
Cu(II)-catalyzed ‘in-water’ N-arylation of electron-deficient NH-heterocycles†
Steeva
Sunny‡
 ,
Mohit
Maingle‡
,
Loddipalle
Sheeba
,
Firojkhan Rajekhan
Pathan
,
Gowri Sankar
J.
,
Harika
Juloori
,
Sainath Ganesh
Gadewar
and
Kapileswar
Seth
*
,
Mohit
Maingle‡
,
Loddipalle
Sheeba
,
Firojkhan Rajekhan
Pathan
,
Gowri Sankar
J.
,
Harika
Juloori
,
Sainath Ganesh
Gadewar
and
Kapileswar
Seth
*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER)-Guwahati, Sila Katamur, Changsari, Kamrup, 781101, Assam, India. E-mail: kapileswar@niperguwahati.in; kapilseth.niper@gmail.com
First published on 23rd February 2024
Abstract
Cu(II)-catalyzed N-arylation of electron-deficient NH-heterocycles ‘in-water’ at room temperature is described. A wide substrate scope with respect to structural and electronic variation of NH-heterocycles plus arylboronic acids allows product diversity. The functional group tolerance, ‘gram-scale’ synthesis, synthetic elaboration of N-arylated products and late-stage arylation are the distinct advantages.
Introduction
N-containing heterocycles are omnipresent as subunits in many natural products, are present as backbones of several bio-active molecules and exhibit diversified biological activities.1 Several prescribed approved drugs and marketed pharmaceutical agents are enriched with N-heterocycles as essential structural cores.2 Typically, the substitution of a CH-group by an N-atom in (hetero)arene systems may induce vital features in terms of physicochemical properties and molecular interactions of a new therapeutic lead, which can be translated into enhancing pharmacological profiles during the identification of safe and efficacious drug candidates as well as exploring the space of high-quality drugs.3 Thus, synthesis and/or chemical modifications of small molecule N-based heterocycles have received significant attention in drug design, discovery medicinal chemistry and drug development efforts.4Moreover, the N-arylated analogues of NH-heterocycles are prevalent components in numerous medicinal compounds and key building blocks of biologically active natural products and have found numerous applications in medicinal chemistry, pharmaceutical research, and drug discovery.5 Due to a broad range of biological activities and extensive applications in medicinal and pharmaceutical sciences, the N-arylated heterocycles have attracted considerable interest from synthetic organic/medicinal chemists, and the C–N bond constructions have been under continuous investigation in the synthetic chemistry community.6
The copper-mediated oxidative cross-coupling reaction of NH-(hetero)arenes with arylboronic acids, popularly known as Chan–Lam coupling,7 offers a straightforward synthetic method for (hetero)arene N-arylation.8 Recently, copper-catalyzed Chan–Lam coupling has also been explored as a prominent and broadly applicable powerful tool for coupling of N-nucleophiles to access N–C(aryl) bonds.9 The copper-catalyzed Chan–Lam coupling has been extensively studied with electron-rich heteroarenes (e.g., imidazole, benzimidazole, pyrazole, indazole, 1,2,3-benzotriazole),5b,6d,8c,9a,10 primarily because of their high nucleophilicity, which facilitates coordination of N-atom to copper centre. In contrast, the participation of moderate/strong electron-deficient NH-heterocycles in copper-catalyzed Chan–Lam coupling remains elusive10e,11 and only one or two examples have been included for each structural class of NH-heterocycles, such as cyclic amide, imide, cyclic urea, cyclic sulfonamide, cyclic carbamate, and 2,4-thiazolidinedione, under copper-promoted conditions using undesirable hazardous halogenated CH2Cl2 solvent.7a,c,8a,c Perhaps poor nucleophilicity of electron-deficient NH-heteroarenes inhibits easy coordination of N-atom to copper centre and prevents easy reductive elimination of the copper centre at the elementary steps during catalytic turnover. The slow reaction rate of electron-deficient NH-heterocycles eventually necessitates a long duration of reaction (12 h–72 h) under both Cu-catalyzed and Cu-mediated conditions7c,8a,11 and the participation of electron-rich arylboronic acids as coupling partners.8a,11 Furthermore, non-reactivity or poor conversion,7a,c or inadequate examples of moderate or strong electron-deficient arylboronic acids,7a,c,11a lead to a lack of generality in substrate scope, and use of undesirable high-boiling (DMSO)11a or hazardous halogenated solvents (CH2Cl2)7a,c,8a,c,11b further highlight the notable limitations of electron-deficient NH-heterocyclic scaffolds for Chan–Lam coupling. Therefore, the Chan–Lam coupling of electron-deficient NH-heterocycles typically represents a formidable challenge. Thus, there is a need to develop a mild, robust, efficient, and more generalized strategy for Chan–Lam coupling of electron-deficient NH-heterocycles.
The ever-growing demand for the application of green chemistry tools in medicinal chemistry research and chemistry-based organizations urges the use of green reaction media.12 The improvement of reaction methodologies, which is associated with deficiencies in atom economy, cost-effectiveness, safety, poor reactivity, and scalability, drives the working philosophy of medicinal chemistry research to find a better synthetic approach to enrich the medicinal chemist's toolbox.13 In an endeavour toward sustainable practices as part of our synthetic medicinal chemistry program to identify new therapeutic leads via the functionalization of heterocyclic scaffolds, recently, we became interested in exploring the Chan–Lam coupling of electron-deficient bio-relevant NH-heterocycles in a water medium. Growing interest in using water as a preferred non-conventional reaction medium12a,14 in designing green chemical syntheses15 is mirrored by significant efforts in the execution of a variety of organic reactions in an aqueous medium.16 The use of water as a reaction medium replacing common volatile organic solvents (VOSs) can circumvent the environmental non-benignity of VOSs, as these are major contributors to environmental pollution because of their frequent, abundant use (on average contributing 85–90% of the overall mass of a chemical process) as well as incomplete recovery efficacy.17 However, most organic compounds are poorly soluble in water, which may restrict the application of water-mediated organic reactions.18 The solubility issue has been cleverly addressed using surfactants in water-mediated organic reactions, wherein the surfactants generate miceller nanoreactors by self-aggregation in an aqueous medium to entrap organic reactants to promote or accelerate organic transformations.19 The catalytic applications of different transition metals in aqueous media benefitting various organic transformations have been well established,19a,c,20 which prompted us to devise an efficient, mild, and robust strategy for the Chan–Lam coupling of electron-deficient NH-heterocycles. Herein, we describe Cu(II)-catalyzed water-mediated Chan–Lam coupling of electron-deficient biorelevant NH-heterocycles, such as isatin, 4(3H)-quinazolinone, and 2-oxazolidinone, in line with the ‘triple bottom line’ philosophy21 of green chemistry practice.
The isatin, 4(3H)-quinazolinone, and 2-oxazolidinone scaffolds have proved to be privileged structural units present in several naturally occurring compounds having divergent biological and pharmacological properties. For example, the isatin core and many of its derivatives display inhibitory activities, such as those against CNS-MAO B (monoamine oxidase B) and SARS protease, act as GAL3 receptor antagonists, and are present in clinically approved drugs (e.g., sunitinib, semaxanib, etc.) (Fig. 1A).22 The 4(3H)-quinazolinone core is widely distributed as a building block in several synthetic and natural product-based drugs to treat various disease conditions and exhibits a broad spectrum of biological functions, including anti-malarial, anti-cancer, anti-convulsant, anti-inflammatory, anti-diabetic, anti-fungal, kinase inhibitory and many more activities.23 The 2-oxazolidinone nucleus is present as the backbone in medicinally significant molecules expressing a broad range of pharmacological activities, such as HIV-1 protease inhibition, anti-bacterial, MAO inhibition, effective agents in the treatment of bone resorption and osteoporosis, etc.24 The functionalization (e.g., N-arylation) of these bio-relevant NH-heterocycles has, therefore, been desired to broaden the chemical and molecular space25 to identify new therapeutic lead molecules for drug discovery efforts in a medicinal chemistry program.
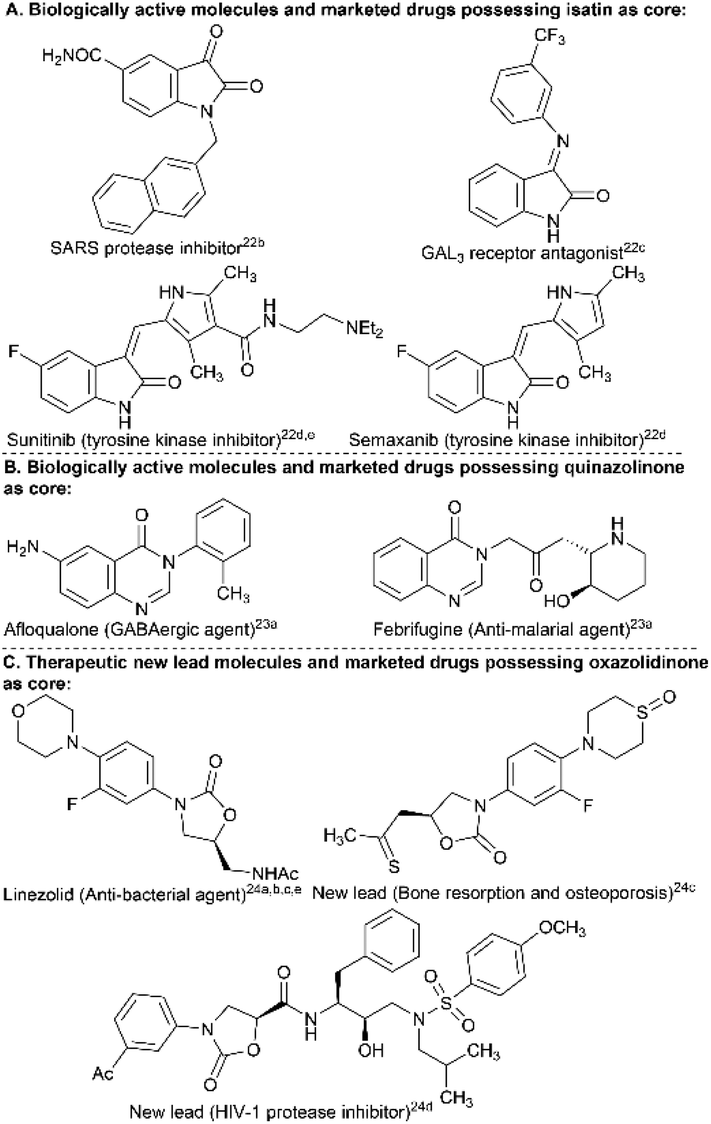 | ||
| Fig. 1 A few selected examples of biologically active molecules, new therapeutic leads and marketed drugs that contain isatin, 4(3H)-quinazolinone, and 2-oxazolidinone as the structural core. | ||
Results and discussion
The popularity of Cu(II)-catalyzed Chan–Lam coupling9–11 prompted us to initiate the investigation by assessing N-arylation in a model study of isatin (1a) (1 equiv.) with phenylboronic acid (2a) (1.5 equiv.) using various Cu-based catalysts applying different conditions in a water medium (Table 1). We started optimization of the reaction conditions by using 1 equiv. of Cu(OAc)2 in the presence of catalytic surfactant sodium dioctylsulfosuccinate (SDOSS) (20 mol%) and Et3N (2 equiv.) as a base in H2O at rt under air, which afforded the desired N-phenylisatin (3a) in 24% isolated yield (entry 1). Reducing the amount of Cu(OAc)2 to a catalytic quantity of 50 and 25 mol% either gave poor yield (15%) of 3a (entry 2) or did not work (entry 3). The N-donor chelating ligands, separately or in combination, are capable of facilitating/accelerating coinage metal-catalyzed N-arylation of NH-heterocycles.9f,26 So, we tested N-chelating ligands, such as ethylenediamine (en) and N,N-dimethyl ethylenediamine (N,N-DMEDA). However, neither en and N,N-DMEDA separately nor a combination of en plus N,N-DMEDA was successful (entries 4 and 5 and ESI†). At this point, we hypothesized enhancing the electrophilicity of the Cu(II) centre, which could have a few advantages. An acidic reaction environment is known to enhance the electrophilic character of a metal centre transiently27 and the application of a strong acid during C–H activation/functionalization to increase the electrophilicity of a metal centre by forming a cationic metal species28via the release of the coordinating acetate ion29 is well documented. Moreover, it was reasoned that tuning the cationicity27–29 of the catalytic Cu(II) centre may endow better reactivity due to the following aspects: (i) the vacant active coordination site around the Cu(II) centre, generated after the dissociation of the coordinating acetate ion, would allow an easy N-atom coordination of poorly nucleophilic 1a circumventing its low reactivity; (ii) the high extent of electronic attraction between the Cu(II) centre and 1a; (iii) accelerated transmetalation with 2a driven by better electronic interaction; (iv) facile reductive elimination at the cationic copper centre for C–N bond formation;30 and (v) potential application of overall mild reaction conditions.29 Attracted by the observations and understanding mentioned above, we added catalytic (20 mol%) CH3CO2H (glacial) to the reaction system, which gave an improved yield (38%) of 3a (entry 6). Increasing the amount of CH3CO2H (40 mol%) was not very effective, and the yield of 3a was 47% (entry 7). However, 60 mol% CH3CO2H further improved the yield of 3a to a decent level (58%) (entry 8). Gratifyingly, replacing CH3CO2H with a stronger acid, CF3CO2H (60 mol%), significantly enhanced the yield of 3a to 86% (entry 9). Poor results were obtained on lowering the quantity of SDOSS and in the absence of it (entries 10 and 11), which suggests a distinct role for SDOSS in the transformation. The reaction failed when the Cu(OAc)2 quantity was decreased to 10 mol% and without it (entries 12 and 13). The catalytic Cu(I) salt (e.g., CuI) and other Cu(II)-based catalysts, such as Cu(acac)2, Cu(BF4)2·xH2O, CuSO4, and CuBr2 either gave poor results (entry 14) or were not effective (entries 15–18). The base Et3N was essential, as otherwise 3a did not form (entry 19). The use of different surfactant, sodium dodecylsulfate (SDS), provided a poor result compared to that with SDOSS (entry 20). Replacing CF3CO2H and Et3N with a weak basic buffer solution NH4OH + NH4Cl (1 equiv. and 2.6 equiv.) proved to be inferior (entries 21 and 22). After careful screening of several reaction parameters, the best reaction conditions was identified as those stated in entry 9, Table 1. The solvent plays a crucial role, and water was the most effective solvent to effectuate the transformation. Other solvents, such as N,N-dimethyl formamide (DMF), N,N-diethyl formamide (DEF), N,N-dimethyl acetamide (DMA), MeOH, nBuOH, and THF were either ineffective or provided inferior results (please see Tables A, B, C, D, E, F, and G in the ESI† for a detailed description of reaction condition optimization).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (X) | Surfactantb (Y) | Additiveb (Z) | Yieldc (%) |
| a 1a (1 mmol) was treated with 2a (1.5 mmol, 1.5 equiv.) under various conditions in the presence of Et3N (2 equiv.) in H2O (2 mL) at rt for 4 h. b The amount used is with respect to 1a. c Isolated yield of 3a. d Both 1a and 2a were recovered. e In the absence of Et3N. f In the absence of CF3CO2H and Et3N. en = ethylenediamnine; N,N-DMEDA = N,N-dimethyl ethylenediamine. | ||||
| 1 | Cu(OAc)2 (100) | SDOSS (20) | None | 24 |
| 2 | Cu(OAc)2 (50) | SDOSS (20) | None | 15 |
| 3 | Cu(OAc)2 (25) | SDOSS (20) | None | 0d |
| 4 | Cu(OAc)2 (25) | SDOSS (20) | en (30) | 0d |
| 5 | Cu(OAc)2 (25) | SDOSS (20) | N,N-DMEDA (30) | 0d |
| 6 | Cu(OAc)2 (25) | SDOSS (20) | AcOH (20) | 38 |
| 7 | Cu(OAc)2 (25) | SDOSS (20) | AcOH (40) | 47 |
| 8 | Cu(OAc)2 (25) | SDOSS (20) | AcOH (60) | 58 |
| 9 | Cu(OAc)2 (25) | SDOSS (20) | CF3CO2H (60) | 86 |
| 10 | Cu(OAc)2 (25) | SDOSS (10) | CF3CO2H (60) | 29 |
| 11 | Cu(OAc)2 (25) | None | CF3CO2H (60) | 19 |
| 12 | Cu(OAc)2 (10) | SDOSS (20) | CF3CO2H (60) | 0d |
| 13 | None | SDOSS (20) | CF3CO2H (60) | 0d |
| 14 | CuI (25) | SDOSS (20) | CF3CO2H (60) | 10 |
| 15 | Cu(acac)2 (25) | SDOSS (20) | CF3CO2H (60) | 0 |
| 16 | Cu(BF4)2·xH2O (25) | SDOSS (20) | CF3CO2H (60) | 0 |
| 17 | CuSO4 (25) | SDOSS (20) | CF3CO2H (60) | 0 |
| 18 | CuBr2 (25) | SDOSS (20) | CF3CO2H (60) | 0 |
| 19 | Cu(OAc)2 (25) | SDOSS (20) | CF3CO2H (60) | 0d,e |
| 20 | Cu(OAc)2 (25) | SDS (20) | CF3CO2H (60) | 63 |
| 21 | Cu(OAc)2 (25) | SDOSS (20) | NH4OH + NH4Cl (100) | 12f |
| 22 | Cu(OAc)2 (25) | SDOSS (20) | NH4OH + NH4Cl (260) | 16f |
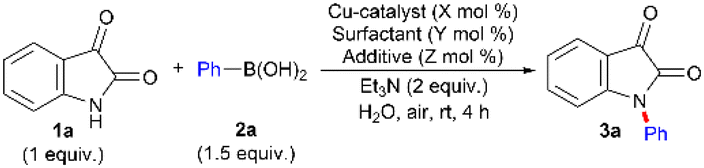
With the optimized reaction conditions in hand, we attempted to generalize the transformation with respect to structural and electronic variations of 1 and 2 (Table 2). Subsequently, the influence of electronic and steric factors of substituents on the aryl ring of both 1 and 2 was assessed. The reaction proceeded smoothly with moderate/strong electron-donating groups (4-Me, 4-OMe, and 3,5-di-OMe), a strong electron-withdrawing group (3-CO2Me), and halogen functionalities (3-Cl, 2-Br) on the aryl unit of 2 affording the corresponding N-arylated products (3b–f, and 3l) in moderate to excellent yields. The survival of sensitive Cl and Br groups on the aryl ring of 2 establishes excellent chemoselectivity of the reaction conditions and provides scope for further synthetic elaboration. Unlike the transition metal-catalyzed amidation of the ester functionality,31 the present catalytic conditions were compatible with the ester group on the aryl ring of 2. The result further highlights the mild, robust, and highly chemoselective nature of the developed catalytic system. Moreover, no ester hydrolysis was observed. The OMe and F groups at the C5-position of 1 also worked well and furnished the N-arylated products (3i, 3k, 3l, and 3n) in synthetically useful yields. Fused π-systems, such as a 2-naphthyl unit on 2, afforded the N-arylated derivative (3g) in an excellent yield. Sterically congested aryl rings of 2, such as 1-naphthyl and ortho-tolyl, participated in the transformation, albeit the product yields being low (3j and 3m). However, the sterically demanding ortho-Br aryl unit of 2 delivered 3e in good yield. Interestingly, the heteroarene, such as the 3-thienyl unit from 2, could also be accommodated as a coupling partner. However, the conversion of the product was less (3h).
a,b
| a Reaction condition: 1 (1 mmol), 2 (1.5 mmol, 1.5 equiv.), Cu(OAc)2 (25 mol%), SDOSS (20 mol%), CF3CO2H (60 mol%), Et3N (2 equiv.) in H2O (2 mL) at rt, 4 h. b Isolated yield of 3. c No proto-debromination was observed. d The figure in parentheses is the total yield based on recovery and reuse of both the unreacted starting materials after the first attempt and further calculated with respect to 1. |
|---|

|
Next, we turned our attention to performing N-arylation of the electron-deficient NH-heterocyclic scaffold quinazolinone (4) with the same optimized reaction condition as stated in entry 9 of Table 1. The Chan–Lam coupling of 4 has been demonstrated under both Cu(II)-promoted32 and Cu(I)-catalyzed33 conditions. The Cu(I)-catalyzed N-arylation of 4 has been performed in high boiling point environmentally non-preferred solvent DMSO,12a,14b,15b requires a long reaction period (16 h–18 h),33 and reveals insufficient substrate scope with limited derivatization.33 The N-phenylation of 4(3H)-quinazolinone (4a) with C-based electrophile iodobenzene has also been reported earlier under Cu(I) catalysis,34 which primarily requires a long period of reaction (24 h) in the presence of environmentally non-benign high boiling point solvent DMF12a,b,14b,15b at 100 °C and demonstrates only one single example of N-arylation of 4a without an attempt to expand the substrate scope study with further derivatization. A wide range of 4 and 2, having variations in their structural and electronic properties, were subjected to N-arylation (Table 3). The reaction conditions worked nicely, irrespective of any prominent electronic bias from the substituents on the aryl ring of both 4 and 2. The corresponding N-arylation products (5a–f, and 5h–j) were obtained in decent yields. Gratifyingly, a heteroarene moiety 3-thienyl ring from 2 also afforded the desired product (5g), although the conversion was less.
a,b
| a Reaction condition: 4 (1 mmol), 2 (1.5 mmol, 1.5 equiv.), Cu(OAc)2 (25 mol%), SDOSS (20 mol%), CF3CO2H (60 mol%), Et3N (2 equiv.) in H2O (2 mL) at rt, 4 h. b Isolated yield of 5. c The figure in parentheses is the total yield based on recovery and reuse of both the unreacted starting materials after the first attempt and further calculated with respect to 4. |
|---|
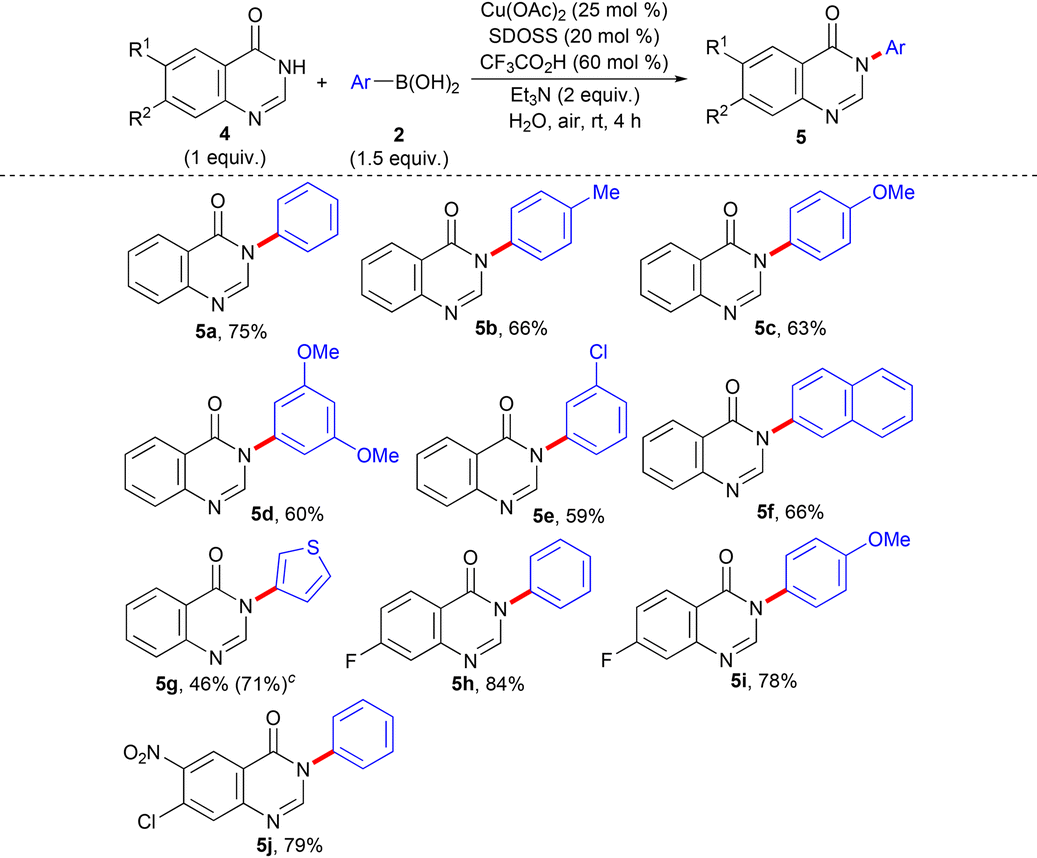
|
Furthermore, we planned to extend the scope of NH-heterocycles, such as the 2-oxazolidinone moiety for N-arylation with 2 under the optimized condition of entry 9, Table 1. An early report describes Cu(I)/1,2-diaminocyclohexane-catalyzed N-arylation of the 2-oxazolidinone scaffold with bromoarenes in refluxing 1,4-dioxane overnight, which exemplifies inadequate substrate scope.5e The Cu(I)-catalyzed N-arylation of structurally related heterocycle 2-pyrrolidinone with iodo-/bromoarenes has also been reported, wherein typically, the 2-oxazolidinone moiety plays a crucial role in the ligand to effectuate the transformation.35 Treating 2-oxazolidinone (6a) with 2a under the established condition of entry 9, Table 1 failed to deliver the desired N-arylation product and both the starting materials 6a and 2a were recovered. Pleasingly, the inclusion of an additional cationic surfactant tetrabutylammonium bromide (TBAB) (20 mol%) with the optimized condition of entry 9, Table 1 provided the expected N-arylation product 7a after 12 h, although the yield was poor (17%). Encouraged by this result we screened different cationic and non-ionic surfactants with varied alkyl chain lengths in combination with equimolar SDOSS to assess their efficiency. After careful screening, we have identified the best optimized condition for N-phenylation of 6a with 2a as Cu(OAc)2 (25 mol%), SDOSS (20 mol%), Tween 80 (20 mol%), CF3CO2H (60 mol%), and Et3N (2 equiv.) in water at room temperature, which afforded 7a in 48% isolated yield after 12 h (please see Table H in the ESI† for the detailed optimization study). Better efficiency of the combination of SDOSS and Tween 80 (20 mol% each) to effectuate the transformation could be attributed to synergistic interactions of anionic and non-ionic binary surfactants driven by the increase in hydrophobicity induced by large alkyl chains (tails), which in turn may enhance the solubility of organic reactants through tuning the property of generated mixed micelles.36 The optimized reaction conditions are viable for the electronically distinct 2 possessing various functionalities for the N-arylation with 6a and afforded the desired products (7a–h) in synthetically acceptable yields (Table 4). The sensitive –OBn group on the aryl unit of 2 survived the reaction conditions. Interestingly, strong electron-deficient aryl rings of 2 having an ester and nitro group participated in the transformation. No hydrolysis was observed for the ester functionality. The results mirror the mild, robust, and dynamic characteristics of the present protocol. Although the reactivity of 6 appears less than 1/4, no homo-coupling of 2 was observed.
a,b,c
| a Reaction condition: 6 (1 mmol), 2 (1.5 mmol, 1.5 equiv.), Cu(OAc)2 (25 mol%), SDOSS (20 mol%), Tween 80 (20 mol%), CF3CO2H (60 mol%), Et3N (2 equiv.) in H2O (2 mL) at rt–50 °C, 12 h. b Isolated yield of 7. c No homo-coupling of 2 was observed. d The figure in parentheses is the total yield based on recovery and reuse of both the unreacted starting materials after the first attempt and further calculated with respect to 6. |
|---|
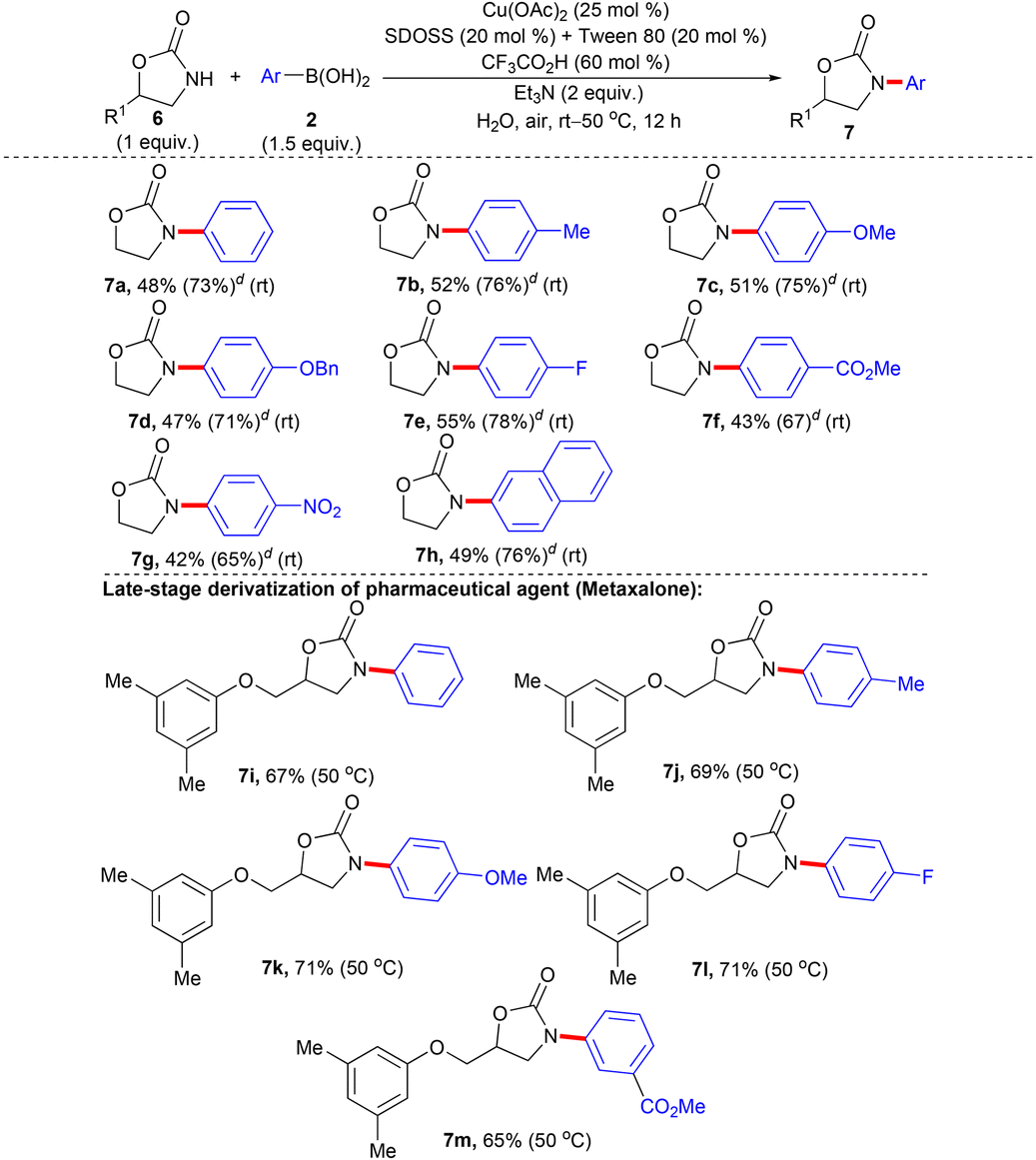
|
Transition metal-catalyzed late-stage functionalization (LSF) of pharmaceutical agents, lead molecules, and bio-active natural products provide an opportunity to rapidly optimize and elaborate their pharmacokinetic and pharmacodynamic properties by synthesizing new analogues and has gained momentum as an increasingly powerful tool for the structural diversification of value-added molecular entities during the drug development program.37,38 Metaxalone, housing 2-oxazolidinone as the core nucleus, has long been identified as a muscle relaxant.39 Gratifyingly, the developed protocol allowed smooth N-arylation of metaxalone with electronically divergent 2 and the corresponding products (7i–m) were delivered in decent yields at 50 °C. The reaction condition were compatible with the ether linkage of metaxalone and the ester group present in the aryl ring of 2. The results reflect the promising scope to streamline late-stage modification of pharmaceuticals, nutraceuticals, and agrochemicals by generating novel valuable derivatives.
The green chemistry metrics, such as atom economy (AE) and environmental factor (E-factor) values of all the synthesized N-arylation derivatives presented in Tables 2–4 have been assessed to evaluate the green credentials (please see Table I, J, and K in the ESI† for detailed descriptions). The AE values were in the range of 78.1–88.6% and the E-factor scores appeared in the range of 2.33–6.03. The calculated high AE value and E-factor count, albeit relatively poor, indicate the present methods as being green and sustainable.40 We observed that the electronic and steric properties of 1, 2, 4, and 6 have been the primary factors controlling their individual reactivity for N-arylation, which along with the homogeneous nature of the catalytic system,40e in turn, governs the AE and E-factor. The relatively poor E-factor score could be mitigated by further customization of the reaction parameters to enhance the reactivity profiles of 1, 2, 4, and 6 in overcoming their kinetic barrier of reactivity.
We have compared the AE value and E-factor score as well as reaction conditions of our method with prior literature reports (please see Tables L–P in the ESI†). Comparable AE values have been obtained in all cases. In most of the cases, our method provided a better E-factor score compared to prior literature reports (Tables L–N, and Table P†), except in one case, in which the E-factor count of our method was inferior (Table O†). However, the prior reports (Tables L–P†) are associated with the requirement of stoichiometric Cu(OAc)2 (1 equiv.), undesirable hazardous halogenated solvent (CH2Cl2), hazardous high-boiling point solvent (DMSO), and a long period of reaction (18 h–72 h). Thus, an overall consideration of all the aspects endorses our method as a more ‘green technology’.41
The catalytic conditions could be readily applied to a ‘gram-scale’ reaction of 1a (1.47 g) with 2a to prepare 3a in 84% isolated yield (Scheme 1A). The result proves the scalability, reproducibility, and potential practical utility of the developed protocol of N-arylation. Furthermore, compound 3e was synthetically modified by introducing a phenyl unit at the ortho C–Br bond following the Suzuki cross-coupling reaction with 2a, which furnished ortho-heterocycle-tethered biphenyl system 8a in 40% isolated yield (Scheme 1B). Moreover, 3a was further derivatized at the C3-position through the nucleophilic addition of the −CH2CN moiety, generated via the sp3C–H activation of acetonitrile 9, under Pd(OAc)2-catalyzed condition42 and the corresponding product 10a was obtained in good yield (69%) (Scheme 1C). These outcomes reflect the possible usefulness of the developed N-arylation method, allowing further synthetic elaboration.
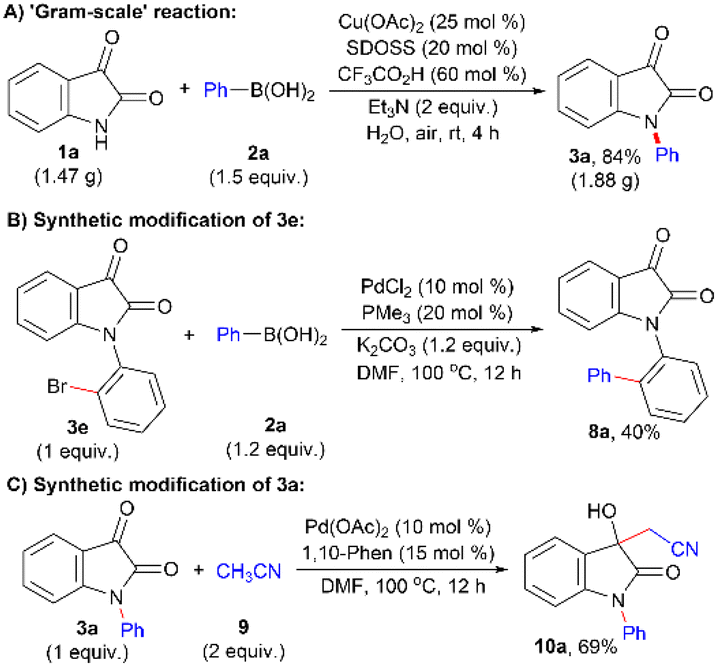 | ||
| Scheme 1 (A) ‘Gram-scale’ production of 3a; (B) derivatization of 3e with 2a to afford 8a; and (C) derivatization of 3a with 9 to afford 10a. | ||
Control experiments were conducted to probe the effect of various oxidants on the reaction outcome because the Chan–Lam coupling between two nucleophiles, N-based and C-based, under the catalytic influence of Cu(II) salts typically relies on an oxidative environment (air or O2).8c,9c,f,10g,11b,43 SDOSS in catalytic quantities is well known to activate aerial oxygen through the oxygen reuptake mechanism facilitating the oxidation process in water-mediated organic transformation44 to promote green aerobic oxidation practice. Therefore, we first investigated the effect of aerial oxidation, O2 balloon and N2 atmospheres during the reaction of 1a with 2a (entries 1–3, Table 5). While aerial oxidation and the use of an O2 balloon gave comparable results having higher conversions of 3a, the use of an N2 balloon provided poor results and established the indispensable role of the oxidative environment (aerial oxidation) for the transformation. We tested several other metal-based and non-metal-based oxidants (entries 4–7, Table 5 and Table G in the ESI†). Water-soluble oxidant, oxone, completely converted 2a to phenol45 (isolated in 96% yield) and thereby, did not allow 2a to further participate in the catalytic cycle (entry 4). H2O2 and tBuOOH gave inferior results as an oxidant (entries 5 and 6). Other toxic and corrosive stoichiometric non-metallic oxidants or costly metallic oxidants either failed or provided inferior results compared to aerial oxidation. Thus, use of catalytic SDOSS to dissolve aerial O2 in water, effectuating the transformation, displays superior performance and is seemingly competent.
a
|
|
||
|---|---|---|
| Entry | Oxidantb | Yieldc (%) |
| a 1a (1 mmol) was treated with 2a (1.5 mmol, 1.5 equiv.) in the presence of Cu(OAc)2 (25 mol%), SDOSS (20 mol%), CF3CO2H (60 mol%), Et3N (2 equiv.) under different oxidative environments in H2O (2 mL) at rt for 4 h. b The amount used is with respect to 1a. c Isolated yield of 3a. d Performed in a closed vessel. e Phenol was isolated in 96% yield. | ||
| 1 | Aerial oxidation | 86 |
| 2 | O2 balloon | 88 |
| 3 | N2 balloon | 10 |
| 4 | Oxone | 0d,e |
| 5 | H2O2 | 24d |
| 6 | t BuOOH | 21d |
| 7 | Ag2O | 0d |

Based on the control experiments and prior literature report,46 we have presented a plausible mechanism of coupling 1a with 2a to afford 3a (Scheme 2). Cu(OAc)2 may exist in equilibrium with the dimeric state I, which in turn may equilibrate with multinuclear forms as a resting state/reservoir.46 At first, Et3N abstracts the N–H proton from poorly nucleophilic 1a, converting it to the anionic form, which is a better nucleophile and further denucleates I to monomeric Cu(II) complex IIvia N-coordination to the Cu(II) centre.46II then undergoes an easy oxidation to putative Cu(III) species III under aerial oxidation,47 facilitated by a decrease in the reduction potential of the Cu(III)/Cu(II) couple48 following N-coordination from 1a. Transmetalation of the phenyl moiety from the ate species492a-I, generated from 2a aided by Et3N,46 to species III provides another Cu(III) species IV. A facile (Ph)C–N bond-forming reductive elimination from the high-valent Cu(III) centre50 of IV delivers the N-arylation product 3a along with Cu(I) species V. Finally, reoxidation of the Cu(I) centre of species V, assisted by Et3N under aerobic condition and promoted by 1a, regenerates the Cu(II) complex46II, thereby, the catalytic cycle closes and complex II is available for the next run.
 | ||
| Scheme 2 Proposed catalytic cycle of coupling of 1a and 2a providing 3a. | ||
Moreover, the formed B(OH)3 in the elementary step (transmetalation) may further speed up the reoxidation of Cu(I) to Cu(II) in the presence of Et3N and AcOH.46 Perhaps, during the oxidation of Cu(II) to Cu(III), hydrogen peroxide could also be generated in the catalytic step, which may decompose 2avia oxidation,51 and accounts for the requirement of 1.5 equivalents of 2 for a better conversion of the desired product. The better reactivity of the Chan–Lam coupling in the presence of Et3N is well known.7a,c,47 The profound effect of Et3N in the proposed catalytic cycle (Scheme 2) indicates that Et3N eventually could play several fundamental roles: (i) abstracts the N–H proton from 1a, rendering it more nucleophilic and favouring the denucleation of dimeric I by N-coordination to the Cu(II) centre; (ii) abstracts the O–H proton from 2a forming an ate species 2a-I and assisting in the effective transmetalation of the aryl unit; (iii) sequesters AcOH preventing reformation of dimeric I and other multinuclear catalytically inactive species; (iv) the resulting salt (Et3N·AcOH) offers H+ and OAc− boosting the reoxidation of the Cu(I) centre to Cu(II).46 Similarly, the amine substrate 1a helps in (i) the denucleation of dimeric I to initiate the catalytic turnover and (ii) promotes reoxidation of the Cu(I) centre.46 However, AcOH may play dual roles: (i) a detrimental effect by forming dimeric I and related catalytically incompetent species; and (ii) beneficial effects on the reoxidation of Cu(I) to Cu(II); and furthermore (iii) stabilizing the high-valent Cu(III) species52III in the form of an ancillary ligand.53 Possibly, catalytic CF3CO2H (i) promotes denucleation of Ivia the dissociation of the coordinating acetate ion29 and enhances the cationicity27,28 of the Cu(II) centre, enabling the facile coordination of the NH-heterocycle, and (ii) may further stabilize the Cu(III) centre in III.
Water plays an indispensable role in performing this oxidative coupling reaction, otherwise the transformation either fails or gives inferior results in different organic solvents. While the role of water in promoting organic reactions is yet to be fully understood, various factors, such as the high cohesive energy density of water, enforced hydrophobic interactions, and hydrogen bonding (H-bonding) effect, could be envisaged as the unique features of water to accelerate the rate of an organic reaction.54 The H-bonding effect has popularly been referred to as one of the factors for the rate enhancement of organic reactions in water originating from interfacial interactions of organic molecules with free hydroxyl groups of water molecules.55 Thus, the benefit of the water medium could be anticipated to form an H-bond with the –OH group of 2a, rendering the O–H proton more acidic, enabling its facile abstraction by Et3N, and subsequent stabilization of the resulting ate species 2a-I, via an H-bonding interaction with O− atom, which in turn enhances the rate of phenyl unit transfer in the transmetalation step. Overall, the H-bond-promoted supramolecular assembly involving the reactant and water molecule(s) could be inferred as a favourable factor.20b,56
Apart from the solubilizing aid of organic molecules within a miceller environment accounting for ‘in-water’ catalysis,19a,c,54a the specific role of Tween 80 could be correlated with its H-bond donor (through terminal free –OH groups) and H-bond acceptor (through polyoxyethylene ethereal O-atoms and carbonyl O-atom) abilities. Moreover, it could be expected that the Tween 80 molecule may experience H-bonding interactions with the dangling –OH groups55b of the interfacial water molecule(s) and further encapsulate and activate 2/6 through H-bonding interactions, resulting in better reactivity.
The hydrotropic effect of monoalkylglycerol ether molecules, controlled by the non-polar alkyl chain length, governs the hydrophobic interaction between a hydrotrope and solute and is the driving force of strong agglomeration of the hydrotrope around a solute in the water medium.57 The hydrotropic effect of catalytic Tween 80 in solubilizing 2 and 6 in the water medium cannot be excluded.
Conclusions
In summary, this work describes Cu(II)-catalyzed simple, mild, powerful, and efficient N-arylation of poorly nucleophilic electron-deficient NH-heterocycles with arylboronic acids ‘in-water’ at room temperature. The catalytic effects of CF3CO2H and SDOSS/Tween 80, stoichiometric Et3N, along with water molecule(s) have been found crucial for the coupling, and each component plays an indispensable role via synchronous interplay for an effective, sustainable transformation. The versatility and robustness of the protocol are established by accommodating different variations of (i) electron-deficient NH-heterocycles, (ii) arylboronic acids of moderate/strong electron-rich plus moderate/strong electron-deficient characteristics, (iii) execution of the reaction in an environmentally benign water medium, (iv) compatibility of sensitive functional groups, which illustrates notable advantages in diversity generation, (v) excellent chemoselectivity, (vi) high reaction rate, and (vii) operational simplicity mostly at room temperature. The promising practical applicability has been demonstrated through a ‘gram-scale’ synthesis, further synthetic manipulations of the resulting N-arylated scaffolds, and facile late-stage arylation of a pharmaceutical agent. Therefore, this strategy is expected to broaden the chemical, molecular and high-quality drug space by synthesizing small molecule-focused libraries58 following sustainable medicinal chemistry practice.59Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S., M. M., L. S., F. R. P., G. S. J., H. J., and S. G. G. thank the Department of Pharmaceuticals (DoP), Govt. of India for their fellowships.References
- Selected representative examples:
(a) A. N. Kim, A. Ngamnithiporn, E. Du and B. M. Stoltz, Chem. Rev., 2023, 123, 9447 CrossRef CAS PubMed
; (b) S. Chahal, P. Rani, Kiran, J. Sindhu, G. Joshi, A. Ganesan, S. Kalyaanamoorthy, Mayank, P. Kumar, R. Singh and A. Negi, ACS Omega, 2023, 8, 17446 CrossRef CAS PubMed
- Selected representative examples:
(a) J. Shearer, J. L. Castro, A. D. G. Lawson, M. MacCoss and R. D. Taylor, J. Med. Chem., 2022, 65, 8699 CrossRef CAS PubMed
-
(a) H. Valluri, A. Bhanot, S. Shah, N. Bhandaru and S. Sundriyal, J. Med. Chem., 2023, 66, 8382 CrossRef CAS PubMed
-
(a) K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed
- Selected representative examples:
(a) A. Kumar, A. K. Singh, H. Singh, V. Vijayan, D. Kumar, J. Naik, S. Thareja, J. P. Yadav, P. Pathak, M. Grishina, A. Verma, H. Khalilullah, M. Jaremko, A.-H. Emwas and P. Kumar, Pharmaceuticals, 2023, 16, 299 CrossRef CAS PubMed
-
(a) P. Oeser, J. Koudelka, A. Petrenko and T. Tobrman, Molecules, 2021, 26, 5079 CrossRef CAS PubMed
-
(a) D. J. Cundy and S. A. Forsyth, Tetrahedron Lett., 1998, 39, 7979 CrossRef CAS
-
(a) D. N. Shinde, R. Trivedi, J. V. S. Krishna, L. Giribabu, B. Sridhar, P. S. Khursade and R. S. Prakasham, New J. Chem., 2018, 42, 12587 RSC
-
(a) J.-Q. Chen, J.-H. Li and Z.-B. Dong, Adv. Synth. Catal., 2020, 362, 3311 CrossRef CAS
- Selected representative examples:
(a) B. Adhikari, M. Teimouri, J. W. Akin, S. Raju, S. L. Stokes and J. P. Emerson, Eur. J. Org. Chem., 2023, e202300620 CrossRef CAS
-
(a) W. Gao, F. Gao, H. Yang, Z. Wang and Y. Huang, Catalysts, 2023, 13, 1060 CrossRef CAS
-
(a) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31 RSC
-
(a) J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug Discovery, 2018, 17, 709 CrossRef PubMed
-
(a) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237 RSC
-
(a) F. Zhou, Z. Hearne and C.-J. Li, Curr. Opin. Green Sustainable Chem., 2019, 18, 118 CrossRef
- Selected representative examples:
(a) M.-Z. Lu and T.-P. Loh, Acc. Chem. Res., 2024, 57, 70 CrossRef CAS PubMed
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS
- L. Chen, S. Zhang, X. Liu and X. Ge, Curr. Opin. Colloid Interface Sci., 2023, 65, 101691 CrossRef CAS
-
(a) B. H. Lipshutz, S. Ghorai and M. Cortes-Clerget, Chem. – Eur. J., 2018, 24, 6672 CrossRef CAS PubMed
- Selected representative examples:
(a) D. P. Satpute, G. N. Vaidya, S. K. Lokhande, S. D. Shinde, S. M. Bhujbal, D. R. Chatterjee, P. Rana, A. Venkatesh, M. Nagpure and D. Kumar, Green Chem., 2021, 23, 6273 RSC
- P. Tundo, P. Anastas, D. S. Black, J. Breen, T. Collins, S. Memoli, J. Miyamoto, M. Polyakoff and W. Tumas, Pure Appl. Chem., 2000, 72, 1207 CrossRef CAS
- CNS-MAO B:
(a) S. N. Pandeya, S. Smtha, M. Jyoti and S. K. Sridhar, Acta Pharm., 2005, 55, 27 CAS
-
(a) P. S. Auti, G. George and A. T. Paul, RSC Adv., 2020, 10, 41353 RSC
-
(a) C. Foti, A. Piperno, A. Scala and O. Giuffrè, Molecules, 2021, 26, 4280 CrossRef CAS PubMed
-
(a) T. Hoffmann and M. Gastreich, Drug Discovery Today, 2019, 24, 1148 CrossRef CAS PubMed
-
(a) K. K. Gurjar and R. K. Sharma, Heliyon, 2020, 6, e03233 CrossRef PubMed
-
(a) M. Maingle, S. Sunny, L. Sheeba, F. R. Pathan and K. Seth, Synthesis, 2024, 56, 312 CrossRef CAS
- C. E. Houlden, M. Hutchby, C. D. Bailey, J. G. Ford, S. N. G. Tyler, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 1830 CrossRef CAS PubMed
- T. Nishikata, A. R. Abela, S. Huang and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 4978 CrossRef CAS PubMed
- Redox ligand assisted reductive elimination: J. Jacquet, P. Chaumont, G. Gontard, M. Orio, H. Vezin, S. Blanchard, M. D.-E. Murr and L. Fensterbank, Angew. Chem., Int. Ed., 2016, 55, 10712 CrossRef CAS PubMed
-
(a) J. Buchspies, M. M. Rahman and M. Szostak, Molecules, 2021, 26, 188 CrossRef CAS PubMed
- K. Sreeramamurthy, E. Ashok, V. Mahendar, G. Santoshkumar and P. Das, Synlett, 2010, 721 CAS
- K. A. Kumar, P. Kannaboina, C. K. Jaladanki, P. V. Bharatam and P. Das, ChemistrySelect, 2016, 3, 601 CrossRef
- G. N. Vaidya, A. Khan, H. Verma, S. Kumar and D. Kumar, J. Org. Chem., 2019, 84, 3004 CrossRef CAS PubMed
- H. C. Ma and X. Z. Jiang, Synlett, 2008, 1335 CAS
-
(a) X. Cui, Y. Jiang, C. Yang, X. Lu, H. Chen, S. Mao, M. Liu, H. Yuan, P. Luo and Y. Du, J. Phys. Chem. B, 2010, 114, 7808 CrossRef CAS PubMed
- LSF via cross-coupling:
(a) T. Zhou, P.-P. Xie, C.-L. Ji, X. Hong and M. Szostak, Org. Lett., 2020, 22, 6434 CrossRef CAS PubMed
- Selected representative examples of LSF via C–H functionalization:
(a) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692 CrossRef CAS PubMed
- J. F. Kurtzke and J. Gylfe, Neurology, 1962, 12, 343 CrossRef CAS PubMed
-
(a) N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680 RSC
- P. Jessop, Green Chem., 2020, 22, 13 RSC
-
(a) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163 CrossRef CAS PubMed
-
(a) M. T. Wentzel, J. B. Hewgley, R. M. Kamble, P. D. Wall and M. C. Kozlowski, Adv. Synth. Catal., 2009, 351, 931 CrossRef CAS
- N. Parikh, D. Kumar, S. Raha Roy and A. K. Chakraborti, Chem. Commun., 2011, 47, 1797 RSC
- K. S. Webb and D. Levy, Tetrahedron Lett., 1995, 36, 5117 CrossRef CAS
- J. C. Vantourout, H. N. Miras, A. Isidro-Llobet, S. Sproules and A. J. B. Watson, J. Am. Chem. Soc., 2017, 139, 4769 CrossRef CAS PubMed
- D. A. Evans, J. L. Katz and T. R. West, Tetrahedron Lett., 1998, 39, 2937 CrossRef CAS
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1 CrossRef CAS
- Pd-catalysis:
(a) K. Seth, P. Purohit and A. K. Chakraborti, Org. Lett., 2014, 16, 2334 CrossRef CAS PubMed
-
(a) K. L. Vikse and P. Chen, Organometallics, 2015, 34, 1294 CrossRef CAS
- M. F. Lappert, Chem. Rev., 1956, 56, 959 CrossRef CAS
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed
- B. S. Billow, T. J. McDaniel and A. L. Odom, Nat. Chem., 2017, 9, 837 CrossRef CAS PubMed
-
(a)
V. M. Kolb, Green organic chemistry and its interdisciplinary applications, CRC Press, Boca Raton, Taylor and Francis, 2016 Search PubMed
-
(a) Y. Zheng and J. Zhang, ChemPhysChem, 2010, 11, 65 CrossRef CAS PubMed
- Selected representative examples:
(a) D. N. Kommi, D. Kumar, K. Seth and A. K. Chakraborti, Org. Lett., 2013, 15, 1158 CrossRef CAS PubMed
-
(a) D. O. Abranches, J. Benfica, B. P. Soares, A. Leal-Duaso, T. E. Sintra, E. Pires, S. P. Pinho, S. Shimizu and J. A. P. Coutinho, Chem. Commun., 2020, 56, 7143 RSC
- T. W. J. Cooper, I. B. Campbell and S. J. F. Macdonald, Angew. Chem., Int. Ed., 2010, 49, 8082 CrossRef CAS PubMed
-
(a) C. T. A. Moermond, N. Puhlmann, A. R. Brown, S. F. Owen, J. Ryan, J. Snape, B. J. Venhuis and K. Kümmerer, Environ. Sci. Technol. Lett., 2022, 9, 699 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc05163c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |