
“On-water” synthesis of thioxoimidazolidinone-isatin/ninhydrin conjugates, followed by temperature-induced dehydration by a ZnMnO3@Ni(OH)2 nano-catalyst†
Soumitra
Rana
 a,
Soumyadip
Basu
a,
Aswini
Bera
b,
Pinaki
Saha
c,
Prasanta
Ghosh
c,
Bhanu Bhusan
Khatua
b and
Chhanda
Mukhopadhyay
*a
a,
Soumyadip
Basu
a,
Aswini
Bera
b,
Pinaki
Saha
c,
Prasanta
Ghosh
c,
Bhanu Bhusan
Khatua
b and
Chhanda
Mukhopadhyay
*a
aDepartment of Chemistry, University of Calcutta, 92 APC Road, Kolkata-700009, India. E-mail: cmukhop@yahoo.co.in
bMaterials Science Centre, Indian Institute of Technology Kharagpur, Kharagpur, West Bengal 721302, India
cDepartment of Chemistry, R. K. Mission Residential College, Narendrapur, Kolkata-700103, India
First published on 11th January 2024
Abstract
A novel and efficient “on water” mediated one-pot ZnMnO3@Ni(OH)2 catalyzed straight-forward synthesis of 3-hydroxy-3-(3-methyl-5-oxo-2-thioxoimidazolidin-4-yl)oxindole via a thiazolidinedione–isatin conjugate has been explored. Concurrently, temperature-regulated dehydrated adducts of 3-hydroxy-3-(3-methyl-5-oxo-2-thioxoimidazolidin-4-yl)oxindole were also demonstrated. The use of a recyclable ZnMnO3@Ni(OH)2 nano-catalyst in the aqueous medium makes the protocol sustainable and green. The synthesis of ZnMnO3@Ni(OH)2 nanopowder has been meticulously characterized using powdered XRD, HRTEM, EDX and BET analyses. Moreover, the choice of readily available and readily made starting materials under mild and operationally simple reaction conditions is the major advantage of this strategy. The catalyst exhibits high water compatibility and demonstrates the ability to be recycled and reused for a minimum of eight cycles. Through a standard leaching experiment, it was confirmed that the reaction proceeded heterogeneously with this recyclable catalyst.
Introduction
Thioxoimidazolidinones (thiohydantoins), belonging to the nitrogen- and sulfur-containing heterocyclic compounds of the imidazolidine family, exhibit a wide array of biological and pharmacological activities.1 Organic chemists and biologists extensively study the substituted thiohydantoin chemistry due to its vital role in drug discovery. N-Substituted thiohydantoins show promise in pharmacology for antiandrogenic2 and anti-prostate cancer effects,3 while the synthesis of 5-arylidene 2-thiohydantoin moieties has gained enormous attention as a valuable constituent for biologically active novel scaffolds due to their antiviral, antimycobacterial,4 NADPH oxidase (NOX) inhibitor,5 antidiabetic, anticonvulsant and antitumor properties and schistosomicidal activities (Fig. 1).6 In this context, we aimed to create a specialized chemical library focusing on the interesting hydantoin moiety in our quest to develop medicinal compounds.7 Although a lot of work has been done for the synthesis of various substituted thiohydantoins,8 no methodology has been developed to date for the incorporation of oxyindene or oxindole moieties in the thiohydantoin core while following a green pathway. Oxindoles are key scaffolds in pharmaceuticals and natural products; notably 3,3-disubstituted oxindoles are present in antitumor and antibacterial drugs (Fig. 1), while heterocycle-fused indanone scaffolds have notable applications in medicinal chemistry.9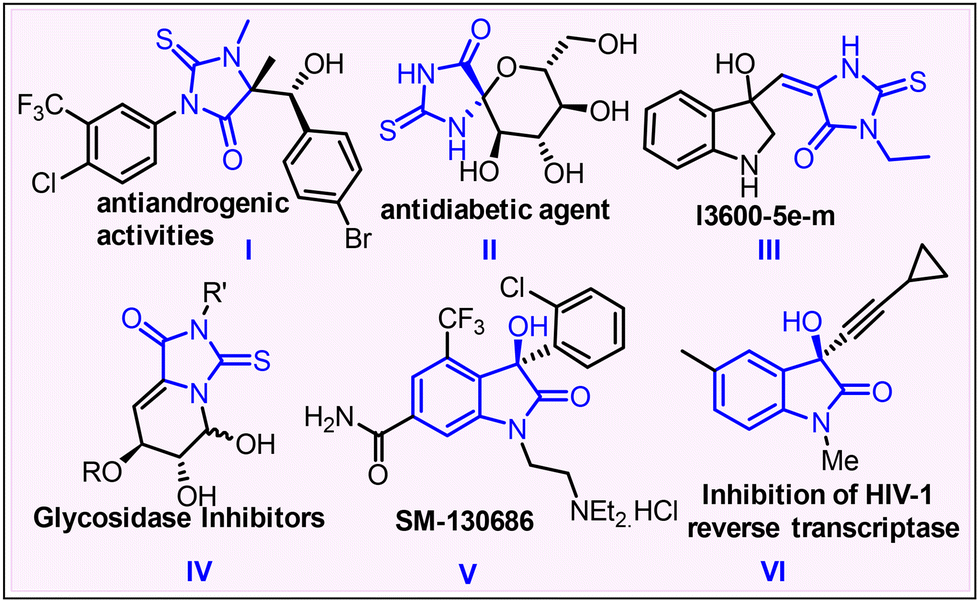 | ||
| Fig. 1 Bioactive natural products and small molecules containing substituted thiohydantoin and 3-hydroxy oxindole core scaffolds. | ||
The reaction is carried out “on water”. The remarkable rate escalation in reactions “on water” is theoretically probed. The emergence of the “on water” concept has served as an inspiration for numerous aqueous organic reactions involving water-insoluble reactants.10 Moreover, the interaction between water and reactants at the oil–water interface is pivotal. The presence of water positively impacts catalysts, enhancing their activity, selectivity and product yields in our chemical reactions.11
This research highlights the possibility of not only environment friendly and green chemistry aspects, but also provides novel possibilities for the effective synthesis of valuable molecules. One-pot multicomponent green reactions in modern science contribute to sustainability and are eco-friendly to society. Additionally, this protocol is strategically efficient due to its step-economy,12 high atom economy, one-pot single-step reaction, improved efficiency and operational simplicity.13 This makes it a desirable alternative for large-scale production because it improves the total yield, at the same time accelerating the synthesis process.14 One of the most remarkable features of this research is its unwavering commitment to green synthesis for minimizing the environmental impact of chemical processes.15
The achievement of success in one-pot green synthesis frequently hinges on meticulous optimization of reaction conditions, the choice of suitable catalysts, and an extensive knowledge of the underlying chemistry. While on the other hand, heterogeneous nanocatalysts, typically made from eco-friendly materials like solid-supported metal nanomaterials, promote eco-conscious transformations in diverse industries.16 Furthermore, the heterogeneous solid support facilitates efficient organic transformations, sometimes even in a chemoselective or regioselective manner.17 The ZnMnO3@Ni(OH)2 nanomaterials are used as heterogeneous “E” catalysts (eco-friendly, economical, and efficient) which makes them highly suitable for numerous catalytic applications.18 We employed ZnMnO3@Ni(OH)2 (ZNO) as the nanocatalyst to enhance the reaction's efficiency in our study. The choice of this nanocatalyst is significant, as it combines the catalytic properties of both ZnMnO3 (as a Lewis acid) and Ni(OH)2 (as a Lewis base) to facilitate the reaction efficiently. The combination of two nanomaterials makes our catalyst have a very stable architecture which shortens the route to produce highly active intermediates, encouraging subsequent spontaneous changes and reducing waste production.19 Furthermore, the extensive surface area of the solid support can absorb a greater quantity of reactants, facilitating rapid interactions with numerous catalytically active sites which exhibit a honeycomb nanostructure with multiple nanosheets and an open-space architecture. In this structure, the ZnMnO3 cluster forms the core, while Ni(OH)2 nanosheets constitute the protective shell,20 and the core–shell structure of our as-synthesized ZNO catalyst gives the structure stability which facilitates the fast reaction.
Considering the elegant approaches accessible for nucleophilic addition, cyclization and temperature-triggered dehydration under environmentally friendly one-pot reaction conditions, we have envisaged the potential versatility in forming C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds at the C5-thiohydantoins with the C3 position of 3-hydroxy oxindole through MTMO catalyst cascade sequences (Scheme 1). The combined effect of the ZNO catalyst and temperature-induced dehydration21 as a subsequent step in the synthesis adds an extra layer of versatility to this approach, enabling product modification and the formation of desired conjugates under recyclable catalytic conditions. The utilization of water serves as a common solvent in our reaction, and zero waste materials were generated during the process and it not only accelerates the reaction rate, but also contributes to the overall environmental sustainability, as documented.22
C bonds at the C5-thiohydantoins with the C3 position of 3-hydroxy oxindole through MTMO catalyst cascade sequences (Scheme 1). The combined effect of the ZNO catalyst and temperature-induced dehydration21 as a subsequent step in the synthesis adds an extra layer of versatility to this approach, enabling product modification and the formation of desired conjugates under recyclable catalytic conditions. The utilization of water serves as a common solvent in our reaction, and zero waste materials were generated during the process and it not only accelerates the reaction rate, but also contributes to the overall environmental sustainability, as documented.22
 | ||
| Scheme 1 Synthetic approach of multisubstituted thiohydantoin. | ||
There are numerous reported traditional techniques for the nucleophilic addition, cyclization and dehydration of C5-thiohydantoins using potentially hazardous reagents such as DMF,23 AcOH,24 and piperidine,25 or by sequential dehydration using an Ir catalyst.26 The approach detailed in the literature (Scheme 1) generates HBr as a byproduct and causes significant waste, demonstrating a clear comparison opportunity. Some of the parameters in the literature do not adhere to the 12 green chemistry principles.27 Various green chemistry parameters28 (atom economy, efficiency, EMY, E-factor, productivity, and RME) demonstrate this. Tables 1 and 7 summarise the entire examination of these indicators (detailed calculation in P10–P24, ESI 1†). Our developed process has a simple operation, high chemical yields, and excellent green chemistry metrics.29
| Parameters | Our work (4ba) | Literature-reported (5) |
|---|---|---|
|
|
|
|
| 1. No. of steps | 1 | 1 |
| 2. % yield | 84% | 78% |
| 3. E factor | 0.37 (g g −1 ) | 0.69(g g−1) |
| 4. Atom economy (AE) | 95.79% | 76.10% |
| 5. Effective mass yield (EMY) | 73.01% | 59.20% |
| 6. Carbon efficiency (CE) | 83.95% | 77.79% |
| 7. Reaction mass efficiency (RME) | 73.01% | 59.20% |
| 8. Waste during reaction | H2O | HBr (hazardous) |

However, their potential applications have faced limitations due to concerns related to safety, cost and ease of operation. Our reaction conditions facilitate thiohydantoin formation at both room temperature and elevated temperatures.25 Previously, a common method involved stepwise addition of reagents for dehydration at varying temperatures, typically for versatile product formation in aldehyde–thiohydantoin conjugation.16
Herein, we present a temperature-induced dehydration process involving a one-pot reaction “on water” catalyzed by ZnMnO3@Ni(OH)2. In reactions utilizing water, the insoluble reactants engage via the hydrophobic effect at the water surface.29 This reaction facilitates a diastereoselective transfer aldol reaction involving the conjugation of both the bioactive heterocyclic core thiohydantoin and 3-hydroxy oxindole, resulting in the formation of C–C and C–N bonds. The reaction takes place in the aqueous medium and yields 3-methyl-5-oxo-2-thioxoimidazolidin indolinone exclusively at 25–30 °C and 3-methyl-5-oxo-2-thioxoimidazolidin-4-ylidene indolinone as the dehydration product under reflux (Scheme 1).
Results and discussion
In this experiment, we successfully adapted and employed an environmentally friendly ZnMnO3@Ni(OH)2 nanocatalyst for a one-pot, “on water” mediated three-component green synthesis. This process led to the formation of highly substituted 3-methyl-5-oxo-2-thioxoimidazolidin indolinone derivatives and 3-methyl-5-oxo-2-thioxoimidazolidin-4-ylidene indolinone, followed by dehydration at different temperatures employing isatins or ninhydrin and methylglycine (sarcosine) and various isothiocyanates.Characterization of the ZNO catalyst
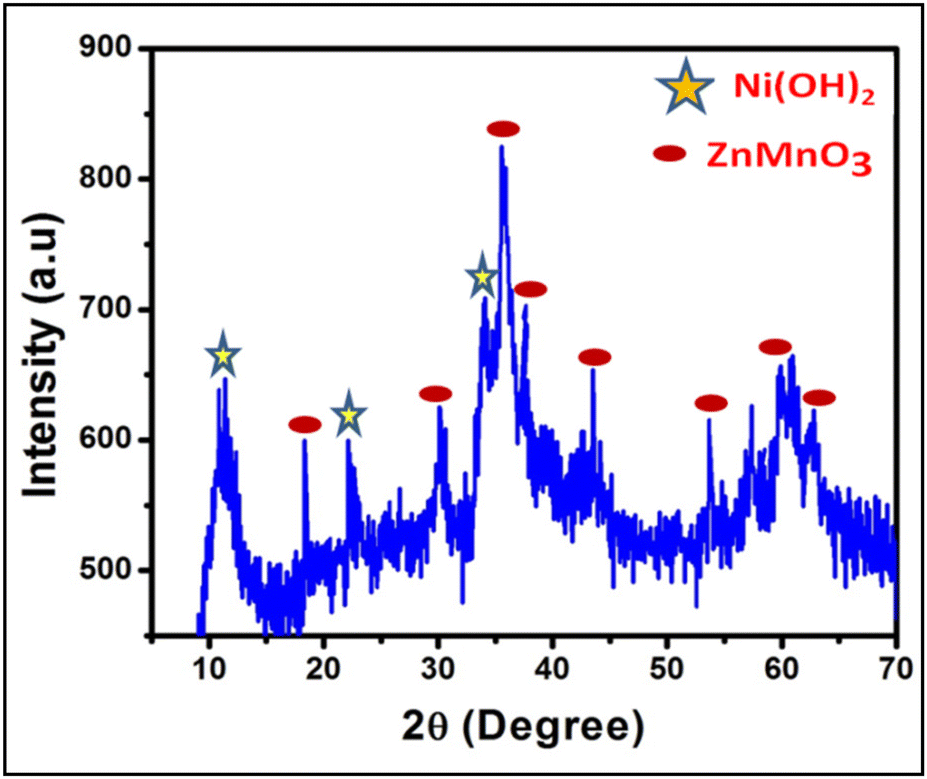 | ||
| Fig. 2 XRD pattern of the as-prepared catalyst ZNO. | ||
 | ||
| Fig. 3 (a) and (b) FESEM images of the ZNO catalyst; (c), (d) and (e) TEM micrographs of the ZNO catalyst and (f) SAED pattern. | ||
From the TEM analysis, the morphological investigation has been further analyzed, as shown in Fig. 3c, d and e. The results of the FESEM analysis are quite similar to those of the surface, shape, and size phenomena from the TEM investigation. The hybrid spherical ZNO core–shell micrographs are effectively shown in Fig. 3e based on the TEM investigation. The core–shell structure (Fig. 3e) has a clear dark/light contrast that indicates integrated nanosheets with open-space nanostructures. Additionally, the SAED pattern (Fig. 3f) illustrates ZNO's polycrystalline nature.
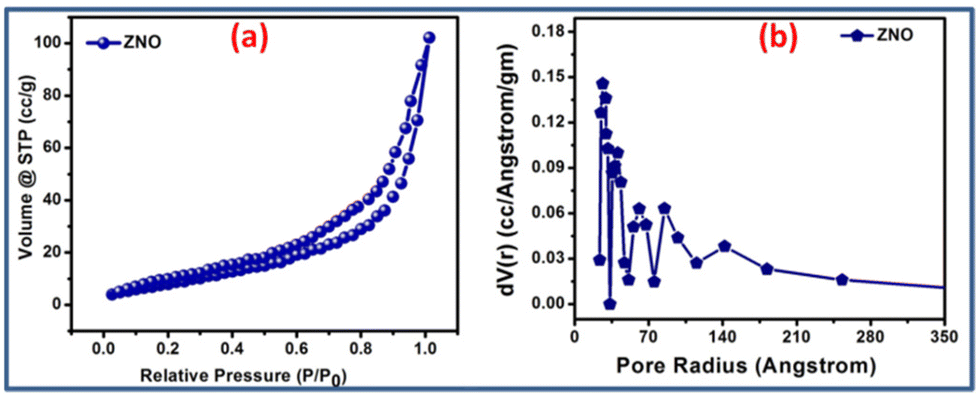 | ||
| Fig. 4 N2 adsorption isotherm and pore size distribution of the ZNO catalyst (a and b) measured at 77 K. | ||
 | ||
| Fig. 5 TGA curve of ZnMnO3@Ni(OH)2. | ||
 | ||
| Fig. 6 IR spectra of (A) the ZnMnO3@Ni(OH)2 catalyst and (B) ZnMnO3. | ||
The Zn–O vibrational band at 599 cm−1 was slightly displaced in the ZnMnO3 due to the change in bond length caused by the presence of manganese, as seen in the spectra (Fig. 6B). Furthermore, another intense signal is found at 772 cm−1 due to the stretching of the Mn–O bond.
In order to standardise the ideal circumstance, we concentrated on the synthesis of 5ab and 10bc (Table 2) as reference reactions. To get the best outcome for these two products, a variety of methods were applied. We observed several reactions using 1-methylindoline-2,3-dione (N-methyl isatin) 4 (0.5 mmol), methylglycine 2a (0.5 mmol) and 1-isothiocyanato-4-methylbenzene 3 (0.5 mmol) as model starting materials to screen the reaction conditions (Table 2). Initially, without a catalyst, only a small amount of the product was produced for both 5ab and 10bc at room temperature for 12 h and under heating conditions for 10 h, respectively (Table 2, entries 1 and 2). A very low percentage of yield for 5ab was produced by a homogeneous acid–base catalyst when stirred at room temperature with NaOH, piperidine, Et3N and AcOH for 8 h (Table 2, entries 3–6). When Lewis acid catalysis [In(OTf)3] is employed in this reaction, there is no predominant increase in yield of 5ab (Table 2, entry 7). The screening of other heterogeneous metal oxide and hydroxide sources, like Ni(OH)2 (basic), ZnMnO3 nanospheres, ZnO, MnO, and ZnMnO3/Ni(OH)2 (acid–base dual nanocatalyst) furnished the desired compound 5ab in low yields (Table 2, entries 8–12). The introduction of the Ni(OH)2 group onto the ZnMnO3 catalyst resulted in a notable increase in the yield percentage, as evidenced in Table 2 (entries 13–29). Surprisingly, when the reaction was extended to a duration of 8 hours under the r.t. (25–30 °C) conditions led to the desired product (5ab) in 91% yield (Table 2, entry 13). In order to optimize the catalyst loading, the concentration of ZnMnO3@Ni(OH)2 was reduced from 20 mol% to 15 mol%, and when further increased to 25 mol%, the reaction yield of 5ab failed to improve (Table 2, entries 14 and 15). After optimising the catalyst, no significant enhancement in yield was detected when different commonly used solvents were tested at room temperature in the presence of the catalyst. These solvents include acetonitrile, dichloromethane, THF, EtOH and DMF (Table 2, entries 16–21). Moderately good yields were obtained when the reactions were conducted in ethanol or a mixed solvent of EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1 v/v) (Table 2, entries 20–23). However, significant yields were obtained for both reactions when conducted in water. After the catalyst and solvent were optimised, on increasing the temperature, there was a notable improvement in the yield percentage of 10bc (Table 2, entries 22–23 and 27–30). Once more, a progressive yield of 5ab was achieved when 20 mol% ZnMnO3@Ni(OH)2 nanomaterials were employed under room temperature (25–30 °C) conditions for a duration of 4 and 15 hours in H2O solvent (Table 2, entries 24–25). Through experimentation, a significant decrease in the formation of 5ab and a corresponding increase in the formation percentage of 10bc were observed when the temperature was raised from room temperature (25–30 °C) to 100 °C in H2O (Table 2, entry 26–30). Therefore, it can be stated that the best optimal circumstances for this reaction approach of 5ab entail the employment of 20 mol% ZnMnO3@Ni(OH)2 nano-powder (50 mg) on an aqueous medium (3 mL) at room temperature for 8 hours, and 10bc at 100 °C for 3 hours is obtained as the minimum reaction time.
:H2O (1:1 v/v) (Table 2, entries 20–23). However, significant yields were obtained for both reactions when conducted in water. After the catalyst and solvent were optimised, on increasing the temperature, there was a notable improvement in the yield percentage of 10bc (Table 2, entries 22–23 and 27–30). Once more, a progressive yield of 5ab was achieved when 20 mol% ZnMnO3@Ni(OH)2 nanomaterials were employed under room temperature (25–30 °C) conditions for a duration of 4 and 15 hours in H2O solvent (Table 2, entries 24–25). Through experimentation, a significant decrease in the formation of 5ab and a corresponding increase in the formation percentage of 10bc were observed when the temperature was raised from room temperature (25–30 °C) to 100 °C in H2O (Table 2, entry 26–30). Therefore, it can be stated that the best optimal circumstances for this reaction approach of 5ab entail the employment of 20 mol% ZnMnO3@Ni(OH)2 nano-powder (50 mg) on an aqueous medium (3 mL) at room temperature for 8 hours, and 10bc at 100 °C for 3 hours is obtained as the minimum reaction time.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Temp.b (°C) | Time (h) | Yieldc (%) | TONf | TOFg (h−1) | |
| 5ab | 10bc (Z/E)e | |||||||
| a Reaction conditions: All reactions were performed with reactants 1-methylindoline-2,3-dione 4 (1 mmol), methylglycine 2a (1 mmol) (water soluble), and 1-isothiocyanato-4-methylbenzene 3 (1 mmol) in the presence of different catalysts (20 mol%), different solvents (3 mL), different times and different temperatures. b Here r.t. corresponds to 25–30 °C and the reflux temperature varies with the solvents in use. c Isolated yield after chromatography purification on silica (bold rows 13 and 29 indicate the optimized reaction conditions). d Isolated yield of 5ab as the nonseparable dr mixture. e Isolated yield of 10bc as the nonseparable Z/E mixture. f Turnover number (TON) units (mol product per mol catalyst). g Turnover frequency (TOF) units ((mol product per mol catalyst) per hour). h THF is tetrahydrofuran. i DMF is dimethylformamide. j EtOH is ethanol. k TON and TOF calculated for the total yield of the products, i.e.5ab and 10bc. | ||||||||
| 1 | — | H2O | R.t. | 12 | 22 | — | — | — |
| 2 | — | H2O | Reflux | 10 | — | 24 | — | — |
| 3 | NaOH (10) | H2O | R.t. | 8 | 45 | — | 4.50 | 0.56 |
| 4 | Piperidine (15) | H2O | R.t. | 8 | 41 | — | 2.74 | 0.34 |
| 5 | Et3N (20) | H2O | R.t. | 8 | 38 | — | 1.90 | 0.24 |
| 6 | AcOH (20) | H2O | R.t. | 8 | 25 | — | 1.25 | 0.16 |
| 7 | In(OTf)3 (15) | H2O | R.t. | 8 | Trace | — | — | — |
| 8 | Ni(OH)2 (20) | H2O | R.t. | 8 | 56 | — | 2.80 | 0.35 |
| 9 | ZnMnO3 (20) | H2O | R.t. | 8 | 45 | — | 2.25 | 0.28 |
| 10 | ZnO (20) | H2O | R.t. | 8 | 40 | — | 2.00 | 0.25 |
| 11 | MnO (20) | H2O | R.t. | 8 | 44 | — | 2.20 | 0.28 |
| 12 | ZnMnO3/Ni(OH)2 (20) | H2O | R.t. | 8 | 48 | — | 2.40 | 0.30 |
| 13 | ZnMnO 3 @Ni(OH) 2 (20) | H 2 O | R.t. | 8 | 91 | — | 4.55 | 0.57 |
| 14 | ZnMnO3@Ni(OH)2 (15) | H2O | R.t. | 8 | 52 | — | 3.47 | 2.89 |
| 15 | ZnMnO3@Ni(OH)2 (25) | H2O | R.t. | 8 | 91 | — | 3.64 | 0.46 |
| 16 | ZnMnO3@Ni(OH)2 (20) | CH3CN | R.t. | 15 | 45 | — | 2.25 | 0.15 |
| 17 | ZnMnO3@Ni(OH)2 (20) | CH2Cl2 | R.t. | 15 | 41 | — | 2.05 | 0.14 |
| 18h | ZnMnO3@Ni(OH)2 (20) | THF | R.t. | 20 | 37 | — | 1.85 | 0.09 |
| 19i | ZnMnO3@Ni(OH)2 (20) | DMF | R.t. | 12 | 42 | — | 2.10 | 0.18 |
| 20j | ZnMnO3@Ni(OH)2 (20) | EtOH | R.t. | 24 | 63 | — | 3.15 | 0.31 |
| 21 | ZnMnO3@Ni(OH)2 (20) | EtOH:H2O (1:1 v/v) |
R.t. | 24 | 68 | 3.40 | 0.41 | |
| 22 | ZnMnO3@Ni(OH)2 (20) | EtOH | Reflux | 4 | — | 55 | 2.75 | 0.69 |
| 23 | ZnMnO3@Ni(OH)2 (20) | EtOH:H2O (1:1 v/v) |
Reflux | 4 | — | 57 | 2.85 | 0.71 |
| 24 | ZnMnO3@Ni(OH)2 (20) | H2O | R.t. | 4 | 70 | — | 3.50 | 0.88 |
| 25 | ZnMnO3@Ni(OH)2 (20) | H2O | R.t. | 15 | 87 | — | 4.35 | 0.29 |
| 26k | ZnMnO3@Ni(OH)2 (20) | H2O | 50 | 3 | 42 | 34 | 3.80 | 1.27 |
| 27k | ZnMnO3@Ni(OH)2 (20) | H2O | 70 | 2 | 5 | 53 | 2.90 | 1.45 |
| 28 | ZnMnO3@Ni(OH)2 (20) | H2O | 80 | 3 | Trace | 59 | 2.95 | 0.98 |
| 29 | ZnMnO 3 @Ni(OH) 2 (20) | H 2 O | Reflux | 3 | — | 68 | 3.4 | 1.13 |
| 30 | ZnMnO3@Ni(OH)2 (20) | H2O | Reflux | 5 | — | 63 | 3.15 | 0.63 |

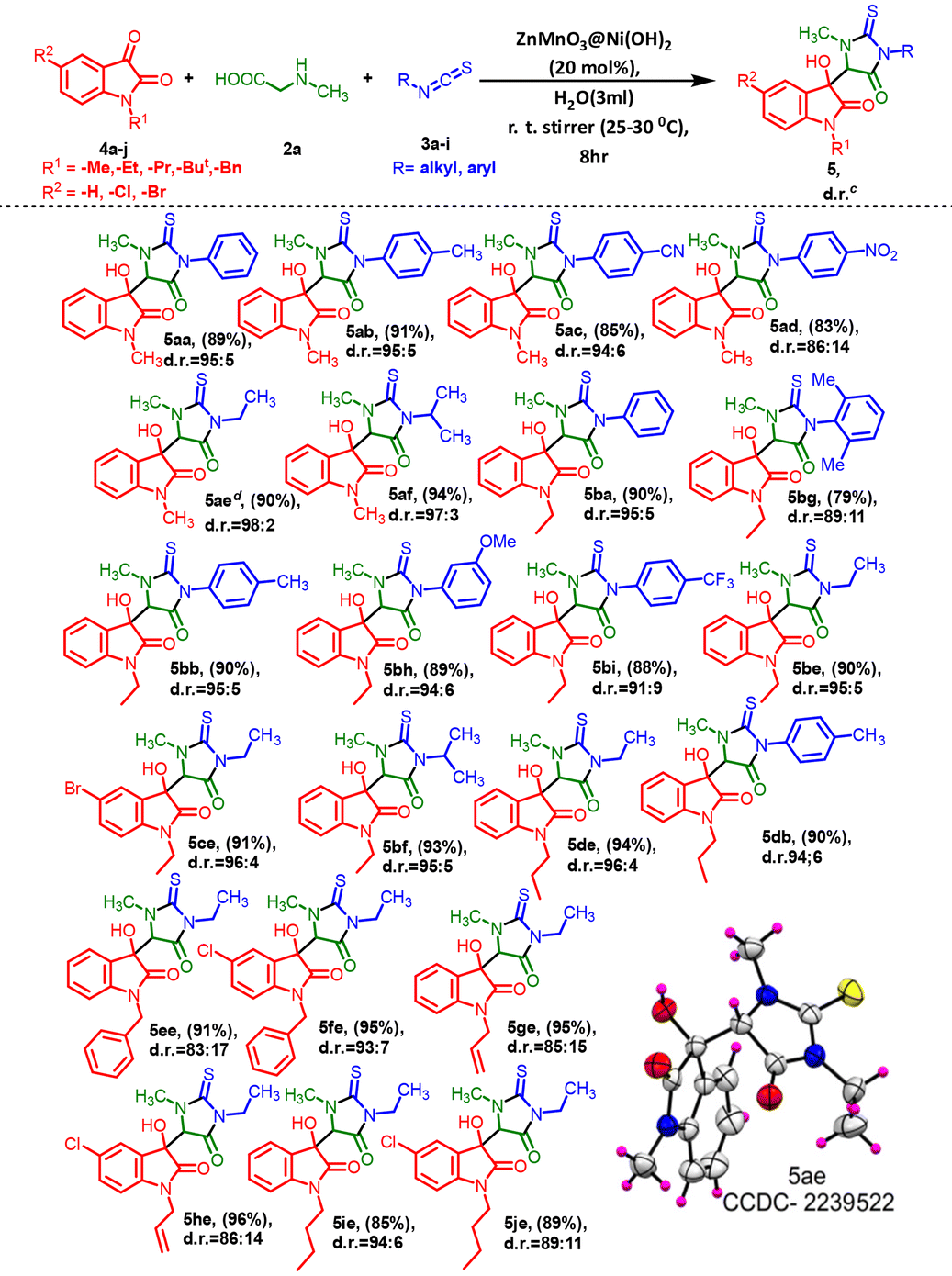
After establishing the optimized reaction conditions, we proceeded to investigate the compatibility of the substituent groups within the reaction. Herein, we successfully synthesized a series of 3-methyl-5-oxo-2-thioxoimidazolidin indolinone (4 and 5) with 3-methyl-5-oxo-2-thioxoimidazolidin indene dione (7) derivatives, and the outcomes are detailed in Tables 3–5. We explored the variety of commercially available isatins to successfully synthesize a series of 3-methyl-5-oxo-2-thioxoimidazolidin indolinone compounds, as illustrated in Table 3. The reactions of isatins (1a–i) with various substituted isothiocyanates were examined first. The results demonstrate excellent yields and diastereoselectivities, as depicted in Table 3. Isatins bearing 5- and 6-substitution with electron-withdrawing groups like –Cl, –Br, and –NO2 exhibited favorable yields ranging from 80% to 86% (4ba–4db and 4ib) along with notably high diastereoselectivities ranging between 92:8 and 95:5. In contrast, the presence of –OMe as an electron-donating group resulted in moderate yield (4eb). In contrast, when unsubstituted isatin was reacted with aromatic isothiocyanates containing electron-donating groups at any position, it yielded relatively well to moderate yields, as compared to those with electron-withdrawing groups in the ring (Table 3, 4ab and 4ac). The alkyl-substituted isothiocyanate provided excellent yields (89%–93%) and diastereoselectivities (97:3) (4ad–4gd), whereas phenyl or unsubstituted isothiocyanates resulted in moderate yields (4aa) and diastereoselectivities (88:12). Nevertheless, despite evident steric hindrance, the introduction of 2,6-di-substituted isothiocyanatobenzene was successfully achieved (4he).
| a Reaction conditions: isatins 1a–1i (1 mmol) and methylglycine 2a (1 mmol) and different isothiocyanates 3a–3e (1 mmol) in the presence of the ZnMnO3@Ni(OH)2 catalyst (20 mol%) and solvents (3 mL) at room temperature for 8 h. b Isolated yields after chromatography purification on silica. c The nonseparable diastereomeric ratios (d.r. ratio) of all molecules were determined by 1H NMR analysis. |
|---|

|
| a Reaction conditions: N-substituted isatins 4a–4j (1 mmol) and methylglycine 2a (1 mmol) and different isothiocyanates 3a–3i (1 mmol) in the presence of ZnMnO3@Ni(OH)2 catalyst (20 mol%) and solvents (3 mL) at room temperature for 8 h. b Yields are reported for isolated products. c The nonseparable diastereomeric ratios (d.r. ratios) of all molecules were assessed through 1H NMR analysis. d Major isomer of 5ae, recrystallization in MeOH and hexane bimixture solvent [Fig S5, P25, (ESI) 1†]. |
|---|
|
| a Reaction conditions: 2,2-dihydroxy-1H-indene-1,3(2H)-diones 6a–b (1 mmol) and methylglycine 2a (1 mmol) and different isothiocyanates 3a–g (1 mmol) in the presence of the ZnMnO3@Ni(OH)2 catalyst (20 mol%) and solvents (3 mL) at room temperature for 8 h. b Yields are reported for isolated products. c Purity of 7aa is determined by HPLC [Fig. S7, P29, (ESI) 1†]. |
|---|
|
The promising yields obtained in these experiments with N-substituted isatins (4a–4j) inspired us to broaden the substrate scope and have no effect on the diastereoselectivity and yield. We extended our exploration to include isatin derivatives containing N-methyl, N-ethyl, N-propyl, N-benzyl, and N-butyl substituents (4a–4j) in combination with substituted isothiocyanatobenzenes (3a–3i), leading to the formation of the desired hydroxyl isatin, i.e. 3-methyl-5-oxo-2-thioxoimidazolidin indolinone (5) derivatives in excellent yields and diastereoselectivities (Table 4).
The reactions were explored using a series of substituted N-methyl isatins. Aliphatic isothiocyanates and para-substituted isothiocyanatobenzenes bearing electron-donating groups (–Me) provided excellent diastereoselectivity ranging between 95:5 and 98:2 and yields in the range of 90–94% (Table 4, 5ab, 5ae, and 5af).
However, the introduction of electron-withdrawing groups (–CN and –NO2) on the isothiocyanatobenzene also gave good yields of the products 5ac (85%) and 5ad (83%), respectively (Table 4). The electronic effects of N-ethyl substituents on isatins appeared to exhibit little consistency in the reaction. A steric effect became apparent when ortho-di-substituted isothiocyanatobenzene was employed, resulting in slightly lower product yield (79%) and diastereoselectivity (Table 4, 5bg). However, both 5bh and 5bi also demonstrated successful reactions, resulting in excellent product yields. Isatins with alkyl substitutions, such as N-propyl or N-butyl, produced the products 5de, 5db, 5ge, and 5he also in excellent yields and diastereoselectivities ranging between 85:15 and 96:4. Furthermore, isatins featuring a diverse array of electron-releasing and -withdrawing groups at the 5-position of the ring readily participated in the cross-coupling process under the current conditions and produced the corresponding products (Table 4, 5fe and 5he, 5je) in good to excellent yields ranging from 89% to 96%. The electronic effects of 5-Cl and N-benzyl substituents on isatins seemed to have minimal impact on the selectivity (Table 4, 5ee and 5fe) of the reaction. Single crystal X-ray diffraction (XRD) provided insights into the stereochemistry of one final compound within the diastereomeric mixture of hydroxy products. Ultimately, using single-crystal X-ray crystallographic analysis, the structures and orientation of 5ae molecules were confirmed (CCDC 2239522†) (Table 4).
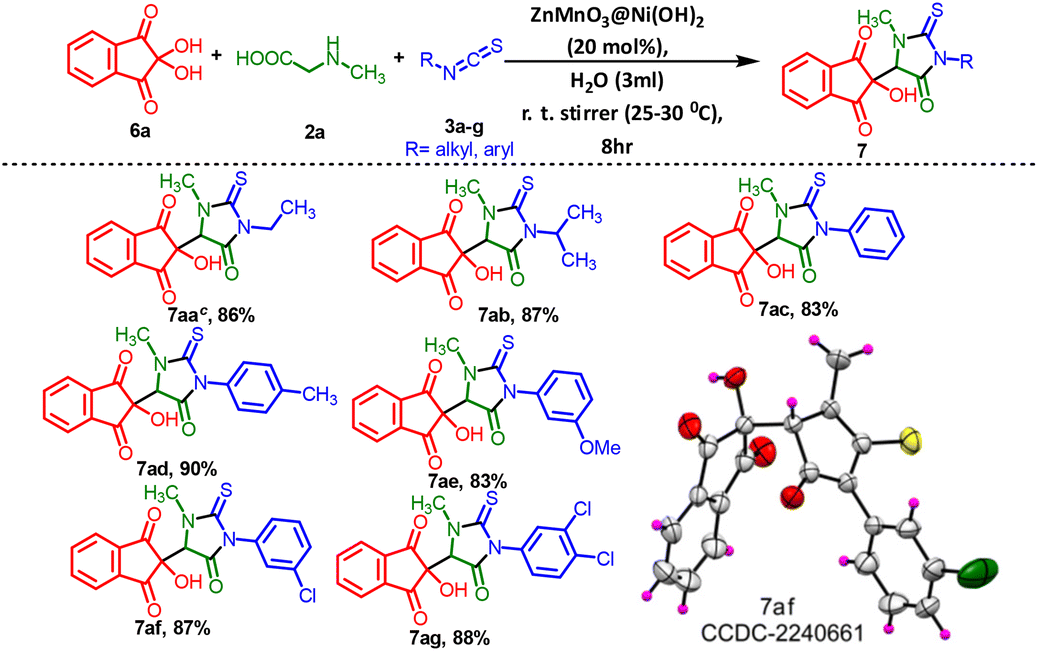
Furthermore, our extended investigations showed that the same catalytic process was compatible with a diverse range of 2-thioxoimidazolidin-4-one fused rings, exhibiting good tolerance for various functional groups (7aa–7ag). This is a very good practical method, for creating various types of 3-methyl-5-oxo-2-thioxoimidazolidin indene diones (7) by using ninhydrin and other accessible materials. Methyl, methoxy, mono and dihalogenated isothiocyanatobenzenes, as well as aliphatic isothiocyanates like ethyl and isopropyl, were very well-tolerated. They yielded the desired products, 7aa–7ag, in very good yields ranging from 83% to 90%. The experimental results also indicated that the reaction of aromatic isothiocyanates bearing electron-donating groups (EDGs) such as –CH3 and –OCH3 at either the 3- or 4-position resulted in a notably excellent yield of the target compounds (Table 5, 7ad and 7ae), achieving 90% and 83% yields, respectively, compared to unsubstituted isothiocyanatobenzenes. Among this series of compounds containing electron-withdrawing groups, the products 7af and 7ag were obtained with notably improved yields of 87% and 88%, respectively. When acenaphthoquinone was employed instead of isatin, phenylglycine was utilized as a substitute for methylglycine, and glycine was used instead of methylglycine [P34–P36, (ESI) 1†], the reaction did not proceed effectively under the different temperature reaction conditions in the presence of the ZnMnO3@Ni(OH)2 catalyst. A very low conversion (15%) was observed, along with a complex mixture that prevented the determination of the target product yield. The use of ninhydrin as a substrate in Table 5 showed no diastereomeric ratio (d.r. ratio) despite all compounds in Table 5 having a single chiral center, confirmed by the corresponding NMR and HPLC data. Specifically, the HPLC data for compound 7aa demonstrated high purity with no presence of isomers [Fig. S7, P29, (ESI) 1†].
The substrate scope of isatin 8a–8g or ninhydrin 9a, methylglycine 2a and isothiocyanates 3a–3i was investigated, as shown in Table 6. The condensation reaction likely occurs through a dehydration step involving the initial addition of the same accessible materials in the presence of ZnMnO3@Ni(OH)2 as a heterogeneous catalyst under reflux conditions. To gain a better understanding of the formation of 5-methylene-2-thioxoimidazolidin-4-one, we conducted stoichiometric reactions involving 8a–8g or 9a, 2a, and 3a–3i with the intermediate generated at elevated temperatures (Scheme 2). Initially, we summarized the results of the reaction using various isatins 8a–8g(Table 6). Unfortunately, electron-rich substituents, such as aryl 4-methyl, 3-methoxy and aliphatic isothiocyanates (10aa–10bc, 10ba–10da and 11ac) resulted in moderate yields (64–75%) and significant diastereoselectivities (Z:E) ranging between 89:11 and 95:5. However, the reaction was significantly impressed with 4-CN, 4-NO2 isothiocyanates (10bd, 10be and 10ed) owing to the strong electron-withdrawing property leading to higher yield (77–82%) and low diastereoselectivities (Z:E). The electron-withdrawing-substituted isatin derivative produced aldol products (10fg–10gh) with yields ranging from 77% to 80%, displaying a remarkable Z:E selectivity between 86:14 and 88:12. When ninhydrin 9a was used instead of isatins 8a–8g, it led to a loss of diastereoselectivity in the reaction due to the generation of symmetry in the product (11ai and 11ac). The structure of the (Z)-3-(1-ethyl-3-methyl-5-oxo-2-thioxoimidazolidin-4-ylidene)indolin-2-one motif was confirmed through single-crystal X-ray diffraction analysis of compound 10aa (CCDC 2289809†).
 | ||
| Scheme 2 A stoichiometric reaction of 5ab was conducted at different temperatures, which included control experiments for comparison. | ||
| a Reaction conditions: substituted isatin 8a–8g (1 mmol) or 2,2-dihydroxy-1H-indene-1,3(2H)-diones 9a (1 mmol), and methylglycine 2a (1 mmol) and isothiocyanates 3a–i (1 mmol) in the presence of the ZnMnO3@Ni(OH)2 catalyst (20 mol%) and solvents (3 mL) under reflux for 3 h. b Yields are reported for isolated products. c Major isomer of 10aa, recrystallization in MeOH and hexane bimixture solvent. d Purity of the Z/E ratio of 10cf determined by HPLC [Fig. S8, P31, (ESI) 1†], whereas the nonseparable Z/E ratios of other molecules were determined by 1H NMR analysis. |
|---|

|
| Sr. no | Green chemistry metrics | Ideal value | Product (5bh) | Product (10cf) |
|---|---|---|---|---|
| 1 | E factor (g g−1) | 0 | 0.17 | 0.48 |
| 2 | Atom economy (AE)% | 100% | 95.81 | 91.61 |
| 3 | Effective mass yield (EMY)% | 100% | 85.29 | 67.11 |
| 4 | Carbon efficiency (CE) % | 100% | 89.02 | 75.18 |
| 5 | Reaction mass efficiency (RME) % | 100% | 85.29 | 67.11 |
Water-insoluble solid reactants undergo reactions on the water surface at both room temperature and refluxing temperature. At room temperature, the water surface stabilizes the final product and catalyst activity via hydrophilic interactions. At refluxing temperature, the water surface accelerates the reaction for dehydration product formation without compromising the catalytic activity of the heterogeneous catalyst.
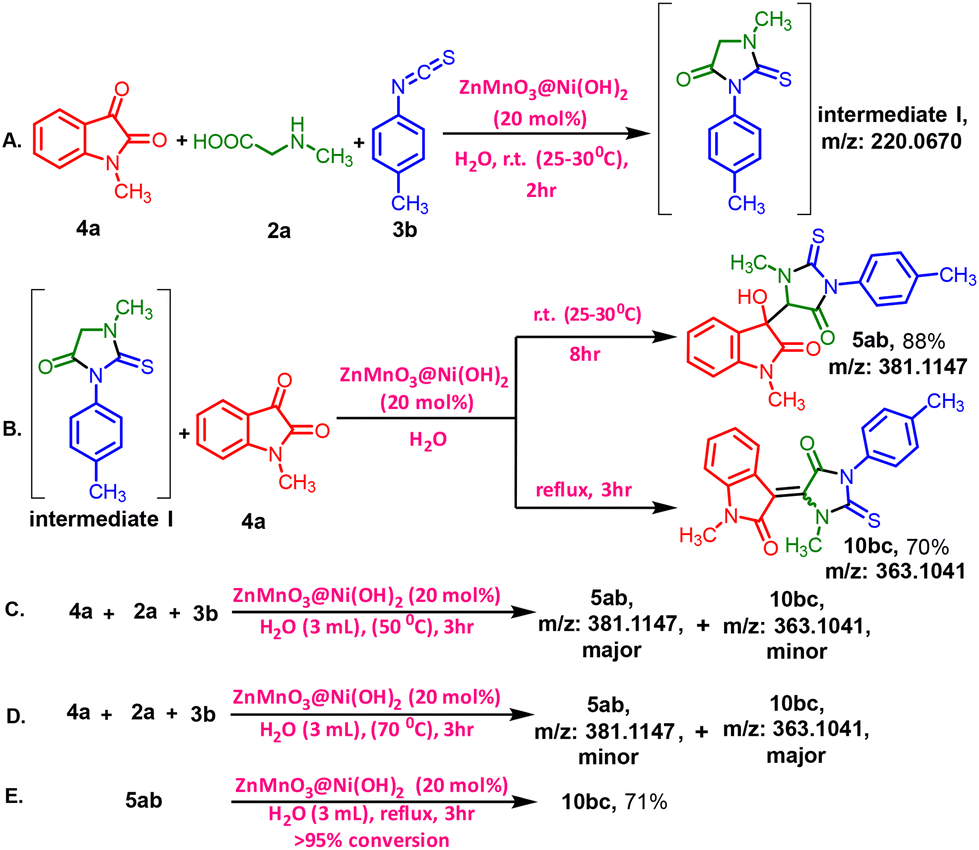
To further investigate the reaction mechanism, we conducted some control experiments (Scheme 2). At first, the intermediate (I) was synthesized by stirring 4a, 2a, and 3b for 2 hours at room temperature in the presence of the catalyst. The formation of the intermediate was confirmed by HRMS data (P32–P34, ESI 1†) (Scheme 2A).
Next, the reaction of 1-methyl-2-thioxo-3-(p-tolyl) imidazoline-4-one (I) with N-methyl isatin (4a) was carried out under the standard conditions for synthesizing compounds 5ab or 10bc (Scheme 2B). 10bc was not detected, but 5ab was obtained in an 88% yield at room temperature, while 10bc was obtained only under reflux conditions, resulting in 5ab in 70% yield. These results indicate that intermediate I, 1-methyl-2-thioxo-3-(p-tolyl)imidazolidin-4-one, is a key intermediate in the reaction. Under the standard reaction conditions at 50 °C for 3 hours, a mixture of both 5ab and 10bc was observed, 10bc being the minor product compared to 5ab, as detected by HRMS (Scheme 2C) [Fig. S10, P32, (ESI) 1†]. Furthermore, an increased percentage of the dehydrated product 10bc was observed when the temperature was raised from 70 °C [Fig. S11, P33, (ESI) 1†] to reflux conditions [Fig. S12, P33, (ESI) 1†] for 3 hours (Scheme 2C and D). In the last experiment (Scheme 2, entry D), only 10bc was detected under reflux conditions [Fig. S12, P33, (ESI) 1†]. The complete transformation of isolated 5ab to 10bc under standard reflux conditions also suggests that the formation of 10bc occurs via5ab and the unique intermediate I was the key intermediate in the reaction (Scheme 2E).
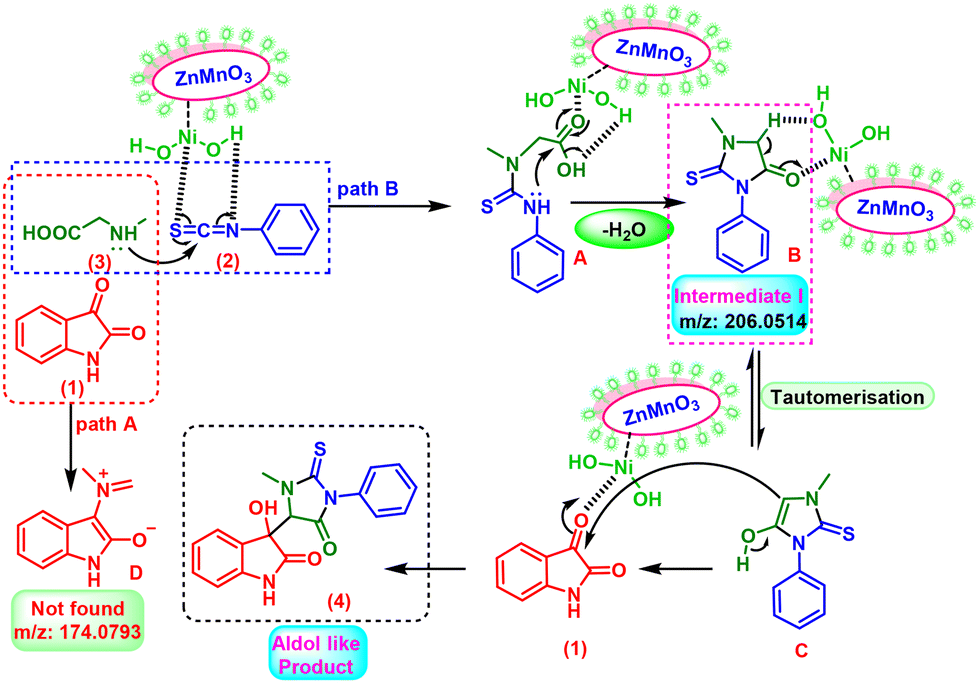
Based on mechanistic research and knowledge from relevant literature.33 In the ZnMnO3@Ni(OH)2 nanocatalyst, OH groups function as a Lewis base, providing Lewis base sites. These OH groups are engaged with electrophilic species, thus stabilizing the intermediates and facilitating specific reactions within the catalyst. On the other hand, “Ni” offers Lewis acid sites capable of coordinating with reactants, initiating specific bond activations, and promoting chemical conversions. It is reasonable to assume that the initial addition of the amine group of sarcosine attacks the carbon center of the isothiocyanate group with the assistance of the ditopic nature34 of the corresponding ZnMnO3@Ni(OH)2 catalyst through the coordination, resulting in the formation of a substituted thiourea moiety (A). Subsequently, the secondary amine moiety of (A), derived from the isothiocyanate group, initiates an attack on the carbonyl center of the acid group. This results in the formation of C–C and C–N bonds, concurrently generating the imidazole-type ring system (B) through the eco-friendly elimination of H2O (Scheme 3, pathway B). This intermediate I, as detected by HRMS [Fig. S13, P34, (ESI) 1†] can undergo further catalyzed reactions at room temperature, enhancing the reactivity of the active methylene group within the ring system. The intermediate I exhibits a propensity for facile tautomerization and subsequent addition to the electrophilic carbonyl group of isatin (1). This process leads to the creation of aldol-like C–C bonds, forming thiohydantoin-isatin/ninhydrin conjugates (4) with a product formed exhibiting diastereoselectivity (Scheme 3, pathway B). In contrast, this specific “D” type intermediate (follow pathway A) is formed when L-proline is utilized instead of methyl glycine.35
 | ||
| Scheme 3 Plausible mechanism for the reaction between isatin, methylglycine (sarcosine) and phenyl isothiocyanate at room temperature. | ||
Based on the above experiments, a plausible mechanism for the second reaction can be depicted as given in Scheme 4. Initially, the formation of the same imidazole-type ring system (B), i.e., intermediate I, occurs. Consequently, the formation of 5-(hydroxymethyl)-1-methyl-2-thioxoimidazolidin-4-one as an intermediate (4) occurred. During this high-temperature reaction, the feasibility of C3–C4′ sigma bond rotation leads to the formation of an intermediate (4′), and a dehydration step occurs simultaneously. At this temperature, the dehydration of the intermediate (4) through syn-elimination generates a less stable ‘E’ isomer due to dipole–dipole repulsion (Scheme 4D). However, such a dipole–dipole repulsion factor is minimized when the ‘Z′ isomer is formed through anti-elimination from the intermediate (4′) (Scheme 4F). Concurrently, the combined influence of the catalyst and temperature-induced dehydration gave rise to diastereomeric (Z/E) products (Table 6).
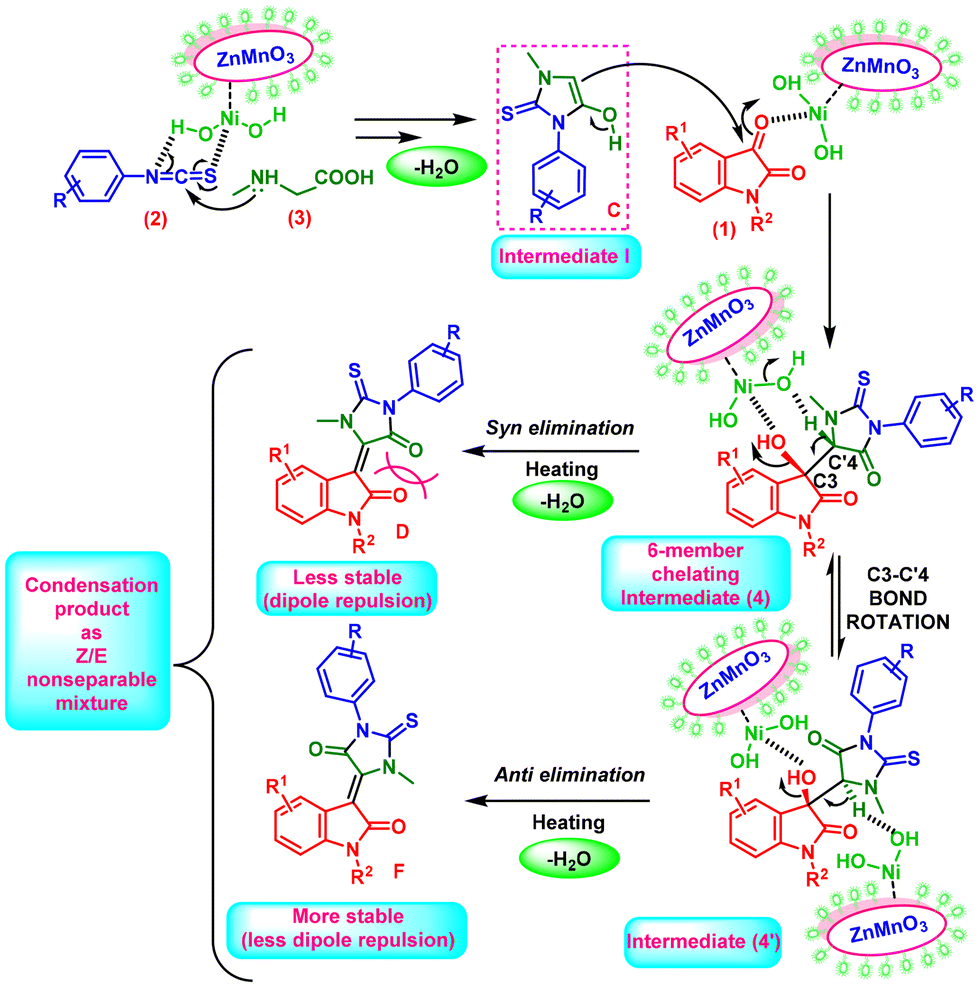 | ||
| Scheme 4 Plausible mechanism for the reaction between isatin, methylglycine (sarcosine) and phenyl isothiocyanate under reflux conditions. | ||
Therefore, the turnover frequency (TOF) quantifies the activity level of a catalytic site, indicating how active it is. Conversely, the turnover number (TON) measures the stability or endurance of this active site (Table 8) [detailed calculation in P8 and P9, ESI 1†].
| Product | TONa | TOFb (h−1) | Product | TON | TOF (h−1) |
|---|---|---|---|---|---|
| a Turnover number (TON) units (mol product per mol catalyst). b Turnover frequency (TOF) units ((mol product per mol catalyst) per hour). | |||||
| 4aa | 4.09 | 0.51 | 5db | 4.49 | 0.56 |
| 4ab | 4.19 | 0.52 | 5ee | 4.56 | 0.57 |
| 4ac | 4.05 | 0.51 | 5fe | 4.75 | 0.59 |
| 4ba | 4.20 | 0.52 | 5ge | 4.75 | 0.59 |
| 4bb | 4.25 | 0.53 | 5he | 4.80 | 0.60 |
| 4cb | 4.20 | 0.53 | 5ie | 4.25 | 0.53 |
| 4db | 4.29 | 0.54 | 5je | 4.45 | 0.56 |
| 4eb | 4.00 | 0.50 | 7aa | 4.29 | 0.54 |
| 4ad | 4.65 | 0.58 | 7ab | 4.35 | 0.54 |
| 4fd | 4.44 | 0.56 | 7ac | 4.15 | 0.52 |
| 4gd | 5.19 | 0.65 | 7ad | 4.50 | 0.56 |
| 4he | 4.13 | 0.52 | 7ae | 4.15 | 0.52 |
| 4ib | 4.25 | 0.53 | 7af | 4.35 | 0.54 |
| 5aa | 4.45 | 0.56 | 7ag | 4.40 | 0.55 |
| 5ab | 4.55 | 0.57 | 10aa | 3.50 | 1.17 |
| 5ac | 4.25 | 0.53 | 10bb | 3.55 | 1.18 |
| 5ad | 4.15 | 0.52 | 10bc | 3.39 | 1.13 |
| 5ae | 4.50 | 0.56 | 10bd | 3.95 | 1.32 |
| 5af | 4.68 | 0.59 | 10be | 4.09 | 1.36 |
| 5ba | 4.49 | 0.56 | 10ba | 3.40 | 1.13 |
| 5bg | 3.95 | 0.49 | 10cf | 3.76 | 1.25 |
| 5bb | 4.50 | 0.56 | 10da | 3.19 | 1.07 |
| 5bh | 4.45 | 0.56 | 10ed | 3.85 | 1.28 |
| 5bi | 4.39 | 0.55 | 10fg | 3.85 | 1.28 |
| 5be | 4.49 | 0.56 | 10gh | 4.00 | 1.33 |
| 5ce | 4.55 | 0.57 | 11ai | 3.95 | 1.32 |
| 5bf | 4.64 | 0.58 | 11ac | 3.70 | 1.23 |
| 5de | 4.69 | 0.59 | |||
Hot filtration test
A hot filtration test was performed with our standard reaction for the synthesis of 10bc under the optimized conditions (Table 1, entry 29) to validate the heterogeneous nature of the catalyst. After 30 min of the reaction, the reaction mixture underwent filtration using a hot frit, and the resulting filtrate was further subjected to reflux for another 4 hours. A high decrease in the yield percentage (68% to 20%) was observed at the end of the experiment. This result supported the heterogeneity of the catalyst and also concluded that after the filtration, the reaction did not proceed to completion, which indicates that no active catalyst remained in the filtrate.Leaching test
The probable leaching of ZnMnO3@Ni(OH)2 into the reaction mixture was assessed using ICP-MS analysis. For the ICP-MS analysis, samples were obtained via a syringe filter (PTFE, Whatman Puradisc 4, 0.45 μm, 4 mm diameter) during standard heterogeneous catalytic reactions (reaction temperature: 25–30 °C), followed by solvent evaporation and residue dissolution in HNO3. ICP-MS analysis revealed that the zinc (Zn) concentration in the reaction solution was less than 0.11% of the initial ZnMnO3@Ni(OH)2 amount (50 mg), while manganese (Mn) and nickel (Ni) concentrations were less than 0.13% and 0.05% of the initial catalyst amount, respectively. These findings strongly suggest that no leaching of Zn, Mn, and Ni from the catalyst surface into the solution occurred.The results from the hot filtration and leaching tests suggested that elevated reaction temperatures do not cause an increase in ZnMnO3@Ni(OH)2 leaching into the reaction solution. Therefore, the lower reaction temperature is assumed to affect solely the obtained yields and not the leaching behavior of ZnMnO3@Ni(OH)2. Moreover, these results also imply the environmental friendliness or “greenness” of the reaction.
To assess the catalyst's reusability, we utilized 50 mg of ZnMnO3@Ni(OH)2 nano-powder in a model reaction with isatins 1a, methylglycine 2a, and various isothiocyanates 3a under optimized conditions to produce the product 5aa. After verifying the reaction's completion via TLC, the crude mixture was diluted with 10 mL of ethanol, and the catalyst was separated by filtration. It was then washed five times with 10 mL of ethanol each time to remove any adhering organic substances and dried under vacuum. Subsequently, we conducted recycling experiments using the recovered catalyst for multiple cycles, even exposing it to ambient atmosphere for 10 days without observing any deterioration in its activity. Over eight cycles, the catalyst demonstrated nearly identical activity (Fig. 7). After the 8th run, we conducted a detailed characterization of the catalyst and found that almost 95% of the catalyst was recovered from each run. After recovery, XRD, SEM, and TEM analyses of ZnMnO3@Ni(OH)2 have been performed. It is found that there is no change in the XRD pattern and SEM and TEM morphologies. It indicates that the catalyst is highly stable [Fig. S3, P7, (ESI) 1†].
 | ||
| Fig. 7 Recycling efficiency of the ZnMnO3@Ni(OH)2 nanocatalyst under optimized conditions. | ||
Conclusions
In conclusion, this study significantly advances medicinal chemistry and drug discovery through the synthesis and conjugation of thioxoimidazolidinones with 3-hydroxy oxindoles occurring “on water” has been developed and implemented. The demonstrated one-pot, green reactions offer an efficient and cost-effective approach with a strong commitment to eco-friendly principles. The success hinges on meticulous optimization, catalyst selection, and chemical understanding. This approach not only enhances total yields, but also accelerates synthesis, making it suitable for large-scale production. The use of heterogeneous nanocatalysts, like ZnMnO3@Ni(OH)2 (ZNO), contributes to eco-conscious transformations. The unique morphology of ZNO combines the catalytic properties of ZnMnO3 and Ni(OH)2, enabling efficient reactions. The reaction demonstrates high atom economy, simplicity in operation, and environmental friendliness (E-factor = 0.5). Key green attributes include the lack of hazardous catalysts and the sole by-product being H2O. Notably, the isatin–thiazolidine conjugate undergoes a stereoselective transfer aldol reaction in an aqueous environment. This study presents a versatile approach for forming C–C and C–N bonds, showcasing the potential of temperature-induced dehydration catalyzed by ZNO in synthesizing bioactive heterocyclic compounds, furthering green and sustainable chemistry principles. This work underscores the importance of sustainable practices in modern chemical research, charting a promising path for future endeavours.Experimental
Synthesis of spherical-like ZnMnO3
Typically, 45 ml of water was mixed with 5 mmol of each of manganese chloride (MnCl2) and zinc sulphate (ZnSO4) to create a transparent solution. The aforementioned aqueous mixture was subsequently mixed with a 30 mmol Na2CO3 solution and 50 mg polyvinylpyrrolidone (PVP) under constant stirring at a temperature of 50 °C for two hours. A 50 ml Teflon-lined stainless steel autoclave was then filled with the solution and heated at 160 °C for 18 hours. The solid result was then cleaned/washed with a 3:1 v/v mixture of water and ethanol before the entire system was progressively cooled to ambient temperature. The solid ZnMnO3 product was then moved and dried under vacuum for 12 hours at 100 °C. Finally, a mortar and a pestle were used to crush the resulting solid. It was then calcined in an air oven at 350 °C for 8 hours before being cooled to room temperature.
Synthesis of the spherical honeycomb like ZnMnO3@Ni(OH)2 (ZNO) catalyst
The catalyst ZnMnO3@Ni(OH)2 (ZNO) was prepared using a straightforward hydrothermal process. Usually, probe sonication was performed for 30 minutes after taking 20 mg of ZnMnO3 in 45 ml of water. The aqueous solution was then added along with 35 mg of nickel chloride and 20 mg of HMTA while being continuously stirred for two hours. The entire aqueous solution was then put into a 50 ml Teflon-lined stainless steel autoclave and heated for 18 hours at 100 °C. The black product was then cleaned with a water and ethanol mixture (4:1 v/v) once the entire system was cooled to ambient temperature. Finally, a black product, the ZNO catalyst, was taken and dried under vacuum conditions for 12 hours at 80 °C.
General procedure for the synthesis of N-substituted thiazolidinediones with isatin and ninhydrin derivatives (4aa–4ib, 5aa–5je, 7aa–7ag and 10aa–11ac)
[ESI 1†].Author contributions
The concept was developed by Soumitra Rana under the guidance of Chhanda Mukhopadhyay. Synthesis and purification of the final compounds were performed by Soumitra Rana and Soumyadip Basu. Aswini Bera and Bhanu Bhusan Khatua contributed to the synthesis of the catalyst and analysis of the experimental results of the catalyst. Pinaki Saha and Prasanta Ghosh assisted in conducting single crystal XRD experiments and analyzing the corresponding results. The manuscript was written by Soumitra Rana under the guidance of Chhanda Mukhopadhyay, who oversaw the overall work.Conflicts of interest
No conflicts of interest have been identified or declared.Acknowledgements
One of the authors (S. R.) thanks the Council of Scientific and Industrial Research (CSIR), New Delhi, for the fellowship (SRF). The authors are also grateful to CAS-V (UGC) (540/3/CASV/2015 (SAP-I)), DST-FIST (SR/FST/CSI-198/2008) and DST-PURSE DST Purse II No. SR/PURSE Phase 2/21(C), Department of Chemistry, University of Calcutta, for funding as departmental projects.References
- (a) M. Krasavin, Eur. J. Med. Chem., 2015, 97, 525–537 CrossRef CAS PubMed; (b) K. Mezgebe, Y. Melaku and E. Mulugeta, ACS Omega, 2023, 8, 19194–19211 CrossRef CAS PubMed.
- G. L. Khatik, J. Kaur, V. Kumar, K. Tikoo, P. Venugopalan and V. A. Nair, Eur. J. Med. Chem., 2011, 46, 3291 CrossRef CAS PubMed.
- K. Tachibana, I. Imaoka, T. Shiraishi, H. Yoshino, M. Nakamura, M. Ohta, H. Kawata, K. Taniguchi, N. Ishikura, T. Tsunenari, H. Saito, M. Nagamuta, T. Nakagawa, K. Takanashi, E. Onuma and H. Sato, Chem. Pharm. Bull., 2008, 56, 1555–1561 CrossRef CAS PubMed.
- K. K. Kononowicz and E. Szymanska, Farmaco, 2002, 57, 909–916 CrossRef PubMed.
- Y. S. Bae, S. Choi, J. J. Park, J. H. Joo, M. Cui, H. Cho, W. J. Lee, and S. H. Lee, Bioorg. Med. Chem., 2016, 24, 4144–4151 CrossRef CAS PubMed.
- (a) H. Elokdah, T. S. Sulkowski, M. Abou-Gharbia, J. A. Butera, S.-Y. Chai, G. R. McFarlane, M.-L. McKean, J. L. Babiak, S. J. Adelman and E. M. Quinet, J. Med. Chem., 2004, 47, 681–695 CrossRef CAS PubMed; (b) M. Aguilar-Moncayo, C. O. Mellet, J. M. G. Fernández and M. I. García-Moreno, J. Org. Chem., 2009, 74, 3595–3598 CrossRef CAS PubMed; (c) Z. Zhang, P. J. Connolly, L. T. Escolar, C. Rocaboy, V. Pande, L. Meerpoel, H.-K. Lim, J. R. Branch, J. Ondrus, I. Hickson, T. L. Bush, J. R. Bischoff and G. Bignan, ACS Med. Chem. Lett., 2021, 12, 1245–1252 CrossRef CAS PubMed; (d) H. R. Kim, H. J. Lee, Y. J. Choi, Y. J. Park, Y. Woo, S. J. Kim, M. H. Park, H.W. Lee, P. Chun, H. Y. Chung and H. R. Moon, MedChemComm, 2014, 5, 1410–1417 RSC; (e) S. M. Khirallah, H. M. M. Ramadan, H. A. A. Aladl, N. O. Ayaz, L. A. F. Kurdi, M. Jaremko, S. Z. Alshawwa and E. M. Saied, Pharmaceuticals, 2022, 15, 1576 CrossRef CAS PubMed.
- S. Aryal, C. A. Hone, M. I. Polson and D. J. Foley, Chem. Sci., 2023, 14, 7905–7912 RSC.
- (a) L. Konnert, F. Lamaty, J. Martinez and E. Colacino, Chem. Rev., 2017, 117, 13757–13809 CrossRef CAS PubMed; (b) R. M. Shaker, H. A. A. El-Naby, E. K. Ahmed, M. A. A. Ibrahim and S. A. Gedamy, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 147–155 CrossRef CAS; (c) Y. J. Huang, X. R. Peng and M. H. Qiu, Nat. Prod. Bioprospect., 2018, 8, 405–412 CrossRef CAS PubMed; (d) P. Vicini, A. Geronikaki, M. Incerti, F. Zani, J. Dearden and M. Hewitt, Bioorg. Med. Chem., 2008, 16, 3714–3724 CrossRef CAS PubMed; (e) A. S. Kumar, P. Ramesh, G. S. Kumar, J. B. Nanubolu, T. P. Rao and H. M. Meshram, RSC Adv., 2015, 5, 51581–51585 RSC; (f) P. B. Thakur and H. M. Meshram, RSC Adv., 2014, 4, 5343–5350 RSC; (g) H. M. Meshram, P. Ramesh, B. C. Reddy, B. Shreedhar and J. S. Yadav, Tetrahedron, 2011, 67, 3150–3155 CrossRef CAS; (h) M. Pastor, M. Vayer, H. Weinstabl and N. Maulide, J. Org. Chem., 2021, 87, 606–612 CrossRef PubMed; (i) G. Zhang, Z. Xia, C. Tian, A. Xia, J. You, J. Liu, S. Yang and L. Li, Bioorg. Med. Chem. Lett., 2023, 92, 129383 CrossRef CAS PubMed.
- (a) M. Hayashi, M.-C. Rho, A. Enomoto, A. Fukami, P. Kim, Y. Kikuchi, T. Sunazuka, T. Hirose, K. Komiyama and S. Ojmura, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14728–14733 CrossRef CAS PubMed; (b) R. Niu, J. Xiao, T. Liang and X. Li, Org. Lett., 2012, 14, 676–679 CrossRef CAS PubMed; (c) K. Omar, A. Geronikaki, P. Zoumpoulakis, C. Camoutsis, M. Sokovic, C. Ciric and J. Glamoclija, Bioorg. Med. Chem., 2010, 18, 426–432 CrossRef CAS PubMed.
- (a) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS PubMed; (b) R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef CAS PubMed; (c) J. Huang, X. Zhang and D. W. Armstrong, Angew. Chem., Int. Ed., 2007, 46, 9073 CrossRef CAS PubMed; (d) L. Zu, H. Xie, H. Li, J. Wang and W. Wang, Org. Lett., 2008, 10, 1211 CrossRef CAS PubMed.
- (a) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed.
- R. Maccari, R. Ettari, I. Adornato, A. Naß, G. Wolber, A. Bitto and R. Ottanà, Bioorg. Med. Chem. Lett., 2018, 28, 278–283 CrossRef CAS PubMed.
- (a) E. Soleimani, S. Ghorbani and H. R. Ghasempour, Tetrahedron, 2013, 69, 8511–8515 CrossRef CAS; (b) P. Verma, S. Pal, S. Chauhan, A. Mishra, I. Sinha, S. Singh and V. Srivastava, J. Mol. Struct., 2020, 1203, 127410 CrossRef CAS; (c) B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS PubMed; (d) H. Bienayme, C. Hulme, G. Oddon and P. Schmitt, Chem. – Eur. J., 2000, 6, 3321–3329 CrossRef CAS PubMed.
- (a) Z. Hosseinidoust, M. Basnet, T. G. van de Ven and N. Tufenkji, Environ. Sci. Nano, 2016, 3, 1259–1264 RSC; (b) P. E. Cardoso-Avila, R. Patakfalvi, C. Rodríguez-Pedroza, X. Aparicio-Fernández, S. Loza-Cornejo, V. Villa-Cruz and E. Martínez-Cano, RSC Adv., 2021, 11, 14624–14631 RSC; (c) Q. Chen and J. E. Beckman, Green Chem., 2008, 10, 934–938 RSC.
- For reviews, see: C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson and A. A. Lapkin, Chem. Rev., 2023, 123, 3089–3126 CrossRef CAS PubMed.
- (a) R. S. Varma, ACS Sustainable Chem. Eng., 2016, 4(11), 5866–5878 CrossRef CAS PubMed; (b) S. R. Attar and S. B. Kamble, Nanoscale, 2022, 14, 16761–16786 RSC; (c) P. Bradu, A. Biswas, C. Nair, S. Sreevalsakumar, M. Patil, S. Kannampuzha and A. V. Gopalakrishnan, Environ. Sci. Pollut. Res., 2022, 1–32 Search PubMed; (d) A. D. Kreuder, T. House-Knight, J. Whitford, E. Ponnusamy, P. Miller, N. Jesse, R. Rodenborn, S. Sayag, M. Gebel, I. Aped, I. Sharfstein, E. Manaster, I. Ergaz, A. Harris and L. N. Grice, ACS Sustainable Chem. Eng., 2017, 5, 2927–2935 CrossRef.
- (a) X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC; (b) A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC; (c) S. Neamani and L. Moradi, ChemistryOpen, 2022, 11, e202200041 CrossRef CAS PubMed.
- (a) R. A. Sheldon, Green Chem., 2023, 25, 1704–1728 RSC; (b) M. Halder, M. M. Islam, P. Singh, A. Singha Roy, S. M. Islam and K. Sen, ACS Omega, 2018, 3, 8169–8180 CrossRef CAS PubMed; (c) X. Zhang, L. Wen, Y. Xu, K. Sun and X. Hao, Inorg. Chem., 2020, 59, 16205–16214 CrossRef CAS PubMed.
- M. M. Elbadawi, A. I. Khodair, M. K. Awad, S. E. Kassab, M. T. Elsaady and K. R. Abdellatif, J. Mol. Struct., 2022, 1249, 131574 CrossRef CAS.
- H. Chen, L. X. Ding, K. Xiao, S. Dai, S. Wang and A. H. Wang, J. Mater. Chem., 2016, 4, 16318–16323 RSC.
- (a) S. Scurti, A. Allegri, F. Liuzzi, E. Rodríguez-Aguado, J. A. Cecilia, S. Albonetti, D. Caretti and N. Dimitratos, Catalysts, 2022, 12, 323 CrossRef CAS; (b) A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418–514 CrossRef CAS PubMed; (c) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed; (d) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- (a) N. L. Le and S. P. Nunes, SM&T, 2016, 7, 1–28 Search PubMed; (b) E. A. Peterson, B. Dillon, I. Raheem, P. Richardson, D. Richter, R. Schmidt and H. F. Sneddon, Green Chem., 2014, 16, 4060–4075 RSC; (c) A. Chanda, and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef PubMed; (d) D. Raabe, Chem. Rev., 2023, 123, 2436–2608 CrossRef CAS PubMed.
- F. Erben, D. Michalik, H. Feist, D. Kleeblatt, M. Hein, A. Matin and P. Langer, RSC Adv., 2014, 4, 10879–10893 RSC.
- Y. Ma, T. T. Ding, Y. Y. Liu, Z. H. Zheng, S. X. Sun, L. S. Zhang and R. L. Wang, Biochem. Biophys. Res. Commun., 2021, 579, 40–46 CrossRef CAS PubMed.
- B. T. Gregg, K. C. Golden, J. F. Quinn, D. O. Tymoshenko, W. G. Earley, D. A. Maynard, D. A. Razzano, W. M. Rennells and J. Butcher, J. Comb. Chem., 2007, 9, 1036–1040 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- E. R. Monteith, P. Mampuys, L. Summerton, J. H. Clark, B. U. W. Maes and C. R. McElroy, Green Chem., 2020, 22, 123–125 RSC.
- (a) K. Mal and C. Mukhopadhyay, J. Mol. Struct., 2022, 1265, 133377 CrossRef CAS; (b) I. Chatterjee, D. Roy and G. Panda, Green Chem., 2023, 25, 9097–9102 RSC; (c) M. B. Bejjam, G. S. Kumar, A. Swetha and H. M. Meshram, RSC Adv., 2016, 6, 13820–13828 RSC.
- (a) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492 CrossRef CAS PubMed; (b) O. Acevedo and K. Armacost, J. Am. Chem. Soc., 2010, 132, 1966 CrossRef CAS PubMed.
- A. Bera, L. Halder, S. K. Si, A. De, S. Ojha, S. Bera, P. Maity, A. Mondal and B. B. Khatua, Results Chem., 2022, 4, 100404 CrossRef CAS.
- X. Liu, C. Zhao, H. Zhang and Q. Shen, Electrochim. Acta, 2015, 151, 56–62 CrossRef CAS.
- A. Bera, A. Maitra, A. K. Das, L. Halder, S. Paria, S. K. Si, A. De, S. Ojha and B. B. Khatua, ACS Appl. Electron. Mater., 2020, 2, 177–185 CrossRef CAS.
- (a) C. Lv, X. Wang, L. Gao, A. Wang, S. Wang, R. Wang, X. Ning, Y. Li, D. W. Boukhvalov, Z. Huang and C. Zhang, ACS Catal., 2020, 10, 13323–13333 CrossRef CAS; (b) J. M. Grill, J. W. Ogle and S. A. Miller, J. Org. Chem., 2006, 71, 9291–9296 CrossRef CAS PubMed.
- S. Barik, S. Shee and A. T. Biju, Org. Lett., 2022, 24, 6066–6071 CrossRef CAS PubMed.
- (a) S. Rana, S. Basu and C. Mukhopadhyay, Mol. Diversity, 2022, 26, 2561–2573 CrossRef CAS PubMed; (b) S. Basu, S. Rana, P. Saha, P. Ghosh and C. Mukhopadhyay, J. Org. Chem., 2022, 87, 9755–9763 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and full spectroscopic data of all the newly synthesised compounds. CCDC 2239522, 2240661 and 2289809. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc03730d |
| This journal is © The Royal Society of Chemistry 2024 |