
Visible light-driven α-sulfonylation of ketone-derived silyl enol ethers via an electron donor–acceptor complex†
Barakha
Saxena
,
Roshan I.
Patel
and
Anuj
Sharma
 *
*
Green Organic Synthesis Laboratory, Indian Institute of Technology Roorkee, Roorkee, 247667, India. E-mail: anujsharma.mcl@gmail.com; anuj.sharma@cy.iitr.ac.in
First published on 18th October 2024
Abstract
The diverse utility of β-ketosulfones in pharmaceuticals and bioactive compounds has generated considerable interest in their synthesis. However, existing synthetic approaches often depend on transition-metal catalysts, which require extensive purification and result in low yields. Herein, we present a cost-effective, metal- and photocatalyst-free, visible light electron donor–acceptor (EDA) complex-mediated sulfonylation of ketone-derived silyl enol ethers with thiosulfonates (acceptor) and DABCO as an electron donor under mild conditions, offering a more efficient and straightforward approach. Our method enables the synthesis of a diverse range of β-ketosulfone derivatives, including biologically active and late-stage molecules, in good yields. Our strategy offers several significant advantages over existing techniques, which include (i) transition-metal and photoredox catalyst-free conditions; (ii) no need for an external SO2 source; (iii) broad substrate scope; (iv) recyclable and reusable by-products; and (v) excellent atom economy, reaction mass efficiency, process mass intensity, and E-factor and EcoScale scores, highlighting its efficiency and economic sustainability. Detailed mechanistic studies confirm the involvement of an EDA-complex-mediated radical process that operates without a catalyst.
Introduction
Organosulfones are essential in synthetic and medicinal chemistry, particularly in modulating and controlling drug metabolism and biotransformation rates.1 Several clinically approved drugs containing sulfone groups are shown in Fig. 1A. Among these, β-ketosulfones have garnered significant attention from synthetic chemists due to their versatile applications in organic synthesis and their considerable pharmaceutical relevance.2 These compounds demonstrate a wide array of biological activities, making them particularly valuable in organic synthesis. As a result, extensive efforts have been devoted to synthesizing β-ketosulfones, underscoring their importance in chemical and biological contexts. Several methods have been developed for the synthesis of β-ketosulfones. These include the acylation of alkyl sulfones with acid chlorides, esters, or N-acyl benzotriazoles,3 the oxidation of β-ketosulfides or β-hydroxysulfones with stoichiometric oxidants,4 and the alkylation of metallic arene sulfinates with α-halo- or α-tosyloxy ketones.5 All these methods have drawbacks such as the use of costly transition metal catalysis, hazardous oxidants, and elevated temperatures. In this regard, radical sulfonylation has gained attention as an effective approach for synthesizing β-ketosulfones. This method typically involves the reaction of alkenes,6 alkynes7 or activated alkenes8 with various radical sulfonylation reagents. Although traditional methods are useful, they often require cumbersome multi-step prefunctionalization or preactivation of the starting materials, have limited substrate scopes, yield low results, or involve harsh reaction conditions, all of which present significant challenges. In the past decade, visible-light-promoted photoredox reactions have achieved significant milestones in organic synthesis.9 Amongst these, three methods involving visible light-promoted synthesis of β-ketosulfones have been reported involving the reaction between silyl enol ethers, an external sulfur dioxide (SO2) source, and an alkyl/aryl radical source. The first of these was disclosed by Ye, Wu, and co-workers in 2019, who reported a visible light Ir-catalyzed sulfonylation of silyl enol ethers using amine-derived Katritzky salts as alkyl radical precursors with K2S2O5 as a SO2 source (Fig. 1Ba).10 However, the substrate scope was limited and did not include aryl radical intermediates and alkyl-substituted O-silyl enol ethers. Moreover, the reaction produced 2,4,6-triphenylpyridine as a non-reusable by-product. The following year, Wu and co-workers developed a Ru-catalyzed synthesis of β-ketosulfones via a three-component reaction using aryldiazonium tetrafluoroborates, Na2S2O5, and 2,2-difluoroenol silyl ethers under photochemical conditions (Fig. 1Bb).11 Additionally, this method also suffers from a narrow substrate scope due to the use of unstable diazonium salts with an expensive photocatalyst. Later in 2022, He, Wu, and co-workers introduced an Ir-catalyzed sulfonylation of O-silyl enol ethers with DABCO·(SO2)2 and arene or alcohol-derived thianthrenium salts (Fig. 1Bc).12 Despite its innovation, the method has limitations, including its restriction to aryl-substituted O-silyl enol ethers, reliance on an expensive photoredox catalyst, the use of a complex and costly SO2 surrogate, and a long reaction time of 48 h. Typically, in these methods, the photoexcited catalyst (*PC) reduces the starting substrate via single electron transfer (SET), generating a radical intermediate (Fig. 1Bd). This radical then reacts with the external SO2 source, forming a sulfonyl radical intermediate, which adds to the silyl enol ethers, followed by SET reduction and subsequent hydrolysis, yielding β-ketosulfones. Consequently, developing a method for β-ketosulfone synthesis that eliminates the need for any transition metals or photocatalysts and directly leads to the sulfonyl radical intermediate would be a significant breakthrough and a major advancement in β-ketosulfone synthesis.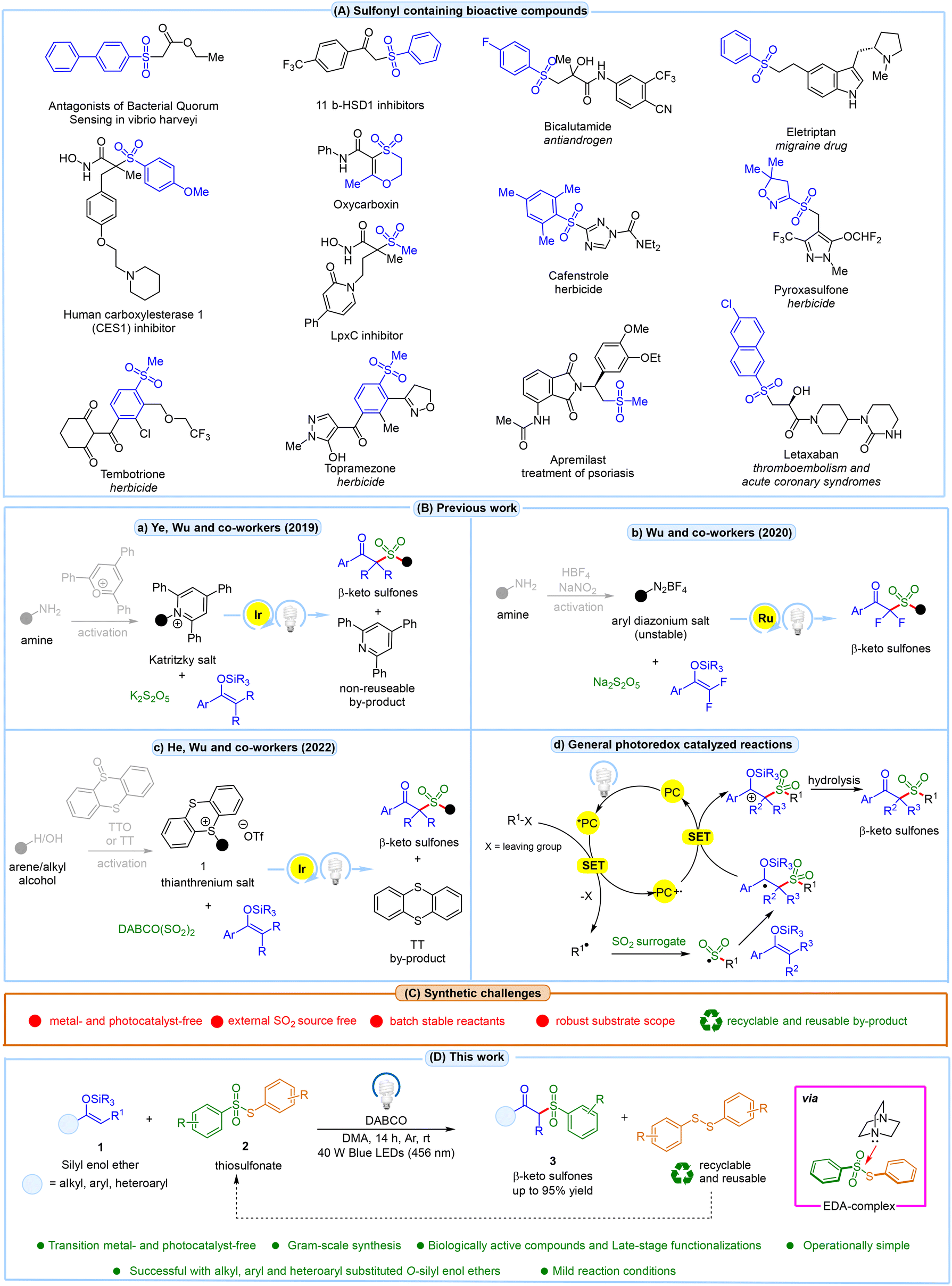 | ||
| Fig. 1 (A) Sulfonyl-containing bioactive drugs; (B) previous work; (C) synthetic challenges; and (D) this work. | ||
In this context, electron donor–acceptor (EDA) complex-based reactions have garnered significant interest in organic synthesis.13 Such EDA complexes, formed in the ground state, can absorb visible light, triggering an intermolecular single electron transfer (SET) event and eliminating the critical need for a customary redox potential matching in such reactions. Building on our ongoing research into green and sustainable photoinduced EDA-complex reactions,14 we present a practical method for the synthesis of β-ketosulfones using silyl enol ethers with thiosulfonates (acceptor) and DABCO (donor) under visible light irradiation (Fig. 1D).
We hypothesize that an EDA-complex forms between thiosulfonates (acceptor) and DABCO (donor), leading to the generation of an S-centered radical intermediate. This intermediate reacts with the silyl enol ether to yield the desired β-ketosulfone product and an aryl disulfide by-product. The use of an inexpensive base as an electron donor and thiosulfonates as a sulfonyl source under photochemical conditions facilitates the synthesis of β-ketosulfones, a challenging feat to achieve without the need for expensive photoredox catalysis and an external SO2 source. Additionally, the aryl disulfide by-product generated in the reaction is recyclable and reusable, offering additional advantages in terms of economic sustainability. Our method also enables the synthesis of various bioactive molecules and late-stage modification of pharmaceutical complexes, thus significantly benefiting industrial and academic research.
Results and discussion
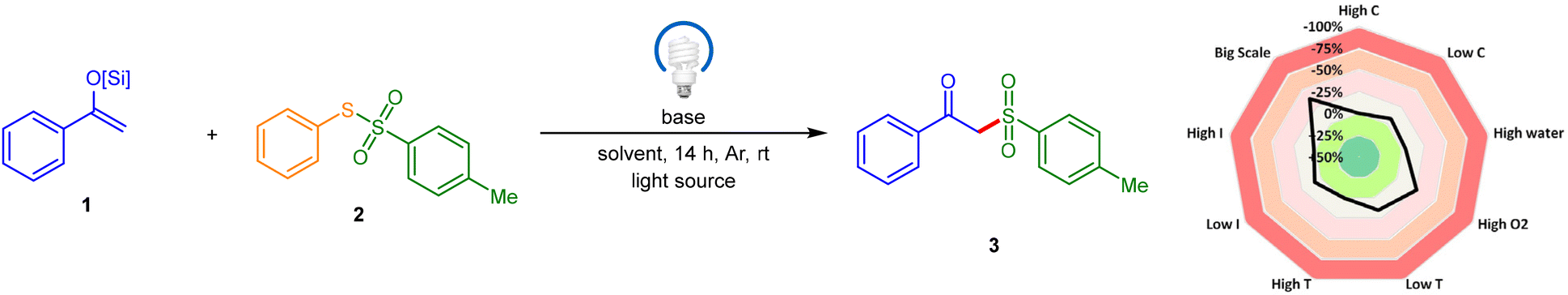
We initiated our study using the trimethylsilyl enol ether of acetophenone 1a and S-phenyl 4-methylbenzenesulfonothioate 2a as model substrates in MeCN solvent, with DBU as a base, under 456 nm Kessil lamp irradiation. To our satisfaction, the desired product was obtained in 52% yield (entry 1). Further screening of other bases proved unproductive (entries 2–6) except for DABCO, which emerged as the optimal base, delivering the highest yield of 67% (entry 6). We then tested various solvents, including DMSO, acetone, DCM, DMF, DCE, EtOH, and MeOH (entries 7–15). Among these, the reaction achieved an 81% yield in DMA solvent (entry 11), likely due to the efficient solubilization of the reaction components (1a and 2a). Notably, mixing 1a and 2a led to an intense yellow color change in the reaction mixture, suggesting the formation of an EDA-complex. The reaction yield was 55% when 1.0 equiv. of DABCO was used (entry 16). However, the yield increased to 89% when 3.0 equiv. of DABCO was used (entry 17). The reaction did not proceed without DABCO, underscoring its critical role in product formation (entry 18). Additionally, the reaction failed without light, confirming the necessity of light irradiation (entry 19). Screening of other light sources resulted in reduced yields (entries 20–23), indicating that 456 nm Kessil lamp irradiation is optimal for product formation. Lastly, we investigated compound 1, which features sterically more demanding triisopropylsilyl (TIPS), triethylsilyl (TES), and tert-butyldimethylsilyl (TBS) groups in place of the trimethylsilyl (TMS) group of the acetophenone-derived enol ether under the optimized conditions, leading to reduced yields or complete failure (entries 24–26) and indicating that the TMS group in enol ethers is crucial for a high yield of this reaction. In a condition-based sensitivity assessment, our EDA-protocol transformation was sensitive to high oxygen concentration, low temperature, and low light intensity. However, it generally exhibited tolerance to variations in substrate concentration, high reaction temperature, high light intensity, and water (see the radar diagram in Table 1 and the ESI† for details).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Base (2.0 equiv.) | Solvent | Light | Yield (%)a | Entry | Base (2.0 equiv.) | Solvent | Light | Yield (%) |
| a Reaction conditions: a mixture of O-silyl enol ether [Si] = TMS (trimethylsilyl) 1a (0.2 mmol), S-phenyl 4-methylbenzenesulfonothioate 2a (0.24 mmol, 1.2 equiv.), base (0.4 mmol, 2.0 equiv.) and solvent (0.5 mL) under visible light irradiation (light source) for 14 h under an Ar atmosphere at rt. b Base (0.2 mmol 1.0 equiv.). c Base (0.6 mmol 3.0 equiv.). d No base. e In the dark. f [Si] = TIPs (triisopropylsilyl). g [Si] = TES (triethylsilyl). h [Si] = TBS (tert-butyldimethylsilyl). | |||||||||
| 1 | DBU | MeCN | 456 | 52 | 14 | DABCO | THF | 456 | 42 |
| 2 | TMEDA | MeCN | 456 | 48 | 15 | DABCO | MeOH | 456 | N.D |
| 3 | DMAP | MeCN | 456 | 30 | 16b | DABCO | DMA | 456 | 55 |
| 4 | Et3N | MeCN | 456 | Trace | 17c | DABCO | DMA | 456 | 89 |
| 5 | Triphenylamine | MeCN | 456 | Trace | 18d | — | DMA | 456 | N.D |
| 6 | DABCO | MeCN | 456 | 67 | 19e | DABCO | DMA | Dark | N.D |
| 7 | DABCO | DMSO | 456 | 72 | 20 | DABCO | DMA | 23 W CFL bulb | 40 |
| 8 | DABCO | Acetone | 456 | 38 | 21 | DABCO | DMA | 427 | 65 |
| 9 | DABCO | DCM | 456 | 52 | 22 | DABCO | DMA | 390 | 55 |
| 10 | DABCO | DMF | 456 | 70 | 23 | DABCO | DMA | Green LED | 72 |
| 11 | DABCO | DMA | 456 | 81 | 24f | DABCO (TIPS) | DMA | 456 | 52 |
| 12 | DABCO | EtOH | 456 | N.D | 25g | DABCO (TES) | DMA | 456 | 0 |
| 13 | DABCO | DCE | 456 | 55 | 26h | DABCO (TBS) | DMA | 456 | 0 |
After establishing the optimized reaction conditions, we applied the protocol to various O-silyl enol ethers of acetophenone bearing alkyl, aryl, and heteroaryl functionalities, using S-phenyl 4-methylbenzenesulfonothioate 2a as the coupling partner with DABCO. The O-silyl enol ether of acetophenone bearing halide groups, such as fluoro-, chloro-, bromo-, and iodo-substituents, reacted smoothly, producing the desired products in moderate to good yields (4–11). Additionally, aryl substituted O-silyl enol ethers with both electron-withdrawing and electron-donating substituents, such as nitro-, cyano-, trifluoromethyl-, methylsulfonyl-, methyl, and methoxy groups, were well tolerated under the optimized conditions, resulting in moderate to excellent yields (12–21). Additionally, biphenyl β-keto sulfone 22 was obtained with a 78% yield. We were pleased to discover that this mild protocol demonstrated good reactivity towards the synthesis of pharmaceutical and bioactive molecules under the optimized conditions. For example, β-keto sulfone 23, known for its excellent anti-analgesic properties, was synthesized with a 79% yield. Furthermore, β-keto sulfones 24 and 25, which function as carboxylesterase 1 and 11 β-hydroxysteroid dehydrogenase type I inhibitors, were obtained with yields of 65% and 69%, respectively. Additionally, β-keto sulfone 26, which exhibits anti-bacterial properties, was synthesized with a 77% yield. Furthermore, a heteroaryl O-silyl enol ether containing a pyridine group reacted well, producing the desired product 27 in 71% yield. To further assess the versatility of this method, a series of alkyl-substituted O-silyl enol ethers was subjected to the EDA-protocol, resulting in the desired products in moderate to good yields (28–31). Additionally, an acyl-substituted O-silyl enol ether was evaluated, resulting in the targeted product 32 in 26% yield. Next, an internal silyl enol ether derivative was successfully applied under the reaction protocol, yielding the desired products 33–34 in 62% and 40% yields, respectively. Finally, the utility of this method was further demonstrated through the late-stage functionalization of silyl enol ether derivatives derived from the hypertriglyceridemia drug clofibrate and the anti-inflammatory drug ibuprofen, yielding the desired products in 63% and 48% yields, respectively (Scheme 1).
 | ||
| Scheme 1 Reaction conditions: O-silyl enol ethers 1 (0.2 mmol, where R = Me), S-phenyl 4-methylbenzene sulfonothioate 2a (1.2 equiv., 0.24 mmol), and DABCO (3 equiv., 0.6 mmol) in 0.5 mL DMA were irradiated with a 456 nm Kessil lamp at room temperature under argon for 14 h. aR = isopropyl. | ||
To further diversify our strategy, we employed various thiosulfonates 2a with alkyl and aryl functionalities under the optimized conditions with O-silyl enol ether 1. These thiosulfonates 2a reacted efficiently with O-silyl enol ether 1, yielding the corresponding β-ketosulfones 38–52 in moderate to excellent yields, as illustrated in Scheme 2. Simple, non-substituted thiosulfonates 2 reacted well with O-silyl enol ether 1 to yield the desired product 38 in 90% yield. Furthermore, thiosulfonates 2 bearing halide groups (F and Cl) were well tolerated under the reaction conditions, yielding the corresponding β-ketosulfones 39–43 in 58–91% yield. Notably, thiosulfonates 2 with a t-butyl group on the benzene ring afforded the β-ketosulfone product 44 in 49% yield. This method was also compatible with polyaromatic thiosulfonates, such as those with a naphthalene ring, affording the required product 45 in 92% yield. Conversely, biphenyl derived β-ketosulfone 46 was obtained in 79% yield. Impressively, heteroaromatic thiosulfonates were also well tolerated under the optimized conditions, resulting in the desired product 47 in 66% yield. Moreover, aliphatic thiosulfonates 2 were subjected to our EDA-complex protocol with O-silyl enol ethers 1. Aliphatic thiosulfonates 2 with primary alkyl chains (C1 to C4) were well tolerated under the reaction protocol, producing the desired products 48–51 in 47–95% yields. Additionally, thiosulfonates 2 with secondary alkyl chains proved compatible, yielding the target product 52 in 77% yield.
 | ||
| Scheme 2 Reaction conditions: O-silyl enol ether 1 (0.2 mmol), thiosulfonate 2a (1.2 equiv., 0.24 mmol), and DABCO (3.0 equiv., 0.6 mmol) in 0.5 mL DMA were irradiated with a 456 nm Kessil lamp at room temperature under argon for 14 h. | ||
Mechanistic insights
Next, a series of mechanistic studies were conducted to elucidate the reaction mechanism, as illustrated in Fig. 2. UV/Vis-absorption analysis of individual components and the reaction mixture in DMA was performed, as shown in Fig. 2a. Neither silyl enol ether 1a nor DABCO displayed an absorption band beyond 400 nm. However, the mixture of 1a and 2a exhibited a significant bathochromic shift (brown line). Additionally, the mixture of 2a and DABCO (green line), as well as the reaction mixture of 1, 2a, and DABCO (purple line), displayed a notable bathochromic shift with visible light absorption in the range of 425–500 nm, indicating the formation of an EDA-complex. The intense yellow color observed in the reaction mixture further suggested the formation of an EDA-complex aggregate, as depicted in Fig. 2a.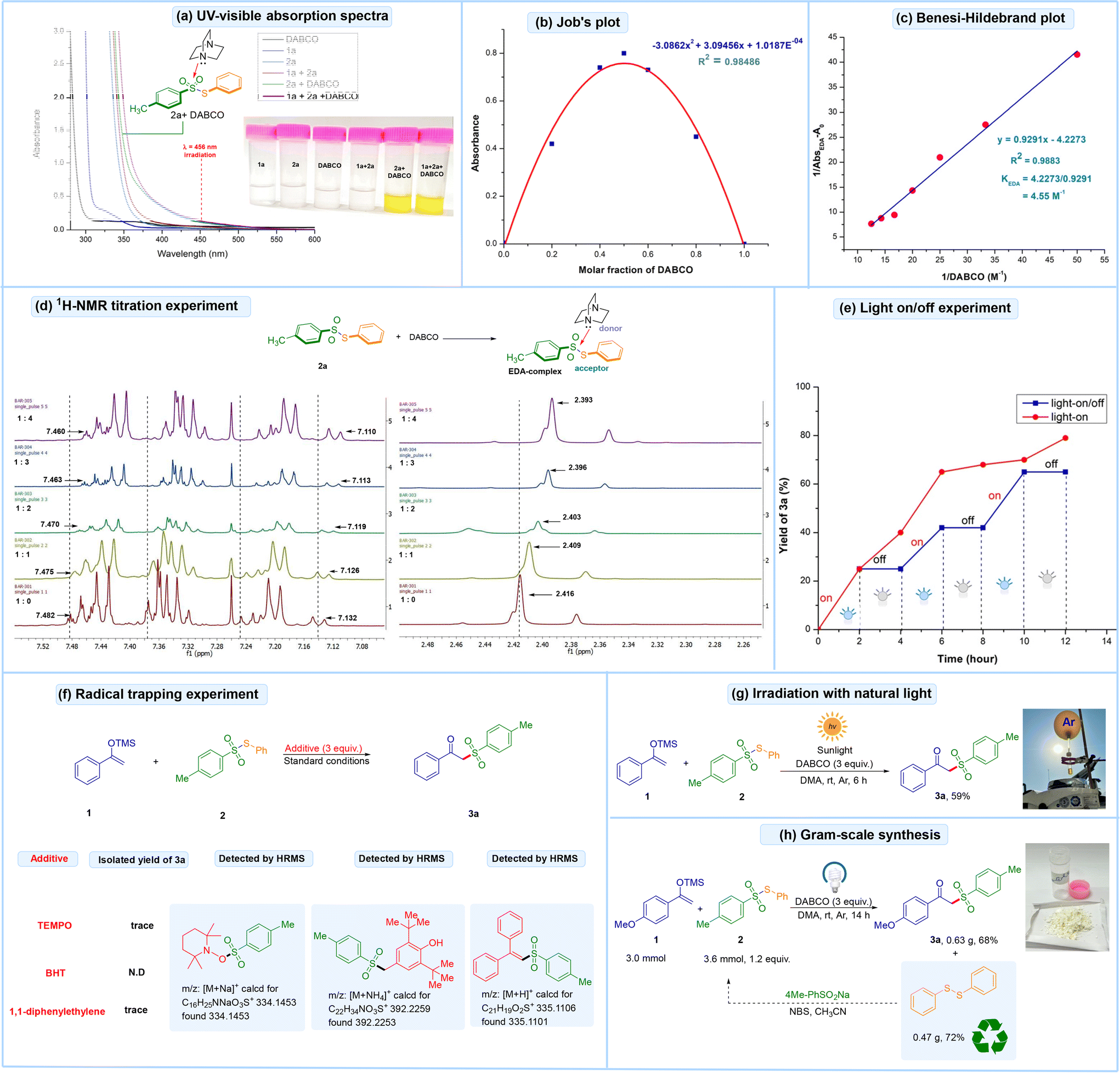 | ||
| Fig. 2 (a) UV-visible absorption spectra; (b) Job's plot; (c) Benesi–Hildebrand plot; (d) 1H-NMR titration experiment; (e) light on/off experiment; (f) radical trap experiment; (g) irradiation with natural light; and (h) gram-scale synthesis. | ||
Additionally, a molar absorption ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for the mixture of 2a and DABCO was demonstrated by Job's plot study (Fig. 2b). Subsequently, a Benesi–Hildebrand experiment15 was performed, yielding an association constant of 4.55 M−1 in DMA, which confirmed the formation of a visible light-active EDA complex (Fig. 2c). Next, a 1H-NMR titration experiment between S-phenyl 4-methylbenzenesulfonothioate 2a and DABCO in CDCl3 revealed that the 1H-proton signals of 2a shifted upfield, thus indicating the formation of an EDA-complex between 2a and DABCO (Fig. 2d). Thereafter, light-on and on/off experiments were performed, demonstrating that continuous visible-light irradiation is necessary for the reaction to proceed (Fig. 2e). Following this, a radical trapping experiment was performed under standard conditions using TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), butylated hydroxytoluene (BHT) and 1,1-diphenylethylene. The reactions were quenched, and the resulting radical trapped adducts were successfully detected by high-resolution mass spectrometry (HRMS), indicating the involvement of a radical pathway (Fig. 2f). Next, the reaction mixture was irradiated with natural sunlight, resulting in the formation of the desired product 3a in 59% yield (Fig. 2g). The reaction was scaled up to the gram scale, achieving a 68% yield of the target product 3a (Fig. 2h). Lastly, a quantum yield experiment was performed and the yield was found to be Φ = 0.82, which indicates a closed-chain pathway (see the ESI† for detailed discussion).
:1 for the mixture of 2a and DABCO was demonstrated by Job's plot study (Fig. 2b). Subsequently, a Benesi–Hildebrand experiment15 was performed, yielding an association constant of 4.55 M−1 in DMA, which confirmed the formation of a visible light-active EDA complex (Fig. 2c). Next, a 1H-NMR titration experiment between S-phenyl 4-methylbenzenesulfonothioate 2a and DABCO in CDCl3 revealed that the 1H-proton signals of 2a shifted upfield, thus indicating the formation of an EDA-complex between 2a and DABCO (Fig. 2d). Thereafter, light-on and on/off experiments were performed, demonstrating that continuous visible-light irradiation is necessary for the reaction to proceed (Fig. 2e). Following this, a radical trapping experiment was performed under standard conditions using TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), butylated hydroxytoluene (BHT) and 1,1-diphenylethylene. The reactions were quenched, and the resulting radical trapped adducts were successfully detected by high-resolution mass spectrometry (HRMS), indicating the involvement of a radical pathway (Fig. 2f). Next, the reaction mixture was irradiated with natural sunlight, resulting in the formation of the desired product 3a in 59% yield (Fig. 2g). The reaction was scaled up to the gram scale, achieving a 68% yield of the target product 3a (Fig. 2h). Lastly, a quantum yield experiment was performed and the yield was found to be Φ = 0.82, which indicates a closed-chain pathway (see the ESI† for detailed discussion).
Green chemistry metrics
To assess the environmental impact of our EDA strategy,16 we evaluated the green chemistry metrics for the synthesis of compound 3a (0.0487 g, 89%), starting from 1a (0.2 mmol, 0.0384 g) and S-phenyl 4-methylbenzenesulfonothioate 2a (0.24 mmol, 0.0634 g), using DABCO (3.0 equiv., 0.0673 g) under visible light conditions. The results are depicted in Fig. 3a. Our method demonstrates outstanding green chemistry metrics. Specifically, the atom economy was calculated to be 96.6%, the reaction mass efficiency was 82.22%, and the process mass intensity was 7.65, all of which indicate a highly efficient process. Additionally, the E-factor was determined to be 6.7, the lowest among comparable methods. The EcoScale score was calculated to be 68 (see the ESI† for detailed calculation), which is acceptable and reflects a favorable balance of safety, economic, and ecological considerations. Overall, the green chemistry metrics of our method indicate a strong alignment with sustainability goals. The comparison between the green chemistry metrics of our strategy and other reports is shown in Fig. 3b (see the ESI† for detailed calculation). | ||
| Fig. 3 Green chemistry metrics analysis: (a) green chemistry metrics evaluation of our method and (b) summary of the green chemistry metrics of our method compared to previous methods (see the ESI† for detailed calculation). Note: atom economy (AE), reaction mass efficiency (RME), process mass intensity (PMI), carbon efficiency, and E-factor. (↑) Higher is better and (↓) lower is better. | ||
Based on the experimental observations and prior literature,17 a plausible mechanism for this photochemical EDA-complex mediated transformation is depicted in Scheme 3. Initially, an EDA-complex aggregate is formed between 2 and DABCO. In the presence of visible light irradiation, this EDA complex undergoes a single-electron transfer (SET) event from DABCO to S-phenyl 4-methylbenzenesulfonothioate 2, generating the thioyl anion intermediate Int-I, the S-centered radical intermediate Int-II, and the DABCO radical cation. The generated sulfonyl radical intermediate Int-II reacts with silyl enol ether 1, leading to the formation of the radical intermediate Int-III, which subsequently undergoes SET with the DABCO radical cation to generate the cation intermediate Int-IV. Lastly, tautomerization, followed by hydrolysis, yields the desired product 3.
 | ||
| Scheme 3 Possible reaction mechanism. | ||
To broaden the synthetic application, various reactions were conducted using compound 3a, as shown in Fig. 4. The reaction of 3a with phenylhydrazine produced the required product 53 in 90% yield.18 Additionally, the reaction with o-phenylenediamine yielded the corresponding product 54 in 76% yield.19 Similarly, the reaction with hydroxylamine hydrochloride gave the oxime product 55 in 93% yield.18 Conversely, the reaction with (2-amino-5-chlorophenyl)(phenyl)methanone yielded a six-membered ring product 56 in 89% yield.20 The carbonyl group of the β-ketosulfone was smoothly reduced to compound 57 using NaBH4 in methanol.21 The resulting hydroxyl group was then dehydrated to produce vinyl sulfone 58, with an approximate yield of 93%.22 Finally, we also demonstrated the propargylation,23 benzylation,24 and allylation25 of compound 3a, resulting in the corresponding products (59–61) in good to excellent yields.
 | ||
| Fig. 4 Diversification of β-Keto sulfones. Reaction conditions: (a) 3a (0.2 mmol), phenylhydrazine (1.0 equiv.), acetic acid (cat. amount), ethanol (1.0 ml), reflux, and 1 h; (b) 3a (0.2 mmol), o-phenylenediamine (1 equiv.), AcOH, 120 °C, and 10 h; (c) 3a (0.2 mmol) hydroxylamine hydrochloride (1.5 equiv.) and NaOAc (1.5 equiv.) ethanol (1 mL), water (3 ml), reflux, and 5 h; (d) 3a (0.2 mmol), (2-amino-5-chlorophenyl)(phenyl)methanone (1.5 equiv.), TfOH (20 mol%), PhCl, 150 °C, and 10 h. (e) 3a (0.2 mmol), NaBH4 (1.5 equiv.), methanol (2.0 mL), 25 °C, and 1 h; (f) 57 (0.1 mmol), BF3·Et2O (0.2 mmol), DCM, 25 °C, and 20 h; (g) 3a (0.2 mmol), K2CO3 (2.0 equiv.), propargylic bromide (1.1 equiv.), acetone (2.0 mL), reflux, and 12 h; (h) 3a (0.2 mmol), K2CO3 (3.5 equiv.), BnBr (1.1 equiv.), acetone, and reflux; and (i) 3a (0.2 mmol), K2CO3 (2.0 equiv.), allyl bromide (1.1 equiv.), acetone (2.0 mL), reflux, and 16 h. | ||
Conclusion
In summary, we reported a transition metal- and photocatalyst-free method for the visible light-induced synthesis of β-ketosulfones via the sulfonylation of ketone-derived silyl enol ethers with thiosulfonates (electron acceptor) and DABCO (electron donor) under mild conditions. This method facilitates the synthesis of a wide range of β-ketosulfone derivatives, including biologically active and late-stage molecules, from structurally diverse silyl enol ethers, including alkyl, aryl, and heteroarylsilyl enol ethers. Extensive mechanistic studies, including UV-visible absorption studies, Job's plot, 1H-NMR titration experiment, Benesi–Hildebrand plot, radical trapping experiment, light on/off experiment, and quantum yield experiment, confirm the involvement of an EDA complex-mediated radical process. Additionally, our method offers several advantages over the existing literature. This includes (i) being transition-metal and photoredox catalyst-free; (ii) being external SO2-source free; (iii) accommodating a diverse range of substrates; (iv) yielding recyclable and reusable by-products; and (v) demonstrating excellent atom economy, reaction mass efficiency, process mass intensity, E-factor, and EcoScale score, highlighting its efficiency and economic sustainability. Moreover, the versatility of β-ketosulfones was demonstrated by their successful conversion into various aromatic and heteroaromatic compounds, showcasing their broad utility as intermediates and making them a practical solution for academic research and industrial applications.Author contributions
B. Saxena optimized the reaction conditions and synthesized all the derivatives. R. I. Patel synthesized the thiosulfonates. B. Saxena and R. I. Patel performed the mechanistic studies and wrote the manuscript with helpful insights from Prof. A. Sharma. Prof. A. Sharma supervised the whole work, interpreted the results, and edited the manuscript. All the authors have given their approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Experimental data have been provided as the ESI.†
Conflicts of interest
The authors declare the following competing interests: one patent has been registered (no: 202411058425) in India.Acknowledgements
Financial support from SERB (CRG/2022/002691), India, is gratefully acknowledged. We also acknowledge DST-FIST (SR/FST/CSII/2018/72(C)) for the NMR and HRMS facilities in the Chemistry Department, IIT Roorkee. B. S. and R. P. thank CSIR and UGC for the SRF Fellowship, respectively.References
-
(a)
N. S. Simpkins, in Sulfones in Organic Synthesis, ed. J. E. Baldwin, Pergamon, Oxford, 1993 Search PubMed
; (b) B. M. Trost, Comprehensive Organic Chemistry, Pergamon, Oxford, 1991 Search PubMed
-
(a) R. E. Swenson, T. J. Sowin and H. Q. Zhang, J. Org. Chem., 2002, 67, 9182–9185 CrossRef CAS
- A. R. Katritzky, A. A. Abdel-Fattah and M. Y. Wang, J. Org. Chem., 2003, 68, 1443–1446 CrossRef CAS
-
(a) B. M. Trost and D. P. Curran, Tetrahedron Lett., 1981, 22, 1287–1290 CrossRef CAS
-
(a) G. E. Vennstra and B. Zwaneburg, Synthesis, 1975, 519–520 CrossRef CAS
-
(a) W. Wei, C.-L. Liu, D.-S. Yang, J.-W. Wen, J.-M. You, Y.-R. Suo and H. Wang, Chem. Commun., 2013, 49, 10239–10241 RSC
-
(a) Q.-Q. Lu, J. Zhang, G.-L. Zhao, Y. Qi, H.-M. Wang and A.-W. Lei, J. Am. Chem. Soc., 2013, 135, 11481–11484 CrossRef CAS PubMed
-
(a) Y. Tang, Y. Zhang, K. Wang, X. Li, X. Xu and X. Du, Org. Biomol. Chem., 2015, 13, 7084 RSC
- For recent reviews on photoredox reaction, see:
(a) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC
- X. Wang, Y. Kuang, S. Ye and J. Wu, Chem. Commun., 2019, 55, 14962–14964 RSC
- F.-S. He, Y. Yao, W. Xie and J. Wu, Chem. Commun., 2020, 56, 9469–9472 RSC
- F.-S. He, P. Bao, Z. Tang, F. Yu, W.-P. Deng and J. Wu, Org. Lett., 2022, 24, 2955–2960 CrossRef CAS
-
(a) A. K. Wortman and C. R. J. Stephenson, Chem, 2023, 9, 1–26 CrossRef
-
(a) R. I. Patel, B. Saxena and A. Sharma, Green Chem., 2024, 26, 10265–10274 RSC
- H. A. Benesi and J. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS
-
(a) N. Mukherjee and T. Chatterjee, Green Chem., 2021, 23, 10006–10013 RSC
-
(a) J. Tong, H. Li, Y. Zhu, P. Liu and P. Sun, Green Chem., 2022, 24, 1995–1999 RSC
- H. A. A. Aziz, K. A. A. Rashood, K. H. El-Tahir and G. M. Suddek, Eur. J. Med. Chem., 2014, 80, 416–422 CrossRef
- M.-Y. Chang, C.-K. Chan and Y.-C. Chen, Heterocycles, 2014, 89, 1237 CrossRef
- C.-K. Chan, C.-Y. Lai, W.-C. Lo, Y.-T. Cheng, M.-Y. Chang and C.-C. Wang, Org. Biomol. Chem., 2020, 18, 305–315 RSC
- Y.-S. Lin, Y.-C. Kuo, C.-H. Kuei and M.-Y. Chang, Tetrahedron, 2017, 73, 1275–1282 CrossRef CAS
- Y. Wang, Y. Zhao, C. Cai, L. Wang and H. Gong, Org. Lett., 2021, 23, 8296–8301 CrossRef CAS PubMed
- M.-Y. Chang, Y.-C. Cheng and Y.-J. Lu, Org. Lett., 2015, 17, 1264–1267 CrossRef CAS
- M.-Y. Chang, Y.-C. Chen and C.-K. Chan, Synlett, 2014, 1739–1744 CrossRef
- M.-Y. Chang, Y.-C. Cheng and Y.-J. Lu, Org. Lett., 2014, 16, 6252–6255 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed optimization, experimental procedure, and 1H, 13C, and 19F NMR spectra of all the synthesized compounds. See DOI: https://doi.org/10.1039/d4gc04554h |
| This journal is © The Royal Society of Chemistry 2024 |