
General electron–donor–acceptor complex mediated thioesterification reaction via site-selective C–H functionalization using aryl sulfonium Salts†
Roshan I.
Patel
,
Barakha
Saxena
and
Anuj
Sharma
 *
*
Green Organic Synthesis laboratory, Indian Institute of Technology Roorkee, Roorkee, 247667, India. E-mail: anujsharma.mcl@gmail.com; anuj.sharma@cy.iitr.ac.in
First published on 3rd September 2024
Abstract
Contemporary methods for synthesizing thioesters often necessitate expensive catalysts and harsh conditions, making their synthesis from chemical feedstocks challenging. Herein, we report a sustainable metal-, photocatalyst-, and oxidant-free electron donor–acceptor (EDA) mediated synthesis of thioesters via site-selective C–H functionalization using aryl sulfonium salts (acceptor) with potassium thioacid salts (donor) under visible light irradiation. Our approach enables rapid access to thioesters from a wide variety of arenes, including pharmaceutical and agrochemical compounds, as well as a diverse range of alkyl, aryl, and heteroaryl potassium thioacid salts with excellent efficiency and regioselectivity. Mechanistic studies supported the formation of an EDA-complex, and radical trapping experiments corroborated the involvement of a radical-based mechanism for the product formation. Moreover, our method demonstrates excellent atom economy and E-factor scores, which are considered excellent in terms of safety, economic and ecological yardsticks.
Introduction
Thioesters are an essential functional group in a wide range of complex natural products, polymers, and therapeutics.1 They serve as high-energy intermediates in numerous biochemical processes, playing a critical role in cellular regulation and biosynthetic pathways such as metabolism, fatty acid synthesis, and the production of esters and polyketides in living organisms.2 Due to their significant biological and pharmaceutical properties, exemplified in Fig. 1a,3 thioesters are essential intermediates in organic synthesis. Consequently, the synthesis of thioesters has attracted considerable interest, prompting the development of various strategies for their preparation.4 Contemporary methods for synthesizing thioesters have expanded to a wide range of starting materials, including alkenes, alkynes, amines, halides, alcohols, aldehydes, and phenol derivatives.5 These methods often employ toxic carbon monoxide (CO) gas as a carbonyl group source and toxic and malodorous thiols, sulfonyl hydrazides, disulfides, or sulfonyl chlorides as S-group sources.5 In addition, these approaches have other notable drawbacks, such as the use of expensive transition metal catalysts, harsh oxidants, and high reaction temperatures.4,5 In recent years, the synthesis of thioesters has focused on developing more efficient techniques that utilize carbonyl compounds such as aldehydes, acyl chlorides, ketoacids, and carboxylic acids or their activated forms with thiols, disulfides, sodium sulfinates, or elemental sulfur (S8) to construct C–S bonds.6 Unfortunately, these methods also invariably require expensive transition metal catalysts under harsh reaction conditions, which result in reduced yields and necessitate extensive purification of the final products. In the past decade, visible light photoredox catalysis has become one of the most exciting fields in research among chemists, harnessing light energy for chemical transformations.7 Given its extensive applications in organic synthesis, several light-induced thioesterification approaches have been developed.8 For instance, Liao and co-workers (2019) demonstrated a visible light-induced base-mediated deaminative thioesterification of amino-acid-derived Katritzky salts (acceptor) using a single example of non-substituted thiobenzoic acid (donor), thus seriously limiting substrate scope of the method (Fig. 1b).9 Additionally, the reaction produced 2,4,6-triphenylpyridine as a non-reusable by-product. Subsequently, the same group published a dual Ru/Cu-catalyzed decarboxylative thioesterification of carboxylic acid-derived N-(acetoxy)phthalimides with again a single thiobenzoic acid as a sulfur source under visible light irradiation.10 In addition to the limited substrate scope, the method suffered in terms of expensive transition metal catalysts. In 2022, independent research groups revealed a visible light dual 4-CzIPN/Ni-catalyzed or photoinduced dehalogenative thioesterification of aryl halides (X = –Br, –I) with potassium thioacid salts.11 However, these methods are limited to simple aryl halides (X = –Br, –I) with a low functional group tolerance and have yet to be tested on more complex molecules, thereby restricting its substrate scope. Additionally, these methods require long reaction times and expensive photocatalysts under light, making them less favorable. Later, Zhang, Wu, and co-workers, in 2023, made a significant advancement in the synthesis of thioesters by developing a tetra-n-butylammonium decatungstate (TBADT)-catalyzed thioesterification of aldehydes with elemental sulfur (S8) and alkenes or alkynes via hydrogen atom-transfer (HAT).12 The method, however, suffers from the use of toxic elemental sulfur (S8) and an expensive transition metal catalyst. Around the same time, Liu, Zhao, and co-workers reported a visible light palladium-catalyzed decarboxylative thiocarbonylation of sulfonium salts with oxalic acid mono-thioethers (OAMs).13 However, the method required an expensive metal catalyst with a complex ligand system for thioester synthesis. In general, a metal-, photocatalyst-, oxidant-, and base-free methods can be a significant addition to thioesterification reactions.6,8 Moreover, the synthesis of thioesters via C–H functionalization is challenging and highly desirable, and achieving this would represent a substantial advancement in thioesterification reactions.5,6,8 In this context, site-selective C–H functionalization via thianthrenium salt synthesis has emerged as a powerful tool in organic synthesis, enabling the introduction of various functional groups into the aromatic compound and providing a streamlined and efficient pathway toward complex molecules.14 Concurrently, EDA-complex photochemistry has garnered significant interest due to its unique capability to harness visible light to activate colorless substances, generate radical intermediates, and drive subsequent reactions without the need for external catalysts.15 In line with our ongoing research into photoinduced EDA-complex reactions,16 herein, we present a practical method for synthesizing thioesters. We hypothesize the formation of an EDA-complex between aryl thianthrenium salts (acceptor) and potassium thioacid salts (donor) to synthesize valuable thioester products (Fig. 1d). Under visible light irradiation, potassium thioacid salt (donor) engage in a single electron transfer (SET) process with aryl thianthrenium salts (acceptor), generating an aryl radical intermediate and recyclable thianthrene. This aryl intermediate subsequently interacts with the resulting sulfur-centered radical to give the desired thioester product. By leveraging the EDA-complexes in our reaction, this method enables regioselective C–H thioesterification of arenes, a challenging feat to achieve without transition metal catalysts, aryl halides as radical precursors, and toxic, foul-smelling sulfur reagents. Additionally, the thianthrene by-product generated during the reaction is reusable and recyclable, offering further benefits in terms of sustainability and efficiency. Moreover, our method facilitates coupling complex agrochemical and pharmaceutical compounds with thioacids, offering substantial benefits to academic research and industrial applications. | ||
| Fig. 1 (a) Thioesters containing bioactive compounds; (b) previous work; (c) synthetic potential challenges; (d) advancement (our strategy). | ||
Results and discussion
Our study commenced with the use of methyl-2-methoxybenzoate-derived thianthrenium tetrafluoroborate 1a and potassium thioacetate 2a as representative substrates in DMSO solvent under 390 nm Kessil lamp irradiation (Table 1). We were delighted to observe the desired product obtained in 69% yield (entry 1). DMSO efficiently solubilized the reaction components (1a and 2a), resulting in an intense yellow color change in the reaction mixture, which may be speculated to be an EDA-complex. Screening other solvents such as MeCN, DCM, THF, DMA, DMF, and EtOAc resulted in lower yields (entries 2–7). We then investigated the effect of different additives (entries 8–15). To our delight, using 4 Å MS as an additive slightly increased the yield to 77% (entry 9). We speculate that 4 Å MS functions as an ion-trap reagent, which captures the potassium ion of thioacetate 2a, which may result in the formation of a stable thiolate anion.17 This can then give rise to a more efficient interaction between the electron accepter 1a and the electron donor 2a. Notably, using an inorganic base such as K2CO3 resulted in reduced product yield (entry 12). Screening of other inorganic bases as additives, such as Na2CO3, K2HPO4, and Cs2CO3 did not improve the reaction yield either (entries 13–15). We next explored using different light sources (entries 16–18). Remarkably, under 427 nm, visible light irradiation led to an 85% yield of the required product (entry 16). No product formation was observed when the reaction was conducted in the dark, underscoring the necessity of light irradiation (entry 19). Moreover, performing the reaction in an open-to-air condition resulted in a lower yield of 72%, indicating the importance of an inert atmosphere for optimal product formation (entry 20). Furthermore, the optimized time of 30 minutes resulted in no additional change in yield (entry 21).|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Additive | Light (nm) | Yield (%) |
| a 1a (0.3 mmol), and 2a (0.6 mmol, 2.0 equiv.), 4 Å MS (60 mg), dry DMSO (1.5 mL). b Open-to-air. c 1a (0.3 mmol), and 2a (0.6 mmol, 2.0 equiv.), dry DMSO (1.5 mL), 4 Å MS (60 mg), 30 min; nr = no reaction. | ||||
| 1 | DMSO | — | 390 nm | 69% |
| 2 | MeCN | — | 390 nm | 58% |
| 3 | DCM | — | 390 nm | 37% |
| 4 | THF | — | 390 nm | 25% |
| 5 | DMA | — | 390 nm | 48% |
| 6 | DMF | — | 390 nm | 43% |
| 7 | EtOAc | — | 390 nm | 54% |
| 8 | DMSO | SiO2 (20 mg) | 390 nm | 72% |
| 9 | DMSO | 4 Å MS (20 mg) | 390 nm | 77% |
| 10 | DMSO | 3 Å MS (20 mg) | 390 nm | 70% |
| 11 | DMSO | Basic alumina (20 mg) | 390 nm | 73% |
| 12 | DMSO | K2CO3 | 390 nm | 12% |
| 13 | DMSO | Na2CO3 | 390 nm | 17% |
| 14 | DMSO | K2HPO4 | 390 nm | 15% |
| 15 | DMSO | Cs2CO3 | 390 nm | 21% |
| 16 | DMSO | 4 Å MS (20 mg) | 427 nm | 85% (83%)a |
| 17 | DMSO | 4 Å MS (20 mg) | 440 nm | 62% |
| 18 | DMSO | 4 Å MS (20 mg) | 456 nm | 50% |
| 19 | DMSO | 4 Å MS (20 mg) | Dark | Nr |
| 20 | DMSO | 4 Å MS (20 mg) | 427 nm | 72%b |
| 21 | DMSO | 4 Å MS (60 mg) | 427 nm | 83%c |
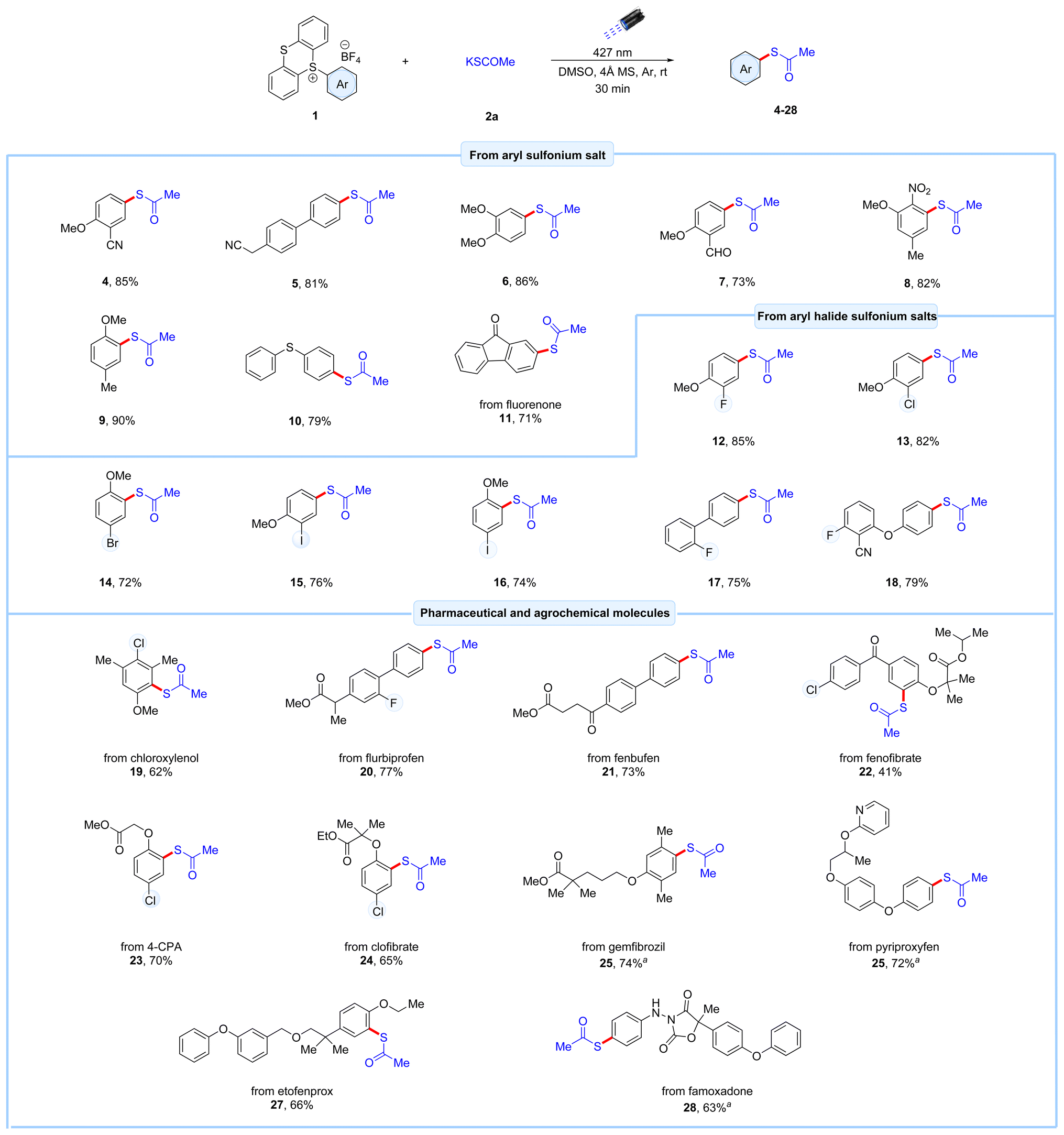
With the optimized reaction conditions established, various aromatic feedstock substrates were subjected to the C–H thioesterification using potassium thioacetate 2a as the coupling partner in the photochemical reaction. Aryl thianthrenium salts bearing electron-donating as well as electron-withdrawing substituents such as cyano-, trifluoromethyl-, nitro-, methoxy-, and methyl groups, reacted well under the optimized conditions, yielding the desired thioester products 4–9 in 73–90% yields (Scheme 1). Additionally, diphenyl sulfide-derived thianthrenium salt reacted efficiently under this protocol, producing the thioacetate product 10 in 79% yield. The bioactive fluorenone-derived thioacetate 11 was also obtained with a 71% yield. The retention of halides is problematic under reported transition metal and light-induced reactions. To evaluate the compatibility of this method, a series of aryl halide-derived thianthrenium salts were subjected to the EDA protocol. Pleasingly, the protocol effectively retained the halide groups to give the corresponding thioester products 12–18. Notably, aryl thianthrenium salts with fluoro- and chloro-group reacted exceptionally well (12–13), giving higher yields than their bromo- and iodo-substituted counterparts (14–16). Additionally, meta-substituted aryl iodo-substituted thianthrenium salts reacted smoothly under this method, achieving a yield of 74% (16). This protocol was also extended to diaryl-fluoro-substituted thianthrenium salts, resulting in the isolation of their corresponding thioester products 17 and 18 in good yields (75 and 79% yield, respectively). With the excellent site-selectivity achieved through the synthesis of thianthrenium salts,14a combined with the effectiveness of our EDA-complex protocol, makes this method a powerful tool for executing precise late-stage thioesterification of complexed agrochemical and pharmaceutical compounds, as illustrated in the Scheme 1. The thioesterification of thianthrenium salt derived from the polysubstituted antiseptic and disinfectant agent chloroxylenol proceeded smoothly, yielding the desired thioacetate product 19 in 62% yield. Moreover, using this protocol, aryl ester bearing thianthrenium salts derived from the anti-inflammatory drugs flurbiprofen and fenbufen underwent successful thioesterification, yielding the desired products 20 and 21 in 77% and 73% yield, respectively. Next, a series of aryl ether-substituted agrochemical and pharmaceutical-derived thianthrenium salts were subjected to the optimized conditions. These included thianthrenium salts derived from the hypertriglyceridemia drugs fenofibrate and gemfibrozil, the plant growth regulator p-chlorophenoxyacetic acid (4-CPA), antilipidemic drug clofibrate, the insecticidal agents pyriproxyfen, and etofenprox, as well as the amide, ester and ether-bearing fungicidal agent famoxadone. These reactions were well tolerated, affording the corresponding thioester products as single regioisomers in satisfactory to good yields (22–28). Notably, the hypertriglyceridemia drug fenofibrate 22, which contains a highly bulky ether group on the corresponding carbon, resulted in a lower yield of 41%, likely due to steric hindrance. In general, the protocol exhibited excellent compatibility with numerous functional groups, including halides (F and Cl), esters, ethers, amides, and heteroarenes.
 | ||
Scheme 1 Reaction conditions: aryl sulfonium salt 1 (0.3 mmol), potassium thioacetate 2a (2 equiv., 0.6 mmol), 4 Å MS (60 mg), in 1.5 mL DMSO, were irradiated with 427 nm Kessil Lamp (40 W) at room temperature under argon for 30 min. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 h. 2 h. | ||
To further diversify our EDA-complex strategy, we employed various potassium thioacid salts with alkyl, aryl, and heteroaryl functionalities with the aryl thianthrenium salt. We were delighted that various potassium thioacid salts reacted smoothly with the aryl thianthrenium salts to yield the corresponding thioester products in moderate to good yields, as illustrated in Scheme 2. Potassium thioacid salts derived from naturally occurring long-chain saturated fatty acids such as pelargonic and capric acid, performed well under the established conditions, yielding the corresponding thioester products 29 and 30 with 64% and 61% yield, respectively. Additionally, potassium thioacid salts derived from the antiproliferative agent veratric acid and antioxidant agent eudesmic acid salts proved to be efficient electron donors in this photochemical strategy, producing the desired thioester products 31 and 32 in 69% and 71% yield, respectively. The potassium thioacid derived from the anti-inflammatory drug ibuprofen was also compatible under this protocol, yielding the thioester product 33 in 56% yield. Aryl potassium thioacid salts bearing alkyl functionalities were found to be suitable coupling partners with aryl thianthrenium salts, yielding the corresponding thioester products 34–36 in yields ranging from 68–74% yield. Furthermore, aryl potassium thioacids bearing trifluoromethyl- and fluoro-, and dichloro-groups were also well tolerated, yielding the required thioester product 37, 38, and 39 with yields of 62%, 67%, and 60% yield, respectively. Additionally, potassium thioacids derived from various O- and S-containing heterocycles were also effective in this photochemical process, yielding the desired thioester products 40–43 in satisfactory to good yields. Moreover, non-substituted potassium benzothioate proved to be a suitable coupling partner under this protocol, efficiently reacting with different aryl thianthrenium salts to produce the required thioester products 44–47 in good yields. Lastly, the compatibility of this method was demonstrated in the thioesterification of the insecticidal agent pyriproxyfen with eudesmic acid-derived potassium thioacid salt, affording the required thioester product 48 in 60% yield.
 | ||
| Scheme 2 Reaction conditions: aryl sulfonium salt 1 (0.3 mmol), potassium thioacid salt 2 (2 equiv., 0.6 mmol), 4 Å MS (60 mg), in 1.5 mL DMSO, were irradiated with 427 nm Kessil Lamp (40 W) at room temperature under argon for 30 min. a2 h. | ||
Mechanistic insights
Next, a series of mechanistic studies were conducted to elucidate the reaction mechanism as illustrated in Fig. 2. The UV/Vis-absorption analysis of individual components and the reaction mixture (1a + 2a) in DMSO is shown in Fig. 2a. The DMSO solutions of methyl-2-methoxybenzoate-derived thianthrenium salt 1a (red line), and potassium thioacetate 2a (green line) displayed a small absorption band in the visible light region (>400 nm). Moreover, a clear bathochromic shift (blue band) was observed of the reaction mixture (1a + 2a) in DMSO, which was visible by the intense yellow color of the reaction mixture, as shown in Fig. 2a. It indicates the formation of an electron donor–acceptor (EDA) aggregate (blue band). Additionally, a Job's plot using UV-visible absorption experiments was performed to determine the stoichiometry of the EDA-complex between 1a and 2a. The maximum absorption of the reaction mixture [1a + 2a] at a 50% molar fraction indicated a 1:1 ratio of 1a and 2a (Fig. 2b). Moreover, 1H NMR titrations were conducted to provide further evidence of an EDA-complex formation in DMSO-d6 (Fig. 2c). The 1H NMR signal of C1–H proton in methyl-2-methoxybenzoate-derived thianthrenium salt 1a, shifted downfield along with the increasing amount of potassium thioacetate 2a. In contrast, C4–H, C3–H, C2–H, and C5–H proton shifted upfield, thus indicating the formation of an EDA-complex between 1a and 2a.18 Under standard conditions, the photochemical reaction was subjected to a radical trap experiment with TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), butylated hydroxytoluene (BHT) and 1,1-diphenylethylene (Fig. 2d). The reactions were quenched, and the radical trapped adducts were analyzed and detected by high-resolution mass spectrometry (HRMS). These results support the generation of an aryl radical as well as thiyl radical intermediate in the photochemical transformation, likely initiated by the photoactive EDA-complex between methyl-2-methoxy benzoate-derived thianthrenium salt 1a and potassium thioacetate 2a.
 | ||
| Fig. 2 (a) UV-visible absorption spectra; (b) Job's plot; (c) 1H NMR titration experiment; (d) radical trap experiment; (e) sunlight irradiation; (f) scale-up; (g) sensitivity assessment of reaction; (h) green chemistry metrics (our method). | ||
A quantum yield experiment was also performed to determine a possible reaction pathway for this transformation. The quantum yield was measured as Φ = 44, indicating a radical chain pathway (see ESI† for detailed discussion). Furthermore, the reaction also proceeded successfully under natural sunlight irradiation, yielding the targeted product 3a in 70% yield (Fig. 2e). A gram-scale synthesis was conducted using methyl-2-methoxy benzoate-derived thianthrenium salt 1a (1.404 g, 3 mmol) and potassium thioacetate 2a (0.685 g, 6.0 mmol, 2 equiv.) (Fig. 2f). The reaction was carried out under visible light irradiation for 2 h, yielding the desired product 3a in 58% yield (0.420 g). Moreover, 0.598 g (92% yield) of thianthrene was also recovered via chromatographic separation and was recycled back to thianthrene-S-oxide (0.620 g, 97%). This thianthrene-S-oxide was further converted back to 1a (0.957 g, 77%) (see ESI† for detailed discussion). Next, the efficiency of our reaction protocol for the synthesis of 3a was investigated under various sensitivity assessment parameters.19 This EDA-complex transformation was found sensitive to water, oxygen concentration, and low light intensity (Fig. 2g). However, the protocol showed good tolerance towards substrate concentration, temperature, and high light intensity variations. Further attempts were made to synthesize the thioester compound via one-pot, but the reaction failed to yield the required product.
To evaluate the eco-friendliness and greenness of our developed strategy, we assessed the green chemistry metrics for the synthesis of compound 3a (0.06 g, 83%) from methyl-2-methoxybenzoate-derived thianthrenium salt 1a (0.3 mmol, 0.141 g) and potassium thioacetate 2a (0.6 mmol, 0.069 g). The reaction was carried out using 4 Å molecular sieve (0.06 g) in DMSO (1.65 g, 1.5 mL) under visible light conditions. The results, as shown in Fig. 2h, demonstrate that the green chemistry metrics are excellent, the E-factor is 14.95, process mass intensity (PMI) is 15.37%, reaction mass efficiency is 57.62%, atom economy (AE) is 78.34%, and carbon efficiency is 100%.20 Additionally, the EcoScale score was calculated to be 70.5, which is considered acceptable in terms of sustainability (see ESI† for detailed discussion).
In light of all the above experimental data, a plausible mechanism for this EDA-complex mediated thioesterification reaction is depicted in Scheme 3. Initially, an EDA-complex aggregate is formed between 1 and potassium thioacid salt 2. Under visible light irradiation, this EDA-complex undergoes a single-electron transfer (SET) event from thiolate anion to the aryl thianthrenium salt 1, generating thiyl radical intermediate Int-I, aryl radical intermediate Int-II, and recyclable thianthrene by-product. The generated thiyl radical intermediate Int-I and aryl radical intermediate Int-II, can undergo subsequent radical–radical coupling, leading to the formation of the desired product 3. Moreover, the aryl radical intermediate Int-II interacts with thiolate anion 2 to give Int-III. This intermediate Int-III undergoes SET with 1 to give the desired product 3, thianthrene by-product, and Int-II, which propagates the chain again.
 | ||
| Scheme 3 Possible mechanism. | ||
Conclusion
In summary, we have developed an EDA-complex mediated thioesterification reaction via site-selective C–H functionalization using thianthrenium salts. This protocol is compatible with a wide range of aromatic feedstock, and a diverse range of alkyl, aryl, and heteroaryl potassium thioacid salts, yielding the desired thioester products in good yields. Additionally, we demonstrated the versatility of the method through late-stage thioesterification of agrochemical and pharmaceutical compounds. Our method offers several key advantages over previously known methods, including (I) C–H functionalization;(II) halide retention; (III) compatibility with alkyl, aryl, and heteroaryl potassium thioacid salts; and (IV) reusable and recyclable by-products, and (V) excellent atom economy, and E-factor scores, which are considered excellent in terms of safety, economic and ecological yardsticks, making it a practical solution for both academic research and industrial applications.Author contributions
R. I. Patel optimized the reaction conditions and synthesized all the derivatives. B. Saxena synthesized the potassium thioacid salts. R. I. Patel and B. Saxena performed the mechanistic studies and wrote the manuscript with the helpful insights of Prof. A. Sharma. Prof. A. Sharma supervised the whole work, interpreted the results, and edited the manuscript. All the authors have given their approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from SERB (CRG/2022/002691), India is gratefully acknowledged. We also acknowledge DST-FIST (SR/FST/CSII/2018/72(C)) for the NMR and HRMS facilities in the Chemistry Department, IIT Roorkee. R. P. and B. S. thank UGC and CSIR for the SRF Fellowship, respectively.References
-
(a) K. Taori, V. J. Paul and H. Luesch, J. Am. Chem. Soc., 2008, 130, 1806–1807 CrossRef
; (b) X.-L. Liu, Y. Shi, J. S. Kang, P. Oelschlaeger and K.-W. Yang, ACS Med. Chem. Lett., 2015, 6, 660–664 CrossRef PubMed
-
(a) K. Chandru, A. Gilbert, C. Butch, M. Aono and H. J. Cleaves, Sci. Rep., 2016, 6, 29883 CrossRef CAS
-
(a) Y. Yang, J. Zhu, M. Hassink, L. M. M. Jenkins, Y. Wan, D. H. Appella, J. Xu, E. Appella and X. Zhang, Emerging Microbes Infect., 2019, 6(1), 1–8 CAS
-
(a) A. Pal, P. P. Mondal, F. Niloofar and B. Sahoo, Eur. J. Org. Chem., 2022, e202201159 CrossRef CAS
- For selected examples see:
(a) V. Hirschbeck, P. H. Gehrtz and I. Fleischer, J. Am. Chem. Soc., 2016, 138, 16794–16799 CrossRef CAS PubMed
- For selected examples see.
(a) S.-H. Guo, X.-L. Zhang, G.-F. Pan, X.-Q. Zhu, Y.-R. Gao and Y.-Q. Wang, Angew. Chem., Int. Ed., 2018, 57, 1663–1667 (
Angew. Chem.
, 2018
, 130
, 1679–1683
) CrossRef CAS
-
(a)
C. R. J. Stephenson, T. Yoon and D. W. C. Macmillan, in Visible light photocatalysis in organic chemistry, Wiley-VCH, German, 2018 CrossRef
- For selected examples, see.
(a) G. Liu, N. Zheng, X. Duan, X Sun and W. Song, Green Chem., 2023, 25, 5035–5040 RSC
- M. Yang, T. Cao, T. Xu and S. Liao, Org. Lett., 2019, 21, 8673–8678 CrossRef CAS PubMed
- T. Xu, T. Cao, M. Yang, R. Xu, X. Nie and S. Liao, Org. Lett., 2020, 22, 3692–3696 CrossRef CAS
-
(a) W. Zheng, Y. Xu and L. Lin, ChemPhotoChem, 2022, 6, e202100264 CrossRef CAS
- H. Tang, M. Zhang, Y. Zhang, P. Luo, D. Ravelli and J. Wu, J. Am. Chem. Soc., 2023, 145, 5846–5854 CrossRef CAS PubMed
- G.-Q. Hu, W.-Y. Zhang, Y.-X. Liu, J.-H. Liu and B. Zhao, J. Org. Chem., 2023, 88, 14351–14356 CrossRef CAS PubMed
- For selected examples see.
(a) F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed
-
(a) A. K. Wortman and C. R. J. Stephenson, Chem, 2023, 9, 1–26 CrossRef
-
(a) B. Saxena, R. I. Patel and A. Sharma, Adv. Synth. Catal., 2023, 365, 1538–1564 CrossRef CAS
- K. Shin, Y. Zheng, F. Zhang, S. Wu and Y. Tang, ACS Appl. Energy Mater., 2020, 3, 7030–7038 CrossRef CAS
- W. Liu, H. Hou, H. Jing, S. Huang, W. Ou and C. Su, Org. Lett., 2023, 25, 8350–8355 CrossRef CAS PubMed
- L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572–8576 CrossRef CAS PubMed
-
(a) K. Van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 3 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed optimization, experimental procedure, 1H, 13C, and 19F NMR spectra of all the synthesized compounds. See DOI: https://doi.org/10.1039/d4gc03768e |
| This journal is © The Royal Society of Chemistry 2024 |