
Mesoporous ceria-supported vanadia catalysts for selective aerobic oxidation of ethylbenzene
Silligandla
Nazeer
ab,
Palli
Sitaramulu
 a,
Kamma
Yogendra
ab,
Shivani
Dalal
a,
Palnati Manoj
Kumar
a,
Bojja
Sreedhar
a,
Benjaram M.
Reddy
ac and
Tumula Venkateshwar
Rao
*ab
a,
Kamma
Yogendra
ab,
Shivani
Dalal
a,
Palnati Manoj
Kumar
a,
Bojja
Sreedhar
a,
Benjaram M.
Reddy
ac and
Tumula Venkateshwar
Rao
*ab
aCatalysis and Fine Chemicals Department, CSIR–Indian Institute of Chemical Technology, Uppal Road, Hyderabad – 500 007, India. E-mail: tvrao@iict.res.in; tvraoiict@gmail.com
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad – 201 002, India
cDepartment of Chemistry and Materials Center for Sustainable Energy & Environment, Birla Institute of Technology & Science (BITS) Pilani, Hyderabad Campus, Hyderabad – 500 078, India
First published on 1st May 2024
Abstract
A series of mesoporous ceria-supported vanadium oxide catalysts were prepared and explored for aerobic oxidation of ethylbenzene to acetophenone without the involvement of any solvent or additive. Catalysts with varying contents of V2O5 (3, 6, and 10 wt%) on a mesoporous CeO2 support were prepared by the wet-impregnation method. Catalysts were characterized using techniques namely, XRD, Raman spectroscopy, BET surface area analysis, ICP-OES, FE-SEM, HR-TEM, XPS, and H2-TPR to establish a correlation between the physicochemical properties and catalytic performance. Among the investigated catalysts, the catalyst with 6 wt% of V2O5 on mesoporous ceria showed better activity with 69% ethylbenzene conversion and 88% acetophenone selectivity. The superior activity of this catalyst was ascribed to a strong synergetic interaction between ceria and vanadia as well as the formation of a CeVO4 phase, and a high specific surface area. The reaction parameters like temperature, pressure, and the amount of catalyst were optimized for possible commercial utilization. The stability of the catalyst was also investigated by the reusability test and was found to be stable and active up to five cycles.
Introduction
Oxidative upgradation of side chains in aromatic hydrocarbons to their corresponding oxygenated products is important as the resulting products from this reaction are useful in a variety of fields in the chemical industry.1 Selective transformation of ethylbenzene (EB) into acetophenone is a very demanding reaction, as it is used in manufacturing many valuable products.2 Acetophenone is employed in the manufacture of medicines (pyrrobutamine, dextropropoxyphene, cycrimine, and others), tear gas, esters, resin, chewing gums, scent, flavouring agents, cellulose ether solvents, and dyestuffs.3 Traditionally, this ketone is produced from benzene using Friedel–Crafts acylation with acyl halide or acid anhydride, and requires homogeneous Lewis acids like MCln (M = Al, Fe, Zn, Sn, and Ti) or strong protic acids, which results in generation of huge amounts of toxic waste.4 The use of inorganic stoichiometric oxidants like permanganate, dichromate, etc. for the oxidation of alkyl arenes is environmentally and economically not favourable due to the generation of significant amounts of harmful inorganic waste and their high cost.5 To overcome these problems, extensive research has been focused on developing various catalysts for oxidation of hydrocarbons employing oxidants like TBHP, nitric acid, halides, ozone, etc.6,7 However, in the industrial context, these oxidants are expensive and unfriendly. Organic peroxides generate significant amounts of organic waste, and hydrogen peroxide spontaneously decomposes into water, potentially reducing the efficiency of catalysts.8 Therefore, in recent past, much attention has been paid in the petrochemical industry to aerobic oxidation of EB to acetophenone.9 However, the reaction of molecular oxygen (O2) with hydrocarbons in the singlet state is hindered by its triplet-state nature.10 As a result, the success of such processes relies heavily on the development of appropriate catalysts. Recently, efforts have been made to develop efficient homogeneous catalysts for the liquid-phase oxidation of EB with O2.11–13On a commercial scale, acetophenone is typically manufactured by oxidation of EB with molecular oxygen in the liquid phase. This process uses cobalt catalysts in acetic acid with promoters like manganese or bromide. The use of homogeneous catalysts complicates the catalyst separation from the reaction mixture, and acidic solvents raise environmental concerns.5 These difficulties led to develop heterogeneous catalysts that are more efficient, and easily separable by simple filtration, facilitating the recycling of the catalyst for subsequent runs.14 On these lines, various catalysts based on the Au–Pd alloy supported on zeolite type MOFs, Pd@N-doped carbon, and Pd on g-C3N4-GO were investigated and found to show reasonably good activity towards aerobic oxidation of EB.15–17 However, the use of noble metal catalysts makes the process more expensive. Therefore, switching to non-noble low-cost metal oxide catalysts is highly recommended. In that direction, SiO2/Al2O3 mixed-oxide supported Mn, Co(II) and Cu(II) metal complexes supported on a polymer–silica hybrid, and the Mn-MCM-41 catalyst were investigated and observed to exhibit better conversion and selectivity with TBHP as the oxidant.4,18,19 Furthermore, Liu et al. reported a Ti–Zr–Co catalyst8 and Gao et al. reported a Co3O4/rGO catalyst,20 achieving a good conversion of EB with a remarkable selectivity towards acetophenone. Carbon nanotube catalysts were also employed for this reaction with moderate conversion and selectivity for acetophenone.5 Notably, all these approaches necessitate the use of reaction initiators and organic solvents.
Ceria-based materials are well-recognized as efficient catalytic systems for three-way catalytic converters in automobiles and superior catalysts in various oxidation reactions, including those involving the oxidation of diesel soot,21 hydrocarbons,22 alcohols,23,24 alkenes,25,26 benzylamine,27etc. This efficacy stems from their exceptional oxygen storage and release capacity (OSC), directly resulting from ceria's easily modulated redox properties (2CeO2 ↔ Ce2O3 + O2). Recently, Subbiah et al. reported that a ceria-based CeAlPO-5 catalyst exhibits better activity and selectivity for EB conversion to acetophenone at high temperatures in the gas phase,28 and a CeO2 nanoparticle-decorated Co3O4 microsphere catalyst was also reported showing better activity and selectivity.16 Pd particles immobilized on a nano CeO2 support were also investigated for this reaction using TBHP as the oxidant,2 and a ceria-supported vanadia catalyst was reported with only 20.5% conversion of EB and 72.2% selectivity towards acetophenone using H2O2 as the oxidant.29 A ceria–cobalt mixed oxide catalyst was also explored for this reaction more recently by our group which showed 60% conversion of EB with 87% selectivity to acetophenone.30
In general, ceria-based vanadia catalysts have gained significant attention owing to their impressive activity and selectivity in various oxidation reactions of commercial significance.31,32 Their catalytic activity depends on various factors such as the quantity of vanadium oxide deposited, support material characteristics including the preparation method, vanadium oxidation state within the support, and other related features. Incorporation of electrons into cerium f orbitals maintains the catalyst in a reduced state and keeps vanadium in its most oxidized state which is crucial in the oxidation reactions.33,34 In this context, it is aimed to find an efficient ceria-based vanadia catalyst for aerobic liquid phase oxidation of EB to acetophenone by preparing high surface area ceria with optimum vanadia content deposition. In this study, different weight percentages (3–10%) of vanadia were impregnated over mesoporous ceria, which was synthesized by a template-assisted approach. The resulting catalysts were evaluated for oxidation of EB to acetophenone using O2 as an oxidant without the use of solvents and promoters or additives. To establish a correlation between the catalytic performance and the physicochemical attributes of the catalyst, a range of characterization techniques were employed. Additionally, in order to achieve optimal conversion and selectivity, various reaction parameters including reaction temperature, pressure, time, and quantity of the catalysts were also systematically fine-tuned.
Experimental
Chemicals
Ethylbenzene, cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, ≥ 99.99%), ammonium metavanadate (NH4VO3), 1-butanol (anhydrous, 99.8%), Pluronic P123 (poly-(ethylene glycol)-block-poly(propylene glycol)-block-poly-(ethylene glycol) (PEO20-PPO70-PEO20)), oxalic acid (H2C2O4), and concentrated nitric acid (HNO3, 68–70%) were purchased from Sigma-Aldrich, and the absolute ethanol (99.7%, AR) was purchased from Merk. All chemicals were used without any further purification.Catalyst preparation
In this, the calculated quantity of the precursor, cerium(III) nitrate hexahydrate, was taken in a beaker and added butane-1-ol as a solvent, stirred for 30 min at room temperature to dissolve the salt completely. The required amount of P123 surfactant was added and stirred until complete dissolution of the surfactant. Then the desired amount of concentrated nitric acid was added dropwise and stirred for 1 h. Later this mixture was heated at 120 °C to obtain a black powder. The powder was washed with absolute ethanol, centrifuged, and dried in an electrical oven overnight at 120 °C. Finally, the synthesized compound was subjected to calcination cycles at different temperatures in an air atmosphere of 150 °C for 12 h, 250 °C for 6 h, 350 °C for 4 h, 450 °C for 2 h, and 550 °C for 1 h.35Catalysts with different wt% of vanadia on ceria (3, 6, and 10%) were prepared by impregnating the ceria support with ammonium metavanadate aqueous solution, which was prepared by dissolving the required quantity of the metal precursor (NH4VO3) in aqueous solution of oxalic acid (1/2 molar ratio of NH4VO3/H2C2O4). This mixture was stirred at room temperature for 6 h, dried at 120 °C with continuous stirring, and kept the sample in an oven at 120 °C for 12 h. The resultant solid compound was finely ground and calcined at 550 °C for 5 h. The prepared catalysts with different wt% of vanadia on ceria (3, 6, and 10 wt%), pristine ceria, and vanadia were denoted as CV3, CV6, CV10, CeO2, and V2O5, respectively.
Catalyst characterization
X-ray diffraction (XRD) patterns of catalyst powders were recorded using a Rigaku Multiflex diffractometer equipped with a nickel-filtered Cu Kα radiation source (wavelength of 1.5418 Å) and a scintillation counter detector on a 10–80° 2θ scale with a step size of 0.021 and 1 point per second. The mean crystallite size was determined by calculating the X-ray line broadening of the most intense peak using the Scherrer equation. The surface areas of catalysts were examined using a BELSORP II (Japan) analyser. Samples were degassed under vacuum at 250 °C before measurements at −196 °C. Surface areas were computed from desorption isotherms using the multipoint Brunauer–Emmett–Teller (BET) method, and average pore diameters were determined using the Barrett–Joyer–Halenda (BJH) method. The metal content in samples is estimated using inductively coupled plasma-optical emission spectroscopy (ICP-OES) (Thermo Jarrell Ash model IRIS Intrepid II XDL, USA). The Raman spectra of the catalysts were obtained using a CCD detector on a Horiba Jobin-Yvon Labram HR Evolution Confocal Raman Spectrometer with spectral deviation less than 2 cm−1. An Ar+ ion laser (632 nm) light was used to stimulate the sample on the slide (1 μm diameter spot) beneath the microscope. The acquisition time was decided to keep the strength of Raman scattering in view. The morphology of the catalyst was investigated using a JEOLJSM-7610F scanning electron microscope and HRTEM (TALOS F200X from FEI) with an 80–200 kV electron beam. A Shimadzu ESCA 3400 with an Mg Kα radiation source was used to record the X-ray photoelectron spectra (1253.6 eV). To reduce spectral noise, samples were vacuumed for several hours (<10−8 Pa). In the spectra, binding energy values were corrected with reference to the carbon 1s peak as 284.5 eV.36 A tubular reactor coupled to a gas chromatograph with a thermal conductivity detector was used to perform the hydrogen temperature programmed reduction (H2-TPR) (GC-TCD) of the samples. Before performing the H2-TPR study, samples were heated to 200 °C with helium gas.Catalytic activity measurements
The efficiency of CeO2, V2O5, CV3, CV6, and CV10 catalysts was evaluated for the oxidation of ethylbenzene with O2 as an oxidizing agent. 25 mL of EB and 50 mg of the catalyst were charged into a 100 mL stainless steel vessel fitted to an autoclave reactor. The vessel was subjected to three rounds of flushing with molecular oxygen to remove atmospheric air before being pressurized to 20 bar with O2. The reaction mixture was stirred at 600 rpm for 8 h at 120 °C. Then the reactor was cooled to room temperature, and the catalyst was separated from the reaction mixture using a centrifuge. Products were identified through GCMS employing a DB-5 column and analyzed using a GC with a BP-20 wax column.Results and discussion
XRD patterns of CeO2, CV3, CV6, CV10, and V2O5 samples are depicted in Fig. 1. XRD peaks at 2-theta values of 28.8, 33.2, 47.6, 56.6, 59.2, 69.6, and 76.9 corresponding to Miller indices (111), (200), (220), (311), (222), (400), and (331), respectively, were noticed for ceria and CV (ceria supported vanadia catalysts) samples. These findings confirm the presence of a characteristic crystal structure consistent with cubic fluorite-type ceria.37,38 The crystallite sizes of all samples were determined using the Scherrer equation and presented in Table 1. | ||
| Fig. 1 XRD patterns of CeO2, CV3, CV6, CV10, and V2O5 samples. | ||
| Entry | Sample | Surface areas (m2 g−1) | Crystallite sizes (nm) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|---|---|
| 1 | CeO2 | 76.96 | 9.63 | 0.138 | 6.69 |
| 2 | V2O5 | 4.71 | 35.2 | 0.016 | 15.93 |
| 3 | CV3 | 43.16 | 13.4 | 0.110 | 12.19 |
| 4 | CV6 | 39.13 | 14.3 | 0.084 | 10.98 |
| 5 | CV10 | 34.86 | 15.6 | 0.114 | 16.75 |
Upon the addition of V2O5, an increase in the particle size was observed in ceria-supported vanadia catalysts compared to pure ceria. The XRD patterns of CV10, a CeO2 supported V2O5 catalyst, revealed distinct peaks at 18.3, 24.2, and 32.7 2θ values and those of CV6 exhibited a small peak at 24.2 2θ with low intensity corresponding to the CeVO4 phase.33,39 This suggests that when vanadia loading is lower, CeVO4 is in the finely dispersed form and more evenly distributed, which can directly impact the catalytic activity. The absence of V2O5 related lines suggests a finely dispersed vanadium oxide on the ceria surface in these samples.
The nitrogen gas adsorption and desorption isotherms of CeO2, V2O5, and ceria supported vanadia catalysts are presented in Fig. 2. The presence of H3-type hysteresis loops within the P/Po range of 0.49 to 0.99 in the type IV isotherms indicates that the CeO2 and CV series samples exhibit mesoporous characteristics.40 BET surface areas derived from N2 physisorption, and crystallite sizes calculated from XRD studies are listed in Table 1. The results show that the surface area of pure ceria is 76.96 m2 g−1, after loading of vanadia onto the ceria support surface areas gradually decreased. This can be attributed to two main factors. The penetration of dispersed vanadia species can block or narrow the pores of the support. Solid-state reactions between ceria and vanadia may lead to the formation of orthovanadate crystals which may further contribute to the blockage of ceria pores.41,42
 | ||
| Fig. 2 N2 adsorption–desorption isotherms of CeO2, CV3, CV6, CV10, and V2O5 samples. | ||
In the Raman spectra of CeO2 and CV samples (Fig. 3), there is a prominent band at 468 cm−1 corresponding to the F2g vibrational mode, indicating the symmetrical coordination of surrounding oxygen atoms around Ce4+ ions in the CeO2 lattice.39 There is another Raman band at 866 cm−1 (A1g) corresponding to the symmetric stretching of oxygen atoms around vanadium in the vanadate, while peaks at 790 and 802 cm−1 (B2g, Eg) signify the antisymmetric stretching of the VO43− ion.43 Notably, CV3 and CV6 samples exclusively exhibit ceria-related peaks, suggesting a high dispersion of vanadium oxide species, and they are molecularly dispersed making them undetectable by the Raman instrument. The V2O5 sample displays characteristic Raman peaks at 1000, 708, 488, 411, 290, and 151 cm−1.35
 | ||
| Fig. 3 Raman spectra of the CeO2, CV3, CV6, CV10, and V2O5 samples. | ||
The contents of Ce and V elements of the synthesized catalysts were analyzed by ICP-OES analysis, as shown in Table 2. The obtained results are close to calculated metal ratio in the preparation of catalysts. These outcomes serve as evidence that the method applied for the preparation of ceria supported vanadia catalysts in this study is indeed efficacious.
| Entry | Sample | Nominal values (%) | ICP-OES analysis values (%) | ||
|---|---|---|---|---|---|
| Ce | V | Ce | V | ||
| 1 | CV3 | 97 | 3 | 97.5 | 2.5 |
| 2 | CV6 | 94 | 6 | 94.5 | 5.5 |
| 3 | CV10 | 90 | 10 | 90.5 | 9.5 |
The FE-SEM technique was used to investigate the surface morphology of the synthesized catalysts. FE-SEM images of CeO2 (a), V2O5 (b), CV3 (c), CV10 (d), and CV6 (e and f) samples are displayed in Fig. 4. Except for V2O5 (b), images for all samples appear with similar textures and having a porous morphology. The CV6 sample's surface morphology was further analyzed using HR-TEM, and the particle sizes were determined through image analysis using the instrument software. The results were then compared with the XRD findings. Fig. 5 displays various regions of HR-TEM and SAED images of the CV6 sample. According to the images, the particles are uniformly dispersed, and sizes of the particles are about 9–15 nm and are in good agreement with the XRD results. The inter-planar distance of the (111) plane is 0.31 nm corresponding to cubic fluorite structured ceria.
 | ||
| Fig. 4 The FE-SEM images of CeO2 (a), V2O5 (b), CV3 (c), CV10 (d), and CV6 (e) and (f) samples. | ||
 | ||
| Fig. 5 The HR-TEM images of the CV6 sample: (a) (100 nm), (b) (50 nm), (c) (10 nm), (d) (5 nm), (e) (SAED image), and (f) (magnified image of ‘d’). | ||
The oxidation states or electronic configuration of elements on the surface of the CeO2, V2O5, and CV6 catalysts were studied using the XPS technique. The obtained Ce 3d XPS spectra of CeO2, CV3, CV6, and CV10 samples are depicted in Fig. 6. The Ce 3d spectra reveal two multiplets of spin–orbit, denoted as ‘u’ and ‘v’, corresponding to the 3d3/2 and 3d5/2 states, respectively. These two spin multiplets produce eight broad peaks. The two peaks labelled as u1 and v1 have resulted from trivalent compounds of Ce (Ce3+ ↔ 3d104f1). On the other hand, the six peaks namely u0, u2, u3, and v0, v2, v3 originated from tetravalent Ce compounds (Ce4+ ↔ 3d104f0). These findings provide evidence for the coexistence of Ce3+ and Ce4+ ions, indicating the redox nature of these catalysts.44,45 Compared to the pure ceria, the CV6 sample shows increased intensities of u1 and v1 peaks associated with the Ce3+ oxidation state or trivalent Ce compounds. The Ce3+ quantity was determined by calculating the ratio of the area occupied by Ce3+ to the combined area of Ce3+ and Ce4+, as outlined in Table 3. This enhanced presence of Ce3+ ions indicates the formation of more oxygen vacancies in the ceria-supported vanadia catalysts, which is a crucial factor influencing the catalyst's activity in oxidation reactions.46,47
 | ||
| Fig. 6 The Ce 3d XPS spectra of CeO2, CV3, CV6, and CV10 samples. | ||
| Sample | Ce3+/(Ce3+ + Ce4+) | Oβ/(Oβ + Oα) |
|---|---|---|
| CeO2 | 0.261 | 0.451 |
| CV3 | 0.288 | 0.583 |
| CV6 | 0.301 | 0.625 |
| CV10 | 0.312 | 0.691 |
Fig. 7 shows the V 2p XPS spectra of the V2O5, CV3, CV6, and CV10 samples. For the V2O5 sample, the peak positioned at ∼513.4 eV binding energy is attributed to the V (2p3/2) state, and another small peak centered at ∼520.9 eV is related to the V (2p1/2) state. These two peaks suggest the presence of the +5 oxidation state of vanadium in the catalyst.48,49 Compared to V2O5, these two peaks are shifted toward a higher binding energy side and broadened. This shifting and broadening of peaks could be attributed to the interaction between the ceria support and deposited vanadia.31
 | ||
| Fig. 7 The V 2p XPS spectra of V2O5, CV3, CV6, and CV10 samples. | ||
Fig. 8 illustrates the O 1s XPS spectra of CeO2, CV3, CV6, and CV10 samples. In the CeO2 sample, a peak at around 527.26 eV is attributed to lattice oxygen (Oα), while another broad peak at around 529.53 eV is associated with surface oxygens (Oβ). As the vanadia loading increases, the peak associated with surface oxygens (Oβ) shifts toward lower binding energies, sharpens and its intensity also increases. The reason for these changes may be due to the formation of a CeVO4 phase on the surface of the ceria support.
 | ||
| Fig. 8 The O 1s XPS spectra of CeO2, CV3, CV6, and CV10 samples. | ||
The catalysts' redox properties were explored through TPR experiments, and the TPR profiles are presented in Fig. 9. The pure ceria support exhibits two reduction peaks corresponding to the Ce4+ to Ce3+ state (CeO2 ↔ Ce2O3). The low-temperature reduction peak at ∼500 °C is assigned to surface oxygen. Another peak at a higher temperature at ∼800 °C is attributed to the reduction of bulk oxygen of the ceria lattice.50 For CV samples, the temperature gap between the two peaks is decreased after the addition of vanadia to the ceria support. The low-temperature reduction peak shifted towards the high-temperature side and sharpened. This decreased reducibility of surface oxygen is due to the stabilization of the Ce3+ state through the interaction between the V2O5 species and the ceria surface. As the loading of vanadia increases the peak shifts more, sharpens, and becomes small. On the other hand, compared to pure ceria, for the CV3 and CV6 samples the high-temperature reduction peak moves towards the low-temperature side implying that the reducibility of bulk oxygen is increased.51,52
 | ||
| Fig. 9 TPR profile of the CeO2, CV3, CV6, CV10, and V2O5 samples. | ||
The performance of synthesized CeO2, V2O5, and ceria-supported vanadia (CV3, CV6, and CV10) catalysts was examined for aerobic oxidation of EB by taking 25 mL of EB and 50 mg of the catalyst in a 100 mL stainless steel reactor vessel by maintaining 20 bar pressure with molecular oxygen at 120 °C, for 8 h (Scheme 1). After the reaction, the catalyst was separated and the liquid contents were analyzed (Table 4). Without using a catalyst very little activity (5% conversion) was observed, which indicates the need of a catalyst for better activity. Pure CeO2 and V2O5 catalysts showed considerable activity with 42 and 38% conversion, respectively. Compared to individual metal oxides, ceria-supported vanadia (CV3, CV6, and CV10) catalysts showed higher catalytic activity. This enhanced catalytic efficiency could be attributed to a strong synergistic interaction between CeO2 and V2O5, as well as the formation of CeVO4. These synergistic interactions serve as a crucial oxygen activation pathway in numerous oxidation reactions. Among the various Ce/V catalyst ratios tested, CV6 demonstrated superior catalytic activity, achieving a 69% EB conversion with 88% selectivity towards acetophenone. The CV6 catalyst exhibited better activity because it contains a greater amount of the CeVO4 phase compared to CV3 and has a higher BET surface area than CV10. This suggests that the optimal combination of the CeVO4 content and a high surface area of the CV6 sample contributes to its superior catalytic performance.29 To check the influence of the CeVO4 phase, we prepared a CeVO4 solid solution catalyst by a facile co-precipitation process.53 This catalyst shows better activity than individual oxides of Ce and V and less activity than ceria supported vanadia catalysts with 51% EB conversion and 90% selectivity to acetophenone.
 | ||
| Scheme 1 Oxidation of ethylbenzene. | ||
| Entry | Catalyst | Conversion of EB (%) | Selectivity (%) | ||
|---|---|---|---|---|---|
| AP | BZ | BA | |||
| Reaction conditions: ethylbenzene (25 mL), O2 (20 bar), catalyst (50 mg), time (8 h), and temperature (120 °C), [EB = Ethyl benzene AP = Acetophenone, BZ = Benzaldehyde, and BA = Benzoic acid]. | |||||
| 1 | Blank | 5 | 100 | — | — |
| 2 | CeO2 | 42 | 90 | 6 | 4 |
| 3 | V2O5 | 38 | 85 | 6 | 9 |
| 5 | CV3 | 56 | 89 | 7 | 4 |
| 6 | CV6 | 69 | 88 | 7 | 5 |
| 7 | CV10 | 61 | 86 | 7 | 7 |
To know the effect of temperature on the reaction under the mentioned reaction conditions, the reaction was carried out over the highly active CV6 catalyst in the temperature range from 80 to 140 °C and the findings are depicted in Fig. 10. At 80 °C, EB conversion was 19% with 92% selectivity to acetophenone. With the increase in temperature, the conversion of EB increased gradually and reached 69% at 120 °C. However, the selectivity to acetophenone is slightly decreased by 4% (88%). A further increase in temperature to 140 °C resulted in an insignificant increase in EB conversion (69–75%) and the selectivity to acetophenone dropped considerably (from 88% to 76%). These results suggest that 120 °C is the optimum temperature for the reaction.
 | ||
| Fig. 10 Effect of temperature on ethylbenzene oxidation. Reaction conditions: ethylbenzene (25 mL), O2 pressure (20 bar), catalyst (50 mg), and time (8 h). | ||
The impact of pressure on the reaction was explored by changing it within the range of 5–25 bar, and the findings are presented in Fig. 11. At 5 bar pressure, EB conversion was 19%, with 93% selectivity to acetophenone. As the pressure increased, conversion gradually rose, reaching 69% at 20 bar, with an 88% selectivity towards acetophenone. However, a further increase to 25 bar pressure led to a significant decline in selectivity due to over-oxidation. These results suggest that 20 bar pressure is sufficient for the optimum product yield.
 | ||
| Fig. 11 Effect of reaction pressure on ethylbenzene oxidation. Reaction conditions: ethylbenzene (25 mL), temperature (120 °C), catalyst (50 mg), and time (8 h). | ||
The impact of the reaction time was examined by conducting the reaction at 2, 4, 6, 8, and 10 h and the results are depicted in Fig. 12. After two hours of reaction, the conversion of EB was 24%, with a selectivity of 92% towards acetophenone. As the reaction continued up to 8 h, the conversion increased to 69%, accompanied by a slight drop in the selectivity to 88%. Beyond eight hours, the rate of conversion was very low, and the selectivity also dropped. This suggests that an 8-hour reaction time is enough for obtaining better results in this process.
 | ||
| Fig. 12 The influence of the reaction time on oxidation of ethylbenzene. Reaction conditions: ethylbenzene (25 mL), temperature (120 °C), O2 pressure (20 bar), and catalyst (50 mg). | ||
The effect of catalyst quantity on the EB oxidation over the CV6 catalyst was investigated by varying the catalyst amount from 15 to 75 mg and the results are shown in Fig. 13. With 15 mg of the catalyst, 25% EB conversion and 90% acetophenone selectivity were observed while 50 mg of the catalyst provided 69% and 88% of conversion and selectivity, respectively. With 75 mg of the catalyst, the increase in the conversion was less and the selectivity was dropped. These findings suggest that 50 mg of the catalyst is sufficient for the reaction under mentioned conditions.
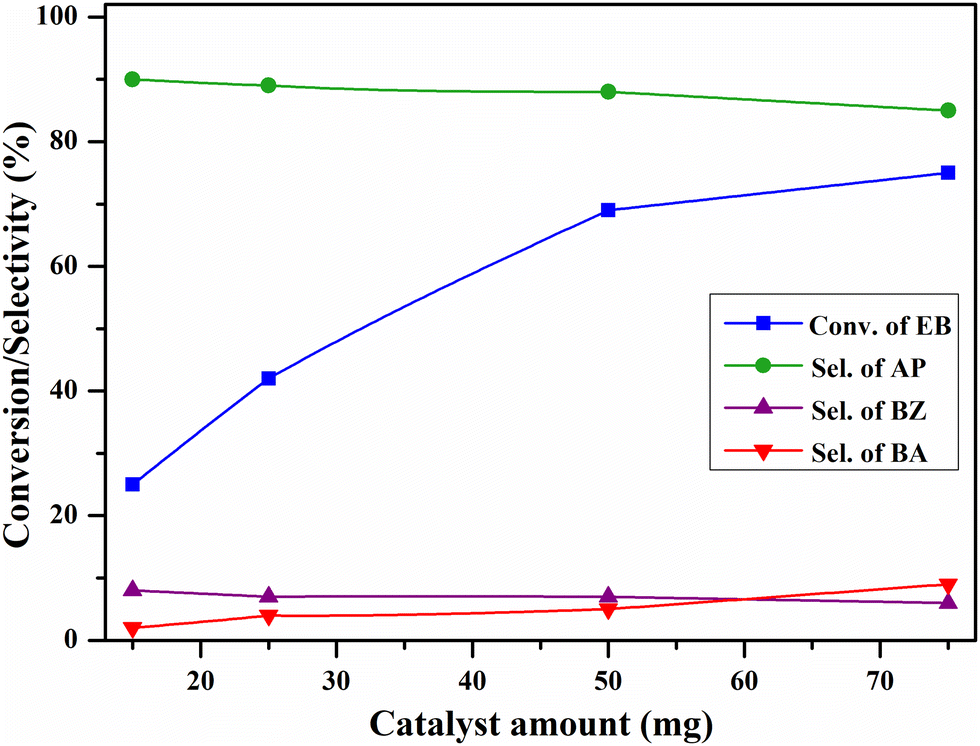 | ||
| Fig. 13 The influence of the catalyst amount on oxidation of ethylbenzene. Reaction conditions: ethylbenzene (25 mL), temperature (120 °C), O2 pressure (20 bar), and time (8 h). | ||
The stability and reusability of the CV6 catalyst were examined up to 5 cycles and the outcomes are displayed in Fig. 14. After completion of the reaction, the catalyst was isolated from the reaction mixture through centrifugation. Any impurities contained in the catalyst were removed by washing with methanol thoroughly and dried for 5 h at 150 °C. The catalyst maintained its activity over the course of five cycles, showing no significant decrease in conversion, and the selectivity towards acetophenone remained constant.
 | ||
| Fig. 14 Reusability study. Reaction conditions: ethylbenzene (25 mL), temperature (120 °C), O2 pressure (20 bar), catalyst (50 mg), and time (8 h). | ||
Conclusions
Mesoporous ceria-supported vanadia catalysts (CV3, CV6, and CV10) were prepared, characterized, and evaluated for aerobic oxidation of ethylbenzene without the involvement of solvents and additives and were investigated systematically. These catalysts were prepared by adopting wet-impregnation and template-assisted methods (for mesoporous ceria). Among different contents of vanadia loaded catalysts, the 6 wt% sample exhibited superior activity with 69% conversion of EB and 88% selectivity towards acetophenone. This exceptional performance of CV6 was ascribed to the robust synergistic interactions between CeO2 and V2O5, coupled with the formation of CeVO4. Notably, CV6 surpassed its counterparts, CV3 and CV10, due to a highly dispersed and abundant CeVO4 phase and a larger BET surface area. The catalytic activity was optimized for the reaction by varying different parameters such as time, oxygen pressure, temperature, and the catalyst amount.Author contributions
Silligandla Nazeer: conceptualization, methodology, data curation, validation, investigation, and writing – original draft. Palli Sitaramulu: data curation. Kamma Yogendra: data curation. Shivani Dalal: formal analysis. Palnati Manoj Kumar: formal Analysis. Bojja Sreedhar: formal analysis. Tumula Venkateshwar Rao: funding acquisition, project administration, supervising, and writing – review and editing. Benjaram M. Reddy: writing – review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors P.S and S.N thank University Grant Commission (UGC), New Delhi and K.Y thank. Council of Scientific and Industrial Research (CSIR), New Delhi, for research fellowships. The authors thank Director, CSIR-IICT for granting permission to communicate this work (IICT/Pubs./2024/030) and Dr. Manorama for XPS characterization.References
- J. S. Kirar, J. Singh and S. Khare, Appl. Organomet. Chem., 2018, 32(8), e4408 CrossRef.
- L. Kalita and L. Saikia, ChemistrySelect, 2020, 5(16), 4848–4855 CrossRef CAS.
- S. Keshipour and K. Adak, Appl. Organomet. Chem., 2017, 31(11), e3774 CrossRef.
- M. Arshadi, M. Ghiaci, A. Rahmaniana, H. Ghaziaskara and A. Gil, Appl. Catal., B, 2012, 119, 81–90 CrossRef.
- J. Luo, F. Peng, H. Yu, H. Wang and W. Zheng, ChemCatChem, 2013, 5(6), 1578–1586 CrossRef CAS.
- L. Chen, B. Li and D. Liu, Catal. Lett., 2014, 144, 1053–1061 CrossRef CAS.
- B. Sarkar, P. Prajapati, R. Tiwari, R. Tiwari, S. Ghosh, S. S. Acharyya, C. Pendem, R. K. Singha, L. N. S. Konathala, J. Kumar, T. Sasaki and R. Bal, Green Chem., 2012, 14(9), 2600–2606 RSC.
- T. Liu, H. Cheng, L. Sun, F. Liang, C. Zhang, Z. Ying, W. Lin and F. Zhao, Appl. Catal., A, 2016, 512, 9–14 CrossRef CAS.
- B. Gao, S. Meng and X. Yang, Org. Process Res. Dev., 2015, 19(10), 1374–1382 CrossRef CAS.
- H. Ma, J. Xu, Q. Zhang, H. Miao and W. Wu, Catal. Commun., 2007, 8(1), 27–30 CrossRef CAS.
- S. K. Jana, P. Wu and T. Tatsumi, J. Catal., 2006, 240(2), 268–274 CrossRef CAS.
- A. P. Unnarkat, S. Singh and S. Kalan, Mater. Today: Proc., 2021, 45, 3991–3996 CAS.
- B. B. Wentzel, M. P. Donners, P. L. Alsters, M. C. Feiters and R. J. Nolte, Tetrahedron, 2000, 56(39), 7797–7803 CrossRef CAS.
- M. Arshadi, M. Ghiaci, A. A. Ensafi, H. Karimi Maleh and S. L. Suib, J. Mol. Catal. A: Chem., 2011, 338(1–2), 71–83 CAS.
- J. Long, H. Liu, S. Wu, S. Liao and Y. Li, ACS Catal., 2013, 3(4), 647–654 CrossRef CAS.
- J. Liu, R. Meng, P. Jian and R. Jian, ACS Sustainable Chem. Eng., 2020, 8(45), 16791–16802 CrossRef CAS.
- S. Nilforoushan, M. Ghiaci, S. M. Hosseini, S. Laurent and R. N. Muller, New J. Chem., 2019, 43(18), 6921–6931 RSC.
- V. Chaudhary and S. Sharma, Catal. Today, 2021, 375, 601–613 CrossRef CAS.
- K. M. Parida and S. S. Dash, J. Mol. Catal. A: Chem., 2009, 306(1–2), 54–61 CrossRef CAS.
- L. Gao, W. Zhuge, X. Feng, W. Sun, X. Sun and G. Zheng, New J. Chem., 2019, 43(21), 8189–8194 RSC.
- S. K. Megarajan, S. Rayalu, Y. Teraoka and N. Labhsetwar, J. Mol. Catal. A: Chem., 2014, 385, 112–118 CrossRef.
- J. Y. Luo, M. Meng, J. S. Yao, X. G. Li, Y. Q. Zha, X. Wang and T. Y. Zhang, Appl. Catal., B, 2009, 87(1–2), 92–103 CrossRef CAS.
- P. Sitaramulu, K. Yogendra, S. Nazeer, B. M. Reddy and T. Venkateshwar Rao, Res. Chem. Intermed., 2022, 48(11), 4579–4599 CrossRef.
- S. Nazeer, P. Sitaramulu, K. Yogendra, P. M. Kumar, B. M. Reddy and T. Venkateshwar Rao, Catalysts, 2023, 13(7), 1058 CrossRef CAS.
- P. Sitaramulu, K. Yogendra, S. Nazeer, R. Kishore, B. M. Reddy and T. Venkateshwar Rao, New J. Chem., 2023, 47(16), 7556–7565 RSC.
- P. Sitaramulu, S. Nazeer, K. Yogendra, A. S. Prasad, P. Chandrashekar and T. Venkateshwar Rao, New J. Chem., 2024, 48, 1932–1942 RSC.
- B. Govinda Rao, P. Sudarsanam, A. Rangaswamy and B. M. Reddy, Catal. Lett., 2015, 145, 1436–1445 CrossRef CAS.
- S. Devika, M. Palanichamy and V. Murugesan, Appl. Catal., A, 2011, 407(1–2), 76–84 CrossRef CAS.
- T. Radhika and S. Sugunan, Catal. Commun., 2007, 8(2), 150–156 CrossRef CAS.
- P. Sitaramulu, K. Yogendra, S. Nazeer, B. M. Reddy and T. Venkateshwar Rao, Res. Chem. Intermed., 2022, 48, 471–490 CrossRef.
- G. Raju, B. M. Reddy and S.-E. Park, J. CO2 Util., 2014, 5, 41–46 CrossRef CAS.
- K. N. Rao, B. M. Reddy, B. Abhishek, Y.-H. Seo, N. Jiang and S.-E. Park, Appl. Catal., B, 2009, 91, 649–656 CrossRef CAS.
- B. Beck, M. Harth, N. G. Hamilton, C. Carrero, J. J. Uhlrich, A. Trunschke, S. Shaikhutdinov, H. Schubert, H. J. Freund, R. Schlogl, J. Sauer and R. Schomacker, J. Catal., 2012, 296, 120–131 CrossRef CAS.
- J. S. Kirar and S. Khare, RSC Adv., 2018, 8(34), 18814–18827 RSC.
- P. R. G. Nallappa Reddy, B. G. Rao, T. Venkateshwar Rao and B. M. Reddy, Appl. Petrochem. Res., 2020, 10, 67–76 CrossRef.
- S. Ramana, B. G. Rao, P. Venkataswamy, A. Rangaswamy and B. M. Reddy, J. Mol. Catal. A: Chem., 2016, 415, 113–121 CrossRef CAS.
- Y. Li, Z. Wei, F. Gao, L. Kovarik, R. A. Baylon, C. H. Peden and Y. Wang, ACS Catal., 2015, 5(5), 3006–3012 CrossRef CAS.
- S. El- Korso, S. Bedrane, A. Choukchou Braham and R. Bachir, RSC Adv., 2015, 5(78), 63382–63392 RSC.
- M. Kurian and C. Kunjachan, J. Environ. Chem. Eng., 2016, 4(1), 1359–1366 CrossRef CAS.
- P. Challa, S. S. Enumula, K. S. Reddy, M. Kondeboina, D. R. Burri and S. R. Rao Kamaraju, Ind. Eng. Chem. Res., 2020, 59(40), 17720–17728 CrossRef CAS.
- V. K. Nguyen, J. H. Park and C. H. Shin, Korean J. Chem. Eng., 2014, 31, 582–586 CrossRef CAS.
- X. Gu, J. Ge, H. Zhang, A. Auroux and J. Shen, Thermochim. Acta, 2006, 451(1–2), 84–93 CrossRef CAS.
- A. Iglesias-Juez, M. V. Martínez-Huerta, E. Rojas-García, J. M. Jehng and M. A. Banares, J. Phys. Chem. C, 2018, 122(2), 1197–1205 CrossRef CAS.
- P. Sudarsanam, B. Hillary, D. K. Deepa, M. H. Amin, B. Mallesham, B. M. Reddy and S. K. Bhargava, Catal. Sci. Technol., 2015, 5(7), 3496–3500 RSC.
- D. Jampaiah, P. Venkataswamy, V. E. Coyle, B. M. Reddy and S. K. Bhargava, RSC Adv., 2016, 6(84), 80541–80548 RSC.
- M. N. Taylor, A. F. Carley, T. E. Davies and S. H. Taylor, Top. Catal., 2009, 52, 1660–1668 CrossRef CAS.
- B. M. Reddy, K. N. Rao, G. K. Reddy, A. Khan and S. E. Park, J. Phys. Chem. C, 2007, 111(50), 18751–18758 CrossRef CAS.
- T. Feng and J. M. Vohs, J. Catal., 2004, 221(2), 619–629 CrossRef CAS.
- Q. Wang, K. L. Yeung and M. A. Banares, J. Catal., 2018, 364, 80–88 CrossRef CAS.
- C. Kunjachan, A. Sreevalsan and M. Kurian, J. Water Process. Eng., 2017, 19, 42–50 CrossRef.
- Z. Wu, A. J. Rondinone, I. N. Ivanov and S. H. Overbury, J. Phys. Chem. C, 2011, 115(51), 25368–25378 CrossRef CAS.
- M. V. Martínez-Huerta, G. Deo, J. L. G. Fierro and M. A. Banares, J. Phys. Chem. C, 2007, 111(50), 18708–18714 CrossRef.
- J. H. Zain, V. Grover, J. Ramkumar, K. Bhattacharyya and A. K. Tyagi, J. Mater. Sci., 2020, 55, 5690 CrossRef.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |