
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flow electrosynthesis of phosphinamides and phosphoramidates through P–N coupling†
Tribani
Boruah
ab,
Ren
Ishizeki
a,
Alberto
Roldan
 b,
Rebecca L.
Melen
*b and
Thomas
Wirth
*a
b,
Rebecca L.
Melen
*b and
Thomas
Wirth
*a
aSchool of Chemistry, Cardiff University, Park Place, Main Building, Cardiff CF10 3AT, Cymru/Wales, UK. E-mail: wirth@cf.ac.uk
bSchool of Chemistry, Cardiff Catalysis Institute, Cardiff University, Translational Research Hub, Maindy Road, Cardiff CF24 4HQ, Cymru/Wales, UK. E-mail: MelenR@cardiff.ac.uk
First published on 18th October 2024
Abstract
A robust flow electrochemical methodology operating under mild reaction conditions for P–N and P–O oxidative coupling has been developed. Potentiostatic and galvanostatic electrolyses were used to investigate reactant redox behaviour, showcasing a broad substrate scope (47 examples, up to 85% yield) across various chemical contexts.
Introduction
Molecules containing P(O)–N bonds such as phosphinamides and phosphoramidates have emerged as essential entities in organic synthesis, showing unparalleled reactivity and structural diversity. Their multifaceted roles span from serving as synthetic intermediates and ligands in coordination chemistry to acting as chelating agents in metal extraction, recovery, and detection. These compounds also serve as biologically active scaffolds for medicines and agrochemicals, demonstrating remarkable antiviral, antibacterial or anticancer effects, such as acelarin (1, Fig. 1).1 Beyond their biological applications, phosphinamides are an integral part of functional materials in flame retardants (2, Fig. 1), polymers, dendrimers, or metal–organic frameworks,2 and have been used as efficient chiral ligands (3, Fig. 1).3 | ||
| Fig. 1 Functional phosphinamides and phosphoramidates. | ||
The stability of phosphinamides, surpassing that of phosphine oxides, positions them as valuable alternatives in various synthetic transformations. Different methodologies exist to produce phosphinamides, each offering distinct advantages tailored to the desired specific structural and functional requirements. Conventional approaches include the phosphorylation of amines using phosphorus trichloride,1 the Michaelis–Arbuzov reaction,4,5 phosphination of imines, reductive amination of phosphinic acids or esters,6 and P–N bond formation through cross-coupling reactions.7 Despite their utility, these conventional methods have limitations such as lengthy synthetic routes or the use of toxic chemicals. Therefore, the synthesis of phosphinamides from various phosphine oxides through direct coupling with amines has been explored. Recently, a method for the synthesis of α-aminoalkyl phosphine oxides in an aqueous medium using Ar2P(O)–H reagents, alcohols, and amines has been developed by Huang et al.8 This approach efficiently facilitates aminophosphinoylation under mild conditions. Although the method is versatile with different amines, it showed limited applicability with alcohols and H-phosphine oxides. Tan et al. contributed to this field with a zinc iodide mediated oxidative coupling using tert-butylhydroperoxide as an oxidant (Scheme 1).9 Very recently, Studer et al. also used a similar approach for heterocycle synthesis.10 Shi et al. developed a Tf2O (trifluoromethanesulfonic anhydride)-mediated amination of P(O)–OH or P(O)–H with diverse amines.11 However, substrate sensitivity to Tf2O and its high reactivity require careful handling, and steric/electronic factors limited the scope. In another effort to address the shortcomings of conventional methods, Yu et al. proposed a photocatalytic approach for phosphinamide synthesis using copper-based catalysts,12 while Qu reported C–H phosphonylations.13 There is growing interest in the development of efficient, sustainable, and industrially scalable methods for phosphinamide synthesis. The exploration of direct coupling strategies of phosphine oxides presents a promising avenue, leading to the exploration of electrochemical synthesis as a sustainable and efficient method.14 Various batch electrochemical methods have been reported; however, they suffer from limitations such as small scale operation, manual intervention requirements, lower productivity, and challenges in maintaining precise control over reaction parameters. The research described herein is built on Wang's work, which involved the use of a batch electrochemical approach.15 The continuous flow electrochemical synthesis described here allows access to phosphorus-containing compounds using aromatic amines, alcohols and thiols as substrates. The flow electrochemical synthesis of phosphinamides and phosphoramidates has not yet been reported. This approach is adaptable to a broad substrate scope, offering reduced reaction times and minimal use of redox mediators. The choice between galvanostatic and potentiostatic conditions significantly impacts the yield and selectivities.16 This research addresses challenges associated with conventional methods and overcomes limitations observed in prior electrochemical batch approaches.
 | ||
| Scheme 1 Challenges and benefits of previous synthetic approaches and current work. | ||
Results and discussion
Our research group is actively engaged in different research areas aiming to achieve organic transformations under flow-electrochemical conditions with a microfluidic reactor.17–19 Herein we describe the synthesis of phosphinamides and phosphoramidates via flow electrochemistry. Diphenylphosphine oxide 4 and aniline 5a were selected as model substrates to explore optimal electrolysis conditions for phosphinamide synthesis. Initially, we investigated batch electrochemical conditions leading to only low yields of the product 6a (Table S1†) after which we moved to flow electrochemical conditions for the above reaction (Table 1). The electrolysis was performed in an undivided commercially available flow electrochemical microfluidic reactor (see the ESI, Fig. S1–S3†),20 utilising a 0.03 M solution of 4 and 5a in acetonitrile. The reaction mixture was pumped at a flow rate of 0.05 mL min−1 with an applied charge of 2.5 F mol−1 and a constant current of 6 mA, with 20 mol% KI serving as the supporting electrolyte. This approach resulted in the desired product 6a in a yield of 43%, (Table 1, entry 8), which encouraged us to further investigate flow electrolysis parameters in detail, including varying the electrode materials, solvents, flow rates, and current densities, to establish optimal reaction conditions. Firstly, different electrodes to determine the optimal combination of cathode and anode materials were explored, as the electrodes significantly impact reaction kinetics, selectivity, and efficiency. While platinum cathodes exhibit high selectivity for reacting 4 (Table 1, entry 1), changing the cathode material to graphite (Gr) decreased the yield to 33% as did other cathodic materials such as glassy carbon (GC, 39%), stainless steel (SS, 55%) or copper (56%) (Table 1, entries 3–7). In contrast, anodes made of inert materials such as graphite proved suitable for oxidative coupling reactions, avoiding undesired reactions or contamination. Under the optimised reaction conditions, platinum as the cathode and graphite as the anode gave 76% yield of 6a (Table 1, entry 1). Subsequent optimisation of the charge provided in the reaction was crucial for achieving an efficient and selective conversion to 6a. Increasing the applied charge from 2.5 to 3 F mol−1 at a constant concentration of 0.03 M with 20 mol% KI increased the yield of 6a from 43% to 71% (Table 1, entries 8 and 9). However, a further increase in the charge led to a decrease in the yield (Table 1, entry 10), emphasising the importance of balancing energy consumption and reaction efficiency. Additionally, the impact of the flow rate on the reaction efficiency was investigated. Increased flow rates from 0.05 to 0.1 mL min−1, while maintaining a constant charge of 3 F mol−1, resulted in decreased yields of 35% and 29% (Table 1, entries 11 and 12), attributed to shorter residence times leading to incomplete transformations and reduced interactions between reactants and electrodes. Conversely, reducing the flow rate to 0.025 mL min-1 also led to a moderate decrease in the yield to 53% (Table 1, entry 13).|
|
||
|---|---|---|
| Entry | Deviation from the reaction conditionsa | 6a Yieldb (%) |
| a Reaction conditions: undivided flow cell, Pt cathode and graphite (Gr) as an anode, interelectrode distance: 0.5 mm, 4 (30 mM) and 5a (30 mM) in CH3CN, KI (30 mol%, 9 mM) as an electrolyte, and flow rate: 0.05 mL min−1, constant current: 7 mA, charge: 3 F mol−1. GC: glassy carbon, SS: stainless steel. b Yield determined by 1H NMR spectroscopy using dodecane as an internal standard. | ||
| 1 | No deviation | 76 |
| 2 | Pt as an anode, 2.5 F mol−1, 20 mol% KI, 6 mA | 43 |
| 3 | GC as an anode, 2.5 F mol−1, 20 mol% KI, 6 mA | 67 |
| 4 | Gr as a cathode, 2.5 F mol−1, 20 mol% KI, 6 mA | 33 |
| 5 | GC as a cathode, 2.5 F mol−1, 20 mol% KI, 6 mA | 39 |
| 6 | SS as a cathode, 2.5 F mol−1, 20 mol% KI, 6 mA | 55 |
| 7 | Cu as a cathode, 2.5 F mol−1, 20 mol% KI, 6 mA | 56 |
| 8 | 2.5 F mol−1, 20 mol% KI, 6 mA | 43 |
| 9 | 3 F mol−1, 20 mol% KI, 7 mA | 71 |
| 10 | 3.5 F mol−1, 20 mol% KI, 8 mA | 40 |
| 11 | 0.08 mL min−1, 20 mol% KI, 12 mA | 35 |
| 12 | 0.1 mL min−1, 20 mol% KI, 14 mA | 29 |
| 13 | 0.025 mL min−1, 20 mol% KI, 3 mA | 53 |
| 14 | Without KI | 10 |
| 15 | 10 mol% KI | 43 |
| 16 | 30 mol% KI | 76 |
| 17 | 35 mol% KI | 65 |
| 18 | 40 mol% KI | 52 |
| 19 | EtOH as a solvent | 0 |

Furthermore, the choice of the supporting electrolyte has a significant role in facilitating the reaction. Different electrolytes revealed that halogen salts facilitated the reaction, with KI providing the best result as already found in the batch electrochemical reaction.15 The absence of KI led to a drastic decrease in the yield (Table 1, entry 14), highlighting the dual role of halogen salts as an electrolyte and a redox mediator.15 Optimising the KI concentration (Table 1, entries 15–18) revealed 30 mol% as the most effective which is significantly lower compared to the amount (330 mol%) of the supporting electrolyte required in batch electrolysis.15 This is attributed to the sufficient conductivity of the homogeneous reaction mixture and the small interelectrode distance of 0.5 mm. The choice of solvent has a significant influence on the reaction. While ethanol has been reported to assist in determining mechanistic pathways for such electrochemical reactions,15 it does not lead to any product (Table 1, entry 19). The substrate concentration was finally investigated (see the ESI, Table S2†). Higher concentrations were found to increase the reaction rates due to the greater availability of reactants at the electrode surface, resulting in higher yields. However, very high concentrations lead to mass transfer limitations and incomplete reactions. Flow electrochemical reactions typically occur efficiently at room temperature, and we observed degradation of reactants and intermediates at higher temperatures (30–50 °C) (ESI, Table S2†). When the optimisations were performed at a different constant voltage under flow and batch conditions, the highest yield of 70% was observed at 2.25 V and a current of 4 mA with a flow rate of 0.05 mL min−1 (see the ESI, Tables S3 and S4†). Similarly, we investigated all the parameters with indole as a substrate, providing the product 7f with 79% yield at constant current, while under potentiostatic reaction conditions, 69% yield was obtained (see the ESI, Tables S5 and S6†).
The flexibility to generate phosphinamides using different amines and diphenylphosphine oxide was subsequently explored (Fig. 2). More electron-rich aromatic amines led to phosphinic amides in moderate to high yields (6a–6f). This was attributed to the more nucleophilic character of the amines, which can stabilise radical intermediates effectively and enhance reaction yields. Conversely, electron-poor aromatic amines resulted in lower yields (6g–6j). A primary amine bearing a phenyl substituent at the ortho-position was investigated, resulting in higher yields (6k, 85%). Additionally, amines with oxidation-sensitive benzyl positions were investigated.21,22 The P–N bond was selectively formed (6l–6q) without C–P coupling, with moderate to excellent yields (68% to 85%). The established protocol was also examined with aliphatic secondary amines,15 resulting in excellent yields (Fig. 2, 7a–7c).
 | ||
| Fig. 2 Substrate scope for phosphinamide synthesis using different amine nucleophiles. | ||
Meanwhile, amines with pyridine functionalities also produced excellent yields (7d, 7e). Notably, despite being a part of an aromatic ring system, the nitrogen atom in indole results in oxidative coupling with good yields (7f–7h).23 Additionally, diphenylphosphine oxide 4 was also employed to examine the addition of aromatic and aliphatic alcohols (Fig. 3), resulting in single products (8a,248b) with excellent yields. Bifunctional substrates containing both an amine and an alcohol were also examined leading to product mixtures 9a/9b and 10a/10b. For 2-hydroxyaniline, N–P coupling (forming 9a) was preferred whereas for 2-chloro-4-hydroxyaniline, O–P coupling (forming 10b) gave slightly higher yields. Additionally, benzene-1,2-diamine and catechol were re-optimised to examine the formation of diphosphorylated products; however, benzene-1,2-diamine was obtained as both mono- (11a) and di-substituted (11b) products. Catechol only produced the monosubstituted product 12 and only trace amounts of a disubstituted product were observed (Tables S7 and S8†). Also, difunctional indole substrates containing both 1H-indole nitrogen substituted with amine or hydroxy functionalities were explored. The N–P coupling product 13 with the amine functionality and the O–P coupling product 14 were formed, demonstrating the chemoselective nature of the protocol as no reaction at the indole N-H group was observed. Furthermore, carbazole and thiophenol were also explored as substrates and products 1525 and 1626 were obtained. In the reaction with thiophenol, diphenyl disulfide was obtained as a side product. Moreover, drug modifications were investigated with benzocaine and paracetamol, resulting in good product yields (68% and 71% for 17 and 18, respectively) (Fig. 3). The protocol was also implemented with various substituted phosphites, which resulted in the formation of P–N coupling phosphoramidate products 19–21 in good to excellent yields (Fig. 4). Unfortunately, diphenylphosphine could not be used as a replacement for 4 under the reaction conditions.
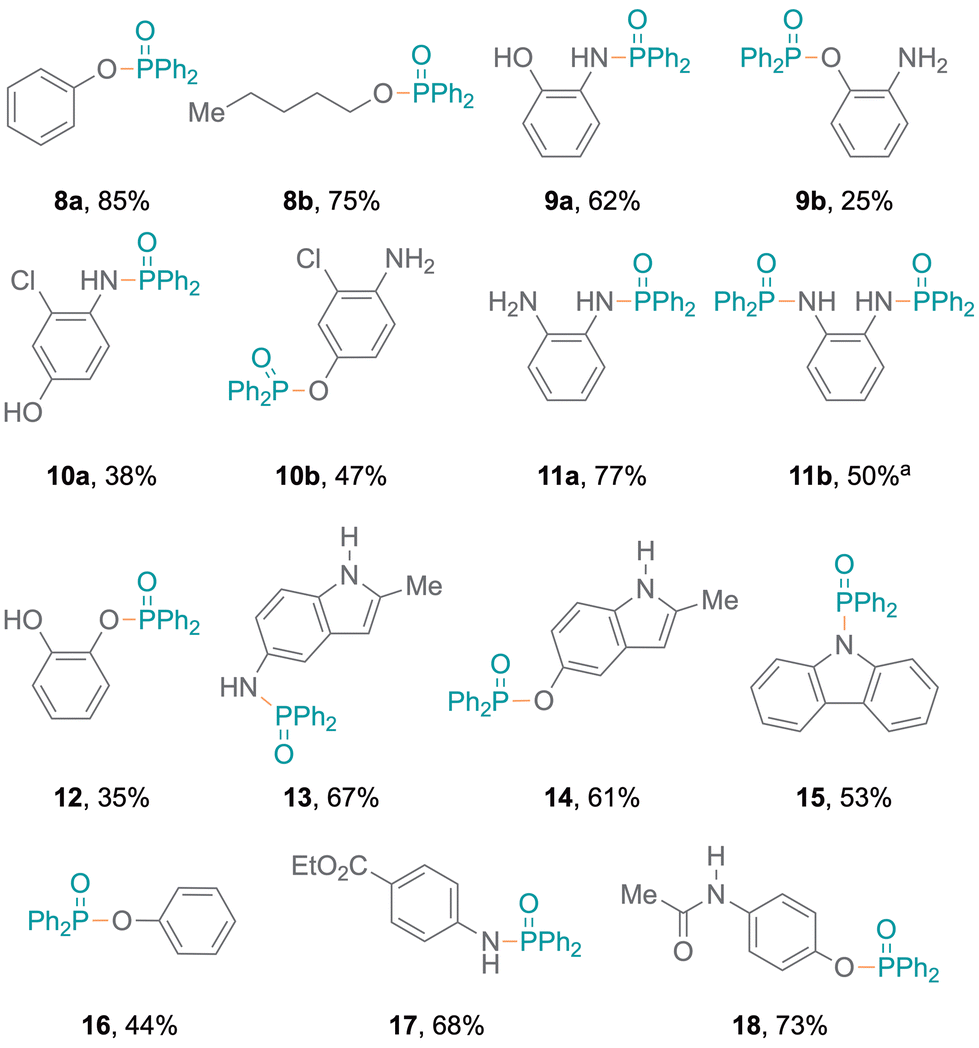 | ||
| Fig. 3 Alcohols and bifunctional substrates as nucleophiles for P–N or P–O coupling. aFormed together with 11a (32%) under re-optimised reaction conditions. | ||
 | ||
| Fig. 4 Structures and yields of the electrochemical synthesis of phosphoramidates 17–19. | ||
The N–P cross-coupling reaction was showcased through a larger-scale reaction of 4 and 5a. The reaction, conducted with a current of 7 mA for 55 h, resulted in the successful generation of 0.893 g of cross-coupled product 6a in 62% yield (see the ESI, Fig. S11†). However, passivation on the surface has been observed, obstructing the outlet and causing an increase in voltage and a lower yield (see the ESI, Fig. S12†). Conversely, at a constant potential of 2.25 V and a flow rate of 0.05 mL min−1 over the same duration, a steady yield (64%) was maintained without electrode passivation (see the ESI, Fig. S13 and S14†). Potentiostatic conditions significantly minimised side product formation, demonstrating superior efficiency and reliability. This underscores the method's efficiency and suitability for larger-scale synthesis. Oxidised species generated selectively under potentiostatic conditions mediate the desired oxidative coupling of aniline and triphenylphosphine oxide without over-oxidising other species or promoting side reactions. The fluctuating potential under galvanostatic conditions can inadvertently cause additional redox processes, leading to undesired by-products.
For more detailed mechanistic insights into the electrochemical P–N cross-coupling reaction, cyclic voltammetry (CV) experiments with potassium iodide, diphenylphosphine oxide 4 and aniline 5a were performed.27 In the cyclic voltammogram of the potassium iodide (KI) electrolyte, the first anodic peak at 0.87 V (A) (vs Ag/Ag+) corresponds to the oxidation of iodide to iodine (2I− → I2 + 2e−) (Fig. 5a) while the second anodic peak at 1.07 V (B) corresponds to the further oxidation of iodine to triiodide (I2 + 4I− → 2I3− + 2e−), followed by a third anodic peak at 2.39 V (C) depicting the further oxidation of triiodide to iodine (2I3− → 3I2 + 2e−).28 The first cathodic peak (D) corresponds to the reduction of iodine to iodide ions (I2 + 2e− → 2I−); however, the cathodic peak (E) might correspond to the reduction of triiodide ions back to iodide (I3− + 2e− → 3I−). This provides valuable insight into the electrochemical behaviour and redox processes of KI. Furthermore, the influence of KI on both substrates 4 and 5a was investigated through the CV measurements of 4 and 5a, respectively, in the presence of KI. Interestingly, when KI is added to 4 and 5a separately during the CV measurements, we observed significant shifts in onset and peak potentials towards zero, reducing the potential window further (Fig. 5b and c).
 | ||
| Fig. 5 Mechanistic investigation with cyclic voltammetry: (a) CV of 4 and KI, (b) CV of aniline and KI, (c) expansion of plot (a), (d) CV at different scan rates (inset: variation in peak current and shift in potential), (e) HOMO–LUMO energy gap with and without KI, and (f) proposed mechanism. | ||
The formation of intermediate species with iodide ions influences the electronic environment close to the electrode surface, affecting the kinetics of the redox reactions involving iodide ions. However, when 5a was added to KI, a significant increase in the peak current of the first anodic peak was observed showing the presence of a larger iodide ion concentration. There is a decrease in the peak current of the second peak, suggesting that the formation of triiodide ions is less favoured or occurs at a lower rate in the presence of 5a, as the iodide ions participate in oxidative coupling forming a complex with aniline 5a, which is further validated by control experiments. Compounds 22–25 were detected by ESI-HRMS as shown in Scheme 2 (see the ESI, Fig. S6–S9†).
 | ||
| Scheme 2 Control experiments and mechanistic investigations. | ||
To investigate whether the process is diffusion or adsorption controlled, a range of CV measurements at different scan rates (Fig. 5d) and plotted as a function of the square root of the scan rate against the peak current (Fig. S4†) resulted in a linear relationship indicating a diffusion controlled process, as described by the Randles–Sevcik equation (ESI†).27 A linear relationship between log (Ip) and log (scan rate) further supports a diffusion controlled process as analysed by the Dunn method (Fig. S5†).29 Further supporting the experimental evidence of KI functioning as a redox mediator, DFT shows the influence of the presence of KI on the HOMO–LUMO energy gap (Fig. 5e). This shows that in PhNHI, the HOMO–LUMO energy gap is smaller compared to aniline, which is also demonstrated for indole and phenol, respectively (Fig. S15†). Based on the above experiments, a plausible mechanistic pathway illustrating the coupling of N–I intermediates or N radicals with diarylphosphine oxides to the desired products is shown in Fig. 5f. The evolution of hydrogen gas at the cathode was also observed in the batch electrolysis as shown in Fig. S10.†
Conclusions
In response to the limitations of traditional synthetic methods for phosphinamides and phosphoramidates, this study introduces a novel approach using flow electrochemistry. Systematic optimisation of reaction parameters in a microfluidic reactor resulted in an efficient, selective, and scalable phosphinamide synthesis method. The versatility of the method is highlighted by its applicability to diverse substrates, including different amines and phosphine oxides. Mechanistic insights gained through cyclic voltammetry experiments, potassium iodide mediation, and diffusion-controlled processes aided our understanding of the P–N cross-coupling reactions. This work not only provides a sustainable alternative to existing methodologies, but also opens new avenues for explorations in the field of flow electrochemical synthesis of phosphinamides and phosphoramidates.Author contributions
Conceptualisation: T. W. and R. L. M.; data curation: T. B. and R. I.; formal analysis: all authors; funding acquisition: T. W., A. R. and R. L. M.; investigation: T. B. and R. I.; DFT calculations: T. B. and A. R.; and writing and editing: all authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank TOK Japan for financial support (PhD studentship to T. B.). We also thank Dr Benson Kariuki, Cardiff University, for X-ray structural analyses and the Mass Spectrometry Facility, School of Chemistry, Cardiff University, for mass spectrometric data. We also acknowledge Supercomputing Wales for access to the Hawk HPC facility, part-funded by the European Regional Development Fund via the Welsh Government.References
- Y.-Y. Zhu, Y. Niu, Y.-N. Niu and S.-D. Yang, Org. Biomol. Chem., 2021, 19, 10296–10313 RSC
.
- X. Zhao, H. V. Babu, J. Llorca and D.-Y. Wang, RSC Adv., 2016, 6, 59226–59236 RSC
- M. Hatano, T. Miyamoto and K. Ishihara, Org. Lett., 2007, 9, 4535–4538 CrossRef PubMed
-
L. Micouin, in Comprehensive Heterocyclic Chemistry III, Elsevier, 2008, vol. 11, pp. 107–131 Search PubMed
- A. Jasiak, G. Mielniczak, K. Owsianik, M. Koprowski, D. Krasowska and J. Drabowicz, J. Org. Chem., 2019, 84, 2619–2625 CrossRef PubMed
- I. Wilkening, G. Signore and C. P. R. Hackenberger, Chem. Commun., 2011, 47, 349–351 RSC
- Handoko, Z. Benslimane and P. S. Arora, Org. Lett., 2020, 22, 5811–5816 CrossRef PubMed
- Q. Huang, X. Jin, L. Wu, J. Li, Q. Xing, X. Wang and C. Zhao, Org. Biomol. Chem., 2024, 22, 3860–3865 RSC
- C. Tan, X. Liu, H. Jia, X. Zhao, J. Chen, Z. Wang and J. Tan, Chem. – Eur. J., 2022, 26, 881–887 CrossRef PubMed
- S. K. Banjare, L. Lezius, E. S. Horst, D. Leifert, C. G. Daniliuc, F. A. Alasmary and A. Studer, Angew. Chem., Int. Ed., 2024, 63, e202404275 CrossRef CAS PubMed
- D.-W. Shi, H.-Q. Yue, M. Li, J. Liu, C.-C. Wang, S.-D. Yang and B. Yang, J. Org. Chem., 2024, 89, 6729–6739 CrossRef
- K. C. Yu, H. Li, Y. H. Tu, H. Zhao and X. G. Hu, Org. Lett., 2022, 24, 9130–9134 CrossRef
- J. Yuan, Y. Liu, Y. Ge, S. Dong, S. Song, L. Yang, Y. Xiao, S. Zhang and L. Qu, Chin. J. Org. Chem., 2021, 41, 4738–4748 CrossRef
- N. Sbei, G. M. Martins, B. Shirinfar and N. Ahmed, Chem. Rec., 2020, 20, 1530–1552 CrossRef PubMed
- Y. Wang, P. Qian, J. H. Su, Y. Li, A.M Bi, Z. Zha and Z. Wang, Green Chem., 2017, 19, 4769–4773 RSC
-
(a) M. Rafiee, M. N. Mayer, B. T. Punchihewa and M. R. Mumau, J. Org. Chem., 2021, 86, 15866–15874 CrossRef PubMed
- M. Elsherbini, B. Winterson, H. Alharbi, A. A. Folgueiras-Amador, C. Génot and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 9811–9815 CrossRef CAS PubMed
- M. Á. G. López, R. Ali, M. L. Tan, N. Sakai, T. Wirth and S. Matile, Sci. Adv., 2023, 9, eadj5502 Search PubMed
- R. Ali, R. Babaahmadi, M. Didsbury, R. Stephens, R. L. Melen and T. Wirth, Chem. – Eur. J., 2023, 29, e202300957 CrossRef CAS PubMed
- Ion electrochemical reactor, https://www.vapourtec.com/products/flow-reactors/ion-electrochemical-reactor-features/, (accessed September 2024).
- M. Huang, J. Dai, X. Cheng and M. Ding, Org. Lett., 2019, 21, 7759–7762 CrossRef CAS PubMed
- A. Ollivier, S. Sengmany, M. Rey, T. Martens and E. Léonel, Synlett, 2020, 31, 1191–1196 CrossRef CAS
- Y. Yuan, X. Liu, J. Hu, P. Wang, S. Wang, H. Alhumade and A. Lei, Chem. Sci., 2022, 13, 3002–3008 RSC
- Deposition numbers 2367670 (6a), 2367671 (8a), 2367669 (9a), 2367667 (17) and 2367668 (22)† contain the supplementary crystallographic data for this paper.
- X. Dong, R. Wang, W. Jin and C. Liu, Org. Lett., 2020, 22, 3062–3066 CrossRef CAS
- R. Wang, X. Dong, Y. Zhang, B. Wang, Y. Xia, A. Abdukader, F. Xue, W. Jin and C. Liu, Chem. – Eur. J., 2021, 27, 14931–14935 CrossRef CAS PubMed
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS
- Y. A. Yaraliyev, Electrochim. Acta, 1984, 29, 1213–1214 CrossRef
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2367667–2367671. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc04450a |
| This journal is © The Royal Society of Chemistry 2024 |