
Mechanochemical-assisted decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sodium sulfinate salts†
Barakha
Saxena
,
Roshan I.
Patel
,
Shruti
Sharma
and
Anuj
Sharma
 *
*
Green Organic Synthesis laboratory, Indian Institute of Technology Roorkee, Roorkee, 247667, India. E-mail: anujsharma.mcl@gmail.com; anuj.sharma@cy.iitr.ac.in
First published on 17th January 2024
Abstract
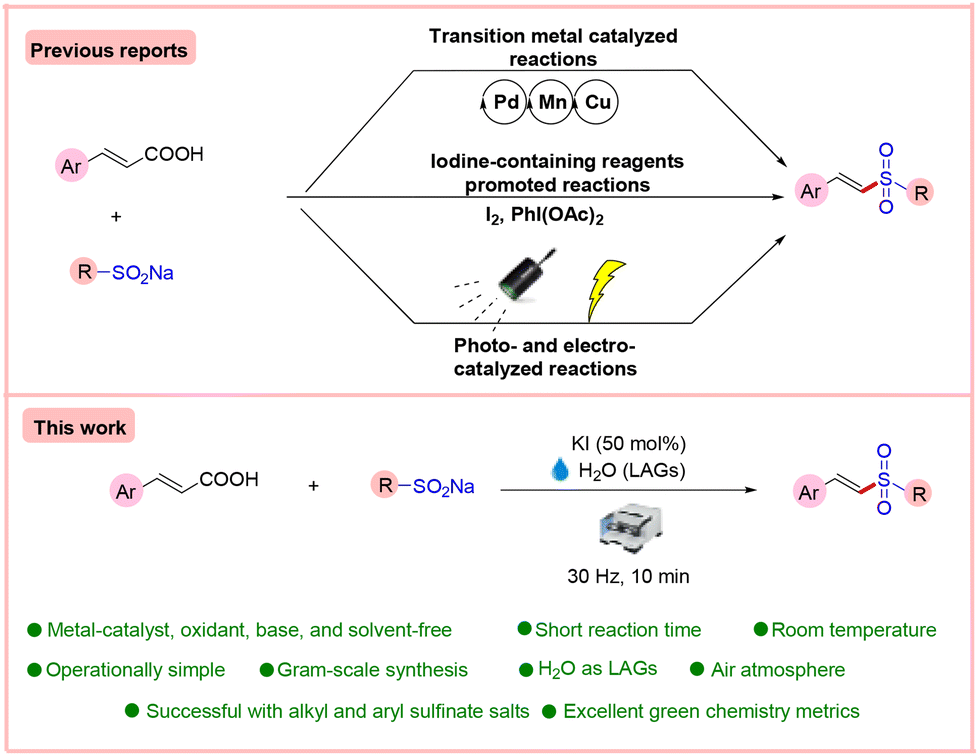
Developing a green and efficient method for synthesizing vinyl sulfones is challenging and highly desirable. We herein report a green, sustainable, and unprecedented mechanochemical-assisted approach for the decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sodium sulfinates using only potassium iodide (50 mol%) as an activator under water-assisted grinding conditions. A library of alkyl and aryl vinyl sulfone derivatives were synthesized successfully in up to 92% yield with excellent functional group compatibility in a short reaction time. This sulfonylation strategy is well tolerated by aryl α,β-unsaturated carboxylic acids and alkyl and aryl sodium sulfinate salts. The method has been successfully applied to the synthesis of neuroprotective agents. Overall, the advantages of this strategy are (i) metal catalyst-, base-, oxidant-, and solvent-free conditions, (ii) operational simplicity with a short reaction time, and (iii) excellent effective mass yield, atom economy, E-factor, and EcoScale score. The practicality of this method is also demonstrated in the gram-scale synthesis of vinyl sulfones.
Introduction
Owing to their easy accessibility, high stability, and low toxicity, decarboxylative cross-coupling reactions with biomass feedstocks of α,β-unsaturated carboxylic acids have emerged as an efficient method for the construction of C(sp2)–C and C(sp2)–X (X = N, P, S, Se) bonds. This can be achieved by utilizing the carbon bond adjacent to the –COOH group in α,β-unsaturated carboxylic acids to obtain synthetically important molecules with the release of non-toxic and easily removable CO2 as the by-product.1 Consequently, in the past few years, the construction of C–S bonds via a decarboxylative coupling strategy despite being an underdeveloped process continues to attract considerable attention from the synthetic community.2Vinyl sulfones (α,β-unsaturated sulfones) are a valuable framework in organic synthesis, displaying excellent biological and pharmaceutical properties, as shown in Fig. 1.3 Thus, the synthesis of vinyl sulfones has garnered a great deal of interest, and different strategies for their preparation have emerged. Classic vinyl sulfone preparations are based on the Knoevenagel condensation of aromatic aldehydes with sulfonyl acetic acids and the Horner–Emmons reaction involving α-sulfonyl phosphonium ylides.4 A promising strategy, however, is the direct sulfonylation of olefins, alkynes, vinyl halides, vinyl tosylate, vinyl triflates and alkenyl boronic acids with sulfonyl sources. Several sulfonyl sources, such as DABSO, thiosulfonates, sulfinic acids/salts, and sulfonyl hydrazides, have been employed in these reactions.5
 | ||
| Fig. 1 Examples of biologically active vinyl sulfones. | ||
In this line, decarboxylative sulfonylation of cinnamic acids with sodium aryl sulfinates is a very promising method for the preparation of vinyl sulfones. This is due to easily available starting materials and CO2 as the only by-product, thereby making this approach sustainable and environmentally benign. However, in the beginning, the decarboxylative sulfonylation of α,β-unsaturated carboxylic acids has been achieved by the use of expensive and hazardous transition metal (TM) catalysts such as Pd, Cu, or Mn with expensive and toxic ligands (Scheme 1).6 Additionally, oxidants such as Ag2CO3, KI, and TBHP have been employed in stoichiometric or over-stoichiometric proportions. In the majority of cases, temperatures are exceedingly high above 100 °C, and environmentally unfriendly solvents such as DMSO and DMF have been employed. Moreover, these reactions have limited substrate scope, as these reactions are unsuccessful in the synthesis of alkyl vinyl sulfones. The products in these methods require extensive purification. From the perspective of green metrics, these methods fare poorly in terms of atom efficiency (AEF), reaction mass efficiency (RME), optimum efficiency (OE), etc.
 | ||
| Scheme 1 Background and summary of research work. | ||
Some metal-free approaches have also come to the fore, such as using iodine-containing reagents like I2 and PhI(OAc)2.7 For example, PhI(OAc)2 has been utilized as an oxidizing/coupling reagent in over-stoichiometric proportions in one method, and the reaction requires 100 °C for completion.7a Despite the non-triviality of PhI(OAc)2, this strategy would necessitate stoichiometric generation of aryl iodides as by-products, limiting atom economy and other green chemistry parameters. Importantly, the synthesis of alkyl vinyl sulfones has not been successful with this method. Likewise, in another example, a combination of stoichiometric amounts of an alkali base and iodine is needed at 60 °C for the product formation which needs 10 hours for completion of the reaction.7b Furthermore, the authors have synthesized only one alkyl vinyl sulfone with merely 45% yield. In yet another method, Shi and co-workers developed an iodine-promoted decarboxylative C–S cross-coupling of cinnamic acids with sodium benzene sulfonates. This method involves an over-stoichiometric amount of TBHP (an F-level organic peroxide) and iodine heated at 90 °C in toluene.7c Like in the previous methods, this method fails with alkyl sulfinate salts.
On similar lines, Wang and co-workers, in 2016, disclosed an electrochemical decarboxylative sulfonylation of cinnamic acid with sodium sulfinates for the synthesis of vinyl sulfones.7e However, this method suffered from issues such as the limited scope and inefficiency of the protocol with alkyl sulfinate salts, the requirement of acetic acid as an additive, and high input electrolysis for its successful execution, resulting in very poor effective mass yield (EMY) and mass productivity (MP). Lately, Wang and co-workers, in 2019, established a photocatalyzed synthesis of vinyl sulfones from cinnamic acid and sodium sulfinates with high regioselectivity.7f This strategy also suffers in terms of the requirement of merrifield resin-supported Rose Bengal as a photocatalyst, over-stoichiometric amounts (2.0 equiv.) of TBHP oxidant, which is environmentally unfriendly, an inert atmosphere (Ar), a long reaction time (12 hours), and poor effective mass yield (EMY). This method, like previous ones, does not work for the synthesis of alkyl-substituted vinyl sulfone derivatives. One major bottleneck of these papers is the failure towards the synthesis of alkyl vinyl sulfones. Overall, a general, environmentally sustainable and mild approach for decarboxylative sulfonylation of cinnamic acids is highly desirable.
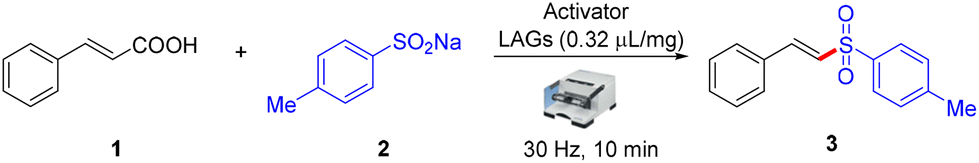
Among the methods of synthetic chemistry, mechanochemistry has become an attractive alternative method for synthesizing organic compounds.8 Mechanochemistry enables chemical synthesis sustained by mechanical forces. It offers a new opportunity for synthesizing organic compounds with solvent-free organic mechanochemical reactions and unlocks new routes towards numerous compounds that are not attainable by other conventional methods.9 In continuation of our interest in sustainable, green, and novel synthetic methodologies,10 we hereby report an unprecedented mechanochemical-assisted decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sodium sulfinates using only potassium iodide as an activator with water (η = 0.32 μL mg−1) as the liquid-assisted grinding additive (LAG)11 at ambient temperature under ball milling in 10 min. The method works equally well on alkyl and aryl sulfinates at room temperature without needing any metal catalyst, oxidant, base, or inert environment. To commence our studies, we chose cinnamic acid 1 and sodium 4-methylbenzenesulfinate 2 as the model substrates to investigate this mechanochemical-mediated decarboxylative sulfonylation reaction, and different conditions were screened, as shown in Table 1. However, the primary attempts did not lead to the desired product on milling of pure reagents (entry 1). Moreover, no desired product was observed when we added NH4I as an iodine-based activator. Interestingly, after the addition of a liquid (η = 0.32 μL mg−1), a so called liquid-assisted grinding additive or LAG, the desired product 3 was formed in 24% yield (Table 1, entry 3). Furthermore, given the significance of LAGs for the chemical transformation, a series of experiments was performed to optimize the best LAGs for the present reaction and the results revealed that H2O was the best among DMA, DMF, DCM, and EtOH (Table 1, entries 4–8), in which the desired product was obtained in 39% yield using NH4I in a sub-stoichiometric amount (50 mol%) with H2O (entry 6). Next, the screening of other iodine-containing reagents, such as TBAI, CuI, KI, I2, and PhI(OAc)2 (entries 9–13), revealed that KI displayed the efficiency to yield the desired product 3 in 92% yield (entry 11). Next, we focused on other halide-based activators, such as NaCl and TBAB. Unfortunately, no product was obtained (entries 14 and 15). Next, changing the loading of KI to 1 equiv., 30 mol%, and 40 mol% had a negative impact on the yield of the desired product (entries 16–18).
|
|
|||
|---|---|---|---|
| Entry | Activator (50 mol%) | LAGs (0.32 μL mg−1) | Yield (%) |
| a Reaction conditions: 1 (0.5 mmol), 2 (0.75 mmol), activator (50 mol%), and LAGs (0.32 μL mg−1). The reaction was milled for 10 min at 30 Hz frequency using a 10 mL Retsch stainless steel jar, and 5 mm, 2.5 g stainless steel grinding balls (5 × 5 mm grinding balls) at room temperature. b KI (1 equiv.). c KI (30 mol%). d KI (40 mol%). | |||
| 1 | — | — | N.R. |
| 2 | NH4I | — | N.R. |
| 3 | NH4I | DMSO | 24 |
| 4 | NH4I | DMA | Trace |
| 5 | NH4I | DMF | Trace |
| 6 | NH4I | H2O | 39 |
| 7 | NH4I | DCM | Trace |
| 8 | NH4I | EtOH | N.R. |
| 9 | TBAI | H2O | 62 |
| 10 | CuI | H2O | 40 |
| 11 | KI | H2O | 92 |
| 12 | I2 | H2O | 76 |
| 13 | PhI(OAc)2 | H2O | 59 |
| 14 | NaCl | H2O | N.R. |
| 15 | TBAB | H2O | N.R. |
| 16 | KI | H2O | 88b |
| 17 | KI | H2O | 65c |
| 18 | KI | H2O | 73d |
Screening of the reaction temperature revealed that 92% yield of the desired product 3 was obtained under standard conditions at room temperature (internal jar temperature, 25.4 °C) (Fig. 2a), while applying an external heat source using a heat gun on the mixing jar for 10 min did not seem to impact the reaction yield (90% yield of 3) (internal jar temperature, 103.0 °C) (Fig. 2b). Furthermore, a hand grinding reaction between 1 and 2 for 10 min with H2O gave access to the desired product 3 in lower yield (42% yield) (Fig. 2c). Finally, a solution-based heating reaction of 1 and 2 in H2O at 80 °C for 24 h resulted in the required product 3 in 29% yield (Fig. 2d). All these parameters established that a combination of inexpensive and readily available KI with H2O as LAGs at 30 Hz frequency for 10 min at ambient temperature was optimal and displayed the highest efficiency in catalyzing the reaction. For other parameters such as the concentration of sodium sulfinate salt, jar size, grinding balls, time and frequency, see the ESI† for more details.
 | ||
| Fig. 2 Various reaction setups: (a) reaction at room temperature; (b) heat gun-based reaction; (c) hand grinding-based reaction; (d) solution-based reaction. | ||
Substrate scope
With the optimized reaction conditions in hand, we explored the substrate scope of this novel mechanochemical-assisted decarboxylative sulfonylation reaction. As shown in Scheme 2, a library of aryl α,β-unsaturated carboxylic acids 1, bearing electron-donating and electron-withdrawing functionalities, successfully underwent the mechanochemical-mediated decarboxylative sulfonylation reaction with sodium benzenesulfinate 2, thus providing an ample opportunity for further derivatization of the products 4–53 in moderate to excellent yields. However, the experimental results suggested that both electronic and steric features of the substituted aryl α,β-unsaturated carboxylic acids 1 affected the efficacy of the decarboxylative sulfonylation reaction. | ||
Scheme 2 Substrate scope for the decarboxylative sulfonylation reaction. Reaction conditions: aryl α,β-unsaturated carboxylic acid 1 (0.5 mmol), sodium sulfinate 2 (0.75 mmol), KI (50 mol%), and H2O (η = 0.32 μL mg−1). The reaction was milled using a 10 mL Retsch stainless steel jar for 10 min at 30 Hz frequency with 5 mm, 2.5 g stainless steel grinding balls (5 × 5 mm grinding balls) at room temperature. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 min, 30 Hz. b40 min, 30 Hz. 30 min, 30 Hz. b40 min, 30 Hz. | ||
Aryl α,β-unsaturated carboxylic acids 1 bearing weak electron-donating groups (4–9) showed higher reactivity than those with strong electron-donating ones (10–12) albeit less efficiently. Additionally, a para-substituted α,β-unsaturated carboxylic acid 4 furnished the desired product in higher yield than meta- and ortho-substituted α,β-unsaturated carboxylic acids (5, 6). Next, aryl α,β-unsaturated carboxylic acids 1, bearing remote electron-withdrawing groups, were subsequently evaluated. Various functional groups such as chloro, fluoro, bromo, nitro, and cyano functionalities at the ortho-, meta-, and para-positions smoothly reacted under this protocol to form the targeted products (13–25) in moderate to good yields. Moreover, the applicability of this method was demonstrated in the synthesis of some potent neuroprotective agents 32, 33 and 34 in 63–78% yields, with (E)-1-chloro-2-(2-(phenylsulfonyl)vinyl)benzene 33 being the most potent one.3f The reaction also worked well with biphenyl and 1,3-benzodioxole derived α,β-unsaturated carboxylic acids to afford the required products 26 and 27 in 74% and 88% yield, respectively. Interestingly, aryl α,β-unsaturated carboxylic acids having extended conjugation, also reacted well and afforded the desired product 28 in 77% yield. This strategy could also be expanded to other heteroaryl such as thiophene, furan, and pyridine derived α,β-unsaturated carboxylic acids, affording the corresponding products (29–31) in 69%, 30%, and 79% yield respectively. Unfortunately, aliphatic α,β-unsaturated carboxylic acids could not participate in this decarboxylative sulfonylation reaction. We presume that the low stability of alkyl radical intermediates may be the reason for failure compared to that of the benzyl radical intermediate in other cases. Next, the potential of this methodology with aryl sodium sulfinate salts 2 was investigated. Aryl sodium sulfinate derivatives 2 bearing electron-donating and electron-withdrawing functionalities such as methyl, methoxy, t-butyl, nitro, chloro, fluoro, trifluoromethyl, and trifluoromethoxy groups were well tolerated affording the corresponding products (35–45) in moderate to excellent yields. Besides, naphthyl-substituted sulfinate salt also reacted well with cinnamic acid 1 to afford the desired product 46 in 42% yield.
The compatibility of this protocol was further demonstrated by the reaction between aliphatic sodium sulfinate salts 2 and cinnamic acid 1 under our KI-promoted decarboxylative sulfonylation strategy. Methane-, ethane-, 1-propane-, 1-butane, 1-pentane-, and 1-hexane sulfinic acid sodium salts 2 were well tolerated under this protocol to afford the desired products (47–52) in 30–80% yield.
Moreover, cyclopropane-sulfinic acid sodium salt also reacted smoothly and afforded the desired vinyl sulfone (53) in 70% yield. Notably, this is the first report on the decarboxylative sulfonylation synthesis of 1-propane-, 1-butane-1-pentane-, and 1-hexane-bearing vinyl sulfones in good yields.
Scale-up
To demonstrate the practicality of this method, a gram-scale synthesis was performed with cinnamic acid 1 (0.444 g, 3 mmol) and 4-methylbenzenesulfinate 2 (0.801 g, 4.5 mmol) to afford the desired product 3 in 72% yield (0.557 g).Mechanistic studies
To glean further insights into the mechanism, a series of control experiments and studies were performed and analyzed (Scheme 3). A radical trapping experiment with (2,2,6,6-tetramethylpiperidin-1-yl) oxy (TEMPO), butylated hydroxytoluene (BHT) or 1,1-diphenylethylene completely inhibited the reaction, and no sulfonylated product was obtained (Scheme 3a). These results indicated that the sulfonylation reaction may involve a radical process. The tosyl–TEMPO adduct 54 was detected by GC-MS (M = 311) and the 1,1-diphenylethylene adduct 55 was produced, isolated (47% yield) upon reaction of cinnamic acid 1 with 4-methylbenzenesulfinate 2, and confirmed by NMR, corroborating the intermediacy of a sulfonyl radical species (see the ESI†). Moreover, a starch–iodine test was performed, which indicated the formation of in situ molecular iodine (I2) in the reaction mixture (see the ESI†). Next, the 4-methylbenzenesulfonyl iodide substrate (X = –SO2I) was subjected to ball mill conditions with H2O and the desired product 3 was obtained in 87% yield, establishing the involvement of a possible sulfonyl iodide intermediate in the reaction medium (Scheme 3c).12 However, the same reaction with aryl sulfinic acid (X = –SO2H) did not yield the expected desired product under ball mill conditions. The above result indicates that the key sulfonyl iodide intermediate may be formed in the reaction medium and sulfinic acid may not be the intermediate. | ||
| Scheme 3 Mechanistic studies. | ||
Next, a series of controlled reactions with styrene and ethyl cinnamate were performed under standard conditions (Scheme 3d). The results revealed that a 59% yield of the desired vinyl sulfones 3 was obtained when the reaction was performed with styrene, whereas ethyl cinnamate derivatives did not yield the required products, which shows the importance of the carboxyl group in the reaction protocol.
Diversification of vinyl sulfones
Compound 3 was subjected to a reaction with pyrrolidin-2-one in DMA at 110 °C with (NH4)2S2O8 as an oxidant under air, resulting in the required product 56 in 74% yield (Scheme 4).13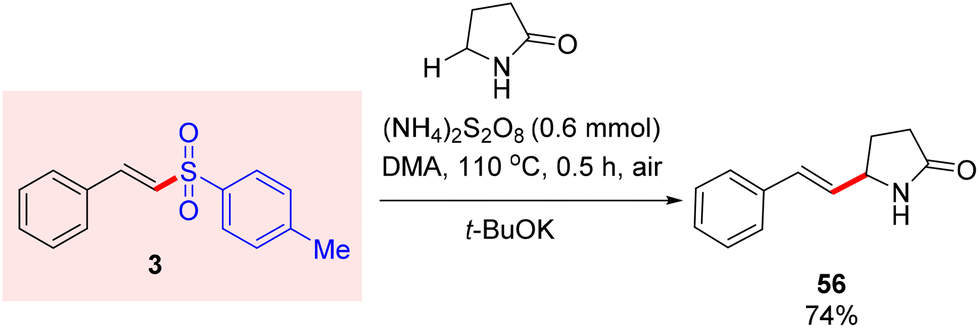 | ||
| Scheme 4 Synthetic applications. | ||
To determine the eco-friendliness and greenness of our developed strategy, the green chemistry metrics were evaluated for the synthesis of 3 (0.118 g, 92%) from cinnamic acid 1 (0.5 mmol, 0.074 g) and 4-methylbenzenesulinate 2 (0.75 mmol, 0.133 g) using KI (50 mol%, 0.0414 g) under ball mill conditions. The results are presented in Fig. 3a. For our method, the green chemistry metrics were found to be top-notch. In particular, effective mass yield (285.02%), atom economy (79.17%), atom efficiency (72.84%), and reaction mass efficiency (47.50%) were found excellent.14–17 The E-factor is calculated to be 1.105, which is the lowest compared to those of other reported methods. Moreover, the EcoScale score was calculated to be 79, which is excellent in terms of safety, economic, and ecological features.18 Besides, the advantage of a short reaction time of our method results in an excellent turnover frequency (TOF) and a satisfactory turnover number (TON), exhibiting the high catalytic ability of KI in the reaction system (see the ESI†).
 | ||
| Fig. 3 Green chemistry metric analysis (a) Green chemistry metric evaluation of synthesis of vinyl sulfones by our method. (b) Summary of green chemistry metrics of our method compared to previous methods (see the ESI† for detailed calculation). Note: atom economy (AE), atom efficiency (AEf), effective mass yield (EMY), reaction mass efficiency (RME), optimum efficiency (OE), process mass intensity (PMI), mass intensity (MI), mass productivity (MP), E-factor, turnover number (TON), and turnover frequency (TOF). (↑), higher is better; (↓), lower is better. aSee the ESI† for detailed calculations. | ||
In general, we observed that the green chemistry metrics of our method are exceptionally tailored towards sustainability. The green chemistry metrics of our strategy are compared with those of other decarboxylative sulfonylation methods as shown in Fig. 3b.6,7 (see the ESI† for detailed calculation).
In light of all experimental data and previous literature reports, the plausible mechanistic pathways for this mechanochemical-mediated decarboxylative sulfonylation reaction are proposed and illustrated in Scheme 5. Initially, KI is oxidized in the presence of atmospheric air and H2O to generate molecular iodine (I2) which is supported by the starch–iodine test. It is easy to generate aryl sulfonyl iodide intermediate A from sodium sulfinate salt and iodine, which undergoes homolysis to give a sulfonyl radical B and an iodine radical.19 The addition of sulfonyl radical intermediate B to α,β-unsaturated carboxylic acids 1 affords the radical intermediate C. Two possible mechanistic pathways for product formation is shown in Scheme 5. In path-A, the benzylic radical intermediate C undergoes hydrogen atom transfer reaction (HAT) with the iodine radical to give diradical intermediate D and HI. Finally, the final product 3 is obtained via decarboxylation of intermediate D. On the other hand (path-B), the intermediate C undergoes deprotonation followed by decarboxylation to give a radical anion intermediate G.
 | ||
| Scheme 5 Possible mechanistic pathway. | ||
Finally, the intermediate G can interact with iodine via a single electron transfer event to give the final product 3. The excellent E/Z selectivity might originate from stereoelectronic and steric effects of the radical anion.20
Conclusions
In summary, we have successfully demonstrated a mechanochemical-mediated decarboxylative sulfonylation reaction to synthesize vinyl sulfones under ball milling conditions. The striking features of this method include: (a) the use of readily available α,β-unsaturated carboxylic acids and sodium sulfinates; (b) use of inexpensive KI as an activator and H2O as a LAG; (c) operational simplicity in terms of room temperature reaction and a short reaction time; (d) metal catalyst-, oxidant-, additive-, and solvent-free conditions; (e) being successful with both alkyl and aryl sulfinate salts; (f) compatibility on a gram scale. Moreover, the green chemistry parameters were found to be excellent in terms of safety, economic, and ecological considerations. We believe that the current method is applicable to late-stage functionalization and in the synthesis of valuable intermediates in organic synthesis at both academic and industrial levels.Author contributions
B. Saxena optimized the reaction conditions and synthesized all the derivatives including gram-scale synthesis. R. Patel synthesized the sodium sulfinate salts. S. Sharma synthesized cinnamic acids. B. Saxena and R. Patel performed the mechanistic studies and wrote the manuscript with the helpful insights of Prof. A. Sharma. Prof. A. Sharma supervised the whole work, interpreted the results, and edited the manuscript. All the authors have given their final approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from UCOST (UCS & T/R & D-35/20-21), Govt. of Uttarakhand, and SERB (CRG/2022/002691), India is gratefully acknowledged. We also acknowledge DST-FIST (SR/FST/CS-II/2018/72(C)) for the NMR facility in the Chemistry Department, IIT Roorkee. B. S. and R. P. thank CSIR and UGC for the SRF, respectively.References
- (a) N. Zhang, D. Yang, W. Wei, L. Yuan, F. Nie, L. Tian and H. Wang, J. Org. Chem., 2015, 80, 3258–3263 CrossRef CAS PubMed; (b) X. Wang, S.-Y. Li, Y.-M. Pan, H.-S. Wang, Z.-F. Chen and K.-B. Huang, J. Org. Chem., 2015, 80, 2407–2412 CrossRef CAS PubMed; (c) N. Chakraborty, K. K. Rajbongshi, A. Dahiya, B. Das, A. Vaishnani and B. K. Patel, Chem. Commun., 2023, 59, 2779–2782 RSC; (d) S. K. Xu, Z. Tan, H. Zhang, J. Liu, S. Zhang and Z. Wang, Chem. Commun., 2017, 53, 10719–10722 RSC; (e) S. Chowdhury and S. Roy, J. Org. Chem., 1997, 62, 199–200 CrossRef CAS PubMed; (f) J. Hu, N. Zhao, B. Yang, G. Wang, L.-N. Guo, Y.-M. Liang and S.-D. Yang, Chem. – Eur. J., 2011, 17, 5516–5521 CrossRef CAS PubMed; (g) J. P. Das, U. K. Roy and S. Roy, Organometallics, 2005, 24(25), 6136–6140 CrossRef CAS.
- (a) C. G. Na, D. Ravelli and E. J. Alexanian, J. Am. Chem. Soc., 2020, 142, 44–49 CrossRef CAS PubMed; (b) Z. Wu and D. A. Pratt, J. Am. Chem. Soc., 2020, 142, 10284–10290 CrossRef CAS PubMed; (c) M. Liu, Z. Zhang, B. Chen, Q. Meng, P. Zhang, J. Song and B. Han, Chem. Sci., 2020, 11, 7634–7640 RSC; (d) Y. Dong, P. Ji, Y. Zhang, C. Wang, X. Meng and W. Wang, Org. Lett., 2020, 24, 9562–9567 CrossRef PubMed; (e) K.-A. Green and J. M. Hoover, ACS Catal., 2020, 10(3), 1769–1782 CrossRef CAS; (f) S. Ranjit, Z. Duan, P. Zhang and X. Liu, Org. Lett., 2010, 12, 4134–4136 CrossRef CAS PubMed; (g) K. Yan, D. Yang, W. Wei, J. Zhao, Y. Shuai, L. Tian and H. Wang, Org. Biomol. Chem., 2015, 13, 7323–7330 RSC; (h) P.-F. Wang, X.-Q. Wang, J.-J. Dai, Y.-S. Feng and H.-J. Xu, Org. Lett., 2014, 16, 4586–4589 CrossRef CAS PubMed; (i) M. Li and J. M. Hoover, Chem. Commun., 2016, 52, 8733–8736 RSC.
- (a) I. D. Kerr, J. H. Lee, C. J. Farady, R. Marion, M. Rickert, M. Sajid, K. C. Pandey, C. R. Caffrey, J. Legac, E. Hansell, J. H. McKerrow, C. S. Craik, P. J. Rosenthal and L. S. Brinen, J. Biol. Chem., 2009, 284, 25697–25703 CrossRef CAS PubMed; (b) D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793–814 CrossRef CAS PubMed; (c) S. Y. Woo, J. H. Kim, M. K. Moon, S.-H. Han, S. K. Yeon, J. W. Choi, B. K. Jang, H. J. Song, Y. G. Kang, J. W. Kim, J. Lee, D. J. Kim, O. Hwang and K. D. Park, J. Med. Chem., 2014, 57, 1473–1487 CrossRef CAS PubMed; (d) T. Aiebchun, P. Mahalapbutr, A. Auepattanapong, O. Khaikate, S. Seetaha, L. Tabtimmai, C. Kuhakarn, K. Choowongkomon and T. Rungrotmongkol, Molecules, 2021, 26, 2211 CrossRef CAS PubMed; (e) R. Ahmadi and S. Emami, Eur. J. Med. Chem., 2022, 234, 114255 CrossRef CAS PubMed; (f) Z.-L. Song, Y. Hou, F. Bai and J. Fang, Bioorg. Chem., 2021, 107, 104520 CrossRef CAS PubMed.
- (a) S. Chodroff and W. F. Whitmore, J. Am. Chem. Soc., 1950, 72, 1073 CrossRef CAS; (b) H. Dressler and J. E. Graham, J. Org. Chem., 1967, 32(4), 985–990 CrossRef CAS; (c) P. B. Hopkins and P. L. Fuchs, J. Org. Chem., 1978, 43, 1208 CrossRef CAS; (d) M. Mikolajczyk, W. Perlikowska, J. Omelanczuk, H. J. Cristau and A. Perraud-Darcy, J. Org. Chem., 1998, 63, 9716–9722 CrossRef CAS; (e) J. H. van Steenis, J. J. G. S. van Es and A. van der Gen, Eur. J. Org. Chem., 2000, 2787–2793 CrossRef CAS; (f) S. V. Ley and N. S. Simpkins, J. Chem. Soc., Chem. Commun., 1983, 1281 RSC.
- For selected examples, see. (a) P. Das, S. Das and R. Jana, Chem. – Asian J., 2022, 17, e202200085 CrossRef CAS PubMed; (b) W. Fan, J. Su, D. Shi and B. Feng, Tetrahedron, 2015, 71, 6740–6743 CrossRef CAS; (c) S. Liang, R. Y. Zhang, G. Wang, S. Y. Chen and X. Q. Yu, Eur. J. Org. Chem., 2013, 7050–7053 CrossRef CAS; (d) A. U. Meyer, S. Jäger, D. P. Hari and B. König, Adv. Synth. Catal., 2015, 357, 2050–2054 CrossRef CAS; (e) G. Rong, J. Mao, H. Yan, Y. Zheng and G. Zhang, J. Org. Chem., 2015, 80, 4697–4703 CrossRef CAS PubMed; (f) H. Jiang, X. Chen, Y. Zhang and S. Yu, Adv. Synth. Catal., 2013, 355, 809–813 CrossRef CAS; (g) X. Gao, X. Pan, J. Gao, H. Huang, G. Yuan and Y. Li, Chem. Commun., 2015, 51, 210–212 RSC; (h) X. Li, Y. Xu, W. Wu, C. Jiang, C. Qi and H. Jiang, Chem. – Eur. J., 2014, 20, 7911–7915 CrossRef CAS PubMed; (i) S. Li, X. Li, F. Yang and Y. Wu, Org. Chem. Front., 2015, 2, 1076–1079 RSC; (j) S. Tang, Y. Wu, W. Liao, R. Bai, C. Liu and A. Lei, Chem. Commun., 2014, 50, 4496–4499 RSC.
- (a) R. Guo, Q. Gui, D. Wang and Z. Tan, Catal Lett., 2014, 144, 1377–1383 CrossRef CAS; (b) Q. Jiang, B. Xu, J. Jia, A. Zhao, Y.-R. Zhao, Y.-Y. Li, N.-N. He and C.-C. Guo, J. Org. Chem., 2014, 79, 7372–7379 CrossRef CAS PubMed; (c) B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2014, 79, 8110–8117 CrossRef CAS PubMed; (d) N. Xue, R. Guo, X. Tu, W. Luo, W. Deng and J. Xiang, Synlett, 2016, 2695–2698 CAS.
- (a) P. Katrun, S. Hlekhlai, J. Meesin, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, Org. Biomol. Chem., 2015, 13, 4785–4794 RSC; (b) J. Gao, J. Lai and G. Yuan, RSC Adv., 2015, 5, 66723–66726 RSC; (c) J. Chen, J. Mao, Y. Zheng, D. Liu, G. Rong, H. Yan, C. Zhang and D. Shi, Tetrahedron, 2015, 71, 5059–5063 CrossRef CAS; (d) Y. Xu, X. Tang, W. Hu, W. Wu and H. Jiang, Green Chem., 2014, 16, 3720–3723 RSC; (e) P. Qian, M. Bi, J. Su, Z. Zha and Z. Wang, J. Org. Chem., 2016, 81, 4876–4882 CrossRef CAS PubMed; (f) P. Li and G.-W. Wang, Org. Biomol. Chem., 2019, 17, 5578–5585 RSC.
- (a) J.-L. Doand and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef PubMed; (b) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC; (c) X. Liu, Y. Li, L. Zeng, X. Li, N. Chen, S. Bai, H. He, Q. Wang and C. Zhang, Adv. Mater., 2022, 34, 2108327 CrossRef CAS PubMed; (d) A. C. Jones, J. A. Leitch, S. E. Raby-Buck and D. L. Browne, Nat. Synth., 2022, 1, 763–775 CrossRef.
- (a) K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504 CrossRef CAS PubMed; (b) C. G. Vogt, S. Grätz, S. Lukin, I. Halasz, M. Etter, J. D. Evans and L. Borchardt, Angew. Chem., Int. Ed., 2019, 58, 18942 CrossRef CAS PubMed; (c) T. Seo, K. Kubota and H. Ito, J. Am. Chem. Soc., 2023, 145, 6823–6837 CrossRef CAS PubMed; (d) R. R. A. Bolt, S. E. Raby-Buck, K. Ingram, J. A. Leitch and D. L. Browne, Angew. Chem., Int. Ed., 2022, 61, e202210508 CrossRef CAS PubMed; (e) Q. Cao, J. L. Howard, E. Wheatley and D. L. Browne, Angew. Chem., Int. Ed., 2018, 57, 11339–11343 CrossRef CAS PubMed; (f) A. C. Jones, M. T. J. Williams, L. C. Morrill and D. L. Browne, ACS Catal., 2022, 12, 13681–13689 CrossRef CAS PubMed; (g) A. C. Jones, W. I. Nicholson, J. A. Leitch and D. L. Browne, Org. Lett., 2021, 23, 6337–6341 CrossRef CAS PubMed; (h) R. Takahashi, T. Seo, K. Kubota and H. Ito, ACS Catal., 2021, 11, 14803–14810 CrossRef CAS; (i) I. Priestley, C. Battilocchio, A. V. Iosub, F. Barreteau, G. W. Bluck, K. B. Ling, K. Ingram, M. Ciaccia, J. A. Leitch and D. L. Browne, Org. Process Res. Dev., 2023, 27, 269–275 CrossRef CAS; (j) R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed; (k) V. S. Pfennig, R. C. Villella, J. Nikodemus and C. Bolm, Angew. Chem., Int. Ed., 2022, 61, e202116514 ( Angew. Chem. , 2022 , 134 , e202116514 ) CrossRef CAS PubMed; (l) K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed; (m) E. Colacino, V. Isoni, D. Crawford and F. García, Trends Chem., 2021, 3, 335–339 CrossRef CAS; (n) O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustainable Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS; (o) C. Patel, E. André-Joyaux, J. A. Leitch, X. M. de Irujo-Labalde, F. Ibba, J. Struijs, M. A. Ellwanger, R. Paton, D. L. Browne, G. Pupo, S. Aldridge, M. A. Hayward and V. Gouverneur, Science, 2023, 381, 302–306 CrossRef CAS PubMed; (p) H. K. Singh, A. Kamal, S. K. Maury, A. K. Kushwaha, V. Srivastava and S. Singh, Org. Biomol. Chem., 2023, 21, 4854–4862 RSC; (q) G. Brahmachari, I. Karmakar and P. Karmakar, Green Chem., 2021, 23, 4762–4770 RSC.
- (a) A. Monga, S. Bagchi, R. K. Soni and A. Sharma, Adv. Synth. Catal., 2020, 362, 2232–2237 CrossRef; (b) A. Monga, A. P. Pandey and A. Sharma, Adv. Synth. Catal., 2019, 361, 3554–3559 CrossRef CAS; (c) R. I. Patel, A. Sharma, S. Sharma and A. Sharma, Org. Chem. Front., 2021, 8, 1694–1718 RSC; (d) R. I. Patel, J. Singh and A. Sharma, ChemCatChem, 2022, 14, e202200260 CrossRef CAS; (e) B. Saxena, R. I. Patel and A. Sharma, Adv. Synth. Catal., 2023, 365, 1538–1564 CrossRef CAS; (f) R. I. Patel, S. Sharma and A. Sharma, Org. Chem. Front., 2021, 8, 3166–3200 RSC; (g) B. Saxena, R. I. Patel, J. Tripathi and A. Sharma, Org. Biomol. Chem., 2023, 21, 4723–4743 RSC.
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- M. R. Mutra, J. Li, Y.-T. Chen and J.-J. Wang, Chem. – Eur. J., 2022, 28, e202200742 CrossRef CAS PubMed.
- M. Li, L. Zheng, L. Ma and Y. Chen, J. Org. Chem., 2021, 86, 3989–3998 CrossRef CAS PubMed.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- (a) L. Wei, J. Zhang and L. Xu, ACS Sustainable Chem. Eng., 2020, 8, 13894–13899 CrossRef CAS; (b) P. Yan, R. Zeng, B. Bao, X.-M. Yang, L. Zhu, B. Pan, S.-L. Niu, X.-W. Qi, Y.-L. Li and Q. Ouyang, Green Chem., 2022, 24, 9263–9268 RSC.
- N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. Garcia and E. Colacino, ChemRxiv, DOI:10.26434/chemrxiv-2022-b370p.
- (a) K. Van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 3 Search PubMed; (b) A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Green Chem., 2018, 20, 964–968 RSC.
- (a) W. E. Truce and G. C. Wolf, J. Org. Chem., 1971, 36, 1727 CrossRef CAS; (b) P. Katrun, C. Mueangkaew, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, J. Org. Chem., 2014, 79, 1778 CrossRef CAS PubMed; (c) E. Truce, D. L. Heuring and G. C. Wolf, J. Org. Chem., 1980, 45, 406 CrossRef; (d) L. M. Harwood, M. Julia and G. L. Thuiller, Tetrahedron, 1980, 36, 2483 CrossRef CAS; (e) D. C. Craig, G. L. Edwards and C. A. Muldoon, Synlett, 1977, 1441 Search PubMed.
- L. Zhang, Z. Hang and Z.-Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 236–239 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed optimization, the experimental procedure, calculation of green metrics, and 1H and 13C NMR spectra of all the synthesized compounds. See DOI: https://doi.org/10.1039/d3gc04954j |
| This journal is © The Royal Society of Chemistry 2024 |