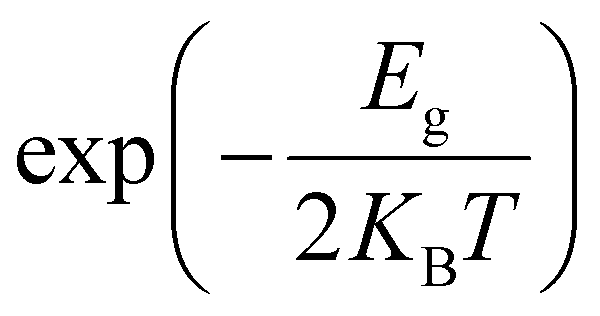 where Eg is the band gap energy of the system, KB is the Boltzmann constant, and T is the temperature. Thus, this exponential term constitutes a good approximation for determining the electrical conductivity, as has been shown by other authors.12,56–58 The Quantum ATK computational package59,60 was used for this first part of the study.
where Eg is the band gap energy of the system, KB is the Boltzmann constant, and T is the temperature. Thus, this exponential term constitutes a good approximation for determining the electrical conductivity, as has been shown by other authors.12,56–58 The Quantum ATK computational package59,60 was used for this first part of the study.
The optical properties of the MOFs have been studied using the electronic absorption spectra calculated by the time-dependent DFT (TD-DFT) method.61,62 For this purpose, clusters extracted from the optimized crystal geometry are used. Considering the proximity of each C60 to four porphyrin units within the MOFs, we selected a cluster consisting of four porphyrin units and one fullerene unit for the C60@MOF. We performed calculations TD-DFT to generate the absorption spectra for these cluster and then compared it with the absorption spectrum of a single porphyrin unit. This allowed us to investigate the effect of having four porphyrin units in the cluster on the absorption spectrum. To illustrate, the Fig. 1c–f show the optimized crystal geometry as bulk of Rpy and C60@Rpy from which the clusters were obtained to study optical properties (Fig. 1g–i). However, the methodology was applied to the other systems with and without fullerene. We performed TD-DFT calculations for the cluster models using the TPSSh functional63 and the 6-31G basis set, in a solvent phase consisting of dimethylformamide with dielectric constant of 36.7. This choice of solvent agrees with the solvent that was used by Son et al.49 for the measurement of the experimental spectra of the parent MOF. The choice of the functional was based on the results of our previous work48 where, after comparing the experimental UV-Vis spectra of structure of porphyrin, fullerene and porphyrin–fullerene complex reported in the literature with the theoretical UV-Vis spectra calculated with different functionals, we found that the TPSSh functional is the one that best reproduces the optical properties of these systems. The solvent was simulated with the conductor-like polarizable continuum model (CPCM).64,65 The singlet–singlet vertical excitations of the Franck–Condon type were modeled considering 60, 400, and 600 excited states of the porphyrin unit for the isolated system (Fig. 1g) and MOF cluster without (Fig. 1h) and with fullerene (Fig. 1i), respectively. These calculations were performed using the Gaussian 09 program suite.66
We designed theoretical molecular junctions67–70 with clusters of MOFs as the active molecular component within the scattering zone. Due to the size of the clusters and the computational effort required to obtain the charge transport properties of these junctions, we studied only the charge transport properties of Rsulf and C60@Rsulf MOFs. These clusters were obtained from the corresponding optimized crystal structures and consist of several MOF elements, mainly ligands and fullerenes in the case of the doped system. The linkers and the metal node were not considered because they affect the electron flow of the electronic devices.48 After optimizing the clusters in their molecular configurations, we sandwiched them between gold electrodes modeled as semi-infinite Au (111) wires. These junctions were optimized as electronic devices (Fig. S2†) from which we calculated the charge transport properties. Our calculations followed the Keldysh non-equilibrium Green's function formalism combined with density functional theory (DFT-NEGF),71,72 implemented using the Quantum ATK package. All calculations on this part of work were performed at the PBE/DZP/PseudoDojo theoretical level. The current–voltage (I–V) characteristics through the junction are given by the Landauer–Büttiker formula:73
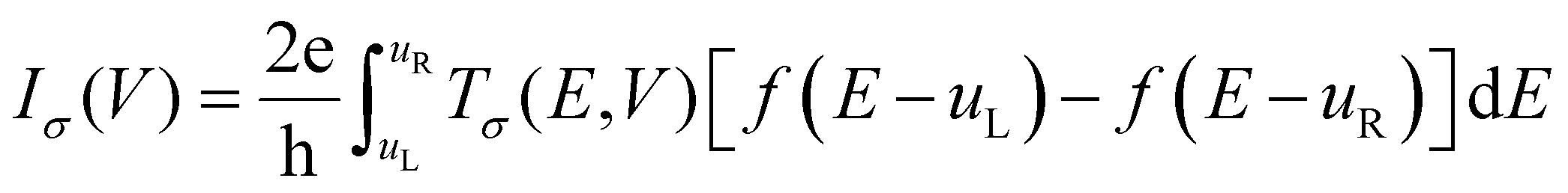
3. Results and discussion
3.1. Structural and electronic properties
The lattice parameters of the MOFs and C60@MOFs are shown in the ESI (Table S1†). The structures functionalized with the biphenyl moiety (Ramine,biph, Roxy,biph, Rsulf,biph) (Scheme 1), with and without fullerene, show a significant difference in the crystallographic axis c with respect to the rest of the MOFs. This difference is due to the size of the biphenyl group, which causes an increase in the unit cell volume produced by increasing the length of the crystallographic axis where the structure has been modified (Fig. S1†). As expected, no significant difference was observed between the MOFs without fullerene in the a and b crystallographic axes as well as in the unit cell angles, since the functionalization in the MOFs is in the z-plane. A similar behavior was found for C60@MOFs. After C60 incorporation, the optimized lattice parameters change significantly with respect to the MOFs without C60. This is an indication that there may be a kinetic barrier to the incorporation of fullerenes; however, the calculated interaction energy at the PBE-D3 level for C60@Rpy, C60@Rpyr, C60@Rthiopyr, C60@Ramine, C60@Rphen, C60@Rsulf, C60@Ramine,biph, C60@Roxy,biph, and C60@Rsulf,biph is −1.25, −1.08, −1.34, −1.95, −8.55, −1.50, −1.23, −7.35 and −6.71 eV, respectively, indicating that incorporation is quite favorable in all cases. C60@Rphen and C60@Roxy,biph MOFs are the materials with the most stable interactions, which may be due to dipole-induced dipole attraction van der Waals interactions between the –O– moiety and the fullerene.We have shown the density of states (DOS) of MOFs with C60 in several figures to analyze the crystalline-phase orbitals of the materials. The DOS of nitrogen-functionalized MOFs are shown in Fig. 2 while Fig. S3 and S4† show those oxygen-functionalized and sulfur-functionalized MOFs, respectively. Fig. S5† shows the band structures for MOFs before and after the introduction of C60. For MOFs functionalized with substituents where the heteroatom is inside the phenyl ring (Rpy, Rpyr, Rthiopyr), there is a noticeable decrease in the band gap for the systems when the fullerene is incorporated. It can be explained in terms of a possible contribution of the lone pairs of the heteroatom toward the electronic resonance of the phenyl ring. The lowest energy of the unoccupied crystalline-phase orbitals (LUCOs) corresponds mainly to the p orbitals of fullerene (Fig. 2, S3 and S4†), which is reflected in one peak closer to the Fermi level. Therefore, the conduction band (CB) of MOFs stabilizes, and the band gap decreases with the incorporation of C60. In the cases of MOFs functionalized with the other substituents (Fig. S5†), where the heteroatom is not part of the resonance path within the phenyl ring, the incorporation of the fullerene causes the destabilization of the CB, leading to an increase in the band gap energy of the MOFs. The LUCOs orbitals mainly correspond to the p orbitals of the ligand and therefore the conduction band of the MOF is not stabilized by the incorporation of fullerene. Although C60 causes an increase in the band gap of these MOFs, their unoccupied degenerate bands close in energy to the CB would stimulate the charge transfer (CT) processes of the system.74 In general, sulfur-functionalized MOFs have the lowest band gap energy when comparing Rthiopyr with Rpy and Rpyr, Rsulf with Ramine and Rphen, Rsulf,biph with Ramine,biph and Roxy,biph. This is because the sulfur atom is soft and electron-rich, which is advantageous for designing polar and electron-rich structures with soft guest interaction sites. However, these MOFs can be limited because the sulfur atom is heavier than oxygen and nitrogen, resulting in structures with smaller surface area, and the –S– moiety is also partially oxidized at higher temperatures.75 In crystalline materials, atom size, and bond length affect packing efficiency and surface area. Due to larger atomic radius and longer bonds, sulfur-containing compounds often have more open crystal structures. This reduces the total surface area of the structure, limiting the ability to form tightly packed arrangements.
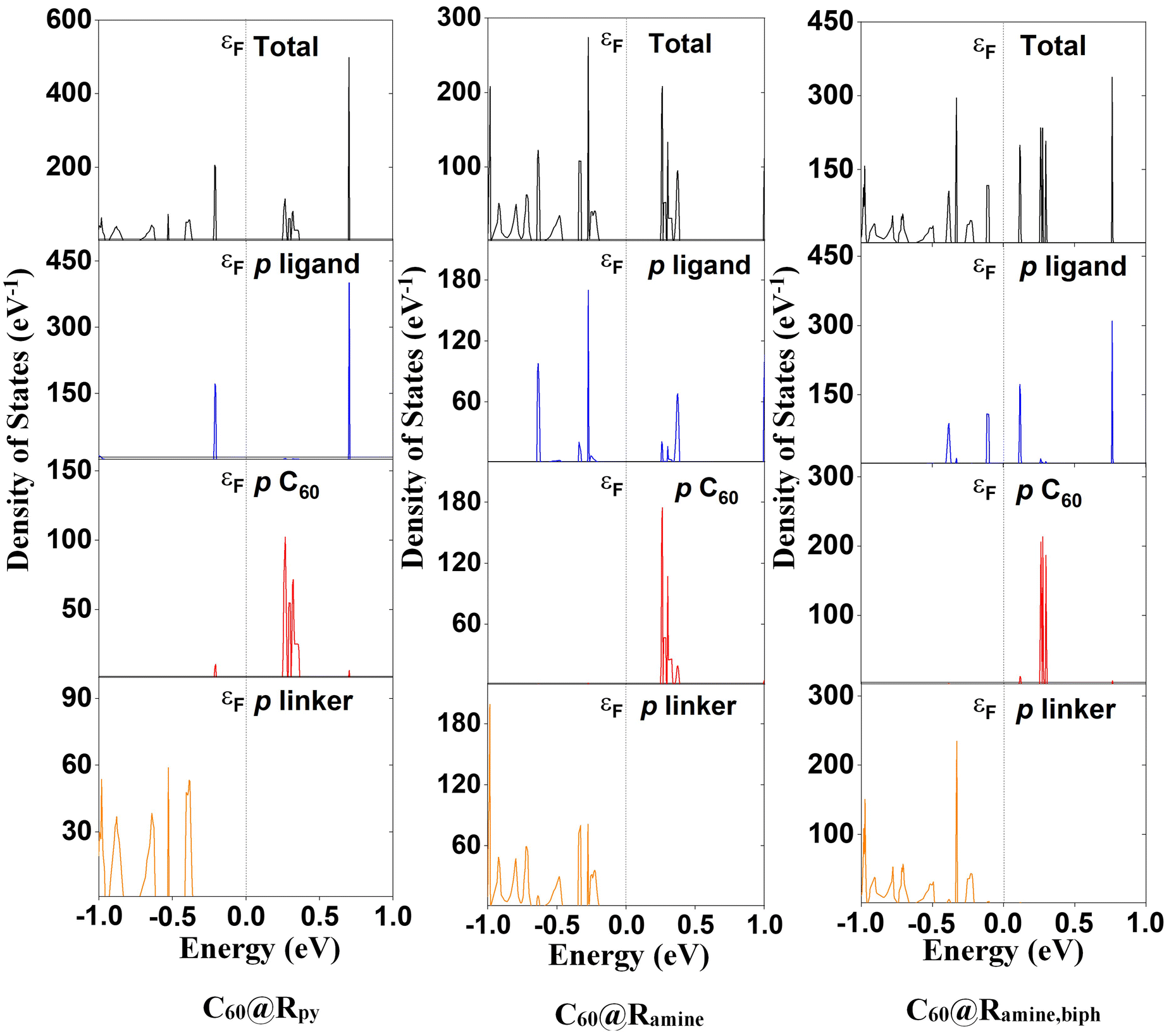 | ||
| Fig. 2 Density of states (DOS) of C60@Rpy, C60@Ramine and C60@Ramine,biph (nitrogen-functionalized materials) MOFs calculated using the PBE-D3 level. Fermi level energy is indicated by εF. | ||
C60@Rpy MOF (Fig. 2) may have a greater potential for charge transfer processes from porphyrin to fullerene, because the higher energy occupied crystal orbitals (HOCOs) correspond mainly to the p orbitals of the ligand, whereas the LUCOs, as we have already mentioned, are mainly attributed to the p orbitals of C60. The electronic behavior of the MOF changes significantly when the heteroatom is outside the phenyl ring (C60@Ramine). The HOCOs and LUCOs orbitals mainly correspond to the p orbitals of the linker and ligand, respectively. Therefore, the charge transfer processes from linker to the ligand could be favored. Lastly, the HOCOs and LUCOs orbitals are due to the p orbitals of ligand when the MOF is functionalized with the biphenyl moiety (C60@Ramine,biph), which favors ligand–ligand CT processes. A similar trend is observed for MOFs functionalized with oxygen- and sulfur-based substituents (Fig. S3 and S4,† respectively). In all cases, a very important contribution of the p orbitals of C60 in the unoccupied states of the MOFs is seen. However, the conduction band corresponds to the fullerene orbitals only in MOFs C60@Rpy, C60@Rpyr and C60@Rthiopyr.
On the other hand, we also estimated the electrical conductivity (σ) of the MOFs before and after incorporating the fullerene (Table 1). The incorporation of C60 increases the σ of MOFs C60@Rpy, C60@Rpyr and C60@Rthiopyr, while it slightly decreases in the other systems. As expected from the smallest band gap, Rsulf,biph and C60@Rsulf,biph are the MOFs with better electrical conductivity. Although is not observed a clear trend concerning to the heteroatom, we found that a higher conductivity is favored for C60@MOFs where the heteroatom is outside of the phenyl ring (O- and S-functionalization). These results demonstrate that the electronic properties of porphyrin-based conductive MOFs can be tuned by modifying the substituents present at 5,15 meso-positions of the ligand.
| MOF | E g (eV) | σ (S cm−1) | MOF | E g (eV) | σ (S cm−1) |
|---|---|---|---|---|---|
| Rpy | 0.94 | 1.13 × 10−8 | C60@Rpy | 0.45 | 1.57 × 10−4 |
| Rpyr | 0.72 | 8.16 × 10−7 | C60@Rpyr | 0.33 | 1.62 × 10−3 |
| Rthiopyr | 0.65 | 3.19 × 10−6 | C60@Rthiopyr | 0.36 | 9.03 × 10−4 |
| Ramine | 0.41 | 3.41 × 10−4 | C60@Ramine | 0.44 | 1.90 × 10−4 |
| Rphen | 0.28 | 4.29 × 10−3 | C60@Rphen | 0.35 | 1.10 × 10−3 |
| Rsulf | 0.28 | 4.29 × 10−3 | C60@Rsulf | 0.33 | 1.62 × 10−3 |
| Ramine,biph | 0.16 | 4.44 × 10−2 | C60@Ramine,biph | 0.21 | 1.68 × 10−2 |
| Roxy,biph | 0.18 | 3.01 × 10−2 | C60@Roxy,biph | 0.20 | 2.04 × 10−2 |
| Rsulf,biph | 0.10 | 1.43 × 10−1 | C60@Rsulf,biph | 0.14 | 6.55 × 10−2 |
3.2. Optical properties
The simulation of the electronic absorption spectra for MOF clusters with and without C60 and the isolated porphyrin unit was calculated using time-dependent DFT methodology. Fig. 3 shows the spectra of C60@Rpy, C60@Rpyr, and C60@Rthiopyr, while Fig. S6† shows the spectra of the six remaining clusters with and without C60 and the respective porphyrin units. Table 2 shows the first absorption band of a larger wavelength with non-zero oscillator strengths. An in-depth analysis of the main transitions of these absorption spectra is presented in Tables S2–S4.† In all C60@MOFs, the bathochromic effect generated by the incorporation of C60 was demonstrated by a strong increase in the absorption wavelength with respect to the corresponding cluster without fullerene. The wavelength of porphyrin–porphyrin CT transitions is larger for MOFs clusters without fullerenes, except for Rpyr and Rthiopyr MOFs. In the latter, the absorption bands are observed at lower excitation wavelengths due to the presence of many dark states. A dark state (DS) is a state that has a transition dipole moment of zero with respect to the ground state, i.e., the state cannot be reached by absorption of a photon from the ground state and is therefore “dark” to electromagnetic radiation.76–81 However, it can play an important role in photochemistry, photophysics, and spectroscopy of molecules.76–81 The oscillator strength (f), which is a measure of the probability of an electronic transition between two states, is one way to identify DS in TD-DFT calculations. A dark state will have a zero-oscillator strength because f is proportional to the square of the transition dipole moment.76,82–84 The presence of DS was observed for all MOFs clusters without C60; Rpy, Ramine, Rphen, Rsulf, Ramine,biph, Roxy,biph and Rsulf,biph MOFs contain four dark states, while Rpyr and Rthiopyr MOFs show 14 and 15 DS, respectively. The incorporation of fullerenes leads to the elimination of dark states in MOFs, which also cause the absorption at longer wavelengths for the doped materials.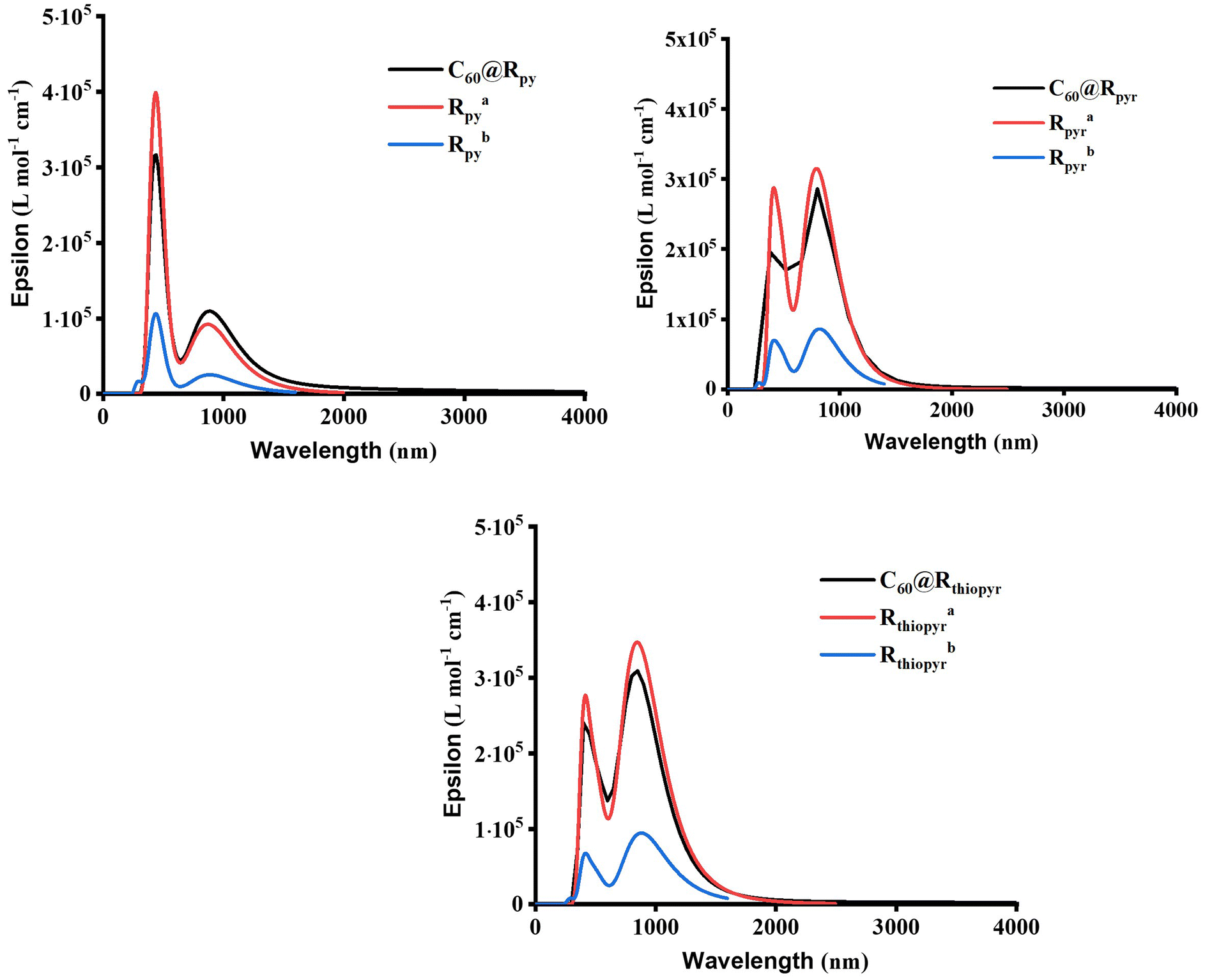 | ||
Fig. 3 Electronic absorption spectra of MOFs clusters calculated at the TPSSh/6-31G level of theory. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MOF cluster without C60. bPorphyrin unit. MOF cluster without C60. bPorphyrin unit. | ||
| MOF | Excited State | λ (nm) | f | MOF | Excited State | λ (nm) | f |
|---|---|---|---|---|---|---|---|
| a MOF cluster without C60. b Porphyrin unit. | |||||||
| C60@Rpy | 1 | 2139 | 0.0001 | C60@Rsulf | 1 | 2809 | 0.0042 |
| Rpy | 5 | 1009 | 0.0003 | Rsulf | 5 | 1146 | 0.0004 |
| Rpy | 1 | 889 | 0.6189 | Rsulf | 1 | 955 | 0.8722 |
| C60@Rpyr | 1 | 7161 | 0.0015 | C60@Ramine,biph | 1 | 2780 | 0.0006 |
| Rpyr | 15 | 819 | 0.0004 | Ramine,biph | 5 | 1078 | 0.0005 |
| Rpyr | 1 | 820 | 2.0541 | Ramine,biph | 1 | 930 | 1.2742 |
| C60@Rthiopyr | 1 | 4545 | 0.0001 | C60@Roxy,biph | 1 | 2597 | 0.0117 |
| Rthiopyr | 16 | 849 | 8.2807 | Roxy,biph | 5 | 1082 | 0.0003 |
| Rthiopyr | 1 | 883 | 2.3060 | Roxy,biph | 1 | 936 | 1.2366 |
| C60@Ramine | 1 | 3549 | 0.0017 | C60@Rsulf,biph | 1 | 2706 | 0.0005 |
| Ramine | 5 | 1157 | 0.0002 | Rsulf,biph | 5 | 1082 | 0.0003 |
| Ramine | 1 | 945 | 0.8701 | Rsulf,biph | 1 | 937 | 1.1446 |
| C60@Rphen | 1 | 4053 | 0.0001 | ||||
| Rphen | 5 | 1109 | 0.0002 | ||||
| Rphen | 1 | 924 | 0.8461 | ||||
Tables S2–S4† show the main transitions of the calculated absorption spectra of C60@MOFs clusters, MOFs clusters without C60, and isolated porphyrin unit, respectively. TD-DFT is a quantum mechanical method that can simulate a multi-electron description of the transitions between the occupied and unoccupied energy levels.85,86 It can also provide information about vibrational transitions, some molecules can exhibit electronic excitations coupled to molecular vibrational excitations, resulting in absorption bands extending into the infrared region. This can occur in highly conjugated systems or when electron–vibrational interactions are strong. To visualize how the electrons move from one energy level to another, we can use natural transition orbitals (NTOs). NTOs are a simplified representation of electronic transitions in which each excited state is described by occupied (NTOocc) and unoccupied (NTOvirt) orbitals and help us to understand the nature and character of electronic excitations. For these reasons we use NTOs calculations to describe each of the reported transitions. By exhibiting configurations consistent with strong π–π interactions, multiple donor–acceptor conjugates promote charge transfer and photoinduced electron transfer (PET) processes from porphyrin to C60. These phenomena are widely known in analogous molecular and supramolecular structures.87,88 In this context, Table S2† reports a series of CT and PET processes from ligand to fullerene for all studied MOFs. Low energy mid-IR (∼2.5–25 μm wavelength) transitions from occupied porphyrin orbitals to unoccupied C60 orbitals occur in C60@Rpyr, C60@Rthiopyr, C60@Ramine, C60@Rphen, C60@Rsulf, C60@Ramine,biph, C60@Roxy,biph, and C60@Rsulf,biph MOFs. For all C60@MOFs, we also observe low-energy porphyrin–C60 transitions in the near-IR (∼800–2500 nm wavelength). These results are consistent with the periodic calculations that predict an important contribution of porphyrin-based HOCOs and C60-based LUCOs. Transitions with higher energy levels are associated with intramolecular π → π* transitions that occur within the porphyrin moiety and involve absorption of near-IR and visible light (∼400–800 nm wavelength). According to Kasha's rule,89 these higher energy states must undergo efficient internal transformations to reach the lowest excited state based on the porphyrin. The subsequent transfer of electrons to the LUMO of C60 would therefore represent a PET process. Appropriate spatial and geometric alignment within the donor–acceptor conjugate system makes this possible.
Upon exposure to light-induced oxidation, molecular and supramolecular aggregates of porphyrin and fullerene are known to generate persistent radical cations and radical anion.88,90 Chen et al.91 showed that the spatial confinement of C60 by covalent anchoring within the nanochannels of a porphyrin-based covalent organic framework ([C60]y–ZnPc–COFs) effectively facilitates photoinduced electron transfer. This process allows charge separation leading to the formation of ZnPc˙+ and C60˙− species. This ultimately results in the observed photoelectric conductivity of the COF. In a similar way, the formation of ionized species, which could contribute to the photoelectric conductivity, may be possible in the C60@MOFs reported here.
Finally, to demonstrate that the models used are representative approximations of MOFs, we built a new C60@Rpy cluster model, named C60@Rpy-new, consisting of eight porphyrin units, two fullerenes, four metal nodes, and one linker (Fig. S7†), and calculated the UV-Vis absorption spectrum. The main transitions of the spectrum are listed in Table 3. The first excitation appears at 1986 nm and corresponds to a CT band from the porphyrins to the fullerene in the near-infrared region. Excited states 3, 6, and 7, which occur in 1929, 1878, and 1873 nm, are also CT bands from the porphyrins to the fullerene. Concerning the Q band of the porphyrin, this band appears at 923 nm (1.34 eV). Comparing these results with the data obtained for the C60@Rpy cluster model (Table S2†), the deviations are small, with the porphyrin Q band (889 nm, 1.39 eV) showing the largest deviation (0.05 eV). These deviations are considered small for TD-DFT calculations, with errors of up to 0.6 eV accepted in the literature.92,93 Therefore, we can conclude that the clusters chosen and shown in Fig. 1 are well suited to calculate the optical properties of the MOFs. Moreover, since porphyrins are the chromophores molecules and we are mainly interested in the interaction with C60, where porphyrins play an important role, the choice of clusters is well justified. In the literature, cluster models have been widely applied to study the optical properties of MOFs.47,94–98
| Excited state | λ (nm) | f | Transition involved |
|---|---|---|---|
| 1 | 1986 | 0.0001 |
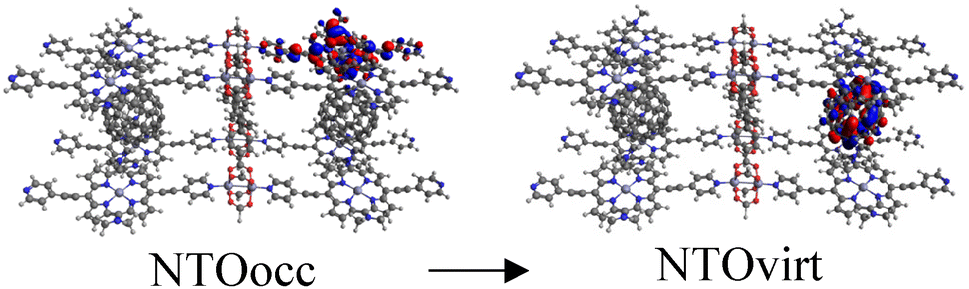
|
| 3 | 1929 | 0.0080 |
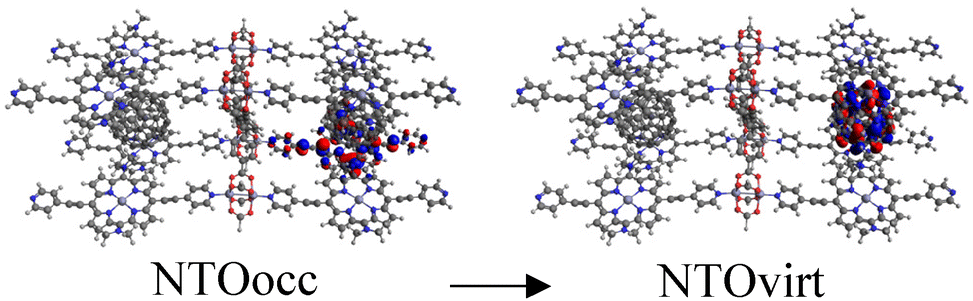
|
| 6 | 1878 | 0.0228 |

|
| 7 | 1873 | 0.0485 |
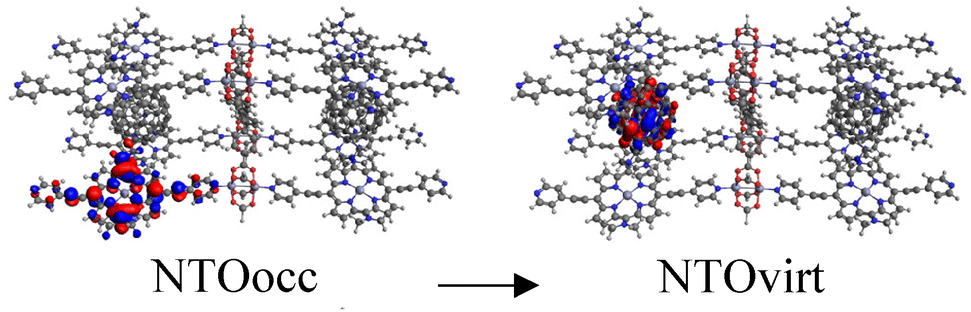
|
| 111 | 923 | 6.1809 |

|
3.3. Charge transport properties
In this section, we will only focus on Rsulf and C60@Rsulf MOFs due to high computational cost. To study the charge transport properties of Rsulf and C60@Rsulf MOFs, each cluster was attached to two semi-infinite Au(111) wires as electrodes using the sulfur atoms of the MOFs as shown in Fig. S2.† In the hollow position of the Au(111) surface, the sulfur atoms are bonded to gold atoms corresponding to adatoms. This contact design was selected based on its established use in numerous previous studies designed to investigate molecular transport properties using semi-infinite gold electrodes.67–70 For the two studied systems the conductance values at zero bias obtained through DFT-NEGF methods is considerably high with values of 0.42 and 0.72 G0 for Rsulf and C60@Rsulf, respectively. The fullerene moiety slightly increased the conductance of the system, possibly due to donor–acceptor interactions between porphyrins and C60, favoring electron flow between the electrodes.Transmission plots (Fig. 4) show strong signals at energies in the range of −0.3–0.3 eV very close to the Fermi level (εF), below and above εF for both systems studied, along with small bands in the PDOS at corresponding energy values. Resonances with the highest occupied and lowest unoccupied molecular orbitals may be responsible for these transmission peaks. A significant increase in the unoccupied states around 0.3–1.2 eV is observed with the incorporation of C60. Furthermore, the number of available occupied states increase significantly in the range −1.5 to −1.2 eV. Charge transport processes can be favored by increasing the number of occupied and unoccupied states due to the incorporation of C60. The current through of the studied molecular junctions was calculated to confirm the effect of C60 on the transport properties. The I–V results are shown in Fig. 5. It can be observed that the current values calculated in the systems reproduce the trends obtained for the conductance. In units of μA, the currents obtained for Rsulf andC60@Rsulf at each voltage point are quite significant. A slight increase in C60@Rsulf current was observed compared to Rsulf, which is consistent with the transmission spectra, PDOS, and calculated conductance, suggesting an increase in charge transport processes for the doped MOF.
 | ||
| Fig. 4 PDOS (top) and transmission spectra (bottom) of Rsulf andC60@Rsulf molecular junctions. | ||
 | ||
| Fig. 5 Calculated current versus applied bias voltage on Rsulf (blue) and C60@Rsulf (red) molecular junctions. | ||
4. Conclusions
Our research involved an in-depth investigation of the effects of introducing fullerene doping on the electronic properties of porphyrin-based conductive MOFs using density functional theory in bulk modeling calculations. We also investigated the optical and transport properties using cluster models of the materials. Our study shows that the electronic properties of porphyrin-based conductive MOFs can be finely tuned by modifying the substituents at the 5,15 meso-positions of the porphyrin. MOFs functionalized with biphenyl substitutions show lower band gap energy and higher electrical conductivity. TD-DFT calculations indicate that electron transfer from porphyrin to C60 can occur directly via mid/near-infrared charge transfer transitions or via photoinduced electron transfer upon visible light excitation. Finally, the electronic devices studied suggest that the C60 incorporation enhances charge transport processes through π-mediated donor–acceptor interactions for materials with and without C60. Our results demonstrate that an effective strategy for designing new photoconductive MOFs is to induce donor–acceptor interactions in the material's pores by incorporating C60.Author contributions
Kevin Granados-Tavera: Methodology, investigation, writing – original draft. Gloria Cárdenas-Jirón: Writing – review & editing, supervision, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank ANID/CHILE for its financial support under Project FONDECYT No. 1221072 (G. C.-J.). Powered@NLHPC: This research was partially supported by the NLHPC (ECM-02) supercomputing infrastructure of the Universidad de Chile. It is grateful for the support of ANID/Chile by Program Doctorado Becas Nacionales 2021/21210404 (K. G.-T.).References
- R.-B. Lin, S. Xiang, H. Xing, W. Zhou and B. Chen, Coord. Chem. Rev., 2019, 378, 87–103 CrossRef CAS
.
- R. Hou, M. Miao, Q. Wang, T. Yue, H. Liu, H. S. Park, K. Qi and B. Y. Xia, Adv. Energy Mater., 2020, 10, 1901892 CrossRef CAS
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS
- J. Liu, Y. Zhou, Z. Xie, Y. Li, Y. Liu, J. Sun, Y. Ma, O. Terasaki and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 1081–1086 CrossRef CAS
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed
- M. Wang, R. Dong and X. Feng, Chem. Soc. Rev., 2021, 50, 2764–2793 RSC
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC
- Y. He, F. Yan, X. Zhang, C. Zhu, Y. Zhao, B. Geng, S. Chou, Y. Xie and Y. Chen, Adv. Energy Mater., 2023, 13, 2204177 CrossRef CAS
- X.-L. Wang, L.-Z. Dong, M. Qiao, Y.-J. Tang, J. Liu, Y. Li, S.-L. Li, J.-X. Su and Y.-Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 9660–9664 CrossRef CAS
- H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Büchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS
- W.-H. Li, W.-H. Deng, G.-E. Wang and G. Xu, EnergyChem, 2020, 2, 100029 CrossRef
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed
- G. Wu, J. Huang, Y. Zang, J. He and G. Xu, J. Am. Chem. Soc., 2017, 139, 1360–1363 CrossRef CAS
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS
- C. Li, L. Zhang, J. Chen, X. Li, J. Sun, J. Zhu, X. Wang and Y. Fu, Nanoscale, 2021, 13, 485–509 RSC
- U. Khan, A. Nairan, J. Gao and Q. Zhang, Small Struct., 2023, 4, 2200109 CrossRef CAS
- Y. Wang, B. Liu, X. Shen, H. Arandiyan, T. Zhao, Y. Li, M. Garbrecht, Z. Su, L. Han, A. Tricoli and C. Zhao, Adv. Energy Mater., 2021, 11, 2003759 CrossRef CAS
- W. Xu, Y. Zhang, J. Wang, Y. Xu, L. Bian, Q. Ju, Y. Wang and Z. Fang, Nat. Commun., 2022, 13, 2068 CrossRef CAS
- W. Cheng, Z.-P. Wu, D. Luan, S.-Q. Zang and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 26397–26402 CrossRef CAS
- W.-H. Li, J. Lv, Q. Li, J. Xie, N. Ogiwara, Y. Huang, H. Jiang, H. Kitagawa, G. Xu and Y. Wang, J. Mater. Chem. A, 2019, 7, 10431–10438 RSC
- L. Zhao, B. Dong, S. Li, L. Zhou, L. Lai, Z. Wang, S. Zhao, M. Han, K. Gao, M. Lu, X. Xie, B. Chen, Z. Liu, X. Wang, H. Zhang, H. Li, J. Liu, H. Zhang, X. Huang and W. Huang, ACS Nano, 2017, 11, 5800–5807 CrossRef CAS PubMed
- H. Meng, Y. Han, C. Zhou, Q. Jiang, X. Shi, C. Zhan and R. Zhang, Small Methods, 2020, 4, 2000396 CrossRef CAS
- L. Lin, Q. Zhang, Y. Ni, L. Shang, X. Zhang, Z. Yan, Q. Zhao and J. Chen, Chem, 2022, 8, 1822–1854 CAS
- X. Deng, J.-Y. Hu, J. Luo, W.-M. Liao and J. He, Top. Curr. Chem., 2020, 378, 27 CrossRef CAS PubMed
- M. J. Golomb, J. Calbo, J. K. Bristow and A. Walsh, J. Mater. Chem. A, 2020, 8, 13160–13165 RSC
- M. Souto, K. Strutyński, M. Melle-Franco and J. Rocha, Chem. – Eur. J., 2020, 26, 10912–10935 CrossRef CAS
- J. G. Park, M. L. Aubrey, J. Oktawiec, K. Chakarawet, L. E. Darago, F. Grandjean, G. J. Long and J. R. Long, J. Am. Chem. Soc., 2018, 140, 8526–8534 CrossRef CAS
- R. Chen, Y. Wang, Y. Ma, A. Mal, X.-Y. Gao, L. Gao, L. Qiao, X.-B. Li, L.-Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS
- J. Jing, J. Yang, W. Li, Z. Wu and Y. Zhu, Adv. Mater., 2022, 34, 2106807 CrossRef CAS
- C. Lin, C. Han, H. Zhang, L. Gong, Y. Gao, H. Wang, Y. Bian, R. Li and J. Jiang, Inorg. Chem., 2021, 60, 3988–3995 CrossRef CAS PubMed
- M. I. Menéndez, N. Montenegro-Pohlhammer, R. Pino-Rios, R. Urzúa-Leiva, S. Morales-Lovera, M. Borges-Martínez, K. Granados-Tavera, R. López and G. Cárdenas-Jirón, J. Chem. Phys., 2023, 158, 164305 CrossRef
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS
- M. C. So, S. Jin, H.-J. Son, G. P. Wiederrecht, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 15698–15701 CrossRef CAS PubMed
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC
- Z.-W. Huang, K.-Q. Hu, X.-B. Li, Z.-N. Bin, Q.-Y. Wu, Z.-H. Zhang, Z.-J. Guo, W.-S. Wu, Z.-F. Chai, L. Mei and W.-Q. Shi, J. Am. Chem. Soc., 2023, 145, 18148–18159 CrossRef CAS
- G. Ding, S.-T. Han, C.-C. Kuo, V. A. L. Roy and Y. Zhou, Small Struct., 2023, 4, 2200150 CrossRef CAS
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. Chen, T. Islamoglu, A. J. Howarth, Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615 CrossRef CAS PubMed
- L. Feng, K.-Y. Wang, E. Joseph and H.-C. Zhou, Trends Chem., 2020, 2, 555–568 CrossRef CAS
- X. Feng, L. Liu, Y. Honsho, A. Saeki, S. Seki, S. Irle, Y. Dong, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2012, 51, 2618–2622 CrossRef CAS
- S. S. Rajasree, X. Li and P. Deria, Commun. Chem., 2021, 4, 47 CrossRef CAS PubMed
- S. M. Shaikh, A. Chakraborty, J. Alatis, M. Cai, E. Danilov and A. J. Morris, Faraday Discuss., 2019, 216, 174–190 RSC
- W. Yu, W. Zhen, Q. Zhang, Y. Li, H. Luo, J. He and Y. Liu, ChemMedChem, 2020, 15, 1766–1775 CrossRef CAS
- X. Liu, M. Kozlowska, T. Okkali, D. Wagner, T. Higashino, G. Brenner-Weiß, S. M. Marschner, Z. Fu, Q. Zhang, H. Imahori, S. Bräse, W. Wenzel, C. Wöll and L. Heinke, Angew. Chem., Int. Ed., 2019, 58, 9590–9595 CrossRef CAS PubMed
- S. M. Pratik, L. Gagliardi and C. J. Cramer, J. Phys. Chem. C, 2020, 124, 1878–1887 CrossRef CAS
- K. Granados-Tavera, N. Montenegro-Pohlhammer and G. Cárdenas-Jirón, Surf. Interfaces, 2023, 40, 103002 CrossRef CAS
- H.-J. Son, S. Jin, S. Patwardhan, S. J. Wezenberg, N. C. Jeong, M. So, C. E. Wilmer, A. A. Sarjeant, G. C. Schatz, R. Q. Snurr, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 862–869 CrossRef CAS
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed
- M. J. van Setten, M. Giantomassi, E. Bousquet, M. J. Verstraete, D. R. Hamann, X. Gonze and G. M. Rignanese, Comput. Phys. Commun., 2018, 226, 39–54 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS
- D. Ray, S. Goswami, J. Duan, J. T. Hupp, C. J. Cramer and L. Gagliardi, Chem. Mater., 2021, 33, 1182–1189 CrossRef CAS
- C.-W. Kung, S. Goswami, I. Hod, T. C. Wang, J. Duan, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2020, 53, 1187–1195 CrossRef CAS
- R. Saha, K. Gupta and C. J. Gómez García, Cryst. Growth Des., 2024, 24, 2235–2265 CrossRef CAS
- S. Smidstrup, T. Markussen, P. Vancraeyveld, J. Wellendorff, J. Schneider, T. Gunst, B. Verstichel, D. Stradi, P. A. Khomyakov, U. G. Vej-Hansen, M.-E. Lee, S. T. Chill, F. Rasmussen, G. Penazzi, F. Corsetti, A. Ojanperä, K. Jensen, M. L. N. Palsgaard, U. Martinez, A. Blom, M. Brandbyge and K. Stokbro, J. Phys.: Condens. Matter, 2020, 32, 015901 CrossRef CAS PubMed
- QuantumATK version Q-2021.06, Synopsys QuantumATK. (https://www.synopsys.com/manufacturing/quantumatk.html).
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS
-
M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, 2009 Search PubMed
- P. A. Derosa and J. M. Seminario, J. Phys. Chem. B, 2001, 105, 471–481 CrossRef CAS
- N. Montenegro-Pohlhammer, R. Urzúa-Leiva, D. Páez-Hernández and G. Cárdenas-Jirón, Dalton Trans., 2019, 48, 8418–8426 RSC
- A. Yamada, Q. Feng, Q. Zhou, A. Hoskins, K. M. Lewis and B. D. Dunietz, J. Phys. Chem. C, 2017, 121, 10298–10304 CrossRef CAS
- Q. Zhou, A. Yamada, Q. Feng, A. Hoskins, B. D. Dunietz and K. M. Lewis, ACS Appl. Mater. Interfaces, 2017, 9, 15901–15906 CrossRef CAS PubMed
- J. Taylor, H. Guo and J. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245407 CrossRef
- M. Brandbyge, J.-L. Mozos, P. Ordejón, J. Taylor and K. Stokbro, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165401 CrossRef
- M. Büttiker, Y. Imry, R. Landauer and S. Pinhas, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 6207–6215 CrossRef
- M. Borges-Martínez, N. Montenegro-Pohlhammer, X. Zhang, D. E. Galvez-Aranda, V. Ponce, J. M. Seminario and G. Cárdenas-Jirón, Spectrochim. Acta, Part A, 2022, 269, 120740 CrossRef
-
S. A. A. R. Ali Morsali, in Functional Metal–Organic Frameworks, 2020, pp. 133–164. DOI:10.1002/9781119640998.ch7
-
P. Lambropoulos and D. Petrosyan, Fundamentals of Quantum Optics and Quantum Information, 2007. DOI:10.1007/978-3-540-34572-5
- C. Emary, J. Phys. B: At., Mol. Opt. Phys., 2013, 46, 224008 CrossRef
-
A. M. Fox, Quantum optics: an introduction, Oxford University Press, USA, 2006 Search PubMed
- P.-A. Yin, Q. Wan, Y. Niu, Q. Peng, Z. Wang, Y. Li, A. Qin, Z. Shuai and B. Z. Tang, Adv. Electron. Mater., 2020, 6, 2000255 CrossRef CAS
- R. M. Godun, M. B. d'Arcy, M. K. Oberthaler, G. S. Summy and K. Burnett, Opt. Commun., 1999, 169, 301–308 CrossRef CAS
- M. Auzinsh, N. N. Bezuglov and K. Miculis, Phys. Rev. A, 2008, 78, 053415 CrossRef
- W. Zeng, S. Gong, C. Zhong and C. Yang, J. Phys. Chem. C, 2019, 123, 10081–10086 CrossRef CAS
- V. Dharaniprabha, P. Saravanakumar, A. Kalavathi, K. Satheeshkumar, K. N. Vennila and K. P. Elango, J. Mol. Struct., 2024, 1296, 136816 CrossRef CAS
- A. Weigel, A. L. Dobryakov, M. Veiga and J. L. Pérez Lustres, J. Phys. Chem. A, 2008, 112, 12054–12065 CrossRef CAS
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS
- F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410–1422 CrossRef
- J. Sukegawa, C. Schubert, X. Zhu, H. Tsuji, D. M. Guldi and E. Nakamura, Nat. Chem., 2014, 6, 899–905 CrossRef CAS PubMed
- J. C. del Valle and J. Catalán, Phys. Chem. Chem. Phys., 2019, 21, 10061–10069 RSC
- D. M. Guldi, A. Hirsch, M. Scheloske, E. Dietel, A. Troisi, F. Zerbetto and M. Prato, Chem. – Eur. J., 2003, 9, 4968–4979 CrossRef CAS
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef CAS PubMed
- K. Harmandar, K. Granados-Tavera, M. Gezgin, M. Nebioğlu, İ. Şişman, G. Cárdenas-Jirón, D. Atilla and A. G. Gürek, New J. Chem., 2022, 46, 714–725 RSC
- X. Li, S. Surendran Rajasree, J. Yu and P. Deria, Dalton Trans., 2020, 49, 12892–12917 RSC
- A. Nath, S. Chawla, A. K. De, P. Deria and S. Mandal, Chem. – Eur. J., 2023, 29, e202202978 CrossRef CAS PubMed
- S. Chong, D. T. Park and J. Kim, J. Phys. Chem. C, 2021, 125, 10198–10206 CrossRef CAS
- M. Ingham, A. Aziz, D. Di Tommaso and R. Crespo-Otero, Mater. Adv., 2023, 4, 5388–5419 RSC
- P. Mialane, C. Mellot-Draznieks, P. Gairola, M. Duguet, Y. Benseghir, O. Oms and A. Dolbecq, Chem. Soc. Rev., 2021, 50, 6152–6220 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01459f |
| This journal is © The Royal Society of Chemistry 2024 |