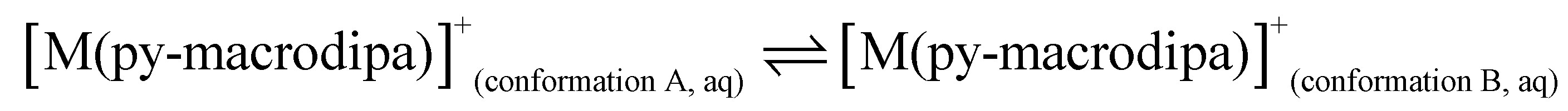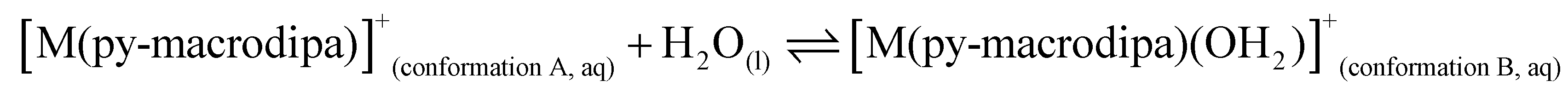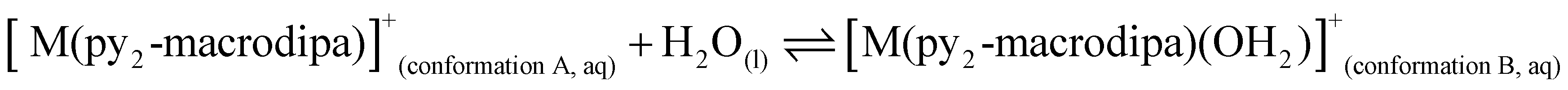DOI:
10.1039/D4DT02146K
(Paper)
Dalton Trans., 2024,
53, 14634-14647
Chelation of [111In]In3+ with the dual-size-selective macrocycles py-macrodipa and py2-macrodipa†
Received
25th July 2024
, Accepted 13th August 2024
First published on 20th August 2024
Abstract
Indium-111 (111In) is a diagnostic radiometal that is important in nuclear medicine for single-photon emission computed tomography (SPECT). In order to apply this radiometal, it needs to be stably chelated and conjugated to a targeting vector that delivers it to diseased tissue. Identifying effective chelators that are capable of binding and retaining [111In]In3+in vivo is an important research area. In this study, two 18-membered macrocyclic chelators, py-macrodipa and py2-macrodipa, were investigated for their ability to form stable coordination complexes with In3+ and to be effectively radiolabeled with [111In]In3+. The In3+ complexes of these two chelators were characterized by NMR spectroscopy, X-ray crystallography, and density functional theory calculations. These studies show that both py-macrodipa and py2-macrodipa form 8-coordinate In3+ complexes and attain an asymmetric conformation, consistent with prior studies on this ligand class with small rare earth metal ions. Spectrophotometric titrations were carried out to determine the thermodynamic stability constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KML) of [In(py-macrodipa)]+ and [In(py2-macrodipa)]+, which were found to be 18.96(6) and 19.53(5), respectively, where the values in parentheses are the errors of the last significant figures obtained from the standard deviation from three independent replicates. Radiolabeling studies showed that py-macrodipa and py2-macrodipa can quantitatively be radiolabeled with [111In]In3+ at 25 °C within 5 min, even at ligand concentrations as low as 1 μM. The in vitro stability of the radiolabeled complexes was investigated in human serum at 37 °C, revealing that ∼90% of [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ remained intact after 7 days. The biodistribution of these radiolabeled complexes in mice was investigated, showing lower uptake in the kidneys, liver, and blood at the 24 h mark compared to [111In]InCl3. These results demonstrate the potential of py-macrodipa and py2-macrodipa as chelators for [111In]In3+, suggesting their value for SPECT radiopharmaceuticals.
KML) of [In(py-macrodipa)]+ and [In(py2-macrodipa)]+, which were found to be 18.96(6) and 19.53(5), respectively, where the values in parentheses are the errors of the last significant figures obtained from the standard deviation from three independent replicates. Radiolabeling studies showed that py-macrodipa and py2-macrodipa can quantitatively be radiolabeled with [111In]In3+ at 25 °C within 5 min, even at ligand concentrations as low as 1 μM. The in vitro stability of the radiolabeled complexes was investigated in human serum at 37 °C, revealing that ∼90% of [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ remained intact after 7 days. The biodistribution of these radiolabeled complexes in mice was investigated, showing lower uptake in the kidneys, liver, and blood at the 24 h mark compared to [111In]InCl3. These results demonstrate the potential of py-macrodipa and py2-macrodipa as chelators for [111In]In3+, suggesting their value for SPECT radiopharmaceuticals.
Introduction
In recent years, substantial efforts in the pharmaceutical industry have been directed toward the development of new therapeutic radiopharmaceutical agents for the treatment of disease.1–6 Notably, these efforts have resulted in FDA approval of Xofigo® (radium-223 dichloride citrate) and Pluvicto® (lutetium-177 vipivotide tetraxetan, also recognized as 177Lu-PSMA-617) for treating adult patients with metastatic castration-resistant prostate cancer,7–9 and Lutathera® ([177Lu]Lu-DOTA-TATE) for the treatment of gastroenteropancreatic neuroendocrine tumors.10 These radiotherapeutic agents use a radionuclide with a biologically compatible half-life that emits particles with sufficiently high linear energy transfer to damage biomolecules, and are usually coupled to a targeting moiety designed to selectively deliver them to cancer cells. Once properly localized, these compounds cause DNA damage, leading to cell death.11 When implemented correctly, this targeted approach provides precise and effective treatments with fewer adverse effects compared to traditional cytotoxic drug candidates.
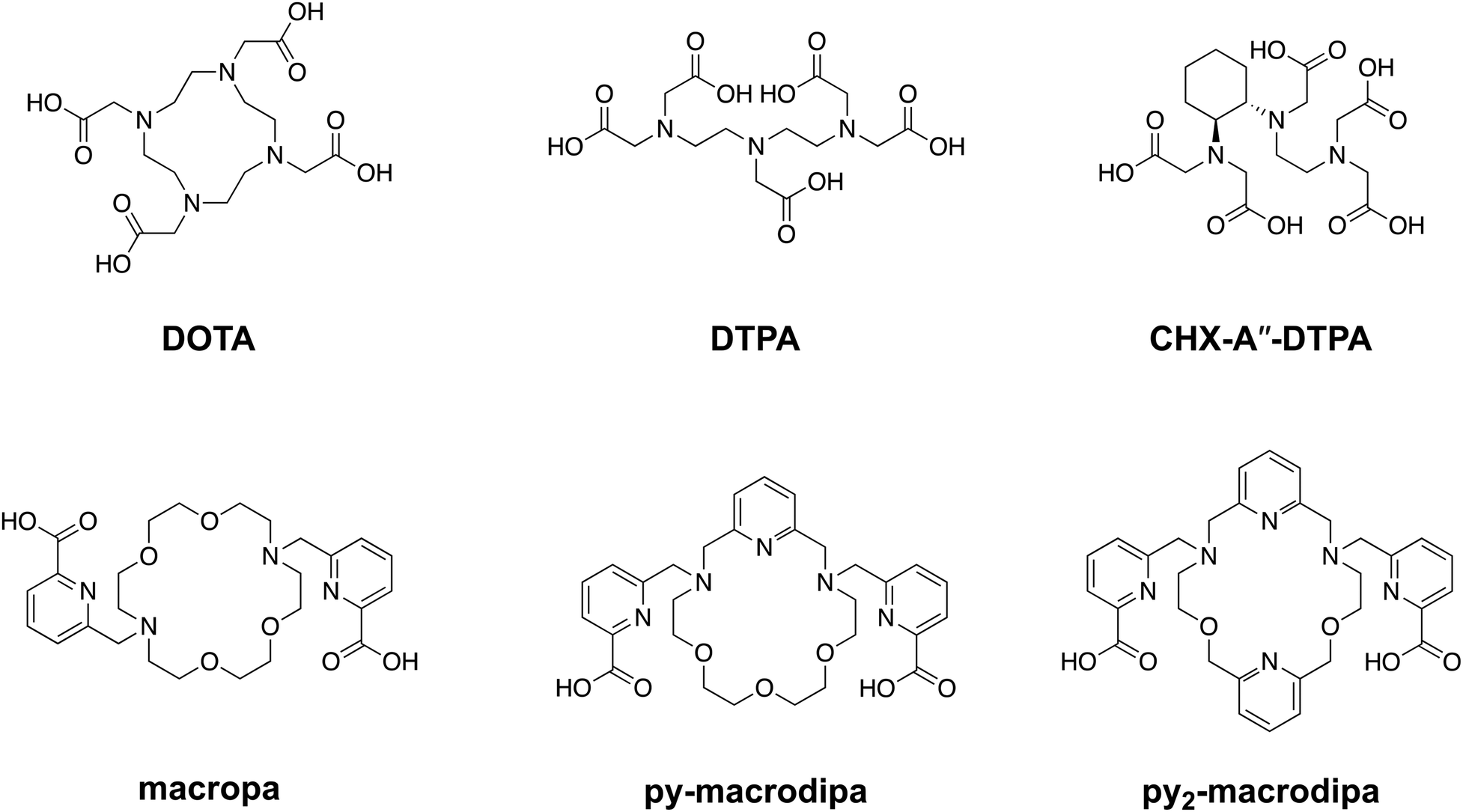
As reflected by this type of FDA-approved drugs, therapeutic radiopharmaceuticals have primarily relied on beta particle (β)-emitting radionuclides, such as 131I, 177Lu, and 90Y.12–18 The investigation of alpha particle (α)-emitters for therapeutic applications, enabled in part by advancements in isotope production and purification, has arisen as a promising alternative to β-emitters.19 For example, α-particles have a substantially higher linear energy transfer, meaning they deposit more of their energy over a shorter path length, giving rise to more damaging biological effects and greater cytotoxicity per decay. Additionally, the short range of α-particles allows for precise absorbed dose delivery to tumors while sparing healthy tissues, a property that may enhance the efficacy of targeted internal radiotherapy.20–23 One of the major challenges in harnessing α-emitting radionuclides for nuclear medicine has been developing chemical strategies to conjugate them to biological targeting vectors. Generally, a bifunctional chelator, which can stably bind radiometal ions and form a covalent linkage to targeting moieties, is needed. The macrocyclic ligand DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid), shown in Chart 1, is an established chelator that works well for many different radiometals.24–27 A key limitation of DOTA is that it is significantly less effective for large metal ions, as reflected by its poorer thermodynamic affinity for larger compared to smaller lanthanide ions.28 This limitation presents challenges associated with its use for promising α-emitting radionuclides, like 223Ra, 225Ac, and 227Th, which are characterized by their large ionic radii.29
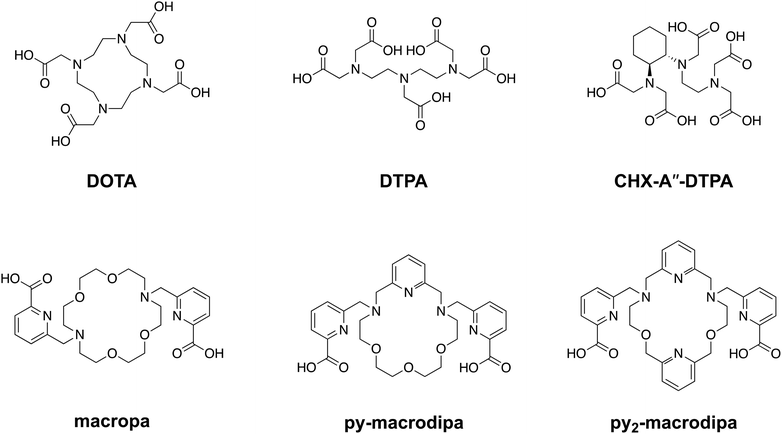 |
| | Chart 1 Structures of ligands discussed in this work. | |
To address this problem, our group has developed chelating agents that possess high affinity and selectivity for large metal ions. These efforts have built upon the success of N,N′-bis[(6-carboxy-2-pyridil)methyl]-4,13-diaza-18-crown-6, or macropa (Chart 1), a ligand that binds preferentially to large over small ions.30 Given its high affinity for large metal ions, macropa has been shown to be effective for chelating the large α-emitting radiometal ions 225Ac, 223Ra, and 213Bi, in addition to the large diagnostic radiometals 203Pb, 132La, 131Ba, and 134Ce.31–43 A limitation of macropa, however, is that it cannot effectively bind to smaller radiometal ions that are widely available and commonly employed in diagnostic applications, including 68Ga, 111In, and 44Sc.44
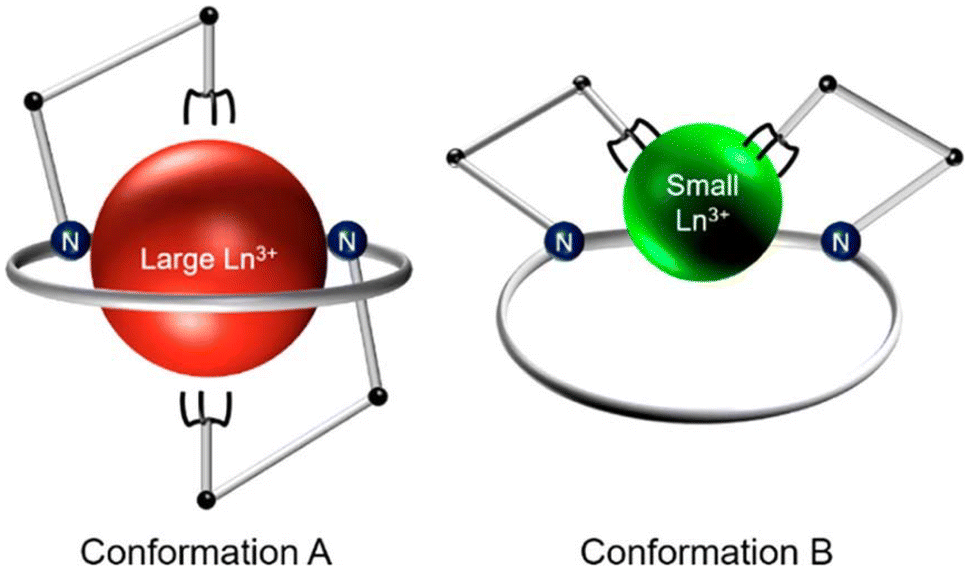
Continuing our investigations on expanded 18-membered macrocyclic chelators, we discovered the “dipa” class of ligands, which includes macrodipa, py-macrodipa, and py2-macrodipa (Chart 1).45–47 A unique feature of this ligand class is its metal ion selectivity pattern that enables it to bind strongly to both large and small metal ions, but relatively poorly to intermediately sized ones. This property arises from the flexibility of this ligand, allowing it to undergo a conformational toggle when binding to small versus large metal ions. Within the realm of nuclear medicine, this ligand class can be applied simultaneously for large therapeutic radiometal ions and small diagnostic radiometal ions. This principle was recently demonstrated using the large radiometal 135La, a therapeutic Auger electron-emitter, and the small radiometal 44Sc, a diagnostic positron-emitter, in conjunction with py-macrodipa. Despite the significant ionic radii differences between La3+ and Sc3+, py-macrodipa could be efficiently radiolabeled with both radionuclides and formed complexes that were stable to human serum over several half-lives.46 Accordingly, a bifunctional version of py-macrodipa was synthesized and conjugated to a PSMA-targeting vector to deliver 135La and 44Sc to tumors in vivo.48 Extending these efforts towards α-emitters, we demonstrated that py-macrodipa was also effective for [213Bi]Bi3+ chelation, and py2-macrodipa could be applied for [225Ac]Ac3+.47 These studies show the versatility of py-macrodipa and py2-macrodipa for theranostic applications because of their abilities to chelate both large α-emitters and smaller diagnostic radiometals.
Among other conventional and highly promising diagnostic radiometals, 111In has been extensively used within the clinic for SPECT imaging and has therapeutic potential as a robust Auger electron emitter.49–51 This cyclotron-produced isotope undergoes decay via electron capture with a half-life of 2.8 d, releasing γ rays (245 and 172 keV) that are well-suited for diagnostic applications.52 Although [111In]In3+ can be effectively chelated with the common bifunctional chelators DOTA and CHX-A′′-DTPA, its small 6-coordinate ionic radii of 0.80 Å (ref. 53) makes it unamenable for chelation by macropa. Thus, using 111In as a partner diagnostic radionuclide for macropa-based bioconjugates for α therapy is not possible. In this work, we investigate the potential of the dual-size-selective chelators py-macrodipa and py2-macrodipa for 111In. Our results demonstrate that these chelators can effectively be used with this diagnostic radionuclide, thereby opening the possibility of pairing it with the larger α therapy isotopes 225Ac and 213Bi.
Results and discussion
Ligand synthesis
The syntheses of py-macrodipa and py2-macrodipa have previously been reported.46,47 Py-macrodipa can be accessed through a 3-step procedure, with the key reaction being the construction of the pyridine-containing macrocycle via a Ca2+-templated reductive amination. Following the macrocycle formation, installation of the picolinate pendent arms through alkylation and acidic deprotection affords py-macrodipa. By contrast, the prior synthesis of py2-macrodipa required a more extensive route to form the key dipyridine-containing macrocycle (4). To access 4, a procedure involving the construction of a benzyl-protected macrocycle was previously reported. This method requires deprotection of the benzyl group through reduction with H2 over Pd/C catalysts, followed by the incorporation of picolinate pendent arms via alkylation (Scheme 1, top). Despite the success of this initial approach, the deprotection step is particularly time-consuming and low-yielding, thus prompting the pursuit of more efficient synthetic strategies. As an alternative approach, we explored the metal ion template-driven synthesis that was used successfully for py-macrodipa (Scheme 1, bottom). The key macrocycle-forming step in these new reactions involves the condensation of diamine 6 with the dialdehyde 7 in the presence of the templating Ca2+ ion, followed by the direct in situ reduction with NaBH4. The subsequent addition of picolinate arms to 4 was accomplished as previously reported.47 The identity and purity of both intermediate and final products were confirmed via NMR spectroscopy and mass spectrometry (Fig. S1–S8, ESI†). This new synthetic strategy is advantageous over the previously reported one with a significantly shorter reaction time of 8 h compared to 2 weeks, which is required for benzyl deprotection. Additionally, the template-driven approach substantially increases the yield, achieving an overall yield of 79%.
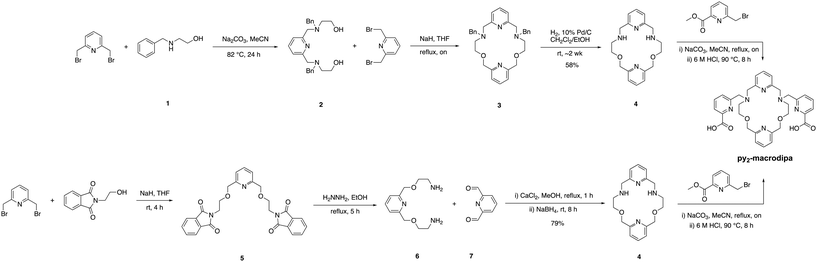 |
| | Scheme 1 Previously reported synthetic approach of dipyridine macrocyclic backbone (top) and improved synthetic approach of dipyridine macrocyclic backbone by using metal ion template synthesis (bottom). | |
Indium complex characterization
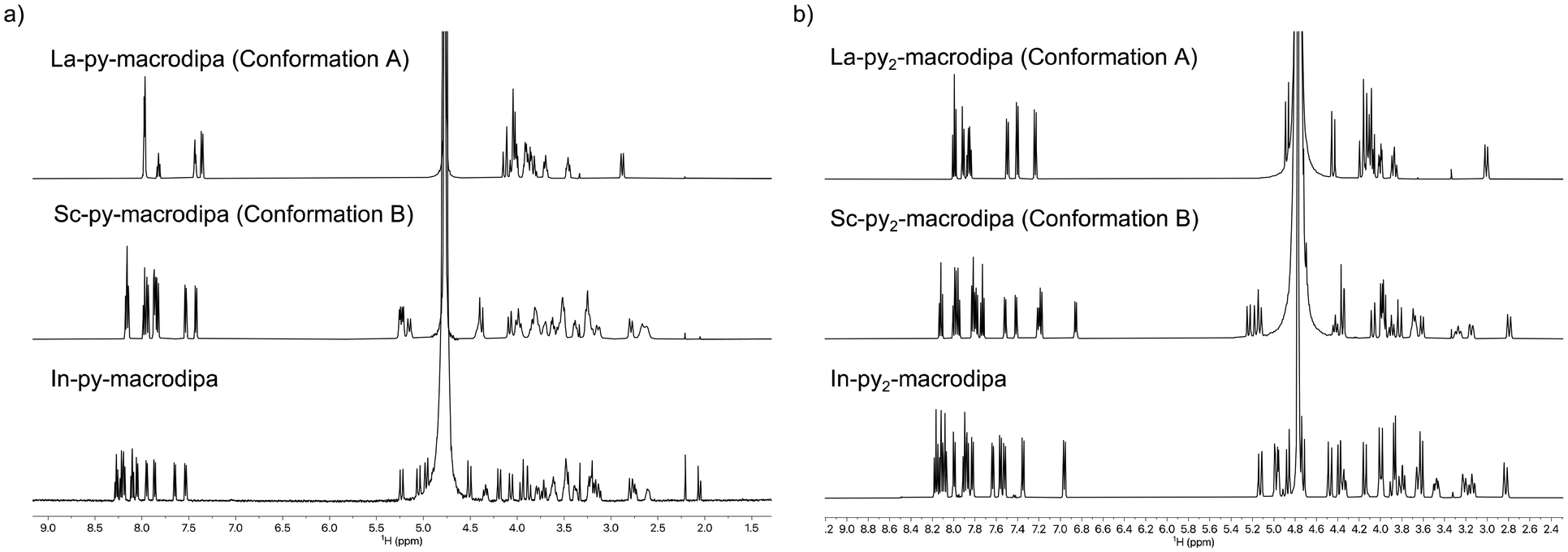
To assess the suitability of py-macrodipa and py2-macrodipa for 111In SPECT imaging applications, we characterized the In3+ complexes of these ligands. Because In3+ is diamagnetic, its complexes can be readily investigated via NMR spectroscopy. The 1H NMR spectra of [In(py-macrodipa)]+ and [In(py2-macrodipa)]+, obtained in D2O, show sharp well-resolved resonances within the aromatic region and display diastereotopically split signals for the aliphatic protons. Notably, for both complexes, there are two distinct sets of signals for the picolinate arms, indicating that the complexes lack symmetry. In the context of prior studies with these ligands, it is well known that they form complexes of distinct symmetries depending on the size of the metal ion. We have previously described these different conformations as Conformation A and Conformation B (Scheme 2).46,47 The symmetric Conformation A is attained by large ions, like La3+, whereas the asymmetric Conformation B occurs in complexes of small ions, like Sc3+. Fig. 1 compares the 1H NMR spectra of the In3+ complexes with those of La3+ and Sc3+. The number of distinct resonances, as well as their chemical shift values, within the spectra of the In3+ complexes closely mirror those of the Sc3+. Thus, like Sc3+, In3+ attains the asymmetric Conformation B with both py-macrodipa and py2-macrodipa. Because the six-coordinate ionic radius of In3+ (0.80 Å) is comparable to that of Sc3+ (0.745 Å), this result is expected.53
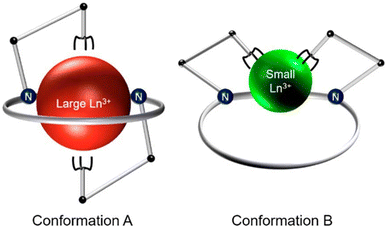 |
| | Scheme 2 Depiction of the conformational toggle present in Ln3+-py-macrodipa and Ln3+-py2-macrodipa complexes. Reproduced with permission from A. Hu, S. N. MacMillan, J. J. Wilson, Macrocyclic Ligands with an Unprecedented Size-Selectivity Pattern for the Lanthanide Ions, J. Am. Chem. Soc. 2020, 142, 13500–13506. Copyright 2020 American Chemical Society. | |
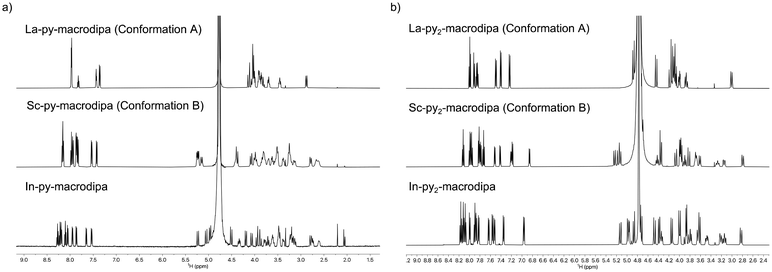 |
| | Fig. 1 (a) 1H NMR spectra of [La(py-macrodipa)]+, [Sc(py-macrodipa)]+ and [In(py-macrodipa)]+ (500 MHz, D2O, pD 5, 25 °C). (b) 1H NMR spectra of [La(py2-macrodipa)]+, [Sc(py2-macrodipa)]+, and [In(py2-macrodipa)]+ (500 MHz, D2O, pD 5, 25 °C). | |
Further characterization of these In3+ complexes was achieved through single-crystal X-ray diffraction. The crystal structures of these complexes are depicted in Fig. 2. Consistent with the NMR spectroscopy results described above, these structures show both In3+ complexes to adopt the asymmetric Conformation B. For [In(py-macrodipa)]+, the immediate coordination sphere of the ion is provided by the pyridyl group, two tertiary amines of the macrocycle, and the two picolinate arms. An eighth donor is provided by a macrocyclic ether oxygen atom. The In–O distance within this interaction is 2.64 Å, notably longer than In–O distances found in related In-crown ether complexes.54,55 Within [In(py2-macrodipa)(OH2)]+, a similar coordination sphere is observed. However, in place of the macrocycle ether oxygen atom, an inner-sphere water molecule is present. The In–O distance afforded by this interaction is 2.21 Å, longer than the In–OH2 distances observed in other crystal structures.56–58 The presence of this inner-sphere water molecule leads to a more expanded conformation of the macrocycle that is stabilized by intramolecular hydrogen-bonding interactions. These two structures highlight some of the variability that is possible within Conformation B. The differential presence and absence of inner-sphere water within related Conformation B structures of [Lu(macrodipa)]+, [Sc(py-macrodipa)(OH2)]+, [Lu(py-macrodipa)]+, and [Sc(py2-macrodipa)(OH2)]+ has also been previously observed.45–47 These results suggest a fluxionality associated with the exchange of the inner-sphere solvent molecules for the ether oxygen of the macrocycle. Slightly different crystallization conditions may lead to the isolation of these different forms.
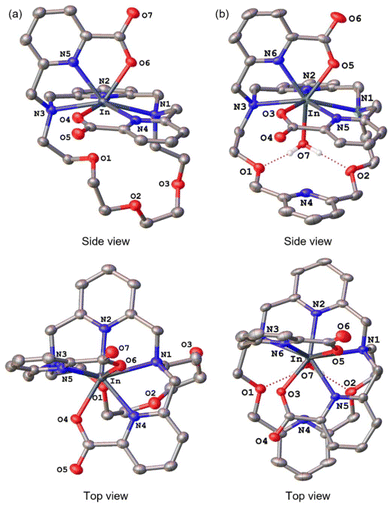 |
| | Fig. 2 Crystal structures of (a) [In(py-macrodipa)]+ and (b) [In(py2-macrodipa)(OH2)]+. Thermal ellipsoids are drawn at the 50% probability level. Solvent, counterions, and non-acidic hydrogen atoms are omitted for clarity. | |
DFT calculations
To further investigate the conformational preferences of the In3+ complexes of py-macrodipa and py2-macrodipa, DFT calculations were undertaken. The geometries of the In3+ complexes of these two ligands in both Conformations A and B were optimized using the ωB97XD functional, the LANL2DZ effective core potential and basis set for In, and the 6-31+G(d,p) basis set for other atoms. This method has been previously employed for Ln3+-py-macrodipa and Ln3+-py2-macrodipa complexes.46,47 Subsequently, their standard free energies (G°) were calculated. Given our observation of Conformation B with and without inner-sphere water molecules, both forms of this conformation were investigated in these calculations. The equilibria between conformations A and B in an aqueous solution can be represented by balanced chemical reactions (eqn (1)–(4)), with the standard free energy changes denoted as ΔG° and the metal ions denoted as M. Our previous studies with Ln3+-py-macrodipa and Ln3+-py2-macrodipa complexes have shown the ΔG° values are positive for the large La3+ ion, but negative for the small Sc3+ ion, indicating a thermodynamically driven transition from conformation A to B.46,47 For the equilibria that do not invoke an inner-sphere water molecule (eqn (1) and (3)), the ΔG° for [In(py-macrodipa)]+ and [In(py2-macrodipa)]+ complexes are −17.1 and −20.5 kJ mol−1, respectively, as detailed in Table 1. When an inner-sphere water is included in Conformation B (eqn (2) and (4)), these ΔG° values become substantially more negative, attaining −74.6 and −84.6 kJ mol−1 for [In(py-macrodipa)]+ and [In(py2-macrodipa)]+, respectively. The negative values, which indicate a thermodynamic preference for Conformation B, are expected due to the relatively small size of the In3+ ion, which is comparable to that of Sc3+.53 Thus, both In3+ complexes favor Conformation B, regardless of the presence or absence of an inner-sphere water molecule.| |  | (1) |
| | 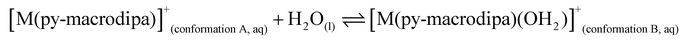 | (2) |
| | 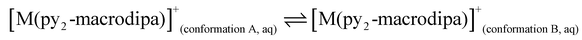 | (3) |
| | 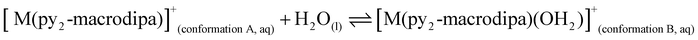 | (4) |
Table 1 Changes of standard free energy in aqueous solution of DFT-optimized py-macrodipa and py2-macrodipa complexes at 298 K
| |
ΔG°(A, B) (kJ mol−1) |
| |
La3+ |
Sc3+ |
In3+ |
|
Ref. 46.
Ref. 47.
|
| (1) |
+55.4a |
–56.6a |
–17.1 |
| (2) |
+24.6a |
–80.5a |
–74.6 |
| (3) |
+61.2 |
–49.2 |
–20.5 |
| (4) |
+27.0b |
–87.4b |
–84.6 |
In3+ complex thermodynamic stability
To determine the thermodynamic stability of the In3+ complexes of py-macrodipa and py2-macrodipa, UV-vis spectrophotometric titrations were carried out. The values for the protonation constants (Ki) of py-macrodipa and py2-macrodipa were obtained from previous pH potentiometric titrations of these ligands.46,47 Although pH potentiometric titrations of [In(py-macrodipa)]+ and [In(py2-macrodipa)]+ were initially attempted, their stability constants (KML) were too large to be determined over the pH range accessible to this method. Consequently, we conducted spectrophotometric titrations over an acidic pH (0.2–2.4) to determine these KML values. The UV-vis spectra of these titrations are shown in Fig. 3, and the data fitting analysis is shown in Fig. S9 and S10, ESI.† As shown in Table 2, the logKInL values for both py-macrodipa and py2-macrodipa (18.96 and 19.53, respectively) are several orders of magnitude larger than those of other metal ions. Notably, these values are even larger than those of the Sc3+ complexes of these ligands. Compared to the conventional chelators DOTA and DTPA, however, the KInL values are still over 10 orders of magnitude smaller.59,60 Although the In3+ complexes of py-macrodipa and py2-macrodipa do not attain the same level of thermodynamic stability as those of DOTA and DTPA, this property does not preclude their use for radiopharmaceutical applications. For example, the stability constant of macropa with La3+ is only 14.99, but this ligand has proven to be highly effective for in vivo applications using 225Ac, 132/135La, and 134Ce. Thus, in addition to thermodynamic stability, the inertness of relevant metal ion complexes needs to be considered under biologically relevant conditions.
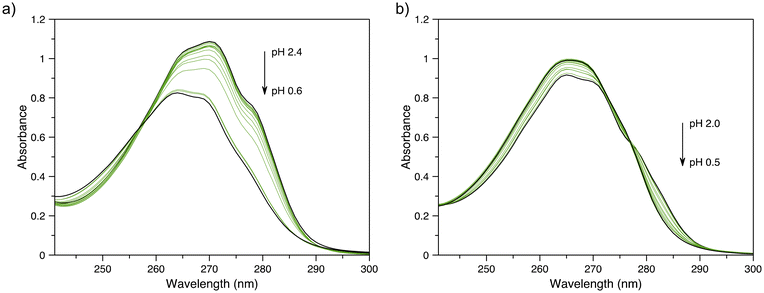 |
| | Fig. 3 (a) Representative UV-vis spectra of solutions containing py-macrodipa and InCl3 in spectrophotometric titrations over the pH range of 0.6–2.4. (b) Representative UV-vis spectra of solutions containing py2-macrodipa and InCl3 in spectrophotometric titrations over the pH range 0.5–2.0. | |
Table 2 Stability constants of complexes formed with py-macrodipa, py2-macrodipa, DOTA, and DTPA
| |
py-macrodipa |
py2-macrodipa |
DOTA |
DTPA |
|
Ref. 59 (0.1 M KCl, 25 °C).
Ref. 46 (0.1 M KCl, 25 °C).
Ref. 47 (0.1 M KCl, 25 °C).
Ref. 60 (0.1 M KNO3, 25 °C).
Ref. 61 (0.1 M KCl, 25 °C).
Ref. 62 (0.1 M KNO3, 25 °C).
Ref. 63 (0.1 M NMe4Cl, 25 °C).
pIn = –log [In3+]free. Calculated for 10 μM total ligand and 1 μM total metal at pH 7.4 and 25 °C.
|
| logKInL |
18.96(6) |
19.53(5) |
23.9a |
29.0d |
| logKLaL |
14.31b |
16.68c |
22.9e |
18.02f |
| logKCeL |
14.65b |
17.13c |
23.4e |
19.06f |
| logKPrL |
14.81b |
17.28c |
23.0e |
19.64f |
| logKNdL |
14.51b |
17.11c |
23.0e |
20.23f |
| logKSmL |
13.66b |
16.56c |
23.0e |
20.79f |
| logKEuL |
13.29b |
15.93c |
23.5e |
20.74f |
| logKGdL |
12.63b |
15.25c |
24.7e |
21.15f |
| logKTbL |
11.95b |
14.76c |
24.2e |
21.15f |
| logKDyL |
11.47b |
14.04c |
24.8e |
21.23f |
| logKHoL |
10.69b |
12.68c |
24.5e |
21.43f |
| logKErL |
10.60b |
12.17c |
24.4e |
21.41f |
| logKTmL |
10.92b |
11.98c |
24.4e |
21.10f |
| logKYbL |
11.31b |
11.82c |
25.0e |
20.96f |
| logKLuL |
11.54b |
11.90c |
25.4e |
21.14f |
| logKScL |
15.83b |
16.28c |
30.79g |
27.43g |
| pIn |
19.3h |
19.7h |
18.8h |
25.7h |
Radiolabeling experiments
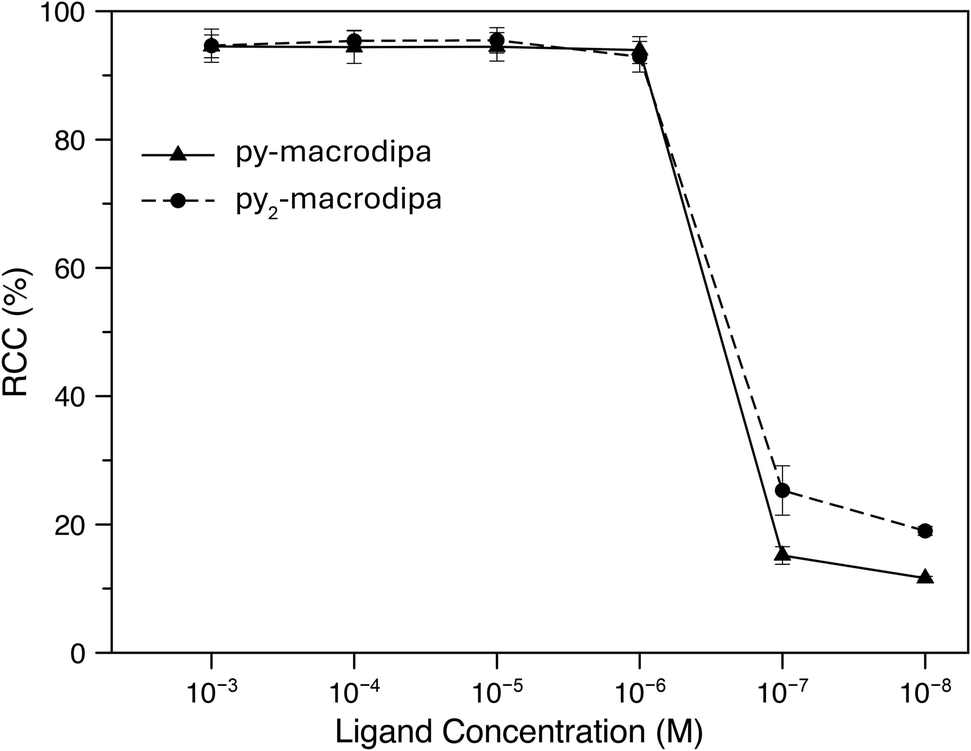
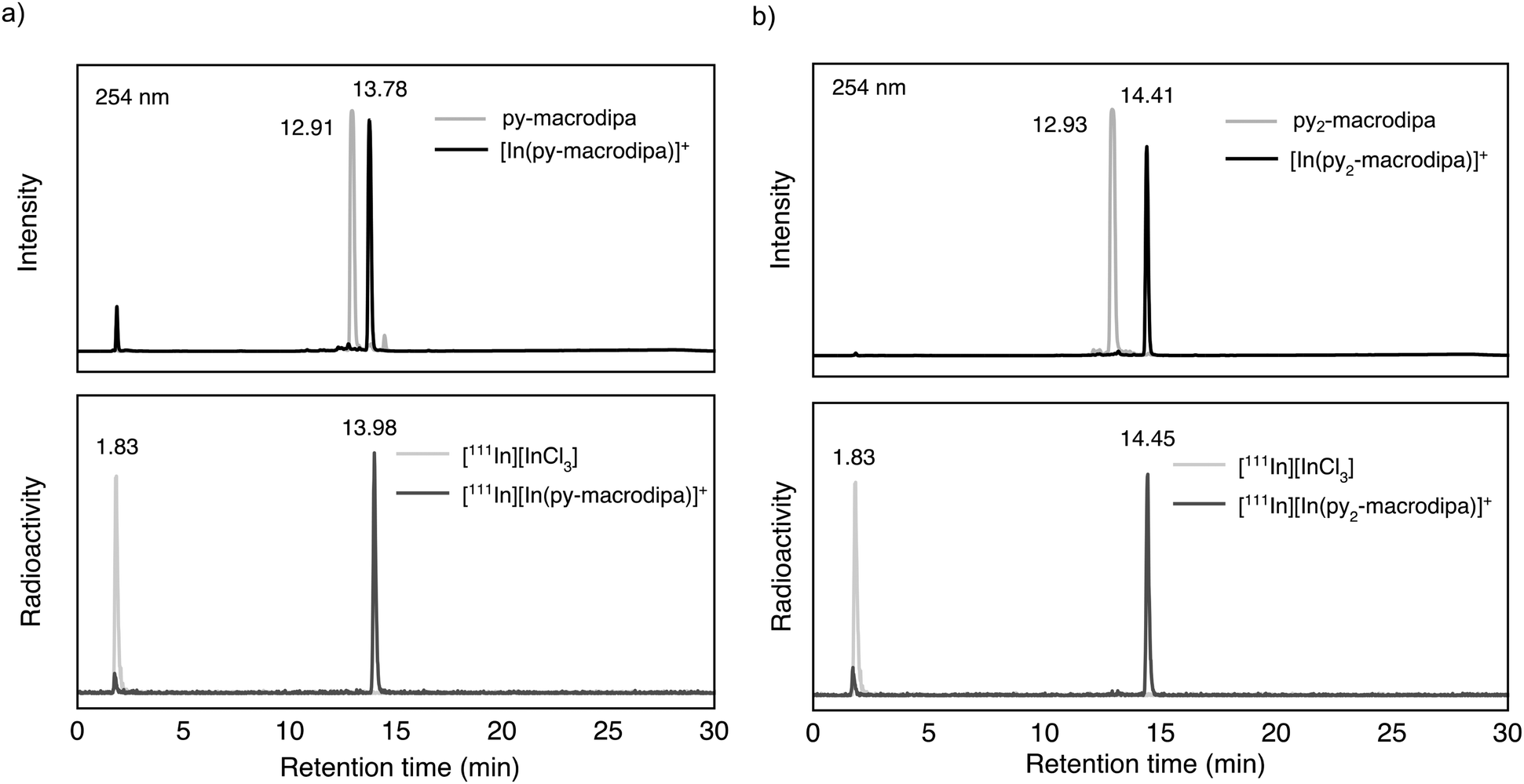
Based on the promising In3+ coordination chemistry of these ligands, we next evaluated their ability to be labeled with radioactive [111In]In3+. Concentration-dependent radiolabeling studies were conducted to ascertain the minimum concentration necessary for achieving high radiochemical conversions (RCCs). Varied concentrations of py-macrodipa and py2-macrodipa, ranging from millimolar to nanomolar, were incubated with ∼668 kBq of [111In]In3+ in a pH 5.5 ammonium acetate buffer at 25 °C for 5 min. The RCCs of the radiolabeled complexes were quantified using radio-thin-layer chromatography (radio-TLC). Remarkably, the RCCs of both py-macrodipa and py2-macrodipa 111In complexes were greater than 95% at ligand concentrations as low as 10−6 M (Fig. 4). By contrast, macropa was unable to incorporate [111In]In3+ under the same conditions, even with ligand concentrations as high as 1 mM (Fig. S13, ESI†). To achieve quantitative RCCs of macropa with [111In]In3+, 4 mM of macropa was needed to incubate with [111In]In3+ at 25 °C for 1 h. (Fig. S14, ESI†). This result highlights how the preference of macropa for large ions limits its use for the smaller diagnostic [111In]In3+ radiometal ion. In addition, the abilities of py-macrodipa and py2-macrodipa to radiolabel at 25 °C marks an advantage over DOTA, for which the [111In]In3+ complexes are most effectively formed by heating at 100 °C for 30 min.64 The effective and mild 111In-radiolabeling capabilities of py-macrodipa and py2-marodipa are particularly important in the context of the fact that these chelators can also similarly radiolabel the large therapeutic [225Ac]Ac3+, [135La]La3+, and [213Bi]Bi3+ radiometal ions. Lastly, the identities of the radiolabeled [111In]In3+ complexes were confirmed with radio-high-performance liquid chromatography (radio-HPLC). In these studies, HPLC retention times of the non-radioactive In3+ complexes of py-macrodipa and py2-macrodipa were used as reference values. As shown in Fig. 5, the UV-vis-HPLC chromatograms of the cold In3+ complexes display peaks with nearly identical retention times as the radio-HPLC chromatogram of the 111In-labeled complexes. Therefore, the [111In]In3+ complexes are most likely identical in structure and conformation to their well-characterized non-radioactive analogs.
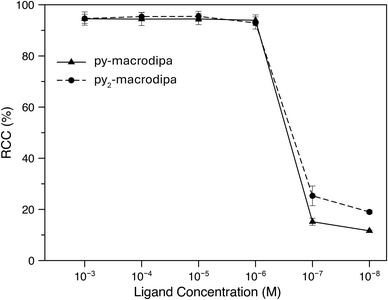 |
| | Fig. 4 Radiochemical conversions of [111In]In3+ with py-macrodipa and py2-macrodipa at different ligand concentrations (25 °C; pH 5.5; 5 min). | |
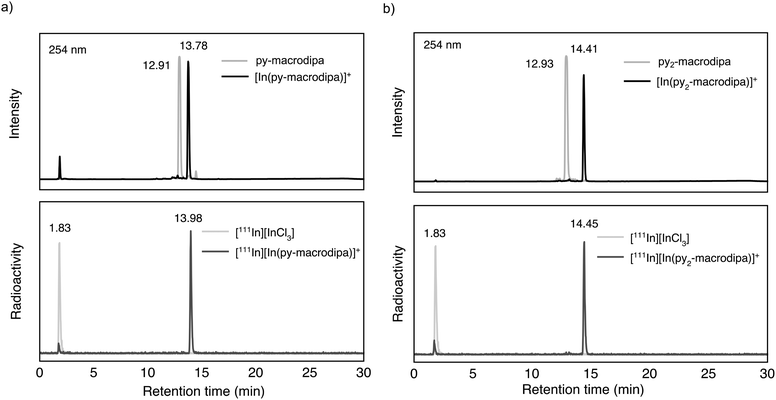 |
| | Fig. 5 (a) UV-vis-HPLC chromatogram (254 nm) of nonradioactive [In(py-macrodipa)]+ and py-macrodipa (top) compared with radio-HPLC chromatogram of [111In][In(py-macrodipa)]+ and [111In]InCl3 (bottom). (b) UV-vis-HPLC chromatogram (254 nm) of nonradioactive [In(py2-macrodipa)]+ and py2-macrodipa (top) compared with the radio-HPLC chromatogram of [111In][In(py2-macrodipa)]+ and [111In]InCl3 (bottom). | |
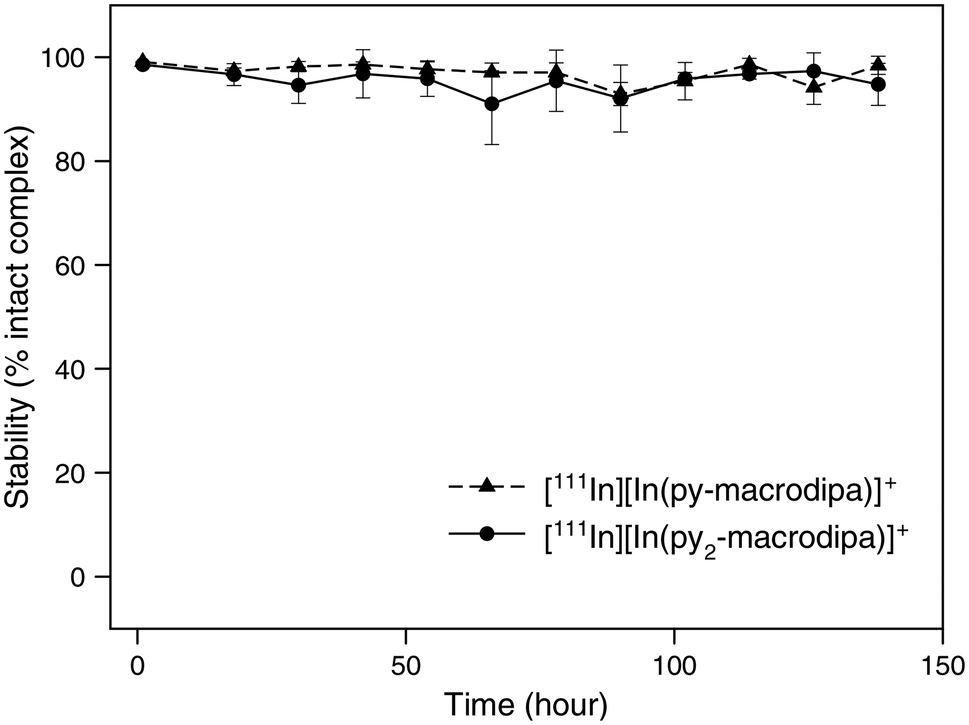
Human serum stability
After the successful formation of the [111In]In3+ complexes of py-macrodipa and py2-macrodipa, their stabilities under biologically relevant conditions were assessed. The complexes were formed using 8.77 MBq of [111In]InCl3 and a 1 μM ligand solution, with radiochemical purity confirmed by radio-TLC and radio-HPLC, and subsequently incubated in human serum at 37 °C. As shown in Fig. 6, both [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ remained 98.4 ± 1.8% and 94.8 ± 4.7% intact, respectively, after 1 week based on radio-TLC. Radio-HPLC was also conducted at each time point to further confirm the identity of the activity (Fig. S15 and S16, ESI†). Despite the gradual decrease in the signal-to-noise ratio of the chromatograms due to the physical decay of the radionuclides, only a single peak corresponding to the [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ complexes were observed throughout the experiment. These results indicate that both [111In]In3+ complexes are inert under these biologically relevant conditions, making them promising for radiopharmaceutical applications. In comparison, 73.2 ± 10.2% of [111In][In(macropa)]+, formed with 4 mM of macropa and 8.77 MBq [111In]In3+, remained intact in human serum after 12 h at 37 °C (Fig. S17, ESI†). Furthermore, the similarly high stability of [225Ac][Ac(py2-macrodipa)]+ in human serum underscores the potential for using 225Ac and 111In as a theranostic pair with py2-macrodipa.
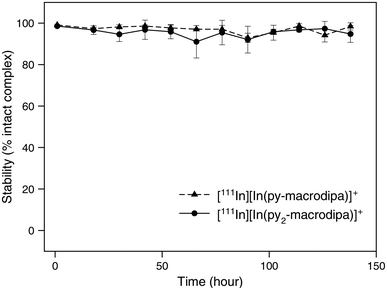 |
| | Fig. 6 Stability of [111In][In(py-macrodipa)]+ and [111In][In(py2-marodipa)]+ in human serum at 37 °C. | |
Biodistribution studies
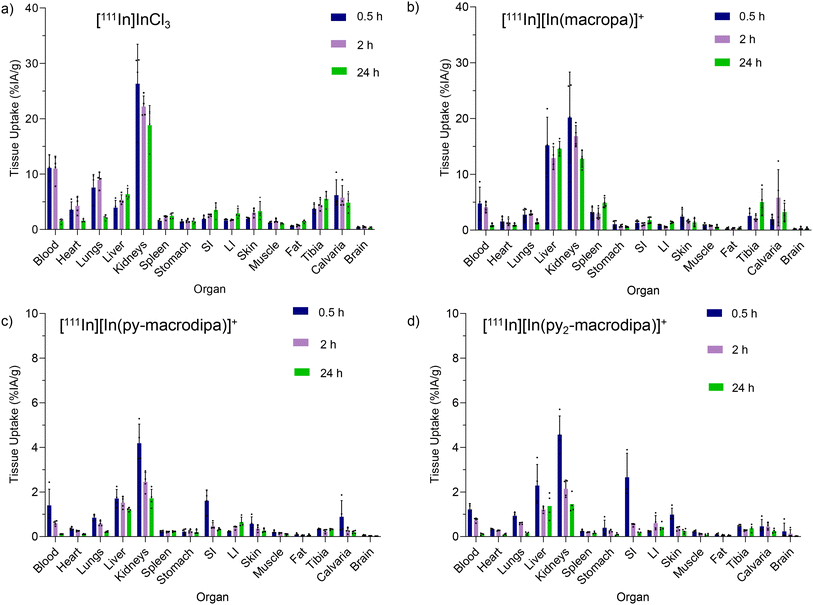
To evaluate the in vivo stabilities of [111In][In(macropa)]+, [111In][In(py-macrodipa)]+, and [111In][In(py2-macrodipa)]+, their biodistribution in mice was investigated in comparison to [111In]InCl3. The complexes were formed using 17 mM of each chelator and 11.8 MBq of [111In]In3+ after incubation for 5 min at 25 °C in pH 5.5 NH4OAc buffer. The RCCs of all [111In]In3+ complexes were confirmed with radio-TLC. Swiss Webster mice were retro-orbitally (RO) administered with 0.703 MBq of complexes or [111In]InCl3 and sacrificed at time points of 0.5, 2, and 24 h for biodistribution analysis. These results are shown in Fig. 7. For [111In]InCl3, most of the injected activity is excreted through the kidneys with a smaller amount clearing through the liver. The activity persisted in the blood, heart, lungs, liver, kidneys, tibia, and calvaria. After 24 h post-injection, relatively high activity levels were observed in the kidneys, and moderate uptake was found in the liver and bone (Fig. 7a). These results are consistent with prior [111In]InCl3 biodistribution studies.65 [111In][In(macropa)]+ exhibited a similar biodistribution and clearance profile as [111In]InCl3 with high uptake in the kidney and slow blood clearance (Fig. 7b). The organs with the greatest amount of activity were the kidneys and liver. Low activities were observed in the heart, lungs, stomach, small intestine (SI), large intestine (LI), skin, fat, muscle, and brain, and moderate levels were found in blood, spleen, and bone. By contrast, the biodistribution profiles of [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ differ significantly from [111In]InCl3 (Fig. 7c and d). Low accumulation in the kidney, liver, and SI was observed, and a very low accumulation of activity in blood was found for both complexes 2 h post-injection, indicating rapid clearance. During this study, the complexes exhibited negligible uptake by other organs (<1% IA g−1 at 30 min post-injection), and activity did not accumulate over time, indicating no release of [111In]In3+in vivo. These biodistribution studies reveal that [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ are highly stable in vivo, a property that is crucial for their use as radiopharmaceutical agents.
 |
| | Fig. 7
In vivo biodistribution of (a) [111In]InCl3, (b) [111In][In(macropa)]+, (c) [111In][In(py-macrodipa)]+, and (d) [111In][In(py2-macrodipa)]+ following retro-orbital injection in mice. Four Naive Swiss Webster females of 4–6 weeks old were sacrificed after 0.5, 2, and 24 h. Values for each time point are given as percent injected activity per gram tissue (%IA g−1). | |
Conclusions
In this study, we assessed the potential of macropa, py-macrodipa, and py2-macrodipa as chelators for the widely used SPECT radioisotope 111In. Through computational and experimental analyses, we characterized their coordination chemistry with In3+ and observed that both py-macrodipa and py2-macrodipa ligands form stable complexes with this ion, adopting asymmetric Conformation B upon complexation. Furthermore, radiolabeling experiments demonstrated effective complexation with [111In]In3+ under mild conditions, and the resulting radiolabeled complexes were stable in human serum. This excellent stability was also verified in mice, which showed that [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ did not accumulate in the kidneys, liver, and blood at the 0.5 h or 24 h mark, unlike [111In]InCl3 and [111In][In(macropa)]+. In the context of earlier studies on the suitability of these chelators for large therapeutic radiometals like 135La, 213Bi, and 225Ac, their ability to be used with the small diagnostic 111In radionuclide portend well for use in theranostic applications. The conformational flexibility provided by this ligand class presents an interesting opportunity to chelate useful radiometal ions of different ionic radii. Thus, py-macrodipa and py2-macrodipa join several other recently reported chelators that have mutually high affinity for large α-emitting and small diagnostic radionuclides.66–73 Ongoing efforts are focused on synthesizing a bifunctional analog of py2-macrodipa to enable its use for SPECT imaging with 111In and α therapy with 225Ac and 213Bi.
Experimental
General
All chemicals were purchased from commercial suppliers and used without further purification. py-Macrodipa, macropa, pyridine-2,6-dicarbaldehyde, and 6-bromomethylpyridine-2-carboxylic methyl ester were synthesized as previously described.46 The compounds 2,6-bis(bromomethyl)pyridine and N-(2-hydroxyethyl) phthalimide were purchased from VWR (Radnor PA). Anhydrous solvents were obtained following storage over 3 Å molecular sieves, which were activated under heat. 1D-NMR (1H and 13C {1H}) spectra were acquired at 25 °C on a 500 MHz Bruker AV 3HD spectrometer equipped with a broadband Prodigy cryoprobe (Bruker, Billerica, MA). Water (18 MΩ cm) was purified using an ELGA PURELAB flex 2 (High Wycombe, U.K.). High-resolution mass spectra (MS) were recorded on an Exactive Orbitrap mass spectrometer (Thermo Fisher) in positive ionization mode with sample analysis in real-time (DART) mode. Elemental analyses (C, H, N) were carried out by Atlantic Microlab Inc. (Norcross, GA). High-performance liquid chromatography (HPLC) consisted of a CBM-20A communications bus module, an LC-20AT pump, and an SPD-20AV UV-Vis detector monitoring at 254 nm (Shimadzu, Japan). Analytical chromatography was carried out using a Luna C18 column, 100 Å, 5 μm, 150 mm × 3 mm (Phenomenex, Torrance, CA) at a flow rate of 1.0 mL min−1. [111In]InCl3 (in 0.01M HCl with radionuclide purity >99.9%) was purchased from BWXT Medical Ltd (Vancouver, Canada). Activity amounts of [111In]InCl3 solution were measured with a dose calibrator (CAPINTEC, CRC-15R) and 2480 automated γ-counter (WIZARD2 PerkinElmer). Glass microfiber chromatography paper impregnated with silica gel (iTLC-SG) was purchased from Agilent Technologies (Folsom CA). Female Swiss Webster mice (4–6 weeks, Charlies River Laboratories) were purchased for in vivo biodistribution studies. All radioactive material handling and animal experiments were conducted in compliance with institutional regulations and approved by Environmental Health and Safety Radioactive Materials at Washington University in St. Louis under protocol D16-00245 and Institutional Animal Care and Use Committee protocol #22-0023.
Chelator synthesis
2,2′-(((Pyridine-2,6-diylbis(methylene))bis(oxy))bis(ethane-2,1-diyl))bis(isoindoline-1,3-dione) (5).
To a solution of N-(2-hydroxyethyl)phthalimide (4.00 g, 20.9 mmol) in anhydrous tetrahydrofuran (30 mL) was added NaH (60% dispersion in mineral oil) (1.68 g, 41.0 mmol) at 0 °C over 1 h. Commercially available 2,6-bis(bromomethyl)pyridine (2.63 g, 10.5 mmol) was added to the mixture portion-wise over 30 min and allowed to stir at room temperature for 4 h. The mixture was then filtered, and the filtrate was concentrated under reduced pressure to give a yellow solid. The crude solid was then washed with 100 mL of methanol on filter paper to yield 5 (2.51 g, 52%) as an off-white solid. 1H NMR (500 MHz, CDCl3) δ = 7.84 (dq, J = 6.0, 3.2 Hz, 4H), 7.71 (dq, J = 5.1, 3.0 Hz, 4H), 7.55 (t, J = 7.7 Hz, 1H), 7.21 (d, J = 7.7 Hz, 2H), 4.58 (s, 4H), 3.97 (t, J = 5.7 Hz, 4H), 3.79 (t, J = 5.7 Hz, 4H). 13C{1H} NMR (126 MHz, CDCl3) δ = 168.39, 157.61, 134.08, 132.26, 123.42, 119.89, 6812, 67.83, 37.71. DART-MS m/z: 486.17; calcd for [M + H]+: 486.17.
2,2′-((Pyridine-2,6-diylbis(methylene))bis(oxy))bis(ethan-1-amine) (6).
To a solution of 5 (2.00 g, 4.12 mmol) in absolute ethanol (30 mL) was added 80% hydrazine monohydrate (0.60 mL, 10.1 mmol). The reaction mixture was heated at 80 °C for 4 h. A quantity of 10 mL of 6 M HCl was added, and the mixture was stirred at 80 °C for an additional 1 h. After cooling to room temperature, the mixture was filtered, and the collected solid was washed with water (10 mL). The combined filtrate was concentrated under reduced pressure to give a yellow oil. Ethanol was added to this crude oil and the precipitates were collected by filtration to afford 6 (0.94 g, 68%) as a white solid. 1H NMR (500 MHz, D2O, pD ≈ 5) δ = 8.58 (t, J = 8.0 Hz, 1H), 8.02 (d, J = 8.0 Hz, 2H), 5.04 (s, 4H), 3.94 (t, J = 5.0 Hz, 4H), 3.34 (t, J = 4.9 Hz, 4H). 13C{1H} NMR (126 MHz, DMSO-d6) δ = 158.26, 137.82, 120.24, 79.63, 73.43, 73.31, 41.77. DART-MS m/z: 226.15; calcd for [M + H]+ : 226.15.
3,13-Dioxa-6,10-diaza-1,8(2,6)-dipyridinacyclotetradecaphane (4).
Anhydrous methanol (10 mL) was added to a vial containing pyridine-2,6-dicarbaldehyde (0.20 g, 1.49 mmol) and CaCl2·2H2O (0.24 g, 1.57 mmol). The mixture was stirred at room temperature until pyridine-2,6-dicarbaldehyde was fully dissolved. A solution of 6 (0.50 g, 1.49 mmol) in anhydrous methanol (10 mL) was then added dropwise. The mixture was stirred at room temperature for 30 min and subsequently heated at 65 °C for 1 h. After cooling on ice to 0 °C, solid NaBH4 (0.28 g, 7.47 mmol) was added in small portions over 15 min. The reaction mixture was allowed to slowly return to RT and stirred for 8 h. The reaction was quenched with H2O (10 mL), and methanol was removed under reduced pressure. The mixture was then extracted with 3 × 10 mL of CH2Cl2. The combined organic layers were dried over Na2SO4 and removed under reduced pressure. The crude product was purified by column chromatography (25 g silica gel, eluted with a gradient of 100% DCM to 75% DCM/MeOH) to afford 4 (0.39 g, 79%) as a clear oil. 1H NMR (500 MHz, MeOD-d4) δ = 7.80 (t, 1H, J = 7.7 Hz), 7.69 (t, 1H, J = 7.7 Hz), 7.35 (d, 2H, J = 7.7 Hz), 7.22 (d, 2H, J = 7.7 Hz), 4.56 (s, 4H), 3.80 (s, 4H), 3.73 (t, 4H, J = 4.8 Hz), 2.86 (t, 4H, J = 4.8 Hz). DART-MS m/z: 329.20; calcd for [M + H]+: 329.20.
X-ray crystallography
Low-temperature X-ray diffraction data of [In(py-macrodipa)]+ and [In(py2-macrodipa)]+ were collected on a Rigaku XtaLAB Synergy diffractometer coupled to a Rigaku Hypix detector with Cu Kα radiation (λ = 1.54184 Å), from a PhotonJet micro-focus X-ray source at 100 K. The diffraction images were processed and scaled using the CrysAlisPro software (Oxford Diffraction LTD, TX). The structures were solved through intrinsic phasing using SHELXT74 and refined against F2 on all data by full-matrix least squares with SHELXL75 following established refinement strategies.76 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms bound to carbon were included in the model at geometrically calculated positions and refined using a riding model. Hydrogen atoms bound to oxygen were located in the difference Fourier synthesis and subsequently refined semi-freely with the help of distance restraints. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the Ueq value of the atoms they are linked to (1.5 times for methyl groups). [In(py2-macrodipa)]+ contains disordered solvent molecules of Et2O and MeOH but could not be satisfactorily modeled. Therefore, those solvents were treated as diffuse contributions to the overall scattering without specific atom positions using the solvent mask routine in Olex2 with a volume of 327.6 Å3 corresponding to 81.2 electrons.77 The X-ray crystallographic data is deposited in the Cambridge Structural Database (CCDC) as crystallographic information files (cif) under deposition numbers 2373568 and 2373569.†
Stability constant measurements
The stability constants of [In(py-macrodipa)]+ and [In(py2-macrodipa)]+ complexes were measured by UV-vis spectrophotometric titrations. Ligand stock solutions (17.83 mM py-macrodipa and 19.57 mM py2-macrodipa) were made by dissolving the solid ligand in 18 MΩ·cm H2O. An In3+ stock solution (21.18 mM) was made by dissolving InCl3·4H2O (99.9% purity) in standardized HCl (0.1 M). The exact concentrations were determined by complexometric titrations with a standardized Na2H2EDTA solution (Alfa Aesar). The complexometric titrations were performed at pH 5.4 maintained by a sodium acetate buffer, and the endpoint was indicated by xylenol orange.
UV-vis spectra were recorded on a Shimadzu UV-1900 UV-vis spectrometer with a 1 cm quartz cuvette. The stability constants were determined with UV-vis spectrophotometric titrations using a batch method. Specifically, a set of solutions (around 15 samples) containing In3+ and py-macrodipa, and py2-macrodipa (1:1 metal to ligand molar ratio, 45–80 μM) at different pH values were prepared. All samples were maintained at low pH (1.1 ≤ pH ≤ 2.4), which was adjusted with standardized HCl (0.1 M). Under these conditions, pH cannot be accurately measured with the glass electrode; pH values were calculated directly from the concentration of HCl used in each sample. The ionic strength was fixed at 0.1 M with KCl when possible. The solutions were incubated at 25 °C overnight after which all samples had reached equilibrium. The pH-dependent spectrophotometric data were analyzed with HypeSpec 2014 software.78 The absorption band from 240–300 nm was used for analysis and the spectra for free ligand species were included in the fitting. The stability constants were calculated from the average of three independent titrations.
Radiolabeling
Py-macrodipa and py2-macrodipa were dissolved in ammonium acetate buffer solution (pH 5.5) to give 10 mM stock solutions from which serial dilutions were used to prepare ligand solutions of concentrations ranging from 10−3 to 10−7 M. For [111In]In3+ labeling studies, a 5 μL aliquot of the respective ligand solution was further diluted with 44 μL of 0.5 M NH4OAc buffer solution (pH = 5.5), and 1 μL of [111In]InCl3 solution in 0.01 M HCl (0.668 MBq) was added. RCCs were determined with radio-iTLC using a mobile phase of 50 mM EDTA (pH 5.5). Under these conditions, chelated [111In]In3+ remained at the baseline (Rf = 0), and uncomplexed [111In]In3+ migrated with the solvent front (Rf ∼1).79 At 5 min, ∼5 μL aliquots were spotted on the plates and developed. Once dried, RCCs were measured using a Dual Scan-Ram Scanner (Lablogic, UK) equipped with a NaI detector and quantified with Laura software. Radio-HPLC was further used to confirm the identity of the [111In]In3+ complex with elution conditions: A: H2O + 0.1% TFA, B: MeOH; 0 to 90% B linear gradient 20 min. [111In][In(py-macrodipa)]+ (tR = 13.98 min), [111In][In(py2-macrodipa)]+ (tR = 14.45 min), and [111In]InCl3 (tR = 1.83 min).
Human serum stability
The compounds [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ were prepared with the radiolabeling protocol as described above. A quantity of 1.5 mL of 0.33 mM ligand solutions of py-macrodipa and py2-macrodipa were incubated with 9 μL of [111In]InCl3 (8.77 MBq) solution in 114.5 μL of 0.5 M NH4OAc buffer (pH = 5.5) for 5 min at 25 °C. A 375 μL portion of human serum, purchased from Sigma (Sera human Lot# SLCF9666), was added, and the mixture was incubated at 37 °C. At each time point, 10 μL of each solution was analyzed by radio-TLC to determine the % intact complex, and the values were tabulated as shown in Table S2, ESI.† At each time point, 50 μL of the mixture was added to 250 μL of MeOH to precipitate the proteins. After centrifugation, 10 μL of the supernatant liquid was used for radio-HPLC to identify the activity.80
Biodistribution study
For preparation of [111In][In(macropa)]+, 20 μL of [111In]InCl3 (11.8 MBq) in 0.01 M HCl (Sigma-Aldrich, trace-metal grade, 99.999%) was added to ammonium acetate buffer (250 μL, 0.5 M, pH 5.5) to adjust the pH to 5.5, and then 100 μL of 17 mM of macropa chelator was added. The mixture was vortexed and incubated at 25 °C for 5 min. Radiochemical purity was evaluated using iTLC-SG with a 50 mM EDTA pH 5.5 mobile phase. Radio-TLC was developed using a gas proportional scanner (Bioscan AR-2000) with a 1 min acquisition time. For subsequent in vivo evaluation, the labeled material was then diluted with 1.5 mL of normal saline (0.9% NaCl in deionized H2O; Hospira), and activities were prepared in 30G syringes for administration. [111In][In(py-macrodipa)]+ and [111In][In(py2-macrodipa)]+ were also prepared following the same labeling conditions. Healthy Swiss webster female mice from Charlies River Laboratories of 4–6 weeks old, were injected via the RO sinus with 0.703 MBq/100 μL [111In]InCl3, [111In][In(macropa)]+, [111In][In(py-macrodipa)]+, or [111In][In(py2-macrodipa)]+ complexes for biodistribution studies. Four animals for each complex at each time point 0.5, 2, and 24 h post-injection were euthanized by CO2 asphyxiation before tissue dissection. A blood sample was acquired, and the organs of interest were collected (heart, lungs, liver, kidneys, spleen, stomach, small intestine, large intestine, skin, muscle, fat, tibia, calvaria, and brain), rinsed of excess blood, weighed, and measured by γ counting. Data were corrected for background and decay, and the activity of each tissue was normalized to an injection standard. The organ uptake values were calculated as percent of injected activity per gram of tissue (%IA g−1).
DFT calculations
DFT calculations were carried out with the Gaussian 16 software package.81 The molecular geometries of [In(py-macrodipa)]+ and [In(py2-macrodipa)]+ (Conformation A and B) were optimized employing the ωB97XD functional,82,83 and the basis set 6-31+G(d,p) was used for the lighter elements (C, N, H, and O).84,85 For the heavier indium atom, the LANL2DZ effective core potential, as well as its associated basis set, was applied.86 In addition, frequency calculations were carried out on optimized geometries to confirm, via the absence of imaginary frequencies, that these structures are local minima on the potential energy surface. Zero-point energies and thermochemical correction factors were also afforded by the frequency calculations. The effect of the aqueous environment was considered using the polarizable continuum model (PCM) by a single-point calculation on the optimized structures.87 The initial Conformation B geometries of [In(py-macrodipa)]+ and [In(py2-macrodipa)]+ were taken from the crystal structures. The initial Conformation A geometries were taken from the previously obtained crystal structure of La3+ complexes with the metal center replaced with In3+.46,47 For all aqueous species (everything except H2O), a correction factor of 7.9 kJ mol−1 was applied to adjust the free energy values from the software state default (298 K, 1 atm) to standard state in aqueous solution (298 K, 1 M). For H2O(l), this correction factor is 17.9 kJ mol−1, accounting for its larger standard state concentration in the liquid phase of 55.5 M.
Data availability
The data supporting this article have been included as part of ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the National Institutes of Biomedical Imaging and Bioengineering of the National Institutes of Health under the award number 2R56EB029259 (J.J.W.), as well as the National Cancer Institute at the National Institutes of Health for support under award numbers R01CA240711 and R01CA229893 (D.L.J.T.). T.K. thanks the German Academic Exchange Council (DAAD) for the postdoctoral fellowship. This work made use of the Cornell University NMR facility, which is supported by the NSF under award number CHE-1531632.
References
- J. Czernin, I. Sonni, A. Razmaria and J. Calais, J. Nucl. Med., 2019, 60, 3S–12S CrossRef CAS PubMed
 .
.
- D. A. Mulford, D. A. Scheinberg and J. G. Jurcic, J. Nucl. Med., 2005, 46, 199S–204S Search PubMed .
- D. Delbeke and G. M. Segall, J. Nucl. Med., 2011, 52, 24S–28S CrossRef PubMed .
- M. Dondi, R. Kashyap, D. Paez, T. Pascual, J. Zaknun, F. M. Bastos and Y. Pynda, J. Nucl. Med., 2011, 52, 16S–23S CrossRef PubMed .
- G. Sgouros, L. Bodei, M. R. McDevitt and J. R. Nedrow, Nat. Rev. Drug Discovery, 2020, 19, 589–608 CrossRef CAS PubMed .
- G. Kramer-Marek and J. Capala, Tumor Biol., 2012, 33, 629–640 CrossRef CAS PubMed .
- J. Fallah, S. Agrawal, H. Gittleman, M. H. Fiero, S. Subramaniam, C. John, W. Chen, T. K. Ricks, G. Niu, A. Fotenos, M. Wang, K. Chiang, W. F. Pierce, D. L. Suzman, S. Tang, R. Pazdur, L. Amiri-Kordestani, A. Ibrahim and P. G. Kluetz, Clin. Cancer Res., 2023, 29, 1651–1657 CrossRef CAS PubMed .
- M. Benešová, M. Schäfer, U. Bauder-Wüst, A. Afshar-Oromieh, C. Kratochwil, W. Mier, U. Haberkorn, K. Kopka and M. Eder, J. Nucl. Med., 2015, 56, 914–920 CrossRef PubMed .
- P. G. Kluetz, W. Pierce, V. E. Maher, H. Zhang, S. Tang, P. Song, Q. Liu, M. T. Haber, E. E. Leutzinger, A. Al-Hakim, W. Chen, T. Palmby, E. Alebachew, R. Sridhara, A. Ibrahim, R. Justice and R. Pazdur, Clin. Cancer Res., 2014, 20, 9–14 CrossRef CAS PubMed .
- U. Hennrich and K. Kopka, Pharmaceuticals, 2019, 12, 114 CrossRef CAS PubMed .
- J. Nonnekens, K. L. S. Chatalic, J. D. M. Molkenboer-Kuenen, C. E. M. T. Beerens, F. Bruchertseifer, A. Morgenstern, J. Veldhoven-Zweistra, M. Schottelius, H.-J. Wester, D. C. van Gent, W. M. van Weerden, O. C. Boerman, M. de Jong and S. Heskamp, Cancer Biother. Radiopharm., 2017, 32, 67–73 CrossRef CAS PubMed .
- M. S. Kaminski, K. R. Zasadny, I. R. Francis, A. W. Milik, C. W. Ross, S. D. Moon, S. M. Crawford, J. M. Burgess, N. A. Petry, G. M. Butchko, S. D. Glenn and R. L. Wahl, N. Engl. J. Med., 1993, 329, 459–465 CrossRef CAS PubMed .
- C. T. Sawin and D. V. Becker, Thyroid, 1997, 7, 163–176 CrossRef CAS PubMed .
- T. E. Witzig, L. I. Gordon, F. Cabanillas, M. S. Czuczman, C. Emmanouilides, R. Joyce, B. L. Pohlman, N. L. Bartlett, G. A. Wiseman, N. Padre, A. J. Grillo-López, P. Multani and C. A. White, J. Clin. Oncol., 2002, 20, 2453–2463 CrossRef CAS PubMed .
- A. Imhof, P. Brunner, N. Marincek, M. Briel, C. Schindler, H. Rasch, H. R. Mäcke, C. Rochlitz, J. Müller-Brand and M. A. Walter, J. Clin. Oncol., 2011, 29, 2416–2423 CrossRef CAS PubMed .
- D. J. Kwekkeboom, J. J. Teunissen, W. H. Bakker, P. P. Kooij, W. W. de Herder, R. A. Feelders, C. H. van Eijck, J.-P. Esser, B. L. Kam and E. P. Krenning, J. Clin. Oncol., 2005, 23, 2754–2762 CrossRef CAS PubMed .
- J. R. Strosberg, M. E. Caplin, P. L. Kunz, P. B. Ruszniewski, L. Bodei, A. Hendifar, E. Mittra, E. M. Wolin, J. C. Yao, M. E. Pavel, E. Grande, E. Van Cutsem, E. Seregni, H. Duarte, G. Gericke, A. Bartalotta, M. F. Mariani, A. Demange, S. Mutevelic and E. P. Krenning, Lancet Oncol., 2021, 22, 1752–1763 CrossRef CAS PubMed .
- O. Sartor, J. de Bono, K. N. Chi, K. Fizazi, K. Herrmann, K. Rahbar, S. T. Tagawa, L. T. Nordquist, N. Vaishampayan, G. El-Haddad, C. H. Park, T. M. Beer, A. Armour, W. J. Pérez-Contreras, M. DeSilvio, E. Kpamegan, G. Gericke, R. A. Messmann, M. J. Morris and B. J. Krause, N. Engl. J. Med., 2021, 385, 1091–1103 CrossRef CAS PubMed .
- V. Radchenko, A. Morgenstern, A. R. Jalilian, C. F. Ramogida, C. Cutler, C. Duchemin, C. Hoehr, F. Haddad, F. Bruchertseifer, H. Gausemel, H. Yang, J. A. Osso, K. Washiyama, K. Czerwinski, K. Leufgen, M. Pruszyński, O. Valzdorf, P. Causey, P. Schaffer, R. Perron, S. Maxim, D. S. Wilbur, T. Stora and Y. Li, J. Nucl. Med., 2021, 62, 1495–1503 CrossRef CAS PubMed .
- Y.-S. Kim and M. W. Brechbiel, Tumor Biol., 2012, 33, 573–590 CrossRef CAS PubMed .
- F. D. C. Guerra Liberal, J. M. O'Sullivan, S. J. McMahon and K. M. Prise, Cancer Biother. Radiopharm., 2020, 35, 404–417 CrossRef PubMed .
- C. F. Ramogida and C. Orvig, Chem. Commun., 2013, 49, 4720–4739 RSC .
- M. W. Brechbiel, Dalton Trans., 2007, 43, 4918–4928 RSC .
- S. N. M. Chilla, C. Henoumont, L. V. Elst, R. N. Muller and S. Laurent, Isr. J. Chem., 2017, 57, 800–808 CrossRef CAS .
- G. J. Stasiuk and N. J. Long, Chem. Commun., 2013, 49, 2732–2746 RSC .
- R. E. Mewis and S. J. Archibald, Coord. Chem. Rev., 2010, 254, 1686–1712 CrossRef CAS .
- Z. Baranyai, G. Tircsó and F. Rösch, Eur. J. Inorg. Chem., 2020, 2020, 36–56 CrossRef CAS .
- S. L. Wu and W. D. Horrocks Jr, J. Chem. Soc., Dalton Trans., 1997, 9, 1497–1502 RSC .
- H. Yang, J. J. Wilson, C. Orvig, Y. Li, D. S. Wilbur, C. F. Ramogida, V. Radchenko and P. Schaffer, J. Nucl. Med., 2022, 63, 5–13 CrossRef CAS PubMed .
- A. Roca-Sabio, M. Mato-Iglesias, D. Esteban-Gómez, É. Tóth, A. de Blas, C. Platas-Iglesias and T. Rodríguez-Blas, J. Am. Chem. Soc., 2009, 131, 3331–3341 CrossRef CAS PubMed .
- E. Aluicio-Sarduy, N. A. Thiele, K. E. Martin, B. A. Vaughn, J. Devaraj, A. P. Olson, T. E. Barnhart, J. J. Wilson, E. Boros and J. W. Engle, Chem. – Eur. J., 2020, 26, 1238–1242 CrossRef CAS PubMed .
- N. A. Thiele, V. Brown, J. M. Kelly, A. Amor-Coarasa, U. Jermilova, S. N. MacMillan, A. Nikolopoulou, S. Ponnala, C. F. Ramogida, A. K. H. Robertson, C. Rodríguez-Rodríguez, P. Schaffer, C. Williams Jr, J. W. Babich, V. Radchenko and J. J. Wilson, Angew. Chem., Int. Ed., 2017, 56, 14712–14717 CrossRef CAS PubMed .
- A. P. King, N. T. Gutsche, N. Raju, S. Fayn, K. E. Baidoo, M. M. Bell, C. S. Olkowski, R. E. Swenson, F. I. Lin, S. M. Sadowski, S. S. Adler, N. A. Thiele, J. J. Wilson, P. L. Choyke and F. E. Escorcia, J. Nucl. Med., 2023, 64, 549–554 CrossRef CAS PubMed .
- J. M. Kelly, A. Amor-Coarasa, S. Ponnala, A. Nikolopoulou, C. Williams, N. A. Thiele, D. Schlyer, J. J. Wilson, S. G. DiMagno and J. W. Babich, J. Nucl. Med., 2019, 60, 649–655 CrossRef CAS PubMed .
- K. N. Bobba, A. P. Bidkar, N. Meher, C. Fong, A. Wadhwa, S. Dhrona, A. Sorlin, S. Bidlingmaier, B. Shuere, J. He, D. M. Wilson, B. Liu, Y. Seo, H. F. VanBrocklin and R. R. Flavell, J. Nucl. Med., 2023, 64, 1076–1082 CrossRef CAS PubMed .
- K. N. Bobba, A. P. Bidkar, A. Wadhwa, N. Meher, S. Drona, A. M. Sorlin, S. Bidlingmaier, L. Zhang, D. M. Wilson, E. Chan, N. Y. Greenland, R. Aggarwal, H. F. VanBrocklin, J. He, J. Chou, Y. Seo, B. Liu and R. R. Flavell, Theranostics, 2024, 14, 1344–1360 CrossRef CAS PubMed .
- M. K. Blei, L. Waurick, F. Reissig, K. Kopka, T. Stumpf, B. Drobot, J. Kretzschmar and C. Mamat, Inorg. Chem., 2023, 62, 20699–20709 CrossRef CAS PubMed .
- B. J. B. Nelson, S. Ferguson, M. Wuest, J. Wilson, M. J. M. Duke, S. Richter, H. Soenke-Jans, J. D. Andersson, F. Juengling and F. Wuest, J. Nucl. Med., 2022, 63, 584–590 CrossRef CAS PubMed .
- D. S. Abou, N. A. Thiele, N. T. Gutsche, A. Villmer, H. Zhang, J. J. Woods, K. E. Baidoo, F. E. Escorcia, J. J. Wilson and D. L. J. Thorek, Chem. Sci., 2021, 12, 3733–3742 RSC .
- B. J. B. Nelson, J. D. Andersson and F. Wuest, Nucl. Med. Biol., 2022, 110–111, 59–66 CrossRef CAS PubMed .
- F. Reissig, D. Bauer, M. Ullrich, M. Kreller, J. Pietzsch, C. Mamat, K. Kopka, H.-J. Pietzsch and M. Walther, Pharmaceuticals, 2020, 13, 272 CrossRef CAS PubMed .
- D. Abou, N. Thiele, A. Villmer, N. Gustche, F. Escorcia, J. Wilson and D. Thorek, J. Nucl. Med., 2020, 61, 587–587 Search PubMed .
- D. J. Fiszbein, V. Brown, N. A. Thiele, J. J. Woods, L. Wharton, S. N. MacMillan, V. Radchenko, C. F. Ramogida and J. J. Wilson, Inorg. Chem., 2021, 60, 9199–9211 CrossRef CAS PubMed .
- A. Hu and J. J. Wilson, Acc. Chem. Res., 2022, 55, 904–915 CrossRef CAS PubMed .
- A. Hu, S. N. MacMillan and J. J. Wilson, J. Am. Chem. Soc., 2020, 142, 13500–13506 CrossRef CAS PubMed .
- A. Hu, E. Aluicio-Sarduy, V. Brown, S. N. MacMillan, K. V. Becker, T. E. Barnhart, V. Radchenko, C. F. Ramogida, J. W. Engle and J. J. Wilson, J. Am. Chem. Soc., 2021, 143, 10429–10440 CrossRef CAS PubMed .
- A. Hu, M. E. Simms, V. Kertesz, J. J. Wilson and N. A. Thiele, Inorg. Chem., 2022, 61, 12847–12855 CrossRef CAS PubMed .
- A. Hu, K. E. Martin, D. Śmiłowicz, E. Aluicio-Sarduy, S. J. Cingoranelli, S. E. Lapi, J. W. Engle, E. Boros and J. J. Wilson, Eur. J. Inorg. Chem., 2023, 26, e202300457 CrossRef CAS PubMed .
- J. Sörensen, D. Sandberg, M. Sandström, A. Wennborg, J. Feldwisch, V. Tolmachev, G. Åström, M. Lubberink, U. Garske-Román, J. Carlsson and H. Lindman, J. Nucl. Med., 2014, 55, 730–735 CrossRef PubMed .
- D. Rosik, A. Orlova, J. Malmberg, M. Altai, Z. Varasteh, M. Sandström, A. E. Karlström and V. Tolmachev, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39, 693–702 CrossRef CAS PubMed .
- D. Wild, M. Fani, M. Behe, I. Brink, J. E. F. Rivier, J. C. Reubi, H. R. Maecke and W. A. Weber, J. Nucl. Med., 2011, 52, 1412–1417 CrossRef CAS PubMed .
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed .
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef .
- L. A. Kloo and M. J. Taylor, J. Chem. Soc., Dalton Trans., 1997, 15, 2693–2696 RSC .
- B. F. T. Cooper, C. G. Andrews and C. L. B. Macdonald, J. Organomet. Chem., 2007, 692, 2843–2848 CrossRef CAS .
- D. G. Samsonenko, M. N. Sokolov, A. V. Virovets, N. V. Pervukhina and V. P. Fedin, Eur. J. Inorg. Chem., 2001, 2001, 167–172 CrossRef .
- L. Zhang, F. Wang, X. Zhang, F. Liang, Z. Hu and Y. Wu, Cryst. Growth Des., 2024, 24, 627–631 CrossRef CAS .
- R. L. Davidovich, A. V. Gerasimenko, E. I. Voit and V. B. Logvinova, J. Fluorine Chem., 2019, 226, 109343 CrossRef CAS .
- E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1991, 190, 37–46 CrossRef CAS .
- C. F. G. C. Geraldes, R. Delgado, A. M. Urbano, J. Costa, F. Jasanada and F. Nepveu, J. Chem. Soc., Dalton Trans., 1995, 13, 327–335 RSC .
- W. P. Cacheris, S. K. Nickle and A. D. Sherry, Inorg. Chem., 1987, 26, 958–960 CrossRef CAS .
- T. S. Grimes and K. L. Nash, J. Solution Chem., 2014, 43, 298–313 CrossRef CAS .
- M. Pniok, V. Kubíček, J. Havlíčková, J. Kotek, A. Sabatie-Gogová, J. Plutnar, S. Huclier-Markai and P. Hermann, Chem. – Eur. J., 2014, 20, 7944–7955 CrossRef CAS PubMed .
- S. Liu, J. Pietryka, C. E. Ellars and D. S. Edwards, Bioconjugate Chem., 2002, 13, 902–913 CrossRef CAS PubMed .
- R. A. Fawwaz, T. S. T. Wang, A. Estabrook, J. M. Rosen, M. A. Hardy, P. O. Alderson, S. C. Srivastava, P. Richards and S. Ferrone, J. Nucl. Med., 1985, 26, 488–492 CAS .
- P. Cieslik, M. Kubeil, K. Zarschler, M. Ullrich, F. Brandt, K. Anger, H. Wadepohl, K. Kopka, M. Bachmann, J. Pietzsch, H. Stephan and P. Comba, J. Am. Chem. Soc., 2022, 144, 21555–21567 CrossRef CAS PubMed .
- I. Kopp, P. Cieslik, K. Anger, T. Josephy, L. Neupert, G. Velmurugan, M. Gast, H. Wadepohl, S. A. Brühlmann, M. Walther, K. Kopka, M. Bachmann, H. Stephan, M. Kubeil and P. Comba, Inorg. Chem., 2023, 62, 20754–20768 CrossRef CAS PubMed .
- L. Wharton, M. D. G. Jaraquemada-Peláez, C. Zhang, J. Zeisler, C. Rodríguez-Rodríguez, M. Osooly, V. Radchenko, H. Yang, K.-S. Lin, F. Bénard, P. Schaffer and C. Orvig, Bioconjugate Chem., 2022, 33, 1900–1921 CrossRef CAS PubMed .
- L. Wharton, C. Zhang, J. Zeisler, C. Rodríguez-Rodríguez, M. Osooly, V. Radchenko, H. Yang, K.-S. Lin, F. Bénard, P. Schaffer and C. Orvig, Bioconjugate Chem., 2022, 33, 2381–2397 CrossRef CAS PubMed .
- L. Wharton, H. Yang, M. D. G. Jaraquemada-Peláez, H. Merkens, G. Engudar, A. Ingham, H. Koniar, V. Radchenko, P. Kunz, P. Schaffer, F. Bénard and C. Orvig, J. Med. Chem., 2023, 66, 13705–13730 CrossRef CAS PubMed .
- L. Wharton, C. Zhang, H. Yang, J. Zeisler, V. Radchenko, C. Rodríguez-Rodríguez, M. Osooly, B. O. Patrick, K.-S. Lin, F. Bénard, P. Schaffer and C. Orvig, Bioconjugate Chem., 2022, 33, 505–522 CrossRef CAS PubMed .
- D. Fiaccabrino, T. Masvikeni, M. D. G. Jaraquemada-Peláez, C. Orvig and P. Schaffer, Inorg. Chem., 2024, 63, 13911–13923 CrossRef CAS PubMed .
- M. E. Simms, Z. Li, M. M. Sibley, A. S. Ivanov, C. M. Lara, T. C. Johnstone, V. Kertesz, A. Fears, F. D. White, D. L. J. Thorek and N. A. Thiele, Chem. Sci., 2024, 15, 11279–11286 RSC .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed .
- P. Müller, Crystallogr. Rev., 2009, 15, 57–83 CrossRef .
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS .
- P. Gans, A. Sabatini and A. Vacca, Ann. Chim., 1999, 89, 45–49 CAS .
- B. L. McNeil, K. J. Kadassery, A. W. McDonagh, W. Zhou, P. Schaffer, J. J. Wilson and C. F. Ramogida, Inorg. Chem., 2022, 61, 9638–9649 CrossRef CAS PubMed .
- C. Müller, H. Struthers, C. Winiger, K. Zhernosekov and R. Schibli, J. Nucl. Med., 2013, 54, 124–131 CrossRef PubMed .
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01 2016 Search PubMed .
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed .
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC .
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS .
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS .
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS .
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data, titration curves, radio-HPLC and radio-TLC chromatograms, biodistribution data, and X-ray diffraction data. CCDC 2373568 and 2373569. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02146k |
| ‡ Present Address: Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, California, 93117, USA. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Mou
Chakraborty
bc,
Aohan
Hu
a,
Thines
Kanagasundaram
a,
Daniel L. J.
Thorek
bcd and
Justin J.
Wilson‡
a,
Mou
Chakraborty
bc,
Aohan
Hu
a,
Thines
Kanagasundaram
a,
Daniel L. J.
Thorek
bcd and
Justin J.
Wilson‡