
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable electrochemical synthesis of dry formaldehyde from anhydrous methanol†
Florian
Schwarz
 ,
Elizabeth
Larenz
and
Anna K.
Mechler
*
,
Elizabeth
Larenz
and
Anna K.
Mechler
*
Electrochemical Reaction Engineering (AVT.ERT) RWTH Aachen University, Forckenbeckstr. 51, Aachen, Germany. E-mail: anna.mechler@avt.rwth-aachen.de; Tel: +49 241 80 47695
First published on 8th March 2024
Abstract
Formaldehyde is a platform chemical used for example in the syntheses of polymers and complex molecules. The current formaldehyde synthesis relies on high temperatures to produce an aqueous solution, which requires energy intensive water removal for its use as a C1 building block. In this work, we report the successful electrification of the direct synthesis of anhydrous formaldehyde from methanol. Sustainable formaldehyde is produced with Faraday efficiencies of 80% in an H-cell and even up to 90% and elevated current densities in a scaled-up flow reactor. Comparing different reaction conditions, we furthermore show an impact of the current density and electrolyte concentration on the prevalent reaction mechanism. Our study demonstrates a selective and efficient electrochemical synthesis of dry formaldehyde at relevant scales, paving the way for green industrial production processes.
1. Introduction
The European Union strives for net-zero greenhouse gas (GHG) emissions by 2050 with a reduction of 55% by 2030. The GHG emissions can be reduced by making existing processes more efficient or by capturing and recycling of the GHG as it is attempted with carbon dioxide (CO2). For example, CO2 can be converted to the versatile platform chemical methanol (MeOH).1–5 Methanol, currently obtained from fossil resources, is oxidized to formaldehyde on a large scale with a worldwide annual production of 45.6 × 106 t in 2020.6 There are three main industrial processes for the formaldehyde synthesis: the silver contact process at 600–720 °C, with either full or incomplete conversion; and the iron-molybdenum oxide catalyzed Formox process at 250–400 °C. All processes use excess air and produce aqueous formaldehyde solutions.7 In case of the silver catalyst, water steam is added to the methanol–air feed to help the surface restructuring process and catalyst activation.8,9 The two main reaction pathways leading to formaldehyde have hydrogen (eqn (1)) and water (eqn (2)) as side products.4,7,10–12 Both reactions occur in the silver contact processes, whereas only the oxidation reaction to water (eqn (2)) occurs in the Formox process.7,13,14| CH3OH(g) → HCHO(g) + H2(g) ΔH = +84 kJ mol−1 | (1) |
 | (2) |
The presence of water is required for the methanol oxidation on silver catalysts and produced in the Formox process, which produces an aqueous formaldehyde solution for all processes. Yet, water-free formaldehyde is required for the production of resins, glues and polymers, such as polyoxymethylenes;7,15 and the removal of water requires a high energy input.6 For example, formaldehyde can be dried and handled as 1,3,5-trioxane, a cyclic trimer of formaldehyde. The conversion of aqueous formaldehyde solution to trioxane requires an energy input of 65 MJ per kg trioxane.16 Hence, it would be preferable to produce anhydrous (non-aqueous) formaldehyde. An additional advantage of the anhydrous methanol dehydrogenation (eqn (1)) is that the produced hydrogen can be recycled in the upcoming hydrogen economy or used for further reduction of CO2 to methanol.1–5
In the past decades, several materials were investigated as thermally active methanol dehydrogenation catalysts. Early success was found with transition metal catalysts, such as silver, copper, zinc and indium at operation temperatures of 250–760 °C.6,17,18 A silver/copper catalyst converted 85% of the methanol to formaldehyde with a selectivity of 92% at 620 °C.17 Sodium was tested successfully as vapor and as a salt-cation at temperatures between 450–900 °C, yet these materials struggle with stability.6,15,17 Experiments with a Na-vapor catalyst converted 95% of the methanol to formaldehyde with a selectivity of 83% with CO as the major side product.19 More recently, a highly active catalyst based on sulfated Ce2(MoO4)3 loaded on SiO2 was reported, which yielded 100% conversion and 100% selectivity for small scale experiments with 0.5 g catalyst at 325 °C.20
The electrochemical oxidation of methanol to formaldehyde was investigated as part of the fuel cell research, in which it occurs as an unwanted side reaction.21,22 Thus, the research often focused on the occurring reaction mechanisms and not on high yields.23–25 A recent study proposed the synthesis of formaldehyde from methanol in an electrochemical flow reactor as an alternative hydrogen source to the more common water electrolysis. They managed to produced formaldehyde with a Faraday efficienciey of 51% after 4 h and a production rate of 7 mol h−1 m−2.26 However, all these systems work in aqueous methanol solutions and produce aqueous formaldehyde.
Early studies in the 1960s and 70s have reported the electrocatalytic oxidation of anhydrous methanol to formaldehyde.27–30 First results were obtained by Sasaki et al., who reported a Faraday efficiency to formaldehyde of 70% with 0.1 mol L−1 NaOMe in MeOH after 2 h at a current density of 3.3 mA cm−2.27 Sundholm reported high current densities of up to 100 mA cm−2 when applying potential/time square waves, during which the electrode spent 0.3 s at 0.0 V every 3 s, but they did not provide a product analysis of the experiment.28 Belanger also investigated methanol oxidation to formaldehyde with current densities of up to 10 mA cm−2 and reported a Faraday efficiency of up to 77%.30 Iwakura et al. tested a variety of metals (Pt, Ni, Ag, Fe, Cu, Ti and Hg), but found that only platinum was active for the methanol oxidation.29 They also investigated different electrolytes and NaOMe appeared to be the most active for the formaldehyde formation.27,29 Besides the lower temperatures required for the electrochemical process, it also yields the possibility to obtain (pure) hydrogen on the cathode, which might be beneficial with respect to the heterogeneous catalytic pathway.
The few reported studies achieved high Faraday efficiencies for the electrochemical formaldehyde synthesis, but only with small current densities in setups that are far from industrial applicability. Here, we not only successfully reproduce these studies from more than 50 years ago, but also report the scaling-up of the electrocatalytic oxidation of liquid anhydrous methanol to a flow cell reactor and elevated current densities. The scale up is vital in the transition of this technology to an industrial application.
2. Experimental part
The electrochemical measurements were performed with a Gamry Interface 1010E. Current densities above 50 mA cm−2 were measured with a Gamry Reference 3000. Two experimental setups were used, an H-cell and an electrochemical flow cell. In the H-cell, a Pt-wire (0.25 mm, 99.99+%, Goodfellow) was used as the working and counter electrode. The geometric surface area of the working electrode in contact with the electrolyte was estimated as 0.7 cm2. A reference electrode was used in the H-cell consisting of a AgCl-coated Ag-wire, which was immersed in a saturated KCl methanol solution. The potential was referenced vs. the ferrocene/ferrocenium (Fc/Fc+) redox couple (0.1 mol L−1 LiClO4, 5 mmol L−1 Fc) and the potentials are given vs. Fc. The Ag/AgCl reference electrode potential was measured before and after each experiment. The halfway redox potential of the Ag/AgCl reference electrode vs. Fc was 401 ± 5 mV over 30 days. The H-cell had a reaction volume of 150 mL, which was not stirred and no separator was used.The electrolyte solutions were prepared by diluting 5.4 mol L−1 sodium methoxide (NaOMe) in methanol (30 wt%, thermoscientific) with methanol (MeOH, 99.9%, Emsure).
The flow cell experiments were carried out in a flex-E-cell, provided by flex-X-cell (Fig. S1†). The electrodes are parallel with an electrolyte gap of 8 mm, which was filled with an additional 3D printed turbulence promoter prepared with a high temperature resin (Formlabs) (Fig. S2†). The turbulence promoter assists the tightness of the cell and helps to prevent leakages. It also guaranteed a defined electrolyte channel width between the electrodes. No separator was used. A polycrystalline platinum foil (0.25 mm, 99.99+%, Goodfellow) was used as the working electrode with a geometric surface area of 12 cm2. A platinum coated titanium plate (2.5 μm Pt coating, Metakem) was used as the counter electrode (12 cm2). All data points for the flow cell are based on the average of 2–3 repeated experiments and the error bars display the standard deviation.
The flow rate was adjusted with a Masterflex L/S pericular pump with a Masterflex L/S Easy-Load pump head. The 40 mL electrolyte was recirculated in a glass electrolyte reservoir.
Samples were taken after the measurement and analyzed via high-performance liquid chromatography (HPLC) for formaldehyde and formic acid. An HPLC Agilent 1260 was used with an organic acid resin column. The eluent (5 mmol L−1 H2SO4 in ultra pure water) was fed with a flow rate of 0.5 mL min−1. The column was heated to 40 °C. The conductivity was measured with a conducotometer 712 by Metrohm, which was calibrated with 11.67 mS cm−1 KCl solution. The water content was determined with a Karl Fischer coulometer 831 by Metrohm. It was calibrated with a water standard of 0.1 wt% and seven repeated measurements averaged 0.102 wt% water with a standard deviation of 0.0054 wt%.31
3. Results and discussion
3.1. H-cell experiments
As a first proof-of-concept of the early studies, the experimental conditions from Sasaki et al. were adapted for our H-cell setup. They used a 30 cm2 platinum working electrode, a platinum counter electrode and as reference a saturated calomel electrode (SCE). Their electrochemical cell had an electrolyte volume of 100 cm3, which was filled with 0.1 mol L−1 electrolytes: ammonium halides (fluoride, chloride, bromide, iodide) and sodium methoxide. In their experiments, they applied a current density of 3.3 mA cm−2 for 2 h during which they cooled the reaction with an ice bath.27 Our H-cell setup consisted of two Pt-wires as working and counter electrodes. The aqueous SCE was replaced with a non-aqueoues Ag/AgCl reference electrode and the potential was measured vs. Fc. We used ambient temperature 0.1 mol L−1 NaOMe as the electrolyte as it performed best among their tested electrolytes.A cyclic voltammetry (CV) was measured in this setup (Fig. 1). The curve shows two distinct oxidation peaks in the anodic sweep at 0.7 V and 1.3 V vs. Fc with peak current densities of 10.4 mA cm−2 and 16.1 mA cm−2, respectively. The current response increases further with higher potentials to a maximum current density of 22.1 mA cm−2 at 2.4 V vs. Fc. An oxidation peak in the cathodic CV sweep roughly coincides with the first oxidation peak of the anodic sweep. The occurrence of multiple oxidation peaks indicates that different reactions occur at the different potentials.
 | ||
| Fig. 1 Cyclic voltammetry of MeOH on Pt in 0.1 mol L−1 NaOMe in MeOH between −0.6 to 2.4 V vs. Fc with a scan rate of 100 mV s−1. Two oxidation peaks are visible in the anodic sweep with a further increasing current response at higher potentials. During the cathodic sweep, an overlapping oxidation peak occurs at 0.7 V vs. Fc. | ||
A CV of 1 mol L−1 NaOMe in MeOH was measured by Iwakura et al. The curve shows two oxidation peaks in the anoidc sweep at 0 V and 1.0 V vs. SCE. A small oxidation peak is visible on the cathodic sweep at −0.45 V vs. SCE. A more detailed comparison is unfeasible due to incomplete documentation.29
The shape of the measured CV (Fig. 1) is similar to the methanol oxidation on Pt/C in aqueous media.32–34 The methanol CV of Pt/C shows an oxidation peak at 1 V vs. RHE in the anodic sweep. With increasing potential the current response drops until it increases again at potentials above 1.2 V vs. RHE. The CV also shows an oxidation peak in the cathodic sweep, which is attributed to CO oxidation.34 In aqueous media, the oxidation peaks of the anodic and cathodic sweep do not overlap as the peak of the cathodic sweep is shifted to lower potentials. A chronoamperometry (CA) was conducted at the potential of each of the different oxidation peaks of the measured CV in 0.1 mol L−1 NaOMe (Fig. 2). HPLC analysis showed that formaldehyde was produced at all three potentials, but the amount of formaldehyde and the Faraday efficiency vary vastly (Table 1). The only liquid side product detected was formic acid.
 | ||
| Fig. 2 Chronoamperometry at peak potentials vs. Fc of the measured CV on Pt in 0.1 mol L−1 NaOMe in MeOH. | ||
| E vs. Fc (V) | Charge (C) | c(HCHO) (mg L−1) | c(HCHO) (mmol L−1) | FE (%) |
|---|---|---|---|---|
| 0.7 | 39.1 | 17 | 0.53 | 49 |
| 1.3 | 1.8 | 6 | 0.17 | n.a. |
| 2.4 | 88.0 | 71 | 6.20 | 78 |
At 0.7 V and 2.4 V, stable currents were achieved during the experiments. The comparison of the total charge and the formaldehyde yields lead to Faraday efficiencies of 49% and 78%, respectively. The measurement at 1.3 V vs. Fc was interrupted by the potentiostat due to the internal resistance of the system becoming too high. The calculated Faraday efficiency at this potential was beyond 100%. This is ascribed to the low formaldehyde concentration, which was close to the detection limit of the HPLC. Hence, the HPLC analysis would lead to an exaggeration of the Faraday efficiency. The differing current responses and Faraday efficiencies also indicate that the applied potential influences the occurring reaction pathway. The increase in the internal resistance at 1.3 V might be caused by CO formation on the electrode surface. At 0.7 V, current was constantly flowing and even slightly increasing over the 2 h measurement, which indicates that the electrode was not poisoned or degrading. The highest applied potential of 2.4 V yielded the highest Faraday efficiency and also the highest total current. The high Faraday efficiency suggests that less CO is formed than at 1.3 V instead of CO formation followed by its oxidation. The Faraday efficiencies obtained at 2.4 V vs. Fc are comparable to the ones described in the literature 50 years ago,27–30 and demonstrate the viability of our approach.
More recently, the reaction of pure methanol to formaldehyde in an electric field was also predicted by Cassone et al.35 In a sufficiently strong electrical field of 0.55 V Å−1, methanol can theoretically disproportionate to formaldehyde, methane and water. No additional catalytic activity is required at these conditions. Such field strengths can possibly be reached locally at the electrode–liquid interface.36 To investigate whether the proposed disproportionation is occurring, the water content was determined prior and after the CAs via a Karl Fischer coulometry (Table 2). The analysis shows that the water amounts remain nearly stable, while minor changes can also be caused by exchange with the atmosphere. According to the disproportionation by Cassone et al., formaldehyde and water form in equal amounts.35 As the change in the water content is multiple orders of magnitude below the formation of formaldehyde (Table 1), the disproportionation is not the dominant reaction mechanism. Hence, we assume that the detected formaldehyde forms during a catalytic conversion.
| E vs. Fc (V) | wt%prior (H2O) | wt%after (H2O) | Δc(H2O) (μmol L−1) |
|---|---|---|---|
| 0.7 | 0.132 | 0.183 | 21.72 |
| 1.3 | 0.190 | 0.207 | 7.12 |
| 2.4 | 0.107 | 0.107 | — |
The experiments have so far been carried out in 0.1 mol L−1 NaOMe solution. However, the low electrolyte concentration causes a low overall electrolyte conductivity (Fig. 3). The conductivity of NaOMe in MeOH increases with increasing NaOMe concentration up to maximum of 21.4 mS cm−1 at a concentration of 1.25 mol L−1, at which the conductivity reaches a small plateau after which it slowly decreases. The conductivity of NaOMe in MeOH was also measured by Iwakura et al. Their conductivity curve mirrors the one shown in Fig. 3, yet they reported the conductivity in S cm−1, which is three orders of magnitude above our measurement.29
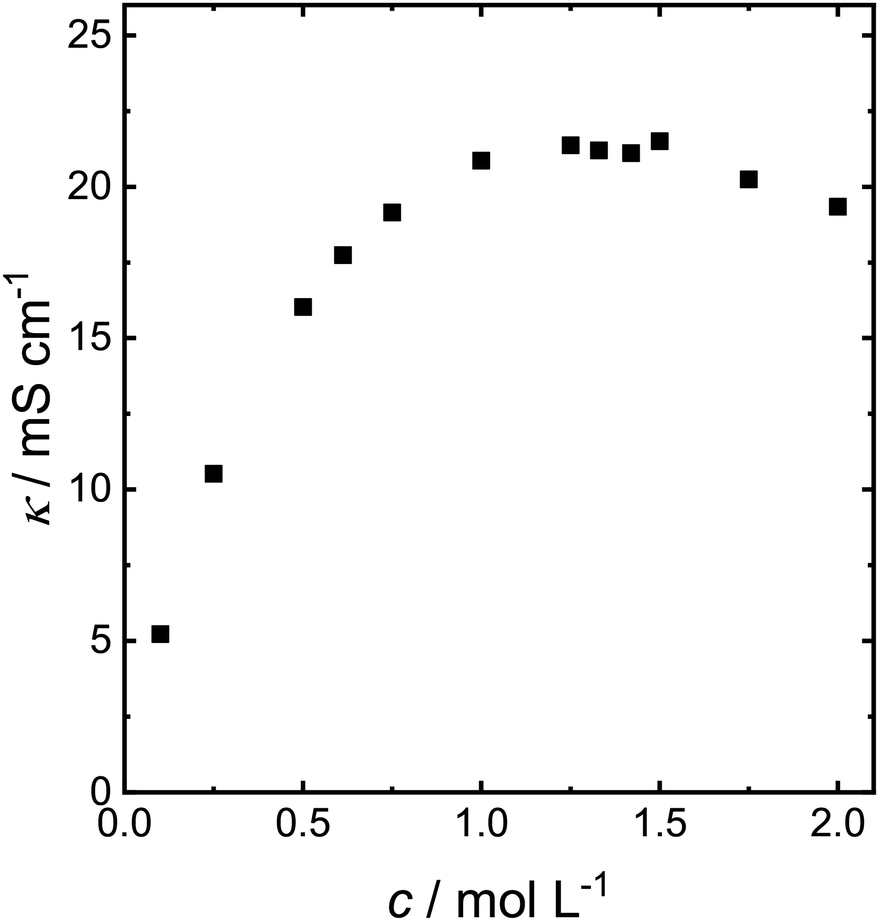 | ||
| Fig. 3 Electrical conductivity of different concentrations of NaOMe in MeOH. | ||
In order to lower the electrolyte resistance, the CA at 2.4 V was repeated with 0.5 and 1.0 mol L−1 NaOMe in MeOH. The Faraday efficiencies and formaldehyde concentration after 2 h are shown in Fig. 4. The obtained concentrations of formaldehyde and formic acid are shown in Table S1.† The Faraday efficiency decreases from 78% to 50% with increasing electrolyte concentration, whereas as the overall yield increases from 71 to 199 mg L−1 and 176 mg L−1 for 0.5 and 1.0 mol L−1 NaOMe, respectively. The increasing electrolyte conductivity allows higher currents at fixed potentials, as the transferred charge increase from 88 C to 345 C from 0.1 to 0.5 mol L−1 and 400 C for 1.0 mol L−1 (CAs shown in Fig. S3†). The lower Faraday efficiency in the 1.0 mol L−1 electrolyte is caused by larger amounts of produced formic acid.
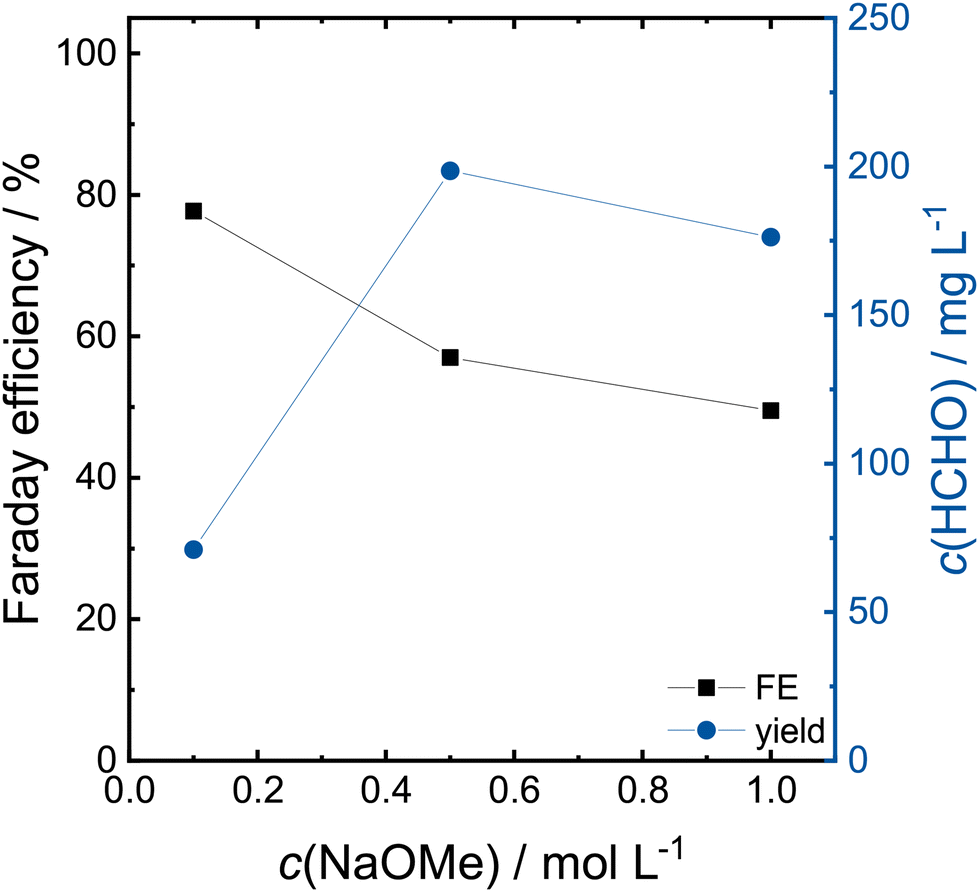 | ||
| Fig. 4 Faraday efficiency and concentration of formaldehyde at different NaOMe concentrations in a H-cell on a Pt working electrode after a 2 h CA at 2.4 V. | ||
3.2. Flow cell experiments
The proof-of-concept was successful in a small scale setup with static batch conditions, which are far from industrial application. However, the transition to larger electrodes and dynamic operation conditions are not necessarily straight forward. Thus, we investigated the scale up to a 12 cm2 electrode in a flow reactor with a recirculating electrolyte. An increase in the electrolyte concentration strongly reduced the Faraday efficiency in the H-cell. However, high Faraday efficiencies are vital for an industrial process. Therefore, the effect of the electrolyte concentration was reinvestigated in the flow cell. Furthermore, the impact of mass transport was investigated by varying the flow rate. Chronopotentiometries (CP) were carried out for 30 min at an applied current density of 3 mA cm−2 and the resulting yields analyzed via HPLC (Fig. 5).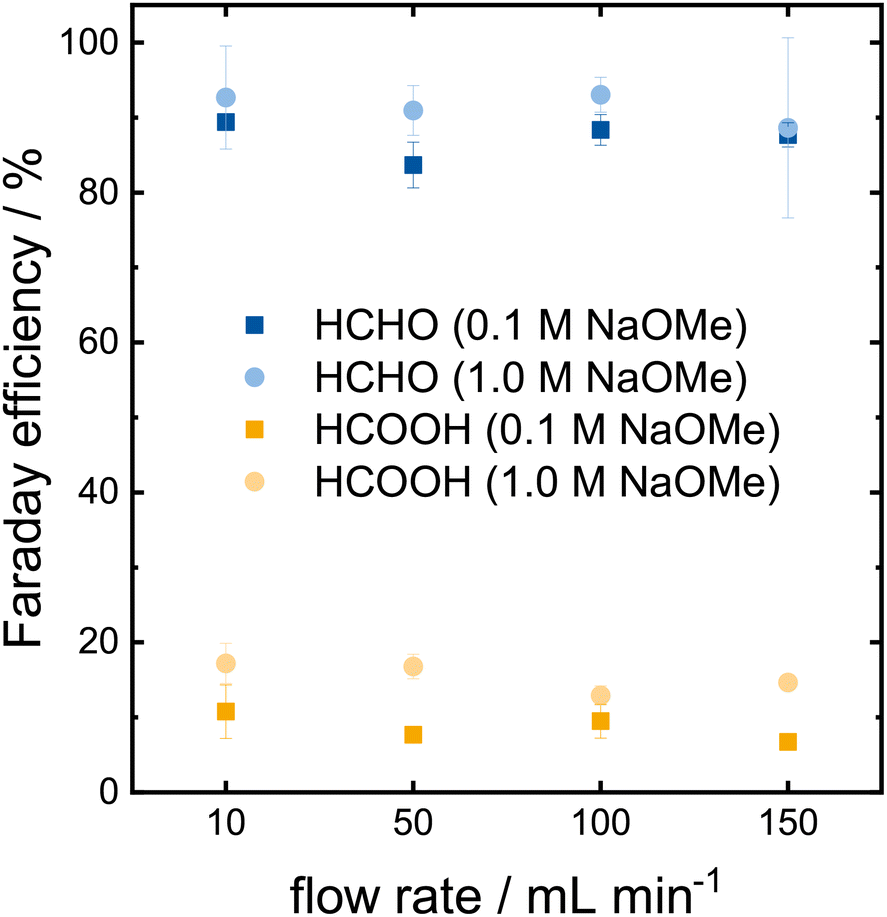 | ||
| Fig. 5 Faraday efficiency of formaldehyde and formic acid at different electrolyte concentrations and flow rates on Pt after 30 min CP at 3 mA cm−2. | ||
The Faraday efficiencies in the 0.1 mol L−1 electrolyte (darker colors in Fig. 5) vary between 84–89% for formaldehyde (blue) and 7–11% for formic acid (yellow) across the different flow rates. The product concentrations determined via HPLC are shown in Table S2.† No trend is observed for the Faraday efficiency or the total formaldehyde yield with respect to the tested flow rates. The indifference of the Faraday efficiency to the flow rate indicate that the mass transport is not the limiting reaction factor at the chosen current density. Increasing the flow rate increases the mass transport on the electrode surface, yet the product formation remains unaffected.
The Faraday efficiencies in the 1.0 mol L−1 electrolyte (lighter colors) are slightly higher compared to the lower concentrated electrolyte as they vary between 89–93% for formaldehyde and 13–17% for formic acid. The Faraday efficiency for formic acid increased slightly more, which lowers the overall selectivity of the reaction by ∼3%. One needs to keep in mind that the mode of operation changed from an applied potential in the H-cell to an applied current in the flow cell. In the H-cell, the current quadrupled from 20 to 80 mA cm−2 when we increased the electrolyte concentration from 0.1 to 1.0 mol L−1. To properly compare these two setups, we need to perform flow cell experiments with higher current densities. Higher currents are required to produce more product, which is also a crucial criteria for industrial electrochemical applications. Thus, we investigated how the methanol oxidation performs at current densities of 10–100 mA cm−2 at a flow rate of 100 mL min−1 (Fig. 6a). The measurement at 3 mA cm−2 from Fig. 5 is shown for comparison. The determined concentrations of formaldehyde and formic acid are shown in Table S3.†
 | ||
| Fig. 6 Faraday efficiency for formaldehyde and formic acid and the required potential for different current densities on Pt in (a) 0.1 mol L−1 and (b) 1.0 mol L−1 NaOMe in MeOH at a flow rate of 100 mL min−1 after 30 min. | ||
High Faraday efficiencies (79–90%) were obtained for current densities of 10–100 mA cm−2 in 0.1 mol L−1 NaOMe. At 100 mA cm−2, a formaldehyde product concentration of 7.7 ± 0.2 g L−1 was achieved within 30 min. This equals a production rate of 17 mol h−1 m−2. We observe a dip of the Faraday efficiency for formaldehyde at 10 and 20 mA cm−2, which increases again at higher current densities. This might be related to the observed CO-poisoning in the H-cell experiments at medium potentials (Fig. 2, orange curve), which we might have reached with these medium current densities, but surpassed thereafter (green curve). Noticeably, the Faraday efficiency for formic acid drops off with increasing current. As no other liquid reaction products were detected, CO or CO2 might have evolved under these conditions. As the electrolyte is recycled repeatedly and no separator is used, it is also possible that formaldehyde is reduced again on the platinum cathode with hydrogen to methanol. The required cell potential for the current densities increases linearly until 90 mA cm−2 after which the potential levels off. The linear increase of the potential indicates a poor electrolyte conductivity. Due to this ohmic resistance, the electrolyte heated up at current densities >50 mA cm−2 up to 40 °C at 100 mA cm−2.
To tackle the poor electrolyte conductivity, the experiments were repeated with a 1.0 mol L−1 NaOMe electrolyte solution (Fig. 6b). The required potential at 100 mA cm−2 is reduced from 18 V to 6.6 V by increasing the electrolyte concentration from 0.1 to 1.0 mol L−1 NaOMe. Unfortunately, we observed a drop in the fromaldehyde Faraday efficiency from 93% at 3 mA cm−2 to 62% at 10 mA cm−2. The Faraday efficiency at 10 mA cm−2 is also 16% lower in the higher concentrated electrolyte and further decreases with increasing current densities down to 44% at 100 mA cm−2. In contrast to the 0.1 mol L−1 electrolyte, formic acid does not disappear as the side product, but is formed in increasing amounts from 18% to 44%. After 30 min at 100 mA cm−2, concentrations of 3.8 ± 0.1 g L−1 for formaldehyde and 2.9 ± 0.1 g L−1 and formic acid, were obtained.
In the higher concentrated electrolyte, more formic acid is produced, which is the higher oxidized product compared to formaldehyde. Subsequent oxidation may be caused by the increased viscosity of the higher concentrated solution, which slows the mass transport on the electrode surface. However, the difference in product distribution only occurs at current densities ≥10 mA cm−2 as it was not observed at 3 mA cm−2.
A comparison of the H-cell experiments with the flow cell setup further yields interesting insights. The 0.1 mol L−1 electrolyte in the H-cell resulted in a current density of 20 mA cm−2 and a Faraday efficiency for formaldehyde of 78%. This matches the Faraday efficiency of 81% the flow cell at 20 mA cm−2. By increasing the electrolyte concentration to 1 mol L−1, the current flow in the H-cell increased to 80 mA cm−2 while lowering the Faraday efficiency to 50%, which matches the 46% of the flow cell measurement for 80 mA cm−2. Thus, the results from the static H-cell are also valid in a flow cell reactor.
Reflecting on the distinctively different behavior in the two electrolyte concentrations at very low vs. elevated current densities, we postulate that this can be correlated to the observed changes in reaction mechanisms in the H-cell. At low current densities (3 mA cm−2), the reaction might perform according to the reaction observed at 0.7 V vs. Fc, whereas at current densities beyond 20 mA cm−2 the reaction mechanism observed at 2.4 V vs. Fc prevails. At 10 mA cm−2, CO-poisoning might (partially) occur and lower the overall cell performance as it was observed in the H-cell at 1.3 V. The different reaction behaviour at varying electrolyte concentrations indicates that the mechanism at 2.4 V vs. Fc is dependent on the electrolyte concentration unlike the one at lower potentials/current. As the higher concentrated electrolyte is more viscous, the mechanism at 2.4 V vs. Fc might be mass transport dependent. It was furthermore postulated in the literature that the NaOMe itself might be a reactant.29 In that case, we would expect the Faraday efficiency to increase with increased NaOMe concentration. Yet, our observations contradict this proposed reaction mechanism at elevated potentials/current densities.
Although the Faraday efficiency dropped in 1.0 mol L−1 NaOMe, the cell potential significantly decreased, leading to a potentially lower energy consumption. To compare the flow cell performance across the different electrolytes (Fig. 7), we calculated the specific energy consumption in kW h kg−1 based on the cell voltage Ecell, the number of transferred electrons z, the Faraday constant F, the Faraday efficiency φ and the stoichiometric factor νp.
 | (3) |
 | ||
| Fig. 7 Specific energy consumption for the formaldehyde synthesis in NaOMe on a 12 cm2 Pt electrode in an electrochemical flow cell at 100 mL min−1. | ||
The specific energy consumption for both electrolyte concentrations is similar for current densities of up to 10 mA cm−2 (Fig. 7). At higher current densities, the specific energy consumption is higher in the 0.1 mol L−1 NaOMe electrolyte, and the gap increases further with increasing current densities. Although the Faraday efficiency for formaldehyde is halved at 100 mA cm−2 from the 0.1 to the 1.0 mol L−1 electrolyte, the required cell potential was decreased by two thirds. Thus, at elevated current densities, the overall formaldehyde synthesis is more efficient in the higher electrolyte concentration by about 10 kW h kg−1. However, subsequent process steps to separate the produced formaldehyde from the formic acid might require additional energy.
4. Conclusion
In this study, we investigated the selective electrochemical oxidation of anhydrous methanol to formaldehyde. The direct synthesis circumvents the energy intensive water removal, which is required after the conventional formaldehyde synthesis for several further production processes.The reaction was first verified in a static batch system with adapted conditions from the literature. We were able to reproduce the 50-years-ago reported Faraday efficiencies of 75–85% and identified formic acid as the main side product. Furthermore, we observed different current response behaviors at different potentials, which indicates that the occurring reaction pathways are possibly potential dependent. Introducing a higher electrolyte concentration significantly increased the current densities at a fixed potential and, hence, tripled the amount of produced formaldehyde but lowered the Faraday efficiency to around 50%.
For more industrially relevant investigations, we then transferred and scaled up our system to a flow cell. At low current densities, even higher Faraday efficiencies beyond 90% could be achieved, which proofed to be independent of the electrolyte concentration and flow rate. Interestingly, at elevated current densities the electrolyte concentration plays again a more significant role. In 0.1 mol L−1 NaOMe, the Faraday efficiency remains between 80–90% when the current density is increased from 10 to 100 mA cm−2 and the formic acid formation almost vanishes at elevated current densities. In contrast, in 1.0 mol L−1 NaOMe the Faraday efficiency decreases dramatically already at 10 mA cm−2 and further drops to 43% at 100 mA cm−2. Along with this decrease, the production of formic acid increases, leading finally to a ca. 4:3 weight ratio of formaldehyde and formic acid. As intended, the overall cell potential is decreased by 66% due to the better conductivity of the electrolyte. This leads to a lower specific energy consumption for the formaldehyde formation in higher concentrated electrolytes despite the lower Faraday efficiencies. We postulate that the drop of Faraday efficiencies at elevated current densities in the higher concentrated electrolyte is proof for a different reaction mechanism. However, further efforts are needed to understand the intricate occurring reaction mechanisms.
The anhydrous oxidation of methanol achieves a higher production rate of 17 mol h−1 m−2 formaldehyde compared to the 7 mol h−1 m−2 of the aqueous system. However, the higher production rate requires a significantly higher energy input due to the lower electrolyte conductivity.
In conclusion, the electrochemical pathway to form formaldehyde from anhydrous methanol shows great potential in terms of efficiency and selectivity. The reaction appears to be robust with regards to the applied currents, but sensitive towards the electrolyte concentration. The total yields are respectable for an electrode area of 12 cm2 and might be further improved with another scale-up step. The required energy input is still high, but the variation in the electrolyte concentration shows that it can be lowered and also optimizations of the cell geometry can contribute to obtain lower potentials. Considering the already prominent results of this initial study, the process has a high potential to produce anhydrous formaldehyde directly and efficiently using electricity at ambient temperature.
Author contributions
F. S. and E. L. conducted the experiments and curated the data. F. S. analyzed the data and wrote the original draft. A. K. M. supervised the project. F. S. and A. K. M. conceptualized the experiments and reviewed and edited the manuscript. All authors contributed to the discussion of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the contributions of Prof. Robert Schloegl to the original project idea and transfer of the project to A. K. M. This work at the RWTH Aachen University was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy - Cluster of Excellence 2186 “The Fuel Science Center”, ID: 390919832.References
- C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS PubMed.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- F. Mantei, R. E. Ali, C. Baensch, S. Voelker, P. Haltenort, J. Burger, R.-U. Dietrich, N. von der Assen, A. Schaadt and J. Sauer, et al. , Sustainable Energy Fuels, 2022, 6, 528–549 RSC.
- M. Deitermann, Z. Huang, S. Lechler, M. Merko and M. Muhler, Chem. Ing. Tech., 2022, 94, 1573–1590 CrossRef CAS.
- A. W. Franz, H. Kronemayer, D. Pfeiffer, R. D. Pilz, G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, Formaldehyde in Ullmann’s Encyclopedia of Industrial Chemistry, 2016, pp. 1–34 Search PubMed.
- X. Bao, M. Muhler, B. Pettinger, R. Schlögl and G. Ertl, Catal. Lett., 1993, 22, 215–225 CrossRef CAS.
- X. Bao, J. V. Barth, G. Lehmpfuhl, R. Schuster, Y. Uchida, R. Schlögl and G. Ertl, Surf. Sci., 1993, 284, 14–22 CrossRef CAS.
- M. Qian, M. Liauw and G. Emig, Appl. Catal., A, 2003, 238, 211–222 CrossRef CAS.
- G. I. Waterhouse, G. A. Bowmaker and J. B. Metson, Appl. Catal., A, 2004, 265, 85–101 CrossRef CAS.
- E. Jones and G. Fowlie, J. Appl. Chem., 1953, 3, 206–213 CrossRef CAS.
- H. Schubert, U. Tegtmeyer and R. Schlögl, Catal. Lett., 1994, 28, 383–395 CrossRef CAS.
- J. Thrane, U. V. Mentzel, M. Thorhauge, M. Høj and A. D. Jensen, Catalysts, 2021, 11, 1329 CrossRef CAS.
- S. Ruf, A. May and G. Emig, Appl. Catal., A, 2001, 213, 203–215 CrossRef CAS.
- J. Burre, D. Bongartz and A. Mitsos, Ind. Eng. Chem. Res., 2019, 58, 5567–5578 CrossRef CAS.
- S. Su, P. Zaza and A. Renken, Chem. Eng. Technol., 1994, 17, 34–40 CrossRef CAS.
- F. Eichner, E. Turan, J. Sauer, M. Bender and S. Behrens, Catal. Sci. Technol., 2023, 13, 2349–2359 RSC.
- S. Ruf and G. Emig, Appl. Catal., A, 1997, 161, L19–L24 CrossRef CAS.
- A. E.-A. A. Said and M. N. Goda, Catal. Lett., 2019, 149, 419–430 CrossRef CAS.
- C. L. Childers, H. Huang and C. Korzeniewski, Langmuir, 1999, 15, 786–789 CrossRef CAS.
- T. Haisch, F. Kubannek, C. Haisch, D. W. Bahnemann and U. Krewer, Electrochem. Commun., 2019, 102, 57–62 CrossRef CAS.
- K.-I. Ota, Y. Nakagawa and M. Takahashi, J. Electroanal. Chem. Interfacial Electrochem., 1984, 179, 179–186 CrossRef CAS.
- E. Batista, G. R. P. Malpass, A. d. J. Motheo and T. Iwasita, Electrochem. Commun., 2003, 5, 843–846 CrossRef CAS.
- S. Lai, N. Lebedeva, T. Housmans and M. Koper, Top. Catal., 2007, 46, 320–333 CrossRef CAS.
- J. Baessler, T. Oliveira, R. Keller and M. Wessling, ACS Sustainable Chem. Eng., 2023, 11, 6822–6828 CrossRef CAS.
- K. Sasaki and S. Nagaura, Bull. Chem. Soc. Jpn., 1965, 38, 649–653 CrossRef CAS.
- G. Sundholm, J. Electroanal. Chem. Interfacial Electrochem., 1971, 31, 265–267 CrossRef CAS.
- C. Iwakura, T. Hayashi, S. Kikkawa and H. Tamura, Electrochim. Acta, 1972, 17, 1085–1093 CrossRef CAS.
- G. Belanger, J. Electrochem. Soc., 1976, 123, 818 CrossRef CAS.
- D. M. Roth, M. Haas, A. Echtermeyer, S. Kaminski, J. Viell and A. Jupke, J. Chem. Eng. Data, 2023, 68, 1397–1410 CrossRef CAS.
- A. M. Hofstead-Duffy, D.-J. Chen, S.-G. Sun and Y. J. Tong, J. Mater. Chem., 2012, 22, 5205–5208 RSC.
- D. Y. Chung, K.-J. Lee and Y.-E. Sung, J. Phys. Chem. C, 2016, 120, 9028–9035 CrossRef CAS.
- P. S. Deshpande, V. R. Chaudhari and B. L. Prasad, Energy Technol., 2020, 8, 1900955 CrossRef CAS.
- G. Cassone, F. Pietrucci, F. Saija, F. Guyot and A. M. Saitta, Chem. Sci., 2017, 8, 2329–2336 RSC.
- F. Che, J. T. Gray, S. Ha, N. Kruse, S. L. Scott and J.-S. McEwen, ACS Catal., 2018, 8, 5153–5174 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04978g |
| This journal is © The Royal Society of Chemistry 2024 |