
A catalytic collaboration: pairing transition metals and Lewis acids for applications in organic synthesis
A. Dina
Dilinaer†
,
Gabriel
J. Jobin†
and
Marcus W.
Drover
 *
*
Department of Chemistry, Western University, 1151 Richmond Street, London, ON N8K 3G6, Canada. E-mail: marcus.drover@uwo.ca
First published on 2nd July 2024
Abstract
The use of metal catalysts to accelerate an organic transformation has proven indispensable for access to structural motifs having applications across medicinal, polymer, materials chemistry, and more. Most catalytic approaches have cast transition metals in the “leading role”; these players mediate important reactions such as C–C cross coupling and the hydrogenation of unsaturated bonds. These catalysts may require collaboration, featuring Lewis acidic or basic additives to promote a desired reaction outcome. Lewis acids can serve to accelerate reactions by way of substrate stabilization and/or activation, and as such, are valuable in optimizing catalytic transformations. A burgeoning area of chemical research which unifies these concepts has thus sought to develop transition metal complexes having ambiphilic (containing a Lewis basic and acidic unit) ligands. This approach takes advantage of metal–ligand cooperativity to increase the efficiency of a given chemical transformation, leveraging intramolecular interactions between a transition metal and an adjacent secondary ligand site. While this has shown significant potential to facilitate challenging and important transformations, there remains unexplored depth for creativity and future advancement. This Frontier highlights inter- and intramolecular combinations of transition metals and Lewis acids that together, provide a collaborative platform for chemical synthesis.
 A. Dina Dilinaer | A. Dina Dilinaer completed her B.Sc. (Hons.) chemistry degree at the University of Windsor in 2018. In 2022, she joined Prof. Marcus Drover's group at Western University where she is currently a Ph.D. student. Dina's research project involves the synthesis of new ligands featuring customized secondary coordination spheres for C–H fluorination. |
 Gabriel Jobin | Gabriel J. Jobin is a Ph.D. candidate under the supervision of Prof. Marcus Drover at Western University. He obtained a B.Sc. (Hons.) Degree from Wilfrid Laurier University (2022) in Waterloo, Canada before beginning his graduate studies. His work focuses on the design of coordination compounds having aluminum secondary coordination spheres (SCSs). |
 Marcus W. Drover | Marcus W. Drover is an Assistant Professor at Western University. He obtained a PhD from the University of British Columbia with Laurel Schafer and Jennifer Love (2016) and performed postdoctoral work at Caltech with Jonas Peters (2017–2019). During his training, Drover was recipient of a Vanier scholarship (2013–2016), MSFSS Supplement (Oxford with Andrew Weller), and Banting/Resnick Postdoctoral Fellowships (2017–2019). Drover's independent program began in July 2019 and intersects the fields of main group/organometallic chemistry. Drover is member of the Editorial Advisory Boards of Organometallics and Journal of Coordination Chemistry, and the Early Career Advisory Boards of Inorganic Chemistry Frontiers and JACS Au. |
1. Introduction
The discovery and production of new chemicals and materials are vital to scientific advancement. As such, transition metal (TM) catalysts are invaluable, providing new avenues for structural diversification and development.1 From the Sabatier process to palladium-catalyzed cross-coupling to copper catalyzed aza-alkyne click chemistry, transition metal catalysis has remained a versatile method for structural diversification.2–7 This has led to advances in the synthesis of new pharmaceutical ingredients as well as materials, among others.8–10 A goal of the chemistry community has been to discover and optimize new chemical transformations. A now common protocol for reaction optimization relies on either the design of superior catalysts or improving reaction conditions through the addition of functional additives (or both).A popular route to enhancing catalytic transformations has been through the use of additives, often giving rise to improved efficiency (yield and/or selectivity).11–13 This has been an ongoing area of research that has invoked intense interest, with commonly used additives including Lewis acids/bases, crown ethers, and metal-containing salts, to name a few.13–16 Of these, Lewis acids (LAs) are easily tailored structural units that are electron-deficient and in some cases, air/water tolerant. Lewis acid additives have been used in a number of catalytic transformations, including organic synthesis, controlled polymerization, and CO2 reduction.17–19 For the latter, s-block metal cations have been used to assist with CO2 reduction (though these will not be discussed in detail here). Kojima and coworkers, for example, reported that Mg2+ can be used in conjunction with a nickel catalyst to reduce CO2-to-CO.20 Complementary to this Frontier, we also recognize a 2019 review by Becica and Dobereiner that provides an excellent overview of the role of intermolecular Lewis acids in organotransition metal catalysis.13
Due to the electron-richness of later (and low-valent) transition metals and the inherent electrophilicity of Lewis acids, inter- or intramolecular pairings offer the potential for catalytic synergy through a disparity in polarity. This concept is complementary to the notion of (frustrated) Lewis pairs (FLPs).21–23 Classical definitions of Lewis acids (LA) and bases (LB) predict neutralization through adduct formation, where the nucleophilic LB donates electron density to an electrophilic LA. When the pair is positioned such that they are unable to neutralize one another (often due to steric hindrance), they form what is known as an FLP. A notable example of this concept was demonstrated by Stephan and co-workers who showed that an intramolecular phosphino–borane could activate H2.24 This “push–pull” concept has also been employed in coordination chemistry where electron-rich metal centers serve as the Lewis basic design component, proving fruitful for both inter- and intramolecular pairings with Lewis acids. For the latter, continual advances have been made in the field of secondary coordination sphere (SCS) ligand design.25,26 Using these design attributes, there has been great progress in the field of catalysis in which synergy between a metal, substrate, and Lewis acid allows for interaction and subsequent turnover. Some examples of such Lewis acid-mediated reactions are provided in Fig. 1A – these offer a broad scope of Lewis acid/transition metal types, while also targeting otherwise difficult to access organic products.27–30 In addition, these examples highlight how a Lewis acid can be used to not only interact with a substrate, but also activate a given catalyst by either interacting with the metal or a coordinated ligand; in these cases, the LA offers a lower energy pathway.13
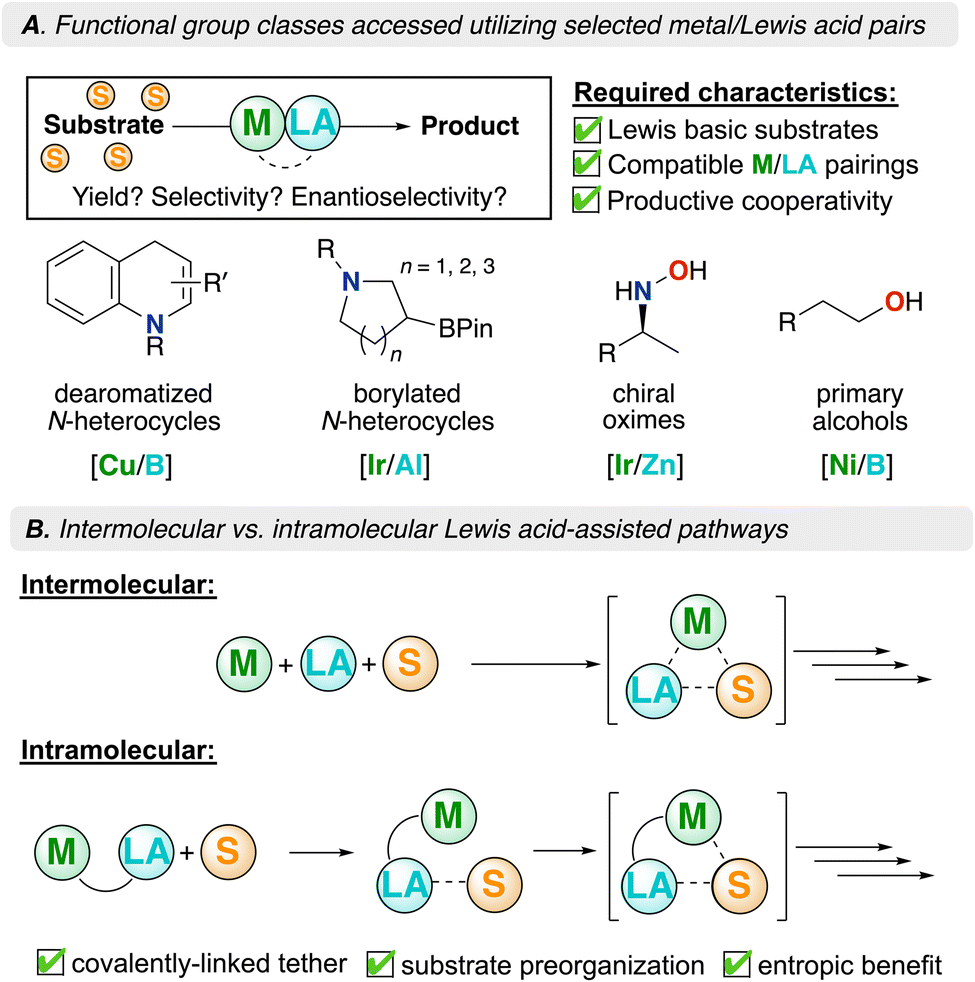 | ||
| Fig. 1 (A) Functional group classes accessed using Lewis acid/transition metal combinations and (B) intermolecular vs. intramolecular Lewis acid-mediated reactivity. M = metal, LA = Lewis acid, S = substrate. | ||
This Frontier emphasizes the importance of transition metal/Lewis acid cooperativity and provides insight for practitioners wishing to contribute to this burgeoning field. In this context, we examine recent advancements where intermolecular Lewis acid/transition metal pairings have improved catalytic outcomes relevant to organic synthesis (Fig. 1A). Additionally, we explore intramolecular Lewis acid/transition metal chemistry and the advantages afforded by these systems; in closing, a summary and future opportunities are provided (Fig. 1B).
2. Lewis acid-assisted organic transformations: intermolecular pairings
The outcome, and success, of a catalytic transformation is largely determined by variables of the catalyst system. These variables, including metal reagent, choice of ligand, temperature, solvent, reaction time, or the use of additives, are tunable. Many recent developments in reaction design have benefited from the use of Lewis acid additives.17,31–36 The addition of an exogenous Lewis acid is convenient since direct modification of the metal or ligand scaffold is unneeded, obviating complicated syntheses. That said, such additives are often used in a stoichiometric or subcatalytic (when compared to metal catalyst loading) amounts. Lewis acids can take on many forms including alkali metal cations and group 13 compounds (ER3; E = group 13 element).37,38 For the latter, ER3 is trigonal planar, leaving an empty and often accessible p-orbital. The presence of such an orbital renders these compounds Lewis acidic, the extent of which can be modified to engender higher reactivity, through the altering of R substituents. In this section, we highlight transition metal/Lewis acid pairings that have been used to achieve challenging organic transformations – asymmetric pyridine dearomatization, hydrosilylation-induced epoxide ring-opening, N-heterocycle C–H borylation, and asymmetric imine reduction, as examples.27–302.1. Substrate activation by a B–N interaction
Dearomatization offers an asymmetric route towards the generation of various chiral N-heterocycles. Earlier examples of dearomatization have been explored,39–41 however suffer from harsh reaction conditions and limited scope. Transition metal catalyzed dearomatizations have received increased attention and offer expanded scope, allowing for comparatively mild conditions with broad functional group tolerance. Despite the high stability of aromatic compounds, a transition metal catalyst can reduce the overall activation energy barrier, making dearomatization more thermodynamically favourable.42 As an additional axis for optimization, Lewis acids have been found to further reduce associated energy barriers through the binding and stabilization of charged intermediates during key steps of the catalytic cycle. Harutyunyan and co-workers demonstrate this merging concept wherein a chiral Cu catalyst, Lewis acid – BF3·OEt2, and Grignard reagent work in synergy to C4-alkylate a suite of quinolines with nearly absolute regio- and stereoselectivity (Fig. 2A).27 In the absence of Lewis acid, this reaction does not proceed. Molecular modelling provided insight into the role of the Lewis acid. BF3 not only activated the quinoline substrate towards added nucleophile, but also subtly influenced the regiochemical outcome of the reaction by providing steric hindrance and preventing C2 alkylation. Combination of Cu and BF3 resulted in a highly effective alkylative dearomatization system with a catalyst turnover number (TON) of up to 1000.27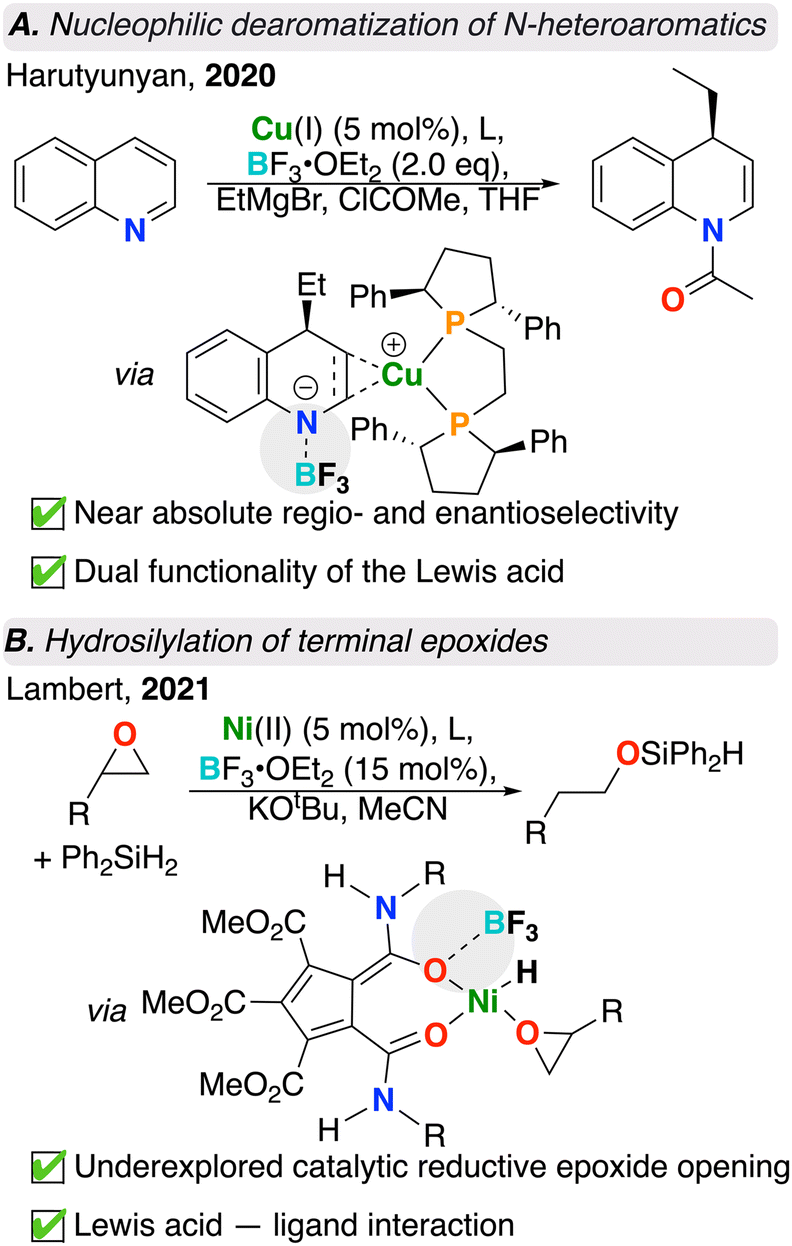 | ||
| Fig. 2 (A) N-heteroaromatic dearomatization and (structure shown after “via” refers to computationally obtained transition state); (B) epoxide hydrosilylation catalyzed by intermolecular transition metal/Lewis acid catalysts (structure shown after “via” is from the proposed catalytic cycle). | ||
2.2. Catalyst activation via a Lewis acid–ligand interaction
Epoxide hydrosilylation also benefits from the addition of a borane-based Lewis acid. Epoxide ring-opening is a versatile reaction that is ubiquitous in organic synthesis, however, despite applications in converting terminal epoxides to primary alcohols, reductive epoxide ring-openings are underexplored. A recent study performed by Lambert and co-workers showcased a Ni catalyst and BF3·OEt2 for the terminal hydrosilylation of terminal monosubstituted epoxides with a silane reagent (Fig. 2B).28 In the absence of Lewis acid, this reaction did not occur, though under optimized conditions, the authors were able to achieve 100% conversion with 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 selectivity for the linear product. Here, BF3 was proposed to increase the Lewis acidity of the metal complex by coordinating to one of the two carbonyl oxygens of the pentacarboxycyclopentadienyl (PCCP) ligand. An overall increase in Lewis acidity weakens the epoxide C–O bond and renders it to be more susceptible to nucleophilic attack by the nickel hydride. It was also proposed by the authors that the site selectivity arises from having a smaller ligand scaffold surrounding the nickel centre. This favoured the hydride transfer at the more sterically hindered site, resulting in the formation of the linear ring-opened product. This system was expanded to a variety of multi-substituted epoxide substrates with high efficiency.28
:15 selectivity for the linear product. Here, BF3 was proposed to increase the Lewis acidity of the metal complex by coordinating to one of the two carbonyl oxygens of the pentacarboxycyclopentadienyl (PCCP) ligand. An overall increase in Lewis acidity weakens the epoxide C–O bond and renders it to be more susceptible to nucleophilic attack by the nickel hydride. It was also proposed by the authors that the site selectivity arises from having a smaller ligand scaffold surrounding the nickel centre. This favoured the hydride transfer at the more sterically hindered site, resulting in the formation of the linear ring-opened product. This system was expanded to a variety of multi-substituted epoxide substrates with high efficiency.28
2.3. Expansion within group 13: from boron to aluminum
Moving down group 13, alanes have also been used as Lewis acid partners.43–46 C(sp3)–H functionalization is a highly sought after transformation due to its atom and step economy.47 Amongst C(sp3)–H functionalization reactions, C(sp3)–H borylation offers a versatile handle for further synthetic manipulation following installation of a borane group.48 As substrates, cyclic amines are important building blocks in synthetic chemistry. Despite amine α-functionalization as being prominent in organic synthesis, β-functionalization remains relatively underexplored.49–51 Recent work published by Nakao and co-workers used methylaluminum-bis(2,6-di-tert-butyl-4-methylphenoxide) (MAD) as a Lewis acid in combination with an iridium catalyst and bis(pinacolato)diboron (B2Pin2) as the borylation reagent to perform site selective C(sp3)–H borylation at the β-H of saturated cyclic amines (Fig. 3A).29 As above, in the absence of a Lewis acid, the reaction had near-zero conversion. Upon addition of MAD, an acid/base interaction was formed between it and a pivaloyl group oxygen, preventing unwanted reactivity at the α-site due to steric repulsion between the Ir catalyst and the bulky Lewis acid masking the α-site. The authors were able to achieve not only high site selectivity, but also high enantioselectivity by means of reacting trialkyl aluminum species and sterically bulky chiral auxiliaries, which forms chiral aluminum Lewis acids in situ. As a result of using this chiral Lewis acid/achiral iridium catalytic system, the authors demonstrated an alternative approach to conventional asymmetric catalysis where a chiral ligand is no longer required. Further computational studies showed that the Lewis pair reduced the energy of the reductive elimination step by approximately 5 kcal mol−1, which is key to the success of this transformation, as reductive elimination is rate-limiting.29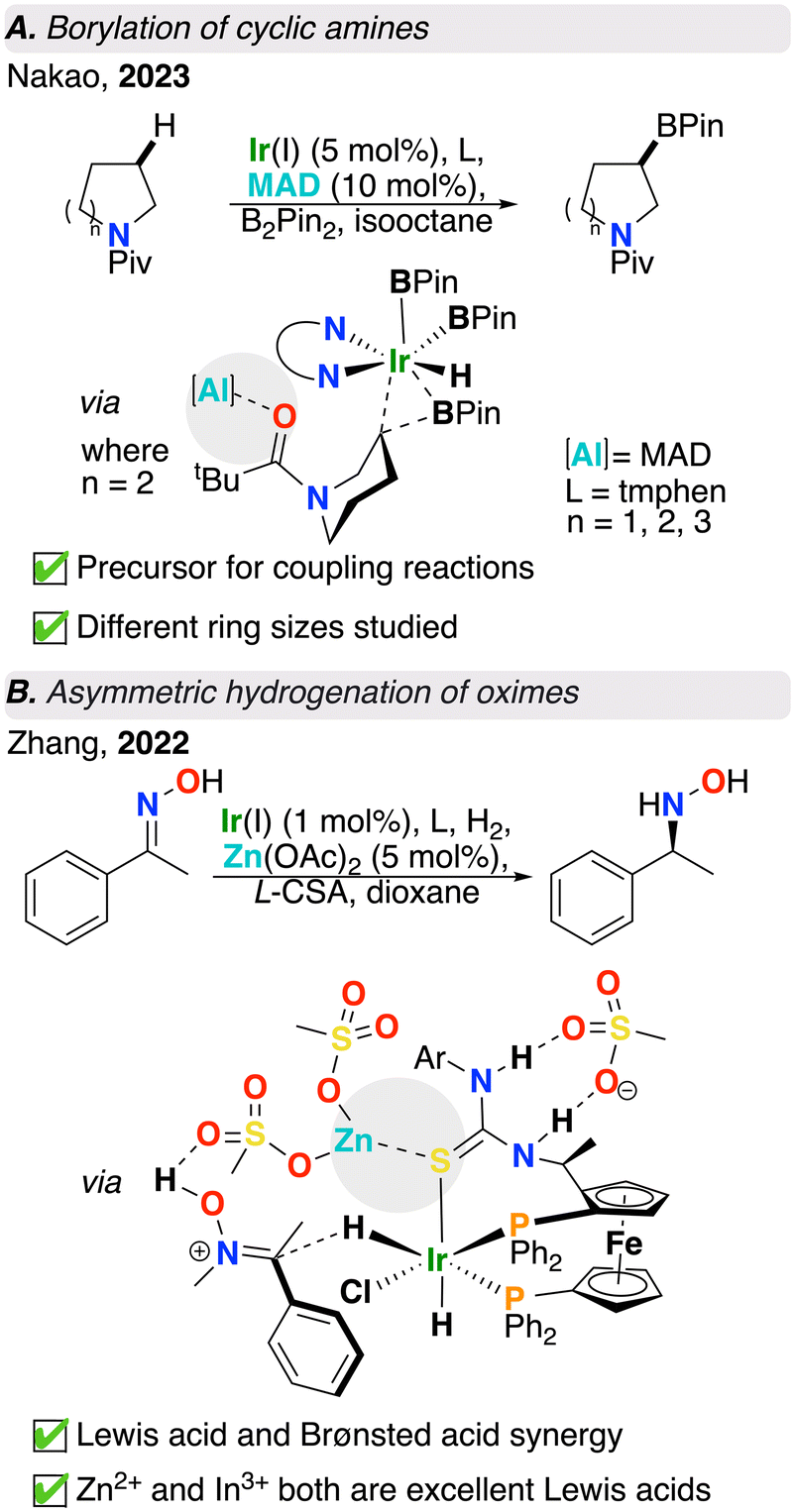 | ||
| Fig. 3 (A) Amine borylation and (B) oxime hydrogenation catalyzed by intermolecular transition metal/Lewis acid catalysts (structures shown after “via” refer to computationally obtained transition states). | ||
2.4. Moving beyond group 13: zinc as a Lewis acid
Besides the main group elements discussed above, metal ions such as Zn2+ are also used as Lewis acids. Asymmetric hydrogenation is a powerful tool to establish stereogenic centres.52 Although there has been steady advances in asymmetric hydrogenation, the asymmetric hydrogenation of oximes has proven to be particularly challenging. This is due to charge delocalization owing to resonance, E/Z-isomerization, and N–O bond splitting due to the presence of lone pairs on both oxime N and O atoms. Tackling this challenge, Zhang and co-workers used a Lewis acid, Brønsted acid, Ir catalyst, and chiral diphosphine ligand in tandem to access hydroxylamines in high yield and enantioselectivity from oximes using H2(g) (Fig. 3B).30 Following optimization, the authors found that the Lewis acid, Zn(OAc)2 and Brønsted acid, (L)-camphorsulfonic acid (L-CSA) were both vital for reaction success. L-CSA activates the oxime via hydrogen bonding and causes the substrate to be more susceptible to nucleophilic attack by the iridium hydride, whereas Zn(OAc)2 fine tunes the chiral environment of the system and enhances enantioselectivity. Density functional theory (DFT) calculations revealed the mechanism for this reaction is most likely to proceed via outer sphere hydride transfer where the Brønsted acid promotes formation of an [Ir]–H species. Additionally, the ligand thiourea fragment was computed to be of crucial importance since the pre-organization of thiourea with the Lewis acid is vital for the high enantioselectivity achieved. Although both L-CSA and methanesulfonic acid (MsOH) produced excellent yield and enantiomeric excess, MsOH was used as a computational model due to its simpler structure. The authors also used In(OTf)3 which was shown to give an even better yield and enantioselectivity compared to Zn(OAc)2. Notably, high yield (93%) and enantioselectivity (93% ee) were achieved on gram scale.In conclusion, various transition metal/Lewis acid cooperative catalysis strategies have been developed in recent years. The simple addition of exogenous Lewis acids has been shown to enhance yield, regioselectivity, and enantioselectivity of these otherwise challenging organic transformations. Chemists have long been harnessing the power of Lewis acids in organic transformations; however, these systems frequently demand high loading of the Lewis acid due to its nature as an additive, lacking the structural rigidity that would optimize its positioning and effectiveness. Hence, developing methods to lower additive loading while maintaining high selectivity and yield remains an ongoing challenge in the field of Lewis acid-assisted catalysis.
3. Lewis acid-assisted organic transformations: intramolecular pairings
Transition metals, when coupled with exogenous Lewis acids, promote synergistic reactivity. Moreover, decreasing the entropic penalty of catalyst/substrate binding can additionally lower activation barriers in a catalytic mechanism. When an intermolecular reaction takes place, several components often forge a single catalytic intermediate, reducing system disorder. As such, intramolecular transition metal/Lewis acid systems mitigate this unfavorable entropic barrier. In these systems, the transition metal, Lewis acid, and substrate must be considered (Fig. 1B). Consequently, there is vested interest in fusing transition metal and Lewis acidic components into a single molecule, thereby reducing entropy costs, leading to more favourable thermodynamics.Ambiphilic ligands feature Lewis acidic and basic moieties which are intramolecularly contained. Binding modes for such ligands often feature the Lewis base coordinating to a metal, while the Lewis acid is left pendant in the secondary coordination sphere (SCS)25,26 to support reactivity.53–63 This approach aims to exploit metal–ligand cooperativity, which leverages intramolecular interactions between a transition metal and an adjacent secondary ligand site.64 The concept of a reactive SCS has been exploited in nature, where peripheral amino acids promote functional bonding to facilitate important biological transformations in the active sites of enzymes.65 Leveraging the synergistic relationship between transition metals and group 13 Lewis acids, many research teams have adopted this strategy to improve the efficiency of a given chemical transformation.
3.1. Substrate direction through Lewis acid–base interactions
The ability to selectively activate challenging bonds is one of the main benefits of transition metal complexes having peripheral Lewis acidic groups. Lewis acids can promote selective activation at a metal centre by coordinating to Lewis basic substrates. This concept is highlighted by Kimura and co-workers where the authors found that when paired with ambiphilic phosphine–borane ligands, an iridium complex was effective in catalyzing the ortho-C(sp2)–H silylation of 2-arylpyridine derivatives (Fig. 4A).60 Varying the tether length between phosphorus and boron, the authors found that a four-carbon linker was optimal. The proposed catalytic cycle features the ligand borane group coordinating to a Lewis basic pyridine substate. The selectivity of subsequent C–H bond activation, following generation of a reactive iridium–silyl intermediate, is promoted by a pyridine–borane coordination. For this system, the authors showed the reaction time could be reduced from 24 to 5 h, without a substantial loss in yield.60 This contrasts with the current state-of-the-art, which requires longer reaction times.66 Pyridine moieties are prevalent in biologically relevant molecules, as such, methodologies that aim to selectively modify this scaffold are important to drug design. | ||
| Fig. 4 (A) C–H silylation and (B) C–H alkenylation catalyzed by intramolecular transition metal/Lewis acid catalysts (structures shown after “via” are from the proposed catalytic cycle). | ||
3.2. Lewis acidic aluminum in the SCS for substrate direction
For pyridine alkenylation, C2 or C4 substitution is common. However, using a nickel/aluminum system, Yu and co-workers demonstrated a C3-selective process (Fig. 4B).67 This reaction benefits from TM/LA cooperativity – a concept that has been applied to several related catalytic transformations.59,61,68–74 Here, the authors capitalized on an asymmetric N-heterocyclic carbene (NHC) ligand having a pendant hydroxyl moiety. Upon addition of Al(iBu)3, this proligand readily forms the active species in situ, resulting in formation of a peripheral Al–O bond, generating a Lewis acidic aluminum site which promotes substrate binding during catalysis. The tether length again proved critical for the selectivity of this process, where a two-carbon alkoxy side arm was shown to be ideal. This intramolecular Lewis acid-assisted directing was integral to achieving C3 selectivity (33% yield C3). When ligands lacking the ability to ligate aluminum were employed, formation of the C3 product was not observed even in the presence of an external Lewis acid (1% C3/16% C4). Notably, substrates with electron-withdrawing functional groups (weaker Lewis bases) exhibited reduced selectivity due to weaker Lewis acid/base interactions. This catalytic system served as the first example of a C3-selective pyridine alkenylation featuring the pyridine substrate as the limiting reagent. This method was applied to the diversification of complex pyridine scaffolds that were derived from biologically- and medicinally-active compounds, demonstrating potential for use in late-stage drug functionalization. Similarly, Ye and co-workers employed a phosphine oxide ligand for the Ni-catalyzed hydroarylation of alkynes with unactivated β-C(sp2)–H bonds (84% yield).75 This ligand form has the ability to tautomerize to phosphinous acid to give intramolecular Al–O bonds in situ after the addition of AlR3. Substituting the phosphine oxide with other commonly used phosphines and NHCs, which rendered the system intermolecular, was ineffective (0% yield).3.3. Altered selectivity through Lewis acid stabilization
The use of intramolecular Lewis acids can alter product selectivity in a given chemical transformation. This concept has been illustrated by Werlé and co-workers and applied to catalytic organic transformations utilizing an ambiphilic triazine ligand coordinated to rhodium76 and cobalt77 (Fig. 5A). Here, they performed careful control experiments with related inter- and intramolecular systems, specifically using a borane with and without a tether connected to the ligand framework. In both cases, the authors found improved catalytic performance for the intramolecular analogues. Furthermore, in studies of the catalytic reduction of nitroarenes to aniline and hydroxylamine, the researchers discovered that intramolecular utilization of the Lewis acid significantly altered reaction selectivity, enabling the selective synthesis of aniline derivatives. Control experiments employing external boranes only resulted in the formation of hydroxylamine products. This work illustrates how making use of intramolecular Lewis acids can have an influence – not only on thermodynamics, but also product selectivity. | ||
| Fig. 5 (A) Controlled nitroarene hydrogenation (structure shown after “via” is the catalyst structure); (B) nitrile dihydroboration catalyzed by intramolecular transition metal/Lewis acid catalysts (structure shown after “via” was obtained crystallographically). | ||
3.4. Small molecule activation through SCS interactions
Favorable interactions between pendant Lewis acids and metal-bound ligands (i.e., B–X (X = N, O, S, halide)) can further stabilize reactive structures. To this end, transition element/Lewis acid synergy has been used in small-molecule activation. Bercaw and co-workers demonstrated the benefits of an intramolecular borane in facilitating C–C bond formation through the reductive coupling of two Re-bound CO ligands.78,79 This concept took advantage of Lewis acid stabilization, where a hydride source was used to reduce CO, which was then stabilized by favorable B–O bonding interactions. This reactivity was juxtaposed against the use of an external trialkyl borane, where C–C coupling was not observed.By designing ligands that can host both a transition metal and group 13 Lewis acid, researchers have unlocked pathways to numerous important chemical transformations. The versatile nature of these ligands is highlighted by their ability to cooperatively bind and direct substrates during catalysis, which promotes selective reactivity. While not without synthetic challenges/drawbacks, the diversity of Lewis acid role highlights the malleability of this approach, allowing tailorability to achieve desired outcomes. The inclusion of boron and, to a lesser extent, aluminum, continues to be a significant focus of many chemists seeking to bridge the fields of organometallic and main group reactivity. This has led to a growing field of transition metal complexes featuring intramolecular Lewis acids, where ligand design has proven imperative to achieve a targeted outcome.
4. Creative ligand design
The productive catalytic chemistry shown for transition metal/Lewis acid pairings has led to a growing crop of new ambiphilic ligands that aim to take advantage of the cooperation exemplified above. Coordination complexes and stoichiometric reactivity can lend insight into new mechanisms of action which may benefit future catalytic systems, thereby providing proof of concept.63,80–86 Drover and co-workers have developed a suite of phosphine ligands hydrofunctionalized with electrophilic borane groups in the SCS (Fig. 6A).55,56,87,88 These ligands are prepared from the functionalization of allyl-appended diphosphines, either pre- or post-metal coordination. In a 2023 study, the group studied the formation of nitrile-bound dinickel dimers. The stabilizing effect inherent to these motifs led to a hindrance in catalytic nitrile dihydroboration, highlighting an inverse relationship between catalytic turnover frequency and number of SCS boranes.56,88 This effect suggests a balancing act – pendant Lewis acids are important modifiers to chemical reactivity that can also serve to over-stabilize bound substrate, rather than enable its productive turnover (Fig. 5B). Ambiphilic ligands can demonstrate exotic coordination chemistry, wherein the Lewis acid, instead of remaining pendant, acts as a Z-type ligand, engaging with the electron-rich metal center. In these instances, the Lewis acid, through its empty orbital, accepts electron density from the metal center. This process notably increases the electrophilicity of the transition metal, proving as a useful design element in catalysis.89 In 2019, Bourissou and co-workers installed boranes into phosphine complexes by employing a PBP pincer ligand.57 Remarkably, this complex was air-stable, where the Lewis acidic borane afforded stability to the Pd(0) complex by decreasing electron density at the reactive metal centre (Fig. 6B). In this example, the co-stabilization afforded to palladium by boron was shown to be an important design element for arene dehalogenation.57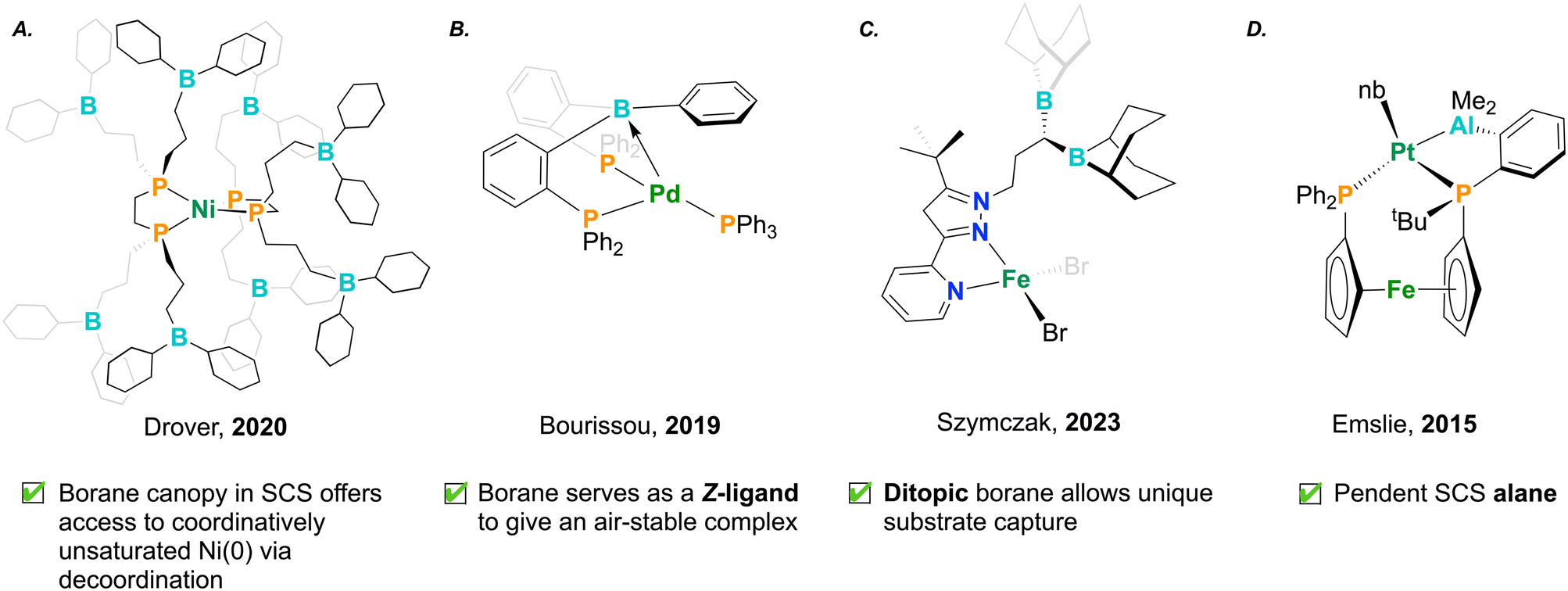 | ||
| Fig. 6 Recent advances in ambiphilic ligand design for transition metal complexes; nb = norbornene. | ||
Lewis acids have also been appended to hard nitrogen donor ligands. Szymczak and co-workers have developed a pyridine–pyrazole ligand which has been modified to include fused monotopic or ditopic borane functionality (Fig. 6C).53,54,90 These have been shown to have a strong affinity for cooperatively binding nucleophilic small molecules. In the case of the latter, the two ditopic boranes in the SCS show additive Lewis acidity, rendering increased activation of dibasic substrates such as hydrazine. The authors found that the activation of hydrazine was further increased upon coordination of the pyridine–pyrazole ligand to Zn, which displayed cooperative stabilization of the highly reactive hydrazido unit by both zinc and the SCS boranes.54
Moving down group 13, in 2015 Emslie and co-workers synthesized an alane-appended ferocenyl phosphine. The alane present in the ligand was coordinated to platinum, delivering a persistent Pt–Al bond. This Pt–Al interaction was strong and indifferent to changes in platinum oxidation state and geometry (Fig. 6D).86 The behaviour of aluminum was contrasted against an analogous ligand framework containing a borane,91 highlighting the distinct behaviour of ambiphilic ligands depending on the identity of the Lewis acid.
It is important to note that while ambiphilic ligands are designed with an emphasis on cost-savings in chemistry i.e., via activation energy lessening in a catalytic reaction, they require significant energy input to prepare. In nearly all cases, syntheses must be conducted in water/air-free environments, as most organyl boranes and alanes are highly susceptible to degradation. Moreover, these reactions also require multiple steps cf., an intermolecular reaction where both components are effectively “tossed” into a reaction vessel. Ambiphilic ligands can also form stable Lewis acid/base pairs, which under some circumstances, can render metal coordination difficult. Fortunately, consistent advancements in the adjacent fields of organic and main group chemistry22,23,92–95 provide inspiration for the generation of new ligand scaffolds, offering opportunities for refinement and further optimization. Given the breadth of ligand scaffolds used in organometallic chemistry, one can begin to think about new ways of incorporating Lewis acidic moieties in innovative ways.
5. Future directions
While the number of Lewis acid-appended transition metal complexes continues to grow, there remain avenues for future development. Importantly, careful determination of reaction characteristics such as kinetic data enable careful comparison between systems that operate via intra- or intermolecular mechanisms. As mentioned earlier, while boron has been the primary focus of ambiphilic ligand design (due to ease of access by hydrofunctionalization), aluminum has been relatively neglected. Aluminum is among the most abundant elements on the planet,96 and has low toxicity.97 Thus, a greater focus on aluminum as a contributor in these chemical transformations falls in line with its high natural abundance. While boron is typically introduced into ligand scaffolds through hydroboration across a terminal double bond, hydroalumination remains challenging. Consequently, most aluminum containing compounds are formed through protonolysis reactions, forming an Al–E (E = O, N etc.) bond, which often has negative effects on resulting Lewis acidity. Additionally, while much work has gone into studying the fundamental properties of monotopic Lewis acid systems, there remains untapped space in developing multitopic systems, which could offer enhanced Lewis acidity through adjacent p-orbital/p-orbital interactions or bind multi-basic substrates.Furthermore, while upper-row group 13 elements such as boron and aluminum have been the most prevalent Lewis acids seen in literature, there is certainly space for exploring and expanding the Lewis acid tool box, including the use of Zn2+, for example. Furthermore, considering the sheer scope of ligands available in organometallic chemistry, it may well be of interest to assess other primary donor compositions, with some backbones likely being more amenable to modification than others. This myriad of possibilities offers a blank canvas for customization to tune systems for desired reaction outcomes.
6. Summary and outlook
Lewis acids have been used as additives for a wide range of catalytic applications, wherein a push–pull dynamic (with a transition metal) allows for controlled and facile substrate activation. The addition of exogenous Lewis acids remains prevalent in cooperative systems, however interest in intramolecular Lewis acid incorporation continues to rise. Although exogenous Lewis acids can be added straightforwardly, without modifying the ligand scaffold, these are usually required in stoichiometric or sub-stoichiometric amounts with respect to substrate. On the other hand, intramolecularly incorporated Lewis acids are often employed in catalytic quantity, as they are appended directly to the catalyst, allowing for intimate control of reactivity. Nonetheless, the synthesis of such ligands can be multistep, generating greater quantities of waste.With an eye towards the design of synergistic systems using bifunctional ligands, we believe there is untapped potential for further enhancing reaction efficiency and product selectivity. We hope this Frontier serves as a launching pad for further advancement and innovation, where, exploring unique combinations of metal, ligand, and Lewis acid could offer novel solutions to pressing synthetic challenges.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Western University, the Council of Ontario Universities for a John C. Polanyi award to M. W. D., the Ontario Early Researcher Award Program, and the Natural Sciences and Engineering Research Council of Canada for program funding (Discovery Grant, RGPIN-2020-04480).References
- R. Luque and F. L.-Y. Lam, Sustainable catalysis: energy-efficient reactions and applications, John Wiley & Sons, 2018 Search PubMed.
- P. Styring, S. McCord and S. Rackley, in Negative Emissions Technologies for Climate Change Mitigation, ed. S. Rackley, G. Andrews, D. Clery, R. De Richter, G. Dowson, P. Knops, W. Li, S. Mccord, T. Ming, A. Sewel, P. Styring and M. Tyka, Elsevier, 2023, pp. 391–413 Search PubMed.
- P. Devendar, R.-Y. Qu, W.-M. Kang, B. He and G.-F. Yang, J. Agric. Food Chem., 2018, 66, 8914–8934 CrossRef CAS PubMed.
- D. Bauer, S. M. Sarrett, J. S. Lewis and B. M. Zeglis, Nat. Protoc., 2023, 18, 1659–1668 CrossRef CAS PubMed.
- B. Zhang, M. C. Aguilera, N. Cajiao and M. L. Neidig, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'hare, Elsevier, Oxford, 2022, pp. 185–209 Search PubMed.
- I. Shafiq, S. Shafique, P. Akhter, W. Yang and M. Hussain, Catal. Rev., 2022, 64, 1–86 CrossRef CAS.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- Ó. López and J. M. Padrón, Catalysts, 2022, 12(2), 164 CrossRef.
- D. Hughes, P. Wheeler and D. Ene, Org. Process Res. Dev., 2017, 21, 1938–1962 CrossRef CAS.
- S. Kumar, B. Z. Dholakiya and R. Jangir, J. Organomet. Chem., 2021, 953, 122066 CrossRef CAS.
- S. Ghosh, S. Shilpa, C. Athira and R. B. Sunoj, Top. Catal., 2022, 65, 141–164 CrossRef CAS.
- A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
- J. Becica and G. E. Dobereiner, Org. Biomol. Chem., 2019, 17, 2055–2069 RSC.
- B. Pérez-Saavedra, Á. Velasco-Rubio, E. Rivera-Chao, J. A. Varela, C. Saá and M. Fañanás-Mastral, J. Am. Chem. Soc., 2022, 144, 16206–16216 CrossRef PubMed.
- L. I. Matienko and L. A. Mosolova, Kinet. Catal., 2003, 44, 221–228 CrossRef CAS.
- R. L. De Carvalho, E. B. T. Diogo, S. L. Homölle, S. Dana, E. N. Da Silva Júnior and L. Ackermann, Chem. Soc. Rev., 2023, 52, 6359–6378 RSC.
- P. Fernández, P. Alonso, F. J. Fañanás and F. Rodríguez, Eur. J. Org. Chem., 2018, 3957–3964 CrossRef.
- C. S. Durfy, J. A. Zurakowski and M. W. Drover, ChemSusChem, 2024, e202400039 CrossRef PubMed.
- H. Suo, Z. Zhang, R. Qu, Y. Gu and Y. Qin, Catalysts, 2023, 13(4), 670 CrossRef CAS.
- D. Hong, T. Kawanishi, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- M. W. Drover, Chem. Soc. Rev., 2022, 51, 1861–1880 RSC.
- J. A. Zurakowski, B. J. H. Austen and M. W. Drover, Trends Chem., 2022, 4, 331–346 CrossRef CAS.
- X. Yan, L. Ge, M. Castiñeira Reis and S. R. Harutyunyan, J. Am. Chem. Soc., 2020, 142, 20247–20256 CrossRef CAS PubMed.
- K. A. Steiniger and T. H. Lambert, Org. Lett., 2021, 23, 8013–8017 CrossRef CAS PubMed.
- Y. Kuroda, K. Park, Y. Shimazaki, R.-L. Zhong, S. Sakaki and Y. Nakao, Angew. Chem., Int. Ed., 2023, 62, e202300704 CrossRef CAS PubMed.
- F. Wang, Y. Chen, P. Yu, G.-Q. Chen and X. Zhang, J. Am. Chem. Soc., 2022, 144, 17763–17768 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Chem. Sci., 2017, 8, 6439–6450 RSC.
- S. Sharif, J. Day, H. N. Hunter, Y. Lu, D. Mitchell, M. J. Rodriguez and M. G. Organ, J. Am. Chem. Soc., 2017, 139, 18436–18439 CrossRef CAS PubMed.
- B. Wang, M. Liang, J. Tang, Y. Deng, J. Zhao, H. Sun, C.-H. Tung, J. Jia and Z. Xu, Org. Lett., 2016, 18, 4614–4617 CrossRef CAS PubMed.
- Y. Li, C. Topf, X. Cui, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 5196–5200 CrossRef CAS PubMed.
- Z. Pan, S. Wang, J. T. Brethorst and C. J. Douglas, J. Am. Chem. Soc., 2018, 140, 3331–3338 CrossRef CAS PubMed.
- S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed.
- E. I. Davydova, T. N. Sevastianova and A. Y. Timoshkin, Coord. Chem. Rev., 2015, 297–298, 91–126 CrossRef CAS.
- A. Y. Timoshkin, Chem. – Eur. J., 2024, 30, e202302457 CrossRef CAS PubMed.
- A. J. Birch, Pure Appl. Chem., 1996, 68, 553–556 CrossRef CAS.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- H. Wynberg, Chem. Rev., 1960, 60, 169–184 CrossRef CAS.
- C. Zheng and S.-L. You, ACS Cent. Sci., 2021, 7, 432–444 CrossRef CAS PubMed.
- P. Kelley, G. A. Edouard, S. Lin and T. Agapie, Chem. – Eur. J., 2016, 22, 17173–17176 CrossRef CAS PubMed.
- M.-S. Yu, W.-C. Lee, C.-H. Chen, F.-Y. Tsai and T.-G. Ong, Org. Lett., 2014, 16, 4826–4829 CrossRef CAS PubMed.
- Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428–2429 CrossRef CAS PubMed.
- Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS PubMed.
- S. Xu, Y. Ping, W. Li, H. Guo, Y. Su, Z. Li, M. Wang and W. Kong, J. Am. Chem. Soc., 2023, 145, 5231–5241 CrossRef CAS PubMed.
- D. G. Hall, in Boronic Acids, John Wiley & Sons, Ltd, 2011, pp. 1–133 Search PubMed.
- K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- D. B. Antermite and A. James, Synthesis, 2019, 3171–3204 CAS.
- P. Behera, D. S. Ramakrishna, M. M. Chandrasekhar and S. R. Kothakapu, Chirality, 2023, 35, 477–497 CrossRef CAS PubMed.
- J. J. Kiernicki, E. E. Norwine, M. Zeller and N. K. Szymczak, Inorg. Chem., 2021, 60, 13806–13810 CrossRef CAS PubMed.
- D. M. Beagan, J. J. Kiernicki, M. Zeller and N. K. Szymczak, Angew. Chem., Int. Ed., 2023, 62, e202218907 CrossRef CAS PubMed.
- M. W. Drover, M. C. Dufour, L. A. Lesperance-Nantau, R. P. Noriega, K. Levin and R. W. Schurko, Chem. – Eur. J., 2020, 26, 11180–11186 CrossRef CAS PubMed.
- M. L. Clapson, H. Sharma, J. A. Zurakowski and M. W. Drover, Chem. – Eur. J., 2023, 29, e202203763 CrossRef CAS PubMed.
- H. Kameo, J. Yamamoto, A. Asada, H. Nakazawa, H. Matsuzaka and D. Bourissou, Angew. Chem., Int. Ed., 2019, 58, 18783–18787 CrossRef CAS PubMed.
- B. Chatterjee, W.-C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
- R. Malacea, F. Chahdoura, M. Devillard, N. Saffon, M. Gómez and D. Bourissou, Adv. Synth. Catal., 2013, 355, 2274–2284 CrossRef CAS.
- K. Anoyama, G. Onodera, T. Fukuda and M. Kimura, Adv. Synth. Catal., 2022, 364, 1223–1227 CrossRef CAS.
- G. Onodera, H. Kumagae, D. Nakamura, T. Hayasaki, T. Fukuda and M. Kimura, Tetrahedron Lett., 2020, 61, 152537 CrossRef CAS.
- C. A. Theulier, Y. García-Rodeja, K. Miqueu, G. Bouhadir and D. Bourissou, J. Am. Chem. Soc., 2023, 145, 10800–10808 CrossRef CAS PubMed.
- M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- E. A. Span, D. L. M. Suess, M. C. Deller, R. D. Britt and M. A. Marletta, ACS Chem. Biol., 2017, 12, 1095–1103 CrossRef CAS PubMed.
- G. Choi, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2013, 135, 13149–13161 CrossRef CAS PubMed.
- T. Zhang, Y.-X. Luan, N. Y. S. Lam, J.-F. Li, Y. Li, M. Ye and J.-Q. Yu, Nat. Chem., 2021, 13, 1207–1213 CrossRef CAS PubMed.
- R. Jana, S. Chakraborty, O. Blacque and H. Berke, Eur. J. Inorg. Chem., 2013, 2013, 4574–4584 CrossRef CAS.
- Y.-X. Luan and M. Ye, Chem. Commun., 2022, 58, 12260–12273 RSC.
- F.-G. Fontaine and D. Zargarian, J. Am. Chem. Soc., 2004, 126, 8786–8794 CrossRef CAS PubMed.
- G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065–1079 RSC.
- S. N. MacMillan, W. Hill Harman and J. C. Peters, Chem. Sci., 2014, 5, 590–597 RSC.
- M. Boudjelel, O. Sadek, S. Mallet-Ladeira, Y. García-Rodeja, E. D. Sosa Carrizo, K. Miqueu, G. Bouhadir and D. Bourissou, ACS Catal., 2021, 11, 3822–3829 CrossRef CAS.
- Y. Saito, J. Kikuchi, C. Wang and N. Yoshikai, Angew. Chem., Int. Ed., 2023, 62, e202301006 CrossRef CAS PubMed.
- S.-L. Qi, Y.-P. Liu, Y. Li, Y.-X. Luan and M. Ye, Nat. Commun., 2022, 13, 2938 CrossRef CAS PubMed.
- V. Chugh, B. Chatterjee, W.-C. Chang, H. H. Cramer, C. Hindemith, H. Randel, T. Weyhermüller, C. Farès and C. Werlé, Angew. Chem., Int. Ed., 2022, 61, e202205515 CrossRef CAS PubMed.
- S. Jena, L. Frenzen, V. Chugh, J. Wu, T. Weyhermüller, A. A. Auer and C. Werlé, J. Am. Chem. Soc., 2023, 145, 27922–27932 CrossRef CAS PubMed.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2008, 130, 11874–11875 CrossRef CAS PubMed.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, Organometallics, 2010, 29, 4499–4516 CrossRef CAS.
- T. G. Ostapowicz, C. Merkens, M. Hölscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2013, 135, 2104–2107 CrossRef CAS PubMed.
- E. E. Norwine, J. J. Kiernicki, M. Zeller and N. K. Szymczak, J. Am. Chem. Soc., 2022, 144, 15038–15046 CrossRef CAS PubMed.
- T. Komuro, Y. Nakajima, J. Takaya and H. Hashimoto, Coord. Chem. Rev., 2022, 473, 214837 CrossRef CAS.
- D. Facchinato, J. A. Zurakowski and M. W. Drover, J. Coord. Chem., 2022, 75, 1929–1939 CrossRef CAS.
- K. Paskaruk, D. J. H. Emslie and J. F. Britten, Dalton Trans., 2022, 51, 15040–15048 RSC.
- B. E. Cowie and D. J. H. Emslie, Organometallics, 2015, 34, 2737–2746 CrossRef CAS.
- B. E. Cowie, F. A. Tsao and D. J. H. Emslie, Angew. Chem., Int. Ed., 2015, 54, 2165–2169 CrossRef CAS PubMed.
- J. A. Zurakowski, M. Bhattacharyya, D. M. Spasyuk and M. W. Drover, Inorg. Chem., 2021, 60, 37–41 CrossRef CAS PubMed.
- B. J. H. Austen, M. L. Clapson and M. W. Drover, RSC Adv., 2023, 13, 19158–19163 RSC.
- D. You and F. P. Gabbaï, Trends Chem., 2019, 1, 485–496 CrossRef CAS.
- J. J. Kiernicki, E. E. Norwine, M. Zeller and N. K. Szymczak, Chem. Commun., 2019, 55, 11896–11899 RSC.
- B. E. Cowie and D. J. H. Emslie, Chem. – Eur. J., 2014, 20, 16899–16912 CrossRef CAS PubMed.
- R. Roesler, B. J. N. Har and W. E. Piers, Organometallics, 2002, 21, 4300–4302 CrossRef CAS.
- F. Schäfer, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2021, 2021, 3265–3271 CrossRef.
- F. Ritter, L. J. Morris, K. N. McCabe, T. P. Spaniol, L. Maron and J. Okuda, Inorg. Chem., 2021, 60, 15583–15592 CrossRef CAS PubMed.
- P. Niermeier, K. A. M. Maibom, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. Sci., 2021, 12, 7943–7952 RSC.
- Aluminum- Elelment Information, Properties and Uses- RSC, 2023.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |