
Valorization of waste biomass for the fabrication of isocyanate-free polyurethane foams†
Dagmara
Trojanowska‡
abc,
Florent
Monie‡
bd,
Giovanni
Perotto
 *a,
Athanassia
Athanassiou
a,
Bruno
Grignard
be,
Etienne
Grau
d,
Thomas
Vidil
d,
Henri
Cramail
*d and
Christophe
Detrembleur
*bf
*a,
Athanassia
Athanassiou
a,
Bruno
Grignard
be,
Etienne
Grau
d,
Thomas
Vidil
d,
Henri
Cramail
*d and
Christophe
Detrembleur
*bf
aIstituto Italiano di Tecnologia, Smart Materials Group, 16163 Genova, Italy
bCenter for Education and Research on Macromolecules (CERM), CESAM Research Unit, University of Liege, Sart-Tilman B6a, 4000 Liege, Belgium. E-mail: christophe.detrembleur@uliege.be
cDepartment of Materials Science, University of Milano-Bicocca, 20125 Milan, Italy
dUniversity of Bordeaux, CNRS, Bordeaux INP, LCPO, 16 Avenue Pey-Berland, 33600 Pessac, France
eFRITCO2T Platform, CESAM Research Unit, University of Liege, Sart-Tilman B6a, 4000 Liege, Belgium
fWEL Research Institute, Wavre 1300, Belgium
First published on 13th June 2024
Abstract
Polyurethane (PU) foams are key players within the large foam market, with applications such as thermal insulating materials, cushioning, protective equipment, etc. With the current regulatory constraints on the use of toxic isocyanates and the ambitious goals to increase the renewable content of plastics while valorizing waste, isocyanate-free liquid formulations containing biofillers that are able to easily self-foam are needed for more sustainable PU foams in the future. Herein, we incorporate various abundant waste stream-sourced biofillers (proteins, lignin derivatives, and polysaccharides) into isocyanate-free PU formulations composed of CO2-based poly(cyclic carbonate)s, diamines and a catalyst. The formulations containing up to 30 wt% of biofillers are foamed at 100 °C without adding any external foaming agent. Moisture naturally present in the biofillers partially hydrolyses the cyclic carbonates, which generates the blowing agent (CO2). The biofiller, even at a low content (1 wt%), stabilizes the growing cells, providing homogeneous foams. Although the nature of the biofiller does not significantly affect the foams’ density and morphology, their mechanical properties are strongly affected, for example from a rigid foam with 10 wt% keratin (compression modulus (E) = 21.9 MPa) to a flexible one with chitosan (E = 0.2 MPa). Preliminary studies indicate that the biofiller does not prevent the foam recycling into polymer films by hot pressing. Virtually any kind of moisture-containing biowaste can be used as a water reservoir to foam the formulations while increasing the bio-based content of the material, which reaches 97% when selecting bio-based monomers. This process valorizes abundant waste stream-sourced biofillers for producing more sustainable PU foams.
Introduction
In 2019, the worldwide use of polyurethane (PU) represented 18 million tons (Mt), accounting for roughly 4% of the global polymer market (460 Mt).1 About 65% of the PU business is dedicated to producing foams that are found in multiple items such as thermal or sound insulating panels, protective equipment, cushioning (mattresses and transportation seats), etc.2–5 The production of PU foams usually involves liquid formulations containing (poly)isocyanates and polyols, combined with a foaming agent (water or pentane, depending on the targeted application) and other additives (foam stabilizers, flame retardants, etc.). Conveniently, in the presence of water, the formulation is self-blown by the CO2 released during the partial hydrolysis of the isocyanates. Although this isocyanate chemistry has been used for decades, current European regulations have prompted people to find greener and safer alternatives for more sustainable PU foams in the future.6 Various isocyanate-free routes are currently being explored in the preparation of so-called non-isocyanate polyurethanes (NIPUs).7–11 Among them, the polyaddition of poly(cyclic carbonate)s with polyamines that can deliver poly(hydroxyurethane)s (PHUs)9–11 is the most investigated process, especially for producing foams. Until now, much effort has been devoted to physical and chemical blowing methodologies using exogenous foaming additives to promote the polymer expansion (e.g. Solkane fluorocarbon,12 CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13,14 or inorganic salts15,16). The only concepts directed towards endogenous foaming (i.e. self-foaming) relied on poorly controlled side reactions17 or hazardous blowing agents (e.g. H2).18–21 In this context, mimicking the PU self-blowing process for PHUs is highly desirable. It is difficult to endow cyclic carbonates with the same dual function as isocyanates, i.e. comonomers for constructing the polymer matrix and the precursor of the foaming agent (CO2). This challenge was first addressed by adding some (masked) thiols to the formulations to promote the decarboxylation of the cyclic carbonate via an S-alkylation reaction.22–26 Later on, it was demonstrated that the partial hydrolysis of the cyclic carbonate groups can be used to generate CO2 in a very similar fashion to isocyanate-based PU foams.27,28 In both cases, the decarboxylation is catalyzed by the organobase DBU at 80–100 °C.
13,14 or inorganic salts15,16). The only concepts directed towards endogenous foaming (i.e. self-foaming) relied on poorly controlled side reactions17 or hazardous blowing agents (e.g. H2).18–21 In this context, mimicking the PU self-blowing process for PHUs is highly desirable. It is difficult to endow cyclic carbonates with the same dual function as isocyanates, i.e. comonomers for constructing the polymer matrix and the precursor of the foaming agent (CO2). This challenge was first addressed by adding some (masked) thiols to the formulations to promote the decarboxylation of the cyclic carbonate via an S-alkylation reaction.22–26 Later on, it was demonstrated that the partial hydrolysis of the cyclic carbonate groups can be used to generate CO2 in a very similar fashion to isocyanate-based PU foams.27,28 In both cases, the decarboxylation is catalyzed by the organobase DBU at 80–100 °C.
A major advantage of these PHU foams is that they behave as covalent adaptable networks29–32 thanks to the hydroxyl groups they contain. For instance, they can be reprocessed by hot pressing. Thus, PHU foams were recycled into polymer films, structural coatings or hot-melt adhesives, offering attractive end-of-life scenarios difficult to achieve with conventional PU foams.23,25–27
The valorization of waste streams, in particular those coming from biomass transformation processes into value-added products is a long-standing goal for more efficient waste management as well as for reducing our carbon footprint.33 Many natural by-products (e.g. lignocellulosic waste from the paper industry, polysaccharides from seafoods, proteins from inedible feather or wool waste, etc.) are available on a large scale and low cost, and some of them were incorporated into conventional PU foam formulations.34–36 However, their valorization for isocyanate-free self-foaming processes exploiting the partial cyclic carbonate decarboxylation has never been explored.
Keratin is a structural protein found in many parts of living organisms (e.g., nails, hairs, feathers, wool, etc.). In the livestock industry, large keratin-rich waste streams composed of chicken feathers or sheep wool contain more than 90% of this protein.37,38 Notably, the worldwide annual chicken consumption generates about 65 Mt of waste feathers each year. Wool production (2.5 Mt) is associated with a significant amount of short and crude fibers that are not valorized, ending up as waste streams too.38,39 Particularly relevant for our study, keratin is rich in cysteine (7–20 mol%, depending on the source) that creates inter- and intra-protein disulfide bridges, essential to the proteins’ properties. By selecting an appropriate extraction strategy, keratin proteins with free pendant thiols could be retrieved from the biomass and were used in PHU formulations.39
Following recent reports on thiol-assisted self-blowing PHU formulations,22–26 we hypothesized that the pendant thiol groups of keratin might be exploited to self-blow liquid isocyanate-free PHU formulations without requiring any additional blowing agent. Unexpectedly, our studies showed that the main action mode for the foaming was the hydrolysis of the cyclic carbonate rather than S-alkylation. Moisture naturally present in this bio-waste was enough to generate the blowing agent. This discovery pushed us to utilize cysteine-free proteins (gelatin, zein), as well as other abundant and very cheap bio-waste such as a lignin derivative (lignosulfonate) or polysaccharides (cellulose, chitosan). In all cases, homogeneous PHU foams were obtained under similar foaming conditions.
Preliminary experiments also indicate that, for all biofillers, thermoset foams can be efficiently recycled into polymer films by hot pressing. This study demonstrates that virtually any moisture-containing biofillers could be used to activate the foaming of the PHU reactive formulations while increasing the bio-based content of the material. It opens an attractive valorization option for abundant bio-waste streams through the facile production of more sustainable foams.
Results and discussion
PHU self-foaming with protein fillers
In this work, a liquid poly(cyclic carbonate) (trimethylolpropane triglycidyl carbonate, TMPTC) and a liquid diamine (m-xylylenediamine, m-XDA) were polymerized in the presence of DBU as a catalyst and keratin as a bio-based filler to obtain self-blown PHUs (Fig. 1). The original idea was to use this thiol-containing protein as a bio-based promotor of the cyclic carbonate (5CC) decarboxylative S-alkylation. Keratin was extracted from wool waste in a recently reported study.40 Among the different protocols, the extraction using metabisulfite was selected to obtain the keratin used here, because sodium metabisulfite disproportionates the disulfide into a free thiol and a sulfonate group. Therefore, this keratin was identified as a promising candidate as it contains a significant amount of free thiol moieties in its structure (∼0.78 mmol g−1). Conveniently, keratin was homogeneously dispersed in a mixture of TMPTC and m-XDA (NH2:5CC = 0.6:1, optimized ratio, Fig. S1† and related discussion) with 10 wt% keratin after mechanical stirring at room temperature for 5 minutes. Once a dense material was obtained by keeping the sample at room temperature (i.e. no foaming), no particles were visible in the images obtained through scanning electron microscopy (Fig. S2†). Keratin was successfully incorporated with ratios ranging from 1 wt% up to 30 wt%. Table S1† summarizes the estimation of the corresponding number of thiol equivalents relative to 5CC, based on the known number of cysteine units present in keratin. Then, liquid reactive formulations of various compositions were foamed using a standard procedure, i.e. curing at 100 °C for 5 hours in an open silicon mold. Fig. 2 illustrates the produced bio-hybrid PHU foams. Clearly, the protein allowed for the in situ release of CO2 as the blowing agent, with a maximum expansion for the formulation containing 20 wt% of keratin. This was well reflected by the foam densities (Fig. 2 and Table S1†) that varied in between 188 and 318 kg m−3.
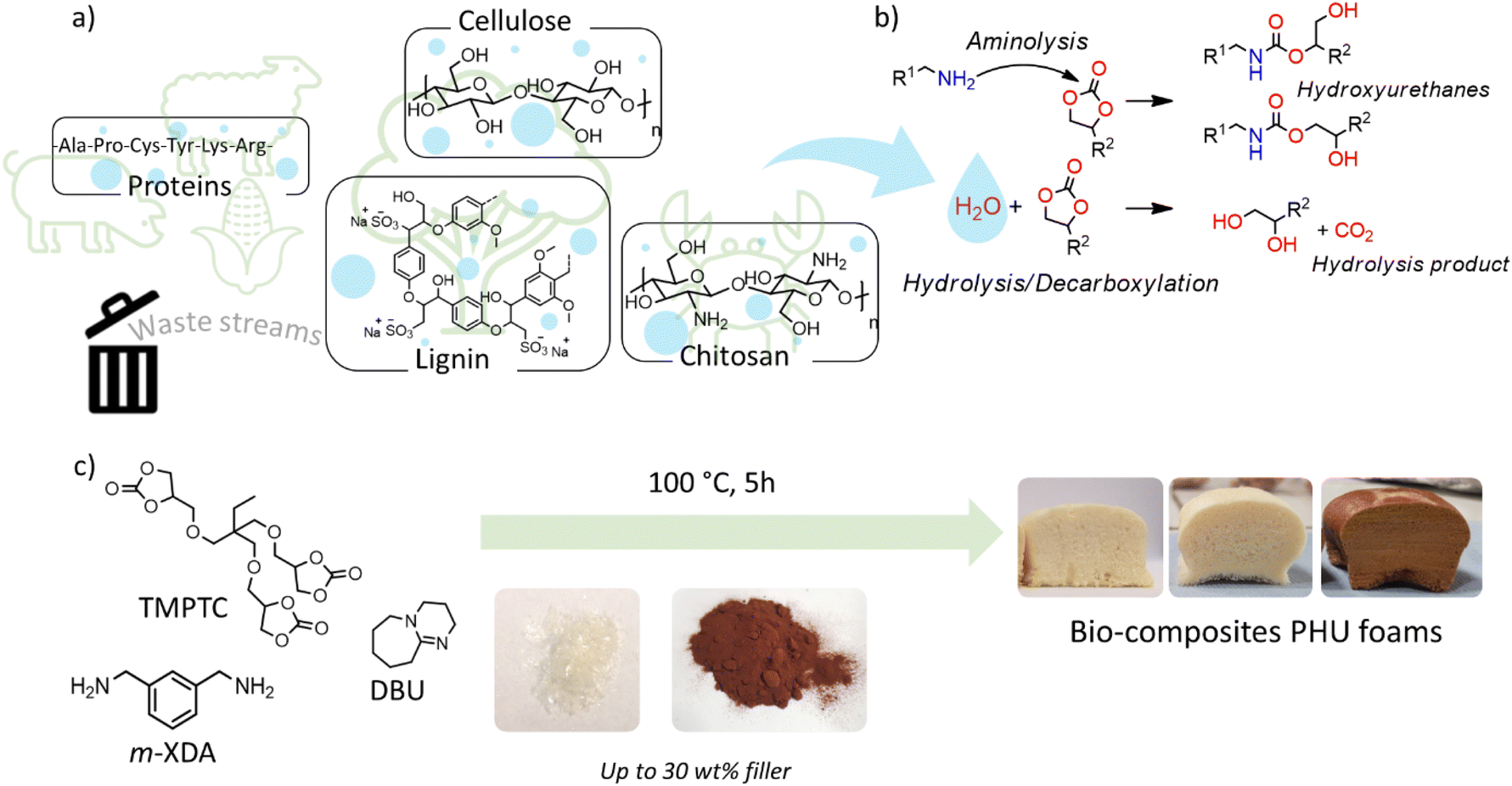 | ||
| Fig. 1 General strategy for the incorporation of waste-biomass into self-foaming isocyanate-free formulations: (A) general chemical structures of the biofillers, (B) reactions for the PHU matrix formation and foaming, (C) example of a typical liquid formulation and its foaming with some representative foams. | ||
 | ||
| Fig. 2 Self-blown NIPU foams prepared from liquid formulations based on TMPTC, m-XDA, DBU and increasing contents of various biofillers: photography, density and gel content of the foams. Foaming was performed at 100 °C for 5 hours. | ||
Surprisingly, even the formulation containing a minimal amount of keratin, i.e. 1 wt%, was effectively expanded, and gel contents (GC) were high in all cases (88–92.5%), in line with a crosslinked polymer matrix. Without keratin, the PHU matrix presented multiple bubble defects leading to a material with a density of 477 kg m−3 (Fig. 3A and B). In contrast, only 1 wt% of the protein was sufficient to deliver a homogeneous foam with a density of 252 kg m−3 (Fig. 3C and D).
 | ||
| Fig. 3 SEM micrographs of foam samples prepared without a filler (A and B), with 1 wt% keratin (C and D) or 1 wt% sodium lignosulfonate (E and F). Standard formulations composed of TMPTC, m-XDA (NH2/5CC = 0.6), DBU (5 mol% with respect to 5CC moieties), foamed at 100 °C for 5 h. | ||
For all formulations, the actual number of thiol equivalents was very small (0.001–0.061 eq. vs. 5CC; Table S1†) and insufficient to explain the successful expansion of the foams via decarboxylative S-alkylation. Moreover, FT-IR analyses (Fig. S3†) suggest that the molecular composition of all the PHU materials is the same. For comparison purposes, in their seminal work dedicated to the expansion of PHU foams via S-alkylation, Detrembleur et al. used 0.25 eq. of thiol vs. CC, i.e. more than 4 times higher.22 In order to better understand the actual origin of the blowing mechanism, cysteine-free proteins were used as alternative biofillers. Commercial zein and gelatin were successfully incorporated in the liquid formulations of TMPTC and m-XDA. By repeating the foaming procedure under identical conditions, all the formulations provided foams with densities comprised between 258 and 328 kg m−3, suggesting that cysteine residues in the keratin-containing foams were not responsible for the decarboxylation of the 5CC. Fig. 2 presents the foam obtained with the various weight fractions of zein and gelatin, and their respective densities and gel contents. All these experiments showed that, within the experimental errors, the maximum expansion was almost reached with 1 wt% of biofiller only, whatever the protein used. Additionally, the gel contents were >86% for all the proteins and loading values (Fig. 2).
The tested proteins have very different amino acid compositions, and a multitude of reactive sites could be involved in reactions with 5CC. They include nitrogen centers (guanidine, imidazole), phenols or carboxylic acids prone to alkylation by cyclic and linear carbonates. However, these functional moieties usually display significant reactivity with cyclic carbonates at high temperatures only (above 100–150 °C).41,42 Despite numerous trials, we were unable to demonstrate a potential involvement of these functions during the foaming process (see the model reactions in ESI, Section 6†).
To elucidate the foaming mechanism, we considered an alternative hypothesis. Common to all proteins is the presence of water trapped in their polyamide 3D structures. Indeed, purified proteins are extracted following operative protocols involving multiple water-based treatments. Despite extensive heat- or freeze-drying, it is well known that a non-negligible amount of water remains attached to the protein at the end of the purification process. For the three proteins, the residual water content was quantified via isothermal thermogravimetric analysis (TGA) at 100 °C under a nitrogen flux (Fig. S5†). Although keratin, gelatin and zein were dried at 50 °C for 24 hours prior to use, they contained 3.7 wt%, 2.3 wt% and 2.0 wt% of residual water, respectively. Knowing that the hydrolysis of 5CC can be used to produce self-blown PHUs via the in situ release of CO2, it might be hypothesized that residual water contained in proteins was the actual promoter of the foaming process.
In order to verify this hypothesis, a sample of keratin was dried at 100 °C under vacuum before incorporation in the liquid formulation of TMPTC and m-XDA. After curing at 100 °C, a limited expanded material similar to the one obtained without any filler was obtained, suggesting that water was the main species responsible for the cyclic carbonate decarboxylation during the self-foaming process. This discovery pushed us to evaluate whether other hydrophilic biofillers/waste might act as dispersed water reservoirs for PHU foam production.
PHU self-foaming with lignosulfonate and polysaccharide fillers
Sodium lignosulfonate (Na-Lign) is an abundant by-product of the paper mill industry resulting from the so-called sulfite process to produce wood pulp (cellulose) that is available on a large scale (1.8 Mt per year) and at a very low cost.43,44 Lignin is considered as the most important source of aromatics beside fossil fuels. Among other applications, lignosulfonate is commonly valorized as an environmentally friendly and readily available dispersant, notably in concretes for the building sector.45 Lignin derivatives are also of great interest for their incorporation in polyurethanes.46 Similar to keratin, this biofiller was also successfully incorporated at different contents (from 1 wt% to 30 wt%) in the liquid TMPTC/m-XDA formulation. After curing under identical conditions that were described previously, all formulations provided foams with densities between 146 and 370 kg m−3 and high gel contents (∼90%) (Fig. 2). The foam with the lowest density was obtained at a high biofiller loading of 30 wt%. The chemical structure of lignosulfonate is quite complex, where the main functional groups are alcohols, phenols and sulfonates. None of these groups were able to react with cyclic carbonates to generate carbon dioxide under the investigated conditions as evidenced by our model reactions (see model reactions in ESI, Section 6†). In contrast, this biofiller contained about 7 wt% of water as determined by isothermal TGA (Fig. S5†). Here again, only 1 wt% of Na-Lign was enough to induce a homogeneous foaming (Fig. 3E and F).Polysaccharides like cellulose or chitosan are important thiol-free bio-waste that are particularly attractive to reuse as well. Cellulose is also obtained from the paper mill industry as the main component of wood pulp.47 It accounts for roughly 70% of the dry weight of lignocellulosic biomass and contains primary and secondary OH functions that do not react with cyclic carbonates under our operative conditions.
Chitosan can be obtained by deacetylation of chitin, one of the main components of crustacean shells. In the seafood industry, it is estimated that 6–8 million tons of crustacean waste is generated annually.48,49 Therefore, it is of great interest to valorize this waste.50 Besides the unreactive alcohol functions, chitosan bears primary amine groups that could ring-open cyclic carbonate functions to form hydroxyurethanes. Therefore, chitosan might also increase the crosslinking degree of the foam. The two types of polysaccharides were successfully dispersed in the liquid formulation with an identical loading of 10 wt%. After curing, foams of similar density (270 kg m−3) and gel content (88%) were obtained in the two cases. Once again, isothermal TGA showed the presence of water in the biofillers (4.3 wt% and 7.1 wt% for cellulose and chitosan, respectively). Surprisingly, the two biofillers contained different amounts of water, and similar foam densities were measured.
All the foams prepared from amine-free biofillers (lignosulfonate and cellulose) and amine-containing ones (keratin, gelatin, zein and chitosan) presented similar gel contents. Together with no significant difference in cyclic carbonate conversion (see IR spectra in Fig. S3 and S4†), this observation suggested that the amine-containing biofillers did not contribute significantly to increasing the crosslinking density of the PHU matrix.
Discussion of the PHU self-foaming process
Table S2† summarizes the number of equivalents of water in the liquid formulations containing the various biofillers at different loadings. The content of water, obtained from the isothermal TGA analysis carried out on the biofillers, was utilized to calculate the theoretical amount of generated CO2 by cyclic carbonate hydrolysis, and therefore estimate the theoretical foams’ density (Table S2, see Section 5 in the ESI† for details). Clearly, for all fillers, the experimental foam density was much lower than the theoretical one where a 1 wt% loading was used, suggesting that the water content was underestimated.In order to understand these discrepancies, we reacted the liquid formulations composed of TMPTC, DBU (0.05 eq. vs. 5CC) and the biofillers (10 wt%) under identical foaming conditions (100 °C, 5 h) without the diamine.
In this way, no aminolysis and no crosslinking reactions were expected, which should facilitate the estimation of the hydrolysis level of the cyclic carbonate groups by 1H-NMR spectroscopy. During the reaction, gas bubbles were observed in the reaction medium. Fig. 4A shows the overlay of the 1H-NMR spectra of TMPTC and of the crude reaction products with the different biofillers. Comparison of the integration of the signals at (a) 0.8 ppm of the –CH3– group and (b) 4.9 ppm associated with the –CH– group of the cyclic carbonate function of TMPTC before and after the reaction enabled us to estimate the content of the hydrolysed product (Table 1, see Section 7 in the ESI† for details). Between 14% and 26% of the cyclic carbonates were hydrolyzed, which now accounted for enough blowing agent generation for producing the previous foams (Table 1). This overall water content accounts for the water present in both the biofiller and the formulation. When considering the foaming, the diamine might also bring some water, further contributing to the formation of the blowing agent. Note that the presence of water in the biofiller (i.e. lignosulfonate as the representative filler) and in the formulation (TMPTC/lignosulfonate (10 wt%)/DBU) was also confirmed by Karl-Fischer titration (Section 5 in the ESI† for details).
 | ||
| Fig. 4 (A) Comparison of the NMR spectra of the TMPTC hydrolysis in the presence of the different biofillers (10 wt%) and DBU for 5 h at 100 °C, and the reference NMR spectrum of TMPTC (bottom spectrum). (B) Overlay of FT-IR spectra of TMPTC (blue), TMPTC added by Na-Lign (10 wt%) and DBU before (green) and after reaction at 100 °C for 5 h (grey). | ||
| Biofiller (10 wt%) | Hydrolyzed 5CCa (%) | CO2 volumeb (mL) | Theoretical foam densityc (kg m−3) | Experimental foam density (kg m−3) |
|---|---|---|---|---|
| a Determined by 1H-NMR spectroscopy on the 5CC hydrolysis experiments with the various fillers. b Determined from the amount of hydrolyzed 5CC. c Determined from the estimated generated volume of CO2 from TMPTC hydrolysis experiment (see Section 7 in the ESI† for details). d Shouldering on the 0.8 ppm signal may lead to underestimation of the hydrolyzed 5CC content. | ||||
| Keratin | 14d | 148 | 48 | 244 ± 12 |
| Gelatin | 22 | 233 | 31 | 258 ± 23 |
| Zein | 23d | 243 | 29 | 318 ± 10 |
| Na-Lign | 20 | 211 | 34 | 319 ± 21 |
| Cellulose | 20 | 211 | 34 | 270 ± 16 |
| Chitosan | 26 | 275 | 26 | 276 ± 15 |
| — | 5 | 52.9 | 123 | 477 ± 41 |
FT-IR spectra of the formulations containing Na-Lign before and after the reaction are also shown in Fig. 4B. The well-defined band of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching mode of DBU at 1610 cm−1 was fully shifted at 1648 cm−1, characteristic of the DBUH+/HCO3− adduct,51,52 which further confirmed the in situ formation of CO2. A broad band between 3100 cm−1 and 3650 cm−1 was also observed, accounting for the vicinal alcohols generated by the cyclic carbonate hydrolysis. The same observations were made for all other biofillers (Fig. S13†). For the sake of comparison, when TMPTC and DBU were reacted under identical conditions without a biofiller, only 5 mol% of the cyclic carbonates were hydrolysed from the residual water present in this mixture. Also, most of the DBU was carbonated as evidenced by IR spectroscopy with the presence of the characteristic band at 1648 cm−1 (Fig. S13†). These experiments demonstrated that water was responsible for the generation of CO2 as the blowing agent by partial hydrolysis of the cyclic carbonates.
N stretching mode of DBU at 1610 cm−1 was fully shifted at 1648 cm−1, characteristic of the DBUH+/HCO3− adduct,51,52 which further confirmed the in situ formation of CO2. A broad band between 3100 cm−1 and 3650 cm−1 was also observed, accounting for the vicinal alcohols generated by the cyclic carbonate hydrolysis. The same observations were made for all other biofillers (Fig. S13†). For the sake of comparison, when TMPTC and DBU were reacted under identical conditions without a biofiller, only 5 mol% of the cyclic carbonates were hydrolysed from the residual water present in this mixture. Also, most of the DBU was carbonated as evidenced by IR spectroscopy with the presence of the characteristic band at 1648 cm−1 (Fig. S13†). These experiments demonstrated that water was responsible for the generation of CO2 as the blowing agent by partial hydrolysis of the cyclic carbonates.
As the foaming occurred at the boiling point temperature of water, it might be hypothesized that steam was responsible for the foaming. The same foaming experiments with keratin or sodium lignosulfonate in the formulation (5 wt%) were carried out at 90 °C for various NH2/5CC molar ratios. Fig. S1B† clearly shows that foams were obtained in all cases, although their densities were slightly higher than those obtained at 100 °C (Fig. S1A†). These experiments further confirmed that CO2 was the main blowing agent responsible for the polymer matrix expansion.
Table 1 summarizes the data collected for the hydrolysis with and without the different biofillers. The comparison of the water content and TMPTC hydrolysis with and without the biofillers showed that the overall water content was mainly coming from the biofiller. Interestingly, it must be noted that, without a biofiller, the water content in the formulation was (in theory) sufficient to generate enough blowing agent for providing a foamed material of rather low density (Table 1). However, the material only contained bubble defects (Fig. 3A and B). This is quite different from the homogeneous foams produced with only 1 wt% biofiller (Fig. 3C–F). This suggested that the biofiller, besides contributing to the generation of the blowing agent via the water it contains, is also likely to improve its trapping within the polymer matrix and to stabilize the growing cells to deliver high quality homogeneous foams.
It is important to note that it is impossible at this stage to draw general trends regarding the water content in the biofiller/formulation and the foam density. As demonstrated in our seminal work,27 there is an optimal content of water for maximal foam expansion. Above this content, the quantity of produced CO2 increased. However, this blowing agent could not be efficiently trapped as the crosslinking degree of the polymer matrix was decreased (the hydrolysis of cyclic carbonate consumed one crosslinking node). This resulted in poorly foamed or collapsed materials. Moreover, water is not the only factor affecting the density of the foams. The efficient trapping of the blowing agent strongly depends on the formulation viscosity when CO2 is generated and on the comonomer mixture and temperature. It also depends on the type of filler and on its interaction with the polymer matrix. The content of hydrolyzed cyclic carbonate groups, the quality of the biofiller dispersion as well as non-covalent interactions (e.g. hydrogen bonding) between the fillers, the comonomers and the polymer network under construction may all play a significant role in the formulation viscosity and foam expansion. Based on this, it is not surprising to observe similar foam densities from formulations containing different amounts of water (Table 1).
PHU foam characterization
We then assessed the influence of the biofiller on the foam morphology and thermo-mechanical properties. This study was realized on foams produced with 10 wt% biofiller. Fig. 5A and B show the foams prepared from the different biofillers as well as their scanning electron microscopy (SEM). All foams were highly homogeneous and presented an open cell morphology with a pore size mainly in the range of 0.2 mm to 0.5 mm. In general, all the foams displayed a broad distribution of the cell size between 0.1–0.2 mm and 1 mm for most samples (Fig. S14†). The foams were then characterized by differential scanning calorimetry (DSC) to determine their glass transition temperature (Tg) by TGA to evaluate their degradation temperature (Td) and by compression tests for calculating their compression modulus (E) and yield stress (σ) from their stress–strain curves. The collected data are shown in Table 2 and stress–strain curves are shown in Fig. 5C. The impact of the biofiller content (from 1 to 30 wt%) on the Tg values was also investigated and the results are reported in Fig. S15.† As a general trend, Tg values for dried foams were not significantly affected by the biofiller content or the type. The Tg values ranged from 27 °C to 34 °C, which is close to that of the unfoamed PHU matrix (Tg = 33 °C) (Table 2 and Fig. S15A–D†). The Tg values remained in the same range regardless of the biofiller loading. When equilibrated under ambient atmosphere, their Tgs dropped to 5–9 °C as a result of the well-known PHU hydroplasticization (Fig. S16†).23,27,53,54 TGA analysis of these equilibrated foams showed about 2.8 wt% to 4 wt% water uptake, slightly higher than the PHU matrix without a biofiller (2.5 wt%) (Fig. S17†). Foams prepared from different biofillers at an identical loading (10 wt%) were characterized by similar gel contents (88–91%). Their densities slightly varied from 320 kg m−3 (with Na-Lign and zein) to 244–276 kg m−3 (with keratin, gelatin, cellulose and chitosan). These biofiller-loaded PHU foams presented similar densities, pore sizes and thermal behavior to those produced by the water-induced self-foaming process using hydrotalcite or Cloisite Na as the filler.27,28 However, no fair comparison could be done with conventional PU foams loaded with biofillers because significantly different formulations, which also contain surfactants, are used to reach lower foam densities (see Table S3†). Interestingly, all foams loaded with proteins (keratin, gelatin, zein) presented a much higher compression modulus (E = 21.9, 20.3, 18.1 MPa, respectively) compared to the other foams (Na-Lign, 8.4 MPa; cellulose, 1.6 MPa; chitosan, 0.2 MPa). Particularly impressive was the difference in compression modulus between foams loaded with proteins (20–22 MPa) and polysaccharides (0.2–1.6 MPa), despite similar densities (Table 2 and Fig. 5C). Noteworthily, normalizing the compression modulus with respect to the foam density led to similar trends, i.e., high normalized modulus values for protein (56.9–89.7 kPa kg−1 m3) and lignin-based (26.3 kPa kg−1 m3) foams vs. low values for polysaccharide-based ones (0.72–5.93 kPa kg−1 m3) (Table 2). | ||
| Fig. 5 Self-foamed NIPUs with 10 wt% of biofillers. (A and B) Pictures and SEM micrographs, and (C) compressive stress–strain curves. See Table 2 for the detailed properties. | ||
| Filler | Density (kg m−3) | T g (°C) |
T
d,5%c (°C) |
GC (%) | Compression modulus (MPa) | Normalized modulusd (kPa kg−1 m3) |
|---|---|---|---|---|---|---|
| a Dry Tg. b T g of the equilibrated sample under ambient atmosphere for 24 hours. c Determined after the water evaporation plateau (see the ESI† for details). d Compression modulus normalized by the density. | ||||||
| Na-Lign | 319 ± 21 | 32a (9)b | 243 | 90 ± 0.4 | 8.4 ± 4.2 | 26.3 |
| Chitosan | 276 ± 15 | 34a (5.5)b | 244 | 87.8 ± 0.1 | 0.2 ± 0.09 | 0.72 |
| Cellulose | 270 ± 16 | 28a (5)b | 243 | 88.4 ± 0.4 | 1.6 ± 1.8 | 5.93 |
| Keratin | 244 ± 12 | 29a (8)b | 241 | 91 ± 1.5 | 21.9 ± 3.8 | 89.7 |
| Gelatin | 258 ± 23 | 31a (4.5)b | 248 | 88.5 ± 2.3 | 20.3 ± 1.8 | 78.7 |
| Zein | 318 ± 10 | 27a (5)b | 247 | 87.9 ± 0.1 | 18.1 ± 1.5 | 56.9 |
Thus, the protein-loaded foams were rigid, whereas the polysaccharide-loaded ones were flexible. These observations showed that for identical comonomer compositions and reaction conditions, the mechanical properties of the PHU foams could be modulated over a broad range by the nature of the biofiller. Many parameters might explain the different mechanical properties observed, for example the quality of the biofiller dispersion, the protein conformation, specific interactions (e.g., hydrogen bonding) between the PHU matrix and the biofiller, and the interaction of moisture with PHU and the biofiller. However, further experiments under a controlled atmosphere are needed to better understand the system, which will be the topic of a forthcoming paper. All of the characterized foams had a similar degradation profile to the unloaded PHU matrix, with a temperature at 5% weight loss (Td,5%) of about 240–250 °C.
In order to provide preliminary insights into the aging of the prepared foams, some samples were placed in a climatic chamber (25 °C, 80% relative humidity – RH) for 1 week, and the moisture uptake as well as Tg values were monitored over time (see Section 10 in the ESI† for details). Fig. 6A shows a fast moisture uptake for all biofiller-loaded foams, reaching between 6.8% and 10% after 24 h, depending on the biofiller introduced in the formulation (10 wt% biofiller content). The increase of the lignosulfonate loading from 10 wt% to 30 wt% significantly raised the moisture uptake from 10 wt% to 17.2% after 24 hours. For all biofiller-loaded foams, the water uptake induced a strong hydroplasticization of the PHU matrix, with an impressive decrease of the Tg of the foam by about 20 °C to 30 °C after equilibration for 24 hours at 80% relative humidity (RH) (Fig. 6B).
 | ||
| Fig. 6 Moisture uptake (A) and Tg values (B) over aging time at 80% RH and 25 °C for various PHU foams loaded with biofillers. | ||
PHU foam reprocessing
Typical PU foams are made of a crosslinked material that hinders their recycling. In contrast to standard PU, PHU pendant hydroxyl groups are accountable for transcarbamoylation reactions upon heating and the reversing of the aminolysis equilibrium. Both of these reactions are known to enable PHU reprocessing.30–32 Hence, reprocessing of the foams (containing 10 wt% biofillers) into polymer films was evaluated by compression for 1 hour at 160 °C under 1 ton pressure. All recycled foams yielded homogeneous films (Fig. S18†).Freeze-fractured surface SEM images showed a uniform morphology with no evidence of phase separation, proving the compatibility between the PHU matrix and the biofiller after compression. The thermo-mechanical properties and gel content of the repurposed foams were analyzed. These results are presented in Table 3. Gel contents were similar to the foams before compression and ranged between 88.9% and 92%. For all the biofillers, the Tg values of the reprocessed samples (measured on the second heating cycle) were slightly higher than the Tg of the corresponding foams (Table 3 and Fig. S19†). This was attributed to post-curing during hot-compression at 160 °C. Indeed, Torkelson suggested that the residual cyclic carbonate groups of the PHU networks plastified the polymer matrix. Their consumption during reprocessing was associated with a Tg increase. In line with this work, we evidenced that the decrease of the cyclic carbonate group content in the foams during reprocessing (as seen by FT-IR analysis) was linked to a decrease in the intensity of the characteristic carbonyl peak of the cyclic carbonate at 1790 cm−1 (Fig. S20†).26 The reprocessed samples were further analyzed through tensile testing, without prior drying treatment (except for being stored in a desiccator prior to analysis), meaning that they were possibly plasticized by water. Under these conditions, it was not possible to fully rationalize the actual impact of the biofillers on the mechanical properties of the reprocessed networks. However, it is interesting to note that all samples containing protein fillers (keratin, zein and gelatin) exhibited a ductile fracture with an elongation at break between 26% and 85% (Table 3 and Fig. S21†). On the other hand, the samples containing lignin and polysaccharide fillers showed brittle fractures (elongation at break between 2% and 7%). These differences suggested that the incorporation of the waste biofillers could be further valorized to tune the mechanical properties of dense PHU thermosets. This will be investigated in future studies under a controlled atmosphere.
| Filler | T g (°C) | Gel content (%) | Young's modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|---|---|
| None | 38 | 90.3 ± 0.1 | 510 ± 167 | 7.7 ± 1.2 | 9.2 ± 4.6 |
| Na-Lign | 39 | 91.1 ± 0.2 | 643 ± 127 | 12.7 ± 0.5 | 2.7 ± 0.3 |
| Keratin | 41 | 91 ± 0.6 | 67 ± 19 | 5 ± 0.6 | 48.9 ± 20.9 |
| Gelatin | 37 | 92 ± 2.3 | 416 ± 40 | 11.8 ± 1.7 | 26.4 ± 6.9 |
| Zein | 39 | 91.4 ± 1.5 | 166 ± 63 | 5.8 ± 0.4 | 84.7 ± 19.5 |
| Cellulose | 34 | 90.1 ± 1.2 | 407 ± 94 | 12.3 ± 0.9 | 5.7 ± 0.7 |
| Chitosan | 37 | 88.9 ± 0.6 | 263 ± 61 | 8.7 ± 2.7 | 6.2 ± 1.4 |
Conclusions
In summary, this work shows that self-blown isocyanate-free polyurethane foams containing biowaste used as biofillers (proteins, lignosulfonate, polysaccharides) were easily produced from a liquid formulation composed of a CO2-based tricyclic carbonate, a diamine and DBU as the catalyst, without requiring any external blowing agent or additional surfactant. For the first time, we demonstrated that water, naturally contained in the biofiller together with water present in the comonomer mixture, was enough to partially hydrolyze the cyclic carbonate groups, delivering CO2 that expanded the PHU matrix. Homogeneous foams were produced for all tested biofillers with loading contents ranging from 1 wt% to 30 wt%. Although the nature of the biofiller did not significantly affect the density and morphology of the foams, their mechanical properties were strongly affected. This is particularly relevant as foams characterized with very different mechanical properties were obtainable from a single formulation, simply by adapting the nature of the biofiller. Preliminary investigations also indicated that the thermoset foam recycling into polymer films by hot pressing is still possible in the presence of the biofiller, offering them an attractive end-of-life scenario. This work demonstrates that virtually any kind of moisture-containing biowaste could be used to increase the bio-based content of the material while favoring the formulation foaming and its stabilization to deliver homogeneous foams. This process constitutes an attractive valorization option for abundant waste biomass through the production of more sustainable, yet recyclable isocyanate-free PU foams, with mechanical properties that can be easily tuned by the nature of the biofiller.A significant green advance in the field was further achieved when substituting the petro-based monomers (TMPTC and m-XDA) of the formulations by bio-based ones, i.e. a tricyclic carbonate (GTC) derived from glycerol, bio-based epichlorydrin and CO2, and hexamethylenediamine (HMDA) that can be now purely plant-based.55 A flexible PHU foam (222 ± 27 kg m−3, Tg = −2 °C 1st DSC cycle after equilibration under ambient conditions) with an unprecedented theoretical total bio-based content of 97% was obtained in the presence of 20 wt% sodium lignosulfonate (Fig. 7). Compared to previous work claiming almost 100% bio-based content,56 our formulation significantly improves sustainability (i.e. E factor) due to the CO2-based synthetic pathway for the cyclic carbonate monomer (5CC), which has recently been identified as more sustainable than the dimethylcarbonate pathway.57 Moreover, the bio-based content was still higher than previously reported water-induced self-blown NIPU obtained from CO2-based 5CC.28 Therefore, our work represents a significant step toward sustainable biomass-rich self-blown isocyanate-free polyurethane foams.
 | ||
| Fig. 7 PHU foam of high bio-based content prepared with GTC, HMDA and lignosulfonate catalyzed by DBU (5 mol% vs. 5CC). (A) Chemical structures of the precursors, (B) picture of the obtained foam and (C) SEM micrographs. | ||
Experimental
Materials
m-Xylylenediamine (99%, m-XDA), 1,8-diazabicyclo[5.4.0]undec-7-ene (98%, DBU), propylene carbonate, sodium 1-dodecanesulfonate, propionic acid, gelatin from porcine skin with gel strength ∼ 300, type A, trimethylolpropane triglycidylether (TMPTE), glycerol triglycidylether (kindly provided by Ipox; IPOX CL 12), chitosan (low molecular weight, 50 kDa to 190 kDa, 75–85% deacetylated chitin) and zein were purchased from Sigma-Aldrich. Tetrabutylammonium iodide was obtained from TCI. Sodium lignosulfonate (product name ARBON18) was provided by RYAM. Microcrystalline cellulose was obtained from Alfa Aesar. Phenol was ordered from Fisher Scientific. All reagents were used without any purification.Methods
For the PHU foam of high bio-based content (Fig. 7), TMPTC and m-XDA were substituted by GTC and HMDA, respectively. HMDA was preliminary molten to be incorporated in the formulation that was cured for 2 h at 100 °C. The same ratio between components was used (i.e. NH2/5CC = 0.6, 5 mol% DBU vs. 5CC, 20 wt% Na-Lign) based on 15 g of GTC.
The thermal degradation of the PHU foams was obtained on a TGA2 instrument from Mettler Toledo and a TA Instrument Q50. Around 10 mg of sample was heated at 10 °C min−1 until 600 °C under a nitrogen atmosphere (50 mL min−1). Td,5% was measured as the temperature at 5% mass loss from the mass plateau at 160 °C.
Differential scanning calorimetry (DSC) was performed on a TA DSC250 apparatus. About 4 mg of sample was sealed in an Al pan for analysis. The equilibrated Tg was determined on the sample left for 24 h in an open atmosphere before sealing the pan and extracted from the first heating cycle between −50 and 100 °C with a heating ramp of 10 °C min−1 under a N2 flow. The dry Tg was then determined after drying the sample (50 °C under vacuum for 24 h) before sealing in the pan and extracted from the second heating cycle between −50 and 100 °C with a heating ramp of 10 °C min−1 under a N2 flow.
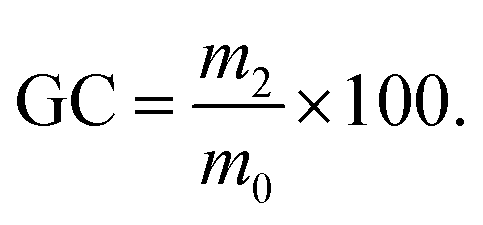
The uniaxial tensile tests were measured using a dual-column Instron 3365 universal testing machine. Dog-bone-shaped samples (width w = 4 mm, useful length l = 25 mm) were stretched at a rate of 2 mm min−1. The Young's modulus, tensile strength, and elongation at break values were evaluated from the stress–strain curves.
For all these experiments, at least three measurements were conducted for each sample and the results were averaged to obtain a mean value. The tests were performed on the samples stored in a desiccator for at least 24 h. The measurements were recorded at 25 °C.
The cell size distributions were determined by averaging the diameter measurements of 100 cells of 6 SEM images (100 measures for each sample).
The top and cross-section views from reprocessed foams have been imaged using a JEOL JSM-6490LA scanning electron microscope equipped with a tungsten thermionic electron source working in a high vacuum, with an acceleration voltage of 10 kV. The samples were freeze-fractured in liquid nitrogen and mounted on aluminum stubs with carbon tape. Prior to imaging, the samples were sputter-coated with a 10 nm layer of gold using a Cressington 208HR high-resolution sputter coater.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
C. D., B. G. and H. C. conceived the idea and wrote the project, and supervised the research. C. D. and H. C. directed the project. T. V. and E. G. co-supervised the research. D. T. prepared the keratin samples under the supervision of G. P. and A. A. F. M. and D. T. designed and carried out the foaming and reprocessing experiments and characterized the products. F. M. and D. T. furnished the first paper draft that was first corrected by C. D. and T. V. All co-authors then corrected the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Scklodowska-Curie grant agreement no. 955700. D. T. and G. P. acknowledge the support of Fondazione Cariplo's grant no. 2018-1005 (PROTHEIFORM project). The authors thank Prof. Stéphane Grelier and Dr Frédérique Ham-Pichavant for their insights on lignocellulosic biomass. RYAM is gratefully acknowledged for providing sodium lignosulfonate. The authors thank Gregory Cartigny, Valérie Colard and Luca Narducci for their technical support. The authors thank Maja Lopandic and Bryan Robb for proofreading the manuscript. C. D. is F.R.S.-FNRS Research Director and thanks FNRS for financial support, as well as the Region Wallonne for funding the Win2Wal project “ECOFOAM” (convention 2010130).Notes and references
- OECD, Global Plastics Outlook: Policy Scenarios to 2060, OECD, 2022.
- J. Peyrton and L. Avérous, Mater. Sci. Eng., R, 2021, 145, 100608 CrossRef.
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- N. V. Gama, A. Ferreira and A. Barros-Timmons, Materials, 2018, 11, 1841 CrossRef PubMed.
- F. Monie, T. Vidil, B. Grignard, H. Cramail and C. Detrembleur, Mater. Sci. Eng., R, 2021, 145, 100628 CrossRef.
- Official Journal of the European Union, L252, 4 August 2020, https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=OJ:L:2020:252:FULL&from=EN.
- C. Ngassam Tounzoua, B. Grignard and C. Detrembleur, Angew. Chem., Int. Ed., 2022, 61, e202116066 CrossRef CAS PubMed.
- B. Grignard, S. Gennen, A. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, c48, 4466–4514 RSC.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 CrossRef PubMed.
- H. Blattmann, M. Lauth and R. Mülhaupt, Macromol. Mater. Eng., 2016, 8, 944–952 CrossRef.
- B. Grignard, J.-M. Thomassin, S. Gennen, L. Poussard, L. Bonnaud, J.-M. Raquez, P. Dubois, M.-P. Tran, C. B. Park, C. Jerome and C. Detrembleur, Green Chem., 2016, 18, 2206–2215 RSC.
- H.-I. Mao, C.-W. Chen, H.-C. Yan and S.-P. Rwei, J. Appl. Polym. Sci., 2022, 139, e52841 CrossRef CAS.
- T. Dong, E. Dheressa, M. Wiatrowski, A. P. Pereira, A. Zeller, L. M. L. Laurens and P. T. Pienkos, ACS Sustainable Chem. Eng., 2021, 9, 12858–12869 CrossRef CAS.
- C. Amezúa-Arranz, M. Santiago-Calvo and M.-Á. Rodríguez-Pérez, Eur. Polym. J., 2023, 197, 112366 CrossRef.
- J. H. Clark, T. J. Farmer, I. D. V. Ingram, Y. Lie and M. North, Eur. J. Org. Chem., 2018, 4265–4271 CrossRef CAS.
- A. Cornille, C. Guillet, S. Benyahya, C. Negrell and S. C. B. Boutevin, Eur. Polym. J., 2016, 84, 873–888 CrossRef CAS.
- G. Coste, C. Negrell and S. Caillol, Eur. Polym. J., 2020, 140, 110029 CrossRef CAS.
- A. Cornille, S. Dworakowska, D. Bogdal, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 66, 129–138 CrossRef CAS.
- G. Coste, D. Berne, V. Ladmiral, C. Negrell and S. Caillol, Eur. Polym. J., 2022, 176, 111392 CrossRef CAS.
- F. Monie, B. Grignard, J.-M. J. Thomassin, R. Mereau, T. Tassaing, C. Jerome and C. Detrembleur, Angew. Chem., Int. Ed., 2020, 59, 17033–17041 CrossRef CAS PubMed.
- F. Monie, B. Grignard and C. Detrembleur, ACS Macro Lett., 2022, 11, 236–242 CrossRef CAS PubMed.
- G. Coste, C. Negrell and S. Caillol, Macromol. Rapid Commun., 2022, 43, 2100833 CrossRef CAS PubMed.
- N. S. Purwanto, Y. Chen and J. M. Torkelson, ACS Appl. Polym. Mater., 2023, 5, 6651–6661 CrossRef CAS.
- N. S. Purwanto, Y. Chen, T. Wang and J. M. Torkelson, Polymer, 2023, 272, 125858 CrossRef CAS.
- M. Bourguignon, B. Grignard and C. Detrembleur, Angew. Chem., Int. Ed., 2022, 61, e202213422 CrossRef CAS PubMed.
- M. Bourguignon, B. Grignard and C. Detrembleur, J. Am. Chem. Soc., 2024, 146(1), 988–1000 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- C. Bakkali-Hassani, D. Berne, P. Bron, L. Irusta, H. Sardon, V. Ladmiral and S. Caillol, Polym. Chem., 2023, 14, 3610–3620 RSC.
- C. Bakkali-Hassani, D. Berne, V. Ladmiral and S. Caillol, Macromolecules, 2022, 55, 7974–7991 CrossRef CAS.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- D. Merino, A. I. Quilez-Molina, G. Perotto, A. Bassani, G. Spigno and A. Athanassiou, Green Chem., 2022, 24, 4703–4727 RSC.
- H. Haridevan, D. A. C. Evans, A. J. Ragauskas, D. J. Martin and P. K. Annamalai, Green Chem., 2021, 23, 8725–8753 RSC.
- P. Zakrzewska, B. Zygmunt-Kowalska, M. Kuźnia, A. Szajding, T. Telejko and M. Wilk, Energies, 2023, 16, 1354 CrossRef CAS.
- S. M. Husainie, X. Deng, M. A. Ghalia, J. Robinson and H. E. Naguib, Mater. Today Commun., 2021, 27, 102187 CrossRef CAS.
- B. Mu, F. Hassan and Y. Yang, Green Chem., 2020, 22, 1726–1734 RSC.
- A. Shavandi, T. H. Silva, A. A. Bekhit and A. E.-D. A. Bekhit, Biomater. Sci., 2017, 5, 1699–1735 RSC.
- A. Aluigi, C. Tonetti, F. Rombaldoni, D. Puglia, E. Fortunati, I. Armentano, C. Santulli, L. Torre and J. M. Kenny, J. Mater. Sci., 2014, 49, 6257–6269 CrossRef CAS.
- D. J. Trojanowska, G. Suarato, C. Braccia, A. Armirotti, F. Fiorentini, A. Athanassiou and G. Perotto, ACS Appl. Nano Mater., 2022, 5, 15272–15287 CrossRef CAS PubMed.
- G. Galletti, P. Prete, S. Vanzini, R. Cucciniello, A. Fasolini, J. De Maron, F. Cavani and T. Tabanelli, ACS Sustainable Chem. Eng., 2022, 10, 10922–10933 CrossRef.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van Den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- T. Wang, H. Li, X. Diao, X. Lu, D. Ma and N. Ji, Ind. Crops Prod., 2023, 199, 116715 CrossRef CAS.
- Y. Yang, Y. Wang, M. Zhu, J. Zhao, D. Cai and H. Cao, Mater. Today Sustainability, 2023, 22, 100367 CrossRef.
- A. Yousuf, D. Pirozzi and F. Sannino, Lignocellulosic Biomass to Liquid Biofuels, Elsevier, 2020, pp. 1–15 Search PubMed.
- J. L. Vidal, T. Jin, E. Lam, F. Kerton and A. Moores, Curr. Res. Green Sustainable Chem., 2022, 5, 100330 CrossRef CAS.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- I. S. Arvanitoyannis and A. Kassaveti, Int. J. Food Sci. Technol., 2008, 43, 726–745 CrossRef CAS.
- D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338 CrossRef CAS PubMed.
- K. N. Onwukamike, T. Tassaing, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2018, 6, 1496–1503 CrossRef CAS.
- F. Magliozzi, A. Scali, G. Chollet, D. Montarnal, E. Grau and H. Cramail, ACS Sustainable Chem. Eng., 2020, 8, 9125–9135 CrossRef CAS.
- C. Pronoitis, M. Hakkarainen and K. Odelius, ACS Sustainable Chem. Eng., 2022, 10, 2522–2531 CrossRef CAS.
- Covestro 2022, Covestro and Genomatica produce important chemical raw material using biotechnology, accessed March 28th 2024, https://www.covestro.com/press/covestro-and-genomatica-produce-important-chemical-raw-material-using-biotechnology/.
- J. Sternberg and S. Pilla, Green Chem., 2020, 22, 6922–6935 RSC.
- F. Mundo, S. Caillol, V. Ladmiral and M. A. R. Meier, ACS Sustainable Chem. Eng., 2024, 12(17), 6452–6466 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01547a |
| ‡ These authors equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2024 |