
PAL 2.0: a physics-driven bayesian optimization framework for material discovery†
Maitreyee Sharma
Priyadarshini‡
 a,
Oluwaseun
Romiluyi‡
a,
Yiran
Wang
a,
Kumar
Miskin
b,
Connor
Ganley
a and
Paulette
Clancy
*a
a,
Oluwaseun
Romiluyi‡
a,
Yiran
Wang
a,
Kumar
Miskin
b,
Connor
Ganley
a and
Paulette
Clancy
*a
aDepartment of Chemical and Biomolecular Engineering, Johns Hopkins University, 3400 North Charles Street, Baltimore, 21218, Maryland, USA. E-mail: pclancy3@jhu.edu
bDepartment of Materials Science and Engineering, Johns Hopkins University, 3400 North Charles Street, Baltimore, 21218, Maryland, USA
First published on 13th November 2023
Abstract
The lack of efficient discovery tools for advanced functional materials remains a major bottleneck to enabling advances in the next-generation energy, health, and sustainability technologies. One main factor contributing to this inefficiency is the large combinatorial space of materials (with respect to material compositions and processing conditions) that is typically redolent of such materials-centric applications. Searches of this large combinatorial space are often influenced by expert knowledge and clustered close to material configurations that are known to perform well, thus ignoring potentially high-performing candidates in unanticipated regions of the composition-space or processing protocol. Moreover, experimental characterization or first principles quantum mechanical calculations of all possible material candidates can be prohibitively expensive, making exhaustive approaches to determine the best candidates infeasible. As a result, there remains a need for the development of computational algorithms that can efficiently search a large parameter space for a given material application. Here, we introduce PAL 2.0, a method that combines a physics-based surrogate model with Bayesian optimization. The key contributing factor of our proposed framework is the ability to create a physics-based hypothesis using XGBoost and Neural Networks. This hypothesis provides a physics-based “prior” (or initial beliefs) to a Gaussian process model, which is then used to perform a search of the material design space. In this paper, we demonstrate the usefulness of our approach on three material test cases: (1) discovery of metal halide perovskites with desired photovoltaic properties, (2) design of metal halide perovskite-solvent pairs that produce the best solution-processed films and (3) design of organic thermoelectric semiconductors. Our results indicate that the novel PAL 2.0 approach outperforms other state-of-the-art methods in its efficiency to search the material design space for the optimal candidate. We also demonstrate the physics-based surrogate models constructed in PAL 2.0 have lower prediction errors for material compositions not seen by the model. To the best of our knowledge, there is no competing algorithm capable of this useful combination for materials discovery, especially those for which data are scarce.
New conceptsMaterials discovery is currently in a state of renaissance of importance, thanks to the acceleration possible through the application of machine learning tools. This paper presents a novel materials discovery algorithm that is based on a Bayesian optimization framework. The key novelty of our method, PAL 2.0, is the construction of a physics-based prior mean for the Gaussian process surrogate model. We achieve this in two steps: first, using XGBoost to select the physical descriptors most correlated to the target property being optimized. Second, we use those selected physical descriptors as the input encoding vector to a neural network model that predicts the target property. This combination of XGBoost with neural networks provides a physics-based prior model of the material space to inform a Gaussian process model. The two most compelling contributions of PAL 2.0 are that we demonstrate superior optimization performance by finding the optimal target within the lowest number of iterations when compared to state-of-the-art models such as a representative off-the-shelf Bayesian optimization package, SMAC, as well as one-hot-encoded Gaussian process models for material discovery, and that we provide a predictive physics-based model for the material space capable of offering valuable chemical insights. Overall, PAL 2.0 offers great potential to advance the field of materials discovery, offering researchers and practitioners a powerful and easy-to-use tool to accelerate the development of materials for critical applications in energy, health, and sustainability. |
1 Introduction
Discovery of new and advanced materials with desirable properties is pivotal for driving technological advancements that can address contemporary challenges in global health, energy, and sustainability. The discovery process invariably involves a search for the optimal material composition (a combinatorial optimization problem) and synthesis conditions (a continuous variable optimization problem). A major bottleneck in finding optimal material compositions and processing conditions is the lack of efficient discovery tools that can search very large material spaces, which sometimes contain on the order of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 materials. A traditional Edisonian experimental search for optimal materials relies on expert knowledge, focusing on materials closely aligned with configurations known to perform well. Further, molecular simulations, by offering insights into microscopic behaviors and accurately predicting macroscopic properties, could potentially obviate the need for expensive experimental measurements.1–3 However, molecular simulation approaches, of which density functional theory (DFT) and molecular dynamics (MD) are two prime examples, can also be prohibitively expensive for an exhaustive search of the large combinatorial search space that often characterizes material systems. As such, there remains a need to design tools that can accelerate materials discovery by exploring only a fraction of the possible combinatorial space. For research lab-scale studies, there is also a need to have a machine learning tool that is adept at handling small data sets, often containing less than 50 data points, for which a neural network approach is untenable.
000 materials. A traditional Edisonian experimental search for optimal materials relies on expert knowledge, focusing on materials closely aligned with configurations known to perform well. Further, molecular simulations, by offering insights into microscopic behaviors and accurately predicting macroscopic properties, could potentially obviate the need for expensive experimental measurements.1–3 However, molecular simulation approaches, of which density functional theory (DFT) and molecular dynamics (MD) are two prime examples, can also be prohibitively expensive for an exhaustive search of the large combinatorial search space that often characterizes material systems. As such, there remains a need to design tools that can accelerate materials discovery by exploring only a fraction of the possible combinatorial space. For research lab-scale studies, there is also a need to have a machine learning tool that is adept at handling small data sets, often containing less than 50 data points, for which a neural network approach is untenable.
Computational tools that can accelerate material discovery generally fall under one of the following three paradigms: (i) feature engineering, (ii) predictive models for molecular properties, and (iii) optimization algorithms, Fig. 1. Feature engineering refers to the extraction of correlations between variables using raw data. Such methods enable us to gain physical and chemical insights into material systems that can then inform material discovery tools. For example, in ref. 4, the authors used a modified version of the SISSO5 method to extract features that assist in classification of solid-state materials into classes such as perovskites, spinels and rare-earth intermetallics. On the other hand, availability of large data sets through sources like the Materials Genome Initiative6,7 and high-throughput quantum chemistry frameworks has led to the development of several neural network and machine learning models for molecular property prediction.8,9 Such models can be used to predict properties of unknown materials and hence inform the material discovery process. However, while feature engineering and predictive modeling offer tools for material discovery, the challenge of navigating potential molecular combinations persists. That is where the third paradigm of methods lies that we call “optimization algorithms” or, more commonly, material discovery methods. These methods provide efficient search and optimization strategies to navigate the often overwhelming combinatorial space of materials candidates.
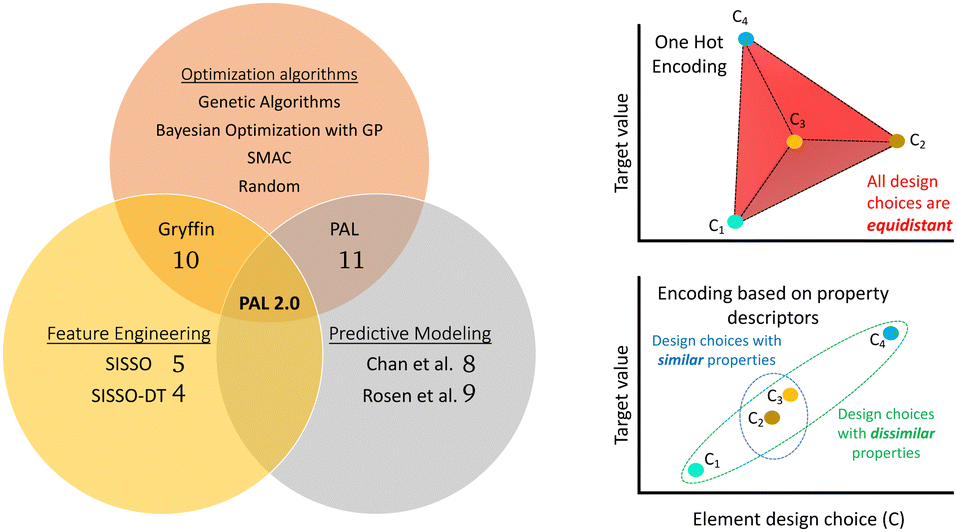 | ||
| Fig. 1 LHS: Schematic depiction of the categories of methods currently used for material modeling and discovery, showing some example approaches. PAL 2.0 lies at the intersection of physics-informed and predictive models used in material discovery methods. RHS:[top] Representation of the commonly used “one-hot-encoding” for design choices and [below] a similarly inspired representation of the concept of “similarity” obtained by using physical descriptors instead. | ||
A brute-force approach to finding the optimal elemental combination of a material for a given target (e.g., the best solar cell efficiency) involves randomly and exhaustively exploring the material space. This approach does not leverage information from previously explored candidates nor expert domain knowledge to improve its search strategies. Additionally, it is typically infeasible unless examining smaller combinatorial spaces. Likewise, evolutionary methods, like Genetic Algorithms,12 have also been used for categorical domain optimization. However, such evolutionary methods are locally exploitative and therefore, can get trapped in locally (rather than more globally) optimal regions.
In recent years, BayesOpt has become a widely adopted algorithm for global optimization of black-box functions that are expensive to evaluate.13–16 It has been used to optimize a wide range of problems, including automatic algorithm configuration, automatic machine learning toolboxes, and optimization of combinatorial spaces for materials and drug discovery.10,17–24 A BayesOpt algorithm essentially requires two sets of functions: (i) a “surrogate model” for the objective function and (ii) an “acquisition function” that is updated, based on the surrogate model, to provide a recommendation for the next candidate to explore. Some typical examples of surrogate models include Gaussian Processes,25 random forests26 and Bayesian Neural Networks.27 The most commonly used acquisition functions include probability of improvement,28 expected improvement,29 and upper confidence bound.30
The application of BayesOpt in material discovery requires additional considerations. Unlike categorical optimization in machine learning and hyperparameter optimization, the notion of similarity and representation of candidate choices becomes important in material science applications. Commonly used “one-hot-encoding” representations for chemical and material domains fail to capture the true physical and chemical similarity between candidate choices, as depicted schematically in Fig. 1 (top right). The drawback arises from using binary variables to depict a design choice such that all design choices are equally similar to each other since they all differ by one Hamming distance. Another representation for materials involves providing compositional information and structural information in terms of graphs.17 However, training models to accurately encode structural data require large training data sets that are frequently unavailable. In reality, there is an obvious similarity between materials that have similar chemical and physical properties. Optimization strategies that can leverage physical and chemical information to determine similarity between candidate choices are expected to accelerate the material discovery process, as was demonstrated by Hase et al.10 through the innovative Gryffin method.
In this paper, we have developed a new materials discovery algorithm, Physical Analytics pipeLine 2.0 (PAL 2.0), in which we leverage domain knowledge, appearing in the guise of chemical and physical properties, to develop surrogate models that are then used within a BayesOpt framework. A description of the construction and workflow of PAL 2.0 are discussed in the following sections and in the ESI.†
2 Results
2.1 Physical analytics pipeLine, PAL 2.0
Addressing the directions highlighted above, we present a new BayesOpt algorithm, Physical analytics pipeLine 2.0 (PAL 2.0) in this work that is intended to be a successor to our earlier version.11 PAL11 was specifically developed to optimize the solution chemistry of solution-processed metal halide perovskites by finding optimal pairing of solvent and metal halide perovskite compositions that are the precursors to producing high-quality thin films1,31–33 and reduce the appearance of crystalline intermediates.2 PAL is based on a Gaussian process (GP) model with a linear prior mean function. In PAL, the combinatorial space of the perovskite constituents is encoded using one-hot-encoded vectors and the solvent is represented by its dielectric constant and density, where the descriptors are chosen based on expert domain knowledge. Although PAL used physics-based GP models, the method is specific to the material system and relies on expert knowledge to choose the physical descriptors that optimally encode the input space.The new approach, PAL 2.0, presented in this paper, is a generalization of that original version of PAL.11 Both involve a Bayesian optimization framework that uses a physics-informed Gaussian process (GP) model. However, there are three key improvements and novel capabilities of PAL 2.0 compared to its progenitor PAL version. The new proposed framework involves: (i) descriptor selection for the search space based on decision trees, (ii) construction of a physics-based prior mean function using neural network (NN) models, and (iii) construction of a GP model using the NN prior mean function and subsequent use of this model in BayesOpt. Mathematical details of the model construction are provided in the Methods section (Section 4) and the overall workflow of the method can be seen in Fig. 2. Note that details of the nomenclature used in this work are given in the ESI† (Section S1).
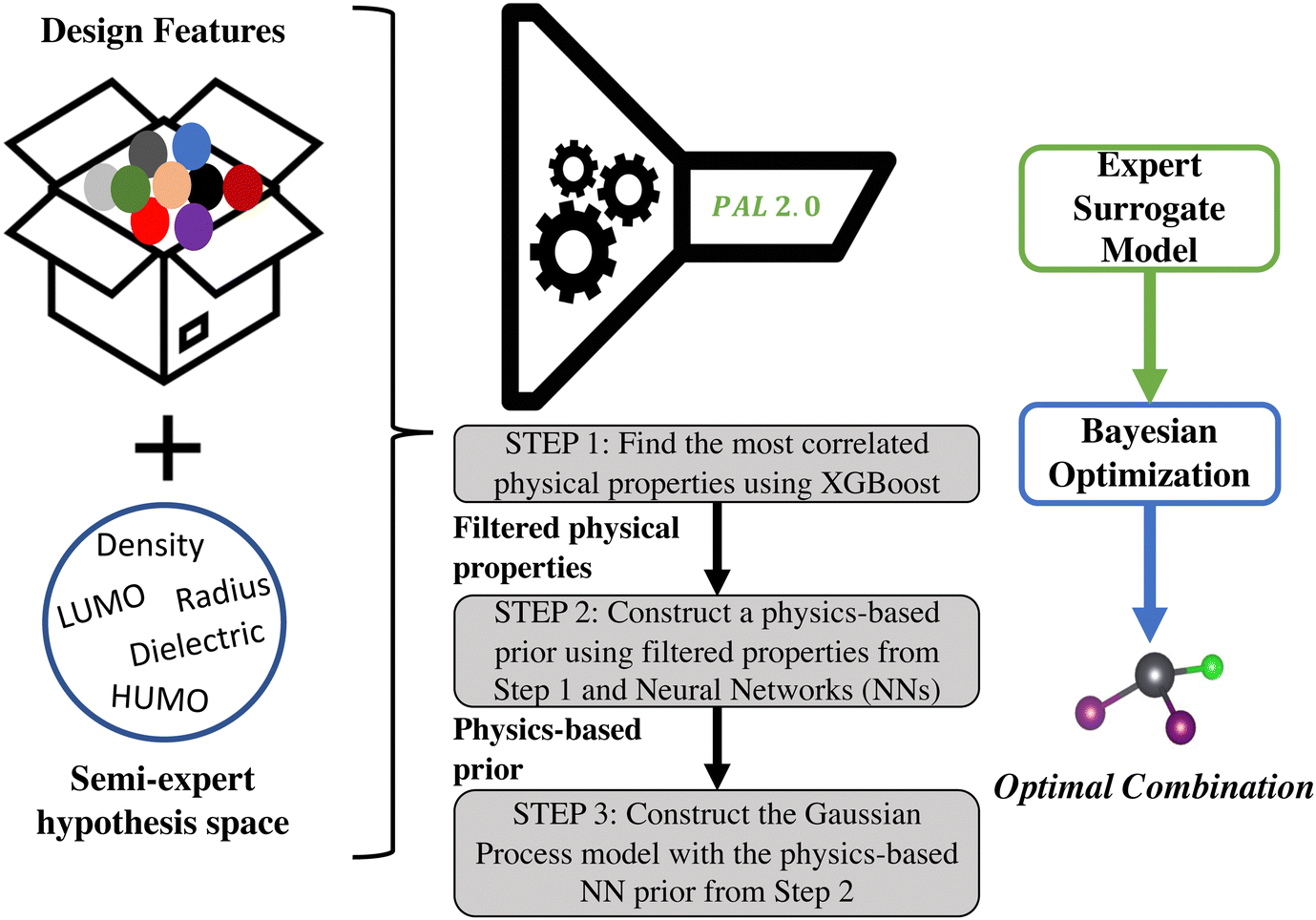 | ||
| Fig. 2 Overall workflow of the PAL 2.0 framework. The inputs to the framework include (i) design choices shown as the colored balls and (ii) a semi-expert hypothesis space that includes physical properties that the user suggests may be correlated with the target property being optimized. The PAL 2.0 methodology itself can be split into three steps shown in the gray rectangles in the figure. The Gaussian process-neural network model is trained with an initial dataset to create the expert surrogate model which is then used in the Bayesian optimization framework with an expected improvement acquisition function to determine the optimal material. | ||
As mentioned earlier, every material can be characterized in terms of its physical and chemical properties, but a priori knowledge of which properties are more important in optimizing the target variable is often lacking. By using XGBoost as part of the PAL 2.0 framework, we pick out the physical descriptors that are most representative of the material domain, making the search essentially unbiased toward expert knowledge, which, in many cases, is unknown. The algorithm typically finds a small number of important properties that correlate with the chosen target (rather than just one) and, importantly, can autonomously determine their relative weighting in a manner that even an expert might be unable to do. In the PAL 2.0 workflow, the physical descriptors chosen by XGBoost become the input variable set for the GP surrogate model.
When fitting GP models on scarce data such as those encountered in materials discovery, the main challenge is to obtain suitable prior knowledge and encode it into the model either through the kernel function or the mean function. In the machine learning literature, research has mainly focused on approximating the kernel function of the GP model using NNs.34 Training deep kernel functions, however, poses two main constraints: (i) they require large training datasets and (ii) they have to be positive definite in order to define an inner product on the material search space. The mean function, in comparison, has no such constraints and therefore, can be trained more easily to create a predictive GP prior. Furthermore, the assumption that all prior information can be encoded in the kernel function of the GP model when using a zero mean function (m(xD) = 0) does not always hold. For example, if the optimization landscape is such that in some regions we have a non-zero objective function value and in other regions the objective function value is zero, we can easily prescribe a prior to the mean function that will encode this information exactly but we cannot ensure the same with a kernel function. Therefore, obtaining an informed prior mean function allows more flexibility and guarantee in encoding prior knowledge. The contribution of our method is to create such a physics-based prior mean function using neural networks (NN). Having a predictive and accurate description of the optimization landscape allows the acquisition function to quickly find the optimal material in a few iterations. As a result, the NN ‘prior mean function’ ultimately boosts the performance of the Bayesian optimization step, as seen in the results.
We demonstrate the performance of the PAL 2.0 methodology on three material data sets relevant to energy applications.
1. Organic semiconductors:
(a) Target: electron affinity of the semiconducting polymer.
(b) Objective: maximize electron affinity.
(c) Possible combinations: 64.
2. Mixed B-site perovskites:8
(a) Target: band gap of perovskite crystal.
(b) Objective: minimize band gap.
(c) Possible combinations: 244.
3. Perovskite molecule–solvent binding energy:11
(a) Target: intermolecular binding energy of perovskite complex and solvent.
(b) Objective: maximize perovskite–solvent binding energy.
(c) Possible combinations: 240.
We also stress test the method by assessing the effect of varying amounts of initial training datasets and by running PAL 2.0 on a very large dataset of approximately 70000 COF structures that find applications in methane storage.35 These stress tests are added in the ESI† (see Section S6). In the subsequent sections, we describe each data set, the data set source and the BayesOpt performance of different methods. For each dataset, the prediction accuracy of the surrogate models (GP-0 and GP-NN) is compared using mean squared error which is computed as:
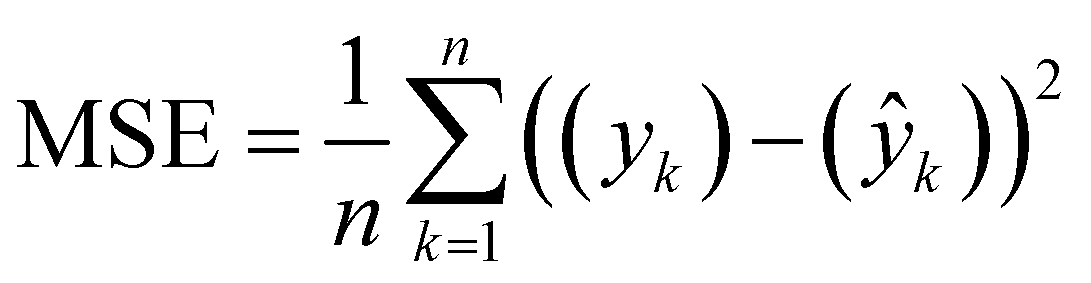 | (1) |
2.2 Discovery of doped p-type organic semiconducting polymers
Organic semiconductors, composed of small molecules or polymers, offer flexible, lightweight, and adaptable optoelectronic properties distinct from their inorganic counterparts like silicon. Within this class, p-type organic materials, which are made by introducing acceptor impurities into the framework of the semiconductor, specialize in transporting positive charge carriers, or holes. In this study, we explore the electronic properties of doped p-type organic semiconducting polymers, which have applications in organic light-emitting diodes (OLEDs), organic solar cells (OSCs), and thermoelectrics, and can be manufactured via highly scalable solution processing protocols.36,37 For doping to occur in solution, the conducting polymer and dopant must experience a dative bond-based interaction (forming a “doped complex”) in a solvent medium. Thus, there are three distinct chemical species in the solution: the polymer, dopant, and solvent. Processing via a solution-based approach can create a large combinatorial space generated from a chemically diverse set of polymer, dopant, and solvent design choices.We analyzed a DFT-generated small subset of this design space of four design choices for each species, leading to 64 unique combinations of the resulting p-doped semiconducting material. This data originates from a detailed study on three polymer segments, each differentiated by its Lewis basicity, backbone functionality, and solid-state microstructural attributes.38 Within this data set, PAL 2.0 was leveraged to identify which properties, if any, from a set of physical constants available from open-source databases39 and DFT calculations, are most important when optimizing a polymer–dopant–solvent system for the EA of the doped complex. We selected the electron affinity as the target metric because p-doping is known to enable charge transfer if there is a sufficient offset between the polymer's HOMO/ionization potential (IP) and the dopant's LUMO/EA.40 Details on the DFT methods used to calculate the electron affinity for each combination are given in the ESI† (Section S3A).
PAL 2.0 achieved an optimal target value while only exploring, on average, roughly 30% of the design space, a modest improvement over existing optimization algorithms (see Fig. 3). The fraction of the space explored is determined as
 | (2) |
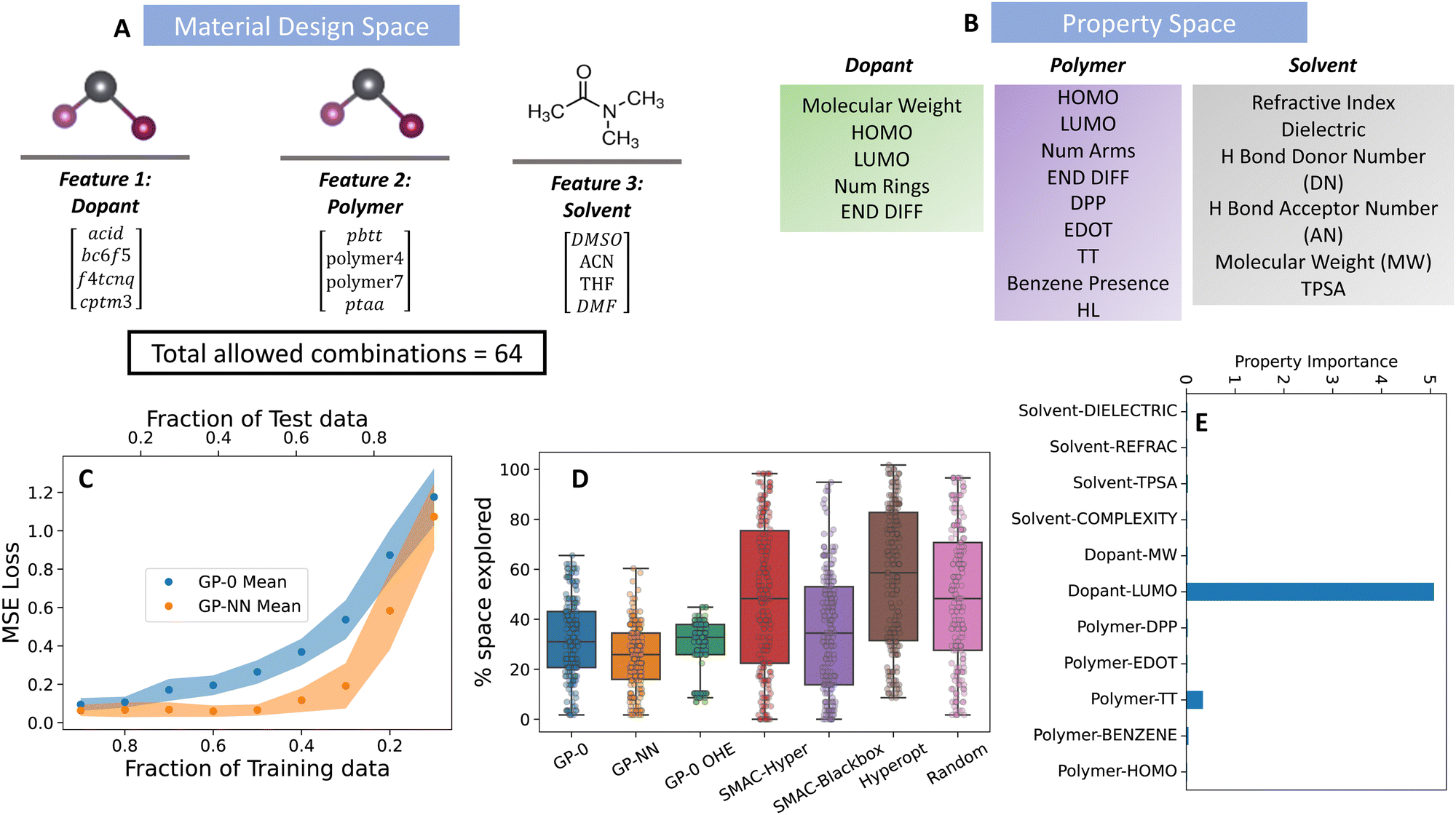 | ||
| Fig. 3 Performance of the GP-NN model on the material discovery of doped p-type organic semiconducting polymers. (A) Material design space, (B) physical property representation of material design features shown in (A). Details of the physical properties are given in Table S1 (ESI†). (C) Predictive accuracy of GP-NN model against state-of-the-art GP models commonly used in material discovery, (D) superior Bayesian optimization performance of the GP-NN model (orange box) compared to state-of-the-art models (indicated by needing less of the parameter space to explore before successfully reaching its target) and (E) Selection of important (well-correlated) physical properties selected by the algorithm from the property space in (B). | ||
2.3 Discovery of optimal metal halide perovskite combinations
MHP have garnered attention due to their exceptional electronic and optical characteristics. These properties position them as useful materials in applications like photovoltaic devices, LEDs, and X-ray detectors. A notable advantage of perovskites is the tunability of their composition and processing methods, which has yielded solar cell efficiencies exceeding 25%.41,42 Furthermore, they can be processed at room temperature using commonly available elements. Within the realm of solar cells, the adaptability of perovskites presents both promise, due to the witnessed boost in efficiencies and stabilities, and challenges stemming from the multitude of design choices. We leverage PAL 2.0 to identify perovskite compositions that (i) possess strong photovoltaic capabilities, and (ii) pair with solvents to yield the best quality of solution-processed thin films.The data set features 244 unique formulations, sourced from specific mixtures of three halide ions, four A-site cations, and six B-site cations. In terms of halides, the selection is made up of the routinely utilized chloride, bromide, and iodide ions. For the B-site, while the most commonly adopted species are lead (Pb) and tin (Sn), the data set also incorporates options to consider germanium (Ge), calcium (Ca), barium (Ba), and strontium (Sr). On the A-site front, the options consist of formamidinium (FA), methylammonium (MA), cesium (Cs), rubidium (Rb), and potassium (K). The design choices for each feature are depicted in Fig. 4. The “property basket” consists of the descriptor choices selected by Mannodi-Kanakkithodi et al., which were utilized in predicting the bandgap of the mixed perovskite species.8 Since the perovskites consist of B-site alloys, properties for the B-site are given as a weighted average of the elemental physical properties. The weights are given by the elemental composition at the B-site.
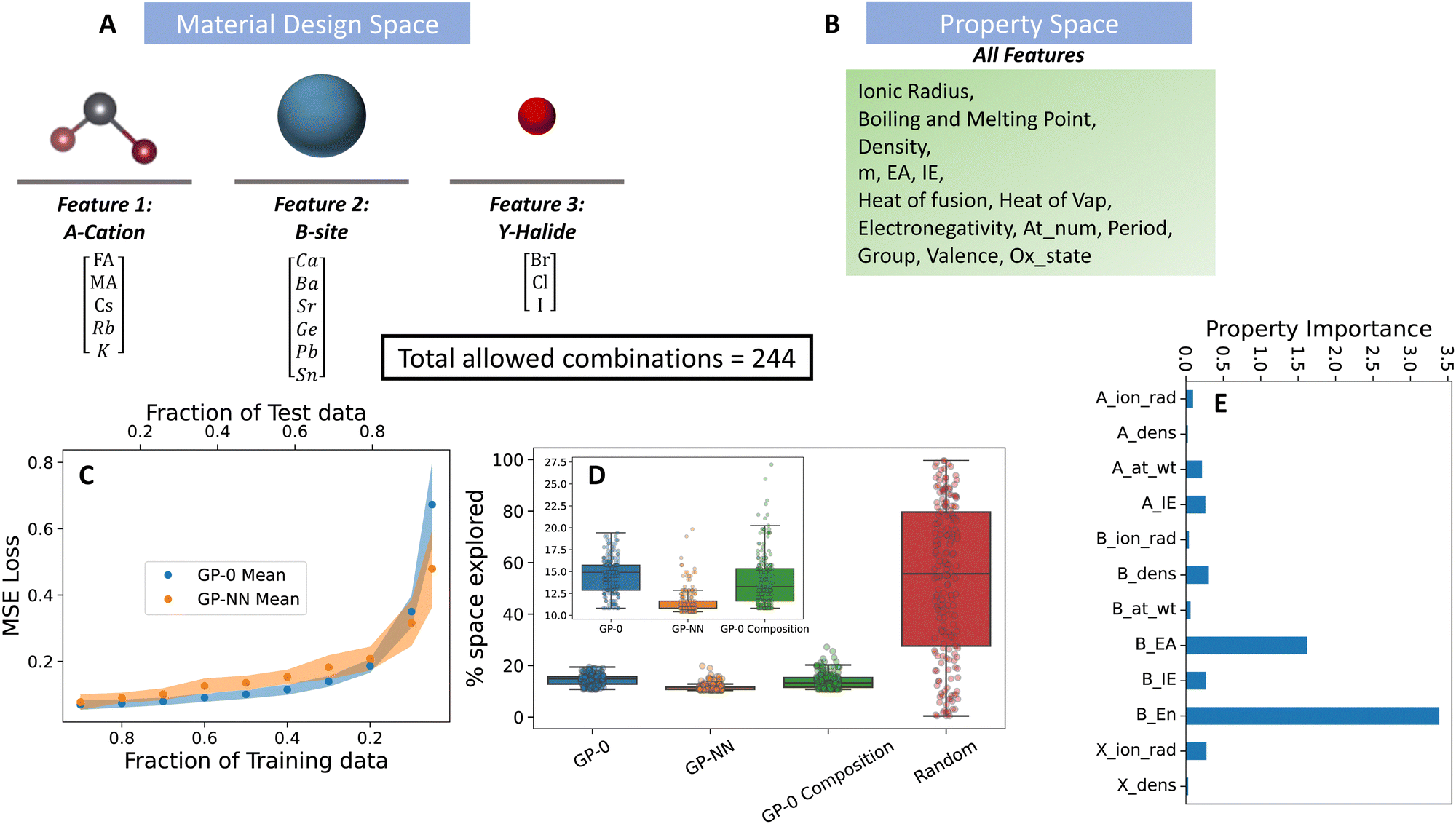 | ||
| Fig. 4 Performance of the GP-NN model on the material discovery of metal halide perovskite solar cell materials. (A) Material design space, (B) physical property representation of material design features shown in (A), (C) predictive accuracy of GP-NN model against state-of-the-art GP models commonly used in material discovery, (D) superior Bayesian optimization performance of the GP-NN model (orange box) compared to state-of-the-art models (indicated by needing less of the parameter space to explore before successfully reaching its target), and (E) selection of important (well-correlated) physical properties selected by the algorithm from the property space in (B). | ||
To pinpoint the combination with the lowest bandgap (MA, Pb, I), PAL 2.0 (GP-NN) proved highly efficient. On average, it explored just 11% of the available design space after 200 BayesOpt trials (see Fig. 4). In contrast, the random search on average explored 50% and a GP model with a 0-prior mean choice (GP-0) searched 15% of the space. These trials used randomly initialized input data from 24 combinations, representing 10% of the entire space. Our feature engineering approach identified the electron affinity of the A-site cation as the most important descriptor in representing the design choices of this feature. For the B-site cation, the electron affinity and ionization energy of the ion were identified as important descriptors, but their importance paled in comparison to the electronegativity – which was identified as the most important differentiator between B-site cation design choices. Lastly, for the halide ions, ionic radius and density were identified as the most relevant properties.
The examined data set consists of five key features: the solvent molecule, choice of A-site cation (A), and choices of the three halide ions (X, Y, Z). Together with a central lead ion, these form the Pb-A-XYZ perovskite structure. The A-site design choices includes cesium (Cs), methylammonium (MA), and formamidinium (FA), while halide options consist of iodide (I), bromide (Br), and chloride (Cl). We provided eight solvent options based on a list of commonly used solvents for perovskite processing. They include: tetrahydrothiophene 1-oxide (THTO), dimethyl sulfoxide (DMSO), dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), gamma-butyrolactone (GBL), acetone, methacrolein (METHA), and nitromethane (NITRO).
We selected properties for each feature (X/Y/Z-halides, A-site cation and solvent) based on prior physical knowledge of their potential impact on the binding energy (our target variable). We explored four basic properties for the halide features: electronegativity, electron affinity, ionization energy, and ionic radius of the halide. For the A-site cations, we considered the ionic radius, binding enthalpy of DMF towards the A-site cation,33 the dipole moment and the number of potential hydrogen bonding atoms of the A-cation. Finally, for the solvent feature, we considered six properties: the Gutmann donor number (DN),1,44,45 Lewis acceptor number (AN),46 lithium cation affinity (LCA),47 dielectric constant,48 dipole moment and molar volume (MV) of the solvent molecule.
Using PAL 2.0's GP-NN, we identified the combination with the highest binding energy (Br, Cl, Cl, FA and THTO) by exploring 11% of the available design space. Comparatively, it took GP-0 with filtered property descriptors 13% percent of the design space to locate the optimum combination. A OHE GP-0 model took 16 percent of the space to do so (with large variability in its convergence results), see Fig. 5. Other methods, like SMAC and HyperOpt explored over 20–40% of the design space before they were able to locate the best combination. Additionally, these benchmark methods had large variability in their results over the BayesOpt trials.
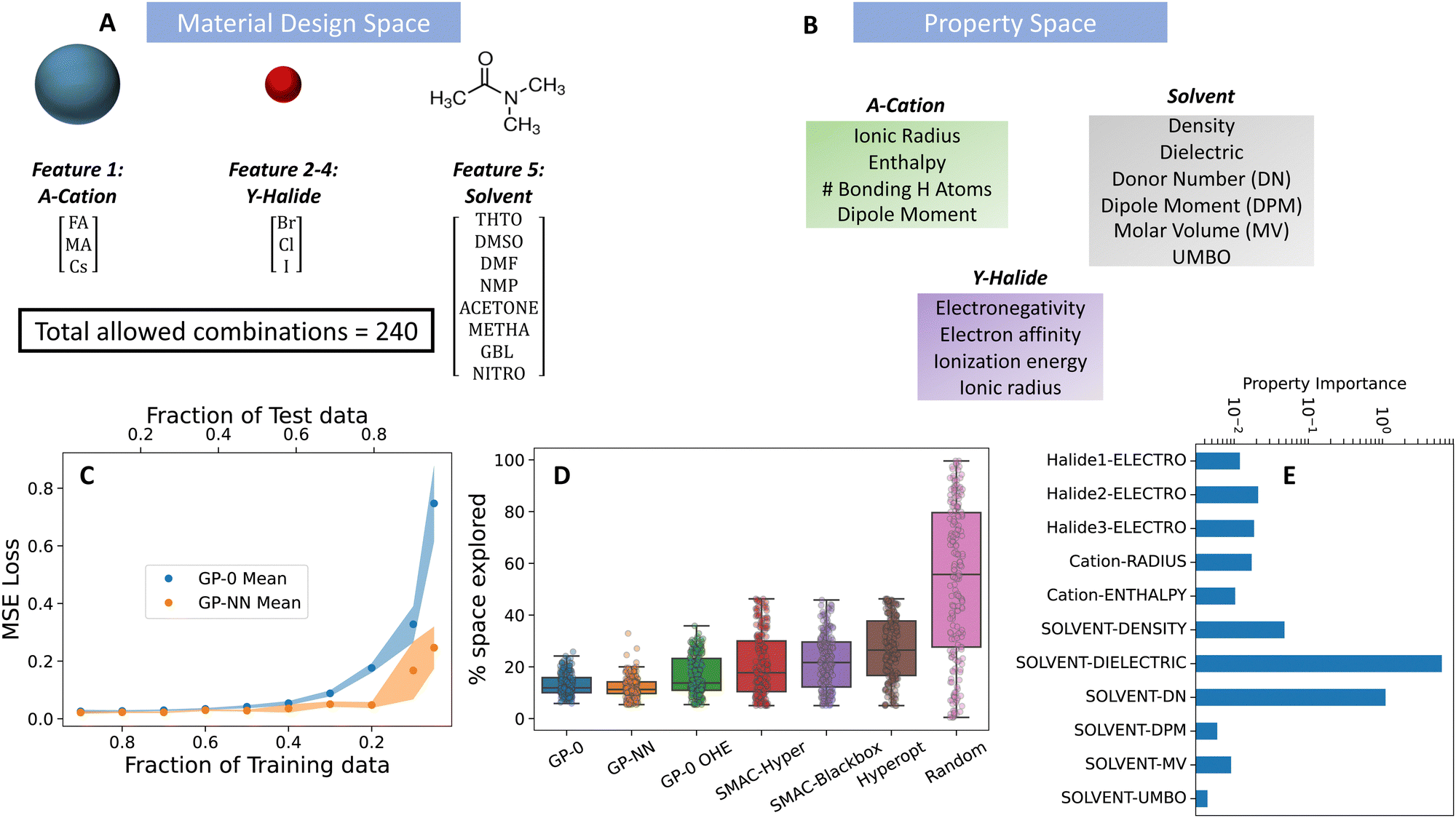 | ||
| Fig. 5 Performance of the GP-NN model on the material design of metal halide perovskite and solvent pairs for best solution processed films for solar cells. (A) Material design space, (B) physical property representation of material design features shown in (A), (C) predictive accuracy of GP-NN model against state-of-the-art GP models commonly used in material discovery, (D) superior Bayesian optimization performance of the GP-NN model (green box) compared to state-of-the-art models (indicated by needing less of the parameter space to explore before successfully reaching its target), and (E) selection of important (well correlated) physical properties selected by the algorithm from the property space in (B). | ||
For property descriptors, there were no standout properties selected to represent the halide and cation features, each selected property having a similar level of importance. On the other hand, two standout properties (dielectric constant and donor number) were identified for the solvent feature to differentiate between the various solvent design choices. The choice of these properties is significant since the simulations that created this dataset are run with an implicit solvent thereby making the dielectric constant an important differentiator for the solvent choices. Additionally, the ability of PAL 2.0 to discern significant features for the solvent through its XGBoost component exemplifies the system's capability to surpass expert selection. The initial version of PAL, as presented in Herbol et al.,11 utilized a physics-based prior reliant on expert-derived factors such as the dielectric constant and solvent density. In contrast, PAL 2.0 independently recognized and attributed significance to these same features, underscoring the advanced feature selection capabilities of XGBoost and its effectiveness in this context.49 Leveraging this capability, PAL 2.0 astutely pinpointed the Gutmann donor number (DN) as a critical property—a measure acknowledged for its efficacy in isolating potent solvents for the solution processing of metal halide perovskites.1,31,45,48,50–53
3 Discussion
In this paper, we have described the construction of a physical analytics pipeLine algorithm, PAL 2.0, that builds on a Gaussian process-based Bayesian optimization framework to accelerate optimization of the large combinatorial spaces that are inherent in many material discovery problems.The novelty of our work lies, firstly, in the incorporation of important physical descriptors selected by the XGBoost algorithm to enhance the physical realism of our surrogate model. Secondly, in the construction of a physics-based prior mean using a neural network approach. The net result of these novel approaches is to leverage physical domain knowledge specific to the system of interest. However, it should be noted that another advantage is that the descriptor selection done by PAL 2.0 dispenses with the need/requirement to be an expert with an understanding of which descriptors/features are the most informative for the system. A semi-expert user is free to provide a list of descriptors that might be important and the method will-autonomously-choose the most appropriate ones from that list. As a result, PAL 2.0 is able to find the optimum target objective faster (i.e., in fewer iterations) than many state-of-the-art optimization methods, including SMAC,54 Hyperopt,55 and Genetic Algorithms.12
The performance of PAL 2.0 is demonstrated on three material data sets which include doped p-type organic semiconductors and perovskites. Both these classes of materials show immense promise for the next generation of solar cells, but the algorithm itself is completely materials-agnostic and indeed can be used for applications well outside the realm of materials. Any application for which the parameter set is either large enough to discourage a systematic search or the data are sparse and/or expensive (i.e., tackling both ends of the data set size), and the features that are most closely correlated with the objective are largely unknown is a suitable candidate for exploration using PAL 2.0. In each of the material cases we studied here, we have shown that PAL 2.0 outperforms all other methods we tested. Within the PAL 2.0 framework, the GP-NN model that combines some neural network assistance in concert with BayesOpt exhibits the best convergence and predictive capabilities.
Furthermore, the surrogate model constructed by PAL 2.0 provides valuable chemical insight into the material system, which can be transferred to learning domains outside of the training set. For example, in the doped p-type semiconducting polymers, of all the descriptors provided, LUMO was selected by the method as the most important physical descriptor when optimizing for EA. This is consistent with previous findings which show that the dopant's EA is most correlated to its LUMO40 and that the EA of the dopant dictates the EA of the entire polymer–dopant complex.38 Additionally, earlier studies research have underscored the significance of the Gutmann donor number (DN) and dielectric constant as pivotal descriptors for distinguishing solvents in the solution processing of metal halide perovskites.1,31,45,48,50–53 These findings show that the physics-based surrogate model, embedded with necessary property descriptors, could be a great starting point to find material candidates in similar domains with scarce data.
In summary, the PAL 2.0 approach exhibited the following advantages:
1. It outperforms or, at the very least, is competitive with, the optimization performance of other BayesOpt approaches that we tested.
2. It has the ability to select physically relevant descriptors for the surrogate model and their relative weighting.
3. The test errors (MSE values) of the GP-NN surrogate model are lower than other models, implying that GP-NN is more predictive.
4. Having a model that is predictive opens up the possibility of optimization in different ranges of target values for different applications where data are scarce, and finally.
5. Can initiate material discovery for a material system with as few as 25 observations from experiments or computation.
4 Materials and methods
This section provides details of the PAL 2.0 methodology.4.1 PAL 2.0 methodology
PAL 2.0 is a physics-based Bayesian optimization framework. The PAL 2.0 logic flow is shown in Algorithm 1.| Algorithm 1 PAL 2.0 methodology |
|---|
| Require: Initial data samples and physical descriptors list for each design choice |
| 1: Estimate most important set of descriptors (D) using XGBoost |
| 2: Estimate hyperparameters of the prior to the GP mean function, i.e. the neural network model (m(xD)) |
| 3: Estimate hyperparameters of the prior for the kernel function of the GP model |
4: Compute the posterior probability distribution based on the prior (m(xD) and 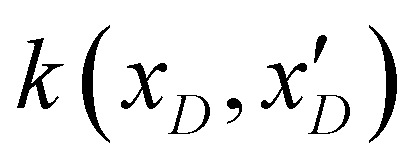 ) and initial data samples ) and initial data samples |
| 5: repeat |
| 6: Select new observation (x(t)D) based on the acquisition function |
| 7: Obtain objective function value at (x(t)D), f(x(t)D) |
| 8: Update posterior with (x(t)D) |
| 9: Every ‘n’ iterations, update the GP model hyperparameters |
| 10: until ‘N’ candidates are explored |
| 11: return Best material explored |
Each step of the algorithm is discussed in the following subsections.
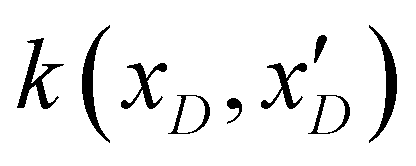 . The posterior probability distribution on the mean function (μi) and covariance (vi) of the GP model is evaluated based on new observations (x(t)D) and the prior using the following equations,
. The posterior probability distribution on the mean function (μi) and covariance (vi) of the GP model is evaluated based on new observations (x(t)D) and the prior using the following equations,| μi = m(xD) + k(xD,x(t)D)(k(xD,x(t)D) + η2I)−1(y(xD) − m(xD)) |
| vi = k(x(t)D,x(t)D) − k(xD,x(t)D)(k(xD,x(t)D) + η2I)−1k(xD,x(t)D)T | (3) |
A popular prior mean function choice is the 0-mean function, m(xD) = 0. This prior mean function is useful when we have very few, or no, observations of our system. It helps in preventing ad hoc assumptions and biases that might be included in the model by forcing a functional prior mean function choice. However, in the case of material discovery, we often start with some observations of the space. In this work, we leverage these observations to pick out the most relevant physical descriptors, as discussed in Section 4.1.1, and construct a prior mean function for the GP model. The novelty of our approach lies in the construction of a physics-based prior mean function (m(x)) using NN for the GP model. The NN prior mean function takes the selected descriptors (D) as the input vector and predicts the optimization target as the output. We use a mean squared error loss function and the Adam optimizer61 to train the NN. Additionally, we use L1 and L2 regularization on the weights of the NN to prevent overfitting the model. Once the NN is trained, we obtain the prior mean function for the GP. The NN is trained using the initial set of observations and then kept fixed through the material discovery process. We refer to this physics-based GP model as the “GP-NN” model. An advantage of employing a NN prior mean function over a 0-prior mean function is the ability to harness available information through a model trained on the input data. Additionally, the NN prior creates a predictive model of the space with just a few observations and therefore, improves the BayesOpt performance of the GP-NN surrogate model. However, the limitation of such a model is that it requires some amount of initial data to train off of.
Finally, we consider that two materials are similar if their physical descriptors (D) are similar. Here, we measure the similarity using a 5/2 Matérn kernel. Formally, a 5/2 Matérn kernel is used to estimate the covariance (k(x(1)D,x(2)D)) for the property descriptors representing the material design choices (see eqn (4)).
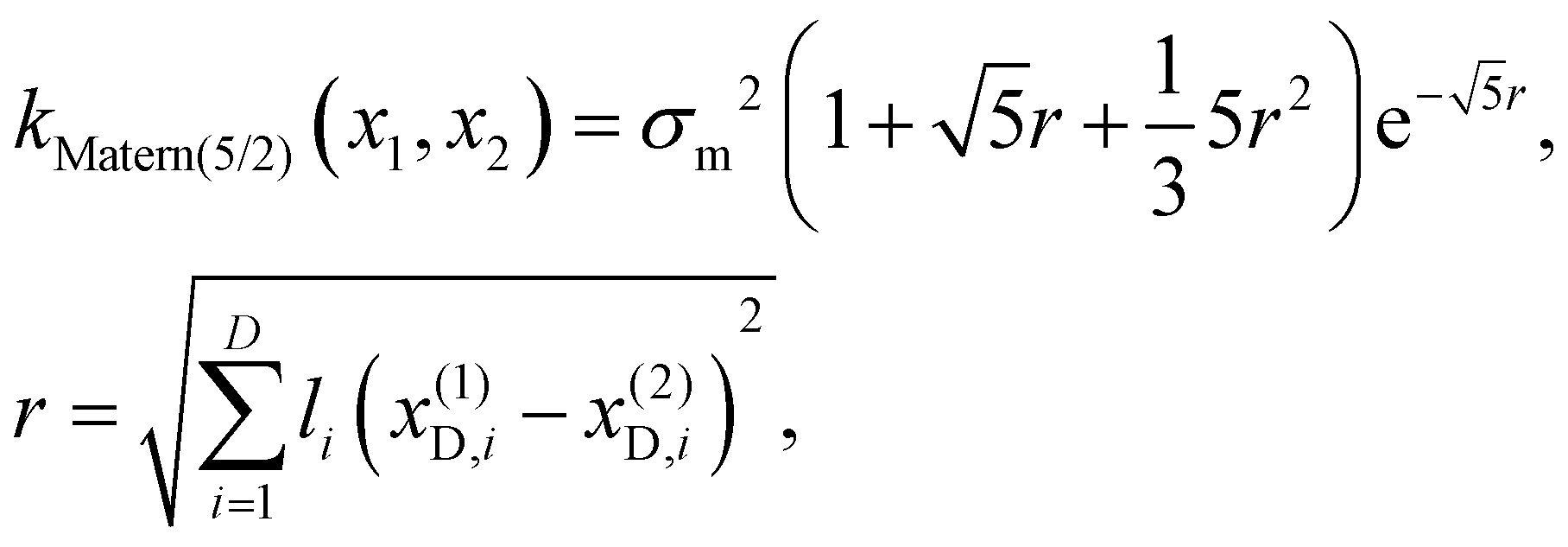 | (4) |
Data availability
The code for this article is made publicly available at https://github.com/ClancyLab/PAL2.Author contributions
MSP and OR worked on the conceptualization, data curation, methodology, software, visualization and writing. YW worked on data curation, benchmarking, software and writing. KM worked on the visualization and writing. CG worked on data curation and writing and PC worked on the conceptualization, funding acquisition, project administration, resources, supervision and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been primarily supported by the DOE, Office of Science, BES, under Award #DE-SC0022305 (formulation engineering of energy materials via multiscale learning spirals). This work has also been supported by NSF grant #2107360 and the HEMI Seed Grant. Computing resources were provided by the ARCH high-performance computing (HPC) facilities, which is supported by National Science Foundation (NSF) grant number OAC 1920103. The authors would also to like to thank Mr. Nihaar Thakkar for his work on streamlining the PAL 2.0 code and creating documentation for the same.Notes and references
- O. Romiluyi, Y. Eatmon, R. Ni, B. P. Rand and P. Clancy, J. Mater. Chem. A, 2021, 9, 13087–13099 RSC.
- A. G. Ortoll-Bloch, H. C. Herbol, B. A. Sorenson, M. Poloczek, L. A. Estroff and P. Clancy, Cryst. Growth Des., 2020, 20, 1162–1171 CrossRef CAS.
- J. Stevenson, B. Sorenson, V. H. Subramaniam, J. Raiford, P. P. Khlyabich, Y. L. Loo and P. Clancy, Chem. Mater., 2017, 29, 2435 CrossRef CAS.
- B. Selvaratnam, A. O. Oliynyk and A. Mar, Inorg. Chem., 2023, 62(28), 10865–10875 CrossRef CAS PubMed.
- R. Ouyang, S. Curtarolo, E. Ahmetcik, M. Scheffler and L. M. Ghiringhelli, Phys. Rev. Mater., 2018, 2, 083802 CrossRef CAS.
- J. J. De Pablo, B. Jones, C. L. Kovacs, V. Ozolins and A. P. Ramirez, Curr. Opin. Solid State Mater. Sci., 2014, 18, 99–117 CrossRef.
- J. J. de Pablo, N. E. Jackson, M. A. Webb, L.-Q. Chen, J. E. Moore, D. Morgan, R. Jacobs, T. Pollock, D. G. Schlom and E. S. Toberer, et al. , npj Comput. Mater., 2019, 5, 41 CrossRef.
- A. Mannodi-Kanakkithodi and M. K. Chan, Energy Environ. Sci., 2022, 15, 1930–1949 RSC.
- A. S. Rosen, S. M. Iyer, D. Ray, Z. Yao, A. Aspuru-Guzik, L. Gagliardi, J. M. Notestein and R. Q. Snurr, Matter, 2021, 4, 1578–1597 CrossRef CAS.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch and A. Aspuru-Guzik, Appl. Phys. Rev., 2021, 8, 031406 Search PubMed.
- H. C. Herbol, W. Hu, P. Frazier, P. Clancy and M. Poloczek, npj Comput. Mater., 2018, 4, 51 CrossRef.
- P. C. Jennings, S. Lysgaard, J. S. Hummelshøj, T. Vegge and T. Bligaard, npj Comput. Mater., 2019, 5, 46 CrossRef.
- B. Shahriari, K. Swersky, Z. Wang, R. P. Adams and N. de Freitas, Proc. IEEE, 2016, 104, 148–175 Search PubMed.
- S. Greenhill, S. Rana, S. Gupta, P. Vellanki and S. Venkatesh, IEEE Access, 2020, 8, 13937–13948 Search PubMed.
- J. Snoek, H. Larochelle and R. P. Adams, Advances in Neural Information Processing Systems, 2012 Search PubMed.
- Q. Liang, A. E. Gongora, Z. Ren, A. Tiihonen, Z. Liu, S. Sun, J. R. Deneault, D. Bash, F. Mekki-Berrada, S. A. Khan, K. Hippalgaonkar, B. Maruyama, K. A. Brown, J. Fisher III and T. Buonassisi, npj Comput. Mater., 2021, 7, 188 CrossRef.
- Y. Zuo, M. Qin, C. Chen, W. Ye, X. Li, J. Luo and S. P. Ong, Accelerating Materials Discovery with Bayesian Optimization and Graph Deep Learning, Mater. Today, 2021, 51, 126–135 CrossRef CAS.
- V. Korolev, A. Mitrofanov, A. Eliseev and V. Tkachenko, Mater. Horiz., 2020, 7, 2710–2718 RSC.
- P. I. Frazier and J. Wang, Information science for materials discovery and design, Springer, 2016, pp. 45–75 Search PubMed.
- A. E. Siemenn, Z. Ren, Q. Li and T. Buonassisi, npj Comput. Mater., 2023, 9, 79 CrossRef.
- D. E. Graff, E. I. Shakhnovich and C. W. Coley, Chem. Sci., 2021, 12, 7866–7881 RSC.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- R. Liang, X. Duan, J. Zhang and Z. Yuan, React. Chem. Eng., 2022, 7, 590–598 RSC.
- D. Frey, J. H. Shin, C. Musco and M. A. Modestino, React. Chem. Eng., 2022, 7, 855–865 RSC.
- C. E. Rasmussen, in Gaussian Processes in Machine Learning, ed. O. Bousquet, U. von Luxburg and G. Rätsch, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 63–71 Search PubMed.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- J. Snoek, O. Rippel, K. Swersky, R. Kiros, N. Satish, N. Sundaram, M. M. A. Patwary, Prabhat and R. P. Adams, Scalable Bayesian Optimization Using Deep Neural Networks, 2015 Search PubMed.
- H. J. Kushner, J. Basic Eng., 1964, 86, 97–106 CrossRef.
- D. R. Jones, M. Schonlau and W. J. Welch, J. Glob. Optim., 1998, 13, 455–492 CrossRef.
- N. Srinivas, A. Krause, S. M. Kakade and M. W. Seeger, IEEE Trans. Inf. Theory, 2012, 58, 3250–3265 Search PubMed.
- J. C. Hamill, J. Schwartz and Y.-L. Loo, ACS Energy Lett., 2018, 3, 92 CrossRef CAS.
- M. Yang, Z. Li, M. O. Reese, O. G. Reid, D. H. Kim, S. Siol, T. R. Klein, Y. Yan, J. J. Berry and M. F. A. M. van Hest, Nat. Energy, 2017, 2, 17038 CrossRef CAS.
- Y. Eatmon, O. Romiluyi, C. Ganley, R. Ni, I. Pelczer, P. Clancy, B. P. Rand and J. Schwartz, J. Phys. Chem. Lett., 2022, 6130–6137 CrossRef CAS.
- A. G. Wilson, Z. Hu, R. Salakhutdinov and E. P. Xing, Artificial Intelligence and Statistics, 2016, pp. 370–378 Search PubMed.
- R. Mercado, R.-S. Fu, A. V. Yakutovich, L. Talirz, M. Haranczyk and B. Smit, Chem. Mater., 2018, 30, 5069–5086 CrossRef CAS.
- A. Brown, C. Jarrett, D. De Leeuw and M. Matters, Synth. Met., 1997, 88, 37–55 CrossRef CAS.
- W. Zhao, J. Ding, Y. Zou, C.-A. Di and D. Zhu, Chem. Soc. Rev., 2020, 49, 7210–7228 RSC.
- T. Mukhopadhyaya, T. Lee, C. Ganley, P. Clancy and H. E. Katz, ACS Appl. Polym. Mater., 2022, 4, 2065–2080 CrossRef CAS.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2022, 51, D1373–D1380 CrossRef PubMed.
- S. N. Patel, A. M. Glaudell, K. A. Peterson, E. M. Thomas, K. A. OHara, E. Lim and M. L. Chabinyc, Sci. Adv., 2017, 3, e1700434 CrossRef.
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem Rev., 2020, 120, 7867–7918 CrossRef CAS.
- T. N. R. E. Laboratory, The National Renewable Energy Laboratory Best Research-Cell Efficiency Chart, Accessed 2021-02-24.
- P. P. Khlyabich, J. C. Hamill and Y.-L. Loo, Adv. Funct. Mater., 2018, 28, 1801508 CrossRef.
- I. Wharf, T. Gramstad, R. Makhija and M. Onyszchuk, Can. J. Chem., 1976, 54, 3430–3438 CrossRef CAS.
- J. C. Hamill, O. Romiluyi, S. A. Thomas, J. Cetola, J. Schwartz, M. F. Toney, P. Clancy and Y.-L. Loo, J. Phys. Chem. C, 2020, 124, 14496–14502 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, Chem. Mon., 1975, 106, 1235–1257 CrossRef CAS.
- S. Bourcier, R. X. Chia, R. N. B. Bimbong and G. Bouchoux, Eur. J. Mass Spectrom., 2015, 21, 149–159 CrossRef CAS PubMed.
- B. A. Sorenson, L. U. Yoon, E. Holmgren, J. J. Choi and P. Clancy, J. Mater. Chem. A, 2021, 9, 3668–3676 RSC.
- T. Chen and C. Guestrin, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, pp. 785–794.
- Y. Cui, S. Wang, L. Ding and F. Hao, Adv. Energy Sustainability Res., 2020, 2, 2000047 CrossRef.
- B. J. Foley, J. Girard, B. A. Sorenson, A. Z. Chen, J. Scott Niezgoda, M. R. Alpert, A. F. Harper, D. M. Smilgies, P. Clancy, W. A. Saidi and J. J. Choi, J. Mater. Chem. A, 2017, 5, 113 RSC.
- J. Kim, B.-w Park, J. Baek, J. S. Yun, H.-W. Kwon, J. Seidel, H. Min, S. Coelho, S. Lim, S. Huang, K. Gaus, M. A. Green, T. J. Shin, A. W. Y. Ho-baillie, M. G. Kim and S. I. Seok, J. Am. Chem. Soc., 2020, 142, 6251–6260 CrossRef CAS PubMed.
- E. Radicchi, E. Mosconi, F. Elisei, F. Nunzi and F. De Angelis, ACS Appl. Energy Mater., 2019, 2, 3400–3409 CrossRef CAS.
- M. Lindauer, K. Eggensperger, M. Feurer, A. Biedenkapp, D. Deng, C. Benjamins, T. Ruhkopf, R. Sass and F. Hutter, J. Mach. Learn. Res., 2022, 23, 1–9 Search PubMed.
- J. Bergstra, D. Yamins and D. Cox, International Conference on Machine Learning, 2013, pp. 115–123.
- G. H. John, R. Kohavi and K. Pfleger, Machine learning proceedings 1994, Elsevier, 1994, pp. 121–129.
- Q. Gu, Z. Li and J. Han, arXiv, 2012, preprint, arXiv:1202.3725 DOI:10.48550/arXiv.1202.3725.
- B. Azhagusundari and A. S. Thanamani, et al. , Int. J. Eng. Innov. Technol. Expl. Eng., 2013, 2, 18–21 Search PubMed.
- R. Tibshirani, J. R. Stat. Soc. Series B Stat. Methodol., 1996, 58, 267–288 Search PubMed.
- I. Cohen, Y. Huang, J. Chen, J. Benesty, J. Benesty, J. Chen, Y. Huang and I. Cohen, Noise reduction in Speech Processing, 2009, pp. 1–4 Search PubMed.
- D. P. Kingma and J. Ba, arXiv, 2014, preprint, arXiv:1412.6980 DOI:10.48550/arXiv.1412.6980.
- Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 2014.
- H. C. Herbol, M. Poloczek and P. Clancy, Mater. Horiz., 2020, 7, 2113–2123 RSC.
- M. Balandat, B. Karrer, D. R. Jiang, S. Daulton, B. Letham, A. G. Wilson and E. Bakshy, arXiv, 2019, preprint, arXiv:1910.06403 [cs.LG] DOI:10.48550/arXiv.1910.06403.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01474f |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |