
In situ hierarchical self-assembly of NiFeHCF nanoparticles on nickel foam: highly active and ultra-stable bifunctional electrocatalysts for water splitting and their environmental assessment towards green energy†
Arunagiri
Gayathri
 ,
Venkatachalam
Ashok
,
Muthukumaran
Sangamithirai
,
Jayaraman
Jayabharathi
and
Venugopal
Thanikachalam
*
,
Venkatachalam
Ashok
,
Muthukumaran
Sangamithirai
,
Jayaraman
Jayabharathi
and
Venugopal
Thanikachalam
*
Department of Chemistry, Material Science Lab, Annamalai University, Annamalai Nagar, Tamil Nadu 608002, India. E-mail: vtchalam2005@yahoo.com
First published on 12th March 2024
Abstract
A 3D hierarchical nickel–iron hexacyanoferrate electrocatalyst was successfully grown on nickel foam using an energy-efficient in situ self-assembly method. The as-prepared NiFeHCF@NF electrode has good morphology and intimate contact with the NF compared to electrodes from the co-precipitation method. The well-designed NiFeHCF@NF nanostructure delivers prominent performances that require overpotentials as low as 210 and 125 mV@10 mA cm−2 for the OER and HER in 1 M KOH, respectively. Tafel slope and electrochemical impedance studies further revealed favourable kinetics during electrolysis. Hence, an NiFeHCF@NF||NiFeHCF@NF water electrolyser only required 1.56 V@10 mA cm−2 with an ∼2.5% potential loss. Furthermore, the synergistic effect of iron and nickel with ferrocyanide improved the structural stability and promoted the generation of active phases during the OER/HER, resulting in outstanding durability for 150 h. Moreover, the novel all-in-one strategy can be used to explore other bifunctional and cost-efficient electrocatalysts for various applications. The solar-based water electrolysis and environmental assessment confirmed the practical use of NiFeHCF@NF for eco-friendly industrial hydrogen production.
1. Introduction
Hydrogen production via electrocatalytic water splitting (EWS) is being extensively observed as a potential method for addressing the world's energy crisis and related environmental problems. EWS comprises two half-reactions: the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER); both are sluggish even while using effective benchmark catalysts of RuO2 and IrO2 (OER) and Pt/C (HER). However, high price, low abundance and instability limit the global scalability of sustainable energy technologies.1–6 Therefore, efficient, stable, abundant and cost-effective bifunctional electrocatalysts are essential for water splitting. Although numerous studies throughout the past few years have concentrated on transition metal-based chalcogenides, nitrides, oxides and phosphides, more emphasis has been paid to the use of materials derived from metal–organic frameworks (MOFs), which are well-known potential electrocatalysts.7–12 Recently, NiFe-LDH has been demonstrated as the best OER catalyst in alkaline electrolytes.13–15 However, its preparation process requires a high cost chain reaction, which makes it difficult to use in practice, hence, the need for a green synthetic method required to produce non-noble metal electrocatalysts without using additional energy, improving the number of catalytic sites by modifying their shape and structure, and enhancing the reactivity of catalytic sites by including additional components are now the essential approaches for enlightening catalytic performance.16,17Prussian blue analogues (PBAs), a form of perovskite-type MOF with high porosity, large surface area and variable metal catalytic sites have emerged in recent years as catalytic materials for EWS, rechargeable batteries, sensors and hydrogen storage applications.18–24 Andronescu et al. reported NiFeNO3 with excellent electrocatalytic activity (270 mV@10 mA cm−2) for the OER. Poor adhesion of the catalyst powder to the electrode surface is a significant challenge in the use of powder materials as electrocatalysts for energy conversion. Additionally, binder materials (Nafion) do not have adequate electrical conductivity, which prevents rapid electron transport between the catalyst particles and between particles and electrodes. Consequently, the possible current density becomes significantly reduced.25 Zhang et al. synthesized in situ grown iron–nickel nitride nanoparticles on nickel (FeNi3N/NF), which display low overpotentials of 75 and 202 mV for the HER and OER@10 mA cm−2, respectively.18 Zhang et al. synthesized NiFeLDH@NiCoP/NF electrodes with low overpotentials of 220/120 mV@10 mA cm−2 for the OER/HER, respectively.26 Lu et al. synthesized 3D-NiFeLDH film for the OER, which exhibited a small overpotential of 1.46 V.27 The aforementioned methods required extra energy supply and were costly, and hence are unsuitable for green energy production.
To overcome all these issues, we synthesised nickel–iron hexacyanoferrate (NiFeHCF@NF) on NF electrocatalysts by using an all-in-one strategy that includes (i) a binder-free in situ growth method; (ii) modifying their shape and structure; (iii) increasing the active sites by adding metal components; and (iv) NiFeHCF@NF being prepared via a complete green synthetic method. The advantages gained through the synthetic procedure have been thoroughly discussed in the results and discussion. The as-prepared NiFeHCF@NF was used as a bifunctional electrocatalyst and displayed outstanding catalytic performance for the HER (125 mV@10 mA cm−2) and OER (210 mV@10 mA cm−2). Notably, an NiFeHCF@NF electrolyser showed excellent bifunctional activity of 1.56 V in 1 M KOH@10 mA cm−2. The current findings provide a novel path for the development of economical and productive catalysts for electrocatalytic water splitting. Moreover, the easy accessibility of NiFeHCF@NF should facilitate commercial hydrogen production.
2. Results and discussion
Nickel–iron hexacyanoferrate (NiFeHCF) on NF was fabricated as a 3D self-supported electrode with bifunctional activity using an in situ hierarchical self-assembly method for electrocatalytic water splitting. Fig. 1 displays the step-wise synthetic procedure of NiFeHCF@NF (details are given in the ESI†). When utilised as an electrocatalyst for overall water splitting, the 3D hierarchical nanostructure of NiFeHCF@NF has several benefits. The porous 3D structure of NF is beneficial for releasing the H2 and O2 gas bubbles and is employed as the collector of current. The in situ hierarchical self-assembly eliminates the requirement for polymer binders (increased resistance) and creates a smooth channel for electron transport over the whole electrode.11,25,27 The successful in situ growth of NiFeHCF on NF was evidenced by the apparent difference in nickel foam colour (Fig. S1a†). The digital photographs of pure Ni foam, as-prepared samples and post-use materials are presented in Fig. S1b.† After water splitting treatment, the shape of the Ni foam remains unchanged but the colour and surface morphology of the electrocatalyst had changed. | ||
| Fig. 1 Synthetic scheme of NiFeHCF@NF. | ||
The morphology and microstructure of bare NF, NiFeHCF@NF and post-use materials were established by the FESEM technique (Fig. S2†). The surface of bare NF is smooth and free of impurities with a 3D porous structure that offers an abundant surface area to grow active material as a scaffold (Fig. S2a†).28 The FESEM images (Fig. S2b–d†) at high magnification show that the NiFeHCF@NF has a hierarchical rugged surface and that it was changed after its electrocatalytic performance. Numerous interconnected ultrathin nanosheets are vertically grown on NiFeHCF@NF after the HER and the rugged layers of the electrocatalyst become smooth after the OER owing to the generation of active sites during the process. Fig. 1 shows the colour and morphological changes of NiFeHCF@NF during the OER/HER and that the catalysts are well adhered to the nickel foam surface.
The crystalline nature and plane values of the prepared catalyst were confirmed by using XRD (Fig. 2a). NiFeHCF@NF has peaks at 2θ of 15.1° (111), 17.6° (200), 25° (220), 29.6° (311), 35.6° (400), 39.9° (420), 44.3° (422), 51.6° (440), 54.7° (600), 58.2° (620) and 74.9° (800). These peaks lie between the Fe4[Fe(CN)6]3 [JCPDS: 73-0687] and Ni2[Fe(CN)6] [JCPDS: 75-0037] peaks, which may be due to the presence of nickel- and iron-coordinated cyanides in NiFeHCF@NF.20,29 According to the XRD of bare NF (JCPDS No. 87-0712), the peaks at 44.3°, 51.6° and 76.5° have been produced due to the nickel foam substrate (Fig. S3†). The aforementioned results confirmed the successful formation of NiFeHCF in the nickel foam. The functional groups of NiFeHCF@NF were identified using FT-IR (Fig. 2b). The peak at 2087 cm−1 is attributed to the stretching frequency of Fe2+-CN-Ni2+. The bands at 1610 and 3400 cm−1 belong to the distortion vibrations and stretching modes of O–H in H2O, respectively. The peak at 597 cm−1 belongs to the in-plane deformation of Fe–C.30
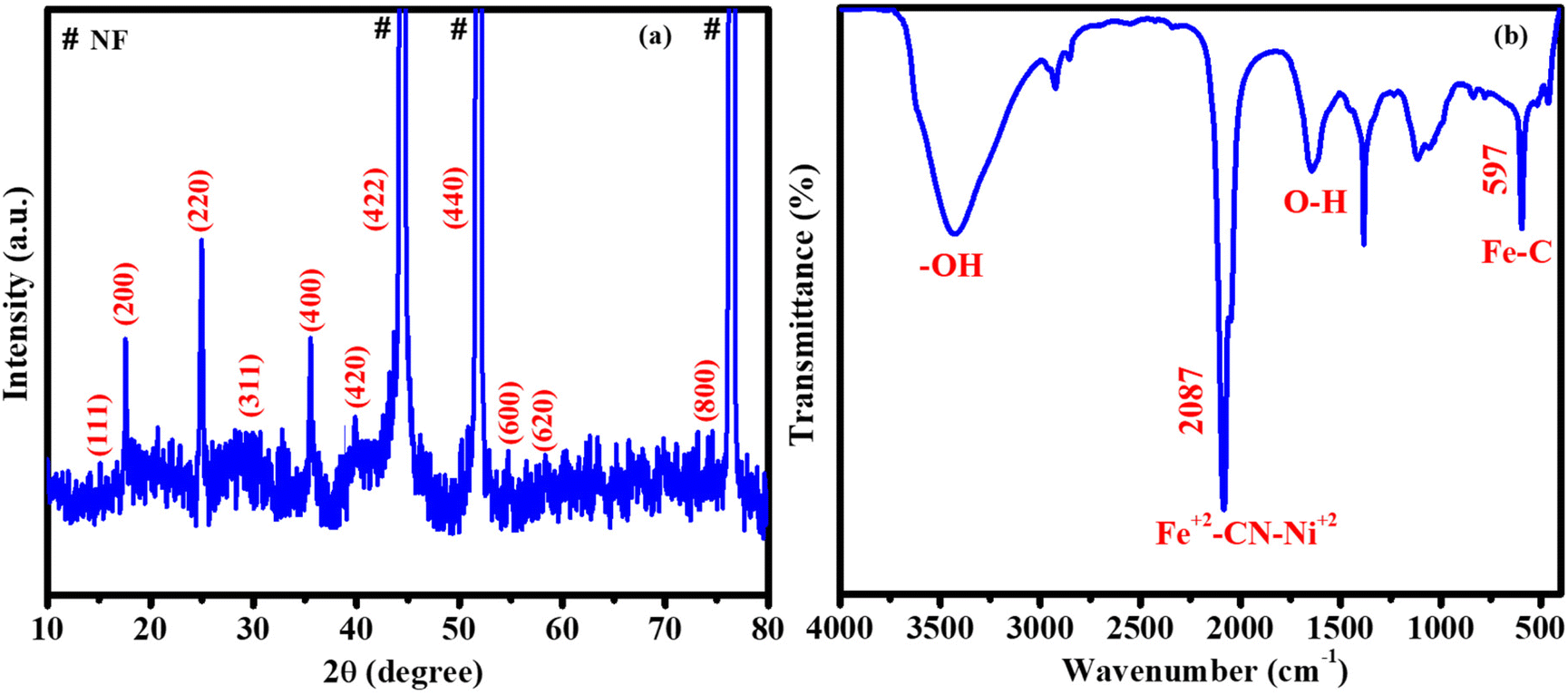 | ||
| Fig. 2 XRD pattern (a) and the FT-IR spectrum (b) of NiFeHCF@NF. | ||
The morphology of NiFeHCF@NF was examined by FESEM at different magnifications. Fig. 3a–d shows the formation of hierarchical coral-like NiFeHCF on the nickel foam. The hierarchical coral-like electrode consists of nanosized units that form a system of nanopores; it generates copious catalytic sites leading to improvement of the catalytic performance per geometric area and enables close contact with electrolytes, thus allowing more efficient utilization of active sites.27 The highly open hierarchical structure facilitates gas release during the HER and OER, and accordingly, this electrode showed high current density (∼500 mA cm−2). The SEM images of NiFeHCF nanoparticles prepared through a co-precipitation method show that they have agglomerated morphology (Fig. 3e and f). Agglomeration of nanoparticles has many disadvantages including reduced surface area, resistance and low catalyst–electrolyte interaction. The SEM images clearly show the morphological improvement of the in situ self-assembly method over the co-precipitation method.31–35 EDX elemental mapping confirmed the homogeneous distribution of component elements and the presence of constituent elements (Fig. S4†). The absence of other elements shows the purity of NiFeHCF@NF, which has the atomic percentages of 28.43% (C), 10.73% (N), 46.21% (O), 11.09% (Ni) and 3.51% (Fe) (Fig. S4g†). The successful in situ growth of NiFeHCF@NF without binder was confirmed by the FESEM images.
 | ||
| Fig. 3 FE-SEM of NiFeHCF@NF; (a–d) the in situ self-assembly method and (e and f) coprecipitation method. | ||
The crystalline structure and morphology of NiFeHCF@NF were further examined by using HRTEM and EDS. The catalysts were scraped from the NF owing to the tight contact of the catalyst with NF. Consequently, it was difficult to retain the original morphology and hence, the hierarchically grown coral-like morphology is inevitably separated.31Fig. 4a and b reveals that together they had a neat shape and smooth coral-like interconnected morphology. Higher magnification HRTEM images show lattice fringes with d spacings of 2.23 Å and 2.5 Å, which are assigned to the FCC (420) and (400) planes, respectively (Fig. 4c). The corresponding SAED (selected area electron diffraction) pattern exhibits concentric circles, which are assigned to the (220), (400), (422) and (440) planes, indicating the crystalline nature of NiFeHCF@NF and this was corroborated by XRD findings (Fig. 4d). The EDX spectrum confirms the existence of constituent elements and the absence of other elements, which shows the purity of the electrocatalyst (Fig. 4e). The atomic percentages of NiFeHCF@NF were determined to be 3.89% (Ni), 2.88% (Fe), 6.79% (O), 9.78% (N) and 76.66% (C). Additionally, inductively coupled plasma optical emission spectroscopy (ICP-OES) was used to analyse the elemental composition of NiFeHCF@NF and the results are shown in Table S1.†
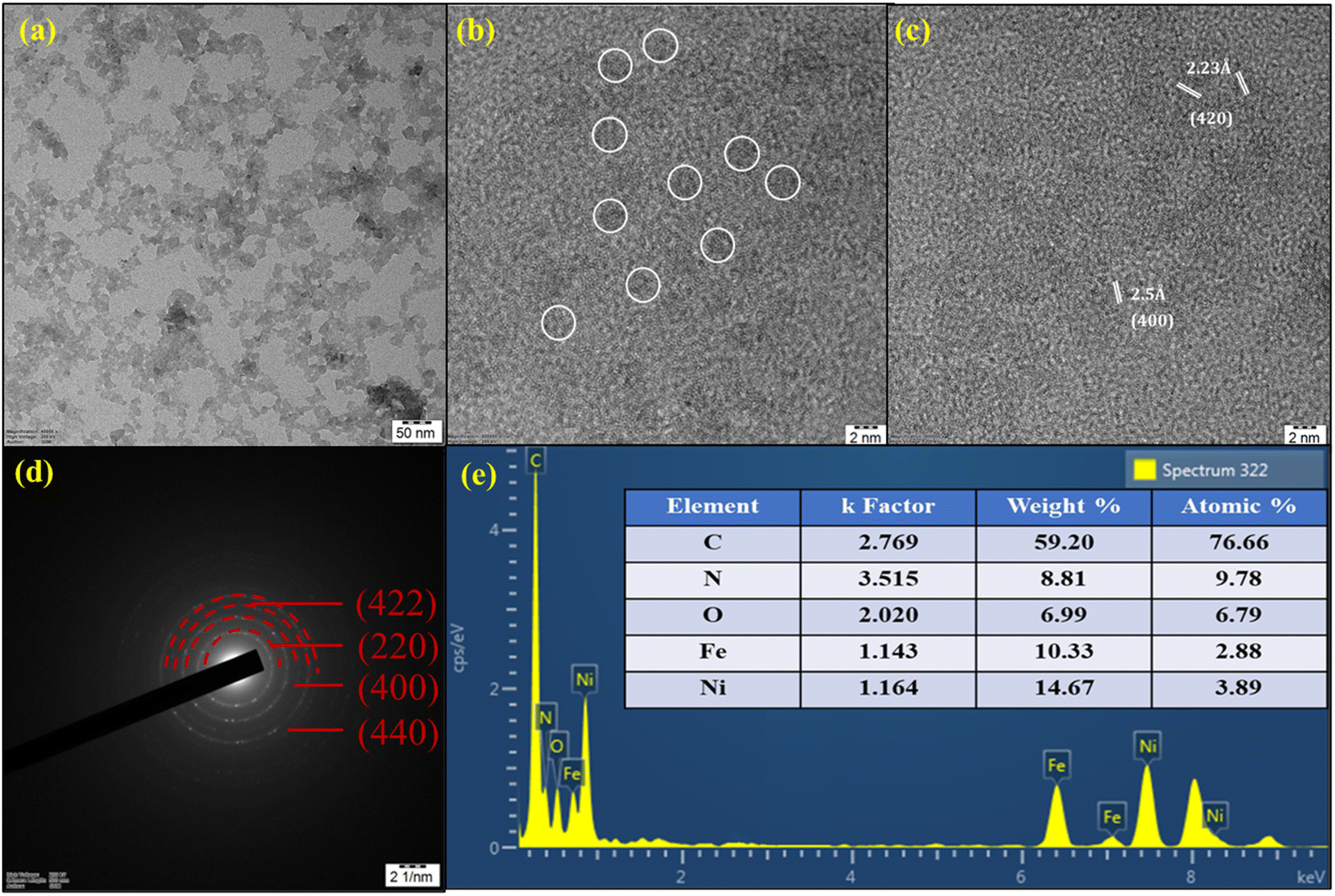 | ||
| Fig. 4 HR-TEM images of NiFeHCF@NF (a and b); lattice fringes (c); SAED pattern (d) and EDX spectrum (e). | ||
The composition and valence state of NiFeHCF@NF were identified by using XPS. The survey spectrum confirmed the presence of Ni, Fe, O, N and C elements in the electrocatalyst (Fig. 5a). The spectrum of C 1s shows peaks at ∼284.8 and ∼285.5 eV attributed to C–C and C–N fragments, respectively (Fig. 5b). Fig. 5c displays the N 1s spectrum corroborating the existence of cyanide content in the catalyst; the peaks at ∼398.1 and ∼398.6 eV are attributed to pristine ferrocyanide (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and metal–N of the ferrocyanide, respectively.36 The O 1s spectrum revealed signals at ∼531.0 and ∼533.1 eV attributed to coordinated water and adsorbed water, respectively (Fig. 5d). The Fe 2p spectrum has peaks at 708.8, 710.1 and 712.5 eV attributed to Fe2+, Fe3+ and satellite peaks of Fe 2p3/2 signals, respectively, and peaks at 721.6, 723.3 and 725.8 eV assigned to Fe2+, Fe3+ and satellite peaks of Fe 2p1/2 signals, respectively (Fig. 5e).37 High-resolution spectra of Ni 2p (Fig. 5f) having peaks at ∼856.0 eV (Ni 2p3/2) and ∼873.9 eV (Ni 2p1/2) and the coexistence of Ni3+ and Ni2+ were confirmed by the binding energy (17.9 eV) difference between the two peaks. Furthermore, it can be fitted into shakeup satellites and spin–orbit doublets using the Gaussian fitting method. The peaks at ∼855.9, ∼873.4 and the satellite at ∼862.1 eV corresponded to Ni2+ and those at ∼857.4, ∼874.8 and the satellite at ∼879.9 eV were assigned to Ni3+.38 Thus, the XPS conclusion further confirmed the formation of NiFeHCF@NF.
N) and metal–N of the ferrocyanide, respectively.36 The O 1s spectrum revealed signals at ∼531.0 and ∼533.1 eV attributed to coordinated water and adsorbed water, respectively (Fig. 5d). The Fe 2p spectrum has peaks at 708.8, 710.1 and 712.5 eV attributed to Fe2+, Fe3+ and satellite peaks of Fe 2p3/2 signals, respectively, and peaks at 721.6, 723.3 and 725.8 eV assigned to Fe2+, Fe3+ and satellite peaks of Fe 2p1/2 signals, respectively (Fig. 5e).37 High-resolution spectra of Ni 2p (Fig. 5f) having peaks at ∼856.0 eV (Ni 2p3/2) and ∼873.9 eV (Ni 2p1/2) and the coexistence of Ni3+ and Ni2+ were confirmed by the binding energy (17.9 eV) difference between the two peaks. Furthermore, it can be fitted into shakeup satellites and spin–orbit doublets using the Gaussian fitting method. The peaks at ∼855.9, ∼873.4 and the satellite at ∼862.1 eV corresponded to Ni2+ and those at ∼857.4, ∼874.8 and the satellite at ∼879.9 eV were assigned to Ni3+.38 Thus, the XPS conclusion further confirmed the formation of NiFeHCF@NF.
 | ||
| Fig. 5 XPS of NiFeHCF@NF: (a) XPS survey; (b) carbon; (c) nitrogen; (d) oxygen; (e) iron; and (f) nickel. | ||
3.1. Electrochemical characterization
The exact half-cell potential of NiFeHCF@NF was investigated by using a three-electrode setup in 1.0 M KOH. The overpotential of electrocatalysts was calculated by LSV in the potential range of 1.1–1.8 V. The overpotentials of FeNiLDH@NF, IrO2, NiFeHCF@NF, NiHCF@NF, FeHCF@NF, HCF@NF and bare NF@10 mA cm−2 are of 230, 341, 210, 290, 330, 370 and 481 mV, respectively (Fig. 6a and b). Compared to FeNiLDH@NF (64 mV dec−1), IrO2 (98 mV dec−1), NiHCF@NF (85 mV dec−1), FeHCF@NF (93 mV dec−1), HCF@NF (128 mV dec−1) and bare NF (150 mV dec−1), NiFeHCF@NF showed the smallest Tafel slope (56 mV dec−1) (Fig. 6c). The long-term durability of a catalytic electrode is another crucial issue for industrial purposes, particularly for these types of porous nanostructured electrocatalysts. The stability of NiFeHCF@NF shows negligible degradation (<2 and <3.5%)@10 and 100 mA cm−2, respectively, after 150 h of testing, revealing its excellent stability (Fig. 6d and S5a†). The OER efficiencies of NiFeHCF@NF electrocatalysts prepared via in situ self-assembly and co-precipitation have been compared with that of bare NF and required overpotential values of 210, 270 and 481 mV, respectively, to attain 10 mA cm−2 (Fig. S6a†). This was ascribed to the tight binding between the active material and the substrate, evidence for which was revealed by movie S1† and post-use OER FESEM (Fig. 10a–c). The turnover frequency (TOF) of NiFeLDH@NF (0.1183 s−1), IrO2 (0.0236 s−1), NiFeHCF@NF (0.2738 s−1), NiHCF@NF (0.0578 s−1), FeHCF@NF (0.0324 s−1), HCF@NF (0.0135 s−1) and NF (0.0029 s−1) was measured at 1.6 V. The benefits afforded by the 3D architecture were confirmed by the high performance and durability of NiFeHCF@NF. Combining the above merits, the 3D NiFeHCF@NF electrode was the most active non-precious 3D metal electrocatalyst (Table S2†). | ||
| Fig. 6 OER polarization curves (without iR-correction) of electrocatalysts: (a) LSV curve; (b) overpotential@10 mA cm−2; (c) Tafel slopes; and (d) the chronopotentiometry durability test (inset: post-use OER image). | ||
The general OER mechanism followed by the electrocatalyst (NiFeHCF@NF) is: M* + OH− → M–OH* + e−: M–OH* + OH− → M–O* + H2O + e−: M–O* + OH− → M–OOH* + e−: M–OOH* + OH− → O2 + H2O + e−. The excellent activity of NiFeHCF@NF was mainly derived from nickel in comparison with iron. During the OER process, Fe ions present as Fe2+/Fe3+ [Fe(OH)2/FeOOH] and Ni ions present as Ni2+/Ni3+ [Ni(OH)2/NiOOH] oxidation states were confirmed by post-use XPS results. The formed FeOOH optimized the electrons of nickel and assisted the development of active Ni(OH)2/NiOOH species, which are the active sites for the OER process. The outstanding durability and active sites during the OER of NiFeHCF@NF were also established by ex situ IR, XRD, SEM and XPS analyses. Both XRD and XPS results changed after the durability test, which confirmed the establishment of Ni(Fe)OOH and Ni(Fe)(OH)2 as catalytic sites. Similarly, the SEM images revealed the well-preserved morphology with an amorphous layer after the OER.26,39–42
The half-cell potential of Pt/C, NiFeHCF@NF, NiHCF@NF, FeHCF@NF, HCF@NF and bare NF for the HER activity was studied by LSV (without iR correction) in a three-electrode system. The LSV was spotted in the potential range of −0.4–0.1 V in 1 M KOH. The LSV and Tafel slope revealed that NiFeHCF@NF has lower overpotential (125 mV) and Tafel value (80 mV dec−1) than those of NiHCF@NF (200 mV; 99 mV dec−1), FeHCF@NF (265 mV; 117 mV dec−1), HCF@NF (295 mV; 126 mV dec−1) and bare NF (318 mV; 150 mV dec−1). Although Pt/C has lower overpotential (51 mV) and Tafel slope (62 mV dec−1) than NiFeHCF@NF, NiFeHCF@NF shows outstanding activity@−10 mA cm−2 compared to the recently stated non-precious metal electrocatalysts (Table S2†). The lower overpotential and Tafel value reveal the fabulous activity and fast kinetics of NiFeHCF@NF (Fig. 7a–c). The calculated TOFs at −0.2 V of 0.1206 s−1 (NiFeHCF@NF), 0.0259 s−1 (NiHCF@NF), 0.0097 s−1 (FeHCF@NF), 0.0071 s−1 (HCF@NF), 0.1496 s−1 (Pt/C) and 0.0067 s−1 (NF) show the extraordinary HER activity of NiFeHCF@NF over a definite period. The NiFeHCF@NF shows constant activity and outstanding stability over 150 h@10 and 100 mA cm−2 with 2.2% and 4.6% potential loss, respectively (Fig. 7d and S5b†).
 | ||
| Fig. 7 HER polarization curves (without iR-correction) of electrocatalysts: (a) LSV curve; (b) overpotential@−10 mA cm−2; (c) Tafel slopes; and (d) the chronopotentiometry durability test (inset: post-use HER image). | ||
In alkaline electrolytes, the overall HER mechanism followed by the NiFeHCF@NF electrocatalyst may be Volmer–Heyrovsky: [Mcat + e− + H2O ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) OH− + McatHchem (Volmer); McatHchem + e− + H2O H2↑ + OH− + Mcat (Heyrovsky)].40 The HER efficiencies of NiFeHCF@NF electrocatalysts prepared via in situ self-assembly and co-precipitation have been compared with bare NF and required overpotentials of 125, 162 and 318 mV, respectively to attain −10 mA cm−2 (Fig. S6b†). The establishment of Ni(Fe)(OH)2 on the surface of NiFeHCF@NF may serve as the catalytic site for the HER, which was supported by the post-use analysis (Fig. 10 and 11).26,33,43–45
OH− + McatHchem (Volmer); McatHchem + e− + H2O H2↑ + OH− + Mcat (Heyrovsky)].40 The HER efficiencies of NiFeHCF@NF electrocatalysts prepared via in situ self-assembly and co-precipitation have been compared with bare NF and required overpotentials of 125, 162 and 318 mV, respectively to attain −10 mA cm−2 (Fig. S6b†). The establishment of Ni(Fe)(OH)2 on the surface of NiFeHCF@NF may serve as the catalytic site for the HER, which was supported by the post-use analysis (Fig. 10 and 11).26,33,43–45
The comparison of OER/HER activity of NiFeHCF@NF, NiHCF@NF, FeHCF@NF and HCF@NF confirms that the excellent activity of NiFeHCF@NF may be due to the synergism of Ni and Fe in the composite. Specifically, the electron redistribution between iron, nickel and ferrocyanide was regulated and optimized the adsorption energy of OER/HER reaction intermediates. Owing to the merits of higher electrical conductivity, more surface-active sites and higher oxidation states of ions, NiFeHCF@NF exhibited excellent OER/HER catalytic performance. Specifically, ultralow overpotential within the large current density region, small Tafel value and consistent stability were included. Current research exposed the observations that iron addition not only caused optimal adsorption energy of intermediates but also induced the partial charge transfer to Ni sites to provide improved electrocatalytic performance.11,31,45–49
Additionally, to reveal the influence of the electrochemically active surface area (EASA) and structure–activity relationship, we calculated the double-layer capacitance by conducting cyclic voltammogram (CV) tests on the electrodes. CV curves have been produced at various scan rates of 10–60 mV s−1 within a range of 1.20–1.30 V vs. RHE, regardless of evident faradaic activities. NiFeHCF@NF exhibits significantly larger linear slope when the current density difference (Δj) at 1.25 V vs. RHE is plotted versus the scan rate (Fig. 8a); a greater Cdl of 5.745 mF cm−2 was observed as related to that of the bare NF substrate (1.617 mF cm−2). NiFeHCF@NF has 3.5 times greater capacitance than bare NF, which may be ascribed to its porous nanostructure.21 The larger capacitance of NiFeHCF@NF compard to Ni foam indicates the large surface area of the catalyst. The large EASA (143.6) and RF (574.5) values of NiFeHCF@NF over those of bare NF (EASA - 40.4 and RF - 161.7) confirmed the large electrochemical surface area of NiFeHCF@NF. To ensure the intrinsic activity of the NiFeHCF@NF catalyst, its EASA-normalized LSV curve was analysed (Fig. S7†). The overpotential needed for a current density of 0.1 mA cm−2EASA was 227 mV for NiFeHCF@NF, confirming its higher intrinsic activity. The higher OER activity of NiFeHCF@NF is not only due to the larger EASA but also due to the enhanced intrinsic activity of the catalyst as a result of the optimised electronic structure.13,50 The hierarchical nature offered copious catalytic sites, which facilitated electrolyte infiltration and assisted fast charge and mass transportation and contributed to the extraordinary activity for the OER and HER.18,19
 | ||
| Fig. 8 The double-layer capacitance (a) and the Nyquist plot (b) of different electrocatalysts. | ||
Electrochemical impedance results are depicted in Fig. 8b accompanied by circuit model fitting analysis (inset: Randles equivalent circuit diagram, where Q1 and Q2 denote the double-layer capacitance; R1 and R2 denote the charge transfer resistance and Rs denotes the electrolyte resistance). The smaller Rct of NiFeHCF@NF (2.5 Ω) than that of NiHCF@NF (3.9 Ω), FeHCF@NF (4.2 Ω), HCF@NF (4.6 Ω) and bare NF (6.4 Ω) may be due to the strong contact between NiFeHCF and conductive nickel foam substrate, which offers a very active network for electron transfer throughout the electrode, resulting in rapid reaction kinetics. The different starting points of each electrocatalyst indicate internal resistance between the catalyst/electrolyte interfaces, and we observe that NiFeHCF has very low internal resistance compared to other electrocatalysts due to its intimate contact with the current collector.51,52 The successful hierarchical self-assembly of NiFeHCF on the NF substrate further enables the synergism of various elements, which boosted both electron and mass transportation, promoted the conductivity and substantially assisted the electrochemical activity.49,53
Encouraged by the easy fabrication, high efficiency and durability of the NiFeHCF@NF electrode toward the OER and HER, we tested the possibility of utilizing it as the cathode and anode (NiFeHCF@NF||NiFeHCF@NF) in a two-electrode system. It was very clear to observe the evolution of H2 and O2 from the cathode and anode, respectively (Movie S1†). Fig. 9a shows the polarization curve of the NiFeHCF@NF||NiFeHCF@NF electrolyser in 1.0 M KOH. A cell voltage of only 1.56 V is needed for achieving 10 mA cm−2, lower than the voltage required for benchmark electrolysers, Pt/C/NF||IrO2/NF (1.62 V) and bare NF||bare NF (1.81 V). The smaller cell voltage reveals that the 3D nano-structured NiFeHCF@NF utilized the advantage of iron, nickel and ferrocyanide components and produced excellent activity for overall water splitting.31 The as-prepared NiFeHCF@NF electrodes achieved excellent bifunctional activity in 1 M KOH compared to that of recently reported Earth-abundant electrocatalysts (Table S2†). Additionally, we investigated the prolonged stability of the fabricated electrolyser, which was extremely excellent. A steady voltage had been preserved for over 150 h@10 mA cm−2 and @100 mA cm−2 (Fig. 9b and S5c†).
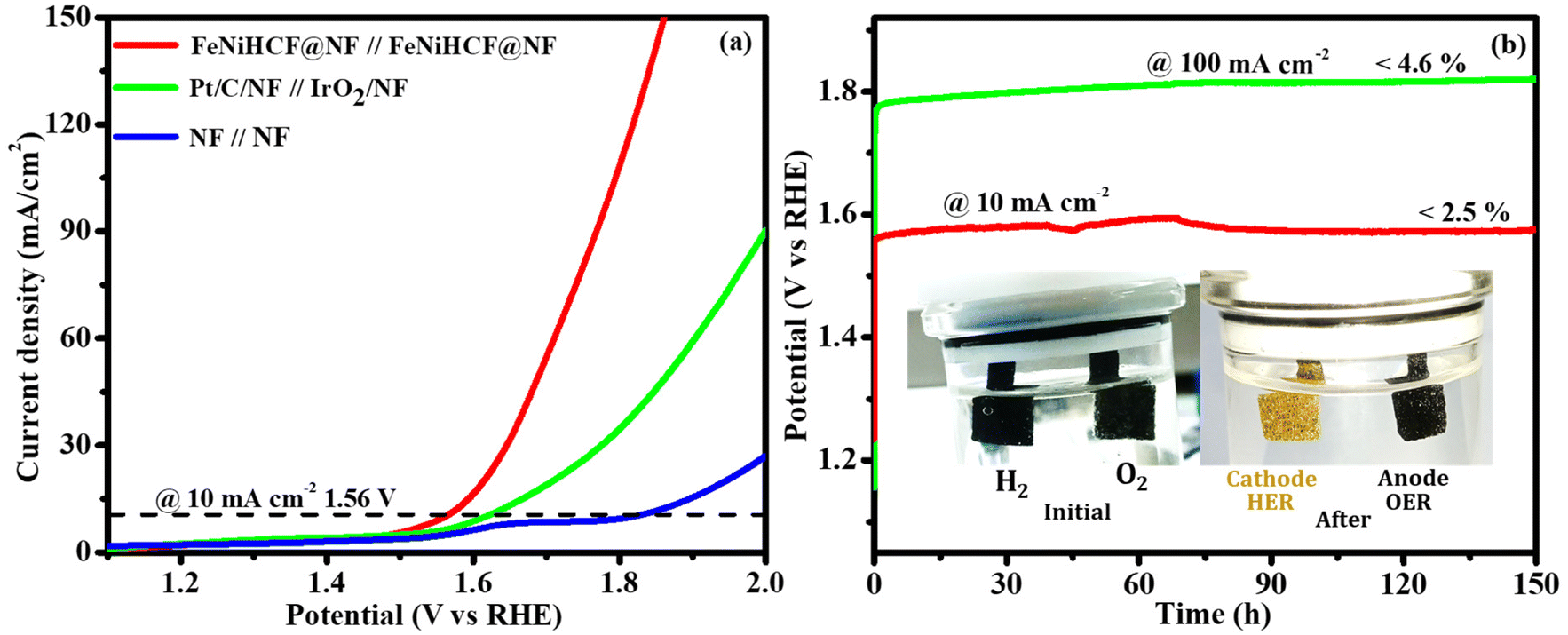 | ||
| Fig. 9 (a) LSV polarization curve (without iR-correction) of NiFeHCF@NF for overall water splitting. (b) Chronopotentiometry durability test (inset: optical photograph of the electrodes). | ||
To further gain insights into the reaction mechanism, the NiFeHCF@NF electrode after the HER and OER was characterized by using FESEM, XRD, FT-IR and XPS analyses. Fig. S8a† presents the post-use XRD of NiFeHCF@NF after the HER process. The crystalline character of the catalyst remained constant and the additional peaks at 19°, 23°, 34°, 59° and 71° are attributable to nickel hydroxide (JCPDS No. 14-0117). Fig. S8b† demonstrates an amorphous nature after the OER analysis with peaks marked “#” denoting the nickel foam substrate (JCPDS No. 87-0712). This may be related to the development of amorphous FeOOH and NiOOH during the OER. Fe3+ and Ni3+ boosted the electrophilicity of adsorbed oxygen, which is predominantly beneficial for the OER. The results are consistent with post-use XPS findings (Fig. 11).43,48,54
The post-use HER-IR spectra of NiFeHCF@NF showed new peaks at ∼890 cm−1 (M–OH) and ∼1278.76 cm−1 belonging to NiFe(OH), which support the formation of metal hydroxides after the HER (Fig. S9a†). The Fe2+–CN–Ni2+ peak at 2087 cm−1 was retained after the HER. The peaks at ∼1356, ∼1635 and ∼3000–3500 cm−1 are attributed to bending, stretching and scissoring of OH, respectively. The post-use OER-IR spectra of NiFeHCF@NF showed a new peak at 677.36 cm−1 ascribed to FeOOH (Fig. S9b†). The peak intensity at 2087 cm−1 was greatly reduced after the OER process. According to post-use IR analyses, the surface of NiFeHCF@NF was enriched with hydroxides (during the HER) and hydroxides/oxyhydroxides (during the OER), these being the electrocatalytically active phases, which is corroborated by the post-use XPS and XRD results.55–57
The post-use FESEM images of NiFeHCF@NF show that the surface of the coral-like structure becomes smooth after the OER process, and it is this that supports the formation of metal oxyhydroxide during the OER process (Fig. 10a–c).58 This agrees with both post-use XRD and post-use XPS findings. EDX elemental mapping confirmed the homogeneous distribution of component elements and the presence of constituent elements after 150 h of stability testing (Fig. S10 and S11†). The absence of other elements shows the purity of NiFeHCF@NF, which has atomic percentages of 27.13% (C), 2.80% (N), 52.14% (O), 16.61% (Ni) and 1.32% (Fe) after the OER (Fig. S10g†). The increment of oxygen in the EDX mapping and the smooth surface confirm the formation of oxyhydroxide during the OER process. Fig. 10d–f shows the morphology change from coral to nanosheet after the HER process, which supports the formation of metal hydroxides during the HER process. After the HER process, NiFeHCF@NF has the atomic percentages of 23.92% (C), 3.61% (N), 50.91% (O), 19.73% (Ni) and 1.83% (Fe) (Fig. S11g†). Movie S1† confirmed the good adhesion of electrocatalyst on the nickel form surface. The higher stability even after 150 h with vigorous H2/O2 gas evolution shows the ultra-sustainability of NiFeHCF@NF. Detachment of the catalyst from the electrode could be the major cause of its faster activity loss. Hence, post-use FESEM analysis and movie S1† confirmed the good adhesion of the electrocatalyst with NF. Comparison of morphology and colour changes of electrocatalyst before and after the OER and HER processes confirmed the development of hydroxides and oxyhydroxides after the HER and OER, respectively (Fig. S1 and S2†). The surface of NiFeHCF@NF was enriched with hydroxides (during the HER) and hydroxides/oxyhydroxides (during the OER), which were transformed from pristine NiFeHCF@NF.25,26,43
 | ||
| Fig. 10 Post-use FE-SEM image of NiFeHCF@NF; after the OER (a–c) and after the HER (d–f). | ||
Post-use XPS was carried out to validate the change of oxidation state and composition after the electrocatalytic performance. However, the surface oxidation state and composition of NiFeHCF@NF were changed substantially after long-term electrocatalytic performance. Compared with the HER operating in a reducing potential environment, the strong anodic oxidation in the OER potential range leads to an evident and irreversible phase transformation of metal ferrocyanide to metal oxide/oxyhydroxide. Here, post-use XRD and FESEM characterization also suggested that the main crystalline phase and morphology of NiFeHCF@NF were changed after long-term OER/HER measurement.
For better understanding, XPS results of post-use OER and HER have together been compared. The post-use OER and HER Ni 2p spectrum revealed that the peak position was the same as that of the pristine electrocatalyst but the domination of peak intensity played an important role (Fig. 11c and f). The peaks at ∼855.9, ∼873.4 eV and the satellite peak at ∼862.1 eV corresponded to Ni2+ and those at ∼857.4, ∼874.8 eV and the satellite peak at ∼879.9 eV were attributed to Ni3+. An evident shape change was found on the nickel spectrum in which the main peak of Ni3+/Ni2+ accounted for 89% due to surface oxidation/reduction during the OER/HER process, which confirms the formation of NiOOH and Ni(OH)2 layers during the OER/HER process.38Fig. 11a and d shows the post-use oxygen XPS spectrum, where new peaks at 529.4, 530.7 and 531.7 eV are ascribed to M–OOH, Ni(Fe)–O and M–OH, respectively, while the new peak at 531.7 eV belongs to M–OH after the OER. The peak intensity of absorbed water at 533.0 eV was reduced after the HER process.26 As shown in the Fe 2p spectrum, two resolved peaks at 2p3/2 (713.5 eV) and 2p1/2 (725.7 eV) and the satellite peak of 2p3/2 at 719.9 eV indicated the presence of iron as Fe3+ after the OER process (Fig. 11b).53 The signal that disappeared at 708.8 eV indicates the conversion of iron to a higher oxidation state after the OER process. The post-use HER Fe 2p spectrum showed two peaks at 708.5 and 721.6 eV for Fe 2p3/2 and 2p1/2 of Fe2+ with satellite peaks at 712.5 and 725.8 eV, respectively (Fig. 11e), which is well consistent with the as-prepared catalyst and demonstrates the existence of Fe2+ after the HER process. These findings demonstrated that the surface of NiFeHCF@NF was enriched by the newly formed Ni(Fe)OOH and Ni(Fe)(OH)2 layers after the OER/HER process, which have been regarded as the electrocatalytically active phases during the OER/HER process.26,56
 | ||
| Fig. 11 Post-use OER-XPS spectra of (a) oxygen, (b) iron, (c) nickel, and post-use HER-XPS spectra of (d) oxygen, (e) iron, and (f) nickel. | ||
The hierarchical NiFeHCF electrode displays highly efficient bifunctional catalytic activity for overall water splitting. The superior catalytic activity of NiFeHCF@NF is due to the following features: (1) close interaction of coral-like nanoparticles directly formed on a highly conductive 3D NF substrate not only enables full utilisation of the electrocatalyst active sites and efficient electron and mass transfer, but also avoids the need for polymer binders, which impart extra resistance;59 (2) synergistic effect of two active metal sites via charge transfer;31 (3) complete coverage of the pristine NF surface by NiFeHCF, which prevents corrosion and enhances the durability of the 3D structure;11 (4) owing to the aggregation-free vertically grown coral nanostructure, the 3D electrode has a larger surface area compared to that of the bare current collector, suggesting a high density of active sites for the OER/HER;18 (5) in situ growth on the metal foam substrate improved the structural stability and provided faster charge transfer as well as hydrogen/oxygen bubbles release;31 and (6) the proposed synthetic procedure provides high reproducibility in terms of both catalytic activity and operational stability of the resulting material.
3.2. Solar-assisted water splitting
The production of hydrogen by solar-assisted water electrolysis has become an economical and sustainable method. Fig. 12 shows a schematic diagram of solar-assisted water splitting using the NiFeHCF@NF||NiFeHCF@NF water electrolyser. Solar energy is fed into the NiFeHCF@NF||NiFeHCF@NF water electrolyser to generate electricity. The electrodes, present in 1 M KOH, and the solar panel (5.63 V) are connected by using crocodile clips. The voltage of the water electrolyser is measured and adjusted using a voltmeter. The solar-based water electrolyser only requires 1.56 V to attain 10 mA cm−2 with continuous evolution of H2 and O2 bubbles at the cathode and anode showing the effectiveness toward H2 production (Fig. S12: Movie S2†). The amount of O2 and H2 generated by using NiFeHCF@NF||NiFeHCF@NF was 1.52 and 3.12 μmol min−1, respectively, which is equal to ∼96.5% faradaic efficiency (Fig. S13†).60 Therefore, the solar-based water electrolyser developed in the present work is highly advised for inexpensive large-scale H2 generation. | ||
| Fig. 12 Schematic diagram of solar-assisted water splitting. | ||
3.3. Environmental impact assessment
The level of the greenness of NiFeHCF@NF and its synthetic process were evaluated quantitatively and qualitatively, utilising presently suggested technological development based on mass-relevant sustainability and socioeconomic elements are compared the environmental effect to past efforts (Table S3†). The corresponding calculations were performed by applying the equations specified in the ESI (SI-S5†).4. Conclusion
In summary, a binder-free bifunctional electrocatalyst with a 3D hierarchical nanostructure of NiFeHCF on NF has been synthesised via a simple in situ growth strategy. The results showed that the NiFeHCF@NF was effectively coupled with robust electronic interaction leading to facilitated charge transfer as well as enhanced reaction kinetics. The enhanced activity of the bifunctional NiFeHCF@NF electrocatalyst was determined by its low overpotential of 210/125 mV@10 mA cm−2 without iR correction for the OER/HER, respectively. An NiFeHCF@NF couple as a water electrolyzer requires 1.56 V to attain 10 mA cm−2 with a durability of 150 h. The solar-based water electrolyser requires 1.56 V@10 mA cm−2. The environmental impact assessments and solar-based water electrolysis respectively confirmed the greenness and effectiveness of NiFeHCF@NF. Hence, NiFeHCF@NF is a potential electrode for industrial hydrogen production.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The author A. Gayathri acknowledges the DST-INSPIRE, Govt. of India, for the award of a Junior Research Fellowship (DST INSPIRE – JRF IF210159/DST/INSPIRE Fellowship/2021).References
- S. Singh, S. Jain, P. S. Venkateswaran, A. K. Tiwari, M. R. Nouni, J. K. Pandey and S. Goel, Renewable Sustainable Energy Rev., 2015, 51, 623–633 CrossRef CAS
.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4(45), 17587–17603 RSC
- J. Wang, X. Yue, Y. Yang, S. Sirisomboonchai, P. Wang, X. Ma, A. Abudula and G. Guan, J. Alloys Compd., 2020, 819, 153346 CrossRef CAS
- A. Raveendran, M. Chandran and R. Dhanusuraman, RSC Adv., 2023, 13(6), 3843–3876 RSC
- H. Jin, J. Xu, H. Liu, H. Shen, H. Yu, M. Jaroniec, Y. Zheng and S. Z. Qiao, Sci. Adv., 2023, 9(42), eadi7755 CrossRef CAS PubMed
- G. Fang, K. Liu, M. Fan, J. Xian, Z. Wu, L. Wei, H. Tian, H. Jiang, W. Xu, H. Jin and J. Wan, Carbon Neutral., 2023, 2(6), 709–720 CrossRef
- G. Kalaiyarasan, K. Aswathi and J. Joseph, Int. J. Hydrogen Energy, 2017, 42(36), 22866–22876 CrossRef CAS
- M. Sangamithirai, S. Mathi, V. Ashok and J. Jayabharathi, ChemistrySelect, 2023, 8(10), e202300102 CrossRef CAS
- M. Vijayarangan, A. Gayathri, V. Ashok, M. Sangamithirai, J. Jayabharathi and V. Thanikachalam, Energy Fuels, 2024, 38(2), 1364–1372 CrossRef CAS
- M. Batool, A. Hameed and M. A. Nadeem, Coord. Chem. Rev., 2023, 480, 215029 CrossRef CAS
- H. Liang, A. N. Gandi, D. H. Anjum, X. Wang, U. Schwingenschlögl and H. N. Alshareef, Nano Lett., 2016, 16(12), 7718–7725 CrossRef CAS PubMed
- J. Liu, D. Zhu, T. Ling, A. Vasileff and S. Z. Qiao, Nano Energy, 2017, 40, 264–273 CrossRef CAS
- S. S. Jeon, P. W. Kang, M. Klingenhof, H. Lee, F. Dionigi and P. Strasser, ACS Catal., 2023, 13(2), 1186–1196 CrossRef CAS
- H. Liu, W. Shen, H. Jin, J. Xu, P. Xi, J. Dong, Y. Zheng and S. Z. Qiao, Angew. Chem., Int. Ed., 2023, 62(46), e202311674 CrossRef CAS PubMed
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan and A. Bergmann, Nat. Commun., 2020, 11(1), 2522 CrossRef CAS PubMed
- A. Hayat, M. Sohail, H. Ali, T. A. Taha, H. I. A. Qazi, N. Ur Rahman, Z. Ajmal, A. Kalam, A. G. Al-Sehemi, S. Wageh and M. A. Amin, Chem. Rec., 2023, 23(2), e202200149 CrossRef CAS PubMed
- T. Tang, W. J. Jiang, S. Niu, N. Liu, H. Luo, Y. Y. Chen, S. F. Jin, F. Gao, L. J. Wan and J. S. Hu, J. Am. Chem. Soc., 2017, 139(24), 8320–8328 CrossRef CAS PubMed
- B. Zhang, C. Xiao, S. Xie, J. Liang, X. Chen and Y. Tang, Chem. Mater., 2016, 28(19), 6934–6941 CrossRef CAS
- X. Zhang, I. U. Khan, S. Huo, Y. Zhao, B. Liang, K. Li and H. Wang, Electrochim. Acta, 2020, 363, 137211 CrossRef CAS
- S. Chong, J. Yang, L. Sun, S. Guo, Y. Liu and H. K. Liu, ACS Nano, 2020, 14(8), 9807–9818 CrossRef CAS PubMed
- C. Chen, D. Xiong, M. Gu, C. Lu, F. Y. Yi and X. Ma, ACS Appl. Mater. Interfaces, 2020, 12(31), 35365–35374 CrossRef CAS PubMed
- B. Singh and A. Indra, Mater. Today Energy, 2020, 16, 100404 CrossRef
- Y. Wu, Y. Li, J. Gao and Q. Zhang, SusMat, 2021, 1(1), 66–87 CrossRef CAS
- Y. Pan, J. Gao, Y. Li, E. Lv, U. Khan, X. Yang, J. Yao, A. Nairan and Q. Zhang, Small, 2024, 20(3), e2304594 CrossRef PubMed
- C. Andronescu, S. Barwe, E. Ventosa, J. Masa, E. Vasile, B. Konkena, S. Möller and W. Schuhmann, Angew. Chem., Int. Ed., 2017, 56(37), 11258–11262 CrossRef CAS PubMed
- H. Zhang, X. Li, A. Hähnel, V. Naumann, C. Lin, S. Azimi, S. L. Schweizer, A. W. Maijenburg and R. B. Wehrspohn, Adv. Funct. Mater., 2018, 28(14), 1706847 CrossRef
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50(49), 6479–6482 RSC
- Z. Lu, Q. Yang, W. Zhu, Z. Chang, J. Liu, X. Sun, D. G. Evans and X. Duan, Nano Res., 2012, 5, 369–378 CrossRef CAS
- S. Yu, Y. Li, Y. Lu, B. Xu, Q. Wang, M. Yan and Y. Jiang, J. Power Sources, 2015, 275, 45–49 CrossRef CAS
- Z. Guo, R. Song, L. Zhang, Z. Li, H. Yao, Q. Liu, J. Wang and Z. Li, J. Colloid Interface Sci., 2022, 613, 796–805 CrossRef CAS PubMed
- X. Xu, T. Wang, L. Su, Y. Zhang, L. Dong and X. Miao, ACS Sustainable Chem. Eng., 2021, 9(16), 5693–5704 CrossRef CAS
- C. Zhang, M. Shao, L. Zhou, Z. Li, K. Xiao and M. Wei, ACS Appl. Mater. Interfaces, 2016, 8(49), 33697–33703 CrossRef CAS PubMed
- M. Li, H. Wang, W. Zhu, W. Li, C. Wang and X. Lu, Adv. Sci., 2020, 7(2), 1901833 CrossRef CAS PubMed
- S. Khairunnisa, V. Wonoputri and T. W. Samadhi, Mater. Sci. Eng., C, 2021, 1(1143), 012006 Search PubMed
- S. Shrestha, B. Wang and P. Dutta, Adv. Colloid Interface Sci., 2020, 279, 102162 CrossRef CAS PubMed
- L. Shen, Q. Zhang, J. Luo, H. C. Fu, X. H. Chen, L. L. Wu, H. Q. Luo and N. B. Li, Appl. Surf. Sci., 2021, 551, 149360 CrossRef CAS
- A. Gayathri, S. Mathi, M. Vijayarangan, J. Jayabharathi and V. Thanikachalam, ChemistrySelect, 2022, 7(45), e202203616 CrossRef CAS
- Z. Liu, B. Tang, X. Gu, H. Liu and L. Feng, Chem. Eng. J., 2020, 395, 125170 CrossRef CAS
- B. K. Barman and K. K. Nanda, Green Chem., 2016, 18(2), 427–432 RSC
- X. Bo, R. K. Hocking, S. Zhou, Y. Li, X. Chen, J. Zhuang, Y. Du and C. Zhao, Energy Environ. Sci., 2020, 13(11), 4225–4237 RSC
- K. Jiang, W. Liu, W. Lai, M. Wang, Q. Li, Z. Wang, J. Yuan, Y. Deng, J. Bao and H. Ji, Inorg. Chem., 2021, 60(22), 17371–17378 CrossRef CAS PubMed
- X. An, Q. Hu, W. Zhu, L. Liu, Y. Zhang and J. Zhao, Appl. Phys. A, 2021, 127, 1–9 CrossRef
- D. R. Chowdhury, L. Spiccia, S. S. Amritphale, A. Paul and A. Singh, J. Mater. Chem. A, 2016, 4(10), 3655–3660 RSC
- A. Gayathri, M. Vijayarangan, M. Sangamithirai, V. Ashok, J. Jayabharathi and V. Thanikachalam, Energy Fuels, 2023, 37(24), 19812–19821 CrossRef CAS
- Z. Wu, Y. Zhao, H. Wu, Y. Gao, Z. Chen, W. Jin, J. Wang, T. Ma and L. Wang, Adv. Funct. Mater., 2021, 31(17), 2010437 CrossRef CAS
- H. Yang, C. Wang, Y. Zhang and Q. Wang, Sci. China Mater., 2019, 62(5), 681–689 CrossRef CAS
- J. R. Swierk, S. Klaus, L. Trotochaud, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2015, 119(33), 19022–19029 CrossRef CAS
- Y. Wu, Y. Li, M. Yuan, Z. Lü, L. Xu and B. Wei, J. Alloys Compd., 2020, 847, 156363 CrossRef CAS
- U. Y. Qazi, C. Z. Yuan, N. Ullah, Y. F. Jiang, M. Imran, A. Zeb, S. J. Zhao, R. Javaid and A. W. Xu, ACS Appl. Mater. Interfaces, 2017, 9(34), 28627–28634 CrossRef CAS PubMed
- Y. Huang, L. W. Jiang, B. Y. Shi, K. M. Ryan and J. J. Wang, Adv. Sci., 2021, 8(18), 2101775 CrossRef CAS PubMed
- X. Yan, W. D. Zhang, Q. T. Hu, J. Liu, T. Li, Y. Liu and Z. G. Gu, Int. J. Hydrogen Energy, 2019, 44(51), 27664–27670 CrossRef CAS
- B. A. Mei, O. Munteshari, J. Lau, B. Dunn and L. Pilon, J. Phys. Chem. C, 2018, 122(1), 194–206 CrossRef CAS
- Y. Zhang, K. Rui, Z. Ma, W. Sun, Q. Wang, P. Wu, Q. Zhang, D. Li, M. Du, W. Zhang and H. Lin, Chem. Mater., 2018, 30(14), 4762–4769 CrossRef CAS
- Y. Yang, H. Yao, Z. Yu, S. M. Islam, H. He, M. Yuan, Y. Yue, K. Xu, W. Hao, G. Sun and H. Li, J. Mater. Chem. A, 2019, 141(26), 10417–10430 CAS
- B. Vishnu, S. Mathi, S. Sriram and J. Jayabharathi, ChemistrySelect, 2022, 7(33), e202201682 CrossRef CAS
- Q. Yan, T. Wei, J. Wu, X. Yang, M. Zhu, K. Cheng, K. Ye, K. Zhu, J. Yan, D. Cao and G. Wang, ACS Sustainable Chem. Eng., 2018, 6(8), 9640–9648 CrossRef CAS
- H. Liao, T. Luo, P. Tan, K. Chen, L. Lu, Y. Liu, M. Liu and J. Pan, Adv. Funct. Mater., 2021, 31(38), 2102772 CrossRef CAS
- S. L. Zhang, B. Y. Guan, X. F. Lu, S. Xi, Y. Du and X. W. Lou, Adv. Mater., 2020, 32(31), 2002235 CrossRef CAS PubMed
- L. Zeng, K. Sun, X. Wang, Y. Liu, Y. Pan, Z. Liu, D. Cao, Y. Song, S. Liu and C. Liu, Nano Energy, 2018, 51, 26–36 CrossRef CAS
- V. Ashok, M. Sangamithirai, M. Vijayarangan, A. Gayathri and J. Jayabharathi, ChemNanoMat, 2023, 9(11), e202300338 CrossRef CAS
- B. Vishnu, S. Sriram and J. Jayabharathi, Sustainable Energy Fuels, 2023, 7(18), 4638–4653 RSC
- J. Pearce, Food Chem. Toxicol., 1994, 32(6), 577–582 CrossRef CAS PubMed
- M. Younes, P. Aggett, F. Aguilar, R. Crebelli, B. Dusemund, M. Filipič, M. J. Frutos, P. Galtier, D. Gott and U. Gundert-Remy, EFSA J., 2018, 16(7), e05374 Search PubMed
- C. Jiménez-González, D. J. Constable and C. S. Ponder, Chem. Soc. Rev., 2012, 41(4), 1485–1498 RSC
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6(1), 32–48 CrossRef CAS
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu and L. N. Pincus, Green Chem., 2018, 20(9), 1929–1961 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, details of instruments used for the characterization of NiFeHCF@NF (XRD, FESEM, HRTEM, XPS, ICP-OES and IR), electrochemical characterization and electrode preparation, OER/HER activity comparison LSV of in situ and co-precipitation methods, EASA normalised LSV, images of post-use analysis (FESEM, XRD and IR), equations for the calculation of TOF, ECSA, RF, faradaic efficiency and environmental impact assessment and comparison table for recently stated non-noble bifunctional electrocatalysts with NiFeHCF@NF. Also, the evolution of O2 (anode) and H2 (cathode) gas using a solar-driven water-splitting device (MP4) has been displayed. See DOI: https://doi.org/10.1039/d4gc00719k |
| This journal is © The Royal Society of Chemistry 2024 |