
Sustainable valorization of cherry (Prunus avium L.) pomace waste via the combined use of (NA)DESs and bio-ILs†
Angelica
Mero
 a,
Andrea
Mezzetta
ab,
Marinella
De Leo
*abc,
Alessandra
Braca
abc and
Lorenzo
Guazzelli
*ab
a,
Andrea
Mezzetta
ab,
Marinella
De Leo
*abc,
Alessandra
Braca
abc and
Lorenzo
Guazzelli
*ab
aDepartment of Pharmacy, Via Bonanno 33, 56126, Pisa, Italy. E-mail: lorenzo.guazzelli@unipi.it; marinella.deleo@unipi.it
bCentre for Instrumentation Sharing, University of Pisa (CISUP), Lungarno Pacinotti 43, 56127 Pisa, Italy
cInterdepartmental Research Center Nutrafood “Nutraceuticals and Food for Health”, University of Pisa, Via del Borghetto 80, 56124 Pisa, Italy
First published on 12th April 2024
Abstract
Sweet cherry pomace waste (CPW) obtained after juice production still contains high amounts of dietary fiber and added value compounds such as polyphenols. For this reason, it can be employed as renewable feedstock to produce novel commodities, bioactive compounds and bio-functional materials. In this study, a sustainable biorefinery approach has been developed combining the use of natural deep eutectic solvents (NADESs) for the recovery of polyphenolic compounds and bio-based ionic liquids (bio-ILs) for the treatment and conversion of the remaining lignocellulosic residue into an ionogel. The extraction efficiency and selectivity of different classes of (NA)DESs were evaluated varying both the hydrogen bond acceptor, HBA (choline chloride (ChCl), betaine (Bet) and L-proline (Pro)) and the hydrogen bond donor, HBD (carboxylic acids and polyols). The extracted phenolic compounds were qualitatively and quantitatively analyzed by UHPLC-HR-ESI-MS leading to the identification of more than 25 compounds, classifiable into four main subclasses (phenolic acids, flavonoid glycosides, flavonoid aglycones, and anthocyanins). The majority of ChCl-based DESs improved the total phenol extraction compared to classical solvents (up to twice). ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylene glycol 1:2 was identified as the best performing system in terms of total phenolic content (total phenol 759 ± 85 μg g−1 of dried CPW) and was employed as the extraction medium for further optimization of the operative conditions and recycling study. The solvent was quantitatively recovered for three cycles with retention of the extraction efficiency towards all the identified subclasses of phenols. Finally, cholinium arginate (ChArg) allowed for obtaining cellulose enriched material (35 wt% of dried biomass) that was subsequently dissolved at 2 wt% in cholinium levulinate (ChLev) to prepare a weak physical ionogel, a functional biomaterial potentially useful in medical and pharmaceutical sectors.
:ethylene glycol 1:2 was identified as the best performing system in terms of total phenolic content (total phenol 759 ± 85 μg g−1 of dried CPW) and was employed as the extraction medium for further optimization of the operative conditions and recycling study. The solvent was quantitatively recovered for three cycles with retention of the extraction efficiency towards all the identified subclasses of phenols. Finally, cholinium arginate (ChArg) allowed for obtaining cellulose enriched material (35 wt% of dried biomass) that was subsequently dissolved at 2 wt% in cholinium levulinate (ChLev) to prepare a weak physical ionogel, a functional biomaterial potentially useful in medical and pharmaceutical sectors.
Introduction
Sweet cherries (Prunus avium L.) with a global production of around 4.0 million tons per year1 are one of the most appreciated and eaten fruits by consumers due to their organoleptic properties and nutritious nature.2 Indeed, cherries contain a wealth of essential nutrients and biologically active molecules such as sugars, organic acids, carotenoids, melatonin, vitamin C, minerals, fibers and, most of all, high amount of phenolic compounds including anthocyanins, flavonoids, catechin and epicatechin, procyanidins and phenolic acids such as hydroxycinnamic acids.3–6 Due to the short life (7–10 days) of sweet cherries, fresh fruits are widely processed into marmalades, juices or fruit cocktails and other food products7 with the concomitant production of large volumes of waste which represent a potential source of high added value bioactive compounds. The most abundant components of these agri-food derived waste are all the typical dietary fibers such as cellulose, hemicellulose, pectin, and lignin.8 Thus, following the concept of the circular economy, these wastes could be also used as valuable renewable feedstock for the production of novel commodities that are alternatives to fossil-based products. Nowadays, the extraction of phenolic compounds with high antioxidant capacity from sour cherry pomace has been much investigated9–11 while there are very few studies about bioactive compounds in processed sweet cherries12 or pomace wastes. Furthermore, the employment of green solvents instead of classical toxic organic solvents outlines one of the main challenges in the field of extraction processes or biomass treatments. Among the different sustainable extraction solvents already proposed in the literature, (natural) deep eutectic solvents ((NA)DESs) play a prominent role. They are obtained by simple mixing of two or more components that are able to establish hydrogen bonds and thus form a homogeneous mixture for which the eutectic temperature is lower than that of an ideal eutectic.13 (NA)DESs possess a lot of interesting features such as very simple preparation without the need of purification steps and the possibility of designing their properties by means of pairing the proper hydrogen bond acceptor (HBA) and hydrogen bond donor (HBD).14–16 The employment of natural or natural-derived compounds such as choline chloride (ChCl), amino acids, sugars, carboxylic acids, and other primary metabolites allows for the preparation of completely natural, biodegradable and generally low toxic solvents.17,18 Another intriguing property of (NA)DESs is related to their great solvation power towards several classes of compounds giving them the applicability as extraction media in several fields. In particular, DESs, especially ChCl-based DESs, have shown great potential towards the extraction of bioactive compounds from disparate food products or wastes.19–24 However, regarding cherry pomace, only one example of polyphenol extraction from sour cherry pomace using NADESs has been proposed to date25 while no works on sweet cherry pomace and DESs have been reported so far. Before the discovery of DESs, ionic liquids (ILs) had been introduced as the first class of designer solvents thanks to the possibility of pairing a wide variety of cations and anions. ILs exhibit several appealing and tunable properties (negligible vapour pressure, high chemical and thermal stability, wide electrochemical range, and non-flammability) which make them suitable for a large range of applications.26 One of the most appreciated properties of ILs is their capability for dissolving a wide range of materials, most importantly polymeric compounds such as cellulose, hemicellulose and lignin that are difficult to solubilize with classical organic solvents. In particular, these solvents are capable of fractionating biomass, selectively dissolving and so extracting one of the components.27,28 This allows for better valorization of each component, especially regarding the cellulosic part, that can be derivatized to yield various useful novel cellulose-based products, i.e. gel-like materials (ionogels, hydrogels, aerogels, etc.).29 Among them, ionogels are gel materials whose liquid phase is an IL while the matrix can be organic, inorganic, or hybrid organic–inorganic possessing a broad range of uses such as in electrochemical devices, energy storage, sensors and biosensors,30,31 biomedical materials, catalysts etc.32,33 In particular, cellulose is an excellent working building block for the preparation of ionogels due to the presence of three hydroxyl groups in each glucose unit. It can be used without any alterations or as modified by introducing functional groups for the formation of both physical or chemical ionogels.34 ILs containing natural or bio-based ions, so-called bio-ILs, have been successfully tested in fractionation processes of lignocellulosic biomass27 or in the preparation of new bio-materials starting from commercial microcrystalline cellulose (MCC).34–36 To the best of our knowledge, while some examples about the preparation of aerogels or hydrogels via ILs starting from waste materials like waste newspapers37 or hybrid poplar and biomass sorghum bagasse samples38 or waste bamboo-based disposable paper cups39 have been reported, only one work showed the preparation of an ionogel using cellulose-rich solids using a biorefinery approach. In this scenario, the development of a two-step biorefinery process for the valorisation of cherry pomace waste was the main objective of the present study (Fig. 1). (i) In the first part of the study, the effect of HBA (choline chloride, betaine, and proline) and HBD (ethylene glycol, glycerol, oxalic acid, malic acid, and citric acid) DES components on the extraction performance of high-value bioactive compounds and on the composition of the extracts was investigated. In this step, operating conditions were optimized for the best performing system in terms of the total amounts of polyphenols extracted. (ii) In the second part, the insoluble residue from the extraction process was fractionated using cholinium arginate (ChArg), and the cellulose-rich part obtained was further used for the preparation of an ionogel using cholinium levulinate (ChLev). The proposed biorefining process aims to valorise a waste product of the food industry exploiting solvents derived from natural sources and endowed with low environmental impact. | ||
| Fig. 1 Scheme of the developed biorefinery approach aimed at the complete valorization of cherry pomace waste. | ||
Experimental
Materials and methods
UHPLC grade acetonitrile, formic acid, and water were purchased from Romil-Deltek (Pozzuoli, Italy). Betaine anhydrous (>97%) and choline hydroxide in water solution (47–50%) were purchased from TCI Europe (Zwijndrecht, Belgium). Choline chloride (98%), L-proline (99%), oxalic acid dihydrate (98%), citric acid (99%), DL-malic acid (98%), ethylene glycol (99%), levulinic acid (98%) were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, USA). L-Arginine (≥98%), methanol, n-BuOH, glycerol (99%) and Amberlite XAD-7 resin were purchased from Merck KGaA (Darmstadt, Germany). Chlorogenic acid (≥95%) and rutin (>98%) used as reference standards were purchased from Extrasynthese (Genay, France), while naringenin (≥95%) and cyanidin 3-O-glucoside (≥98%) were from Merck KGaA (Darmstadt, Germany).The cherry pomace waste (CPW) was provided by “ILVO” (Flanders Research Institute for Agriculture, Fisheries and Food, Merelbeke, Belgium) which destoned the cherries with a cherry destoner of Enotecnica Pillan and pressed them with the VaculIQ for the production of cherry juice. Before using, the pomace was dried at 40 °C in an oven for 48 h and was ground using an MF 10 basic microfine grinder drive (IKA-Werke, Staufen, Germany) with a 0.25 mm sieve hole.
Preparation of classical extracts
The extraction with a classical solvent was performed by dynamic maceration (150 rpm) of 1 g of dried CPW in 10 g of MeOH for 1 h. For LC-MS analyses, the methanolic extract was first partitioned between n-BuOH and H2O to remove unwanted metabolites (e.g., sugars). After centrifugation (2710g for 5 min), the n-butanolic phase was dried under vacuum to obtain a residue, and subsequently used for LC-MS analyses.Anthocyanins were extracted by dynamic maceration (150 rpm) of 1 g dried CPW in 6 mL of 2% HCl methanol solution for 1 h. The extract solutions were then centrifuged (2710g for 5 min) and the obtained supernatants injected into the LC-MS system.
Preparation of (NA)DESs
Before preparation, choline chloride (ChCl), ethylene glycol (EG), and glycerol (Gly) were dried under vacuum for 6 h at 80 °C. The (NA)DESs were prepared by mixing the different HBAs (ChCl, Bet or L-Pro) and the corresponding HBD (carboxylic acid or polyol) at certain molar ratios, as shown in Table 1. The mixtures were stirred at the appropriate temperature (80 °C for DL-malic and citric acid-based DESs, 40 °C for oxalic acid-based DES and room temperature for all the polyol-based DESs) until a homogeneous transparent liquid was formed. After the (NA)DES formation, no purification step was needed, and the solvents were kept at room temperature in sealed vessels until their use. The DESs with a carboxylic acid as the HBD were mixed with water at 25 wt% water in order to reduce their viscosity. 1H-NMR spectra (D2O) were obtained to assess the composition and the purity of the prepared DESs (Fig. S2–S12†).| Name | HBA | HBD | Molar ratio | T (°C) |
|---|---|---|---|---|
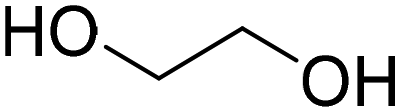
| ChCl:EG (choline chloride:ethylene glycol) |
|
1:2 |
25 | |
| ChCl:Gly (choline chloride:glycerol) |

|
1:2 |
25 | |
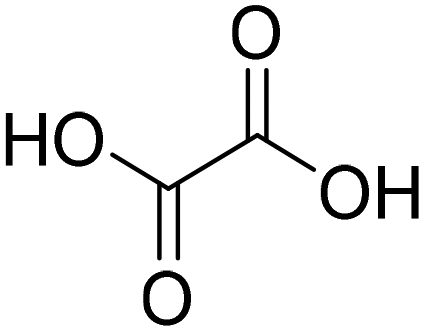
| ChCl:OA + 25% H2O (choline chloride:oxalic acid) |

|
1:1 |
40 | |
| ChCl:MA + 25% H2O (choline chloride:malic acid) |

|

|
1:1 |
80 |
| ChCl:CA + 25% H2O (choline chloride:citric acid) |

|
1:1 |
80 | |
| Bet:EG (betaine:ethylene glycol) |

|

|
1:3 |
25 |
| Bet:Gly (betaine:glycerol) |

|
1:2 |
25 | |
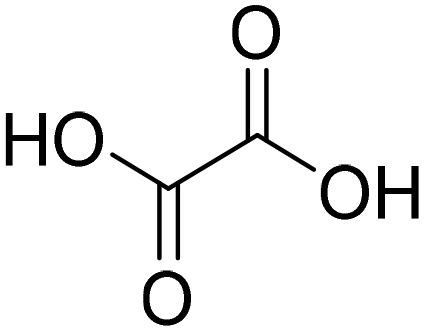
| Bet:OA + 25% H2O (betaine:oxalic acid) |
|
1:1 |
80 | |
|
L-Pro:EG (L-proline:ethylene glycol) |

|

|
1:3 |
25 |
|
L-Pro:Gly (L-proline:glycerol) |

|
1:2 |
25 | |
|
L-Pro:OA + 25% H2O (L-proline:oxalic acid) |
|
1:1 |
80 |
Synthesis of bio-ILs
 | ||
| Fig. 2 Chemical structure of the employed bio-ILs. | ||
Preparation of (NA)DES extracts
The extraction with (NA)DES media was performed in a round-bottomed glass flask by the stirring-heating method as reported in our previous works with slight modifications.22,43 Briefly, for the initial screening of the best performing system, fixed conditions were applied: 1 g of dried CPW was treated with 10 g of (NA)DES for 1 h at 50 °C. Afterwards, the mixture was cooled down to room temperature, diluted with MeOH/H2O (70/30) to reduce the viscosity and facilitate the subsequent filtration step for the separation of the undissolved residue that was collected and dried in an oven at 60 °C. The supernatant was concentrated under reduced pressure to recover the MeOH and then, a polymeric resin, Amberlite XAD-7 (50 g), was used to separate phenolic compounds from the recovered water phase containing the (NA)DES. Prior to use, the resin was washed and activated by stirring it with acidified water (0.01 M HCl) for 30 min. The activated resin was put into a column and the extraction mixture was added. The (NA)DES was collected by washing the absorbed polyphenol–polymeric resin with water. The aqueous fraction thus obtained was evaporated under reduced pressure for recovering the (NA)DES. Subsequently, after drying the resin under airflow, the extracted polyphenols were desorbed from the resin with MeOH (100 mL). Following that, the methanolic extract was evaporated under reduced pressure for recovering the polyphenolic extracts as solid residues, and subsequently used for UHPLC-MS analyses (the method is described in detail in the ESI†).Recovery and reuse of the DES
The possibility of recycling the DES was evaluated for the best performing system studied for the extraction of polyphenols (ChCl:EG 1:2). In particular, at the end of the process, the aqueous fraction obtained after washing the polymeric resin for recovering the DES was evaporated under reduced pressure. Subsequently, the recovered DES was reused without any modification for a new extraction process with fresh biomass waste as described in the previous section. The solvent was recycled three times and it was analyzed by 1H-NMR in D2O before each cycle. The extraction efficiency of the recovered DES was evaluated by determining the phenol content of the methanolic extracts by UHPLC-MS analyses as described in the previous section.
Determination of the chemical composition
The chemical compositions of the starting CPW biomass and of the residue obtained after polyphenol extraction were determined according to the classical methods reported in the literature as described in detail in the ESI.† In particular, free water-soluble sugars, pectin and holocellulose contents were measured and compared.Treatment with bio-ILs for the valorization of the undissolved residue obtained after polyphenol extraction
Results and discussion
Polyphenol extraction with (NA)DES
ChCl-based DESs, were initially selected for polyphenol extraction from CPW since they are reported as the best performing systems in bioactive compound extraction from several biomass materials (plant materials or food wastes) and are by far the most commonly used DESs. Considering the phenolic profile of sweet cherry fruits and pomace which showed high amounts of anthocyanins along with some phenolic acids and flavonoids, three carboxylic acid-based DESs46–50 and two polyol-based DESs51–56 were selected. The molar ratio of the prepared (NA)DESs is reported in Table 1. The high viscosity of the acid-based solvents, that could hamper mass transport thus drastically limiting the efficiency of the extraction process, was reduced by adding water at 25 wt%. The beneficial effect of adding water in extraction processes has been extensively studied and reported in the literature.57,58 To compare the extraction efficiency and the potential selectivity towards specific types of phenols, the extractions were performed under fixed conditions (biomass/solvent 1/10, 1 h and 50 °C). A classical approach using methanol for phenolic acid and flavonoid extraction and acidified methanol for anthocyanin extraction was also carried out and the obtained extracts were used as blank samples. When the methanolic extraction was performed, a subsequent partition between n-butanol and water was necessary, to remove undesired materials such as sugars. Instead, when (NA)DESs were used, the extracted phenolic compounds have been separated from the (NA)DES by using a macroporous resin (Amberlite XAD-7) that is able to adsorb the phenols while the (NA)DES was collected with water. Then, desorption was carried out by passing the minimum amount of methanol through the absorbed polyphenol–polymeric resin. This procedure allowed the quantitative recovery of the employed (NA)DES, which can be recycled and tested as extraction media with fresh biomass. For this purpose, all the recovered (NA)DESs were analyzed through 1H-NMR spectroscopy (some examples are reported in Fig. S16†). All the spectra showed the same signals of pure (NA)DESs along with typical signals of sugar based-compounds, especially for acid-based (NA)DESs. This aspect will be discussed in the following sections. In both (NA)DES and classical solvent extraction, the recovered phenols are dried and obtained as a solid material. To qualitatively and quantitatively characterize the extracted phenols, methanolic solutions of each extract were prepared and analyzed as shown in the following sections.Phenol profiles of cherry pomace waste extracts
All CPW extracts were subjected to UHPLC-HR-Orbitrap/MS/MS analyses to characterize their chemical composition and establish the difference in terms of recovered compounds by both classical and innovative extraction procedures. The attention was focused on phenols reported in cherry fresh fruits to evaluate to which extent cherry industrial waste can be considered a source of high-value substances after juice production. The extracted ion chromatograms obtained by LC-MS showed the phenol profiles of each sample (Fig. 3). Compounds were tentatively identified by a comparison of their elution order and both full and fragmentation HR mass spectra with the literature data (Table 2). A mass error <5 ppm was considered acceptable. All extracts showed a very similar composition, with three main classes of compounds, represented by phenolic acids (peaks 1–3, 5, and 6), flavonoid C- and O-glycosides (peaks 4, 7–13, 15, 17, and 18–22), and flavonoid aglycones (peaks 14, 16, 19–21, 23, and 24). Among phenolic acids, the most representative peak in all samples was represented by dihydroxybenzoic acid in two isomeric forms (1a and 1b), followed by hydroxycinnamic acids previously reported in cherry fruits.59,60 Peaks 3a/3b and 6a/6b were assigned as p-coumaroylquinic acid cis/trans isomers, as deduced by the same deprotonated molecular ion [M − H]− at m/z 337.0932. The position of the p-coumaroyl moiety on quinic acid was established by MS/MS experiments, showing base ion peaks at m/z 163.04 and 173.04 for 3-p-coumaroylquinic acids (3a and 3b) and 4-p-coumaroylquinic acids (6a and 6b), respectively, according to the hierarchical scheme proposed by Clifford et al.61 Similarly, peaks 5a/5b ([M − H]− at m/z 367.1034) were assigned to 3-O-feruloylquinic acids (cis/trans isomers) based on the fragmentation spectra showing a base ion peak at m/z 193.05, due to the loss of the quinoyl moiety. Finally, peak 2 ([M − H]− at m/z 353.0881) was assigned as chlorogenic acid (3-O-caffeoylquinic acid), as deduced by the presence in the MS/MS spectra of a product ion at m/z 191.06 due to the loss of a caffeoyl moiety.61 Flavonoid glycosides were identified as kaempferol, quercetin, apigenin, naringenin, chrysin, and dihydrowogonin derivatives, as deduced by the detection in the MS/MS experiments of aglycone product ions at m/z 285.04, 301.04, 269.05, 271.06, 253.05, and 285.08, respectively. Saccharide chains were composed of one to three sugar units, represented by hexosyl (−162 u) or deoxyhexosyl (−146 u) moieties linked to the aglycones through an O-glycoside bond, except compound 10 that was identified as apigenin C-glucoside ([M − H]− at m/z 431.0989) by the observation in the ESI fragmentation pathway of the diagnostic product ions ([M − H − 90]− at m/z 341.07 and [M − H − 120]− at m/z 311.06) characteristic of flavonoid C-glycosides.62 Flavone C-glycosides have not been previously reported in cherry fruits, but they have been recently found in the bark,63 thus the occurrence of compound 10 could be attributed to woody plant parts in the cherry waste. All aglycones were also detected in the last region of the chromatograms. | ||
| Fig. 3 UHPLC-HR-ESI-Orbitrap/MS/MS phenolic profiles of cherry pomace waste extracts obtained using classical solvents and (NA)DESs (extracted ion chromatograms in negative ionization mode). Peak data are reported in Table 2. | ||
| Peak | Compound | t R (min) | [M − H]− | HR-ESI-MS/MS ions (m/z)a | Molecular formula | Error (ppm) | Ref. |
|---|---|---|---|---|---|---|---|
| a The base ion peak is shown in bold. For peaks 1–24 see Fig. 3. | |||||||
| Phenolic acids | |||||||
| 1a, 1b | Dihydroxy benzoic acid (isomers I and II) | 1.3, 1.4 | 153.0188 | 109.03, 91.02 | C7H6O4 | −2.81 | 60 |
| 2 | Chlorogenic acid (3-O-caffeoylquinic acid) | 1.8 | 353.0881 | 191.06, 179.03 | C16H18O9 | +0.850 | 59 |
| 3a, 3b | 3-p-Coumaroylquinic acid (isomers cis and trans) | 2.1, 2.3 | 337.0932 | 191.06, 163.04, 119.05 | C16H18O8 | +0.890 | 59 |
| 5a, 5b | 3-O-Feruloylquinic acid (isomers cis and trans) | 2.6, 2.7 | 367.1034 | 193.05, 173.04, 134.05, 69.76 | C17H20O9 | −0.272 | 59 |
| 6a, 6b | 4-p-Coumaroylquinic acid (isomers cis and trans) | 2.8, 3.0 | 337.0932 | 173.04, 163.04, 119.05 | C16H18O8 | +0.890 | 59 |
| Flavonoid glycosides | |||||||
| 4 | Kaempferol O-rutinoside | 2.6 | 593.1517 | 284.03, 285.04 | C27H30O15 | +0.843 | 59 |
| 7 | Quercetin O-glucoside O-rutinoside | 3.4 | 771.2001 | 609.15, 463.09, 300.03, 301.04 | C33H40O21 | +1.43 | 59 |
| 8 | Kaempferol O-glucoside O-rutinoside | 3.5 | 755.2052 | 593.15, 447.09, 285.04 | C33H40O20 | +1.59 | 60 |
| 9 | Rutin | 4.2 | 609.1470 | 447.89, 301.04, 300.03 | C27H30O16 | +1.47 | 59 |
| 10 | Apigenin C-glucoside | 4.4 | 431.0989 | 341.07, 311.06, 283.06, 269.05 | C21H20O10 | +1.15 | 63 |
| 11 | Quercetin O-glucoside | 4.5 | 463.0888 | 301.04, 300.03 | C21H19O12 | −0.431 | 64 |
| 12 | Kaempferol O-rutinoside isomer | 4.8 | 593.1520 | 285.04, 255.03 | C27H30O15 | +1.35 | 64 |
| 13 | Kaempferol O-glucoside | 5.0 | 447.0937 | 285.04, 284.03, 255.03 | C21H20O11 | +0.895 | 60 |
| 15a, 15b | Naringenin O-hexoside (isomers I and II) | 5.5, 5.6 | 433.1144 | 271.06, 151.00, 119.05 | C21H22O10 | +0.923 | 60 |
| 17 | Chrysin O-glucoside | 6.9 | 415.1036 | 253.05, 145.03 | C21H20O9 | +0.241 | 60 |
| 18a, 18b | Naringenin O-hexoside (isomers III and IV) | 6.9, 7.0 | 433.1142 | 271.06, 243.07 | C21H22O10 | +0.462 | 60 |
| 22 | Dihydrowogonin 7-O-glucoside | 7.8 | 447.1298 | 432.11, 285.08, 269.05, 241.05 | C22H24O10 | +0.291 | 60 |
| Flavonoid aglycones | |||||||
| 14 | Aromadendrin | 5.2 | 287.0561 | 259.06, 269.05, 243.06, 125.02 | C15H12O6 | −0.349 | 65 |
| 16 | Quercetin | 6.6 | 301.0354 | 273.04, 179.00, 151.00 | C15H10O7 | 0.0 | 65 |
| 19 | Naringenin | 7.5 | 271.0613 | 253.05, 151.00, 119.04 | C15H12O5 | +0.369 | 64 |
| 20 | Apigenin | 7.6 | 269.0457 | 241.05, 181.06 | C15H10O5 | +0.743 | 65 |
| 21 | Naringenin isomer | 7.8 | 271.0613 | 253.05, 151.00, 119.04 | C15H12O5 | +0.369 | 64 |
| 23 | Chrysin | 9.8 | 253.0506 | 209.06, 181.06, 151.50 | C15H10O4 | −0.395 | 64 |
| 24 | Dihydrowogonin | 10.2 | 285.0767 | 270.05, 179.00, 166.00 | C16H14O5 | −0.351 | 60 |
| Anthocyanins | [M] + | ||||||
| 25 | Cyanidin 3-O-rutinoside | 2.9 | 595.1647 | 449.11, 287.05, 241.05 | C27H31O15+ | −1.68 | 59 |
| 26 | Cyanidin 3-O-glucoside | 3.3 | 449.1069 | 287.05, 267.05, 105.04 | C21H21O11+ | −2.00 | 60 |
The LC-MS analyses of the anthocyanin extracts revealed the occurrence of two cyanidin derivatives, cyanidin 3-O-rutinoside (compound 25) and cyanidin 3-O-glucoside (compound 26) in all samples. According to literature data,59,60 full MS showed positive molecular ions [M]+ at m/z 595.1647 and 449.1069, for 25 and 26, respectively, while the base ion peak corresponding to the aglycone cyanidin (m/z 287.05) was generated by the loss of a rutinosyl ([M − 146–162]+) and a glucosyl ([M − 162]+) moiety, respectively.
Quantitative analysis of phenolic compounds in cherry pomace waste extracts
All analyzed extracts were subjected to a LC-MS quantitative analysis using external standards to calculate the amount of each component, thus of each subclass and total phenols. The results (Table S1,†Fig. 4) showed that the amount of total phenol ranged from 245 ± 17 to 759 ± 85 μg g−1 of dried CPW, indicating the waste material as a potential source of bioactive agents. Indeed, these values are higher, also for blank samples, than those reported for different varieties of fresh cherries,59 although in this work dried wastes have been employed and not the frozen biomass which are characterized by a high content of water.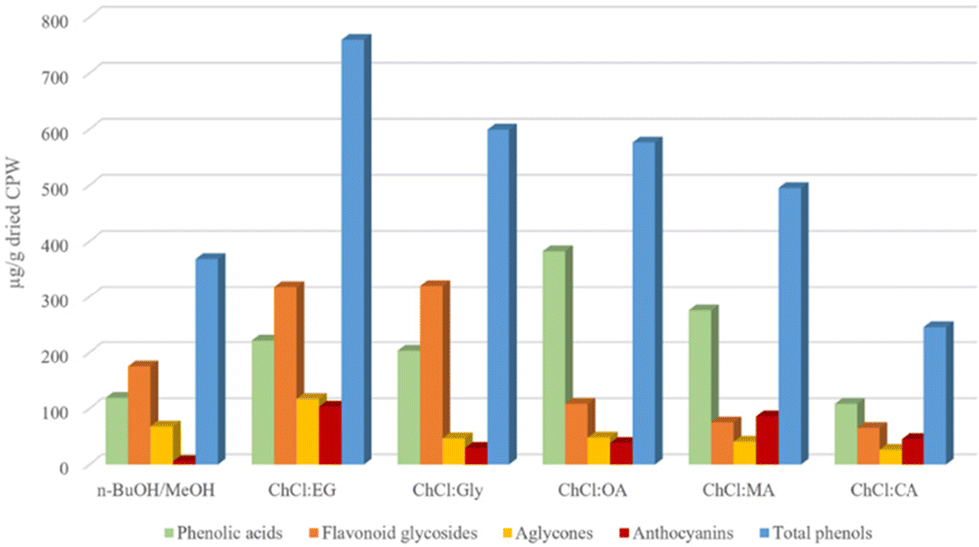 | ||
| Fig. 4 Distribution of different phenol classes in cherry pomace waste (CPW) extracts (μg g−1 of dried CPW) obtained using classical solvents and (NA)DESs. | ||
Compared to classical solvents, all ChCl-based (NA)DESs, except ChCl:CA, showed higher phenol extraction capability. However, among them the most promising solvents as extraction media (in terms of total phenolic content) are those obtained combining ChCl with both the investigated polyols (EG and Gly) and OA among the carboxylic acid, with ChCl:EG being the most efficient (total phenol recovered 759 ± 85 μg g−1 of dried CPW). Besides the total phenolic content of the extracts, it is worth noting the different selectivity towards the main classes of identified phenols as a function of the employed HBD type. Indeed, opposite selectivity was detected when acids or polyol-based DESs were tested. In detail, acid-based DESs extracted higher amounts of total phenolic acids (values ranging from 108 ± 6.9 to 382 ± 59 μg g−1 of dried CPW) compared to flavonoid glycosides (values ranging from 64.9 ± 4.3 to 108 ± 26 μg g−1 of dried CPW). Moreover, ChCl:OA, followed by ChCl:MA, showed the highest values of phenolic acids in the extracts (382 ± 59 and 275 ± 41 μg g−1 of dried CPW, respectively) in comparison with all the investigated systems. Instead, polyol-based DESs showed higher selectivity towards flavonoid glycosides. The values of 318 ± 32 μg g−1 for ChCl:EG and 320 ± 40 μg g−1 for ChCl:Gly were the highest obtained among the tested solvents. ChCl:EG displayed also the highest amounts of flavonoid aglycones (117 ± 13 μg g−1 of dried CPW) and interestingly of anthocyanins (103 ± 7.1 μg g−1 of dried CPW). This latter value is relevant considering that in most of the works reported in the literature on the extraction of anthocyanins from plant material or food industry biomass wastes (especially from wine industry46,47,66), the most efficient DESs usually feature a carboxylic acid acting either as a HBD or a HBA. In particular, among them ChCl:oxalic acid46,49,67 or citric acid-based DESs47,48,50,68–70 are the most frequently used. The great extraction efficiency was explained with the polarity and the acidity of these systems. Indeed, anthocyanins are polar molecules that at low pH (= 1) are predominantly present in the more stable form of red flavylium cation, while at pH values higher than 7 they may be subjected to degradation.46,71 Hence, organic acid-based DESs being more polar solvents than polyol or sugar-based ones and showing very low pH values present the most beneficial features and thus are the best performing extraction media for anthocyanins. However, in this work, the best extractant for anthocyanins was the neutral ChCl:EG. For the acid-based DES series, ChCl:MA was the best performing DES (85.8 ± 6.2 μg g−1 of total anthocyanins in dried CPW) showing also the highest value for the less abundant cyanidin 3-O-glucoside (20.8 ± 2.3 μg g−1 of dried CPW). It is worth noting the great improvement in anthocyanin extraction using (NA)DESs compared to that with the classical solvent (5.71 ± 1.3 μg g−1 of dried CPW). The classical solvent displayed the same selectivity as polyol-based DESs, showing higher amounts of flavonoid glycosides (175 ± 9.9 μg g−1 of dried CPW). Finally, considering the amount of the single extracted phenol, the most abundant compounds in all extracts were dihydroxy benzoic acid (values ranging from 80.2 ± 3.6 to 271 ± 38 μg g−1 of dried CPW) and 3-p-coumaroylquinic acid (values ranging from 11.0 ± 1.2 to 49.8 ± 9.2 μg g−1 of dried CPW, excluding the extract with ChCl:MA) among phenolic acids, rutin (values ranging from 20.9 ± 0.70 to 132 ± 18 μg g−1 of dried CPW) among flavonoid glycosides, chrysin (values ranging from 17.1 ± 0.73 to 75.2 ± 8.0 μg g−1 of dried CPW) among flavonoid aglycones and cyanidin 3-O-rutinoside (values ranging from 29.2 ± to 96.8 ± 6.8 μg g−1 of dried CPW, excluding the extract with the classical solvent) among anthocyanins.
These results showed the influence of the HBD in the extraction ability of different ChCl-based DESs. To get a further insight into the potential of these media, the influence of the HBAs was also investigated and (NA)DESs with Bet and L-Pro instead of ChCl were prepared in combination with the three best HBDs (ethylene glycol, glycerol and oxalic acid). Betaine-based DESs as well as L-proline-based DESs are, after ChCl-based ones, the most investigated natural DESs,72–75 although often they showed worse extraction efficiencies compared to the ChCl containing DESs.76 However, considering the safe profile of these compounds and future potential as DES components, especially in the case of betaine, it was of interest to investigate alternative HBAs.77 The extractions of phenols were performed using the same procedure under the same operative conditions in terms of temperature, time and biomass/(NA)DES ratio. Quantitative data of recovered phenols in all extracts are shown in Table S2† and Fig. 5. All betaine and L-proline-based DESs did not improve the extraction process compared to the classical solvents, showing a total phenolic content ranging from 167 ± 11 to 267 ± 24 μg g−1 of dried CPW, with the lowest values found for the glycerol-based solvents. Obviously, the total phenolic content is also much lower than that obtained by using ChCl-based DESs. However, it is worth noting that polyol-based betaine and proline DESs showed an inverted selectivity towards phenolic acids and flavonoid glycosides compared to ChCl-based DESs. In particular, for Bet:Gly and L-Pro:Gly the total phenolic content was mainly due to phenolic acids. Indeed, 132 ± 2.4 μg g−1 of phenolic acids over 175 ± 4.2 μg g−1 of total phenols was detected when Bet:Gly was employed, while 123 ± 8.4 μg g−1 of phenolic acids over 167 ± 11 μg g−1 of total content was observed for L-Pro:Gly-based extraction. Regarding oxalic acid-based DESs, lower values of all the identified classes of compounds than ChCl:OA were obtained, with the only exception of anthocyanins. For this latter class, similar values were observed, confirming the ability of acid-based DESs to extract thanks to the stabilization of anthocyanins at low pH values. Comparing the amounts of the single phenolic compounds obtained with the betaine- and proline-based DESs, dihydroxy benzoic acid was still the main abundant phenolic acid, showing values significantly higher within this class. Furthermore, chrysin among flavonoid aglycones and cyanidin 3-O-rutinoside among the anthocyanins were the most representative compounds of the respective classes. Rutin was still the most abundant flavonoid glycosides, except for Bet:Gly and L-Pro:Gly extracts, showing higher quantities of apigenin C-glucoside.
 | ||
| Fig. 5 Distribution of different phenol classes in cherry pomace waste (CPW) extracts obtained using (NA)DES (μg g−1 of dried CPW). | ||
Hence, ChCl:EG was selected as the best performing extraction medium in terms of total phenolic content and total content of both flavonoids glycosides and aglycones, and anthocyanins. It only showed lower amounts of phenolic acids compared to ChCl:carboxylic acid DESs. It is worth noting that the yields of the recovered phenols with ChCl:EG were lower than those obtained with other (NA)DESs, especially the acid-based ones. This result can be correlated to a higher selectivity towards phenolic compounds by this system. Conversely, the other (NA)DESs besides polyphenols simultaneously extract other metabolites.
Subsequently, an optimization of the operative conditions was performed. In particular, extractions with ChCl:EG were repeated either at 50 °C varying the extraction time from 30 min to 3 h or increasing the temperature from 25 to 60 °C keeping the extraction time unchanged (1 h).
A relative estimation of phenols recovered by ChCl:EG at different temperatures and extraction times was done by UHPLC-MS data of extracts and then evaluating the percentage variation compared to the highest one (Fig. 6).
 | ||
| Fig. 6 Percentage variation in quantities of phenolic compound classes at different extraction times (A) or temperatures (B) after extraction with ChCl:EG (choline chloride:ethylene glycol). | ||
Unsurprisingly, a detrimental effect was noticed on extending the time over an hour. These findings can be correlated to the degradation of phenols when they are maintained at 50 °C for long times. The amount of phenols increases with time up to reaching a maximum value after which it decreases. This behaviour could be explained by Fick's second law of diffusion which establishes that a final equilibrium between the matrix solid and the extractant is achieved after a certain contact time and the extraction efficiency does not improve with prolonged times.19,52 One hour was selected as the optimal extraction time.
Regarding temperature, it is known that this parameter influences the extraction efficiency since higher temperatures can reduce considerably the viscosity of (NA)DESs enhancing the solubility and diffusion coefficient of the solute thus improving mass transfer. In our study, extractions carried out at different temperatures revealed an increasing recovery of phenols with temperatures up to 50 °C, while at 60 °C a small reduction was observed. Again, this result can be correlated to the thermolability of some phenols. From these preliminary data, 1 h and 50 °C were considered the best extraction conditions. However, further evaluation about the content of anthocyanins is needed and a better comparison will be performed in future studies.
Recovery and reuse of DES
The possibility of recycling the DES was considered for the best performing system applying the optimal operative conditions (ChCl:EG 1:2, 1 h at 50 °C). Indeed, the recovery of the solvent, its reuse in a new extraction cycle and the retention of its extraction efficiency (EE) are all aspects that combine to define the real sustainability of an extraction process. Hence, the aqueous fraction recovered after the separation step with the Amberlite XAD-7 resin was evaporated under reduced pressure to collect the ChCl:EG 1:2. Before use in further extractions with fresh biomass, the mass of the recovered DES was evaluated, and the molar ratio was checked by 1H-NMR. The entire process was repeated for three consecutive cycles. The data related to DES recovery (expressed as the percentage of mass recovered over mass used) and the EE (assessed by means of LC-MS of the third extract) are reported in Fig. 7 and 8. Recovery of DES was practically quantitative; after the first run a value above 100% was obtained. This result can be ascribed to the concomitant extraction of free sugars and presumably of the pectin part, which remains in the DES (NMR spectra are shown in Fig. 7). After each cycle, a slight increase of the intensity of the characteristic signals of sugar compounds (signals between 3 and 4 ppm and the two signals at 4.6 and 5.2 ppm attributable to the anomeric protons) was observed. However, the presence of these compounds in the DES does not affect the extraction capability of the solvent. In Fig. 8, the relative estimation of phenols recovered at the end of the third cycle were determined by UHPLC-MS and the percentage variation with respect to the first cycle, to which the value of 100% was assigned, was evaluated. The total content of phenols as well as the amount of each subclass was similar to that of the first cycle, which proves the retention of the extraction efficiency of the recycled solvent.
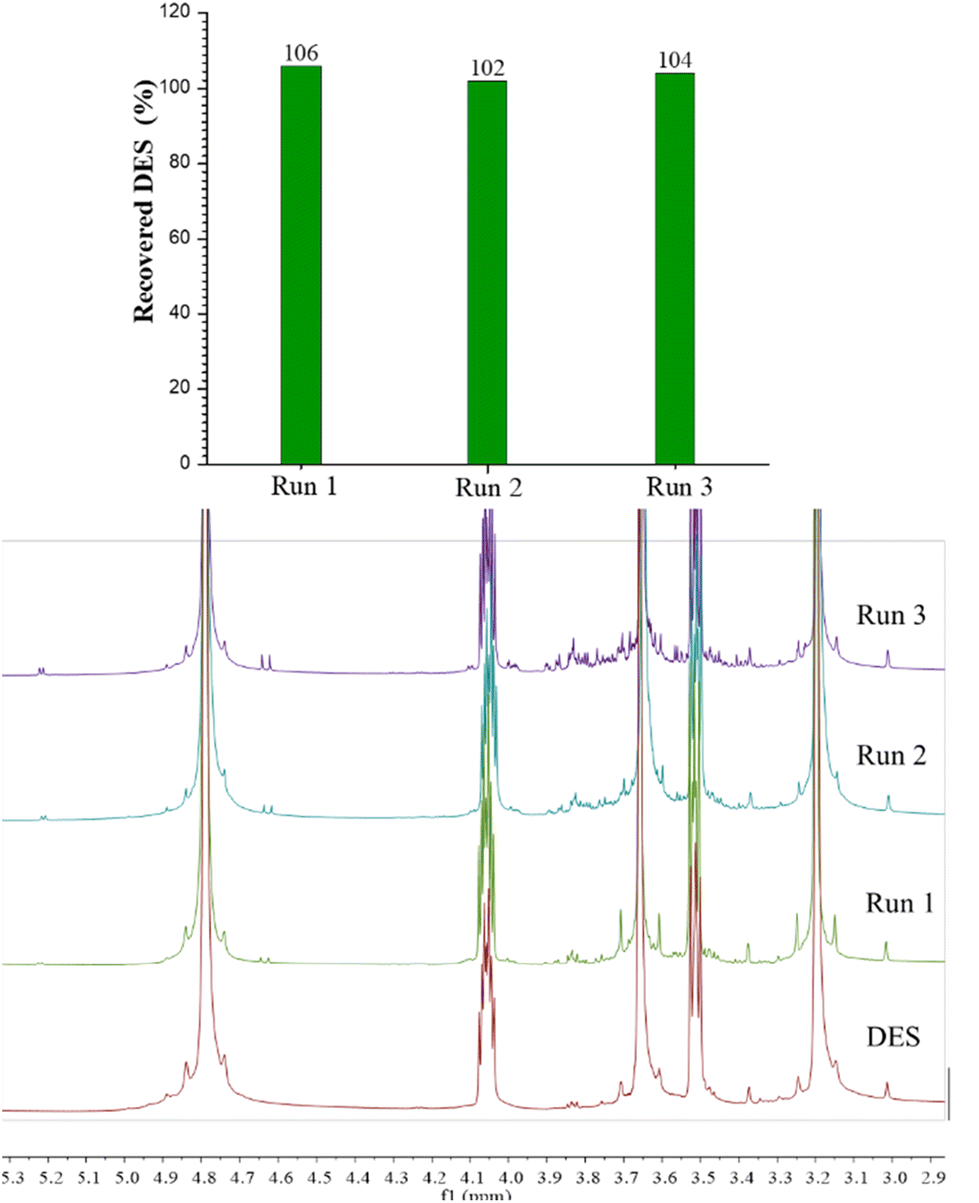 | ||
| Fig. 7 Yields (top) and 1H-NMR (bottom) of the recovered DES after each cycle. | ||
 | ||
| Fig. 8 Percentage variation of phenolic classes at the third cycle compared to the first cycle to which the value of 100% was assigned. | ||
Characterization of the solid residue obtained after polyphenol extraction
The undissolved residues obtained after polyphenol extraction were analyzed to study their composition and further processed to complete the biorefinery process. At first, an evaluation of the amount of the recovered materials was carried out and a clear difference was observed when polyol or acid-based DESs were employed (Fig. 9). For all the tested HBAs, lower amounts of undissolved residue were recovered when the biomass was treated with acid-based DESs. As already discussed in the previous sections, this could be ascribed to the concomitant extraction of free sugars or the pectin fraction with the DES treatment. Indeed, it has been widely reported that acidic media with HCl, H2SO4 or citric acid,78–81 and different NADESs, such as ChCl:MA, ChCl:CA or Bet:CA or different lactic acid-based DESs are able to extract pectin from several biomass materials (e.g. pomelo peel, Averrhoa bilimbi, dragon fruit peels, mango peel powder, sugar beet pulp, apple pomace).82–89 As already shown, this hypothesis was confirmed from the 1H-NMR spectra of the recovered (NA)DESs shown in Fig. S15,† where the signals attributable to sugar based-compounds are evident both in the acid-based (NA)DESs, and in the polyol-based DESs. Future studies will be focused on the recovery of the extracted pectin and sugars from the (NA)DESs.
 | ||
| Fig. 9 Amounts of undissolved residues recovered after polyphenol extraction with (NA)DESs. | ||
Based on the polyphenol content of the extracts, the subsequent valorization of the undissolved residue was performed on the residue obtained after ChCl:EG treatment (test performed with the optimized extraction conditions: 1 h at 50 °C). The residue was initially characterized by FTIR analysis (Fig. S17†). The FTIR spectrum showed all the characteristic absorption bands of typical dietary fibers which are generally present in wastes of processed fruits and vegetables (cellulose, hemicellulose, pectin, and lignin).90 The main bands and the corresponding attributions are reported in Table S3.† These data have been confirmed by TG analysis (Fig. S18†) that showed a main degradation step in the temperature range 200–500 °C due to overlapping phenomena. In detail, the sample started to degrade around 150 °C and it displayed a continuous degradation until the end of the analysis at 800 °C, which behaviour is generally observed for lignin extracted from agro-waste biomass.43,91 Maxima in the derivative curve were around 200–300 °C, which are the typical degradation temperatures of pectin/hemicellulose and cellulose, respectively.
The chemical composition of the residue was ascertained by performing the classical methods reported in the literature (see the ESI†) aimed at the recovery and hence quantification of specific components of a lignocellulosic biomass. This chemical characterization was also performed on the starting material (CPW). At first, the free water-soluble sugar content was obtained. Then, pectin and holocellulose (cellulose and hemicellulose) contents were evaluated leading also to the quantification of lignin content. The obtained results are summarized in Fig. 10, representing the mass balance of the extraction process. The data confirm the hypothesis that the loss of material registered at the end of the process can be related to the concomitant extraction by the DES of free water-soluble sugars and in part of the pectin present in the starting material, along with polyphenols. Conversely, this DES treatment does not practically change the amount of holocellulose and lignin.
 | ||
| Fig. 10 Mass balance of the DES treatment step. | ||
Treatment with bio-ILs (ChArg and ChLev) of the undissolved residue obtained after polyphenol extraction: ionogel formation
After the recovery of polyphenols, the full valorization of the cherry pomace was attempted through the formation of a cellulose ionogel. These kinds of functional materials are attracting increasing attention34,92 since they combine the unique properties of ionic liquids as preferable dispersed liquid phase and cellulose as the solid matrix. Among all the reported ionic liquids for cellulose or lignocellulosic material processing, ChLev has been chosen for its green nature. Indeed, it has been reported as a readily biodegradable and low-toxicity biocompatible ionic liquid.93 At first, an attempt to directly obtain a gel using the whole residue, characterized by cellulose, hemicellulose, pectin, and lignin was carried out. Unfortunately, this strategy did not lead to complete dissolution of the biomass in ChLev, apart when very low amounts of material not sufficient to start the gelification process were used. This could be ascribed to the presence of lignin and hemicellulose, in agreement with the findings reported by Hopson et al. during the formation of an ionogel using EMIM DEP.94 Therefore, before dissolving the material in ChLev, a pretreatment aimed at obtaining a cellulose enriched material through the selective extraction of hemicellulose and lignin fractions was performed. Pectin was not considered as an interfering factor since pectins occurred in the smallest amounts in all the pomace samples analyzed and especially in cherry pomace.90 Moreover, as discussed in detail above, the treatment with (NA)DESs can lead to a concomitant extraction of pectin from the biomass, further reducing its amount in the final undissolved residue. Finally, the importance of pectin in the food sector lies in its ability to form gel,95,96 hence its presence should not influence the gelling process. Therefore, ChArg, a different bio-based ionic liquid able to selectively remove the lignin and hemicellulose from lignocellulosic material41,42 was selected for the pretreatment step which led to two different fractions. The cellulose enriched material, CRM (35 wt% of dried biomass) was obtained via filtration as the undissolved material, while a fraction enriched in lignin and hemicellulose was precipitated from the filtrate acidifying the solution (22 wt% of dried biomass). The two fractions have been analyzed by FTIR spectroscopy and TGA and the obtained spectra and thermograms are compared with the starting material as shown in Fig. 11. Both analyses ascertained the nature of the fractions. Indeed, the FTIR spectrum of CRM (blue line) showed mainly the characteristic bands of cellulose (i.e. 1020, 1142, 1316, 1362 cm−1) while all the bands that were ascribable only to lignin or hemicellulose (i.e. 1730, 1520, 1225 cm−1) were not present anymore. Instead, these bands were clearly visible in the spectrum of HLRM (red line). | ||
| Fig. 11 FTIR spectra (top) and TGA and DTG (bottom) of the two fractions obtained after ChArg (cholinium arginate) treatment compared to the undissolved residue obtained after polyphenol extraction with ChCl:EG (choline chloride:ethylene glycol). | ||
The TGA confirmed these compositions. Indeed, the CRM exhibited a main degradation step at 347 °C, typical behaviour of cellulosic material, while the HLRM showed a main degradation step at 239 °C followed by a continuous mass loss until 800 °C, which are characteristic thermograms of hemicellulose and lignin, respectively.91 It is worth highlighting that it was possible to recover the bio-IL with satisfactory purity at the end of the treatment (1H-NMR spectra of the recovered bio-IL are shown in Fig. S19†). Finally, the result of the holocellulose content measurement, performed on the CRM, confirmed the enrichment in cellulose of the residue (96.3 ± 2.4% of holocellulose).
Subsequently, in order to prepare the ionogel, the CRM was ground to obtain a fine and more manageable material which was dissolved in ChLev at 2 wt%. The CRM was slowly added to the bio-IL and stirred until complete dissolution (24 h). After dissolution, the hot mixture was poured into a suitable mold and kept at room temperature until gel formation (3 d). The excess of ionic liquid was removed and recovered to further enhance the greenness of the process. The recovered ChLev was analyzed by 1H-NMR spectroscopy to verify its purity (Fig. S20†). The obtained gel (Fig. 12) after saturation with water at 30 °C was instead characterized by FTIR spectroscopy, TGA and rheological analyses.
 | ||
| Fig. 12 Image of the obtained ionogel. | ||
As expected, the FTIR spectrum of the ionogel (Fig. 13) showed only the bands attributable to the major component of the gel, the ChLev along with the characteristic bands of the absorbed water (the large band between 3000 and 3500 cm−1 and the band at 1652 cm−1). The characteristic signals of CRM were covered by the bands of ChLev in the registered spectrum. The thermogram of the ionogel (Fig. 13) exhibited two main degradation steps at 66.9 °C and 225.5 °C, attributable to water evaporation and ChLev degradation, respectively. Moreover, the second mass loss was followed by a shoulder at 335 °C, characteristic of the dissolved cellulosic material, as shown in the inset in Fig. 13. From the thermogram, it was also possible to obtain a rough evaluation of the amount of ChLev and water in the final material: 35% water and 58% ChLev in the final gel were determined from each mass loss.
 | ||
| Fig. 13 FTIR spectra (top) and TGA–DTG (bottom) of the obtained ionogel compared to ChLev (cholinium levulinate) and CRM (cellulose enriched material). | ||
Finally, the rheology of the formulated ionogel was studied to get an insight into its internal structure and gel characteristics. Indeed, from dynamic frequency sweeps it is possible to determine if the ionogel is a gel type material or not and also whether it is a physical or a chemical ionogel.34 In our case, the formation of a physical gel is expected since the interactions between the cellulosic material and the ionic liquid are non-covalent (mainly hydrogen bonds). Initially, the amplitude sweep test was performed between 0.1 and 1% at 1 Hz in order to select the correct value of the shear strain within the LVR (linear viscoelastic region). The elastic modulus G′ is almost constant during strain variation suggesting that the internal structure/network of the material does not change with the applied strain. Subsequently, a frequency sweep test was performed at 0.1% (Fig. 14 and Table S4†).
 | ||
| Fig. 14 Rheological characterization of the formulated ionogel. | ||
As observed from the graph, the elastic modulus G′ was higher than the loss modulus G′′ within the plateau region. Indeed, G′ remained constant with the frequency, which is a typical behaviour of a gel like material. These data are similar to that obtained by Mar Villar-Chavero et al.35 for the ionogel prepared dissolving 2 wt% microcrystalline cellulose in cholinium lysinate (ChLys) or to the ionogel obtained by Hopson dissolving 1.7 wt% cellulose rich-solid derived from a biorefinery approach in EMIM DEP.94 Then, to determine the strength of the formulated ionogel and hence to classify it as physical or chemical gel, the dependence of the elastic modulus was calculated applying eqn (1) (see rheological properties in the ESI†). Since the parameter a was >0 (Table 3), the expected physical nature of the gel, due to the hydrogen bonds formed between ChLev and the –OH groups of cellulosic enriched material was confirmed. The nature of the interaction forces was ensured also by FTIR spectrum previously shown, where no modification or new bands were found along with the characteristic bands of the components. Regarding the strength of the ionogel (G0), a similar value to that of the ionogel based on cellulose and ChLys was obtained.35 Moreover, according to the criterion based on the mean relation G′/G′′, the formulated ionogel showing a ratio <10 (Table 3) was classified as weak physical gel97 in contrast to all the strong ionogels composed of EMIM DEP and different concentrations of cellulose rich-solids.94 However, it showed a higher strength than the cellulosic bionogel with ChLys, which showed values ranging from 2.6 to 6.5 at 4 Hz and from 1.2 to 3.4 at 45 Hz.35
| a | G 0 (Pa) | R 2 | (G′/G′′)mean | |
|---|---|---|---|---|
| Ionogel | 0.08173 ± 0.00189 | 11298 ± 1 |
0.99254 | 7.2 |
Finally, the complex viscosity η* of the formulated ionogel was evaluated through the data obtained in the frequency sweep test. The results of η*, reported in Fig. 14, showed that the material exhibited a shear-thinning or pseudoplastic behaviour due to the decrease of η* when the frequency was increased. To further ascertain this behaviour, the complex viscosity was fitted according to eqn (3) (see rheological properties in the ESI†). The obtained data are reported in Table 4. The “p” value was less than the unit which corroborated the shear-thinning behaviour. The parameter q, which is a measure of the consistency of the material, was higher than the values obtained for the bionogel composed of cellulose and ChLys mentioned before35 and more similar to the value obtained from chitosan reinforced cellulosic bionogel prepared by the same authors with ChLys as the ionic liquid.98 However, as widely reported in the literature, this parameter is strongly influenced by the amount and type of cellulose, biopolymer load and post-gelation time. Hence, an in-depth study on the preparation of different ionogels with different CRM loads will be performed in a dedicated work.
| q (Pa sp) | p | R 2 | |
|---|---|---|---|
| Ionogel | 11378 ± 1 |
0.08297 ± 0.00201 | 0.99993 |
Conclusions
The influence of both HBA and HBD components of different (NA)DESs on the extraction efficiency and the selectivity towards phenolic compounds contained in cherry pomace wastes obtained after juice production was evaluated. The extractions were performed under fixed conditions (biomass/solvents of 1/10, extraction time of 1 h and temperature of 50 °C) in order to compare the obtained results. UHPLC-HR-ESI-MS was performed to initially qualitatively identify and then quantify the extracted phenols. More than 25 compounds, classifiable in four main subclasses (phenolic acids, flavonoid glycosides, flavonoid aglycones, and anthocyanins) were detected. The best performing system in terms of total phenolic content was identified as ChCl:EG. The influence of the HBD (carboxylic acids or polyols) on the selectivity performance and then also of the HBA (choline chloride, betaine or L-proline) was evident especially towards phenolic acids and flavonoid glycoside subclasses. All ChCl-based DESs, except ChCl:CA, improved the total phenol extraction compared to classical solvents. Conversely, all Bet and L-Pro-based DESs displayed lower total phenolic content than classical solvents. Optimization studies of the working conditions for the best performing system suggested 1 h and 50 °C as most promising. The DES recyclability was investigated for ChCl:EG 1:2. The solvent was quantitatively recovered, and the extraction efficiency was successfully retained over 3 cycles. Finally, valorization of the undissolved lignocellulosic residue obtained after polyphenol extraction with ChCl:EG was carried out by employing two different bio-based ILs endowed with complementary dissolution properties towards lignocellulosic components. At first, ChArg known to dissolve selectively lignin over cellulose was used to obtain a cellulose enriched material. Then, this was dissolved in ChLev to prepare a weak physical ionogel, a functional biomaterial with potential application in medical and pharmaceutical sectors. The proposed biorefinery approach showcases the valorization of all main components of cherry pomace waste.
Author contributions
Lorenzo Guazzelli: conceptualization; project administration; supervision; funding acquisition; and writing – review and editing. Marinella De Leo: conceptualization; funding acquisition; supervision; methodology; writing – original draft; and writing – review and editing. Alessandra Braca: conceptualization; resources; methodology; and writing – review and editing. Andrea Mezzetta: conceptualization; methodology; data curation; writing – original draft; and writing – review and editing. Angelica Mero: investigation; methodology; data curation; and writing – original draft.Conflicts of interest
There are no conflicts to declare.References
- United States Department of Agriculture Foreign Agricultural Service, Usda, 2021, https://public.govdelivery.com/accounts/USDAFAS/subscriber/new.
- S. Chockchaisawasdee, J. B. Golding, Q. V. Vuong, K. Papoutsis and C. E. Stathopoulos, Trends Food Sci. Technol., 2016, 55, 72–83 CrossRef CAS
.
- M. Serrano, F. Guillén, D. Martínez-Romero, S. Castillo and D. Valero, J. Agric. Food Chem., 2005, 53, 2741–2745 CrossRef CAS PubMed
- B. Mozetic, P. Trebse and J. Hribar, Food Technol. Biotechnol., 2002, 40, 207–212 CAS
- D. Średnicka-Tober, A. Ponder, E. Hallmann, A. Głowacka and E. Rozpara, Antioxidants, 2019, 8, 534 CrossRef PubMed
- V. Usenik, F. Stampar, M. M. Petkovsek and D. Kastelec, J. Food Compos. Anal., 2015, 38, 121–130 CrossRef CAS
- G. Domínguez-Rodríguez, M. L. Marina and M. Plaza, Food Chem., 2021, 339, 128086 CrossRef PubMed
- S. Pathania and N. Kaur, Bioact. Carbohydr. Diet. Fibre, 2022, 27, 100295 CrossRef CAS
- F. M. Yılmaz, M. Karaaslan and H. Vardin, J. Food Sci. Technol., 2015, 52, 2851–2859 CrossRef PubMed
- K. Dziadek, A. Kopeć and M. Tabaszewska, Eur. Food Res. Technol., 2019, 245, 763–772 CrossRef CAS
- S. Hosseini, K. Parastouei and F. Khodaiyan, Int. J. Biol. Macromol., 2020, 158, 911–921 CrossRef CAS PubMed
- A. Chaovanalikit and R. E. Wrolstad, J. Food Sci., 2004, 69, FCT73–FCT83 CAS
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS
- T. El Achkar, H. Greige-Gerges and S. Fourmentin, Environ. Chem. Lett., 2021, 19, 3397–3408 CrossRef CAS
- S. P. Ijardar, V. Singh and R. L. Gardas, Molecules, 2022, 27, 1368 CrossRef CAS PubMed
- S. J. Bryant, A. J. Christofferson, T. L. Greaves, C. F. McConville, G. Bryant and A. Elbourne, J. Colloid Interface Sci., 2022, 608, 2430–2454 CrossRef CAS PubMed
- Y. H. Choi, J. van Spronsen, Y. Dai, M. Verberne, F. Hollmann, I. W. C. E. Arends, G.-J. Witkamp and R. Verpoorte, Plant Physiol., 2011, 156, 1701–1705 CrossRef CAS PubMed
- M. Vieira Sanches, R. Freitas, M. Oliva, A. Mero, L. De Marchi, A. Cuccaro, G. Fumagalli, A. Mezzetta, G. Colombo Dugoni, M. Ferro, A. Mele, L. Guazzelli and C. Pretti, Environ. Sci. Pollut. Res., 2023, 30, 17268–17279 CrossRef CAS PubMed
- P. Gullón, B. Gullón, A. Romaní, G. Rocchetti and J. M. Lorenzo, Trends Food Sci. Technol., 2020, 101, 182–197 CrossRef
- A. Mišan, J. Nađpal, A. Stupar, M. Pojić, A. Mandić, R. Verpoorte and Y. H. Choi, Crit. Rev. Food Sci. Nutr., 2020, 60, 2564–2592 CrossRef PubMed
- M. Jablonský, A. Škulcová, A. Malvis and J. Šima, J. Biotechnol., 2018, 282, 46–66 CrossRef PubMed
- J. González-Rivera, A. Mero, E. Husanu, A. Mezzetta, C. Ferrari, F. D'Andrea, E. Bramanti, C. S. Pomelli and L. Guazzelli, Green Chem., 2021, 23, 10101–10115 RSC
- H. Moni Bottu, A. Mero, E. Husanu, S. Tavernier, C. S. Pomelli, A. Dewaele, N. Bernaert, L. Guazzelli and L. Brennan, Food Chem., 2022, 386, 132717 CrossRef CAS PubMed
- F. S. Costa, L. S. Moreira, A. M. Silva, R. J. Silva, M. P. dos Santos, E. G. P. da Silva, M. T. Grassi, M. H. Gonzalez and C. D. B. Amaral, J. Food Compos. Anal., 2022, 109, 104510 CrossRef CAS
- B. M. Popovic, N. Micic, A. Potkonjak, B. Blagojevic, K. Pavlovic, D. Milanov and T. Juric, Food Chem., 2022, 366, 130562 CrossRef CAS PubMed
- G. Kaur, H. Kumar and M. Singla, J. Mol. Liq., 2022, 351, 118556 CrossRef CAS
- Q. Hou, M. Ju, W. Li, L. Liu, Y. Chen and Q. Yang, Molecules, 2017, 22, 490 CrossRef PubMed
- I. Hasanov, M. Raud and T. Kikas, Energies, 2020, 13, 1–24 CrossRef
- J. Zhang, J. Wu, J. Yu, X. Zhang, J. He and J. Zhang, Mater. Chem. Front., 2017, 1, 1273–1290 RSC
- P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC
- C. Manasa, V. Basavanna and S. Ningaiah, Biointerface Res. Appl. Chem., 2022, 13, 391 Search PubMed
- X. Fan, S. Liu, Z. Jia, J. J. Koh, J. C. C. Yeo, C.-G. Wang, N. E. Surat'man, X. J. Loh, J. Le Bideau, C. He, Z. Li and T.-P. Loh, Chem. Soc. Rev., 2023, 52, 2497–2527 RSC
- Z. Zhang, R. Zhao, S. Wang and J. Meng, Front. Bioeng. Biotechnol., 2022, 11, 1117944 CrossRef PubMed
- C. Hopson, M. M. Villar-Chavero, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, Carbohydr. Polym., 2021, 274, 118663 CrossRef CAS PubMed
- M. M. Villar-Chavero, J. C. Domínguez, M. V. Alonso, V. Rigual, M. Oliet and F. Rodriguez, Int. J. Biol. Macromol., 2019, 133, 262–269 CrossRef CAS PubMed
- M. M. Villar-Chavero, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, Carbohydr. Polym., 2019, 207, 775–781 CrossRef CAS PubMed
- P. Fan, Y. Yuan, J. Ren, B. Yuan, Q. He, G. Xia, F. Chen and R. Song, Carbohydr. Polym., 2017, 162, 108–114 CrossRef CAS PubMed
- R. M. Kalinoski and J. Shi, Ind. Crops Prod., 2019, 128, 323–330 CrossRef CAS
- P. Jia, X. Ji, B. Zheng, C. Wang, W. Hao, W. Han, J. Zhang, G. Xia, X. Ji and J. Zhang, Polymers, 2022, 14, 1589 CrossRef CAS PubMed
- T. Q. To, K. Procter, B. A. Simmons, S. Subashchandrabose and R. Atkin, Faraday Discuss., 2018, 206, 93–112 RSC
- A. Mero, N. R. Moody, E. Husanu, A. Mezzetta, F. D'Andrea, C. S. Pomelli, N. Bernaert, F. Paradisi and L. Guazzelli, Front. Chem., 2023, 11, 1270221 CrossRef CAS PubMed
- Y. X. An, M. H. Zong, H. Wu and N. Li, Bioresour. Technol., 2015, 192, 165–171 CrossRef CAS PubMed
- E. Husanu, A. Mero, J. G. Rivera, A. Mezzetta, J. C. Ruiz, F. D'Andrea, C. S. Pomelli and L. Guazzelli, ACS Sustainable Chem. Eng., 2020, 8, 18386–18399 CrossRef
- X.-D. Hou, J. Xu, N. Li and M.-H. Zong, Biotechnol. Bioeng., 2015, 112, 65–73 CrossRef CAS PubMed
- A. Mezzetta, S. Becherini, C. Pretti, G. Monni, V. Casu, C. Chiappe and L. Guazzelli, New J. Chem., 2019, 43, 13010–13019 RSC
- K. Radošević, N. Ćurko, V. Gaurina Srček, M. Cvjetko Bubalo, M. Tomašević, K. Kovačević Ganić and I. Radojčić Redovniković, LWT–Food Sci. Technol., 2016, 73, 45–51 CrossRef
- K. M. Jeong, J. Zhao, Y. Jin, S. R. Heo, S. Y. Han, D. E. Yoo and J. Lee, Arch. Pharmacal Res., 2015, 38, 2143–2152 CrossRef CAS PubMed
- D. Aslan Türker and M. Doğan, LWT–Food Sci. Technol., 2021, 138, 110775 CrossRef
- X. Fu, D. Wang, T. Belwal, J. Xie, Y. Xu, L. Li, L. Zou, L. Zhang and Z. Luo, LWT–Food Sci. Technol., 2021, 144, 111220 CrossRef CAS
- M. Panić, V. Gunjević, G. Cravotto and I. Radojčić Redovniković, Food Chem., 2019, 300, 125185 CrossRef PubMed
- M. Z. Gao, Q. Cui, L. T. Wang, Y. Meng, L. Yu, Y. Y. Li and Y. J. Fu, Microchem. J., 2020, 154, 104598 CrossRef CAS
- B. Ozturk, C. Parkinson and M. Gonzalez-Miquel, Sep. Purif. Technol., 2018, 206, 1–13 CrossRef CAS
- N. Guo, Y. P. Zou, H. K. Li, P. Kou, Z. M. Liu and Y. J. Fu, Microchem. J., 2020, 159, 105586 CrossRef CAS
- M. E. Alañón, M. Ivanović, A. M. Gómez-Caravaca, D. Arráez-Román and A. Segura-Carretero, Arabian J. Chem., 2020, 13, 1685–1701 CrossRef
- L. Chen, Y. Y. Yang, R. R. Zhou, L. Z. Fang, D. Zhao, P. Cai, R. Yu, S. H. Zhang and J. H. Huang, Anal. Methods, 2021, 13, 1226–1231 RSC
- J. C. López-Linares, V. Campillo, M. Coca, S. Lucas and M. T. García-Cubero, J. Chem. Technol. Biotechnol., 2021, 96, 481–490 CrossRef
- X. Liu, N. Fu, Q. Zhang, S. Cai, Q. Wang, D. Han and B. Tang, J. Chromatogr. Sci., 2019, 57, 272–278 CAS
- A. Ali Redha, J. Agric. Food Chem., 2021, 69, 878–912 CrossRef CAS PubMed
- M. De Leo, A. M. Iannuzzi, M. P. Germanò, V. D'Angelo, F. Camangi, F. Sevi, G. Diretto, N. De Tommasi and A. Braca, Food Chem., 2021, 360, 129999 CrossRef CAS PubMed
- S. Martini, A. Conte and D. Tagliazucchi, Food Res. Int., 2017, 97, 15–26 CrossRef CAS PubMed
- M. N. Clifford, K. L. Johnston, S. Knight and N. Kuhnert, J. Agric. Food Chem., 2003, 51, 2900–2911 CrossRef CAS PubMed
- J. Cao, C. Yin, Y. Qin, Z. Cheng and D. Chen, J. Mass Spectrom., 2014, 49, 1010–1024 CrossRef CAS PubMed
- M. Beszterda and R. Frański, Eur. J. Mass Spectrom., 2020, 26, 369–375 CrossRef CAS PubMed
- L. Agulló-Chazarra, I. Borrás-Linares, J. Lozano-Sánchez, A. Segura-Carretero, V. Micol, M. Herranz-López and E. Barrajón-Catalán, Antioxidants, 2020, 9, 418 CrossRef PubMed
- M. Sanz, E. Cadahía, E. Esteruelas, A. M. Muñoz, B. Fernández De Simón, T. Hernández and I. Estrella, J. Agric. Food Chem., 2010, 58, 4907–4914 CrossRef CAS PubMed
- T. Bosiljkov, F. Dujmić, M. Cvjetko Bubalo, J. Hribar, R. Vidrih, M. Brnčić, E. Zlatic, I. Radojčić Redovniković and S. Jokić, Food Bioprod. Process., 2017, 102, 195–203 CrossRef CAS
- L. Loarce, R. Oliver-Simancas, L. Marchante, M. C. Díaz-Maroto and M. E. Alañón, LWT–Food Sci. Technol., 2021, 149, 111889 CrossRef CAS
- E. Kurtulbaş, A. G. Pekel, M. Bilgin, D. P. Makris and S. Şahin, Biomass Convers. Biorefin., 2022, 12, 351–360 CrossRef
- A. M. G. Maclean, Y. P. A. Silva, G. Jiao and M. S. Brooks, Food Technol. Biotechnol., 2021, 59, 56–62 CAS
- N. Guo, Ping-Kou, Y. W. Jiang, L. T. Wang, L. J. Niu, Z. M. Liu and Y. J. Fu, Food Chem., 2019, 296, 78–85 CrossRef CAS PubMed
- A. Castañeda-Ovando, M. de L. Pacheco-Hernández, M. E. Páez-Hernández, J. A. Rodríguez and C. A. Galán-Vidal, Food Chem., 2009, 113, 859–871 CrossRef
- L. Wu, L. Li, S. Chen, L. Wang and X. Lin, Sep. Purif. Technol., 2020, 247, 117014 CrossRef CAS
- G. Li, Q. Xie, Q. Liu, J. Liu, C. Wan, D. Liang and H. Zhang, Asia-Pac. J. Chem. Eng., 2020, 15, 1–10 CrossRef
- Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed
- J. Bentley, E. K. Olsen, J. P. Moore and J. M. Farrant, Phytochemistry, 2020, 173, 112323 CrossRef CAS PubMed
- M. Ruesgas-Ramón, M. L. Suárez-Quiroz, O. González-Ríos, B. Baréa, G. Cazals, M. C. Figueroa-Espinoza and E. Durand, J. Sci. Food Agric., 2020, 100, 81–91 CrossRef PubMed
- D. O. Abranches, L. P. Silva, M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, ChemSusChem, 2020, 13, 4916–4921 CrossRef CAS PubMed
- M. C. Roy, M. Alam, A. Saeid, B. C. Das, M. B. Mia, M. A. Rahman, J. B. Eun and M. Ahmed, J. Food Process. Preserv., 2018, 42, 1–9 Search PubMed
- E. Hassan, S. Fadel, W. Abou-Elseoud, M. Mahmoud and M. Hassan, Polymers, 2022, 14, 4605 CrossRef CAS PubMed
- T. Mada, R. Duraisamy and F. Guesh, Food Sci. Nutr., 2022, 10, 1222–1238 CrossRef CAS PubMed
- S. Valdivia-Rivera, I. E. Herrera-Pool, T. Ayora-Talavera, M. A. Lizardi-Jiménez, U. García-Cruz, J. C. Cuevas-Bernardino, J. M. Cervantes-Uc and N. Pacheco, Foods, 2021, 10, 2093 CrossRef CAS PubMed
- A. Elgharbawy, A. Hayyan, M. Hayyan, M. Mirghani, H. Salleh, S. Rashid, G. Ngoh, S. Liew, M. Nor, M. Y. Z. bin Mohd Yusoff and Y. Alias, Processes, 2019, 7, 416 CrossRef CAS
- S. Chen, L. Xiao, S. Li, T. Meng, L. Wang and W. Zhang, Ultrason. Sonochem., 2022, 86, 106045 CrossRef CAS PubMed
- M. H. Shafie and C. Y. Gan, Int. J. Biol. Macromol., 2020, 149, 835–843 CrossRef CAS PubMed
- N. N. T. Tien, N. L. Le, T. T. Khoi and A. Richel, J. Food Process. Preserv., 2022, 46, 1–12 Search PubMed
- M. H. Shafie, R. Yusof and C. Y. Gan, Carbohydr. Polym., 2019, 216, 303–311 CrossRef CAS PubMed
- S. Q. Liew, G. C. Ngoh, R. Yusoff and W. H. Teoh, Biocatal. Agric. Biotechnol., 2018, 13, 1–11 CrossRef
- M. Chen and M. Lahaye, Food Hydrocolloids, 2021, 115, 106601 CrossRef CAS
- M. Chen, X. Falourd and M. Lahaye, Carbohydr. Polym., 2021, 266, 118113 CrossRef CAS PubMed
- A. Nawirska and M. Kwaśniewska, Food Chem., 2005, 91, 221–225 CrossRef CAS
- P. Manara, A. Zabaniotou, C. Vanderghem and A. Richel, Catal. Today, 2014, 223, 25–34 CrossRef CAS
- Z. Luo, W. Li, J. Yan and J. Sun, Adv. Funct. Mater., 2022, 32, 2203988 CrossRef CAS
- M. Markiewicz, J. Maszkowska, V. Nardello-Rataj and S. Stolte, RSC Adv., 2016, 6, 87325–87331 RSC
- C. Hopson, V. Rigual, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodríguez, Ind. Crops Prod., 2022, 186, 115230 CrossRef CAS
- B. R. Thakur, R. K. Singh, A. K. Handa and M. A. Rao, Crit. Rev. Food Sci. Nutr., 1997, 37, 47–73 CrossRef CAS PubMed
- W. G. Willats, J. P. Knox and J. D. Mikkelsen, Trends Food Sci. Technol., 2006, 17, 97–104 CrossRef CAS
- S. B. Ross-Murphy and K. P. Shatwell, Biorheology, 1993, 30, 217–227 CAS
- M. M. Villar-Chavero, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, Carbohydr. Polym., 2020, 229, 115569 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00526k |
| This journal is © The Royal Society of Chemistry 2024 |