
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Making chemicals from the air: the new frontier for hybrid electrosyntheses in artificial tree-like devices†
Gabriele
Centi
 * and
Siglinda
Perathoner
*
* and
Siglinda
Perathoner
*
Department ChiBioFarAm – Section of Industrial Chemistry, University of Messina, ERIC aisbl and CASPE–INSTM, V.le F. Stagno d'Alcontres 31, 98166, Messina, Italy. E-mail: gabriele.centi@unime.it
First published on 21st September 2023
Abstract
Making chemicals from the air is a visionary objective that can potentially revolutionise chemical production. This critical review shows that the essential elements to realise this dream exist, even the many challenges, particularly in integrating all the components and operating them in synergy. The production of (i) fertilisers and (ii) food components (carbohydrates, proteins) from the air in artificial tree-like devices is analysed, focusing on the electrosynthesis aspects. Three critical components of these devices were discussed: (i) the system to capture and concentrate small molecules (CO2, H2O, N2) from the air, (ii) the electrocatalytic fixation of CO2 and N2, with the advances in producing directly (one-step) ammonium nitrate solution and/or urea, and (iii) the sustainable production of food from the air, via a first stage of electrocatalytic CO2 fixation to acetate. Although there are advances in these areas, the possibility of combining them is still at an early stage. The concept of hybrid electrosyntheses technologies is crucial to realising and implementing these dream reactions. For this reason, it is indicated as the frontier research in electrosynthesis.
 Gabriele Centi | Gabriele Centi is a Full Professor of Industrial Chemistry at the University of Messina, Italy, and President of the European Research Institute of Catalysis (ERIC). He coordinated the EU Network of Excellence IDECAT and is currently President of the IACS (International Association of Catalysis Societies). He recently initiated and coordinated an ERC Synergy project on plasma catalysis. He is also a member of the board of SUNERGY, the European initiative on solar fuels. He has received numerous awards, including the International Fellowship Initiative of the President of the Chinese Academy of Sciences, PIFI, as a Distinguished Scientist, and the Humboldt Research Award, and is involved in various publishing activities. He is the author of over 600 scientific publications, 12 books and editor of over 20 special issues of journals. The current h-index is 95 with over 37 |
 Siglinda Perathoner | Siglinda Perathoner gained her PhD in Chemical Science in 1988 working on the photophysics and photochemistry of supramolecular systems with V. Balzani and Nobel Laureate J. M. Lehn. In 2001 she joined the University of Messina and is presently a full professor of Industrial Chemistry. She has coordinated many EU projects. She was co-chair of Europacat 2017, an important event in the catalysis community, and chaired several other conferences. Recent recognition includes the 2021 G.M. Levi Medal from the Italian Chemical Society (for innovation leading to industrial realization) and a President's International Fellowship Initiative (PIFI) award from CAS (Chinese Academy). The current h-index is 80 with over 26 |
Introduction
A “grand challenge” for a sustainable future is the possibility of producing chemicals directly from the air, e.g., by capturing N2, H2O and CO2 from the air and transforming them into fertilisers, proteins, carbohydrates, and other chemicals using only sunlight. In other words, develop genuine artificial leaf-type devices beyond those under investigation.1–10 Hybrid electrocatalytic-microbial systems11–16 offer exciting possibilities to meet these highly challenging objectives, although still not systematically explored.Most devices referring to artificial leaves or photosynthesis perform simple reactions, mainly water splitting. Studies on CO2 conversion (CO2RR) or N2 reduction reaction (to ammonia) (NRR) are increasing, even if performances are still low, with solar-to-fuel efficiencies often of 1% or below. In addition, achieving a high current density and efficiency should be realized simultaneously, being current density a measure of productivity. Progress has been significant over the years. For example, a 10% solar-to-fuel efficiency combined with a high current density in CO2 conversion to formic acid and H2 has been reported recently.
However, capturing CO2 or N2 directly from the air (as in natural leaves) is not considered in these studies, if not a long-term possibility. Furthermore, the products of these reactions should generally be further processed to obtain chemicals for the consumer market. A genuine artificial leaf device should be able to capture directly N2, H2O and CO2 from the air and make more complex products for daily life, such as fertilisers and food components, e.g., proteins and carbohydrates. These could be made by combining:
- the capabilities of functionalised membranes (or equivalent systems) able to capture and concentrate these small molecules to the surface of an electrocatalyst while preventing contact with molecules that may inhibit the activity (O2, for example);
- the potentialities of (photo)electrocatalytic devices to use sunlight to fix CO2 and N2, converting them to molecules that can be the feed for microbial conversion;
- the abilities of microbial processes in constructing complex molecules such as carbohydrates or proteins, eventually assisted by photo/electro components to bypass limitations by using co-enzymes (NADH and ATP) and allow process intensification.17–21
A hybrid artificial tree-like device
Combining several artificial-leaf elements would allow the creation of an artificial tree-like device where the “leaf” represents the photoelectrocatalytic elements and the “branches and trunk” are the elements for distribution/collection and to host the other elements, such as the microbial reactor, which cannot be integrated directly in the leaves. A schematic presentation of this conceptual approach to making chemicals from the air in a hybrid artificial-tree-type device is presented in Fig. 1.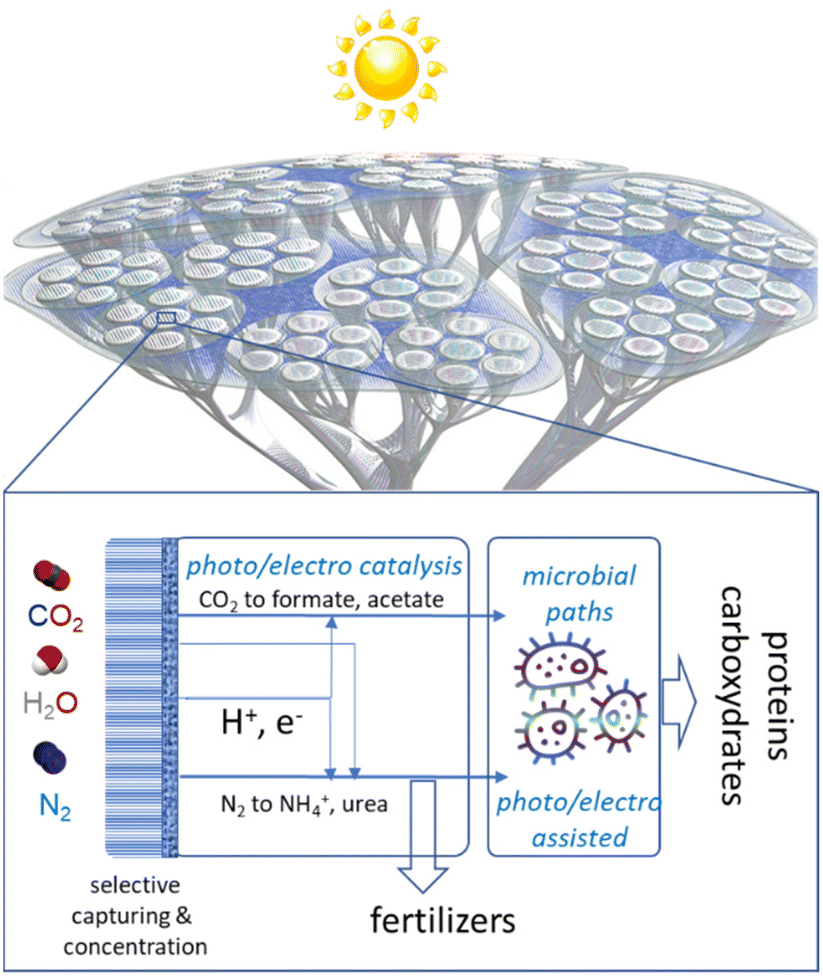 | ||
| Fig. 1 Schematic presentation of the conceptual approach to make chemicals from the air in an hybrid artificial-tree-type integrated device. | ||
These devices will operate in a distributed mode, e.g., producing the chemicals directly at the consumer (or small district) level. The productivity will be met by many of these devices working in parallel. They will directly use solar energy. The advantages are lower environmental impact (see below) and enhanced resilience (avoiding dependence on energy costs and raw materials from external factors). For example, a fertiliser in a diluted aqueous solution can be readily spread on the soil without concentration. On the contrary, centralised productions (due to scale-factor leading to megascale plants)25 require concentration and production of a solid (typically) to be transported. This is an energy-intensive operation with safety issues (for example, the explosivity of ammonium nitrate prills).
The advantages in terms of greener and more sustainable chemical production are multiple: (i) reduce the energy and associated environmental costs of producing the raw materials (energy supply included), (ii) increase resilience, (iii) very low-carbon, and potentially negative footprint, (iv) avoid the costs and environmental impact of transporting/distributing chemicals, (v) decrease time-to-market (once optimised, the scaling occurs by numbers instead by size, drastically reducing time for scale-up), (vi) allows greater adaptability to different applications (enhanced number of technology developers, because significant investments for pilot units are not necessary) and (vii) more extensive number of investors. The change in the production model, from centralised to distributed, thus enables a greener chemical production, avoiding the use of fossil fuels and accelerating the transformative conversion of chemical production. Thus, it is a central element in transformative greener chemistry.
While making chemicals from the air will contribute to only a part of the chemicals we use, even if addressing large-scale chemicals (synthetic food, fertilisers), pushing this possibility has a great societal impact, giving a long-term vision of the novel possibilities empowered by dream chemistry. In addition, the single elements, such as the selective capturing and concentration or the photoelectrocatalytic (PEC) devices and electrodes, impact the technological developments necessary to reduce the carbon footprint of chemical production and related sectors or energy-intensive industries.
Scope and limitations
Electrosynthesis is at the core of this transformation, as outlined in Fig. 1. However, a large part of the studies on electrosynthesis focuses instead on simple reactions and approaches, even in terms of electrodes and reactors, which are not representative of the conditions to progress along the directions needed to realise the conceptual scheme illustrated in Fig. 1. There are, however, uncoordinated advances in the literature on the key technological components necessary to develop the concept presented in Fig. 1.However, the literature studies have a different target; thus, these contributions do not refer to the Fig. 1 conceptual approach with related needs and constraints. This review thus aims to evidence that there are the essential elements to consider feasible, even if challenging, the concept realisation outlined in Fig. 1. This paper will discuss these emerging studies and present them as components for a unitary vision, providing the background to indicate the feasibility of directly producing chemicals from the air. The focus is on the electrosynthesis aspects. Extending current studies in electrosynthesis to new directions by interfacing with other disciplines is necessary.
It is out of the scope to critically analyse all the aspects presented, being available dedicated reviews for each specific topic, although not referring to their use to realize Fig. 1 device/approach. The discussion is limited to the feasibility and state-of-the-art elements concerning the applicability of developing a hybrid artificial-tree-type integrated device.
Besides the scientific and technological, economic feasibility is a key feature. However, such a type of analysis is impossible or even defining metrics for this analysis. An integrated device, as outlined in Fig. 1, is not available even at a prototype level. On the other hand, it is also incorrect to perform a conventional techno-economic analysis comparing with the actual costs of producing fertilizers or food. Many immaterial aspects, from geostrategically to environmental and resilience, related to the change in the production mode are important from a sustainability perspective but not accounted for in the conventional economic assessments. In addition, the conceptual scheme introduced in Fig. 1, compared to the conventional production scheme, has the main advantage of virtually eliminating operative costs (for raw materials and energy) and has mainly fixed costs. The experience with photovoltaic cells shows that it is possible to drastically cut manufacturing costs, while operative costs highly depend on externalities. Furthermore, the production of artificial tree-like devices will be based mainly on parallelized units. Thus, scaling up costs will be minimized, time to market drastically shortened and flexibility in adapting to different requests enhanced. Many novel investors can enter the market. These are the elements which created the actual success in photovoltaics.
Even with these premises, it is important to indicate that very rough preliminary estimations22,23 suggest that the concept presented in Fig. 1 is not a curiosity but a feasible possibility from an economic perspective, confirmed by various companies entering the field (see later).
Selective capturing and concentration of small molecules from the air
Capturing water
In an artificial-tree system, as illustrated in Fig. 1, capturing water directly from the air would reduce the complexity of connecting each of the multiple artificial leaves to a system for water purification/distribution. In addition, from a sustainability perspective, capturing water directly from the air is preferable to decrease the impact on water of water splitting. The stoichiometric minimal amount of water is 9 kg per kg H2, although it is a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in terms of moles. Often, the amount of water used is presented as a main environmental issue of water splitting. While H2 is used in most cases as an energy vector generating then back H2O, capturing water directly from the air would also overcome these issues and societal sensitivity.
:1 ratio in terms of moles. Often, the amount of water used is presented as a main environmental issue of water splitting. While H2 is used in most cases as an energy vector generating then back H2O, capturing water directly from the air would also overcome these issues and societal sensitivity.
Capturing water from the air is a technology already demonstrated to be feasible. In nature (tree frogs in tropical Australia, for example), combining temperature decrease at night and nanohollows is often used to capture water from the air.24 Bio-inspired water harvesting materials can be designed based on these strategies.25 Desert cactuses used a different principle. Water droplets condense at the tops of their spines and move to the stem using capillary and other physical forces.26 Desert beetle uses hydrophilic protrusions on its skin as nucleation sites, while hydrophobic areas cause the condensing of water droplets and funnelling them into the beetle mouth.27
Other principles can also be used to capture and condense water from the air. Deliquescent salts capture water molecules passively through the hydration process. The hygroscopic salt can be confined in composite structures to avoid liquefication. For example, LiCl incorporated in a zwitterionic polymer [as poly [2-(methacryloyloxy)ethyl] dimethyl-(3-sulfopropyl)am-monium hydroxide (PDMAPS)] makes a hygroscopic hydrogel.28 The salt ions remain trapped within the hydrogel because water molecules become part of the zwitterionic polymeric structure. A heater then extracts the bound water. Guo et al.29 reported that a superhygroscopic polymer film containing a hygroscopic salt (LiCl) exhibits high water uptake of 0.64–0.96 g g−1 at 15–30% relative humidity. They use a natural product (Konjac glucomannan) with highly porous structures for active moisture capture and water vapour transport. At the same time, another component (thermoresponsive hydroxypropyl cellulose) enables phase transition at a low temperature to assist the release of collected water via hydrophobic interactions. Other hygroscopic hydrogels also exist.30 An alternative is using materials such as metal–organic frameworks (MOFs) to adsorb water while tailoring the hydrophilicity of the functional groups to facilitate desorption. For example, Zr-based MOF-801 collected and released 0.1 L of water per kilogram MOF daily at 10% relative humidity.31 Rod-like MOF-303 crystal structure has long channels facilitating the uptake and release of water. It delivered 0.7–1 LH2O kgMOF at relative humidity <7%.32,33 A PV panel can provide energy for desorption and water collection.
Several MOF-based prototyping devices have been demonstrated with great practical potential.33–35
Capturing CO2 and N2
More challenging is the capture and concentration of CO2 from the air, avoiding at the same time the contact with the electrodes of undesired air components. Similar issues, although slightly different, are present in separating N2 from O2 for electrocatalytic nitrogen fixation. Nitrogen is present in air with a higher concentration than CO2 but is chemically inert.While effective alternatives (except those discussed later) for CO2 capture and selective transport/concentration to the electrocatalyst are unavailable, membrane (and other) solutions are established for N2 separation. Membrane units for >99% N2 purity are commercially available but require multistep separation stages. The process is too costly in energetic terms36 and especially unsuitable to be integrated into a scheme, as presented in Fig. 1.
It is thus necessary to develop a single-stage membrane unit where N2 selectively diffuses while O2 transport is blocked. Membranes containing metal complexes binding N2 rather than O2 can be imagined. This topic of functional elements for the selective transport of N2 in membranes is an area scarcely investigated. MOF materials containing Cr(III) sites could selectively capture N2 over O2.37 N2-selective group V metallic membranes have also been reported.38 Generally, results are very limited, even proving the conceptual feasibility. There is a rich N2 coordination chemistry supporting this indication.39
In parallel, the poisoning effect of O2 on the electrocatalyst for N2 fixation has also to be reduced by designing O2-tolerant NRR electrocatalysts. This is another area not yet investigated.
The alternative in the case of CO2 to the functionalised membranes outlined in Fig. 1 is the integration of CO2 electrolysis with CO2 capture. The capture of CO2 is an energy-intensive and costly element of carbon capture, utilisation and storage (CCUS) technologies. The overall energy penalty due to a CO2 capture unit depends on the type of solvent and characteristics of the capture process. When using DAC (direct air capture) technologies, the cost of the process and energy intensity are large, ranging from 0.15 to 0.25 Mtoe/MtCO2, e.g., around 0.55 MtCO2 per MtCO2 captured.40 The DAC technology, however, is not well suited for integration into the concept presented in Fig. 1, requiring too large volumes.41
Integrating CO2 electrolysis with its capture provides opportunities for energy reductions by simultaneously removing the energy-demanding regeneration step in CO2 capture and avoiding critical issues faced by CO2 gas-fed electrolysers.42Fig. 2 reports the simplified block diagrams of sequential and integrated routes for amine-based CO2 capture and electrolysis with a more detailed scheme for the integrated CO2 capture and direct CO2 electroreduction from the capture medium. Some elements, such as the compression unit between the stripper and electrolyser are not shown. Classical amine-based CO2 capture requires around half of the energy of DAC, but cannot be applied to capturing CO2 from air, and requires a minimum CO2 concentration over 10%.
 | ||
| Fig. 2 Simplified block diagrams of sequential and integrated routes for amine-based CO2 capture and electrolysis with a more detailed scheme for the integrated CO2 capture and direct CO2 electroreduction from the capture medium. Some elements, such as the compression unit between the stripper and the electrolyser, are not shown. | ||
Although the energy-intensive character of this integrated solution is lower than the sequential solution, it still requires significant energy intensity (estimated energy reduction to sequential operations is around 20–25%).42 Additionally, it cannot be used for directly capturing CO2 from the air. More aspects of the integrated CO2 capture and electrochemical conversion were discussed by Gutiérrez-Sánchez et al.,43 Adamu et al.44 and Sullivan et al.45 The use instead of solid adsorbent was reviewed by Khdary et al.,46 Fu et al.47 and Sun et al.48 However, the latter technology does not adapt to be integrated within artificial tree-type devices as illustrated in Fig. 1.
Li et al.49 proposed integrating an ammine CO2 adsorption step directly in the electrocatalytic cell for CO2 conversion, e.g. to perform the electrocatalytic CO2 conversion directly in the amine solution where CO2 is captured. However, the product will be an amine solution containing the products of CO2 conversion. It is unsuited for directly sending the solution to the integrated microbial unit. The same observation applied to other cases discussed above.
Overcoming the issues given by the electrolyte on the microbial unit is critical. It is necessary to work under a neutral aqueous solution without added salts to promote conductivity (and thus performances) or to use an electrolyte-less (also indicated zero-gap or gas-phase) approach. In this cell design, gas-phase CO2 is adsorbed and concentrated at the electrode surface in a GDL (gas-diffusion layer) electrode. Huang et al.50 modelled using the CFD (computational fluidodynamic) approach, a membrane reactor concept for integrated CO2 capture and conversion. Although their reactor model differs from the membrane/GDL zero-gap electrocatalytic system indicated above, their results support the need to develop the membrane/GDL zero-gap electrocatalytic approach.
Marepally et al.51 showed that it is possible to modify GDL by introducing MOF elements capable of capturing CO2 and enhancing the virtual pressure of CO2 at the electrocatalyst surface. Mg/DOBDC (4-dioxido-2,5-benzenedicarboxylate) MOF (functionalising its open metal coordination sites with pendent amines) results in a material capable of CO2 adsorption at ultra-dilute CO2 partial pressures. A Zr-based UiO-67 MOF functionalised with amino silanes also shows promising performances in selective CO2 adsorption at low pressure.52
The development of membrane/GDL electrodes functionalised by these components has not been investigated, nor was studied how to develop a porous flexible membrane53 permeoselective to CO254,55 which can be integrated into the CO2RR electrode in a zero-gap reactor configuration.56–58 Diluted CO2 sources are fed to the GDL cathode (based on Pt supported on carbon) and O2 deriving from the anode. The CO2 is adsorbed as carbonate, passing first through an anion-exchange membrane (AEM). On the anode side, water is oxidized on a GDL with supported IrO2, with O2 evolving as a gas phase and protons passing through a cation-exchange membrane (CEM). A porous solid-electrolyte (PSE) is present between the two anodic/cathodic and the place for recombination of crossover protons and carbonate to form H2CO3, decomposing then to gaseous CO2. The concept is exciting but unsuited for direct coupling with CO2 electrolyzers.
Furthermore, expensive noble metals electrocatalysts are used on both sides. The capture process is energy-intensive, with productivity limited by two membranes (AEM, CEM) and PSE. They indicated a cost of about $83 per ton of captured CO2, which could be reduced to $58 per ton, remaining thus high.
The above comments remark on properly designing CO2 electrolyzers, particularly in scaling up the results and obtaining industrially relevant processes.59 However, several literature studies of CO2RR and NRR do not have such characteristics. Yuan et al.59 discussed different CO2 electrolyzer designs, including GDE and zero-gap electrolyzers, and their scale-up engineering challenges.
An attractive electrochemical reactor with a continuous carbon capture was presented recently by Zhu et al.60
Fertilisers from the air
Relevance
Fertilisers are the single most significant industrial chemicals, with a global production of ∼260 Mt per y (as nutrients), ∼20% of which are urea (∼235 Mt per y as a product) and ∼6% nitrate. In front of the very spread fertiliser use, production is concentrated in a limited number of plants, often on a megascale (up to 5000–6000 metric tonnes per day). Thus, as commented before, high costs and impacts are associated with their distribution. Critical is that in a highly fluctuating market, these plants do not have the flexibility to adapt production to the needs, negatively impacting economics and having a high local impact on the environment. In addition, their price was subject to huge fluctuations, from less than 300 US$ per ton to over 1200 US$ per ton in the last two years, due to the energy intensity of the process (energy feedstock costs drive 70–80% of the cost of ammonia) and distribution. For this reason, interest in distributed ammonia production to improve energy and food security, reduce costs and deploy ammonia energy solutions is increasing. Various companies, such as Starfire Energy, AmmPower and FuelPositive (with increasing interest also by traditional ammonia plant producers, such as Casale, Stamicarbon, and Topsoe), offer small-scale renewable NH3 solutions based on H2 by hydrolysis, followed by a nearly-traditional Haber–Bosch (HB) process using heterogeneous catalysis operating at high temperatures and pressures.61Direct and indirect (multistep) processes
The processes outlined above are multistep (indirect). Although making H2via electrolysis rather than from fossil fuels significantly reduces the carbon footprint, producing fertilisers is still a complex, multistep and energy-intensive process.62,63 It is thus attracting the possibility of directly making ammonia by electrolysis of N2 + H2O to avoid the energy losses and additional Capex (capital expenses) related to the coupling of the H2 electrolysis (operating typically at near ambient temperature and modest pressures) to the heterogeneous HB step operating typically at 450–500 °C and >200 bar. Furthermore, being an exothermic reversible reaction, very large recycles are necessary.An important aspect is that in the direct N2 + H2O electrolysis to ammonia, the electrocatalytic hydrogenation of coordinated N2 occurs through a concerted proton-coupled electron transfer (PCET) mechanism.64–67 This has relevant consequences. The limiting factor in ammonia synthesis in HB heterogeneous catalysts is the N2 splitting and the formation of strong chemisorbed N species on the surface of the iron catalyst, which hydrogenation requires high temperatures. Thus, high operation pressure (due to thermodynamics) and a large recycle. In addition, thermodynamic limitations associated with the reversible N2 + H2 reaction are no longer present, hydrogenation occurring through the involvement of H+/e− rather than molecular H2. Due to a different, enzymatic-like mechanism, the direct electrocatalytic N2 fixation to ammonia overcomes the limitations in heterogeneous HB catalysts and the consequent impact in terms of cost and energy. Direct ammonia production in a single-stage electrolyser offers advantages of potential cost reduction, process intensification and energy saving, even if the current performances are still quite far from the potential range of interest for possible commercialisation.68–74
Most of the literature data refer to relatively low current densities, often ∼0.1 mA cm−2 or even below, in addition to faradaic efficiencies (FE) still low (typically below 30–40%) due to the parallel formation of H2. In addition, data reported for high performances are made in unsuitable conditions for possible exploitation or using more costly sacrificial agents75,76 to report top performances, as in the widely cited Li-mediated mechanism of N2 fixation to ammonia.77–79 Several of these studies, in addition, are not realised under reaction conditions (electrolytes, type of reactors, and other aspects), which allow the production of an aqueous solution containing ammonium ions to be used as a fertilising solution.
A recent study reported continuous-flow electrosynthesis of ammonia by nitrogen reduction and hydrogen oxidation.79 Although performances are engaging, with a FE up to 61% at a current density of −6 mA cm−2, the method uses H2 as the source of protons rather than H2O, thus missing the advantages of the direct synthesis. The solution cannot be adapted to the direct synthesis of fertilizers. The method is an extension of the Li-mediated electrocatalytic synthesis of ammonia.74,80,81 Despite the high FE, the use of ethanol as a sacrificial agent for supplying protons, the type of electrolyte (LiBF4 in THF), and other issues make these methods unsuitable for direct production of fertilizers in a single electrocatalytic cell.
Therefore, there is still the issue of improving the performance (FE but principally current densities). However, it should be made under conditions and type of reactors (flow, zero-gap reactors, as commented before) that allow the possible exploitation. This is the main challenge, rather than proving mechanistic aspects and data reliability.82
Routes for direct production of fertilisers
While most of the studies in N2 fixation are limited to NH3 synthesis, exploring additional possibilities and analysing whether the direct production of fertilisers is possible represents the next challenge.19 There are two possibilities:1. Exploit the anodic section of the electrocatalytic cell to convert N2 to nitrate and thus produce an ammonium nitrate solution as the results of the two reactions of N2 to NH4+ (cathode) and N2 to NO3− (anode)
2. Electrocatalytic produce urea directly (in one step) from N2, H2O and CO2.
Making fertilisers in a distributed mode using these two approaches, although highly challenging, particularly when the direct use of N2 from the air is integrated into the cell (Fig. 1), can revolutionise fertiliser use and impact. It will (i) cut and stabilise costs, making them independent from the feed and energy variance, (ii) eliminate costs and environmental impact for distribution, (iii) decouple agricultural needs from market dependence and monopolies, (iv) reduce greenhouse gas emissions, (v) favours the creation of energy communities, and (vi) enhance the sustainable management and use of fertilisers. Thus, there are many motivations for greener production.
Ammonium nitrate solutions as fertiliser
Ammonium nitrate is a widely used fertiliser whose production requires a multistep process via ammonia synthesis, then oxidation to NO, absorption in water, and the reaction of nitric acid with ammonia and ammonium nitrate solid production (Fig. 3a). Similar is the process of urea production. Nitrate ions are highly mobile/soluble in soil water and can be assimilated directly by the root system of plants. The ammonium ion is the counter-ion, which may be either assimilated directly by roots or transformed to nitrate in the soil and then assimilated. While a solid is necessary to facilitate storage and transport, an aqueous solution should be made typically for distribution to the soil as fertiliser. Urea is readily converted to ammonium bicarbonate in the soil following assimilation paths as above.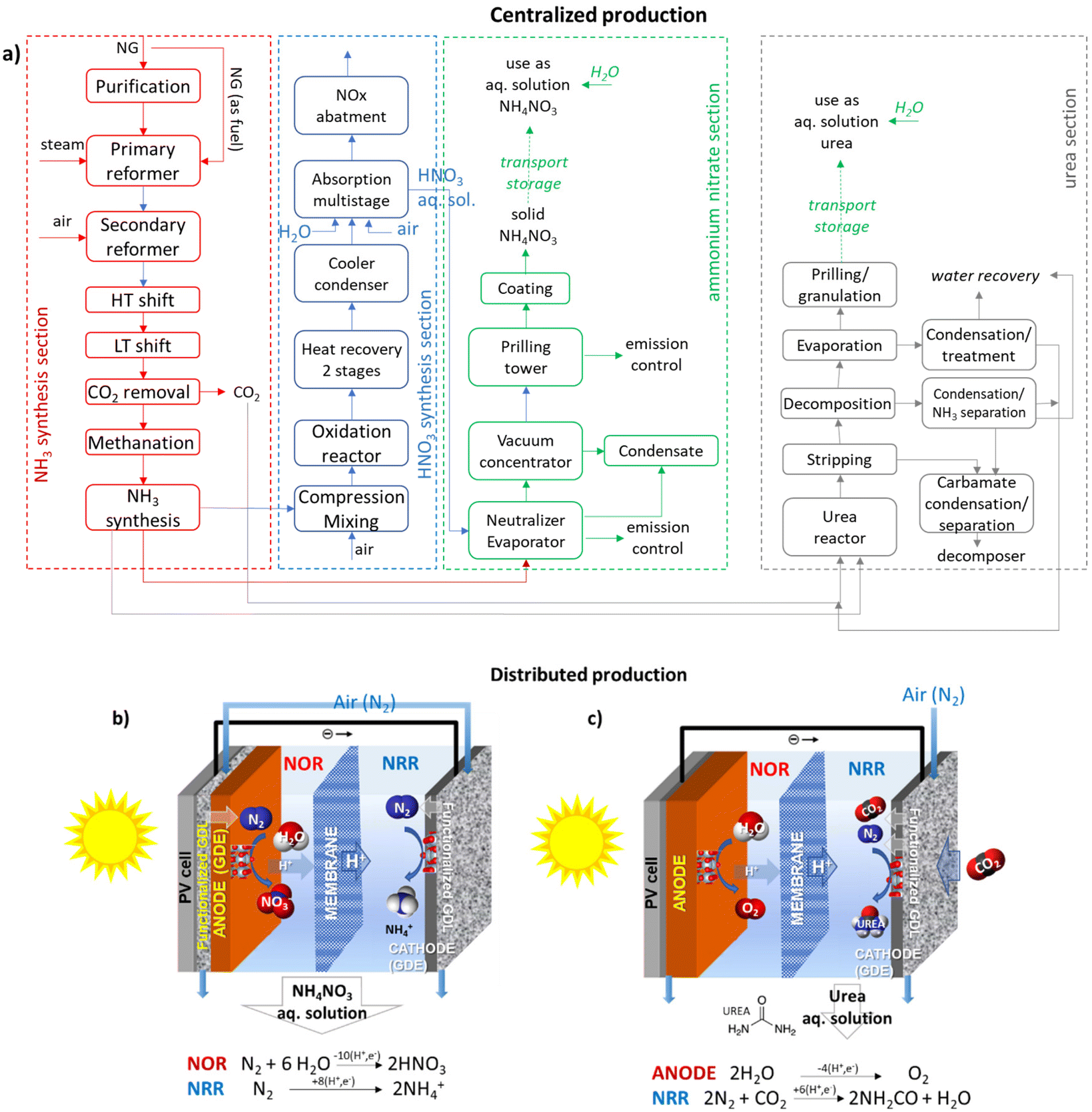 | ||
| Fig. 3 (a) Centralised production of ammonium nitrate and urea: simplified block diagrams of the sequence of unitary operations. NG: natural gas; HT: high temperature; LT: low temperature. (b) and (c) Distributed production: scheme of the photoelectrocatalytic reactors for producing ammonia nitrate and urea from the air. GDL: gas diffusion electrode (functionalised with elements to selectively capture N2 and CO2 and transport them to the electrocatalyst). GDE: gas diffusion electrode. PV: photovoltaic module. | ||
Besides the complexity of the multistep process and the use of fossil fuels (natural gas, NG) as the source of H2 and energy (with thus a significant carbon footprint), ammonium nitrate or urea should be converted to a solid for storage and transport. Making a solid requires concentrating the solution in an evaporator or concentrator and then spraying the concentrated melt into the top of a prilling tower. This step is energy-intensive (around 8500 MJ per ton N) and costly. In contrast, they are often used as an aqueous solution for distribution to the soil, thus avoiding the need for concentration/production of the solid. The scheme outlined in Fig. 3b shows that direct solar energy integration allows for overcoming the above issues. It realises a distributed, resilient and sustainable production of fertilisers with an approach that strongly reduces the current methods’ complexity.
Contrary to the abundant literature on direct N2 reduction to ammonia or ammonium ion (NRR),64,67,68,70,71,83–92 the studies on the N2 direct oxidation to nitric acid or nitrates (NOR) are limited, even if interest is increasing.93–98 Oxygen evolution reaction (OER) competes with NOR, as hydrogen evolution reaction (HER) competes with NRR. The challenge is suppressing these side reactions, thus enhancing faradaic efficiency (FE) while achieving a high rate of N2 activation and conversion, e.g., current density (CD). In general, in NRR and NOR, the FE and CD are still largely unsatisfactory. However, current results as productivities are better for NOR than for NRR, even if, generally, FE is still below 50%.
The challenge is double. Not only is it necessary to increase FE and CD, but also the use of operative conditions (such as electrolytes, avoiding sacrificial agents, continuous operations, type of reactor and other aspects) that are compatible with producing an ammonium nitrate aqueous solution to be used as fertiliser. Several of the best results in NRR or NOR do not respect this indication. Attention is often given to the design criteria of the electrocatalyst, which are not often relevant under practical conditions where other aspects, such as concentration gradients, mass transfer, etc., determine the behaviour and selectivity. For this reason, mechanistic indications (often based on DFT modelling) do not correspond to the right electrocatalytic mechanism. They typically do not include the aspects determining the electrocatalytic behaviour, such as inner- and outer-sphere electron transfer, proton-coupled electron transfer, multi-electron/proton transfer, the dependence of the surface potential on the electrode nanostructure, the role of the electrolyte and concentration gradients due to diffusional limitations, the complex interactions in the electric double layer, and other relevant aspects.64,71,99–113
The current NOR state of the art is based on using Fe single-atom catalysts (SAC) in N-doped carbon nanosheets, giving a FE ∼35% and CD ∼0.2 mA cm−2.96 The applied potential is ∼2.1 V (vs. RHE), and the electrolyte is a 0.05 M K2SO4 aqueous solution. Ru nanoclusters-coupled Mn3O4 electrocatalysts decorated with atomically dispersed Ru atoms103 are also interesting, giving a FE up to ∼29% at low potential (1.6 V vs. RHE) but strongly decreasing at higher potential (around 5% at 2 V) where nitrate production rate becomes more significant (around 35 μg h−1 mg−1). Nanoporous B13C2 gives the highest nitrate yield104 of around 60 μg h−1 mg−1, but at high potential (2.2 V vs. RGE) where FE is about 8%.
In terms of thermodynamic potentials, the reaction:
| N2(g) + 6H2O(l) → 2HNO3(g) + 10H+ + 10e− | (1) |
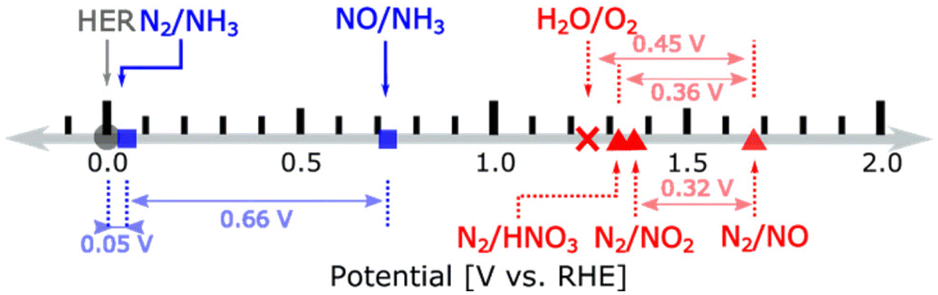 | ||
| Fig. 4 Redox couples for nitrogen reduction (blue) and oxidation (red) with thermodynamic potentials. HNO3 is gas. Reproduced with permission by Wan et al.93 Copyright ACS 2022. | ||
In NOR, nitrate formation from N2 occurs likely through two steps:93 (i) the conversion of N2 into the *NO intermediate (where the asterisk indicates chemisorption on an active site) and (ii) the transformation of *NO to nitrate. The former reaction occurs electrocatalytically and is considered the rate-limiting step.98 The latter is instead a non-electrochemical redox reaction. Design strategies to improve NOR are based on theoretical approaches to improve the first step. For example, how lower the energy of *N2 and the adsorption energy difference between *O and *OH. The latter describes the competition between OER and NOR. Wan et al.,93 using this approach (DFT calculations), concluded that (i) the rate-limiting step is the reaction of N2 with *O forming *N2O and (ii) promoting the reaction requires a weaker *O adsorption together with sites able for a strong N2 adsorption. However, as commented later, such modelling and derived mechanistic conclusions93,97,98 do not consider the intrinsic differences between catalysis and electrocatalysis.
For NRR, the use of inorganic donor–acceptor couples of Ni and Au nanoparticles supported on nitrogen-doped carbon gives a FE ∼60% at low potential applied (−0.14 V vs. RHE) corresponding to a very low CD (−0.1 mA cm−2). The electrolyte is 0.05M H2SO4 electrolyte. Many recent reviews have discussed this reaction, mechanism and type of electrocatalysts.64,65,67–69,106–117 Further discussion is thus unnecessary. However, the CD should be increased by two orders of magnitude to reach more realistic results for coupling with a photoelectrocatalytic (PEC) cell design, as illustrated in Fig. 3b.
Besides studying the single NOR or NRR reactions only, most current studies use very simple H-cell cells and electrodes (not GDE type). These studies do not provide the proper indications concerning electrocatalytic reactors and electrodes (outlined in Fig. 3b) for scaling and exploiting the results. Therefore, it is necessary to go beyond the mechanistic modelling of NOR and NRR from catalysis rather than an electrocatalysis perspective. Instead, the objective should be to perform the studies under conditions realistic regarding the possibility of application and determine the factors determining the behaviour under these conditions.
Reliability and reproducibility of the results and experimental protocols are important92,118–121 but not as crucial as often claimed because it is necessary to increase performance significantly. In these conditions, the impact of contaminations or other artefacts is minimised. The question is thus whether current studies and approaches would be effective in achieving this significant increase in performance (under relevant electrocatalytic conditions for industrial exploitability). We suggested that alternative approaches, including hybrid systems combining electrocatalysis with other methods, should be adopted.
We do not discuss the use of photocatalysis here because the gap in the performances106,122–125 is even larger than in the case of electrocatalysis or PEC approaches. Although photocatalysis is a valuable approach, the performances are too far from an application perspective.
A way to increase the performance is to couple electrocatalysis with non-thermal plasma (NTP). Hawtof et al.126 reported that this hybrid approach, schematically presented in Fig. 5, allows a record-high FE (up to 100%) for NH3 from N2/H2O at ambient conditions. NTP generates upon interaction with a water solution solvated electrons, which react with protons to produce hydrogen radicals that react with vibrationally-excited N2 species (created by plasma) to give ammonia selectively. They indicated the system as a plasma electrolytic system. However, proper mechanistic details are missing, particularly about the nature of the excited species generated in the plasma stream and their lifetime upon interaction with liquid water. In addition, the FE decays fast (Fig. 4b). Nevertheless, the results are interesting. Compared with recent results for similar electrically driven systems, the productivity increases by one to two orders of magnitude.
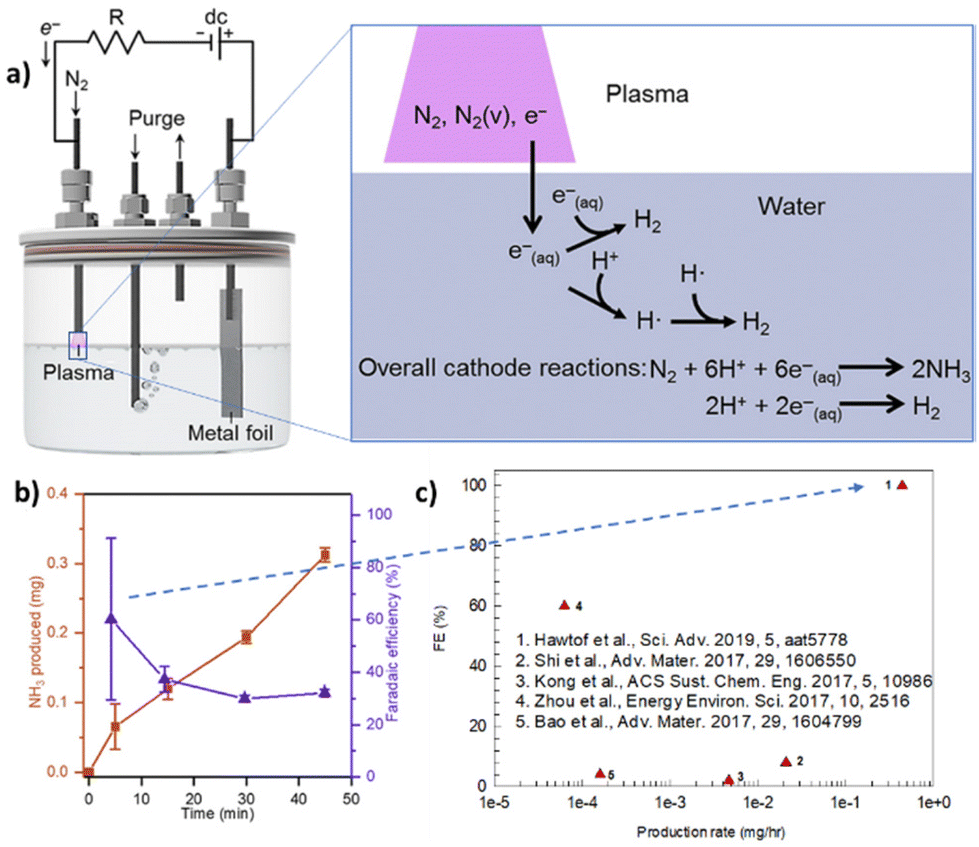 | ||
| Fig. 5 Plasma electrolytic N2 fixation to ammonia. (a) Left: Scheme of the device operated by a dc power supply and galvanostatically controlled using a resistor (R) in series. Right: Mechanism proposed with an indication of the potentially essential species contained in the plasma, such as vibrationally excited N2 [N2(v)], and in the water, such as solvated electrons [e−(aq.)], and their involvement in reactions, such as the generation of hydrogen radicals (H˙), that lead to NH3 formation. The overall reactions for N2 reduction to NH3 and H2 evolution (under acidic conditions) at the cathode are shown. (b) NH3 yield and efficiency in the plasma electrolytic system: total NH3 produced and corresponding faradaic efficiency after different processing times at 6 mA and pH 3.5. (c) Comparison of electro-driven N2 reduction to NH3 demonstrations at ambient temperature and pressure. They were elaborated with permission from Hawtof et al.126 Copyright American Association for the Advancement of Science 2019. | ||
Implementing such an approach in an artificial-like device is challenging but not impossible. However, it requires an entirely new design of the device aimed to maximise the effectiveness of the NTP and simultaneously miniaturing its design to realise the NTP at low applied potentials, those which PV panels can drive.
Several studies are ongoing on plasma-activated N2 fixation,127–132 although based on a different design than that reported in Fig. 5. Plasma-activated electrocatalysis for nitrogen fixation using solid oxide electrolysis cells (SOECs) rather than the scheme presented in Fig. 5 is also feasible.133 Ammonia or nitric oxide could be produced by using oxygen ion or proton conducting SOECs and a radiofrequency plasma (to activate N2), suppressing HER or OER, respectively. In both cases, the concentration of products is orders of magnitude higher than equilibrium without plasma at the same conditions. High selectivity to N2 fixation was observed.
An overview of catalytic and non-catalytic routes (thermal and non-thermal plasma, electrochemical, ultrasonic and photocatalysis) in N2 fixation was presented by Li et al.134 They suggest that coupling multiple processes, as outlined above, is the solution to overcome limitations by only the electrocatalytic approach. At the same time, introducing a further reactant, as shown below for the urea case, e.g. CO2, also offers clues to improve the performances.
Photocatalysis or photoelectrocatalysis is valuable because they use solar energy directly. However, the performances are still too low, and productivity has to be significantly enhanced. Instead, there are exciting opportunities in plasma catalysis or hybrid plasma-electrocatalysis systems. Likely, they will represent an area of future significant improvements.
Advances in direct urea synthesis
Alternatively or eventually, in integration with ammonium nitrate, the direct production of urea is possible (Fig. 3c). There are limited but increasing studies in this direction,135–137 although in part, starting from nitrate solutions rather than N2 because the reaction is faster and more selective.138–141 However, in the light of producing fertilisers, there is no reason to use nitrate solutions to produce urea. Using the nitrate solutions directly is better, eventually concentrating nitrate with established methods.Kayan and Köleli142 were among the first to report the electrocatalytic reduction of N2 and CO2 to urea using conducting polymer electrodes. They operate at high pressure (30 bar N2 + 30 bar CO2), forming ammonia, urea and formic acid in an aqueous 0.1 M Li2SO4/0.03 M H+ solution. However, the maximum FE was about 7%. Several authors have investigated the electrochemical co-reduction of CO2 and N2 for urea.103,116,136,137,143–152 An overview of results is presented in Table 1.
| Electrocatalyst | Urea yield | FEUrea (%) | Ref. |
|---|---|---|---|
| Pd1Cu1/TiO2-400 | 3.36 mmol h−1 g−1 | 9 | 137 |
| Ni3(BO3)2-150 | 9.7 mmol h−1 g−1 | 20 | 143 |
| BiFeO3/BiVO4 | 4.94 mmol h−1 g−1 | 17 | 152 |
| PdCu/TiO2 nanosheets | 3.36 mmol g−1 h−1 | 9 | 137 |
| Bi-BiVO4 | 5.91 mmol h−1 g−1 | 13 | 136 |
| CuPc NTs | 143 μg h−1 mgcat−1 | 13 | 144 |
| Cu-Bi alloy | 0.45 mg L–1 | 9 | 153 |
| Pd1Cu1-TiO2 | 167 molurea molPd−1 h−1 | 23 | 154 |
| Co-PMDA-2mbIM | 14.47 mmol h−1 g−1 | 48 | 155 |
| MoP | 12.4 μg h−1 mgcat−1 | 36 | 156 |
| InOOH nanocrystal with frustrated Lewis pairs | 6.85 mmol h−1 g−1 | 21 | 157 |
Different approaches were used to develop these electrocatalysts. Chen et al.137 based their development on creating oxygen vacancies in TiO2 nanosheets on which PdCu alloy nanoparticles were deposited. Although the mechanism is not well proven, oxygen vacancies likely convert CO2 to CO (the sites for CO2RR), while PdCu NPs activate N2. The critical step is the reaction between *N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N* and CO. The results are not remarkable, as shown in Table 1, although a high urea formation rate and FE were claimed. Cu–Bi alloy catalysts with surface defects153 based on a similar mechanism give comparable results.
N* and CO. The results are not remarkable, as shown in Table 1, although a high urea formation rate and FE were claimed. Cu–Bi alloy catalysts with surface defects153 based on a similar mechanism give comparable results.
To favour the coupling between the surface intermediates in CO2 and N2 electrocatalytic conversion, the presence of surface-frustrated Lewis pairs (FLPs) could be a possibility to favour the coupling. Yuan et al.143 explored this concept using flower-like nickel borate [Ni3(BO3)2]. This concept improves the FE, although performances remain still low. These authors reported that a space-charge region on the heterostructure interface could promote the adsorption and activation of the reactive molecules, reduce the generation of endothermic *NNH intermediates, and suppress CO poisoning. In other words, although not proven, they indicate that the heterostructure promotes a multi-electron/proton transfer (e.g., suppress *NNH intermediate). Yuan et al.157 further developed the concept of FLPs, indicating that InOOH nanocrystals contain electron-deficient Lewis acidic sites (In) and electron-rich Lewis basic (In–OH) able to activate enough closely N2 and CO2 to favour the formation of C–N bond. Reactant molecules’ bonding and antibonding orbitals interact with Lewis acid's unoccupied orbitals and Lewis base's nonbonding orbitals to generate the desired intermediates for urea synthesis in artificial FLPs. The performances are interesting but not significantly different from those of nickel borate electrocatalysts (Table 1).
Two alternative approaches seem preferable. The first is based on transition metal phosphides, emerging electrocatalysts.158,159 In MoP with an exposed (101) surface, Mo atoms can simultaneously absorb and activate CO2 and N2. Nearlylying hydrogenation sites enable the generating of the key reactive intermediate (*NHCOHN*) and form then urea at an ultralow potential of −0.27 V vs. RHE. SAC-based electrocatalysts, a popular topic in electrocatalysis, show better results.160–163 However, whether a single atom can perform the multiple electron transfers necessary for these complex electrocatalytic reactions is often questioned.
Nevertheless, cobalt pyromellitic dianhydride conductive MOF (Co-PMDA-2mbIM) is one of the best-performance electrocatalysts for the coreduction of CO2 and N2 for urea synthesis (Table 1).155 While often SAC electrocatalysts are prepared from MOF and analogous materials by thermal annealing at high temperatures, with thus a whole loss of the MOF structure, in the work of Yuan et al.155 they are deposited (as an ink) on a carbon substrate electrode. They demonstrate that by introducing the 2-mbIM (2-methyl benzimidazole) in the Co-PMDA MOF, the urea synthesis results significantly enhanced over the Co-PMDA MOF non-containing 2-mbIM. Still, performances are low because the current densities are about one mA cm−2. This is also because an H-type electrocatalytic cell was used, which is not ideal for obtaining reliable data, as commented before. The proposed mechanism is that CO2 is activated by 2-mbIM, while cobalt SAC activates N2 as a bicoordinated N2 molecule. CO2 is electro-catalytic reduced to CO, which then reacts (carbonylation) with the nearlying bicoordinated N2, forming an intermediate then reduced to urea (Fig. 6). The mechanism is not proven, and there are several open questions on it, especially regarding N2 coordination and the possibility of carbonylation of this intermediate by nearlying adsorbed CO to form an *NCON-type intermediate.
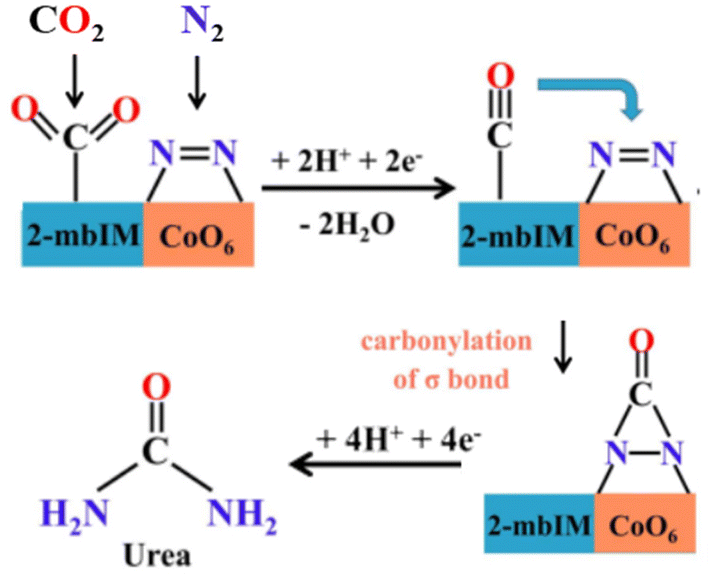 | ||
| Fig. 6 Reaction mechanism of urea electrocatalytic synthesis (at low potential) from CO2 and N2 on Co-PMDA-2mbIM. Adapted with permission by Yuan et al.155 – ESI. Copyright RCS 2022. | ||
Mukherjee et al.144 reported that copper phthalocyanine nanotubes (CuPc NTs), which also contain analogous SAC atoms (although based on Cu), have good performances, even if inferior to those discussed before (Table 1).
As commented before, urea formation is typically significantly enhanced (up to over one order of magnitude higher urea yields) when NOx rather than N2 is used as the nitrogen source. However, this nitrogen feed (NOx) is not motivated from an application perspective. In addition, often, it is not clarified enough that the reaction mechanism depends on the N-source.135,139 The activation of NOx would require sites to break the N–O bond. Thus, not surprisingly, electrocatalysts containing oxygen vacancies are active in the reaction.137,164,165 In N2 activation, it is necessary to have catalysts able to activate selectively molecular N2, which requires an entirely different type of active sites and likely a multi-electron/proton proton-concerted electron transfer mechanism.
Compared to converting N2 to ammonia (NRR), the urea synthesis mechanism requires simultaneous CO2 (CO2RR) activation and C–N bond formation. Several studies have been dedicated to this aspect, which depends on the nitrogen source, e.g., N2, NOx (gas)166 or nitrite/nitrate ions. The formation of the C–N bond is critical, as commented in Fig. 6, but also the various steps to hydrogenate the surface intermediates and transfer the electrons. Their understanding under reliable electrocatalytic conditions. e.g. not using too-simple H-type cells is the challenge to raise the performances to values for possible exploitation.
The solubility of N2 in the electrolytes used is quite low, automatically limiting the performance. The switch to gas-diffusion electrodes and zero-gap-type reactors is thus necessary to intensify the process and move to a better reactor concept, as presented in Fig. 3b. The electrocatalysts should be different under these conditions. Those selected for operations in H-type electrocatalytic cells may not be valid, as the factor determining the behaviour is different.
The critical step to understand and improve is the selective chemisorption of N2 molecules under electrocatalytic conditions. Besides operating under conditions of higher partial pressure of N2 at the electrocatalyst surface (e.g., pass from conventional to zero-gap approach), it is necessary to design the electrocatalysts with specific sites able to coordinate and activate N2. The knowledge of organometallic complexes activating nitrogen would help in this effort.167–170 These results indicate that multimetallic compounds168,171 are necessary to reduce and/or activate N2. In addition, the site for CO2 reduction should be near. Thus, the objective should be to design specific multinuclear sites able to perform all these functions.
The C–N reaction is another critical aspect. Carbonylation of N2 leads to the formation of amides,172 while forming urea would require a μ-η1:η1-N2 end-on bridging coordination.173 Very specific complexes and many d-electrons supplied by the two metals to the π-manifold of M-NN-M are needed to form bridging N2 molecules.
As noted for ammonium nitrate and urea synthesis, it is necessary to develop better specific electrodes and cells for this reaction. The reaction mechanism and the nature of the active sites for urea synthesis are still open. Many aspects, from the role of the operative conditions to the effect of the applied potential, must be studied in more detail.
Even if the studies on direct urea synthesis are only at an initial stage, and thus knowledge is still limited, turning the current approach and focusing better on developing innovative electrocatalysts operating under proper reaction conditions would be necessary rather than focusing studies on mechanistic aspects of catalysts still not having sufficient performances. Their design strategies are not reliable enough to accelerate the area's progress. Nevertheless, the possibilities of improvements are significant by better tuning and fostering research.
Sustainable food from the air
Sustainable food is used here to indicate carbohydrates, proteins, lipids and other food ingredients produced from CO2, H2O and N2 (captured from the air) using solar energy and hybrid electrocatalytic and microbial processes, as outlined in Fig. 1. Food production depends on land intensive and is highly dependent on many factors. It has a massive impact on the environment, requiring extensive water resources and being responsible for around one-quarter of the world's GHG emissions.174–176 Thus, sustainable food production from the air is a key societal objective to mitigate climate change, food security, resilient development, and overcome the burdens of agriculture.This is the objective of the SME “Solar Food”, which produces proteins (Solein®, a mixture of 65–70% protein, 5–8% fat – primarily unsaturated fats, 10–15% dietary fibres and 3–5% mineral nutrient) from CO2 using a bioprocess only. They indicate that their process is 20 times more efficient than photosynthesis (and 200 times more than meat production). However, it still depends on supplying external energy, CO2 and various ingredients.
An artificial tree-like approach, as outlined in Fig. 1, aims instead to maximise microbial capabilities while accelerating the critical steps, simplifying the quite complex machinery of photosynthesis in natural leaves. Thus, the objective is a significant intensification of the biological processes compared to natural leaves and the “solar food” approach mentioned above. Furthermore, selectivity can be tuned by controlling the microbial factory, optimising carbon and energy efficiency while reducing post-treatment costs. The technology aims for a distributed approach (Fig. 1).
These aspects are clarified in Table 2, which compares key metrics associated with industrial agriculture and artificial carbohydrate synthesis23 (as the solar food mentioned above) with indicative estimations for the technology discussed here (sustainable food from the air).
| Industrial agriculture | Artificial carbohydrate synthesis | Sustainable food from the air | |
|---|---|---|---|
| Technology readiness level (TRL) | 9 | 3–4 | 1 |
| Energy intensity (TWh per Gt of carb) | 300–2000 | 14000 |
<100 |
| GHG emissions intensity (Gt of CO2 eq. per Gt of carb) | 2 | ∼1.3 | <0 |
| Land-use intensity (million ha per Gt of carb) | 400 | 0.7 | 1–10 |
| Water intensity (Gt of H2O per Gt of carb) | 1300–1600 | 1 | <1 |
The technology of sustainable food from the air, even if at a very preliminary stage, has the potential to overcome the limitations of both agriculture (in terms of energy intensity, land use, environmental impact on water and GHG emissions) and artificial carbohydrate synthesis (as energy intensity, costs, and still significant impact on GHG). Sustainable food from the air is a carbon-negative technology because it uses only solar energy and only CO2, N2 and H2O from the air. However, it is only at a very preliminary stage of development; thus, the indications in Table 2 are potential values.
Hybrid processes for sustainable foods from the air
Using hybrid integrated (photo)electrocatalytic processes and microbial processes has the advantage over the total natural CO2 fixation (to produce carbohydrates and/or proteins) to make the whole process independent of the natural photosynthesis step and using photo-electro-assisted microbial conversions to accelerate the biosynthetic steps. By eliminating the natural photosynthesis step, it is possible to intensify the process and increase solar use efficiency. It also allows better controlling (through metabolic engineering) of the type of products. Fig. 7 illustrates schematically the scheme of producing sustainable food from the air in artificial-leaf devices. | ||
| Fig. 7 Producing sustainable food from the air in artificial-leaf devices. | ||
There are two possible C-sources to foster the microbial synthesis of carbohydrates, proteins, and lipids: formaldehyde and acetate ions. As commented later, acetate production is possible, while the first cannot be produced effectively by electrocatalytic routes from CO2. In addition, acetate ions are more stable and could be produced in media compatible with the following microbial steps, differently from formaldehyde. Methanol could be used but must be then in situ converted to formaldehyde,177 as commented below. However, metabolic engineering will likely enlarge the range of suitable carbon source substrates derived electrocatalytically from CO2 (CO, methane, formate, methanol acetate and ethanol)178 suitable for producing sustainable foods from the air.
Developing hybrid processes to create “artificial” paths to convert CO2 to food components is an already advanced idea. O'Brien et al.23 presented a perspective on converting CO2 to carbohydrates. However, they suggested converting CO2 catalytically to formaldehyde via methanol, followed by the conversion of formaldehyde to sugar or starch for human consumption, the latter step being indicated to be at a TRL of 3–4. Catalytic production of methanol from CO2 and H2 from electrolysis is an established process at the pilot scale. Converting methanol to formaldehyde is also an established industrial process. This indirect approach is thus well-established but cannot be used to produce food components from the air in a distributed approach (artificial tree-like). O'Brien et al.23 mentioned the possibility of electro-/photo-/plasma catalytic reduction of CO2 to formaldehyde. They cited the results of Kim et al.,179 which reported an 85% FE to formaldehyde using a photoelectrochemical device with a BiVO4 photoanode and a Cu cathode. However, this is achieved for a highly narrow potential (around 0.9 ± 0.05 V vs. RHE), very low current densities (0.4 mA cm−2) and thus very low productivity (∼3 μmol h−1). Thus, improvements would be necessary.
O'Brien et al.23 mentioned that carbon fixation to acetic acid/acetate would be preferable to avoid the many issues related to side reactions of formaldehyde. However, they did not consider the possibility of direct CO2 fixation to acetate. In addition, the possibility of directly capturing CO2 and water from the air is also not considered.
Other authors have reported hybrid chemoenzymatic180 and electro-biocatalytic181,182 systems to convert CO2 to starch, glucose, and other food components. The approach developed by Cai et al.180 is based on the chemo-enzymatic conversion of CO2 to starch using the known formaldehyde to sugars process, e.g. the formose reaction.183,184 After the chemocatalytic conversion of CO2 to formaldehyde via methanol, the production of starch occurs through several enzymatic steps: the first production of D-glyceraldehyde 3-phosphate (GAP), then its conversion to D-glucose-6-phosphate (G-6-P) and finally several steps to convert the latter to starch. This artificial starch anabolic pathway relies on engineered recombinant enzymes.180 It can be tuned to produce amylose or amylopectin at excellent rates and efficiencies (∼410 mg l−1 h−1 from CO2, at an estimated solar-to-starch efficiency of around 7%). This approach could synthesise other carbohydrates, such as cellulose or sugar, by substituting enzymes and modules.
Zheng et al.181 instead developed a hybrid electro-biocatalytic system for converting CO2 to glucose (Fig. 8). The CO2 is electrocatalytically converted to acetate in a two-step process, first to CO and then to acetate in a second electrocatalytic reactor (Fig. 8). The acetate is then converted to glucose by microorganism fermentation in a bioreactor using a genetically engineered Saccharomyces cerevisiae yeast (overexpression of heterologous glucose-1-phosphatase). The CO2 to CO conversion is made using an MEA (membrane electrode assembly) with a Ni–N–C single-atom electrocatalyst.185 CO faradaic efficiency was nearly 100%, and current density was above −150 mA cm−2. A grain-boundary-rich Cu electrocatalyst was used for the second step. An intermediate absorption step in 10 M NaOH (not shown in Fig. 8) is present between the two electrocatalytic reactors. The CO-to-acetate performance was a FE of about 50% at −0.67 V versus RHE at a current density to acetate of around −250 mA cm−2. A 1.0 M KOH electrolyte was used in the second electrocatalytic reactor. Other products of the reaction were 1-propanol, ethanol, ethylene and H2.
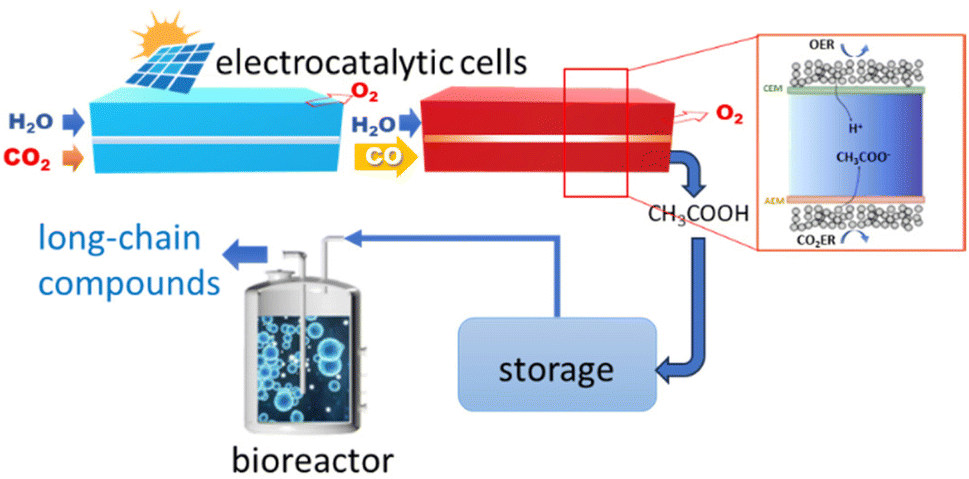 | ||
| Fig. 8 In vitro artificial sugar synthesis system. CO2 was first converted to pure acetic acid through two-step electrolysis; this product was then directly fed for microorganism fermentation in a bioreactor to produce long-chain compounds, for example, glucose. In the inset: schematic illustration of CO reduction (COR) to pure acetic acid in the solid-electrolyte reactor. Original figure based on Zheng et al.181 scheme. | ||
The design of the second electrocatalytic reactor, outlined in the inset of Fig. 8, allows producing an acetic acid solution with a purity of about 90 wt%. A thick anion exchange membrane in the porous solid-electrolyte reactor shown in the inset of Fig. 8 was necessary to slow down the crossover of alcohols from the cathode to the porous solid-electrolyte. Stable performances for 140 h under a current density of −250 mA cm−2 were observed. The porous solid-electrolyte reactor's acetic acid solution was fed to the bioreactor.
The general scheme of conversion of acetate to glucose is shown in Fig. 9. Blue arrows indicate the path suggested by Zheng et al.181 for their engineered yeasts. Alternative paths (indicated with green arrows) are also possible. Lim et al.186 discussed in detail acetate metabolism and its potential for biobased transformation into value-added chemicals. They evidenced that various chemicals can be produced from acetate as the carbon source in a microbial factory, including proteins for the food industry using E. coli BL21 strain.187
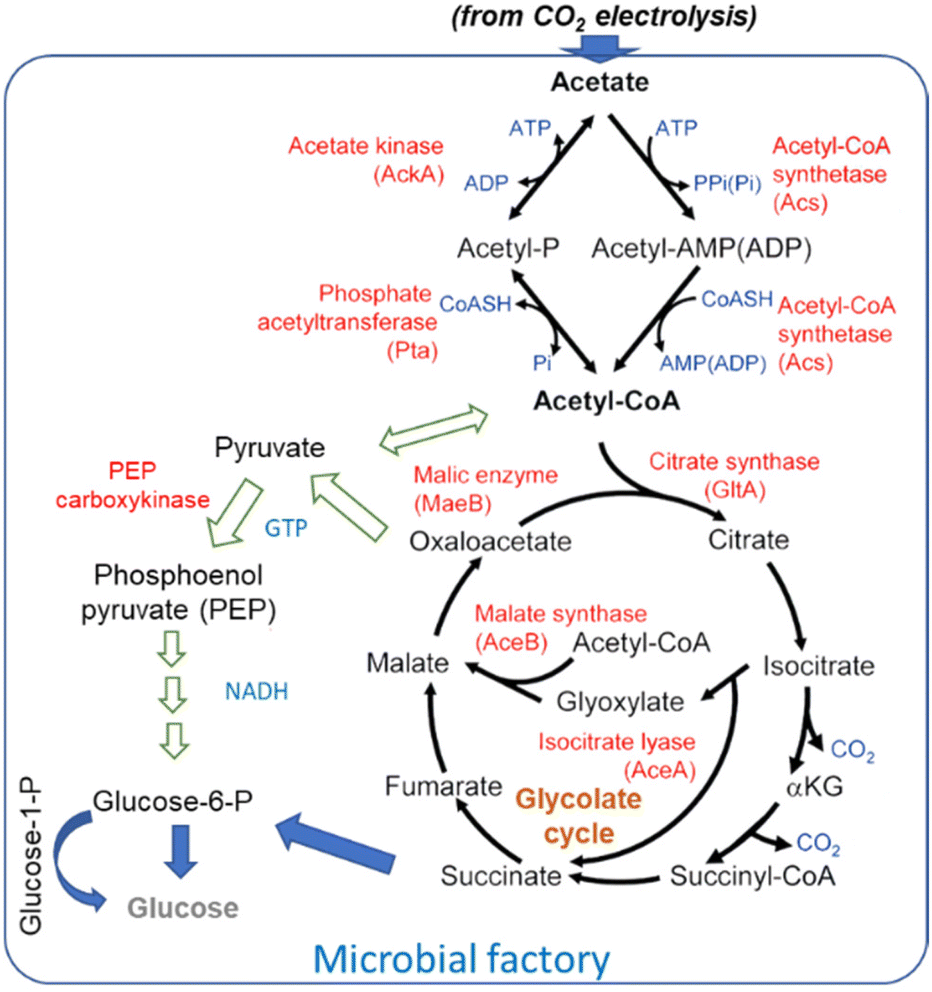 | ||
| Fig. 9 Overall pathway for acetate metabolism in microorganisms. Arrows in blue indicate the path suggested by Zheng et al.181 for their overexpressing enzymes, while the paths indicated by green arrows are alternative paths indicated by Lim et al.186 | ||
Hann et al.182 also used a hybrid approach of a first electrochemical conversion of CO2 to acetate, then used to grow food heterotrophically. The focus of the work, however, was on the second step. Algae, fungi, and crop plants (without the photosynthesis function) could be used in the second step. The solar-to-food energy conversion efficiency is about 4–18 times greater than biological photosynthesis, accelerating the step of carbon fixation through the electrocatalytic first step. An acetate-to-electrolyte ratio >0.4 would be necessary to integrate electrocatalytic carbon fixation with biological food production. The acetate serves as the carbon and energy source for food-producing organisms. Proteins, lipids and carbohydrates could be synthesised by changing the food-producing organisms.
Hann et al.182 used the alga Chlamydomonas (C. reinhardtii), which can grow heterotrophically on acetate in the dark. Using an acetate solution in tris–acetate–phosphate (TAP) buffer (pH 7.2, acetate-to-electrolyte salt ratio of 0.75) as the feed, they observed a yield of 0.28 g algae per g acetate with the utilisation of over 99% of acetate in the media. They also investigated the yeast Saccharomyces cerevisiae.
They also estimated the efficiency of the conversion. The conversion of sunlight and CO2 to food in their system (photovoltaics to electrolysis to acetate to yeast) is almost 18 times more efficient (as solar-to-biomass energy conversion) than typical food production. In addition, it is almost four times more efficient than the biological photosynthesis of crop plants (photosynthesis to crop plants).
The Sankey diagrams of solar energy to plant- and algae-based food production are shown in Fig. 10. The most critical steps (as energy losses) are the photovoltaic (PV) unit to generate green electricity and the electrochemical step of CO2 fixation. By improving these aspects, it is possible to increase the efficiency of the process further and intensify food production to natural processes while alleviating related environmental impacts.
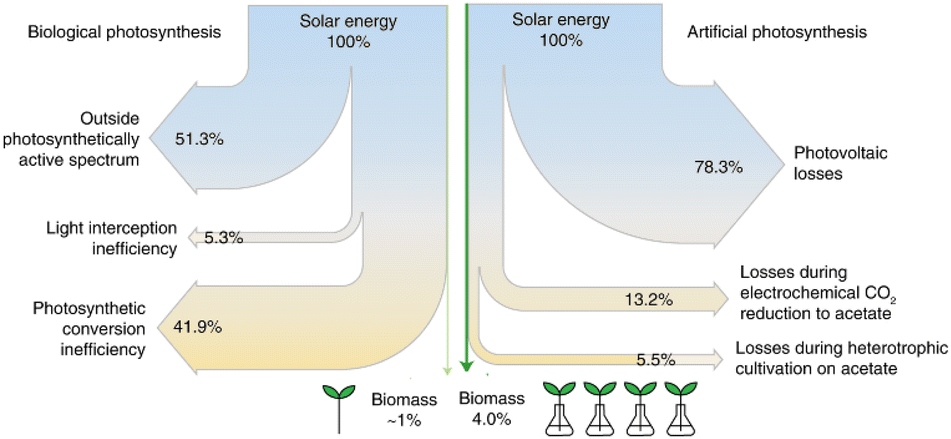 | ||
| Fig. 10 Sankey diagrams of solar energy to plant- and algae-based food production compare the efficiencies of artificial and biological photosynthesis. Adapted from Hann et al.182 Copyright Springer Nature 2022. | ||
Xu et al.188 discussed making edible protein from CO2via hybrid bioinorganic electrosynthesis. They suggest combining the strengths of microbial electrochemistry and microbial metabolisms to convert CO2 and surplus renewable electricity into single-cell protein. In addition, fixing N2 as ammonium ions, as discussed before, will provide the N-source to produce proteins.
Molitor et al.189 presented the concept of “power-to-protein” with a two-stage bioprocessing system: a first stage by anaerobic acetogenic bacteria and growing yeasts or fungi in a second stage under aerobic conditions with acetate as the intermediate metabolite. They thus share the concept of acetate as the intermediate key step but having a first biochemical stage (gas fermentation) rather than electrocatalytic. In addition, a 4:1 H2:CO2 mixture should be fed to the gas fermentation rather than producing onsite the H2 by water electrolysis. In addition, a startup period of 15 days is necessary. The stabilised acetate production is ∼0.6 g L−1 h−1.
An accurate comparison is not possible, being quite different reaction conditions. According to the data of Zheng et al.181 their electrocatalytic system produces ∼0.23 gacetate cm−2 h−1. Thus, productivity is comparable but without the need to feed H2. Note, however, that one order of magnitude lower current densities than those reported by Zheng et al.181 could be obtained when the electrocatalytic process is driven directly from an integrated PV unit. On the other hand, the integration of the PV unit avoids the dependence on an external supply of renewable energy, as commented before.
Therefore, although current studies demonstrate the feasibility of only some aspects of the overall scheme of producing food components from the air, they demonstrate that fixing CO2 by (photo)electrocatalytic conversion would accelerate and intensify the natural process.
Producing acetate by photoelectrocatalytic conversion of CO2 and H2O from the air is preferable, although alternatives (such as formate) or biotechnological are possible. Fixing N2 by photoelectrocatalytic approach would provide the N-source to produce proteins. Several studies exist on the further conversion of acetate (or other C-sources) in a microbial factory to food components or other chemicals.187,190–193
Various SMEs are entering the area. Biotechnology UK startup Deep Branch has designed a biochemical transformation process that turns carbon dioxide (CO2, plus nitrogen sources and mineral salts) into a protein-rich powder for animal food. The Deep Branch process converts carbon dioxide into a powder called Proton (a single-cell protein for the animal feed industry) with around 70% protein content. NovoNutrients startup company also upcycles CO2 into alternative proteins for human and animal food. Finnish startup Solar Foods uses CO2 and microbe to create an edible ingredient, roughly 20–25% carbohydrates, 5–10% fat, and 50% protein. NASA-based technology is used to turn carbon dioxide into food and bio-based products. The startup Kiverdi in California uses this technology to create the world's first ‘air-based meat’. They feed CO2, renewable H2 or syngas to gas carboxydotrophic fermentation micro-organisms.
Fixing CO2 to acetate (photo)electrocatalytically
This is a crucial step, and it is thus relevant to discuss shortly the state of the art in the area. Reviews on CO2 electrocatalytic conversion often do not discuss, or only marginally, the conversion to acetate,194 except a recent review by Wang et al.195 Several studies are attempting to realise, also recently, a direct process196–199 or via intermediate formation of CO.200,201 However, none of them analyze the electrocatalytic production of acetate in relation to the possible coupling with a next microbial step.The electrocatalytic synthesis of acetate from CO2 is an alternative to acetogenesis and the Wood–Ljungdahl pathway of CO2 fixation.202 Although well established, this anaerobic process requires a source of H2 (or the equivalent H+/e−):
| 2CO2 + 4H2 ⇌ CH3COO− + H+ + 2H2O ΔG° = −95 kJ mol−1 | (2) |
From an electrochemical perspective, the acetate synthesis from CO2 could be realized in a one-step or a two-step process via CO, as mentioned above. While the latter allows better performances, it also largely increases the costs associated mainly with electrochemical processes to the fixed costs of manufacturing the cell. The advantage of passing via intermediate CO is that the latter does not present the drawback of CO2 as carbonate formation in basic electrolyte203 besides the possibility of optimizing the single steps (CO2 to CO and CO to acetate). This issue, however, could be overcome by operating at neutral or slightly acid conditions. On the other hand, the CO solubility is lower than that of CO2. Even though the two-step conversion of CO2 to acetate is preferable,182,195,204,205 we indicate the one-step approach as more advisable because the difference in performances does not justify the higher fixed costs. However, more detailed studies are necessary.
There are also possible alternatives, from microbial electrosynthesis (MES),206,207 as also commented later, to new solutions that have to be validated more extensively, such as the piezoelectric reduction of CO2 to acetate with 100% selectivity by SnS nanobelts.208 A high efficiency in solar-to-acetate conversion from CO2 through MES coupled with stable photoanode has been reported.209 However, the efficiency is lower than 1%, and current densities are below 1 mA cm−2. Thus, this route has to be significantly improved. Photocatalytic CO2 fixation to acetate has been scarcely reported,210–212 but the productivities are too low for utilization. A CO2 photoelectrochemical conversion to acetate with 80% faradaic efficiency, using a mixed Fe–Cu oxide catalyst during visible light illumination, has been reported.213 However, products are of the order of μM, and current densities are very low.
The electrocatalysis direct conversion of CO2 (CO2RR) to acetate appears thus the preferable route in a preliminary comparison regarding the objective of air to food artificial trees. No comparative studies exist, which may change this indication, considering the fast scientific advances in this area.
The studies on CO2RR to acetate have been focused mainly on using Cu-based electrocatalysts, assuming that copper is essential to realize the C–C coupling and thus produce acetate or other C2+ chemicals by CO2 electroreduction.214–217 However, this is in contrast with other literature results, as commented before, proving that (i) also other electrocatalysts not based on copper could form C2+ products selectively in CO2RR when there is a sufficient concentration of CO2 adspecies, and (ii) selectivity in C2+ is determined from aspects not related to the electrocatalyst, such as electrode and cell design which determine the local concentration of reactants at the surface.
A mixed mechanism could be possible, e.g., that the electrode determines the critical stage of C–C coupling, while the selectivity is determined from the transport of ketene, a stable (closed shell) intermediate, away from the catalyst surface into solution where it reacts to form acetate.197 Ketene is highly reactive and unlikely to move away from the electrocatalyst. Its presence in the electrolyte is not proven. This mechanism is thus not convincing.
Heenen et al.197 indicate that the acetate selectivity increases at higher pH and depends on the applied potential due to this transport mechanism. At the same time, catalyst specificity remains for the critical step of C–C bond formation. However, this indication contrasts with the observation that (i) on a catalyst such as Pt, the selectivity could be increased up to 60% when enough concentration of CO2 adspecies is present,51 (ii) other mechanisms of C–C coupling not involving the surface reaction between CO adspecies (indicated by DFT studies) can be effective,218 (iii) on the same catalysts C2+ products may or not form depending on electrode design, (iv) the C1/C2 ratio in CO2RR varies insides the electrode profile,219 and (v) other electrocatalysts not containing copper show comparable selectivities to acetate.196,220
In addition, the reaction mechanism and key intermediate are undoubtfully determined.195 Four main paths and critical intermediate steps have been proposed: (i) the generation of *CO and subsequent surface dimerization, (ii) the generation of *CH3 and successive coupling, (iii) the formation and further reduction of an oxalate intermediate, and (iv) the disproportionation of acetaldehyde. Further mechanisms, as commented below, involve other intermediates. The role of species in the electrolyte rather than on the surface in determining the coupling mechanism is further supported by the many evidences on the role of surface nanocavities in enhancing C2+ formation.221,222
Thus, the question on the design criteria for optimal electrocatalysts, including aspects typically indicated as surface, morphology, facets, single or multiple sites, and catalyst design strategies to break the Brønsted–Evans–Polanyi relationship223–225 are not convincing, differently from the opinion of most of the reviews on CO2RR. Even with the increasing attention given to operation conditions (applied overpotential, electrolyte, anodic reaction) or the electrode/cell design, we suggest that the latter aspects dominate the possibility of forming selectively acetate in CO2RR. They also control C1 versus C2+ products and the ratio among the different C2 products (ethylene, ethanol, acetic acid, oxalic acid) or the ratio between C2 to C2+ products.
Regarding general trends in electrocatalysts for CO2RR to acetate, most of the literature studies investigate Cu-based catalysts for the motivations commented above. Minimal effort was made in searching different types of electrocatalysts, thus remaining an open area of investigation.
While initial electrocatalysts were not based on copper, e.g. a nitrogen-doped nanodiamond electrode was used, giving a faradaic efficiency over 90%,220 the later results based on copper, such as monodispersed mixtures of Cu and Ag nanoparticles,226 a 3D dendritic copper-cuprous oxide composite,199 Mo8/Cu heterostructures,227 and a conductive covalent organic framework with isolated Cu active sites228 give significantly worse or at the best comparable results to N-doped nanodiamond electrode. In addition, the nature of the active copper sites is very different in the above-cited examples. The mechanism proposed for each of these electrocatalysts is invalid for the others. Nevertheless, literature still indicates copper's unique role and reaction mechanisms based on specific copper active sites.
Besides these general considerations, analysing more specifically selected literature results is helpful.
Sun et al.229 reported an N-based Cu(I)/C-doped boron nitride (BN-C) electrocatalyst among the best results. The FE to acetic acid was up to 80% but at a medium-low current density (about 14 mA cm−2) when an ionic liquid (IL) containing LiI and water was used as the electrolyte. The compatibility of this type of electrolyte with the following microbial step is questionable, even considering a membrane purification step. The IL was the commercial 1-ethyl-3-methylimidazolium tetrafluoroborate ([Emim]BF4).
De et al.196 reported the electrocatalytic reduction of CO2 to acetic acid by a molecular manganese corrole complex. In a moderately acidic aqueous medium (pH 6), a selectivity of 63% and a turnover frequency of 8.25 h−1 has been shown by this catalyst when immobilised on a carbon paper electrode. This system shows better compatibility with the consecutive microbial step, but stability and absence of leaching must be verified. Furthermore, significant byproducts were CO and H2, which should be avoided. Current densities below one mA cm−2 are also too low.
The mechanism for acetate formation in electrochemical CO2 reduction on Cu was discussed by Heenen et al.,197 combining ab initio simulations, kinetic results and other experiments. They indicate that acetate selectivity (versus other products of CO2 reduction) can be rationalised from variations in electrolyte pH and the local mass transport properties of the catalyst and not from changes in Cu's intrinsic activity. The selectivity mechanism originates from the transport of ketene, a stable (closed shell) intermediate, away from the catalyst surface into solution, where it reacts to form acetate. Acetate selectivity increases with increasing pH, decreasing catalyst roughness and significantly varies with the applied potential.
A recent paper230 showed that on an electrocatalyst formed by atomically dispersed Cu in Ag, a highly selective acetate electrosynthesis from CO at high *CO coverage (10 atm pressure) is possible due to a constrained C2 adsorbate orientation. The CO-to-FE of 91% with an FE of 85% after 820 h was related to the generation of Cu nanoclusters of <4 atoms. The results were obtained in a flow reactor cell at a potential of −0.7 V vs. RHE in 5 M KOH electrolyte, incompatible with a following microbial step. Using a lower KOH concentration (1 M), the FE to acetate decreases to about 50%.
Zhu et al.199 reported a 3D dendritic copper-cuprous oxide composite fabricated by in situ reduction of an electrodeposited copper complex, giving acetic acid and ethanol with a C2 faradaic efficiency of 80% using a 0.1 M KCl aqueous solution and am H-type cell electrocatalytic cell. The FE shows a sharp maximum at around −0.4 V vs. RHE, which could create an issue in managing proper operations.
Zheng et al.231 discussed the formation of acetate/acetic acid as a part of a critical appraisal of the reduction of CO2 to C2 products, focusing on the connection between the fundamentals of reaction and efficient electrocatalysts. Their approach started from an atomistic mechanism of various C2 and C3 products to analyse the factors influencing the behaviour (local pH, overpotential, presence of surface adsorbates). However, indications of these aspects often contradict the first part of atomistic mechanisms. Also, the design principles of C2 electrocatalysts (chemical states, defective sites, nanostructure) are often inconsistent with the experimental results. For example, the experimental evidence shows that the selectivity of acetate/acetic acid depends drastically on the surface coverage by CO2 and cell design. At the same time, this aspect is not accounted for in mechanistic indications. Another example is the review of Wu et al.232 and a recent review on single-atom catalysts (SAC) for producing C2 products from CO2 electroreduction.233
Some other selected results are summarised in Table 3. Guo et al.234 observed good FE attributed to the unique co-coordination of pyridinic N and CO with copper, whose chemical state is between +1 and +2. FE of acetate up to ∼64% is attained at −0.37 V vs. RHE in a 0.5 M KHCO3 electrolyte. However, the current density is rather low.
| Electrode | Electrolyte | CD, mA cm−2 | FE, % | V vs. RHE | Ref. |
|---|---|---|---|---|---|
| a No potential applied, generated by the coupled photoanode (CuO/NtTiO2). | |||||
| Polymeric Cu–ligand complex core–shell microsphere | 0.5 M KHCO3 | ∼1 | 64 | −0.37 | 234 |
| Mo8 clusters@ Cu nanocubes | Saturated NaHCO3 solution | 110 | 49 | −1.13 | 227 |
| Mn-corrole on carbon paper | 0.1 M phosphate | ∼0.5 | 63 | −0.67 | 196 |
| Cu2O/GDL//CuO/NtTiO2 | 0.5 M KHCO3 | 0.03 | 76 | 236 | |
| N-doped nanodiamond | 0.5 M NaHCO3 | ∼0.5–1.0 | 78 | −0.8 | 220 |
| Cu NP/CNT | 0.5 M KHCO3 | 100 | 56 | −1.4 | 218 |
| FeO(OH)/N-CNT | 0.05 M KHCO3 | 0.36 | 61 | −0.5 | 238 |
| 3D dendritic CuO–Cu2O composite | 0.1 M KCl | 11 | 48 | −0.4 | 199 |
Zhang et al.233 studied a Mo8@Cu/TNA electrocatalyst, where TNA indicates a TiO2 nanotube array, while Mo8, the polyoxometalate (POM) Cu2Mo8O26·2H2O. The current densities are relevant but in addition to H2, various other organic products were detected (ethanol, acetate, methane, ethylene, ethane). They suggested that the interface of Cu planes and polyoxometalate clusters with abundant Cu–O–Mo active sites promote the generation of *CH3 and successive coupling with CO2 insertion, highlighting the need to realise a Cu–O–Mo interface for the rational design CO2RR to acetate. However, Giusi et al.235 showed a higher FE to acetic acid (∼62%) using Cu2O deposited over TNA. These tests were realised in a different electrochemical set-up, without a liquid electrolyte and with the gaseous CO2 flowing through a titania nanotube nanomembrane. De Brito et al.236 used a photoelectrocatalytic device, reporting an FE of about 75% to acetate for Cu2O deposited over a TNA electrode. In both these cases, thus, higher FE was reported without the need to realise Cu–O–Mo active sites and nanocomposites with a POM. The work of De Brito et al.236 is one of the few reporting the selective direct formation of acetate from CO2 in a PEC device, although stability has to be improved.
De et al.196 used a molecular manganese corrole complex for the electrocatalytic reduction of CO2 to acetic acid, as commented before. Genovese et al.,218 studying a supported copper electrocatalyst, proved the role of the interface with the electrolyte. A key intermediate generated in this interface is negatively charged CO2 radical species formed by outer-sphere electron transfer.
Liu et al.220 obtained high performances with an N-doped nanodiamond/Si rod array electrode. The FE to acetate was about 80% plus ∼10% to formate. However, the current density is rather low (Table 3). The result was not reproduced later. Finally, as commented before, Zhu et al.199 reported copper-cuprous oxide composite electrocatalysts. At a low overpotential (about 0.5 V), the FE to acetate was 48%, together with 32% FE to ethanol. It may also be noted in Table 3 that good performances could also be obtained without Cu, e.g. using Fe instead.238,239 In most of the studies, it is claimed that Cu has unique characteristics to form C2 products in CO2 electroreduction, while other metals can also give comparable results. Even a metal such as Pt, which is not selective to C2+ products under “normal” testing conditions, can show a FE > 60% when the surface coverage of CO2 on the electrocatalyst is enhanced.51 These results may question the validity of mechanistic indications about the active sites’ nature to form selectivity acetate in CO2 electrocatalytic conversion.
Performance improvement is possible by better understanding the reaction mechanism and nature of the active electrocatalysts. However, there are fundamental issues in the current approaches to these aspects. For example, Guo et al.234 mechanistic indications were based on theoretical calculations. They indicate the need to form Cu–O–Mo sites. However, higher FE could be obtained in different systems based on copper without molybdenum, POM, or even not containing copper. This remarks the general question in electrocatalysis that mechanistic indications can often not be generalised to other electrocatalytic systems. Other theoretical studies remarked on the role of dual heteroatom-sites for efficient electroreduction of carbon dioxide.240 However, the claim is for metal-free electrocatalysts and the dual site necessary is based on (N, B) heteroatoms, which have no relation in terms of characteristics with the Cu–O–Mo sites proposed by Guo et al.234
Zhou and Yeo241 analysed the different mechanisms of forming C–C bonds during CO2RR (including acetate) and the catalysts forming C2+ products, focusing on non-copper electrodes. They demonstrate that (i) there is no need for copper catalysts and (ii) to pass through chemisorbed CO coupling. These are the common assumptions of most theoretical studies on forming C2 products in the electrocatalytic reduction of CO2.231,242–248 Other studies also indicate that the interaction of CO2 with the metal catalyst may not be essential.249
More examples can be presented. It is not the aim to discuss here this question, only to remark that current mechanistic advances are still unable to develop solid bases for a rational and generalised design of the catalysts for electrocatalytic conversion of CO2 to acetate.
A critical analysis of the status in this area
Electrocatalytic direct conversion of CO2 to acetate (CO2RR to acetate) is actually the preferable route to realize artificial tree-like devices for air-to-food conversion. The motivations were discussed at the start of this section, even though specific detailed comparisons are missing and all technologies are at an initial stage of development, with thus expected improvements which may change these preliminary conclusions.Regarding specifically CO2 to acetate, the general initial discussion and the more specific examples analyzed show the many limitations in the literature approaches. Most of the literature studies investigate Cu-based catalysts for the abovementioned motivations. Minimal effort was made in searching different types of electrocatalysts, thus remaining an open area of investigation. In view of realising an artificial tree-like device, the direct (single-step) conversion of CO2 to acetate is preferable. Although FEs of up to 60% have been obtained, performance and stability improvements are necessary based on a better fundamental understanding. However, current studies have limits from this perspective, as commented above.
Maximising FE may not be a significant issue because some byproducts (such as C2–C3 alcohols) may also be co-feed to the following microbial step. However, minimising FE to H2 and CO or byproducts inhibiting microbial activity is instead an issue. However, no specific studies have been dedicated to considering the coupling with a following microbial step, in terms of acetate concentration to reach, compatible byproducts and those to avoid, the influence of the electrolyte, how to eliminate salts, and other relevant aspects commented on before.
Current density has to be analysed concerning the direct coupling with a photoactive element (a photoanode or an external PV cell, e.g. PEC or PV/EC approaches). However, studies on CO2 to acetate in PV/EC or PEC devices are limited.
Fixing CO2 to acetate (photo)electrocatalytically is thus feasible, but more dedicated studies would be necessary. It is mandatory to focus on the need for coupling with subsequent microbial steps. The investigation should go beyond the current limits in the approach both as the class of catalysts, their design and mechanistic approaches, understanding better the role of operative conditions, electrode and cell. How to switch the selectivity to acetate with respect to other C2 products (ethylene, ethanol, oxalic acid) or products with higher carbon numbers (isopropanol and others) is still a question largely unanswered.
Hybrid microbial processes
Chemolithoautotrophic reduction of CO2 to acetic acid in gas and gas-electro fermentation systems allows for significantly enhanced acetate productivity.250 MES can reduce CO2, CO, and water into acetic acid. This is an area of fast development.207,251 Current studies target the reuse of pure CO2 streams, such as from biogas, and co-feed of H2 is typically necessary.252 External electricity sources would also be necessary. Even if MES intensifies the process to cases discussed before, using a PEC approach to produce acetate would be preferable to realize compact distributed devices as outlined in Fig. 1. However, this is an aspect to verify when practical examples of this air-to-food conversion will be available.Bioelectrochemical photoreduction of CO2 is also an emerging possibility,253 but strongly conditioned from the need to operate (as other biotechnological systems) with the CO2 solubilised in the electrolyte, typically low. A design as that commented for gas-diffusion electrodes for the electrocatalysts can overcome these issues but has to be investigated concerning the bioelectrochemical photoreduction of CO2. The electrocatalytic vs. bioelectrocatalytic conversion of CO2 to acetate is apparently a more robust and compact technology and, thus, better suited for the objective discussed here. However, this is an aspect to be verified and clarified.
Although microbial electrosynthesis (MES) has been proven to be advantageous (in terms of process intensification) in various reactions of synthesis of chemicals,20,254,255 including acetate from CO2,256–258 it was not explicitly investigated concerning the conversion of an acetate feed to food components. Several aspects have to be taken into consideration to achieve this objective.192
As outlined in the simplified scheme reported in Fig. 9, the process is complex, with multiple paths that have to be controlled by microbial engineering. Different strategies helped to circumvent the challenges posed by acetate, including the effect of acetate on growth inhibition.192 Engineered E. coli are the most used microbial, with metabolic engineering aimed to improve acetate uptake and tolerance and control the conversion pathways.259
Acetate utilisation remains challenging.260 MES could add benefits, as it was proven for more straightforward microbial paths such as CO2 fixation. However, it is likely a longer-term objective after having consolidated the production of food components from substrates such as acetate produced by electrocatalytic CO2 fixation.
Green metrics
Even if the “air to chemicals” technologies are still at an early stage, with TRL below 3 (proof of the concept), analysing them in terms of green metrics compared to current processes to produce fertilizers or equivalent food components is useful. Green metrics are a collection of indicators that indicate the greenness of the new technology/process compared to the actual ones.261–271 Among them, green metrics often used include:- mass-based metrics of greenness, such as (i) E-factor (E), e.g., total waste/products, (ii) atom economy (AE), e.g. the conversion efficiency of a chemical process in terms of all atoms involved and the desired products or (iii) process mass intensity (PMI), e.g., total mass in a process or process step/mass of product;
- energy efficiency metrics, such as (i) energy efficiency (EE), e.g., the ratio between energy of the products and input energy, (ii) exergy efficiency (ExE), e.g., the ratio of the exergy of products and byproducts over that of input (exergy is the maximum amount of work that a flow of matter or energy can produce as it comes to equilibrium with a reference environment), or (iii) cumulative energy demand (CED), e.g., the total amount of primary energy potential used during the production cycle;
- environmental impact metrics, such as (i) carbon economy (CE), e.g. the carbon in product vs. the input carbon, which is equivalent to the C-factor, e.g. the total mass of CO2 emitted divided by the mass of product formed, (ii) wastewater intensity (WWI), e.g., the total mass of water in the process to mass of the final product, or (iii) the environmental impact factor (cEF), e.g. a comprehensive measures for co-produced waste and accounts for all process materials, including raw materials, reagents, solvents and water;
- green manufacturing metrics, such as (i) innovation green aspiration level (iGAL), e.g. an improvement of GAL (green aspiration level) that quantifies the environmental impact of producing a specific chemical while taking into account the complexity of the ideal synthetic process for producing the target molecule, (ii) green start (GS), e.g., a metric that using a radar diagram evaluates the greenness of a process with respect to targets of the 12 principles of green chemistry or (iii) renewable intensity (RI), e.g. the process intensity in using renewable materials and energy.
Several other metrics have been proposed and used in the literature to assess the greenness of chemical processes. They combine with sustainability indexes related to costs and societal impact and are often integrated with indicators derived by LCA (life cycle assessment) or related methodologies. While green and other metrics are often mixed with LCA indicators without clear distinction, it is useful to clarify that the two approaches are substantially different. LCA evaluate indexes of environmental impact based on assessing the entire value chain and reagents/products life, typically from the cradle to the grave. In green metrics, the boundary limits for the analysis are typically within the plant. Although LCA methods can evaluate comparatively process alternatives, the LCA indexes often refer to global impacts. Examples of LCA indexes include the impact on (i) climate change (kg CO2-eq.), (ii) ozone depletion (kg CFC-11-eq.), (iii) acidification (kg mol H+), and (iv) eutrophication of freshwater (kg PO4-eq.). Other indexes, however, are specific to the process's resource use, such as primary or renewable energy use (MJ). A key question, often unsolved, is whether the databases used to estimate the LCA indexes provide a reliable indication of the specific value chain (how the raw materials are produced, for example, or the life cycle of the products, rather than generic indications). In addition, even if attempts have been made to evaluate emerging technologies (for example, ex-ante LCA272–274), the experience shows several limits in extrapolating the results of ex-ante LCA to validate the technology operating at a larger scale or comparing very early-stage technologies with well-established and optimized industrial processes.
These concise aspects of green metrics and LCA are necessary to compare “air to chemicals” technologies and current solutions from the right perspective. A first attempt is presented in Table 2, where key metrics for industrial agriculture and artificial carbohydrate synthesis are compared with air-to-food technology.
This comparison already shows the limits of using green metrics or LCA approaches to obtain more precise insights on the greenness of the air-to-food approach. Agriculture and food production involves an extremely complex value chain, from producing the chemicals needed to grow the plants, including fertilisers, to water, materials and energy required for cultivation, harvesting, processing, transporting, etc. There are many direct and indirect emissions during each of the above steps, and only a small fraction of the biomass is converted to “food”, but the remaining part is also utilized and is part of the ecosystem cycle of agriculture. Applying green metrics to this sector is not effective. They were applied to biofuels.275,276 Still, the results are questionable277,278 and limited to a specific comparison (use of ethanol or methanol in biodiesel production or optimization of the reaction conditions).
Several attempts have been made to estimate food production and manufacturing using LCA methods.279–281 However, their analysis, discussed, for example, by Ahmad et al.,279 evidence that these results cannot be reliably compared to the “air-to-food” technology, being too different the objectives, the use of resources and energy, the land use and impact on society, the emissions, etc. The results of LCA on food manufacturing and processing indicate the large dependence of the results on countries, boundary limits, type of food, technology of manufacturing, and many other aspects.
The green metrics reported in Table 2 thus represent an attempt to compare “air-to-food” with industrial agriculture and the alternative of artificial carbohydrate synthesis. However, we believe that more precise indications would not be reliable at this stage.
It can be advanced, however, that an important green metric for evaluating the capability of agricultural and food production sustainable development is agricultural eco-efficiency (AEE).282 There is no unique definition, but it is the ratio between the economic value of the product and the environmental impact of the product formation. AEE accounts for the negative impact on the environment (such as fertilizer residue and soil productivity decline) and the positive impact (such as carbon dioxide absorption and noise reduction) in agricultural and food production. The AEE value in the EU ranges from 0.6 to 1, depending on region and type of agricultural/food production, but is below 0.5 in many other parts of the world.283 Although AEE improved in EU28 over the last decade, the average improvement is minimal, below 0.05.
The “air-to-food” technology has virtually no energy and raw materials consumption from sun and air. It does not use fertilizers, pesticides and other chemicals, including fungicides, bactericides, herbicides, etc. There are no emissions to the air but rather the capture of CO2 from the air. There is minimal production of waste. The estimated AEE would be thus >1, representing a significant step improvement in terms of the greenness of producing food components.
The case of fertilizer production is more defined and suitable for applying green and LCA metrics considerations. However, some conventional green metrics cannot be reliably applied due to the completely different objectives. Air-to-fertilizer technology is dedicated to an on-site distributed production where fertilisers are used. The energy for the process derives directly from the sun and raw materials from the air. The latter is the nitrogen source in the conventional fertilizer processes, but the hydrogen source (to make ammonia) and the energy for the process derives from fossil fuels. Large plants are necessary to optimize energy efficiency, with the many issues in terms of ecological and economic impact discussed before. There is a change in terms of societal impact and model of production, which cannot be ignored for a reliable comparison, but there are no green metrics to account for them correctly.
Nevertheless, it can be indicated that a typical value for energy consumption in manufacturing urea is ∼30 GJ per turea (based on LCA methods). This energy consumption translates to an average of about 2.2 t eq. CO2 per turea.284 The air-to-urea process uses only sun energy and captures CO2 from the air. Thus, both these green indicators are (close to) zero. As with most large-scale processes, waste production is minimized; thus, green metrics such as E-factor and AE have limited value. However, by eliminating the use of fossil fuels and with the raw materials captured from the air, air-to-urea technology significantly improves these green metrics. In urea processes, 0.5 tons of wastewater are produced per ton of urea.285 They are virtually eliminated in the air-to-urea technology, thus a clear improvement over green metrics such as WWI and cEF. Being air-to-urea technology based on the direct use of renewable (solar) energy, which is instead not used in conventional urea plants based on fossil fuels, energy efficiency metrics (EE, ExE and CED) are significantly improved, as also commented above regarding energy consumption. Also, indicators such as RI are significantly improved. There are no studies on iGAL or analogous metrics for fertilizer production because these metrics are suited for organic or speciality/pharmaceutical syntheses, even if applied in other industrial cases.286 However, using the GS metric (a holistic green chemistry metric)261,287 to compare air-to-urea technology with the conventional one regarding greenness may be attempted. The results are summarized in Fig. 11. There is an evident improvement in many of the twelve areas of green chemistry. In contrast, in some areas, the apparent absence of impact is mainly connected to the absence of relevance of this criterion to assess industrial urea manufacture.
 | ||
| Fig. 11 Green start (GS) metric to compare air-to-urea technology with the conventional one. P1–P12: Twelve principles of green chemistry. P1, prevention; P2, atom economy; P3, less hazardous chemical synthesis; P4, designing safer chemicals; P5, safer solvents and auxiliary substances; P6, increase energy efficiency; P7, use renewable feedstocks; P8, reduce derivatives; P9, catalysts; P10, design for degradation; P11, real-time analysis for pollution prevention; P12, safer chemistry for accident prevention. | ||
Outlooks and prospects
Even if preliminary results are available in the literature and suggestions to the possibility of producing fertilizers from N2, H2O and eventually CO2 or foods from CO2 in hybrid microbial systems, they do not consider the possibility of capturing these small molecules directly from the air. In this sense, the concept presented initially in Fig. 1 is novel.The principal limit is that no available results demonstrate the feasibility of the whole device, even at a laboratory preliminary scale. However, there are advances in the key components necessary for the whole technology. The main scope of this conceptual review is to show that they can be combined and better focused to address the challenge of an artificial tree-like technology to produce chemicals from the air. Many sustainability, geostrategic and resilience motivations are discussed in each section. They indicate the need to accelerate the development in this area. Many startup companies are entering the area, demonstrating the need for innovation.
The unavailability of a whole technology at a sufficient TRL level (above 5, but at least above 3) prevents the possibility of even preliminary economic estimations. However, as commented at the end of the introduction, this technology is disruptive. It may change the way of production, introducing decentralized production and dependence on externalities. In addition, it will also drastically decrease their impact on the environment and communities. Therefore, traditional economic assessments are unsuited to compare this technology with the current one.
Nevertheless, it may be a question whether this is just a vision or scientific curiosity or may represent an effective building block for a future radically new chemical production and agriculture. It is not easy to give scientific proof of this, and we may even argue that without a vision, there are no scientific advances. Several scientific discoveries, now building blocks of our life (photovoltaic panels as an example), were indicated not to have a possible relevant impact two to three decades ago. The main reason was the use of assessment criteria unable to consider the change in energy scenario, technology advances, and societal changes. In this perspective, we may indicate that the technology proposed and its possible extensions to produce other chemicals or energy carriers will be a key technology for a new production and economy model based on the strong integration with the territory.
This objective would require intense research, especially in integrating the single components in the scheme. Foster this possibility is the scope of this review, which is conceptual rather than a critical review in the traditional sense. Nevertheless, we attempt to evidence the many current limitations and pros/cons in the literature on the single component technologies for the whole artificial tree-like device. Many current studies do not address the demands from a technological perspective. However, this discussion is not the core objective of this contribution. Still, it aims to evidence only the need in several cases to turn the approach to accelerate the progress and exploit the results.
Conclusions
Making chemicals from the air may appear as a visionary, long-term objective. It has a potentially significant impact in intensifying the current food production technologies by one to two orders of magnitude, reducing the related GHG emissions and environmental impact, and overcoming the impact and costs of producing fertilisers.This review demonstrates that the essential bits of knowledge to translate this dream into a reality exist, even with the many challenges. We have discussed two major cases: producing (i) fertilisers and (ii) food components (carbohydrates, lipids, proteins) from the air. They represent relevant case examples for these processes being a new frontier for hybrid electrosyntheses in artificial-like devices. However, other chemicals may be synthesised following the same scheme. Organised in artificial tree-like systems, these devices would allow a distributed production, taking the raw materials from the air (CO2, H2O, N2) and sunlight.
Three key elements of the device were discussed: (i) the system to capture and concentrate these small molecules from the air, (ii) the electrocatalytic fixation of CO2 and N2, with the advances in producing directly (one-step) ammonium nitrate and/or urea solutions, and (iii) the sustainable production of food from the air, via a first stage of electrocatalytic CO2 fixation, particularly to form acetate.
Although there are advances in these areas, the possibility of combining them is still at an early stage. However, fast progress can be expected, and already the preliminary results show interesting perspectives, with a series of SMEs offering their technologies to produce fertilisers or food from the air, although implementing only part of the technological vision presented here.
Thus, it may be expected that when a large scientific community identifies this challenge, faster progress will be made with rapid implementation.
Hybrid electrosyntheses technologies, in terms of combining (i) electrocatalysis with selective functionalised membranes for capturing and concentrating CO2 or N2, (ii) electro- and photo-catalysis in a new type of reactors (such as zero-gap reactors), able to maximise the performances and not depending on external sources of renewable energy, and (iii) electrocatalysis and microbial factory, a crucial integration to develop these artificial-like devices. For this reason, hybrid electrosyntheses are indicated as frontier research to implement the new concepts discussed in this critical review and promote electrosynthesis to the next technological level.
List of acronyms
| ADP | Adenosine di-phosphate |
| AE | Atom economy |
| AEM | Anion exchange membrane |
| ATP | Adenosine triphosphate |
| Capex | Capital expenses |
| CD | Current density |
| CE | Carbon economy |
| CED | Cumulative energy demand |
| cEF | Environmental impact factor |
| CEM | Cation exchange membrane |
| CFD | Computational fluidodynamic |
| CFC-11 | CCl3F (CFC: chlorofluorocarbons) |
| CNT | Carbon nanotubes |
| CoA | Coenzyme A |
| COR | CO reduction |
| CO2RR | CO2 reduction reaction |
| DAC | Direct air capture |
| DFT | Density-functional theory |
| E | E-Factor |
| EE | Energy efficiency |
| ExE | Exergy efficiency |
| FE | Faradaic efficiency |
| FLP | Frustrated Lewis pairs |
| G-6-P | D-Glucose-6-phosphate |
| GAP | D-Glyceraldehyde 3-phosphate |
| GDE | Gas-diffusion electrode |
| GDL | Gas-diffusion layer |
| GHG | Greenhouse gas |
| GS | Green start |
| GTP | Guanosine-5′-triphosphate |
| HB | Haber–Bosch |
| HER | Hydrogen evolution reaction |
| iGAL | Improved green aspiration level |
| IL | Ionic liquid |
| MEA | Membrane electrode assembly |
| MES | Microbial electrosynthesis |
| MOF | Metal–organic framework |
| NADH | Nicotinamide adenine dinucleotide |
| NG | Natural gas |
| NOR | N2 oxidation reaction |
| NP | Nanoparticle |
| NRR | N2 reduction reaction |
| NT | Nanotubes |
| NTP | Non-thermal plasma |
| OER | Oxygen evolution reaction |
| PCET | Concerted proton-coupled electron transfer |
| PEC | Photoelectrocatalytic |
| PEP | Phosphoenolpyruvate |
| PMI | Process mass intensity |
| PSE | Porous solid-electrolyte |
| PV | Photovoltaic |
| RHE | Reversible hydrogen electrode |
| RI | Renewable intensity |
| SAC | Single-atom catalysts |
| SOEC | Solid oxide electrolysis cell |
| TRL | Technology readiness level |
| WWI | Wastewater intensity |
Author contributions
All authors equally contributed to preparing and revising this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The EU supported this work with the ERC Synergy SCOPE project (810182). GC also thanks the Alexander von Humboldt-Stiftung/Foundation (Humboldt Research Award).References
- P. D. Nguyen, T. M. Duong and P. D. Tran, J. Sci.: Adv. Mater. Devices, 2017, 2, 399–417 Search PubMed.
- A. Kumar, V. Hasija, A. Sudhaik, P. Raizada, Q. Van Le, P. Singh, T.-H. Pham, T. Kim, S. Ghotekar and V.-H. Nguyen, Chem. Eng. J., 2022, 430, 133031 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- G. Centi, S. Perathoner, C. Genovese and R. Arrigo, Chem. Comm., 2023, 59, 3005–3023 RSC.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS PubMed.
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed.
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- J. R. Galan-Mascaros, Catal.: Sci. Technol., 2020, 10, 1967–1974 RSC.
- S. Haussener, Sol. Energy, 2022, 246, 294–300 CrossRef CAS.
- J. D. J. Olmos and J. Kargul, Int. J. Biochem. Cell Biol., 2015, 66, 37–44 CrossRef PubMed.
- J. Z. Zhang and E. Reisner, Nat. Rev. Chem., 2020, 4, 6–21 CrossRef CAS.
- J. P. Torella, C. J. Gagliardi, J. S. Chen, D. K. Bediako, B. Colón, J. C. Way, P. A. Silver and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2337–2342 CrossRef CAS PubMed.
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918 RSC.
- R. M. Evans, B. Siritanaratkul, C. F. Megarity, K. Pandey, T. F. Esterle, S. Badiani and F. A. Armstrong, Chem. Soc. Rev., 2019, 48, 2039–2052 RSC.
- N. J. Claassens, C. A. R. Cotton, D. Kopljar and A. Bar-Even, Nat. Catal., 2019, 2, 437–447 CrossRef CAS.
- G. Yi, B. Wang, Y. Feng, D. Fang, L. Yang, W. Liu, Y. Zhang, P. Shao, S. G. Pavlostathis, S. Luo, X. Luo and A. Wang, Resour., Conserv. Recycl., 2022, 181, 106230 CrossRef CAS.
- K. Rabaey and R. A. Rozendal, Nat. Rev. Microbiol., 2010, 8, 706–716 CrossRef CAS PubMed.
- S. Das, L. Diels, D. Pant, S. A. Patil and M. M. Ghangrekar, J. Electrochem. Soc., 2020, 167, 155510 CrossRef CAS.
- P. Dessì, L. Rovira-Alsina, C. Sánchez, G. K. Dinesh, W. Tong, P. Chatterjee, M. Tedesco, P. Farràs, H. M. V. Hamelers and S. Puig, Biotechnol. Adv., 2021, 46, 107675 CrossRef PubMed.
- E. Yang, H. O. Mohamed, S.-G. Park, M. Obaid, S. Y. Al-Qaradawi, P. Castaño, K. Chon and K.-J. Chae, Bioresour. Technol., 2021, 320, 124363 CrossRef CAS PubMed.
- G. Hochman, A. S. Goldman, F. A. Felder, J. M. Mayer, A. J. M. Miller, P. L. Holland, L. A. Goldman, P. Manocha, Z. Song and S. Aleti, ACS Sustainable Chem. Eng., 2020, 8, 8938–8948 CrossRef CAS.
- C. P. O'Brien, M. J. Watson and A. W. Dowling, ACS Energy Lett., 2022, 7, 3509–3523 CrossRef.
- C. R. Tracy, N. Laurence and K. A. Christian, Am. Nat., 2011, 178, 553–558 CrossRef PubMed.
- K. Wan, X. Gou and Z. Guo, J. Bionic Eng., 2021, 18, 501–533 CrossRef.
- C. Liu, Y. Xue, Y. Chen and Y. Zheng, Sci. Rep., 2015, 5, 17757 CrossRef CAS PubMed.
- A. R. Parker and C. R. Lawrence, Nature, 2001, 414, 33–34 CrossRef CAS PubMed.
- C. Lei, Y. Guo, W. Guan, H. Lu, W. Shi and G. Yu, Angew. Chem., Int. Ed., 2022, 61, e202200271 CrossRef CAS PubMed.
- Y. Guo, W. Guan, C. Lei, H. Lu, W. Shi and G. Yu, Nat. Commun., 2022, 13, 2761 CrossRef CAS PubMed.
- P. A. Kallenberger and M. Fröba, Commun. Chem., 2018, 1, 28 CrossRef.
- H. Kim, S. R. Rao, E. A. Kapustin, L. Zhao, S. Yang, O. M. Yaghi and E. N. Wang, Nat. Commun., 2018, 9, 1191 CrossRef PubMed.
- F. Fathieh, M. J. Kalmutzki, E. A. Kapustin, P. J. Waller, J. Yang and O. M. Yaghi, Sci. Adv., 2018, 4, eaat3198 CrossRef PubMed.
- W. Xu and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1348–1354 CrossRef CAS.
- X. Huang, Q. Qin, Q. Ma and B. Wang, Water, 2022, 14, 3487 CrossRef CAS.
- X. Liu, D. Beysens and T. Bourouina, ACS Mater. Lett., 2022, 4, 1003–1024 CrossRef CAS.
- M. Bozorg, B. Addis, V. Piccialli, Á. A. Ramírez-Santos, C. Castel, I. Pinnau and E. Favre, Chem. Eng. Sci., 2019, 207, 1196–1213 CrossRef CAS.
- J. W. Yoon, H. Chang, S.-J. Lee, Y. K. Hwang, D.-Y. Hong, S.-K. Lee, J. S. Lee, S. Jang, T.-U. Yoon, K. Kwac, Y. Jung, R. S. Pillai, F. Faucher, A. Vimont, M. Daturi, G. Férey, C. Serre, G. Maurin, Y.-S. Bae and J.-S. Chang, Nat. Mater., 2017, 16, 526–531 CrossRef CAS PubMed.
- S. Liguori, K. Lee and J. Wilcox, J. Membr. Sci., 2019, 585, 52–59 CrossRef CAS.
- B. A. MacKay and M. D. Fryzuk, Chem. Rev., 2004, 104, 385–402 CrossRef CAS PubMed.
- IEA (International Energy Agency), Specific energy consumption for CO2 capture using current DAC technologies, 2022, https://www.iea.org/data-and-statistics/charts/specific-energy-consumption-for-co2-capture-using-current-dac-technologies Search PubMed.
- N. McQueen, K. V. Gomes, C. McCormick, K. Blumanthal, M. Pisciotta and J. Wilcox, Prog. Energy, 2021, 3, 032001 CrossRef.
- M. Li, E. Irtem, H.-P. Iglesias van Montfort, M. Abdinejad and T. Burdyny, Nat. Commun., 2022, 13, 5398 CrossRef CAS PubMed.
- O. Gutiérrez-Sánchez, B. Bohlen, N. Daems, M. Bulut, D. Pant and T. Breugelmans, ChemElectroChem, 2022, 9, e202101540 CrossRef.
- A. Adamu, F. Russo-Abegão and K. Boodhoo, BMC Chem. Eng., 2020, 2, 2 CrossRef.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- N. H. Khdary, A. S. Alayyar, L. M. Alsarhan, S. Alshihri and M. Mokhtar, Catalysts, 2022, 12, 300 CrossRef CAS.
- H.-C. Fu, F. You, H.-R. Li and L.-N. He, Front. Chem., 2019, 7, 525 CrossRef CAS PubMed.
- S. Sun, H. Sun, P. T. Williams and C. Wu, Sustainable Energy Fuels, 2021, 5, 4546–4559 RSC.
- M. Li, K. Yang, M. Abdinejad, C. Zhao and T. Burdyny, Nanoscale, 2022, 14, 11892–11908 RSC.
- H. Huang, R. C. Samsun, R. Peters and D. Stolten, React. Chem. Eng., 2022, 7, 2573–2581 RSC.
- B. C. Marepally, C. Ampelli, C. Genovese, T. Saboo, S. Perathoner, F. M. Wisser, L. Veyre, J. Canivet, E. A. Quadrelli and G. Centi, ChemSusChem, 2017, 10, 4442–4446 CrossRef CAS PubMed.
- D. K. Yoo and S. H. Jhung, J. Mater. Chem. A, 2022, 10, 8856–8865 RSC.
- B. Nagendra, S. Salman, C. Daniel, P. Rizzo and G. Guerra, Mater. Adv., 2023, 4, 881–889 RSC.
- S. Wang, Z. Zhang, S. Dai and D.-e. Jiang, ACS Mater. Lett., 2019, 1, 558–563 CrossRef CAS.
- Y. Han, Y. Yang and W. S. W. Ho, Membranes, 2020, 10, 365 CrossRef CAS PubMed.
- B. Siritanaratkul, M. Forster, F. Greenwell, P. K. Sharma, E. H. Yu and A. J. Cowan, J. Am. Chem. Soc., 2022, 144, 7551–7556 CrossRef CAS PubMed.
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed.
- P. Senthilkumar, M. Mohapatra and S. Basu, RSC Adv., 2022, 12, 1287–1309 RSC.
- L. Yuan, S. Zeng, X. Zhang, X. Ji and S. Zhang, Mater. Rep.: Energy, 2023, 3, 100177 CAS.
- P. Zhu, Z.-Y. Wu, A. Elgazzar, C. Dong, T.-U. Wi, F.-Y. Chen, Y. Xia, Y. Feng, M. Shakouri, J. Y. Kim, Z. Fang, T. A. Hatton and H. Wang, Nature, 2023, 618, 959–966 CrossRef CAS PubMed.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS.
- D. A. Daramola and M. C. Hatzell, ACS Energy Lett., 2023, 8, 1493–1501 CrossRef CAS.
- M. Ouikhalfan, O. Lakbita, A. Delhali, A. H. Assen and Y. Belmabkhout, Energy Fuels, 2022, 36, 4198–4223 CrossRef CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- B. Wu, Y. Lin, X. Wang and L. Chen, Mater. Chem. Front., 2021, 5, 5516–5533 RSC.
- P. Garrido-Barros, J. Derosa, M. J. Chalkley and J. C. Peters, Nature, 2022, 609, 71–76 CrossRef CAS.
- A. Kaiprathu, P. Velayudham, H. Teller and A. Schechter, J. Solid State Electrochem., 2022, 26, 1897–1917 CrossRef CAS.
- C. Lee and Q. Yan, Curr. Opin. Electrochem., 2021, 29, 100808 CrossRef CAS.
- F. Habibzadeh, S. L. Miller, T. W. Hamann and M. R. Smith, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2849–2853 CrossRef CAS.
- Y. Ren, C. Yu, X. Tan, Q. Wei, Z. Wang, L. Ni, L. Wang and J. Qiu, Energy Environ. Sci., 2022, 15, 2776–2805 RSC.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- Y.-X. Lin, S.-N. Zhang, Z.-H. Xue, J.-J. Zhang, H. Su, T.-J. Zhao, G.-Y. Zhai, X.-H. Li, M. Antonietti and J.-S. Chen, Nat. Commun., 2019, 10, 4380 CrossRef PubMed.
- C. M. Johansen, E. A. Boyd and J. C. Peters, Sci. Adv., 2022, 8, eade3510 CrossRef CAS PubMed.
- K. Li, S. Z. Andersen, M. J. Statt, M. Saccoccio, V. J. Bukas, K. Krempl, R. Sažinas, J. B. Pedersen, V. Shadravan, Y. Zhou, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2021, 374, 1593–1597 CrossRef CAS PubMed.
- J. M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. C. Lin, M. Cargnello, T. F. Jaramillo and J. K. Nørskov, Energy Environ. Sci., 2017, 10, 1621–1630 RSC.
- R. Tort, O. Westhead, M. Spry, B. J. V. Davies, M. P. Ryan, M.-M. Titirici and I. E. L. Stephens, ACS Energy Lett., 2023, 8, 1003–1009 CrossRef CAS PubMed.
- X. Cai, C. Fu, H. Iriawan, F. Yang, A. Wu, L. Luo, S. Shen, G. Wei, Y. Shao-Horn and J. Zhang, iScience, 2021, 24, 103105 CrossRef CAS PubMed.
- N. Lazouski, Z. J. Schiffer, K. Williams and K. Manthiram, Joule, 2019, 3, 1127–1139 CrossRef CAS.
- X. Fu, J. B. Pedersen, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Z. Andersen, A. Xu, N. H. Deissler, J. B. V. Mygind, C. Wei, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2023, 379, 707–712 CrossRef CAS PubMed.
- K. Li, S. G. Shapel, D. Hochfilzer, J. B. Pedersen, K. Krempl, S. Z. Andersen, R. Sažinas, M. Saccoccio, S. Li, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, ACS Energy Lett., 2022, 7, 36–41 CrossRef CAS.
- S. Li, Y. Zhou, K. Li, M. Saccoccio, R. Sažinas, S. Z. Andersen, J. B. Pedersen, X. Fu, V. Shadravan, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Joule, 2022, 6, 2083–2101 CrossRef CAS PubMed.
- J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, ACS Energy Lett., 2019, 4, 2986–2988 CrossRef CAS.
- M. Nazemi and M. A. El-Sayed, Acc. Chem. Res., 2021, 54, 4294–4304 CrossRef CAS PubMed.
- F. Jiao and B. Xu, Adv. Mater., 2019, 31, e1805173 CrossRef PubMed.
- Y. Tanabe and Y. Nishibayashi, Chem. Soc. Rev., 2021, 50, 5201–5242 RSC.
- N. Morlanés, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. Gascon, Chem. Eng. J., 2021, 408, 127310 CrossRef.
- Y. Fu, Y. Liao, P. Li, H. Li, S. Jiang, H. Huang, W. Sun, T. Li, H. Yu, K. Li, H. Li, B. Jia and T. Ma, Coord. Chem. Rev., 2022, 460, 214468 CrossRef CAS.
- C. Cui, H. Zhang, R. Cheng, B. Huang and Z. Luo, ACS Catal., 2022, 12, 14964–14975 CrossRef CAS.
- H. Hosono, Faraday Discuss., 2023, 243, 9–26 RSC.
- I. Garagounis, A. Vourros, D. Stoukides, D. Dasopoulos and M. Stoukides, Membranes, 2019, 9, 112 CrossRef CAS PubMed.
- X. Fu, J. Zhang and Y. Kang, Chem Catal., 2022, 2, 2590–2613 CrossRef CAS.
- Z. Li, M. Li, J. Yang, M. Liao, G. Song, J. Cao, F. Liu, Z. Wang, S. Kawi and Q. Lin, Catal. Today, 2022, 388–389, 12–25 CrossRef CAS.
- H. Wan, A. Bagger and J. Rossmeisl, J. Phys. Chem. Lett., 2022, 13, 8928–8934 CrossRef PubMed.
- Y. Wang, T. Li, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202115409 CrossRef CAS PubMed.
- C. Dai, Y. Sun, G. Chen, A. C. Fisher and Z. J. Xu, Angew. Chem., Int. Ed., 2020, 59, 9418–9422 CrossRef CAS PubMed.
- Y. Guo, S. Zhang, R. Zhang, D. Wang, D. Zhu, X. Wang, D. Xiao, N. Li, Y. Zhao, Z. Huang, W. Xu, S. Chen, L. Song, J. Fan, Q. Chen and C. Zhi, ACS Nano, 2022, 16, 655–663 CrossRef CAS.
- E. Tayyebi, Á. B. Höskuldsson, A. Wark, N. Atrak, B. M. Comer, A. J. Medford and E. Skúlason, J. Phys. Chem. Lett., 2022, 13, 6123–6129 CrossRef CAS.
- M. Anand, C. S. Abraham and J. K. Nørskov, Chem. Sci., 2021, 12, 6442–6448 RSC.
- R. E. Warburton, A. V. Soudackov and S. Hammes-Schiffer, Chem. Rev., 2022, 122, 10599–10650 CrossRef CAS.
- G.-F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L.-X. Ding and H. Wang, Small Methods, 2019, 3, 1800337 CrossRef.
- Y. Ying, K. Fan, J. Qiao and H. Huang, Electrochem. Energy Rev., 2022, 5, 6 CrossRef CAS.
- Q. J. Bruch, G. P. Connor, N. D. McMillion, A. S. Goldman, F. Hasanayn, P. L. Holland and A. J. M. Miller, ACS Catal., 2020, 10, 10826–10846 CrossRef CAS.
- Z. Nie, L. Zhang, X. Ding, M. Cong, F. Xu, L. Ma, M. Guo, M. Li and L. Zhang, Adv. Mater., 2022, 34, 2108180 CrossRef CAS PubMed.
- J. Lan, M. Luo, J. Han, M. Peng, H. Duan and Y. Tan, Small, 2021, 17, 2102814 CrossRef CAS PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- K. Ithisuphalap, H. Zhang, L. Guo, Q. Yang, H. Yang and G. Wu, Small Methods, 2019, 3, 1800352 CrossRef.
- H. Xu, K. Ithisuphalap, Y. Li, S. Mukherjee, J. Lattimer, G. Soloveichik and G. Wu, Nano Energy, 2020, 69, 104469 CrossRef CAS.
- S. Y. Park, Y. J. Jang and D. H. Youn, Catalysts, 2023, 13, 639 CrossRef CAS.
- A. Biswas, S. Bhardwaj, T. Boruah and R. S. Dey, Mater. Adv., 2022, 3, 5207–5233 RSC.
- K. Bhunia, S. K. Sharma, B. K. Satpathy and D. Pradhan, Mater. Adv., 2022, 3, 888–917 RSC.
- J. John, D.-K. Lee and U. Sim, Nano Convergence, 2019, 6, 15 CrossRef PubMed.
- X. Xue, R. Chen, C. Yan, P. Zhao, Y. Hu, W. Zhang, S. Yang and Z. Jin, Nano Res., 2019, 12, 1229–1249 CrossRef CAS.
- A. Braun, D. K. Bora, L. Lauterbach, E. Lettau, H. Wang, S. P. Cramer, F. Yang and J. Guo, Catal. Today, 2022, 387, 186–196 CrossRef CAS PubMed.
- Y. H. Moon, N. Y. Kim, S. M. Kim and Y. J. Jang, Catalysts, 2022, 12, 1015 CrossRef CAS.
- H. Zhu, X. Ren, X. Yang, X. Liang, A. Liu and G. Wu, SusMat, 2022, 2, 214–242 CrossRef CAS.
- S. Zhao, X. Lu, L. Wang, J. Gale and R. Amal, Adv. Mater., 2019, 31, 1805367 CrossRef CAS.
- S. Mukherjee, X. Yang, W. Shan, W. Samarakoon, S. Karakalos, D. A. Cullen, K. More, M. Wang, Z. Feng, G. Wang and G. Wu, Small Methods, 2020, 4, 1900821 CrossRef CAS.
- C. Tang and S. Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- C. Tang and S. Z. Qiao, Joule, 2019, 3, 1573–1575 CrossRef.
- L. F. Greenlee, J. N. Renner and S. L. Foster, ACS Catal., 2018, 8, 7820–7827 CrossRef CAS.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- P.-W. Huang and M. C. Hatzell, Nat. Commun., 2022, 13, 7908 CrossRef CAS PubMed.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- X. Niu, A. Shi, D. Sun, S. Xiao, T. Zhang, Z. Zhou, X. a. Li and J. Wang, ACS Catal., 2021, 11, 14058–14066 CrossRef CAS.
- C. Guo, J. Ran, A. Vasileff and S.-Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- R. Hawtof, S. Ghosh, E. Guarr, C. Xu, R. M. Sankaran and J. N. Renner, Sci. Adv., 2019, 5, eaat5778 CrossRef PubMed.
- H. Chen, D. Yuan, A. Wu, X. Lin and X. Li, Waste Disposal Sustainable Energy, 2021, 3, 201–217 CrossRef PubMed.
- L. R. Winter and J. G. Chen, Joule, 2021, 5, 300–315 CrossRef CAS.
- S. Li, J. A. Medrano, V. Hessel and F. Gallucci, Processes, 2018, 6, 248 CrossRef CAS.
- Z. Huang, A. Xiao, D. Liu, X. Lu and K. Ostrikov, Plasma Processes Polym., 2022, 19, 2100198 CrossRef CAS.
- A. Anastasopoulou, R. Keijzer, S. Butala, J. Lang, G. Van Rooij and V. Hessel, J. Phys. D: Appl. Phys., 2020, 53, 234001 CrossRef CAS.
- E. Vervloessem, M. Aghaei, F. Jardali, N. Hafezkhiabani and A. Bogaerts, ACS Sustainable Chem. Eng., 2020, 8, 9711–9720 CrossRef CAS.
- M. Tsampas, R. Sharma, S. Welzel and R. van de Sanden, ECS Meet. Abstr., 2021, MA2021-01, 878 CrossRef.
- D. Li, L. Zan, S. Chen, Z.-J. Shi, P. Chen, Z. Xi and D. Deng, Natl. Sci. Rev., 2022, 9, nwac042 CrossRef CAS PubMed.
- C. Chen, N. He and S. Wang, Small Sci., 2021, 1, 2100070 CrossRef CAS.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS.
- Y. Wang, Y. Yu, R. Jia, C. Zhang and B. Zhang, Natl. Sci. Rev., 2019, 6, 730–738 CrossRef CAS PubMed.
- M. Jiang, M. Zhu, M. Wang, Y. He, X. Luo, C. Wu, L. Zhang and Z. Jin, ACS Nano, 2023, 17, 3209–3224 CrossRef CAS PubMed.
- J. Leverett, T. Tran-Phu, J. A. Yuwono, P. Kumar, C. Kim, Q. Zhai, C. Han, J. Qu, J. Cairney, A. N. Simonov, R. K. Hocking, L. Dai, R. Daiyan and R. Amal, Adv. Energy Mater., 2022, 12, 2201500 CrossRef CAS.
- X. Wei, X. Wen, Y. Liu, C. Chen, C. Xie, D. Wang, M. Qiu, N. He, P. Zhou, W. Chen, J. Cheng, H. Lin, J. Jia, X.-Z. Fu and S. Wang, J. Am. Chem. Soc., 2022, 144, 11530–11535 CrossRef CAS PubMed.
- D. B. Kayan and F. Köleli, Appl. Catal., B, 2016, 181, 88–93 CrossRef CAS.
- M. Yuan, J. Chen, Y. Xu, R. Liu, T. Zhao, J. Zhang, Z. Ren, Z. Liu, C. Streb, H. He, C. Yang, S. Zhang and G. Zhang, Energy Environ. Sci., 2021, 14, 6605–6615 RSC.
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS.
- Y. Liu, X. Tu, X. Wei, D. Wang, X. Zhang, W. Chen, C. Chen and S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300387 CrossRef CAS PubMed.
- M. Qiu, X. Zhu, S. Bo, K. Cheng, N. He, K. Gu, D. Song, C. Chen, X. Wei, D. Wang, Y. Liu, S. Li, X. Tu, Y. Li, Q. Liu, C. Li and S. Wang, CCS Chem., 2023, 1–11 Search PubMed.
- C. Chen, S. Li, X. Zhu, S. Bo, K. Cheng, N. He, M. Qiu, C. Xie, D. Song, Y. Liu, W. Chen, Y. Li, Q. Liu, C. Li and S. Wang, Carbon Energy, 2023, e345 CrossRef (Early View).
- Y. Huang, Y. Wang, Y. Wu, Y. Yu and B. Zhang, Sci. China: Chem., 2022, 65, 204–206 CrossRef CAS.
- G. Bharath, G. Karthikeyan, A. Kumar, J. Prakash, D. Venkatasubbu, A. K. Nadda, V. K. Gupta, M. A. Haija and F. Banat, Appl. Energy, 2022, 318, 119244 CrossRef CAS.
- X. Zhu, X. Zhou, Y. Jing and Y. Li, Nat. Commun., 2021, 12, 4080 CrossRef CAS PubMed.
- P. Roy, A. Pramanik and P. Sarkar, J. Phys. Chem. Lett., 2021, 12, 10837–10844 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Shi, S. Li, X. Wang and G. Zhang, Chem. Sci., 2021, 12, 6048–6058 RSC.
- W. Wu, Y. Yang, Y. Wang, T. Lu, Q. Dong, J. Zhao, J. Niu, Q. Liu, Z. Hao and S. Song, Chem Catal., 2022, 2, 3225–3238 CrossRef CAS.
- L. Pan, J. Wang, F. Lu, Q. Liu, Y. Gao, Y. Wang, J. Jiang, C. Sun, J. Wang and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216835 CrossRef CAS PubMed.
- M. Yuan, J. Chen, H. Zhang, Q. Li, L. Zhou, C. Yang, R. Liu, Z. Liu, S. Zhang and G. Zhang, Energy Environ. Sci., 2022, 15, 2084–2095 RSC.
- D. Jiao, Y. Dong, X. Cui, Q. Cai, C. R. Cabrera, J. Zhao and Z. Chen, J. Mater. Chem. A, 2023, 11, 232–240 RSC.
- M. Yuan, H. Zhang, Y. Xu, R. Liu, R. Wang, T. Zhao, J. Zhang, Z. Liu, H. He, C. Yang, S. Zhang and G. Zhang, Chem Catal., 2022, 2, 309–320 CrossRef CAS.
- B. Kim, T. Kim, K. Lee and J. Li, ChemElectroChem, 2020, 7, 3578–3589 CrossRef CAS.
- Z. Pu, T. Liu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, P. Ji, W. Hu, J. Liu and S. Mu, Adv. Funct. Mater., 2020, 30, 2004009 CrossRef CAS.
- J. Yang, W. Liu, M. Xu, X. Liu, H. Qi, L. Zhang, X. Yang, S. Niu, D. Zhou, Y. Liu, Y. Su, J.-F. Li, Z.-Q. Tian, W. Zhou, A. Wang and T. Zhang, J. Am. Chem. Soc., 2021, 143, 14530–14539 CrossRef CAS PubMed.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- H. Hu, J. Wang, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, J. Mater. Chem. A, 2022, 10, 5835–5849 RSC.
- J. Su, L. Zhuang, S. Zhang, Q. Liu, L. Zhang and G. Hu, Chin. Chem. Lett., 2021, 32, 2947–2962 CrossRef CAS.
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS.
- N. Cao, Y. Quan, A. Guan, C. Yang, Y. Ji, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2020, 577, 109–114 CrossRef CAS PubMed.
- H. Wan, X. Wang, L. Tan, M. Filippi, P. Strasser, J. Rossmeisl and A. Bagger, ACS Catal., 2023, 13, 1926–1933 CrossRef CAS.
- A. Chauhan, H. S. Karnamkkott, S. M. N. V. T. Gorantla and K. C. Mondal, ACS Omega, 2022, 7, 31577–31590 CrossRef CAS PubMed.
- D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS.
- M. D. Fryzuk, Chem. Rec., 2003, 3, 2–11 CrossRef CAS PubMed.
- M. P. Shaver and M. D. Fryzuk, Adv. Synth. Catal., 2003, 345, 1061–1076 CrossRef CAS.
- M. Keener, F. Fadaei-Tirani, R. Scopelliti, I. Zivkovic and M. Mazzanti, Chem. Sci., 2022, 13, 8025–8035 RSC.
- K. Ueda, Y. Sato and M. Mori, J. Am. Chem. Soc., 2000, 122, 10722–10723 CrossRef CAS.
- L. S. Yamout, M. Ataya, F. Hasanayn, P. L. Holland, A. J. M. Miller and A. S. Goldman, J. Am. Chem. Soc., 2021, 143, 9744–9757 CrossRef CAS PubMed.
- R. Hannah, R. Pablo and R. Max, Our World in Data, 2022 Search PubMed.
- M. Clark, M. Springmann, M. Rayner, P. Scarborough, J. Hill, D. Tilman, J. I. Macdiarmid, J. Fanzo, L. Bandy and R. A. Harrington, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2120584119 CrossRef CAS.
- T. Nemecek, N. Jungbluth, L. M. i Canals and R. Schenck, Int. J. Life Cycle Assess., 2016, 21, 607–620 CrossRef.
- A. Sarwar and E. Y. Lee, Synth. Syst. Biotechnol., 2023, 8, 396–415 CrossRef CAS PubMed.
- C. Zhang, C. Ottenheim, M. Weingarten and L. Ji, Front. Bioeng. Biotechnol., 2022, 10, 874612 CrossRef.
- C. W. Kim, M. J. Kang, S. Ji and Y. S. Kang, ACS Catal., 2018, 8, 968–974 CrossRef CAS.
- T. Cai, H. Sun, J. Qiao, L. Zhu, F. Zhang, J. Zhang, Z. Tang, X. Wei, J. Yang, Q. Yuan, W. Wang, X. Yang, H. Chu, Q. Wang, C. You, H. Ma, Y. Sun, Y. Li, C. Li, H. Jiang, Q. Wang and Y. Ma, Science, 2021, 373, 1523–1527 CrossRef CAS PubMed.
- T. Zheng, M. Zhang, L. Wu, S. Guo, X. Liu, J. Zhao, W. Xue, J. Li, C. Liu, X. Li, Q. Jiang, J. Bao, J. Zeng, T. Yu and C. Xia, Nat. Catal., 2022, 5, 388–396 CrossRef CAS.
- E. C. Hann, S. Overa, M. Harland-Dunaway, A. F. Narvaez, D. N. Le, M. L. Orozco-Cárdenas, F. Jiao and R. E. Jinkerson, Nat. Food, 2022, 3, 461–471 CrossRef CAS PubMed.
- J. B. García Martínez, K. A. Alvarado, X. Christodoulou and D. C. Denkenberger, J. CO2 Util., 2021, 53, 101726 CrossRef.
- I. V. Delidovich, A. N. Simonov, O. P. Taran and V. N. Parmon, ChemSusChem, 2014, 7, 1833–1846 CrossRef CAS PubMed.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- H. G. Lim, J. H. Lee, M. H. Noh and G. Y. Jung, J. Agric. Food Chem., 2018, 66, 3998–4006 CrossRef CAS PubMed.
- S. Leone, F. Sannino, M. L. Tutino, E. Parrilli and D. Picone, Microb. Cell Fact., 2015, 14, 106 CrossRef.
- M. Xu, H. Zhou, R. Zou, X. Yang, Y. Su, I. Angelidaki and Y. Zhang, One Earth, 2021, 4, 868–878 CrossRef.
- B. Molitor, A. Mishra and L. T. Angenent, Energy Environ. Sci., 2019, 12, 3515–3521 RSC.
- G. Lozano Terol, J. Gallego-Jara, R. A. Sola Martínez, M. Cánovas Díaz and T. de Diego Puente, Microb. Cell Fact., 2019, 18, 151 CrossRef PubMed.
- K. Novak, R. Kutscha and S. Pflügl, Biotechnol. Biofuels, 2020, 13, 177 CrossRef CAS PubMed.
- R. Kutscha and S. Pflügl, Int. J. Mol. Sci., 2020, 21, 8777 CrossRef CAS.
- D. Kiefer, M. Merkel, L. Lilge, M. Henkel and R. Hausmann, Trends Biotechnol., 2021, 39, 397–411 CrossRef CAS PubMed.
- G. Feng, W. Chen, B. Wang, Y. Song, G. Li, J. Fang, W. Wei and Y. Sun, Chem. – Asian J., 2018, 13, 1992–2008 CrossRef CAS PubMed.
- H. Wang, J. Xue, C. Liu, Z. Chen, C. Li, X. Li, T. Zheng, Q. Jiang and C. Xia, Curr. Opin. Electrochem., 2023, 39, 101253 CrossRef CAS.
- R. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U.-P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schöfberger, Angew. Chem., Int. Ed., 2020, 59, 10527–10534 CrossRef CAS PubMed.
- H. H. Heenen, H. Shin, G. Kastlunger, S. Overa, J. A. Gauthier, F. Jiao and K. Chan, Energy Environ. Sci., 2022, 15, 3978–3990 RSC.
- A. Saxena, H. Singh and M. Nath, Mater. Renewable Sustainable Energy, 2022, 11, 115–129 CrossRef.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef PubMed.
- P. Zhu, C. Xia, C.-Y. Liu, K. Jiang, G. Gao, X. Zhang, Y. Xia, Y. Lei, H. N. Alshareef, T. P. Senftle and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2010868118 CrossRef CAS PubMed.
- X. Fu, Y. Wang, H. Shen, Y. Yu, F. Xu, G. Zhou, W. Xie, R. Qin, C. Dun, C. W. Pao, J. L. Chen, Y. Liu, J. Guo, Q. Yue, J. J. Urban, C. Wang and Y. Kang, Mater. Today Phys., 2021, 19, 100418 CrossRef CAS.
- S. W. Ragsdale and E. Pierce, Biochim. Biophys. Acta, 2008, 1784, 1873–1898 CrossRef CAS PubMed.
- J. Y. T. Kim, P. Zhu, F.-Y. Chen, Z.-Y. Wu, D. A. Cullen and H. Wang, Nat. Catal., 2022, 5, 288–299 CrossRef CAS.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- N. S. Romero Cuellar, C. Scherer, B. Kaçkar, W. Eisenreich, C. Huber, K. Wiesner-Fleischer, M. Fleischer and O. Hinrichsen, J. CO2 Util., 2020, 36, 263–275 CrossRef CAS.
- J.-N. Hengsbach, B. Sabel-Becker, R. Ulber and D. Holtmann, Appl. Microbiol. Biotechnol., 2022, 106, 4427–4443 CrossRef CAS PubMed.
- B. Bian, S. Bajracharya, J. Xu, D. Pant and P. E. Saikaly, Bioresour. Technol., 2020, 302, 122863 CrossRef CAS PubMed.
- W. Tian, N. Li, D. Chen, Q. Xu, H. Li, C. Yan and J. Lu, Angew. Chem., Int. Ed., 2023, 62, e202306964 CrossRef CAS PubMed.
- B. Bian, L. Shi, K. P. Katuri, J. Xu, P. Wang and P. E. Saikaly, Appl. Energy, 2020, 278, 115684 CrossRef CAS.
- D. Zeng, H. Wang, X. Zhu, H. Cao, W. Wang, Y. Zhang, J. Wang, L. Zhang and W. Wang, Appl. Catal., B, 2023, 323, 122177 CrossRef CAS.
- S. Gong, Y. Niu, X. Liu, C. Xu, C. Chen, T. J. Meyer and Z. Chen, ACS Nano, 2023, 17, 4922–4932 CrossRef CAS PubMed.
- G. Jia, M. Sun, Y. Wang, Y. Shi, L. Zhang, X. Cui, B. Huang and J. C. Yu, Adv. Funct. Mater., 2022, 32, 2206817 CrossRef CAS.
- X. Yang, E. A. Fugate, Y. Mueanngern and L. R. Baker, ACS Catal., 2017, 7, 177–180 CrossRef CAS.
- Y. Lum and J. W. Ager, Nat. Catal., 2019, 2, 86–93 CrossRef CAS.
- L.-J. Zhu, D.-H. Si, F.-X. Ma, M.-J. Sun, T. Zhang and R. Cao, ACS Catal., 2023, 13, 5114–5121 CrossRef CAS.
- W. Ye, X. Guo and T. Ma, Chem. Eng. J., 2021, 414, 128825 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, Green Chem., 2017, 19, 2406–2415 RSC.
- T. Möller, T. N. Thanh, X. Wang, W. Ju, Z. Jovanov and P. Strasser, Energy Environ. Sci., 2021, 14, 5995–6006 RSC.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS PubMed.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS.
- Y.-L. Cheng, C.-T. Hsieh, Y.-S. Ho, M.-H. Shen, T.-H. Chao and M.-J. Cheng, Phys. Chem. Chem. Phys., 2022, 24, 2476–2481 RSC.
- M. Zhang, K. Zhang, X. Ai, X. Liang, Q. Zhang, H. Chen and X. Zou, Chin. J. Catal., 2022, 43, 2987–3018 CrossRef CAS.
- Y. Wang, D. Wang, C. J. Dares, S. L. Marquard, M. V. Sheridan and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 278–283 CrossRef CAS PubMed.
- D. Zang, Q. Li, G. Dai, M. Zeng, Y. Huang and Y. Wei, Appl. Catal., B, 2021, 281, 119426 CrossRef CAS.
- X.-F. Qiu, J.-R. Huang, C. Yu, Z.-H. Zhao, H.-L. Zhu, Z. Ke, P.-Q. Liao and X.-M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202206470 CrossRef CAS PubMed.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, J. Ma, Z. Zhang, G. Yang and B. Han, Green Chem., 2017, 19, 2086–2091 RSC.
- J. Jin, J. Wicks, Q. Min, J. Li, Y. Hu, J. Ma, Y. Wang, Z. Jiang, Y. Xu, R. Lu, G. Si, P. Papangelakis, M. Shakouri, Q. Xiao, P. Ou, X. Wang, Z. Chen, W. Zhang, K. Yu, J. Song, X. Jiang, P. Qiu, Y. Lou, D. Wu, Y. Mao, A. Ozden, C. Wang, B. Y. Xia, X. Hu, V. P. Dravid, Y.-M. Yiu, T.-K. Sham, Z. Wang, D. Sinton, L. Mai, E. H. Sargent and Y. Pang, Nature, 2023, 617, 724–729 CrossRef CAS PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- J. Wu, T. Sharifi, Y. Gao, T. Zhang and P. M. Ajayan, Adv. Mater., 2019, 31, 1804257 CrossRef PubMed.
- J. Zhang, W. Cai, F. X. Hu, H. Yang and B. Liu, Chem. Sci., 2021, 12, 6800–6819 RSC.
- F. Guo, B. Liu, M. Liu, Y. Xia, T. Wang, W. Hu, P. Fyffe, L. Tian and X. Chen, Green Chem., 2021, 23, 5129–5137 RSC.
- D. Giusi, C. Ampelli, C. Genovese, S. Perathoner and G. Centi, Chem. Eng. J., 2021, 408, 127250 CrossRef CAS.
- J. F. de Brito, C. Genovese, F. Tavella, C. Ampelli, M. V. Boldrin Zanoni, G. Centi and S. Perathoner, ChemSusChem, 2019, 12, 4274–4284 CrossRef PubMed.
- G. Centi and S. Perathoner, Green Chem., 2022, 24, 7305–7331 RSC.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, V. Solokha, S. K. Calderon, J. J. Velasco-Velez, C. Ampelli, S. Perathoner, G. Held, G. Centi and R. Arrigo, Nat. Commun., 2018, 9, 935 CrossRef PubMed.
- R. Arrigo, R. Blume, V. Streibel, C. Genovese, A. Roldan, M. E. Schuster, C. Ampelli, S. Perathoner, J. J. Velasco Vélez, M. Hävecker, A. Knop-Gericke, R. Schlögl and G. Centi, ACS Catal., 2022, 12, 411–430 CrossRef CAS.
- C. Jia, W. Ren, X. Chen, W. Yang and C. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 6003–6010 CrossRef CAS.
- Y. Zhou and B. S. Yeo, J. Mater. Chem. A, 2020, 8, 23162–23186 RSC.
- L. Ou, Z. He, H. Yang and Y. Chen, ACS Omega, 2021, 6, 17839–17847 CrossRef CAS PubMed.
- X.-G. Zhang, S. Feng, C. Zhan, D.-Y. Wu, Y. Zhao and Z.-Q. Tian, J. Phys. Chem. Lett., 2020, 11, 6593–6599 CrossRef CAS PubMed.
- Q. Fan, M. Zhang, M. Jia, S. Liu, J. Qiu and Z. Sun, Mater. Today Energy, 2018, 10, 280–301 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- T. Liu, J. Sang, H. Li, P. Wei, Y. Zang and G. Wang, Battery Energy, 2022, 1, 20220012 CrossRef CAS.
- S. Ajmal, Y. Yang, M. A. Tahir, K. Li, A.-U.-R. Bacha, I. Nabi, Y. Liu, T. Wang and L. Zhang, Catal. Sci. Technol., 2020, 10, 4562–4570 RSC.
- L. Ou and Z. He, Surf. Sci., 2021, 705, 121782 CrossRef CAS.
- M. Obst, L. Pavlovic and K. H. Hopmann, J. Organomet. Chem., 2018, 864, 115–127 CrossRef CAS.
- A. Tharak, R. Katakojwala, S. Kajla and S. Venkata Mohan, Chem. Eng. J., 2023, 454, 140200 CrossRef CAS.
- M. Quraishi, K. Wani, S. Pandit, P. K. Gupta, A. K. Rai, D. Lahiri, D. A. Jadhav, R. R. Ray, S. P. Jung, V. K. Thakur and R. Prasad, Fermentation, 2021, 7, 291 CrossRef CAS.
- M. Roy, S. Yadav and S. A. Patil, Front. Energy Res., 2021, 9, 759678 CrossRef.
- P. Gupta, M. T. Noori, A. E. Núñez and N. Verma, iScience, 2021, 24, 102294 CrossRef CAS PubMed.
- H. M. Fruehauf, F. Enzmann, F. Harnisch, R. Ulber and D. Holtmann, Biotechnol. J., 2020, 15, 2000066 CrossRef CAS PubMed.
- X. Christodoulou, T. Okoroafor, S. Parry and S. B. Velasquez-Orta, J. CO2 Util., 2017, 18, 390–399 CrossRef CAS.
- T.-s. Song, K. Fei, H. Zhang, H. Yuan, Y. Yang, P. Ouyang and J. Xie, J. Chem. Technol. Biotechnol., 2018, 93, 457–466 CrossRef CAS.
- T.-s. Song, L. Fu, N. Wan, J. Wu and J. Xie, J. CO2 Util., 2020, 41, 101231 CrossRef CAS.
- X. Christodoulou and S. B. Velasquez-Orta, Environ. Sci. Technol., 2016, 50, 11234–11242 CrossRef CAS PubMed.
- G. Gong, B. Wu, L. Liu, J. Li, Q. Zhu, M. He and G. Hu, Eng. Microbiol., 2022, 2, 100036 CrossRef CAS.
- K. Novak and S. Pflügl, FEMS Microbiol. Lett., 2018, 365, fny226 CAS.
- J. Martínez, J. F. Cortés and R. Miranda, Processes, 2022, 10, 1274 CrossRef.
- R. A. Sheldon, M. L. Bode and S. G. Akakios, Curr. Opin. Green Sustain. Chem., 2022, 33, 100569 CrossRef CAS.
- V. Tulus, J. Pérez-Ramírez and G. Guillén-Gosálbez, Green Chem., 2021, 23, 9881–9893 RSC.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- C. T. Matos, L. Gouveia, A. R. C. Morais, A. Reis and R. Bogel-Łukasik, Green Chem., 2013, 15, 2854 RSC.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- E. R. Monteith, P. Mampuys, L. Summerton, J. H. Clark, B. U. W. Maes and C. R. McElroy, Green Chem., 2020, 22, 123–135 RSC.
- C. Jiménez-González, D. J. C. Constableb and C. S. Pondera, Chem. Soc. Rev., 2012, 41, 1485–1498 RSC.
- F. Roschangar, Y. Zhou, D. J. C. Constable, J. Colberg, D. P. Dickson, P. J. Dunn, M. D. Eastgate, F. Gallou, J. D. Hayler, S. G. Koenig, M. E. Kopach, D. K. Leahy, I. Mergelsberg, U. Scholz, A. G. Smith, M. Henry, J. Mulder, J. Brandenburg, J. R. Dehli, D. R. Fandrick, K. R. Fandrick, F. Gnad-Badouin, G. Zerban, K. Groll, P. T. Anastas, R. A. Sheldon and C. H. Senanayake, Green Chem., 2018, 20, 2206–2211 RSC.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef CAS PubMed.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- S. Cucurachi, C. van der Giesen and J. Guinée, Procedia CIRP, 2018, 69, 463–468 CrossRef.
- H. Röder, K. Kumar, S. Füchsl and V. Sieber, J. Cleaner Prod., 2022, 376, 134329 CrossRef.
- N. Tsoy, B. Steubing, C. van der Giesen and J. Guinée, Int. J. Life Cycle Assess., 2020, 25, 1680–1692 CrossRef.
- E. Martinez-Guerra and V. G. Gud, Appl. Sci., 2017, 7, 869 CrossRef.
- N. Outili, H. Kerras, C. Nekkab, R. Merouani and A. H. Meniai, Renewable Energy, 2020, 45, 2575–2586 CrossRef.
- G. Fiorentino, M. Ripa and S. Ulgiati, Biofpr, 2017, 11, 195–214 CAS.
- H. K. Jeswani, A. Chilvers and A. Azapagic, Proc. R. Soc. A, 2020, 476, 2243 CrossRef PubMed.
- S. Ahmad, K. Y. Wong and R. Ahmad, Procedia Manufacturing, 2019, 34, 49–57 CrossRef.
- S. Cucurachi, L. Scherer, J. Guinée and A. Tukker, One Earth, 2019, 1, 292–297 CrossRef.
- P. Roy, D. Nei, T. Orikasa, Q. Xu, H. Okadome, N. Nakamura and T. Shiina, J. Food Eng., 2009, 90, 1–10 CrossRef.
- G. Wang, R. Shi, L. Mi and J. Hu, Sustainability, 2022, 14, 1051 CrossRef.
- M. Rybaczewska-Błażejowska and W. Gierulski, Sustainability, 2018, 10, 4544 CrossRef.
- L. Shi, L. Liu, B. Yang, G. Sheng and T. Xu, Sustainability, 2020, 12, 3793 CrossRef CAS.
- L. Matijašević, I. Dejanović and H. Lisac, Resour., Conserv. Recycl., 2010, 54, 149–154 CrossRef.
- J. Colberg, K. K. Hii and S. G. Koenig, Org. Process Res. Dev., 2022, 26, 2176–2178 CrossRef CAS.
- M. G. T. C. Ribeiro, D. A. Costa and A. A. S. C. Machado, Green Chem. Lett. Rev., 2010, 3, 149–159 CrossRef CAS.
Footnote |
| † Invited contribution to the Green Chemistry themed collection: advances in electrosynthesis for a Greener Chemical Industry. |
| This journal is © The Royal Society of Chemistry 2024 |