
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-intercalation strategy for simultaneously boosting two-electron conversion and bulk stabilization of Mn-based cathodes in aqueous zinc-ion batteries†
Xuan
Gao‡
a,
Chen
Shen‡
 b,
Haobo
Dong‡
c,
Yuhang
Dai‡
a,
Peie
Jiang
c,
Ivan P.
Parkin
a,
Hongbin
Zhang
b,
Claire J.
Carmalt
*a and
Guanjie
He
*a
b,
Haobo
Dong‡
c,
Yuhang
Dai‡
a,
Peie
Jiang
c,
Ivan P.
Parkin
a,
Hongbin
Zhang
b,
Claire J.
Carmalt
*a and
Guanjie
He
*a
aChristopher Ingold Laboratory, Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: c.j.carmalt@ucl.ac.uk; g.he@ucl.ac.uk
bInstitute of Materials Science, Technische Universität Darmstadt, Darmstadt, 64287, Germany
cDepartment of Chemical Engineering, University College London, London WC1E 7JE, UK
First published on 16th February 2024
Abstract
Zinc-ion batteries (ZIBs) have emerged as a promising candidate due to the abundance, low cost and high energy density. However, the performance of ZIBs needs to be improved to meet the practical requirements of energy storage systems. This work probes the efficacy of co-intercalation as a strategy for enhancing the electrochemical performance of ZIBs through the fabrication of sodium and copper co-intercalated birnessite manganese oxide (NCMO) cathodes. The results show that the co-intercalation of sodium and copper ions catalyzes the activation of copper cations on the surface Mn2+/Mn4+ redox pair, leading to improved specific capacity and cycling stability. The NCMO cathode exhibits a remarkable specific capacity of 576 mA h g−1 after 100 cycles at a low loading of around ∼1 mg cm−2 and a high areal capacity of 2.10 mA h cm−2 at a high loading of ∼10.9 mg cm−2. The mechanism of copper ions to promote the manganese-based cathode conversion reaction is thoroughly investigated. These findings suggest that developing catalytic effects is a promising approach for developing high-performance cathode materials for ZIBs with practical and efficient energy storage systems.
Broader contextIn a significant advancement toward optimizing the electrochemical performance of zinc-ion batteries (ZIBs), our research delves into the innovative strategy of co-intercalation employing sodium and copper. Capitalizing on the abundance, cost-effectiveness, and high energy density of zinc, ZIBs emerge as promising solutions for practical energy storage systems. Despite this potential, enhancing their performance remains a critical pursuit. Our work focuses on the economical fabrication of sodium and copper co-intercalated birnessite manganese oxide (NCMO) cathodes, marking a remarkable breakthrough. By catalyzing the activation of copper cations on the surface Mn2+/Mn4+ redox pair, our findings unveil a substantial improvement in specific capacity and cycling stability. Noteworthy is the NCMO cathode's ultra-high specific capacity of 576 mA h g−1 after 100 cycles, and reaching a high areal capacity of 2.10 mA h cm−2 at a very high cathode mass loading of ∼10.9 mg cm−2. This innovation not only demonstrates an unprecedented high capacity but also highlights the benefits of separately considering surface and bulk phase enhancement strategies, ensuring both high capacity and excellent cycling stability. Extending beyond materials science, our research captures the interest of experts in environmental sustainability, policy development, and energy economics. The meticulously investigated mechanism of copper ions in promoting the manganese-based cathode conversion reaction and enhancing host material stability opens new avenues for high-performance cathode materials. The cost-effective synthesis method and high mass loading capacity presented in our work can significantly reduce the cost of ZIBs, promoting their scale-up and industrialization. Our findings underscore the potential of combining catalytic effects and stable host materials in ZIBs as a promising approach, paving the way for practical and efficient high-mass loading cathodes in energy storage systems within the evolving landscape of ZIBs technology. |
Introduction
The growing demand for efficient and sustainable energy storage solutions has led to the emergence of several energy storage technologies.1 As the world transitions towards a low-carbon economy, there is an urgent need for energy storage systems that can store large amounts of renewable energy and provide reliable and stable power supplies.2 One promising technology for large-scale energy storage applications is Zn-ion batteries (ZIBs).3 ZIBs offer several advantages over traditional battery chemistries, including low cost, high energy density, and excellent safety.4 Moreover, Zn is abundant, widely available, and easily recyclable, making ZIBs an attractive alternative to conventional batteries.5 However, the performance and stability of ZIBs need to be improved for widespread adoption in grid-scale energy storage and other applications.6 The cathode material plays a critical role in determining the capacity, stability, and cycling performance of ZIBs. Manganese dioxide (MnO2) has several advantages as a cathode material for ZIBs. Firstly, MnO2 is abundant and inexpensive.7 Secondly, MnO2 has a high theoretical capacity (308 mA h g−1). Thirdly, MnO2-based cathodes have shown excellent cycling stability, high output plateaus (1.5 V versus Zn/Zn2+) and high-rate capability, which are critical for practical applications of ZIBs.8 It has been demonstrated that various atomic structures of manganese oxides are suitable for ZIBs, such as α- (2 × 2 tunnel), β- (1 × 1 tunnel), γ- (1 × 2 and 1 × 1 tunnel), λ- (spinel) and δ- (layered structure).9 Despite the fact that the tunnel structures offer open paths as well as an abundance of active sites for Zn2+ diffusion and storage, it is always constrained by a significant collapse during the process of discharge. In comparison to other crystalline structures, layered manganese oxide features spacing channels of ∼7 Å, which makes it more suitable for the rapid and reversible intercalation and extraction of Zn ions.10 However, prior investigations have shown that the phase transition of the layered structure of δ-MnO2 during cycling may result in significant changes in volumes as well as structural collapse, which has a significant negative impact on the cycling performance. Moreover, the Jahn–Teller (JT) effect, which arises from the degeneracy of electronic energy levels in [MnO6] octahedra, can lead to the distortion and dissolution of the Mn-based active materials and decrease the cycling stability of the battery.11 On the other hand, the in situ generated zinc hydroxide sulfate (ZHS) on the battery surface is poorly reversible.12 Although it provides electron transfer during cycling, it severely increases the battery polarization and gradually deactivates the cathode active materials.Researchers have contributed much effort to address these challenges, such as material combinations, ion intercalation, electrolyte additives, etc.13 For the research on manganese-based cathode materials, it is possible to obtain stable Zn2+ storage by a process of ion pre-intercalation, which has been used in prior studies with ions such as proton (H+), lithium ion (Li+), sodium ion (Na+), potassium ion (K+), zinc ion (Zn2+), copper ion (Cu2+), and ammonium ion (NH4+). The concept of ion pre-intercalation involves the ion insertion into the host materials before the battery operation.14 This process increases the interplanar spacing of the host materials, creating more space for ion intercalation and improving the electrochemical performances of the materials. As for MnO2-based cathodes, the Na+ pre-intercalation increases the interplanar spacing, leading to improved capacity, and cycling stability. When the ZnSO4 electrolyte gradually replaces the traditional alkaline electrolyte, it can significantly suppress the parasitic reactions and inhibit the formation of passivation by-products, thereby endowing MnO2 with a reversible theoretical specific capacity of 308 mA h g−1 due to the redox pair of Mn4+/Mn3+.15 The single-electron transfer mechanism places a limit on the specific capacity of the battery, despite the fact that it is possible to considerably increase the lifespan of the battery in the mild ZnSO4 electrolyte.16 Because of this, lowering the pH of the electrolyte and raising the charge voltage may result in MnO2 delivering a theoretical capacity of 616 mA h g−1 due to the activation of the Mn4+/Mn2+ redox pair. Despite the fact that MnO2 often demonstrates poor durability in one-electron Mn4+/Mn3+ redox reactions due to the fact that its [MnO6] octahedral close-packed structure is not favorable to the Zn2+ insertion/extraction process, two-electron Mn4+/Mn2+ redox processes have been widely explored under certain conditions.17 For instance, a reversible transition between Mn2+ and MnO2 was demonstrated in MnSO4 electrolytes that were supplemented with 0.1–3.0 M H2SO4.18 This was possible due to the fact that the strongly acidic electrolyte promoted the formation of Mn vacancies, which endowed protons and electrons with fast kinetics and maximized the MnO2 electrolysis process. Strongly acidic electrolytes may be beneficial for the deep reduction of MnO2. However, when ZnSO4 is added to the electrolyte, the whole process is slightly different, and the formation of ZHS also promotes the transformation between Mn2+/Mn4+ redox pair.19 From an industrial point of view, electrolytes that are too acidic do not have industrial significance for use in large-scale energy storage systems owing to the risk of equipment corrosion and other safety problems. Therefore, it is important to construct Zn//MnO2 batteries with a two-electron transfer mechanism under moderate conditions.
Some strategies for boosting Mn2+/Mn4+ two-electron transfer in mild pH environment have been proposed. Liang et al. proposed ion concentration/dilution strategy in the electrolyte for Mn2+/Mn4+ two-electron transfer, which can achieve high specific capacities of 460 mA h g−1.20 Acetate has also been shown to facilitate two-electron transfer for MnO2 to achieve a specific capacity of 556 mA h g−1.16 However, since electrolyte modification is more about surface reactions, most high-capacity or stable cycles are completed under thin electrode conditions, with lower mass loading, usually less than 1 mg cm−2. How to simultaneously stimulate surface Mn2+/Mn4+ two-electron transfer and maintain host material stability is a challenge, especially in the case of high mass loading. Recently, MnO2 intercalated with Cu2+ has been shown to enhance the Mn2+/Mn4+ redox reaction of cathodes in ZIBs, but its cycling stability is still limited, especially in high mass loading.21 It is an interesting topic on how Cu2+ promotes the Mn deposition and dissolution, which is related to several aspects: the JT effect of host materials, the reversibility of by-product ZHS and others.21 Therefore, a better understanding of the interplay between ion pre-intercalation and in situ generation and dissolution of ZHS on the surface of the active materials is needed to improve the performance and stability of Mn-based cathodes in ZIBs.
In this work, we have explored the applicability of co-intercalation as an effective strategy by fabricating sodium (Na) and copper (Cu) ions co-intercalated δ-MnO2 (NCMO) cathode for addressing the key Mn-based cathodes challenges of stable cycling with a high specific capacity at high mass loading in ZIBs, as illustrated in Scheme 1. The activation effect of Cu2+ for the conversion between Mn2+/Mn4+ redox pair is amplified by the pre-intercalation of Na+, leading to enhanced specific capacity and cycling stability. The smaller ionic radius of Cu2+ facilitates faster diffusion into the electrolyte, completing the redox reaction in Cu(0)/Cu2+ during the charge/discharge. The Cu2+ generated during the charge process can accelerate the JT distortion of the mesophase ZnxMnO(OH)y, thereby accelerating the dissolution of ZnxMnO(OH)y on the surface. The co-intercalation of Na+ and Cu2+, combined with the deposition and dissolution of ZnxMnO(OH)y, and ZHS, enables the active materials to exhibit a prolonged continuous capacity increasing process at low mass loadings and a highly reversible Mn deposition and dissolution process at high mass loadings, which is not only due to the dual-ion co-intercalation that amplifies the conversion of Mn on cathode surface, but also beneficial from the stability of the host material during charge and discharge. The NCMO cathode exhibits an intriguing continuous activation process at a low loading of around ∼1 mg cm−2, exhibiting a remarkable specific capacity of 576 mA h g−1 after 100 cycles at a current density of 0.5 A g−1. More importantly, the full cell with the prepared NCMO cathode demonstrates a high areal capacity of 2.10 mA h cm−2 at high mass loading of ∼10.9 mg cm−2 at a current density of 1.09 mA cm−2. These results demonstrate the promising potential of Mn-based cathodes with co-intercalated Na+ and Cu2+ for practical and efficient energy storage systems. Moreover, the mechanism of how Cu2+ promote the deposition and dissolution of manganese-based cathodes during cycling is discussed.
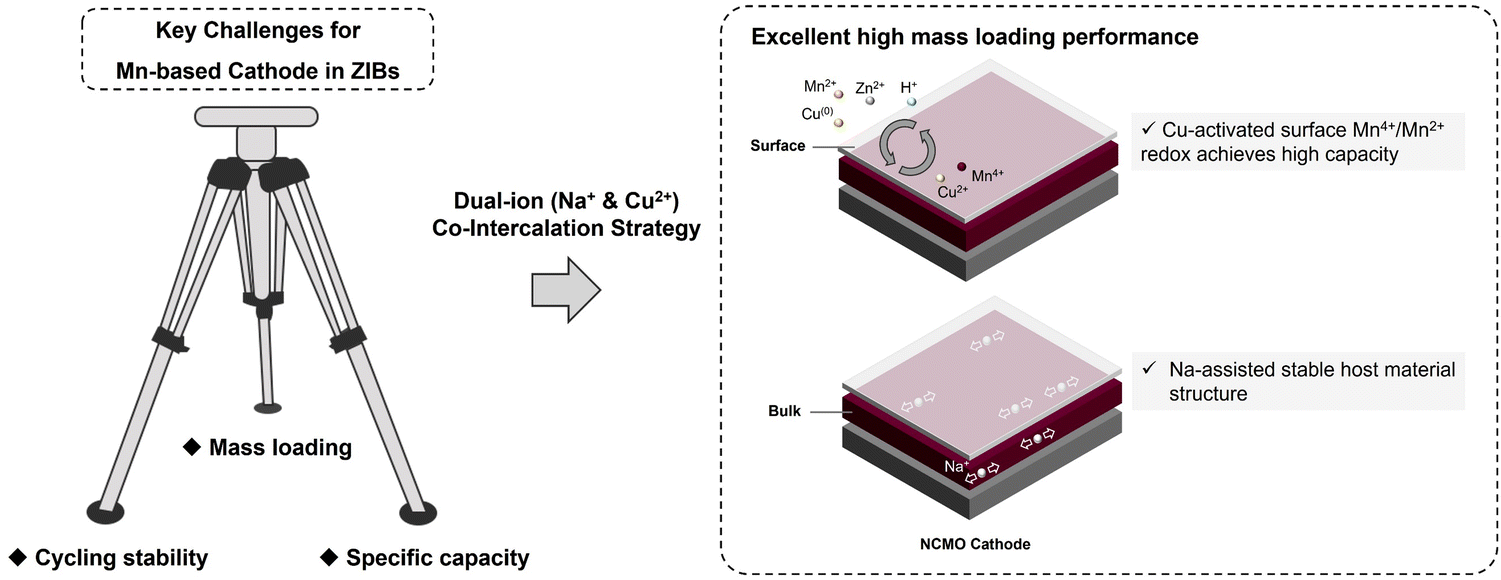 | ||
| Scheme 1 Schematic representation of the manganese oxide cathode based on a dual-ion co-intercalation strategy that simultaneously boosts surface Mn2+/Mn4+ two-electron transfer and maintains the stability of the host material structure to cope with the key challenges of mass loading, cycling stability, and specific capacity in Mn-based cathodes. | ||
Results and discussion
To understand the stacking-selective self-intercalation of δ-MnO2, we performed density functional theory (DFT) calculations. We adopted the δ-MnO2 phase with antiferromagnetic ordering as the host structure.22 As shown in Fig. 1(a), two different structure models of the pristine δ-MnO2 with the intercalation of pure elements (Na, and Cu) and binary elements (NaCu) were considered. The δ-MnO2 phase has the space-group P63/mmc (194) with a hexagonal lattice (see Fig. 1(a)). The relaxed lattice constants from our geometrical optimization are a = 2.925 Å and c = 11.427 Å, which agree with previously calculated results.22 The band structure of pristine δ-MnO2 is displayed in Fig. 1(b), indicating that δ-MnO2 is a semiconductor with a band gap of 1.70 eV. After considering the intercalation, the band gap in δ-MnO2 disappears in the condition of single element (Na or Cu) intercalation, driven by net charges introduced by the intercalants. As seen in Fig. 1(c) and (d), one or two flat bands cross the Fermi energy, suggesting that δ-MnO2 with the intercalation of one element exhibits higher electronic conductivity in comparison to the pristine δ-MnO2, which will facilitate improved electron-transfer capability. For binary (Na and Cu) cation intercalations, even more bands are crossing the Fermi energy, which act as effective charge conduction channels, as shown in Fig. 1(e). The DFT results demonstrate well that the co-intercalation of Na and Cu synergistically modulates the electronic structure of δ-MnO2, which are expected to enable rapid electron transfer processes.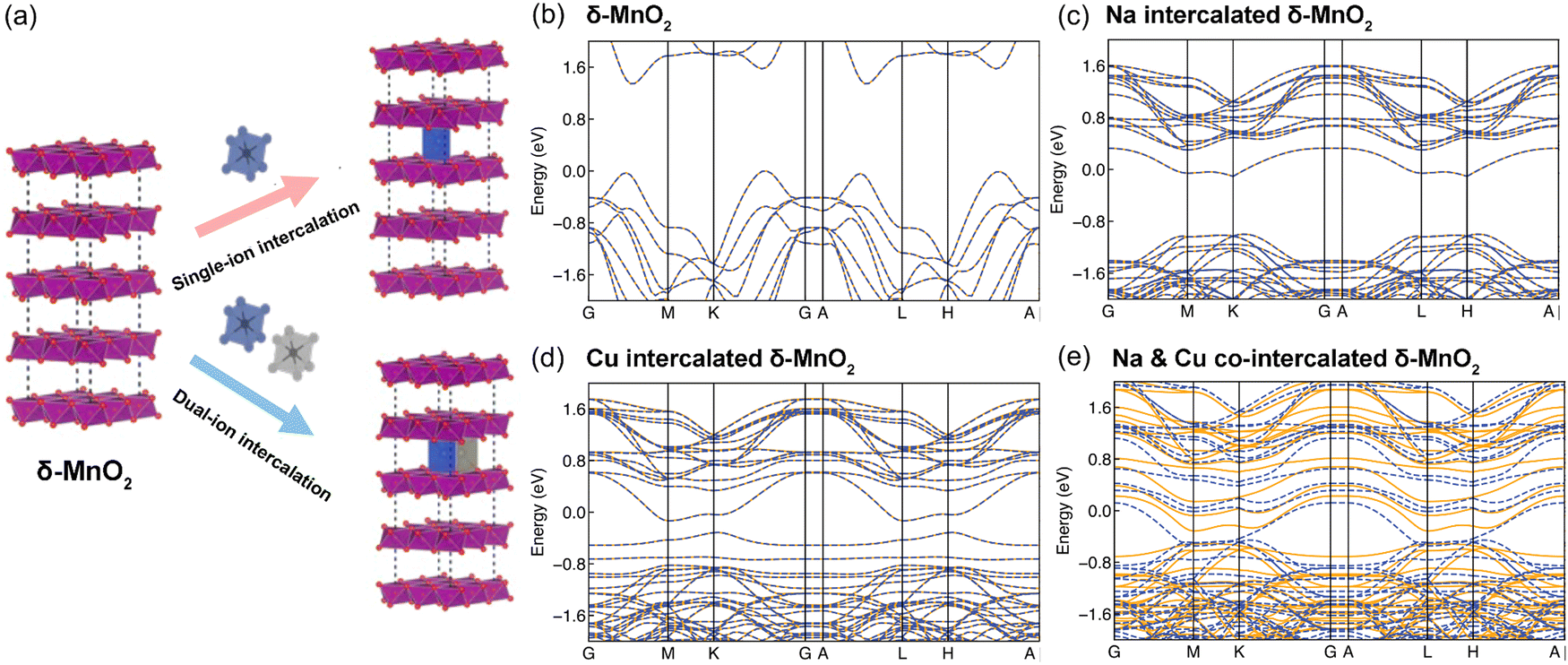 | ||
| Fig. 1 (a) Ion intercalation strategy for high performance δ-MnO2 cathode materials and band structures of (b) δ-MnO2, (c) Na intercalated δ-MnO2, (d) Cu intercalated δ-MnO2 and (e) Na & Cu co-intercalated δ-MnO2 based on DFT modelling. | ||
Based on the advantages of the dual-ion intercalation of cathode materials, a co-intercalation strategy of Na+ and Cu2+ was introduced into δ-MnO2. The morphology of the synthesized NCMO was confirmed by scanning electron microscope (SEM). As shown in Fig. 2(a), NCMO presents a uniform flake shape with a diameter of ∼1–1.5 μm. It can be seen from Fig. 2(b) of the further enlarged SEM image that the thickness of the NCMO flakes was ∼50 nm, which provides a large specific surface area. Fig. 2(c)–(e) show the scanning transmission electron microscopy images to further confirm the crystal structures. The measured interlayer distance between the Na+ pillars and the crystal water were 3.53 Å (see Fig. 2(d)), which is slightly larger than the half value of the (001) crystal plane, indicating easy ion intercalation. Fig. 2(e) shows the top-view structure of the characteristic MnO6 octahedron, which verifies the layered structure. The d-spacing of 2.22 Å corresponds to the (201) lattice plane. Fig. 2(f) illustrates the energy-dispersive X-ray spectroscopy (EDS) mapping diagram, which was used to examine the distribution of each element. Fig. 2(g)–(j) illustrate the distribution of the elements Na, Cu, Mn, and O, respectively. It is clear that both the Na and Cu elements were distributed evenly throughout the structure.
 | ||
| Fig. 2 (a) Low-, (b) high-resolution SEM images of the morphology of the NCMO. (c) low-, (d) high-resolution and (e) atomic resolution STEM images for surface structures of NCMO. (f)–(j) EDS mapping images of NCMO show that Na, Cu, Mn, and O elements are uniformly distributed. | ||
X-ray diffraction (XRD) patterns of NCMO are shown in Fig. 3(a). The 2θ peaks at 5.893°, 11.751°, 15.250°, 16.481°, 27.734° correspond to (001), (002), (200), (111), (001), respectively.9 The pattern exhibited in Fig. 3(a) coincides well with Birnessite (JCPDS no. 43-1456), where a = 5.175(3) Å, b = 2.849(1) Å, c = 7.338(5) Å, α = γ = 90° and β = 103.19(4)°, V = 105.3(6) Å3.23 This is the same as Na-intercalated manganese oxide reported in the previous work, showing that the addition of Cu has no effect on the crystal structures.24 X-ray photoelectron spectroscopy (XPS) was utilized to investigate the atomic ratio and average oxidation state (AOS) of each element on the surface of the material. The relatively high content of O element is due to the surface oxygen absorption. Mn 2p has two separate peaks with a binding energy difference of 11.8 eV, whereas the difference is 5.28 eV for the Mn 3s spectrum. Based on the binding energy difference of the Mn 3s peak, the AOS of Mn was calculated to be 3.01 according to the formula AOS = 8.956–1.126ΔEs.24 Between the two peaks of Mn 2p, a peak of Cu LM3 was observed due to Cu intercalation.25 Meanwhile, the Cu 2p peak in Fig. 3(d) also shows the successful intercalation of Cu. Na was observed as shown in Fig. 3(e) (Na 1s) with a characteristic peak at 1071 eV. In terms of O 1s (Fig. 3(f)), the spectrum was convoluted from three corresponding peaks which are assigned to adsorbed water H–O–H (532.5 eV), surface-adsorbed oxygen Mn–OH (531.2 eV), and lattice oxygen species Mn–O–Mn (529.6 eV), respectively. The macroscopic 3D morphology of the fabricated NCMO electrode was examined using X-ray micro-Computed Tomography (CT), which depicts a homogeneous distribution of NCMO nano particles supported on current collector in 3D space. After extracting the active material phase, carbon fiber phase and pore phase according to the grey scale level using thresholding segmentation, the skeleton of the active materials can be visualized in Fig. S1 (ESI†), from which a complex 3D structure and broad distribution of the pore size are observed. The tortuosity and flux density on three dimensions of segmented figures are simulated based on the reconstructed segmented structures. The porosity ε was measured to be 0.72 and the tortuosity factor was computed to be 1.8–1.9 on X (Fig. S2, ESI†) direction, while the tortuosity was 1.3–1.5 at Y (Fig. S3, ESI†) and Z (Fig. 3(g) and Fig. S4, ESI†) directions, giving an effective diffusion coefficient of 0.40–0.55, as shown in Fig. 3(h), indicating that the mass transfer capabilities in the three directions are within a similar range. The effective diffusion coefficient of NCMO is much higher than previously reported work of ZIBs and lithium-ion batteries (LIBs), which represents a much better reactant transport capabilities.5,26
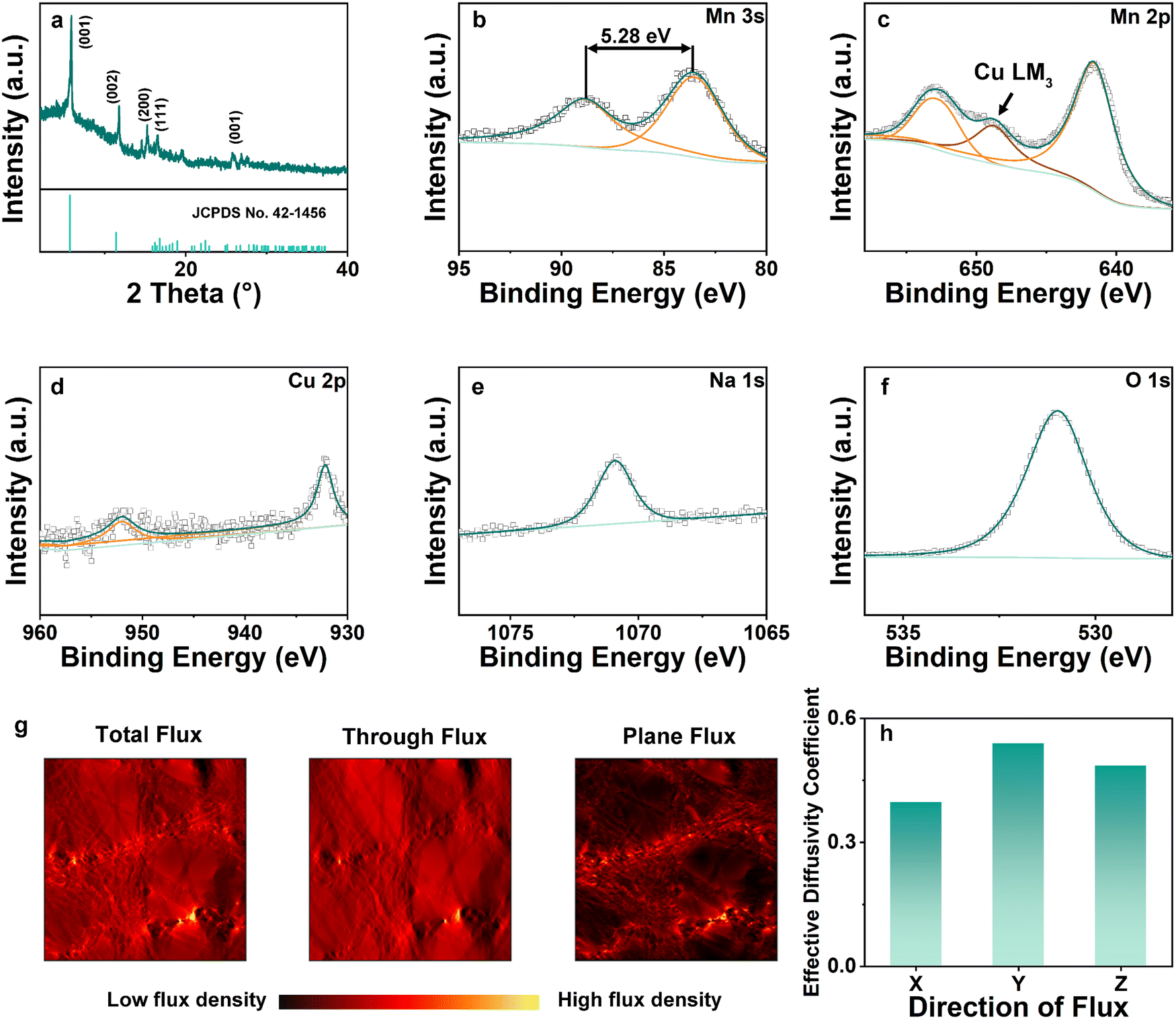 | ||
Fig. 3 (a) XRD pattern of NCMO. XPS patterns of NCMO with a focus on (b) Mn 3s, (c) Mn 2p, (d) Cu 2p, (e) Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1s, and (f) O 1s. (g) The flux density heatmap in Z direction and (h) the effective diffusivity coefficient of 0.40, 0.55, 0.49 in X, Y, and Z directions, respectively. 1s, and (f) O 1s. (g) The flux density heatmap in Z direction and (h) the effective diffusivity coefficient of 0.40, 0.55, 0.49 in X, Y, and Z directions, respectively. | ||
The electrochemical performances of two types of cathodes, namely NCMO and birnessite Na-intercalated manganese oxide (NMO), were investigated by coin cell configurations, which is commonly used in research on ZIBs. As a control material, NMO was also synthesized in a similar method as reported in previous work. To study the effect of Cu intercalation on the electrochemical behavior of cathode, cyclic voltammogram (CV) curves of NCMO and NMO cathodes with the same fabrication method (with binder, carbon agent and carbon paper collector) were measured at different scan rates ranging from 0.1 to 1 mV s−1 in the potential window of 0.8 to 1.9 V. The results, shown in Fig. 4(a) and (d), reveal that at low scan rates, two pairs of reduction and oxidation peaks of NCMO were observed, with reduction peaks at 1.37 and 1.23 V and oxidation peaks at 1.55 and 1.59 V. On the other hand, for the NMO cathode, the two pairs of redox peaks are at 1.36, 1.23, 1.56 and 1.61 V, which exhibit relatively larger polarization than that of NCMO.
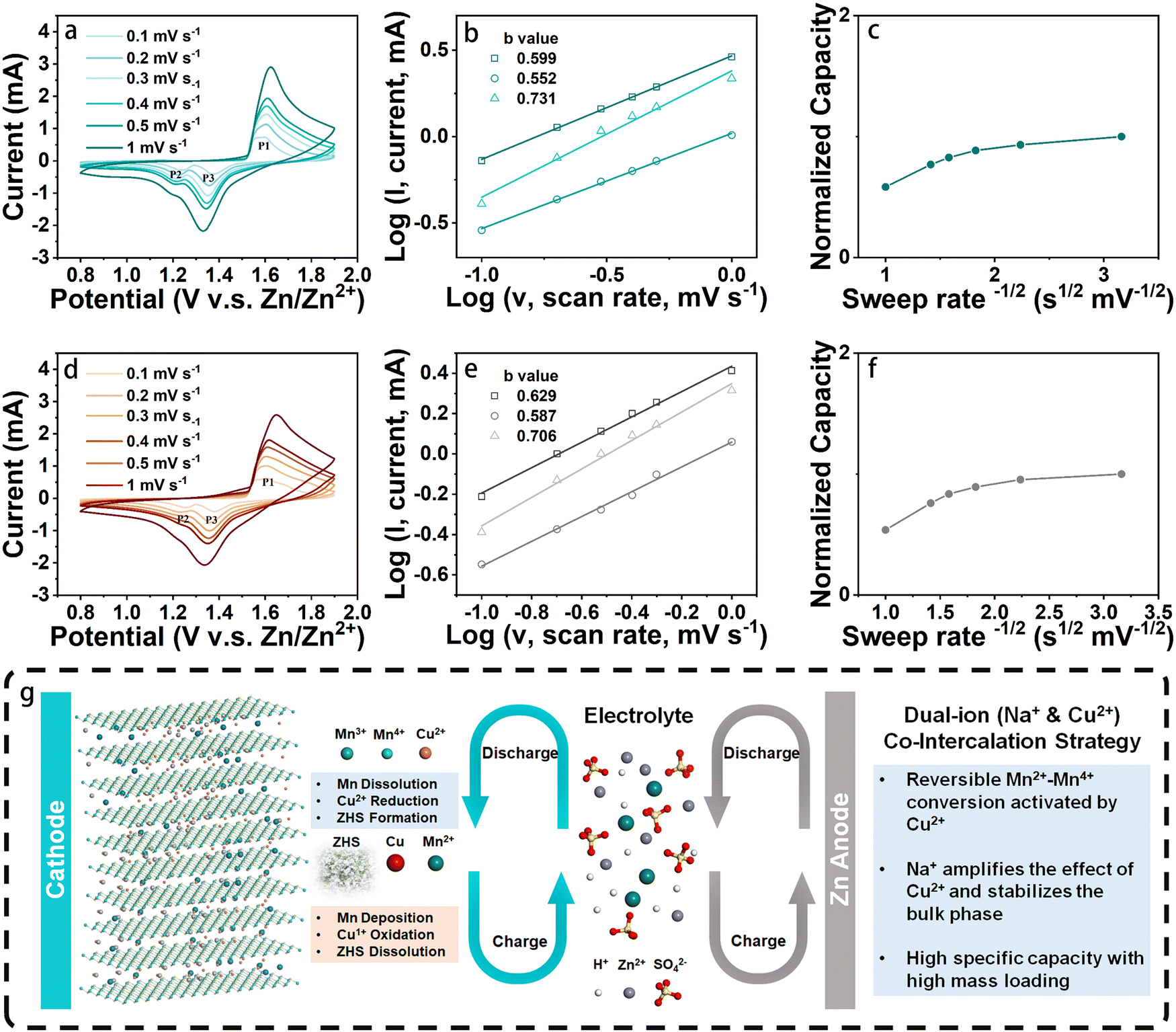 | ||
| Fig. 4 (a) and (d) CV curves with scan rates ranging from 0.1 to 1mV s−1, (b), (e) diffusion-capacitive control contribution, and (c) and (f) the relationship between normalized capacity and sweep rate of NCMO and NMO, respectively. (g) Schematic diagram illustrating the charge and discharge process of NCMO cathodes in ZIBs. | ||
It is worth noting that the coin cell with the NCMO cathode shows a similar redox peak shape to that of NMO, indicating the similarity of the two charge storage mechanisms.27 This further suggests that during cycling, the redox reactions of typical manganese oxide-based cathodes in an aqueous zinc salt (ZnSO4) electrolyte occur. Additionally, the NCMO cathodes exhibit very similar CV curves at different scan rates, suggesting that their reaction behaviors and kinetics are similar at both high and low rates. However, the two pairs of redox peaks of the NMO electrode start to overlap into a single pair from the scan rate of 0.2 mV s−1. When the scan rate reaches 0.3 mV s−1, the two oxidation peaks merge into one and shift to a higher potential. In general, the potential difference (Δφ) between the oxidation peak and the reduction peak of the quasi-reversible electrochemical reaction gradually increases with the increase of the scan rate. However, for a well-reversible electrochemical system, this situation will be alleviated. Fig. 4(d) illustrates that as the scan rate increases, the shifts of P1, P2, and P3 of NMO are 0.049, 0.059, and 0.047 V, respectively. In contrast, the shifts of P1, P2, and P3 of NCMO in Fig. 4(a) are only 0.018, 0.030, and 0.025 V, respectively, and the reduction peak involving Zn2+ remains relatively more obvious, which indicates a more reversible electrochemical system formed by the NCMO cathode.28 These results suggest that the intercalation of Cu not only effectively reduces the polarization of the cathode material but also enhances the reversibility of its redox reaction, with an extremely small difference between the peak positions at high and low scan rates.
To further investigate the kinetic processes of different cathode materials, their diffusion-controlled and capacitive contributions were evaluated based on the 3 peaks in Fig. 4(a) and (d). According to theoretical predictions, the peak current (i) and sweep rate (ν) should follow eqn (1):
| i = aνb | (1) |
Hence, log (i) = log (a) + b·log (ν)where a and b can be calculated by fitting a linear curve to the log–log plot of the peak current and the sweep rates (Fig. 4(b) and (e)). When b is close to 1, the electrochemical reaction is largely controlled by the capacitive behavior, while a value close to 0.5 indicates the dominance of the diffusion process.29 The b values of P1, P2, and P3 for NCMO cathode are 0.60, 0.55, and 0.73, respectively, indicating that the kinetics of the cathode is largely dominated by the diffusion-controlled process. Similarly, the peaks P1, P2, and P3 for NMO cathode exhibited b values of 0.63, 0.59, and 0.71, respectively, suggesting that the kinetic behavior of the NMO cathode is also largely dominated by the diffusion-controlled process. In addition, the relationship between the normalized capacity and the sweep rate (ν) was patterned, as shown in Fig. 4(c) and (f). The normalized capacity of the NMO cathodes showed a more obvious decreasing trend with the increase of scan rates than that of NCMO, indicating that NCMO exhibits better high-rate performance.
Ex situ XRD analysis was carried out to investigate the energy storage mechanism of NCMO cathodes at a specific voltage, as depicted in Fig. S5a (ESI†). We examined the structural evolution of NCMO by performing ex situ XRD measurements at specific states of charge and discharge processes, ranging from 0.8 to 1.9 V, at a current density of 1 A g−1. The XRD patterns indicated that there was no significant phase variation during the charge and discharge processes. However, a 1° peak shift at the characteristic peak (002) is observed during the charge process, and the peak gradually became inconspicuous during the discharge process due to the intercalation and deintercalation of Zn2+, which reduced the intensity of the characteristic peaks during cycling.30 As in other aqueous ZIBs, Zn4SO4(OH)6·nH2O (ZHS, n = 4 or 5) was generated on the cathode during discharge, as evidenced by the disappearance of the deposition peaks during the charge process (Fig. S5a, ESI†).13 This observation suggests that cation intercalation is reversible.31 Specifically, during the discharge process, H+ and Zn2+ ions migrate to the cathode, leading to the formation of ZHS. The reverse process occurs during the charge, resulting in the disappearance of ZHS. The formation of ZHS can be expressed by eqn (2).13
| 6OH− + SO42− + 4Zn2+ + nH2O ⇌ Zn4(OH)6SO4·nH2O | (2) |
During the discharge process from 1.9 V to 0.8 V, the amount of ZHS was increased on the cathode, which is also consistent with other literature. During the charge and discharge process, there will be two different ZHS phases, namely Zn4SO4(OH)6·4H2O and Zn4SO4(OH)6·5H2O. During the cycling of NCMO, as shown in Fig. S5a (ESI†), the phase of ZHS was dominated by Zn4SO4(OH)6·5H2O in the initial full discharge state. Zn4SO4(OH)6·4H2O was gradually produced with the change of potential, which is consistent with the previous literature.32 This also shows that the cycle of the NCMO cathode follows the law of a typical manganese-based cathode, and the intercalation of Cu has no significant effect on the generation of ZHS phases. In addition to the generation of ZHS, the peak of Mn–OH was significantly enhanced in the initial cycle, which corresponds to the H+ intercalation/deintercalation of MnO2-based cathode, as shown in eqn (3),13
| MnO2 + H+ + e− ⇌ MnOOH | (3) |
As the cycle progresses, due to the poor reversibility of ZHS, ZHS will accumulate and cover on the surface of the cathode material after long-term cycling in NMO cathode (100 cycles at a current density of 1 A g−1), resulting in the enhancement of the peaks of ZHS even at the full charge state. It is worth noting that the reversibility of ZHS shows very good reversibility in the full cells of NCMO cathode. In the fully charged state of 100 cycles, the peak of ZHS can hardly be found, but in the fully discharged state, it can still show strong ZHS peaks.
In order to gain an insight into the chemical energy storage mechanism in the Zn//NCMO system, a systematic investigation was conducted on the NCMO electrode's phase, crystal structure, and morphology at various charge/discharge states using XPS techniques. The ex situ XPS patterns of the NCMO cathode were collected at different discharge/charge states in the initial cycle and are presented in Fig. S6a–f (ESI†). The XPS spectra revealed the evolution of valence states for Mn and Cu in NCMO. Fig. S6a and b (ESI†) give the comparison of high resolution XPS spectra of Mn 2p in full discharge and full charge state. The proportion of Mn4+ increase significantly during the charge process. The average valence of Mn in the full charge state and full discharge state is calculated by ΔE between two peaks of Mn 3s (Fig. S6c and d, ESI†). The average valence of Mn was found to be 3.43 and 2.77, respectively, in full charge and full discharge state. During cycling, as shown in Fig. S6e and f (ESI†), the satellite peaks of Cu2+ disappeared during discharge and reappeared when charged back to 1.90 V, indicating Cu2+ reduction and reoxidation.17 The electrochemical cycling process of the NCMO cathode in ZIBs was investigated to explore the mechanism of chemical energy storage.
During the whole cycle, for typical MnO2-based ZIBs, Mn2+ in the electrolyte participate in the cycle of ZHS and produce birnessite-type MnO2, which is related to the joint participation of H+, Zn2+, Mn2+ and ZHS in the cycle system. The introduction of Cu enhanced the intercalation of H+ and Zn2+, which can be demonstrated by Fig. S7 and S8 (ESI†). In the first cycle, NCMO exhibited higher capacity for proton batteries in the electrochemical process without Zn2+, but it was difficult to continue cycling. When Zn2+ was added to the system, the NCMO cathode provided a very long and stable activation process. The cycling process of the NCMO cathode prepared by the dual-ion intercalation strategy can be shown in Fig. 4(g). On the one hand, Cu can promote the intercalation of proton and Zn2+, and this effect was amplified after the introduction of Na+ co-intercalation. On the other hand, the presence of Cu may enhance the effect of ZHS in the circulation system. In NMO, the wide interplanar spacing of the birnessite phase can promote Zn2+ intercalation, but at the same time, the ZHS generated by Zn2+ on the surface of the active material due to proton intercalation also provides electron transfer, which provides most of the capacity. Due to the poor reversibility of ZHS and by-products during cycling, the active material is gradually covered during cycling and weakens the capacity provided by ion intercalation. However, due to the presence of Mn2+ in the electrolyte, the deposited birnessite structure provides more surfaces for the deposition of ZHS, which forms a dynamic balance in terms of capacity performance. When more and more irreversible ZnxMnO2 is formed on the surface and Mn2+ in the electrolyte is used up, the specific capacity of the battery will inevitably decline precipitously, which is also an important factor limiting the lifespan of MnO2-based ZIBs.
The difference in electrochemical behavior between NCMO and NMO during cycling was shown by comparing whether Mn2+ was added to the electrolyte, as shown in Fig. S9–S12 (ESI†). In the Mn2+-free electrolyte, NCMO exhibited higher activity in the first cycle, and no activation process occurred. It can be seen very clearly that the difference between the potential during charge/discharge at the half reversible capacity δ(V(Q/2)) in NCMO is about 0.19 at the 100th cycle, which is significantly smaller than 0.25 in NMO at the 100th cycle, indicating that NCMO has a lower hysteresis degree and better kinetic performance than NMO. In addition, in the first cycle, NCMO exhibits a capacity provided by active Mn dissolution, which is absent in the early stage of NMO. However, the activation process of NMO in the electrolyte without MnSO4 is due to the fact that NMO needs to generate ZHS in the first cycle. In the presence of Mn2+ (from the dissolution of solid MnO2), ZHS is a reactant and induces a deposition reaction that forms layered zinc vernadite (ZnxMnO(OH)y) nanosheets on the cathode surface during charge process, of which plateau voltage is about 1.5 V vs. Zn/Zn2+. During the discharge process, ZHS recombines and simultaneously accelerates the proton reaction with ZnxMnO(OH)y, resulting in the dissolution of ZnxMnO(OH)y nanosheets. This reversible conversion reaction between ZHS and ZnxMnO(OH)y can drive the reversible energy storage reaction of aqueous MnO2-based ZIBs. The reaction can be written as follows,19
During charge process:
| Zn4SO4(OH)6·nH2O + Mn2+ → ZnxMnO(OH)y + (y − 4)H+ + (n − y + 5)H2O + (4 − x)Zn2+ + SO42− + (y − 2x)e− | (4) |
During discharge process:
| ZnxMnO(OH)y + [y + 2]H+ + (y − 2x)e− → Mn2+ + xZn2+ + (y + 1)H2O | (5) |
| 4Zn2+ + SO42− + 6OH− + nH2O → Zn4SO4(OH)6·nH2O | (6) |
The generation of Cu2+ in the charge process promotes the dissolution of ZnxMnO(OH)y during the cycle of Mn-based materials, especially when participating in the process of eqn (5), thereby accelerate the reaction process between ZHS and Mn2+. As shown in Fig. S13b (ESI†), when a scan rate of 0.3 mV s−1 was utilized, the CV curves indicated that Cu(0) was oxidized to Cu2+ during the charge process with good reversibility, which is consistent with the XPS results. Cu2+ has a d9 electron configuration, signifying the occupation of one electron within each of its d orbitals. This results in a degenerate set of d orbitals in a symmetric environment, making Cu2+ more susceptible to experiencing the JT effect. When Cu2+ and the mesophase ZnxMnO(OH)y are generated simultaneously during the charge process, this JT ion will promote the distortion of the [MnO6] octahedral structure (JT effect), thereby promoting the dissolution of ZnxMnO(OH)y for two-electron transfer on the surface.15 On the other hand, Na+ helps the bulk structure to maintain a stable cycle. For NMO cathode, protons and Zn2+ can be intercalated in host materials during discharge because of the particularity of birnessite, which provides early capacity.33 In electrolytes containing Mn2+, NCMO exhibits excellent reversibility and gradually increased cycle capacity. The voltage plateau of NCMO at ∼1.2 V vs. Zn/Zn2+ remains clearly visible after 100 cycles, demonstrating an extremely reversible Mn deposition reaction. In the battery assembled with NMO cathode, after 100 cycles, the platform of Mn cycle is no longer obvious, showing the enhancement of the reversibility of Cu for the deposition and dissolution of Mn. The SEM and EDS mapping of the NCMO cathode after 100 cycles at a current density of 0.1 A mg−1 are shown in Fig. S14 (ESI†), which shows the stability of the NCMO as the host material. After 100 cycles at a low current density, the NCMO cathode maintained a similar morphology to the initial cathode without obvious structural collapse. ZHS was synthesized as the cathode in ZIBs to investigate the effect of Cu2+ on ZHS during charge and discharge. As shown in Fig. S15 (ESI†), when the electrolyte does not contain Mn2+, the ZHS cathode provides almost no capacity. And when MnSO4 is added to the electrolyte, see Fig. S16 (ESI†), ZHS can provide an initial capacity of nearly 200 mA h g−1, and it has been decaying without activation process after the first cycle. This suggests that ZHS is indeed involved in the Mn2+/Mn4+ conversion process, but due to the insufficient reversibility of this process, this limits the possibility of a stable cycle. When the electrolyte containing 0.1 M CuSO4 (the electrolyte contains 2 M ZnSO4, 0.2 M MnSO4, and 0.1 M CuSO4) was introduced into the ZHS cathode ZIBs, high capacities of more than 330 mA h g−1 were miraculously generated in the initial 7 cycles and the cycle curve showed a slight activation process, as shown in Fig. S17 (ESI†), indicating the promotion effect of Cu on the Mn deposition and dissolution process on ZHS cathode. However, its capacity decays rapidly after the 7th cycle, because ZHS cannot be used as a stable host material. It can be confirmed that the introduction of Cu does promote the conversion process of Mn2+/Mn4+ involved in ZHS.
The rate performances of the NCMO and NMO electrodes were measured over a range of current densities from 0.2 to 10 A g−1, as shown in Fig. 5(a), and the specific capacities of both electrodes decreased as the current density increased due to the limited electrochemical reaction kinetics. However, the Zn//NCMO battery demonstrated excellent rate capability, delivering high capacities of 364, 290, 247, 196, 125, and 85 mA h g−1 at current densities of 0.2, 0.5, 1, 2, 5, and 10 A g−1, respectively. Remarkably, the NCMO electrode exhibited ∼1.5 times higher capacities than those of the NMO electrode at the same current densities, and its excellent rate cyclability was demonstrated by recovering the reversible capacity of ∼365 mA h g−1 with no observable loss at a current density of 0.2 A g−1. The galvanostatic discharge/charge curves (Fig. 5(b)) of the NCMO electrode at various current densities showed obvious charge/discharge plateau, indicating superior rate performance resulting from the improved electron transport and ion diffusion kinetics attributed to the modulated electronic structure and ion/charge storage sites due to the Na+ and Cu2+ co-intercalation into NMO. Ex situ SEM images further confirmed the charge storage mechanism and the morphological evolution of the cycled NCMO cathode. The morphology of flakes was maintained throughout the cycling process, indicating a stable host structure. The results of ex situ XRD, XPS, and SEM observations were in good agreement, indicating the reversible formation and disappearance of ZHS, stable host structure of NCMO, and efficient conversion of Mn in different valence during cycling. As shown in Fig. S13a (ESI†), after activation by a small current, the Zn//NCMO full cell was tested for CV at a scan rate of 0.5 mV s−1, and a curve with a high degree of coincidence was obtained, showing a very stable reversible cycle performance.
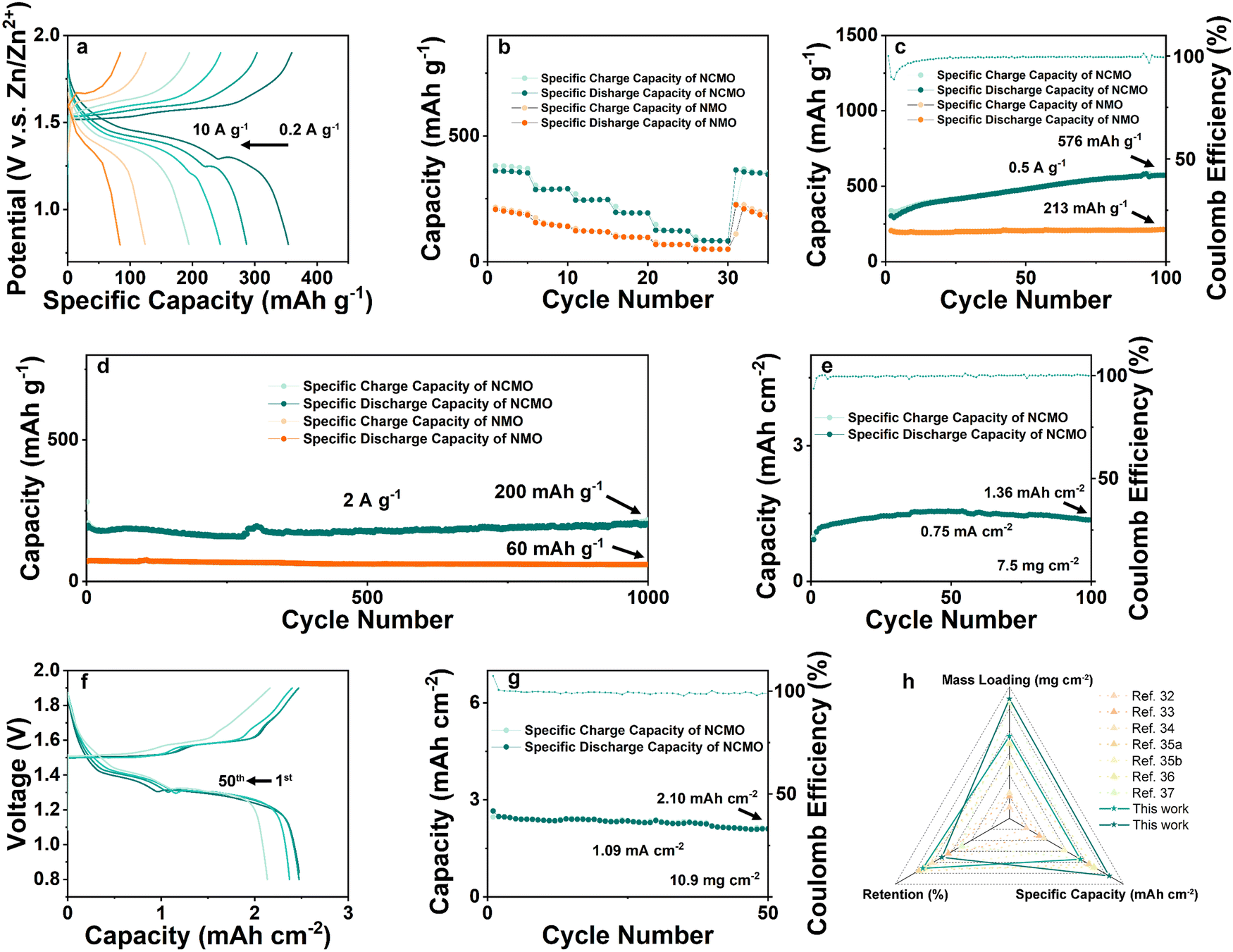 | ||
| Fig. 5 (a) Galvanostatic charge and discharge profiles for NCMO full cell and (b) cycle performance of NCMO compared with NMO at different current densities from 0.2 to 10Ag−1. (c) Low-rate cycle performance of NCMO and NMO at a current density of 0.5Ag−1, which shows that the NCMO can exhibit a reversible specific capacity of 576mA hg−1 after 100 cycles. (d) Long-term cycle performance of NCMO and NMO cathode at a current density of 2Ag−1, which shows that the NCMO can exhibit a reversible specific capacity of 200mA hg−1 after 1000 cycles. (e) Low-rate cycle performance of NCMO with a high active material mass loading of 7.5 mg cm−2 at a current density of 0.1 A g−1 (i.e., 0.75 mA cm−2), which shows that the NCMO exhibits a reversible specific capacity of 1.36 mA h cm−2 (181 mA h g−1) after 100cycles. (f) Galvanostatic charge and discharge profiles for NCMO full cell with a cathode active material mass loading of 10.9 mg cm−2 and the corresponding (g) low-rate cycling performance of NCMO cathodes at a current density of 0.1 A g−1 (i.e., 1.09 mA cm−2), which shows that the NCMO exhibits a reversible specific capacity of 2.10 mA h cm−2 (193 mA h g−1) after 50cycles. (h) Radar chart comparing the performance of this work with recent MnO2-based cathode work from the three dimensions of mass-loading, retention, and specific capacity. | ||
To determine the cycling stability of the as-prepared materials, low-rate tests at a current density of 0.5 A g−1 were carried out on NCMO and NMO, respectively. As shown in Fig. 5(c), the NCMO cathode with an active material mass loading of ∼1 mg cm−2 displays a slow and steady rise in specific capacity and shows a discharge capacity of 576 mA h g−1 after 100 cycles, which is nearly 3 times that of NMO cathode. This is an interesting phenomenon, which is consistent with the gradual deposition of Mn-based material on the cathode by the electrolyte containing Mn2+. It is worth noting that when Cu was introduced into the system, the activation process seemed to be longer and exhibited a surprisingly high reversible specific capacity.
As shown in Fig. 5(d), current density of 2 A g−1 was used to examine the high rate and long cycling performance of NCMO cathode. At a current density of 2 A g−1, the ZIBs with NCMO cathodes achieved ∼100% capacity retention (while NMO exhibited a capacity retention of ∼82%) and a high specific capacity of 200 mA h g−1 at 1000 cycles, which is 3.33 times compared to that of NMO cathodes. In order to verify the industrial feasibility of NCMO, high mass loading cathodes were prepared to test its regulation of Mn deposition and dissolution under high-load conditions. As shown in Fig. 5(e), an NCMO cathode with a moderate active material loading of about 7.5 mg cm−2 was prepared. At a current density of 0.1 A g−1 (i.e., 0.75 mA cm−2), the cathode delivered a specific capacity of 1.36 mA h cm−2 (181 mA h g−1) and exhibited nearly ∼90% retention after 100 cycles. When the loading was further increased to ∼10.9 mg cm−2, a stable reaction process of the electrode could be found in Fig. 5(f). After 50 cycles, the voltage plateau of the full cell is maintained at a stable level, and the capacity retention rate is 81%. As shown in Fig. 5(g), the NCMO cathode with high active material loading exhibited a high specific capacity of 2.60 mA h cm−2 (239 mA h g−1) in initial cycle and a reversible capacity of 2.10 mA h cm−2 (193 mA h g−1) after 50 stable cycles at a current density of 1.09 mA cm−2 (0.1 A g−1). The strategy of simultaneously enhancing the reactivity of surface and stability of the bulk of cathode material seems to have strong reference significance for future manganese oxide cathodes of aqueous ZIBs. On the one hand, the introduction of Cu2+ enhances Mn2+/Mn4+ two-electron transfer on the surface, and on the other hand, Na+ enhances bulk stability. An interesting phenomenon is that the full cells seem to cycle more smoothly as the cathode mass loading increases. Due to the reduced surface ratio, although the specific capacity at high mass loading is lower than that at low mass loading, NCMO still exhibits considerable high mass loading cycle performance due to its stable bulk structure and high surface reactivity. By comparing the work of the cathode in the past three years as shown in Fig. 5(h), such as the mass loading is 1,34 2,35 2.3,36 5,37 6.8,38 10.5 mg cm−2,39etc., the cathode with the dual-ion pre-intercalation strategy in this work has achieved excellent operating capacity, recovery efficiency and high specific capacity.
Conclusion
In conclusion, co-intercalation of Na+ and Cu2+ has been shown to be an effective strategy for enhancing the electrochemical performance of ZIBs through the fabrication of co-intercalated birnessite manganese oxide cathodes. The combination of Na+ and Cu2+ amplifies the activation effect of Cu2+ on the Mn2+/Mn4+ redox pair on the surface and provide a stable host structure for charge and discharge with the help of Na+, thereby leading to improved specific capacity and cycling stability. The co-intercalation of Na+ and Cu2+, along with the deposition and dissolution of ZnxMnO(OH)y, and ZHS, allows for a prolonged continuously capacity increasing process at low mass loadings and a highly reversible manganese deposition and dissolution process at high mass loadings. It is worth noting that the way of achieving high mass loading capacity in this work is achieved through a simple and mature electrode coating process, which is low-cost and scalable. The NCMO cathode exhibited remarkable specific capacity and cycling stability at both low and high mass loadings, which is also the work closest to the theoretical capacity of Mn2+/Mn4+ two-electron transfer in ZIBs among the recent studies. Among the studies of Mn2+/Mn4+ two-electron transfer strategy, this work achieves stable cycling with the highest mass loading, demonstrating the promising potential for practical and efficient energy storage systems. The mechanism of how Cu2+ promote the deposition and dissolution of Mn-based cathodes during cycling has also been discussed. Overall, the results of this study suggest that the strategy of simultaneously promoting surface two-electron transfer while maintaining host material stability is a promising approach for developing high-performance cathode materials for ZIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Xuan Gao thanks University College London and China Scholarship Council for the joint PhD scholarship. The authors acknowledge the Engineering and Physical Sciences Research Council (EPSRC, EP/V027433/3) and UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee (101077226; EP/Y008707/1) for funding support.References
- (a) Y. Liu, X. Lu, F. Lai, T. Liu, P. R. Shearing, I. P. Parkin, G. He and D. J. Brett, Joule, 2021, 5, 2845 CrossRef CAS; (b) G. He, M. Qiao, W. Li, Y. Lu, T. Zhao, R. Zou, B. Li, J. A. Darr, J. Hu and M. M. Titirici, Adv. Sci., 2017, 4, 1600214 CrossRef PubMed; (c) S. Guo, J. Li, B. Zhang, W. Chen, G. Fang, M. Long and S. Liang, Sci. Bull., 2022, 67, 626 CrossRef CAS PubMed.
- (a) X. Gao, H. Wu, C. Su, C. Lu, Y. Dai, S. Zhao, X. Hu, F. Zhao, W. Zhang and I. P. Parkin, Energy Environ. Sci., 2023, 16, 1364 RSC; (b) X. Gao, X. Sun, J. Liu, N. Gao and H. Li, J. Energy Storage, 2019, 25, 100901 CrossRef.
- (a) H. Dong, J. Li, J. Guo, F. Lai, F. Zhao, Y. Jiao, D. J. Brett, T. Liu, G. He and I. P. Parkin, Adv. Mater., 2021, 33, 2007548 CrossRef CAS PubMed; (b) Y. Pan, Z. Liu, S. Liu, L. Qin, Y. Yang, M. Zhou, Y. Sun, X. Cao, S. Liang and G. Fang, Adv. Energy Mater., 2023, 13, 2203766 CrossRef CAS.
- R. Chen, W. Zhang, Q. Huang, C. Guan, W. Zong, Y. Dai, Z. Du, Z. Zhang, J. Li and F. Guo, Nano-Micro Lett., 2023, 15, 81 CrossRef CAS PubMed.
- J. Li, K. McColl, X. Lu, S. Sathasivam, H. Dong, L. Kang, Z. Li, S. Zhao, A. G. Kafizas and R. Wang, Adv. Energy Mater., 2020, 10, 2000058 CrossRef CAS.
- Y. Dai, C. Zhang, W. Zhang, L. Cui, C. Ye, X. Hong, J. Li, R. Chen, W. Zong and X. Gao, Angew. Chem., Int. Ed., 2023, e202301192 CAS.
- X. Gao, K. Liu, C. Su, W. Zhang, Y. Dai, I. P. Parkin, C. J. Carmalt and G. He, SmartMat, 2023, 5, e1197 CrossRef.
- W. Zhang, Y. Dai, R. Chen, Z. Xu, J. Li, W. Zong, H. Li, Z. Li, Z. Zhang and J. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202212695 CrossRef CAS PubMed.
- H. Dong, J. Li, S. Zhao, Y. Jiao, J. Chen, Y. Tan, D. J. Brett, G. He and I. P. Parkin, ACS Appl. Mater. Interfaces, 2020, 13, 745 CrossRef PubMed.
- (a) S. He, Z. Mo, C. Shuai, W. Liu, R. Yue, G. Liu, H. Pei, Y. Chen, N. Liu and R. Guo, Appl. Surf. Sci., 2022, 577, 151904 CrossRef CAS; (b) Y. Hu, Z. Liu, L. Li, S. Guo, X. Xie, Z. Luo, G. Fang and S. Liang, Natl. Sci. Rev., 2023, 10, nwad220 CrossRef PubMed.
- Y. Dai, X. Liao, R. Yu, J. Li, J. Li, S. Tan, P. He, Q. An, Q. Wei and L. Chen, Adv. Mater., 2021, 33, 2100359 CrossRef CAS PubMed.
- R. Chen, C. Zhang, J. Li, Z. Du, F. Guo, W. Zhang, Y. Dai, W. Zong, X. Gao and J. Zhu, Energy Environ. Sci., 2023, 16, 2540 RSC.
- X. Gao, Y. Dai, C. Zhang, Y. Zhang, W. Zong, W. Zhang, R. Chen, J. Zhu, X. Hu and M. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300608 CrossRef CAS PubMed.
- (a) Y. Jiao, L. Kang, J. Berry-Gair, K. McColl, J. Li, H. Dong, H. Jiang, R. Wang, F. Cora and D. J. Brett, J. Mater. Chem. A, 2020, 8, 22075 RSC; (b) X. Guo, J. Zhou, C. Bai, X. Li, G. Fang and S. Liang, Mater. Today Energy, 2020, 16, 100396 CrossRef; (c) Y. Ding, W. Xue, K. Chen, C. Yang, Q. Feng, D. Zheng, W. Xu, F. Wang and X. Lu, Nanomater., 2023, 13, 1075 CrossRef CAS PubMed; (d) M. Manickam, P. Singh, T. B. Issa, S. Thurgate and R. De Marco, J. Power Sources, 2004, 130, 254 CrossRef CAS; (e) G. G. Yadav, J. W. Gallaway, D. E. Turney, M. Nyce, J. Huang, X. Wei and S. Banerjee, Nat. Commun., 2017, 8, 14424 CrossRef PubMed.
- X. Xia, Y. Zhao, Y. Zhao, M. Xu, W. Liu and X. Sun, Nano Res., 2023, 16, 2511 CrossRef CAS.
- X. Zeng, J. Liu, J. Mao, J. Hao, Z. Wang, S. Zhou, C. D. Ling and Z. Guo, Adv. Energy Mater., 2020, 10, 1904163 CrossRef CAS.
- Y. Chen, S. Gu, S. Wu, X. Ma, I. Hussain, Z. Sun, Z. Lu and K. Zhang, J. Chem. Eng., 2022, 450, 137923 CrossRef CAS.
- S. Gao, B. Li, H. Tan, F. Xia, O. Dahunsi, W. Xu, Y. Liu, R. Wang and Y. Cheng, Adv. Mater., 2022, 34, 2201510 CrossRef CAS PubMed.
- H. Chen, C. Dai, F. Xiao, Q. Yang, S. Cai, M. Xu, H. J. Fan and S. J. Bao, Adv. Mater., 2022, 34, 2109092 CrossRef CAS PubMed.
- X. Xie, H. Fu, Y. Fang, B. Lu, J. Zhou and S. Liang, Adv. Energy Mater., 2022, 12, 2102393 CrossRef CAS.
- R. Zhang, P. Liang, H. Yang, H. Min, M. Niu, S. Jin, Y. Jiang, Z. Pan, J. Yan and X. Shen, J. Chem. Eng., 2022, 433, 133687 CrossRef CAS.
- Y. Noda, K. Ohno and S. Nakamura, Phys. Chem. Chem. Phys., 2016, 18, 13294 RSC.
- H. Kawaoka, M. Hibino, H. Zhou and I. Honma, ECS Solid State Lett., 2005, 8, A253 CrossRef CAS.
- X. Gao, C. Zhang, Y. Dai, S. Zhao, X. Hu, F. Zhao, W. Zhang, R. Chen, W. Zong and Z. Du, Small Struct., 2022, 4, 2200316 CrossRef.
- X. Yin, Y. Li, W. Cai, C. Fan, W. Liu, N. Wang, G. Qin, Z. Xie, X. Chen and Y. Han, Appl. Surf. Sci., 2023, 624, 157124 CrossRef CAS.
- S. R. Daemi, C. Tan, T. Volkenandt, S. J. Cooper, A. Palacios-Padros, J. Cookson, D. J. Brett, P. R. Shearing and A. C. S. Appl, Energy Mater., 2018, 1, 3702 CAS.
- V. Srinivasan and J. W. Weidner, J. Power Sources, 2002, 108, 15 CrossRef CAS.
- P. Daubinger, J. Kieninger, T. Unmüssig and G. A. Urban, Phys. Chem. Chem. Phys., 2014, 16, 8392 RSC.
- L. Chen, Z. Yang, H. Qin, X. Zeng and J. Meng, J. Power Sources, 2019, 425, 162 CrossRef CAS.
- A. Moezzi, M. B. Cortie and A. M. McDonagh, Dalton Trans., 2013, 42, 14432 RSC.
- L. Godeffroy, I. Aguilar, J. Médard, D. Larcher, J. M. Tarascon and F. Kanoufi, Adv. Energy Mater., 2022, 12, 2200722 CrossRef CAS.
- (a) J. Wang, B. Zhang, Z. Cai, R. Zhan, W. Wang, L. Fu, M. Wan, R. Xiao, Y. Ou and L. Wang, Sci. Bull., 2022, 67, 716 CrossRef CAS PubMed; (b) I. Aguilar, P. Lemaire, N. Ayouni, E. Bendadesse, A. V. Morozov, O. Sel, V. Balland, B. Limoges, A. M. Abakumov and E. Raymundo-Piñero, Energy Storage Mater., 2022, 53, 238 CrossRef; (c) D. Wu, L. M. Housel, S. T. King, Z. R. Mansley, N. Sadique, Y. Zhu, L. Ma, S. N. Ehrlich, H. Zhong and E. S. Takeuchi, J. Am. Chem. Soc., 2022, 144, 23405 CrossRef CAS PubMed.
- (a) Z. Shang, S. Wang, H. Zhang, W. Zhang, S. Lu and K. Lu, Nanoscale, 2022, 14, 14433 RSC; (b) M. Huang, X. Wang, X. Liu and L. Mai, Adv. Mater., 2022, 34, 2105611 CrossRef CAS PubMed.
- S. Luo, J. Xu, B. Yuan, L. Chen, L. Xu, R. Zheng, Y. Wang, M. Zhang, Y. Lu and Y. Luo, Carbon, 2023, 214, 118334 CrossRef CAS.
- C. Wang, Y. Zeng, X. Xiao, S. Wu, G. Zhong, K. Xu, Z. Wei, W. Su and X. Lu, J. Energy Chem., 2020, 43, 182 CrossRef.
- Y. Zhang, S. Deng, Y. Li, B. Liu, G. Pan, Q. Liu, X. Wang, X. Xia and J. Tu, Energy Storage Mater., 2020, 29, 52 CrossRef.
- (a) Y. Zhao, P. Zhang, J. Liang, X. Xia, L. Ren, L. Song, W. Liu and X. Sun, Energy Storage Mater., 2022, 47, 424 CrossRef; (b) X. Gao, C. Zhang, Y. Dai, S. Zhao, X. Hu, F. Zhao, W. Zhang, R. Chen, W. Zong and Z. Du, Small Struct., 2023, 4, 2200316 CrossRef CAS.
- Y. Zhao, R. Zhou, Z. Song, X. Zhang, T. Zhang, A. Zhou, F. Wu, R. Chen and L. Li, Angew. Chem., Int. Ed., 2022, 61, e202212231 CrossRef CAS PubMed.
- L. Wu, Y. Mei, Y. Liu, W. Xu, M. Zhang, Y. Dong and Z.-S. Wu, J. Chem. Eng., 2023, 459, 141662 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee04492k |
| ‡ Xuan Gao, Chen Shen, Haobo Dong, Yuhang Dai contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |