
Mechanically accelerated catalytic hydrogenation: correlating physical state, reaction rate, and interface area†
Federico
Cuccu
 ,
Francesco
Basoccu
,
Claudia
Fattuoni
and
Andrea
Porcheddu
*
,
Francesco
Basoccu
,
Claudia
Fattuoni
and
Andrea
Porcheddu
*
Dipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Cittadella Universitaria, 09042, Monserrato, Cagliari, Italy. E-mail: porcheddu@unica.it
First published on 14th December 2023
Abstract
This study thoroughly examines the role of mechanochemistry in organic synthesis by analysing the factors affecting the mechanochemical reduction process of unsaturated and oxidized compounds. Hydride species and hydrogen gas are generated in situ by mixing a diboron compound with a stoichiometric amount of water. The process is tested against several substrates to determine how the physical state of the reagents influences the reduction technique. The study aims to thoroughly investigate the correlation between the mixing process, reaction rate, and interface area.
Introduction
In the past 60 years, the study of mechanochemistry has experienced periods of intense growth.1 The investigation of physically and chemically activated inorganic transformations began in the early 1950s.2 However, at the end of the 1960s, when powders’ mechanical processing was successfully applied to metallurgical processes, it received worldwide attention.3 Mechanical alloying and grinding have since expanded to innovative materials, becoming prevalent in materials science from the early 1980s.4The introduction of mechanochemistry within organic chemistry has resulted in significant breakthroughs.5 This innovative combination allows for the synthesis of small molecules through mechanical forces, which has become a widespread approach due to the several issues encountered with solution-based techniques caused by inadequate solubility of reagents.6 Given the growing interest in eco-friendly practices and sustainability in chemical synthesis, it has become crucial to address these problems without relying on solvents and phase-transfer catalysts.7 Mechanochemistry provides a practical solution by lessening or eliminating the solvent phase, making it a desirable alternative for academic and industrial purposes.8
Mechanochemistry offers a broad range of synthetic methods that enable access to various active pharmaceutical ingredients (APIs) and chemicals.9 This success can be attributed to the adequate forced mixing of chemical species induced by severe mechanical deformation on a molecular scale.10,11 However, when mechanochemical conditions are applied to organic systems, it is essential to note some differences from inorganic materials. Unlike covalent, ionic, and metallic solids, organic molecular solids have weaker intermolecular forces and lower cohesive energy, resulting in lower melting points. As a result, molecular solids behave like “soft solid phases” that are prone to flow deformation and melting.11 This response is not directly related to the mechanical activation of covalent bonds but rather to the increased interface area between chemical species resulting from the intensified mixing of solid phases, not to mention the effect of liquid or molten phases. Therefore, data resulting from “mechanically induced” transformations in organic systems must be handled differently, as the role of mechanochemistry in these systems remains unclear.12
On these bases, this study aims to thoroughly investigate the correlation between the mixing process, reaction rate, and interface area. Specifically, we will look at the reactivity of organic compounds against gaseous species to observe how their physical states influence their reactivity during the milling process.
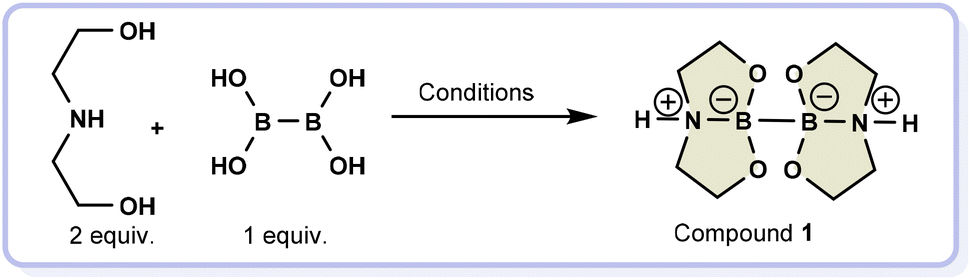
Herein, the selected gaseous reactant is molecular hydrogen, crucial in reducing unsaturated organic compounds to access simple scaffolds and complex molecular motifs like natural products and drugs.13–22 Regarding hydrogen, there are a lot of concerns about how it is transported, stored, and used. Many regulations and safety measures need to be followed to ensure compliance with health regulations. This can make using highly pressurized hydrogen complicated and costly, so researchers are looking for alternative ways to handle this gas. To safely release hydrogen in situ through a water-splitting process, we relied on a highly effective diboron compound (compound 1, Scheme 1), which generates hydrogen from a stoichiometric amount of water during hydrogenation reactions.23
 | ||
| Scheme 1 Similarities and differences of the reported protocol in solution (ref. 23) compared with our solid-state procedure. Novelties and improvements are included in yellow boxes, greener improvements are also marked in green. | ||
To begin with, the solid state's effectiveness in reducing organic compounds was carefully investigated. This involved the preparation of various primary and secondary alcohols by reducing a diverse range of aldehydes and ketones “without metal catalysts”.
We then progressed to catalytic hydrogenation of alkenes and alkynes, extending this methodology to nitro compounds. Lastly, we examined how mechanical processing influences chemical reactivity by studying hydrogenation kinetics.
Results and discussion
Reaction optimization and scope
According to the literature,23 the diboron compound 1 can be prepared starting from the commercially available diethanolamine and tetrahydroxydiboron in dichloromethane (10 mL mmol−1). However, this solvent is a volatile organic compound (VOC), hazardous to the environment and the operator. To address this issue, we have developed a new synthetic route that involves a mechanochemical approach. This process involves mixing diethanolamine (2 mmol) and tetrahydroxydiborane (1 mmol) without solvent. After testing different conditions, the best results were achieved using solvent-free conditions inside a 10 mL stainless steel (SS) jar with one SS ball (8 mm ∅, 2.09 g mass) at 30 Hz for 20 minutes. This solvent-free mechanochemical process resulted in an 84% yield of diboron compound 1 (entry 4, Table 1). Decreasing the reaction time (entries 1–3, Table 1) and the frequency at 25 Hz (entry 6, Table 1) resulted in a lower yield of the product, as occurred when using a smaller ball (entry 5, Table 1). Interestingly, other jar materials are compatible with the reaction, as evidenced in entries 7 and 8 in Table 1.|
|
|||||
|---|---|---|---|---|---|
| Entry | Time | Frequency | Material | Balls | Yieldb |
| Reaction stoichiometry: diethanolamine (2 mmol) and tetrahydroxydiboron (1 mmol), 10 mL jar. If not otherwise stated, the material of the balls employed was the same as the one composing the jar.a ZrO2 ball.b Yields refer to isolated products. | |||||
| 1 | 5 min | 30 Hz | SS | 1 (8 mm) | 7% |
| 2 | 10 min | 30 Hz | SS | 1 (8 mm) | 23% |
| 3 | 15 min | 30 Hz | SS | 1 (8 mm) | 67% |
| 4 | 20 min | 30 Hz | SS | 1 (8 mm) | 84% |
| 5 | 20 min | 30 Hz | SS | 5 (4 mm) | 34% |
| 6 | 20 min | 25 Hz | SS | 1 (8 mm) | 79% |
| 7 | 20 min | 30 Hz | ZrO2 | 1 (8 mm) | 83% |
| 8 | 20 min | 30 Hz | Ertalyte® | 1 (8 mm)a | 81% |
| 9 | 20 min | 30 Hz | SS | None | 5% |
Afterward, we tested the diboron compound 1 in the reduction of a set of aldehydes and ketones. The reaction mechanism for splitting water suggests that the reduction of carbonyl groups involves a hydride intermediate. Therefore, a library of aldehydes was tested to trap the hydride species and validate the reaction mechanism.23 Please refer to Scheme S1 in the ESI† for a complete overview of the reaction mechanism. We made appropriate adjustments to the conditions for the solvent-free water-splitting process. To conduct the screening as reported in Table S1 in the ESI,† we chose benzaldehyde 2a as the model substrate. The best experimental conditions were achieved by ball-milling benzaldehyde 2a (1 mmol), diboron compound 1 (2 mmol), and H2O (2 mmol) inside a 10 mL SS jar, along with one SS ball (8 mm ∅, 2.09 g mass), at a frequency of 30 Hz for 90 min.
These conditions resulted in a 98% yield of benzyl alcohol 3a. It is important to note that shaking the jar vessel without balls only gave 8% of the desired product 3a (see entry 6, Table S1 in the ESI†). This suggests that mechanical deformation is necessary for the reaction to occur correctly.
The reaction process shows a similar trend for benzaldehydes 2b–e and 2g–n in scope, resulting in excellent yields of corresponding alcohols (Scheme 2). Alcohols 3b–e are produced in yields of 93–99%, while alcohols 3g–n are produced in yields of 89–99%. In contrast, the decrease in yield for compound 3f (52%) can be explained by the steric hindrance given by the nitro moiety. Heterocyclic and conjugated aldehydes 2o and 2p also give excellent yields of alcohols 3o and 3p, with 96% and 94% yields, respectively. However, the reaction proceeds more slowly and less efficiently when it comes to aliphatic aldehydes or ketones, requiring one equivalent of LiCl to increase the reaction rate. For instance, ketones 4a–f produce alcohols 5a–f in yields of 42–99% (Scheme 2). LiCl may play a dual role, promoting carbonyl activation as a Lewis acid and forming stronger reducing species with close to lithium borohydride reactivity.24
 | ||
Scheme 2 The reaction scope for the reduction of aldehyde and ketones. Typical procedure: carbonyl compound (1 mmol), compound 1 (2 mmol), and H2O (2 mmol) were placed inside a 10 mL SS jar with one ball of the same material (∅ = 8 mm, 2.09 g). The reaction was conducted for 90 min at room temperature, 30 Hz, and under ball-milling conditions. Yields refer to isolated products if not otherwise stated. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) LiCl (1 mmol) was used as an additive. bThe yield was calculated via GC-MS analysis. LiCl (1 mmol) was used as an additive. bThe yield was calculated via GC-MS analysis. | ||
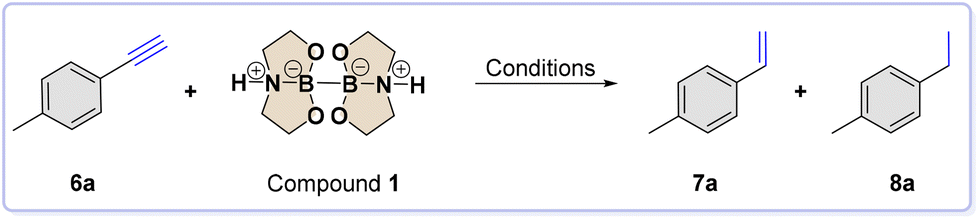
After optimizing the scope of carbonyl reduction, our attention turned to the catalytic hydrogenation reaction of unsaturated substrates. For our experiment, we used alkyne 6a as the substrate model. To conduct the investigation, we mixed 4-methylphenylacetylene 6a (1 mmol), diboron compound 1 (2 mmol), Pd/C (3 mol%), and H2O (2 mmol) in a 10 mL SS jar. We added one SS ball (8 mm ∅, 2.09 g mass) and ball-milled the mixture at a frequency of 30 Hz for 90 minutes. To our great satisfaction, we obtained the corresponding alkane 8a with a 97% yield, meaning the alkyne was completely reduced under the selected conditions. Furthermore, we adjusted the standard conditions and summarized the outcomes in Table 2.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Material | Balls |
6a:7a:8a |
Yielde |
| Reaction stoichiometry: alkyne (1 mmol), 2 mmol of diboron compound 1, H2O (2 mmol), 10 mL jar. If not otherwise stated, the material of the balls employed was the same as the one composing the jar.a Reaction conducted at 25 Hz.b 45 min.c 1 mmol of diboron compound 1 was used.d ZrO2 ball.e Yields refer to GC-MS analysis. | |||||
| 1 | Pd/C | SS | 1 × 8 mm |
0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0:100 :0:100
|
97% |
| 2a | Pd/C | SS | 1 × 8 mm | 0:0:100 |
78% |
| 3 | Pd/C | ZrO2 | 1 × 8 mm | 0:0:100 |
96% |
| 4 | Pd/C | Ertalyte® | 1 × 8 mmd | 0:0:100 |
97% |
| 5b | Pd/C | SS | 1 × 8 mm | 0:0:100 |
43% |
| 6 | None | SS | 1 × 8 mm | — | Traces |
| 7c | Pd/C | SS | 1 × 8 mm | 38:45:17 |
— |
| 8 | Pd/C | SS | 1 × 2 mm | 0:89:11 |
— |
| 9 | Pd/C | SS | None |
0:100:0
|
99% |
| 10a | Pd/C | SS | None | 0:100:0 |
62% |
Prompted by these encouraging results, the optimized conditions were applied (entry 1 in Table 2) to a set of alkynes (6a–c) and alkenes (7a–f) to prepare the corresponding alkanes 8a–c and 8e–f (60–97% yield), as reported in Scheme 3.
 | ||
| Scheme 3 The reaction scope for the catalytic hydrogenation of alkanes, alkenes, and nitro compounds. Typical procedure: alkyne, alkene, or nitro compound (1 mmol), compound 1 (2 mmol), Pd/C (3 mol%), and H2O (2 mmol) were placed inside a 10 mL SS jar with one ball of the same material (∅=8 mm, 2.09 g). The reaction was conducted for 90 min at room temperature, 30 Hz, and under ball-milling conditions. See ESI† for the specific reaction conditions. Yields refers to isolated compounds. aNo grinding media conditions. | ||
In contrast, synthesizing alkenes 7a–d starting from the corresponding alkyne was more challenging. Using a stoichiometric amount of diboron 1 with alkyne 6a led to a mixture of starting material (38%), alkene 7a (45%), and alkane 8a (17%). However, using a smaller ball (2 mm ∅, 0.3 g mass) surprisingly improved the yield of alkene 7a to 89%. The crude sample did not show any starting material, and the alkane product was obtained with a yield of only 11%. This suggests that there may be a connection between the grinding conditions and the selectivity of the product. This claim is backed up by the experiments conducted by shaking the jar without any grinding media. In this case, there was a complete conversion of alkene (99%, as seen in entry 9 of Table 2).
Although several mechanochemical protocols for catalytic hydrogenation of alkynes have been reported,25 herein, we report a robust protocol to selectively interrupt the reduction at the semi-hydrogenation stage.
We used no grinding media conditions and the same alkynes as before for our ball-milling experiments – specifically 6a–d. The results were impressive yields of the corresponding alkenes, specifically 7a–d, with percentages going from 92% to 99%.
Afterward, we broadened the range of reactions to include nitro derivatives 9a–i. We discovered they reacted efficiently under slightly modified conditions, using 3 mmol of diboron compound 1 as stoichiometry demands. This led to successfully preparing anilines 10a-I with outstanding yields (Scheme 3, 89–99% isolated yields).
It is essential to mention that some of the compounds screened resulted in either complicated mixtures (compounds 9j and 9k) or did not react (substrates 11, 12, and 13) under the conditions specified in Scheme 3.
Control experiments and kinetic profiles
We focused on reducing a simple double bond to understand the results better and study the reactivity. For our initial investigation, we used alkene 7a (liquid, Scheme 4) as the starting material. The purpose was to observe the hydrogenation of the substrate depending solely on hydrogen diffusion. We sealed the jar and left it at rest on the bench for 90 minutes – the required reaction time for complete conversion under optimized conditions. The results indicate that the reaction partially proceeded up to 60% conversion, reaching a steady state after only 30 min (Scheme 4, top, black plot). In our second set of control experiments, we achieved similar outcomes by shaking the mixture in the sealed jar at 30 Hz without milling balls (Scheme 4, top, red plot). On these premises, it can be concluded that hydrogen gas interacts fairly well when meeting a starting material in a liquid physical state, even without any external forces. The results remained consistent even when milling balls were used, as seen in the third control experiment, where the reaction was completed. However, in this last case, the steady-state value significantly increased, slightly modifying the shape of the curve (Scheme 4, top, blue plot). The addition of a milling ball somewhat improves the efficiency of the blending and mixing processes.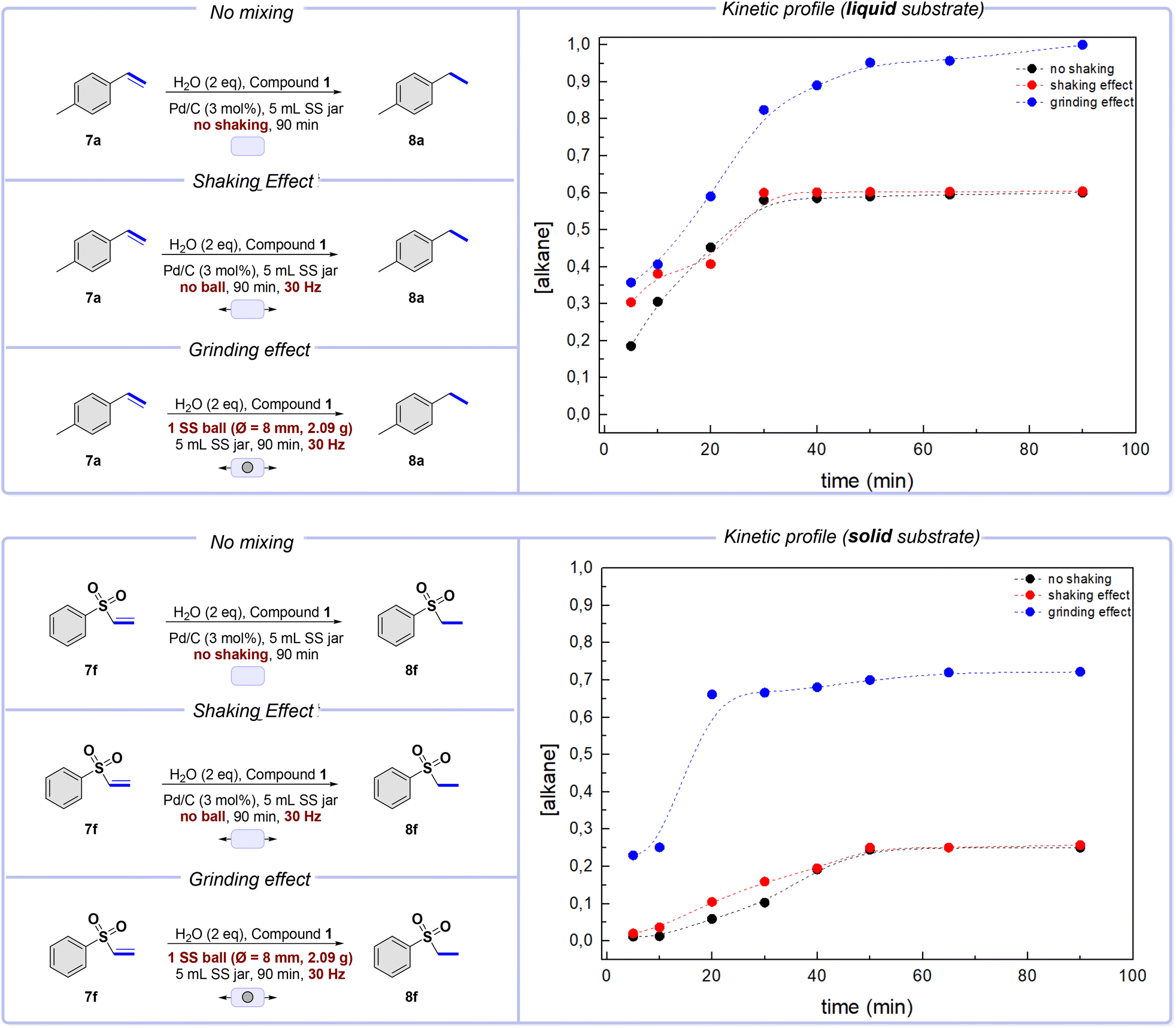 | ||
| Scheme 4 The control experiments and kinetics for liquid (top) and solid (bottom) starting materials. Typical procedure: solid or liquid alkene (0.3 mmol), compound 1 (0.6 mmol), Pd/C (3 mol%), and H2O (2 mmol) were placed inside a 5 mL SS jar. Whenever a ball was used, 1 SS ball (∅ = 8 mm, 2.09 g) was placed inside the reaction vessel. The conversion was calculated via GC-MS analysis. | ||
Intriguingly, the experimental results significantly change when using a solid alkene, as in the case of compound 7f (Scheme 4). Firstly, we verified the responsiveness of the substrate to the reduction under the standard conditions in solution. Under the solvent-based conditions described by Skrydstrup,23 the compound is reduced in a 94% yield.26
Under static conditions, where the reagents are left at rest in a jar, the conversion degree gradually increases, but the final value reached after 50 minutes is relatively low – around 20%. The same trend is observed when the reactant mixture is shaken without a milling ball (Scheme 4, bottom, red plot). However, when a milling ball is used, the conversion degree increases rapidly, and a steady-state value of about 70% yield can be achieved in just 20 minutes (Scheme 4, bottom, blue plot).
According to these results, hydrogenation of substrates occurs spontaneously when hydrogen gas is present, regardless of their physical state, and always at a consistent rate. This suggests that hydrogen can penetrate exposed surfaces and react effectively with molecules at the surface and in its close surroundings.
The physical state during hydrogenation dramatically impacts the overall kinetics of the process. In a liquid phase, molecular species have higher mobility, leading to increased hydrogen gas absorption and diffusion. This allows for creating more contact between the gas and the alkene molecules. The liquid alkene is constantly subjected to loads by the mixing ball, allowing it to flow freely inside the jar and exposing different portions of the bulk. This creates favorable conditions for hydrogen gas to interact with all the reagents upon completion.27
Conversely, in a solid phase, the absorption and diffusion of hydrogen gas through the material is limited, making it difficult for hydrogen to interact with the whole bulk. As a result, only the surface region is efficiently affected, leaving much of the bulk unreacted.28 The mixing process can be made more effective by using a milling ball, which impacts and slides on the surfaces of the jar, leading to better blending of the reactant mixture.29
The milling ball increases the reaction rate by exposing new surfaces from the bulk, as seen in Scheme 4. Additionally, reducing the particle sizes of the solid during the grinding process enhances its surface area, creating new active sites that can interact with activated hydrogen.30 However, the reaction only reaches completion with a liquid alkene. Therefore, a different explanation is needed to understand this behavior.
We hypothesized that diboron compound 1 or activated hydrogen on palladium ([Pd/C + Ha]) can create a cohesive interface with the substrate, which can hinder the complete conversion of the solid substrate. To validate this, we performed SEM analysis on the crude mixture of the three solid components (for SEM images, please see Fig. S3 and S4 in the ESI†). When the powders are milled for 45 minutes, a small size reduction occurs, in line with the reaction kinetic profile of the phenyl vinyl sulfone. Conversely, when the reaction mixture is milled for 90 minutes, the SEM images reveal a sort of cohesive structure. We suggest that the repeated loading and mechanical deformation caused by the grinding ball on the solid mixture can eventually limit the exposure of new interfaces, reducing reactivity within the bulk.30,31 Although the grinding effect results in improved reduction efficiency of solid compounds at first, this phenomenon eventually becomes less effective as the substance becomes trapped within the bulk.29
The literature echoes this theme, notably for the investigation of hydrogenation of CO with a Co50Fe50 catalyst dispersed on TiO2.4d Despite methodological differences, some analogies can be found. The mechanochemical effect is evident through the fine grinding of solid powders, leading to increased surface area and enhanced active sites. In the catalytic hydrogenation of CO, the augmented surface area of Co50Fe50 governs the reaction rate by providing newly formed active sites for proper interaction between the gaseous reagents. This effect is specifically evident in this study when the organic reagent is in its solid physical state, emphasizing the role of its interface area in creating new interaction sites with the gaseous reagent and the catalyst.
“Green Chemistry” assessment
Mechanochemistry has advanced significantly in its application to organic synthesis. The current protocol aims to address many of the common questions surrounding ball-milling processes. While the answers may be partial, they provide a clearer understanding of the role of mechanochemistry in organic synthesis. Another non-negligible aspect of this and many other mechanochemical procedures is their green footprint compared to their traditional solvent-based counterparts.32Recent climate change has made it clear that we can no longer afford to continue with our current practices, and we must explore new ways to prepare organic compounds that are both efficient and sustainable. Mechanochemical processes, including the one disclosed here, certainly go in this direction by eliminating or significantly cutting down on solvent use, especially VOCs and toxic ones.33
Removing solvent from chemical reactions is a complex process that requires careful optimization, often taking up a lot of time and occasionally leading to failure. It is essential to realize that mechanochemistry is more than just a mathematical problem where the solvent can be easily removed from the reaction description in the literature. This is a misconception that needs to be addressed.
The protocol reported here significantly reduces reaction times compared to the solvent-based version (from 18 hours to 90 minutes), leading to a substantial decrease in energy consumption and CO2 emissions. The environmental sustainability of this mechanochemical process has been well-documented and supported by green metrics, as shown in Fig. 1 (E-factor of 1.44 vs. 69.6 in solution). For further details, please refer to the ESI.†
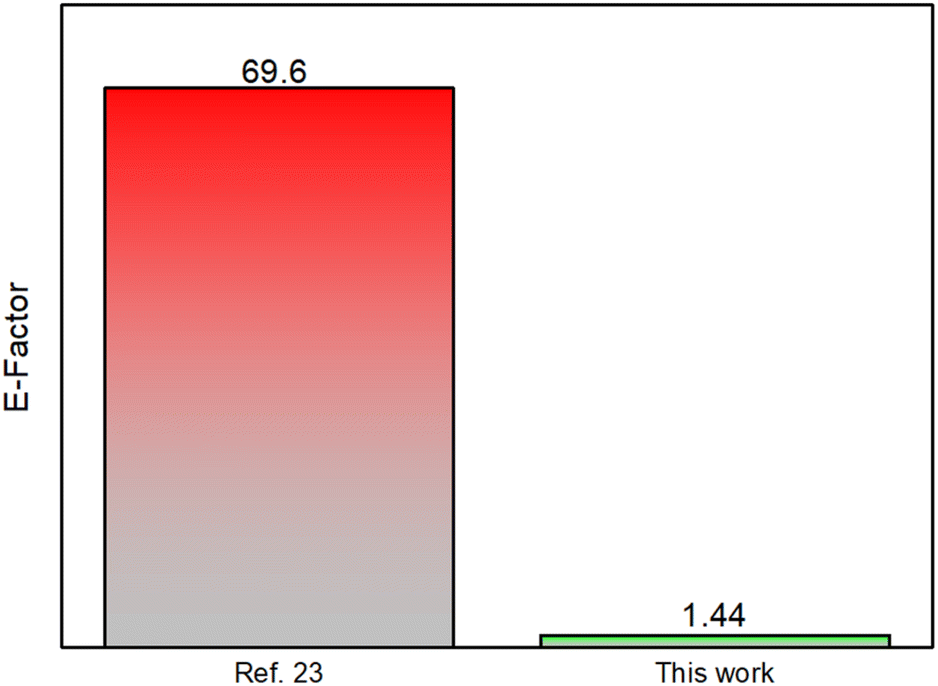 | ||
| Fig. 1 Bar plot comparing the environmental factor of the solution-based protocol (left) relative to this solvent-free methodology (right). | ||
Conclusions
To summarize, we have developed an effective and environmentally friendly procedure for reducing various structurally different chemical compounds by adhering to green chemistry principles and utilizing a mechanochemical process. We successfully prepared various alcohols from aldehydes and ketones with high yields through extensive testing. This confirmed that the reaction mechanism proceeds through water splitting. As part of our protocol, we reduced nitro derivatives (a new feature compared to the counterpart in solution) alongside alkynes and alkenes. When applied to alkynes, this process is effective and selective. We later added control groups to showcase the benefits of mechanochemistry.In our model system, we have concluded that ball-milling conditions improve the efficiency of mixing a liquid substrate and a gaseous species, leading to a complete reaction. When a solid alkene is subjected to the same conditions, mechanical impacts are necessary to increase active sites and interactions with the substrate and the active hydrogen on the Pd catalyst. The solid physical state of the alkene limits the total exposure of the bulk, negatively affecting the yield. We have also included green metrics and catalyst reuse experiments (see the ESI†) in our synthetic protocol to assess the greenness of our ball-milling process.
Author contributions
FC designed the experiments. FC and FB performed the experiments. FC and CF carried out the GC-MS experiments and processed the data. FC and AP wrote the paper. AP conceptualized the idea. All the authors contributed to the paper draft correction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by MIUR Italy, PRIN 2017 project (grant number: 2017B7MMJ5_001) “MultIFunctional polymer cOmposites based on groWn matERials” (MIFLOWER) and PNRR PE2 – NEST – Network 4 Energy Sustainable Transition – (MUR no. PE0000021, CUP no. F53C22000770007). We acknowledge the CeSAR (Centro Servizi Ricerca d'Ateneo) core facility of the University of Cagliari, Dr Sandrina Lampis for assistance with the generation of NMR data, and Gianluigi Corrias for the technical support. We also acknowledge Prof. Francesco Delogu for his expertise in mechanochemistry, which he shared with us during the drafting of this paper.References
- Selected reviews and articles discussing the growth and spread of mechanochemistry in the last 60 years: (a) L. Takacs, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC; (b) V. V. Boldyrev, Herald Russ. Acad. Sci., 2018, 88, 142–150 CrossRef; (c) L. Takacs, J. Mater. Sci., 2018, 53, 13324–13330 CrossRef CAS.
- Selected books, reviews and articles discussing early mechanochemical studies: (a) W. A. Yarbrough and R. Roy, Nature, 1986, 322, 347–349 CrossRef CAS; (b) A. J. Lynch and C. A. Rowland, The History of Grinding, Society of Mining, Metallurgy and Exploration, Inc., Littleton, CO, 2005 Search PubMed; (c) P. A. Rehbinder, Physico-Chemical Mechanics, A New Area of Science, Izd. Znanije, Moscow, 1958 Search PubMed; (d) V. V. Boldyrev, Russ. Chem. Rev., 2006, 75, 177–189 CrossRef CAS; (e) V. V. Boldyrev and E. G. Avvakumov, Russ. Chem. Rev., 1971, 40, 847 CrossRef.
- Selected reviews and articles discussing mechanical alloying: (a) J. S. Benjamin, Metall. Trans., 1970, 1, 2943–2951 CrossRef CAS; (b) J. S. Benjamin and T. E. Volin, Metall. Trans., 1974, 5, 1929–1934 CrossRef CAS; (c) P. S. Gilman and J. S. Benjamin, Annu. Rev. Mater. Sci., 1983, 13, 279–300 CrossRef CAS; (d) Y. Kojima, M. Senna, T. Shinohara, S. Ono, K. Sumiyama and K. Suzuki, J. Alloys Compd., 1995, 227, 97–101 CrossRef CAS.
- Selected books, reviews, and articles discussing mechanochemical treatments in materials science: (a) J. S. Benjamin, in New Materials by Mechanical Alloying Techniques, ed. E. Artz and L. Schultz, DGM Informationsgesellschaft, Oberursel, 1989, p. 3 Search PubMed; (b) P. Baláž, J. Briančin, Z. Bastl, L. Medvecký and V. Šepelák, J. Mater. Sci., 1994, 29, 4847–4851 CrossRef; (c) A. L. Sanna, M. Carta, G. Pia, S. Garroni, A. Porcheddu and F. Delogu, Sci. Rep., 2022, 12, 9445 CrossRef CAS PubMed; (d) M. Carta, A. L. Sanna, A. Porcheddu, S. Garroni and F. Delogu, Sci. Rep., 2023, 13, 2470 CrossRef CAS PubMed; (e) A. L. Sanna, G. Pia and F. Delogu, Adv. Mater. Sci. Eng., 2020, 2020, 8844569 Search PubMed.
- C. Espro and D. Rodríguez-Padrón, Curr. Opin. Green Sustainable Chem., 2021, 30, 100478 CrossRef CAS.
- R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed.
- F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e20220036 CrossRef PubMed.
- Selected reviews and articles discussing the benefits of mechanochemistry for green academic and industrial applications: (a) L. N. Ortiz-Trankina, J. Crain, C. Williams and J. Mack, Green Chem., 2020, 22, 3638–3642 RSC; (b) A. Porcheddu, F. Delogu, L. De Luca, C. Fattuoni and E. Colacino, Beilstein J. Org. Chem., 2019, 15, 1786–1794 CrossRef CAS PubMed; (c) J. Mack, D. Fulmer, S. Stofel and N. Santos, Green Chem., 2007, 9, 1041–1043 RSC; (d) E. Colacino, A. Porcheddu, I. Halasz, C. Charnay, R. Guerra, F. Delogu and J. Fullenwarth, Green Chem., 2018, 20, 2973–2977 RSC; (e) D. E. Crawford, A. Porcheddu, A. S. McCalmont, F. Delogu, S. L. James and E. Colacino, ACS Sustainable Chem. Eng., 2020, 8, 12230–12238 CrossRef CAS; (f) V. V. Boldyrev, J. Mater. Sci., 2004, 39, 5117–5120 CrossRef CAS; (g) F. Basoccu, F. Cuccu and A. Porcheddu, ChemSusChem, 2023, e202301034 CrossRef PubMed.
- Selected reviews and articles discussing organic mechanosynthesis: (a) E. Colacino, M. Carta, G. Pia, A. Porcheddu, P. C. Ricci and F. Delogu, ACS Omega, 2018, 3, 9196–9209 CrossRef CAS PubMed; (b) D. Kong, P. Wu and C. Bolm, ACS Sustainable Chem. Eng., 2022, 10, 2863–2867 CrossRef CAS; (c) T. K. Achar, A. Bose and P. Mal, Beilstein J. Org. Chem., 2017, 13, 1907–1931 CrossRef CAS PubMed; (d) G. W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC; (e) R. R. A. Bolt, J. A. Leitch, A. C. Jones, W. I. Nicholson and D. L. Browne, Chem. Soc. Rev., 2022, 51, 4243–4260 RSC; (f) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed; (g) F. Cuccu, F. Basoccu, C. Fattuoni and A. Porcheddu, Molecules, 2022, 27, 5450 CrossRef CAS PubMed; (h) F. Cuccu, F. Basoccu, C. Fattuoni and A. Porcheddu, Molecules, 2022, 27, 5450 CrossRef CAS PubMed; (i) A. Porcheddu, R. Mocci, M. Brindisi, F. Cuccu, C. Fattuoni, F. Delogu, E. Colacino and M. V. D. Auria, Green Chem., 2022, 24, 4859–4869 RSC; (j) F. Basoccu, F. Cuccu, P. Caboni, L. De Luca and A. Porcheddu, Molecules, 2023, 28, 2239 CrossRef CAS PubMed.
- A. A. L. Michalchuk, E. V. Boldyreva, A. M. Belenguer, F. Emmerling and V. V. Boldyrev, Front. Chem., 2021, 9 DOI:10.3389/fchem.2021.685789.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- J. R. Cabrero-Antonino, R. Adam, V. Papa and M. Beller, Nat. Commun., 2020, 11, 3893 CrossRef CAS PubMed.
- O. A. Kirichenko, E. V. Shuvalova and E. A. Redina, Russ. Chem. Bull., 2019, 68, 2048–2052 CrossRef CAS.
- Y. Kojima, Y. Kawai, M. Kimbara, H. Nakanishi and S. Matsumoto, Int. J. Hydrogen Energy, 2004, 29, 1213–1217 CrossRef CAS.
- T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed.
- L. Wang, G. Kehr, C. G. Daniliuc, M. Brinkkötter, T. Wiegand, A. L. Wübker, H. Eckert, L. Liu, J. G. Brandenburg, S. Grimme and G. Erker, Chem. Sci., 2018, 9, 4859–4865 RSC.
- O. A. Kirichenko, E. V. Shuvalova and E. A. Redina, Russ. Chem. Bull., 2019, 68, 2048–2052 CrossRef CAS.
- H. C. Brown, B. Abraham, A. C. Bond, N. Davidson, A. E. Finholt, J. R. Gilbreath, H. Hoekstra, L. Horvitz, E. K. Hyde, J. J. Katz, J. Knight, R. A. Lad, D. L. Mayfield, L. Rapp, D. M. Ritter, A. M. Schwartz, I. Sheft, L. D. Tuck and A. O. Walker, New Developments in the Chemistry of Diborane and the Borohydrides. I. General Summary1, 1952 Search PubMed.
- C. Li, P. Peng, D. W. Zhou and L. Wan, Int. J. Hydrogen Energy, 2011, 36, 14512–14526 CrossRef CAS.
- S. W. Chaikin and W. G. Brown, J. Am. Chem. Soc., 1949, 71, 122–125 CrossRef CAS.
- Selected reviews and articles discussing the importance of the alcohol moiety and catalytic hydrogenation in medicinal chemistry: (a) O. Robles and D. Romo, Nat. Prod. Rep., 2014, 31, 318–334 RSC; (b) T. Henkel, R. M. Brunne, H. Müller and F. Reichel, Angew. Chem., Int. Ed., 1999, 38, 643–647 CrossRef CAS; (c) A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed; (d) C. R. S. Maior, P. C. S. Costa, C. B. P. Ligiéro, L. S. de Moraes, G. R. Sousa Jr., S. G. Lima, P. M. Esteves and P. C. M. L. Miranda, Chem. – Eur. J., 2023, 29, e202203731 CrossRef CAS PubMed.
- M. Flinker, H. Yin, R. W. Juhl, E. Z. Eikeland, J. Overgaard, D. U. Nielsen and T. Skrydstrup, Angew. Chem., Int. Ed., 2017, 56, 15910–15915 CrossRef CAS PubMed.
- J. Mack, D. Fulmer, S. Stofel and N. Santos, Green Chem., 2007, 9, 1041–1043 RSC.
- (a) T. Portada, D. Margetić and V. Štrukil, Molecules, 2018, 23, 3163 CrossRef PubMed; (b) Y. Sawama, M. Niikawa and H. Sajiki, J. Synth. Org. Chem. Japan, 2019, 77, 1070–1077 CrossRef CAS.
- This experiment was explicitly designed to make the comparison depending only on the physical state of the substrates. Indeed, the reaction outcomes in solution prove that the chemical reactivity of alkene 7f perfectly aligns with that of the liquid alkene 7a. The different response under mechanochemical conditions is likely due to the solid physical state, as mentioned above.
- M. Carta, E. Colacino, F. Delogu and A. Porcheddu, Phys. Chem. Chem. Phys., 2020, 22, 14489–14502 RSC.
- R. Rana, R. Bavisotto, N. Hopper and W. T. Tysoe, Tribol. Lett., 2021, 69, 32 CrossRef CAS.
- M. Carta, F. Delogu and A. Porcheddu, Phys. Chem. Chem. Phys., 2021, 23, 14178–14194 RSC , and references thereof.
- F. Cuccu, D. L. Browne and A. Porcheddu, ChemCatChem, 2023, 15, e202300762 CrossRef CAS.
- (a) M. Carta, S. L. James and F. Delogu, Molecules, 2019, 24, 3600 CrossRef CAS PubMed; (b) E. Avvakumov, M. Senna and N. Kosova, in Soft Mechanochemical Synthesis, Springer New York, Kluwer Academic Publishers, 1st edn, 2002 Search PubMed; (c) I. Tole, K. Habermehl-Cwirzen and A. Cwirzen, Mineral. Petrol., 2019, 113, 449–462 CrossRef CAS.
- (a) K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed, and references thereof; (b) J. Pu, C. Liu, S. Shic and J. Yun, RSC Adv., 2022, 12, 34268–34281 RSC; (c) R. Eivazzadeh-Keihan, F. Radinekiyan, H. Madanchi, H. A. M. Aliabadi and A. Maleki, Carbohydr. Polym., 2020, 248, 116802 CrossRef CAS PubMed; (d) A. Maleki, H. Movahed, P. Ravaghi and T. Kari, RSC Adv., 2016, 6, 98777–98787 RSC.
- V. Fetisov, A. M. Gonopolsky, H. Davardoost, A. R. Ghanbari and A. H. Mohammadi, Energy Sci. Eng., 2023, 11, 1516–1535 CrossRef CAS , and references thereof.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03783e |
| This journal is © The Royal Society of Chemistry 2024 |