
Vanadium(V) arylimido alkylidene N-heterocyclic carbene complexes containing fluorinated alkoxide or halogenated phenoxide ligands for the syndiospecific ROMP of cyclic olefins†
Kotohiro
Nomura
 *a,
Shuko
Kuwahara‡
a,
Jirapa
Suthala‡
a,
Yuta
Kawamoto
a,
Daisuke
Shimoyama
a and
Michael R.
Buchmeiser
*bc
*a,
Shuko
Kuwahara‡
a,
Jirapa
Suthala‡
a,
Yuta
Kawamoto
a,
Daisuke
Shimoyama
a and
Michael R.
Buchmeiser
*bc
aDepartment of Chemistry, Tokyo Metropolitan University, 1-1 Minami Osawa, Hachioji, Tokyo 192-0927, Japan. E-mail: ktnomura@tmu.ac.jp
bInstitute of Polymer Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
cGerman Institutes of Textile and Fiber Research (DITF) Denkendorf, Körschtalstr. 26, D-73770 Denkendorf, Germany
First published on 12th July 2024
Abstract
[V(N-2,6-Cl2C6H3)(CHSiMe3)(OC6X5)(NHC)] [X = F (1), Cl (2), NHC = 1,3-bis-(2,6-dimethylphenyl)-imidazol-2-ylidene], exhibited remarkable catalytic activities [TOF = 133–193 s−1 (1), 90.9–126 s−1 (2)] for ring opening metathesis polymerisation (ROMP) of norbornene (NBE) at 25 °C to afford ring-opened polymers not only with high cis-(93–98%) selectivity, but also with exclusive syndiotactic stereo-regularity. These polymerisations proceeded in a living manner under optimised conditions. The activity improved at 50 °C with decreasing in the cis specificity. Synthesis and the structural analysis of the fluorinated alkoxide complexes, [V(N-2,6-X2C6H3)(CHSiMe3){OC(CF3)3}(NHC)] [X = F (3), Cl (4)] have been studied, and the difluorophenylimdo complex (3) exhibited comparable catalytic activities for ROMP of NBE. The cis selectivities in the resultant polymers prepared by 3 were low (88%) and the selectivities in the ring-opened poly(tetracyclododecene) (TCD) were low (61–66%) compared to those in poly(NBE)s. The resultant ring opened poly(TCD) possessed syndiotactic stereo-regularity confirmed by DSC thermogram of the hydrogenated polymers.
1. Introduction
Olefin metathesis is an efficient method for synthesis of the fine chemicals, and advanced polymers.1–5 Ring opening metathesis polymerisation (ROMP) especially offers access to the functional polymeric materials.2 In this catalysis, metal-carbene (alkylidene) complexes act as the key intermediate,3–5 and development of the alkylidene catalysts with earth abundant metal has thus been the important subject in long term. The PMe3-supported vanadium–alkylidenes, [V(N-2,6-X2C6H3)(CHSiMe3)(OR)(PMe3)2] [X = F, Cl; R = C6F5, C6Cl5, C(CF3)3, Scheme 1, display high potentials in the ROMP of cyclic olefins,4,6 particularly in Z specific ROMP at 80 °C,6a,b in the high temperature controlled ROMP of low strain cyclic olefins (cycloheptene, cis-cyclooctene),6d and in the Z/E specific synthesis of bottlebrush polymers.6g | ||
| Scheme 1 Reported (arylimido)vanadium–alkylidene complexes supported by PMe3 or NHC ligands,6 and cis, syndiospecific ROMP of NBE by catalysts 1 and 2.6f | ||
N-Heterocyclic carbenes (NHC) have been known to stabilise high oxidation state organometallic complexes with early transition metals,7,8 as demonstrated e.g. for molybdenum, tungsten,3g,i,7 and vanadium.6f,8 Recently, we communicated that ROMP of norbornene (NBE) by the vanadium–alkylidene NHC complexes containing perhalogenated phenoxide ligands, [V(N-2,6-Cl2C6H3)(CHSiMe3)(OC6X5)(NHC)] [X = F (1), Cl (2); NHC = 1,3-bis-(2,6-dimethylphenyl)-imidazol-2-ylidene (IXy)] gave cis-, syndiotactic ring-opened polymers (Scheme 1).6f High stereo-control in ROMP has also been reported for cationic molybdenum and tungsten imido or oxo alkylidene NHC initiators.7
We herein report the ROMP of NBE using catalysts 1 and 2 affording highly cis ring-opened polymers with exclusive syndiotactic stereoregularity even at 50 °C. The controlled (living) polymerisation could be demonstrated under optimised (diluted) conditions. Moreover, synthesis and structural analysis of the fluorinated alkoxide complexes, [V(N-2,6-X2C6H3)(CHSiMe3){OC(CF3)3}(NHC)] [X = F (3), Cl (4)], and their use as the catalysts for ROMPs of NBE, tetracyclododecene (TCD) has been explored.
2. Results and discussion
2.1. Ring opening metathesis polymerisation of norbornene (NBE) by [V(N-2,6-Cl2C6H3)(CHSiMe3)(OC6X5)(NHC)] [X = F (1), Cl (2)]
As communicated previously,6f the ring opening metathesis polymerisations (ROMPs) of NBE by the vanadium catalysts (1, 2) reaches completion within 1 or 2 minutes (runs 1, 4, 8, Table 1). Therefore, the ROMPs of NBE were conducted in benzene at 25 °C and 50 °C, respectively, applying different conditions (NBE/benzene = 2.12 mmol/4.8 mL or 4.24 mmol/9.6 mL, initial NBE conc. 0.44 mmol mL−1]. Table 1 summarises the results.| Run | Cat. (μmol) | NBE/mmol | PMe3b/equiv. | Temp./°C | Time/min | Yield/% | TONc | TOFc/h−1 (s−1) | M n × 10−5 | M w/Mnd | cis /% |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: NBE 2.12 mmol, benzene total 4.8 mL, or NBE 4.24 mmol, benzene total 9.6 mL. b Molar ratio to V. c TON (turnovers) = NBE reacted (mol)/mol-V, TOF = TON/time. d By GPC data in THF vs. polystyrene standards. e cis percentage (%) estimated by 1H NMR spectra (selected data are shown in ESI). f Cited from ref. 6f. g Conditions: NBE 200 mg (2.12 mmol) benzene total 9.6 mL (initial NBE conc. 0.22 mmol mL−1). PMe3 was premixed in benzene at 25 °C for 1 min. | |||||||||||
| 1f | 1 (0.3) | 2.12 | — | 25 | 1 | 97 | 6860 | 412![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (114) 000 (114) |
4.72 | 2.26 | 93 |
| 2 | 1 (0.3) | 2.12 | — | 50 | 1 | >99 | 7040 | 423000 (117) |
6.21 | 1.98 | 81 |
| 3 | 1 (0.1) | 2.12 | — | 25 | 1 | 55 | 11600 |
695000 (193) |
6.96 | 2.29 | 94 |
| 4 | 1 (0.1) | 2.12 | — | 25 | 2 | 88 | 18600 |
558000 (155) |
7.51 | 2.89 | 94 |
| 5 | 1 (0.1) | 2.12 | — | 50 | 1 | 59 | 12500 |
752000 (209) |
7.76 | 2.42 | 86 |
| 6 | 1 (0.1) | 2.12 | — | 50 | 2 | 91 | 19300 |
580000 (161) |
8.96 | 3.02 | 86 |
| 7 | 2 (0.5) | 2.12 | — | 25 | 1 | 90 | 3800 | 228000 (63.4) |
3.73 | 1.65 | 97 |
| 8 | 2 (0.5) | 2.12 | — | 50 | 1 | 98 | 4140 | 249000 (69.0) |
4.67 | 1.73 | 91 |
| 9 | 2 (0.3) | 2.12 | — | 25 | 1 | 77 | 5450 | 327000 (90.9) |
4.44 | 1.86 | 97 |
| 10 | 2 (0.3) | 2.12 | — | 50 | 1 | 88 | 6200 | 372000 (103) |
5.44 | 2.07 | 91 |
| 11f | 2 (0.1) | 2.12 | — | 25 | 1 | 42 | 8830 | 530000 (147) |
5.66 | 2.16 | 98 |
| 12f | 2 (0.1) | 2.12 | — | 25 | 2 | 62 | 13300 |
399000 (111) |
6.62 | 2.19 | 97 |
| 13f | 1 (0.5) | 4.24 | — | 25 | 1 | 96 | 8150 | 489000 (136) |
6.17 | 2.15 | 93 |
| 14 | 1 (0.5) | 4.24 | — | 25 | 1 | 94 | 7990 | 479000 (133) |
5.86 | 2.12 | 92 |
| 15 | 1 (0.3) | 4.24 | — | 25 | 1 | 75 | 10500 |
633000 (176) |
8.92 | 2.05 | 95 |
| 16 | 1 (0.3) | 4.24 | — | 50 | 1 | 77 | 10900 |
656000 (182) |
7.11 | 2.52 | 88 |
| 17f | 2 (0.5) | 4.24 | — | 25 | 1 | 75 | 6400 | 384000 (107) |
6.64 | 1.77 | 97 |
| 18 | 2 (0.5) | 4.24 | — | 50 | 1 | 75 | 6390 | 384000 (107) |
6.12 | 1.96 | 89 |
| 19 | 2 (0.3) | 4.24 | — | 25 | 1 | 53 | 7540 | 452000 (126) |
6.55 | 1.76 | 95 |
| 20 | 2 (0.3) | 4.24 | — | 50 | 1 | 60 | 8460 | 508000 (141) |
4.31 | 2.26 | 90 |
| 21g | 2 (0.5) | 2.12 | — | 50 | 3 | 54 | 2290 | 45900 (12.7) |
5.29 | 1.66 | 88 |
| 22g | 2 (0.5) | 2.12 | 1.0 | 50 | 3 | 73 | 3080 | 61600 (17.1) |
6.42 | 1.83 | 76 |
| 23g | 2 (0.5) | 2.12 | 3.0 | 50 | 3 | 70 | 2950 | 59000 (16.4) |
6.29 | 1.95 | 72 |
| 24g | 2 (0.5) | 2.12 | 10 | 50 | 3 | 68 | 2890 | 57800 (16.0) |
5.72 | 2.09 | 64 |
It should be noted that the pentafluoro phenoxide complex (1) showed higher catalytic activities (TOF 193 s−1, run 3) than the pentachloro phenoxide (2, TOF 147 s−1, run 11) when ROMP was conducted at 25 °C. The observed catalytic activities with 1 and 2 were at a similar level than those observed for the PMe3-supported catalyst, [V(N-2,6-Cl2C6H3)(CHSiMe3)(OC6F5)–(PMe3)2] (TOF 153 s−1), conducted under the similar conditions.6a The resultant poly(NBE)s prepared at 25 °C displayed high cis contents [92–94% (by 1), 95–98% (by 2)] and possessed high molecular weights with unimodal molecular weight distributions. The cis selectivity of 2 was slightly higher than that of 1.
It was revealed that the catalytic activity slightly increased when the ROMPs (by 1, 2) were conducted at 50 °C, whereas the cis selectivity apparently decreased in all cases (runs 5, 6, 8, 10). The observed catalytic activity by 2 at 50 °C was improved by in situ addition of small amount of PMe3 (1–3 equiv.), whereas the cis selectivity decreased by addition of PMe3. The further addition suppressed the activity and the cis selectivity (runs 21–24).
Fig. 1a shows 13C NMR spectrum (in CDCl3 at 50 °C) of the hydrogenated polymer sample (run 19) prepared by treating with 4-MeC6H4SO2NHNH2 (excess) in toluene at 130 °C,9,10 and Fig. 1b showed the extended resonances around 31–32 ppm for the estimation of tacticity of the polymers prepared under different conditions (catalyst, temperature); the other selected spectra are shown in ESI.† Note that the resultant hydrogenated polymer prepared by 2 showed a sole resonance at 31.7 ppm, clearly suggesting that the resultant polymer possessed exclusive syndiotactic stereoregularity.7e,10 Similar exclusive selectivity was also found by catalyst 1. Noteworthy from Fig. 1b that the exclusive selectivity was not affected by the polymerisation temperature (25 or 50 °C), since the spectra showed sole resonance irrespective of catalysts (1 or 2) or the polymerisation temperature conducted (25 or 50 °C). The DSC thermograms of the hydrogenated polymers revealed melting temperature of 136.2 °C (run 5, Fig. S17, ESI†) and 137.8 °C (run 9, Fig. S18, ESI†), respectively, in line with a syndiotactic stereobase, as reported previously.10
 | ||
| Fig. 1 13C NMR spectra (in CDCl3 at 50 °C) for hydrogenated ring opened poly(NBE)s prepared by (a) [V(N-2,6-Cl2C6H3)(CHSiMe3)(OC6Cl5)(NHC)] (2) at 25 °C (run 19) and (b) expanded charts (31.7–31.8 ppm) for the polymer samples [from top to bottom: atactic sample for ref. 6b prepared by 2 at 25 °C (run 19), 50 °C (run 18), by [V(N-2,6-Cl2C6H3)(CHSiMe3)(OC6F5)(NHC)] (1) at 25 °C (run 15), 50 °C (run 16), respectively]. | ||
In order to explore the catalyst performance and the polymerisation kinetics in more details, polymerisations were conducted under dilute conditions (initial NBE conc. 0.11 mmol mL−1) at low catalyst loading (0.20 μmol) at 25 °C. The detailed results are summarised in Table S1, ESI.†
It was revealed that (TON) and the Mn values in the resultant polymers increased upon increasing the polymer yields (TON, turnover numbers based on NBE consumed) linearly in both polymerisation runs by catalysts 1 (Fig. 2a) and 2 (Fig. 2b), whereas the Mw/Mn values seemed increasing over the time course probably due to increase in the viscosity of the reaction mixture. The similar trends, linear relationships between the polymer yields and the Mn values, were observed, when these polymerisations were conducted under higher catalyst loadings (0.30 μmol) at 25 and even 50 °C (Fig. S1, and the data are summarised in Table S2, ESI†). These results clearly suggest that these polymerisations by catalysts 1, 2 proceeded in a living manner without significant catalyst deactivation.11
 | ||
| Fig. 2 Plots of Mn values vs. polymer yields (TON, turnover numbers based on polymer yield) in ROMP of norbornene (NBE) by (a) catalyst 1, (b) catalyst 2. Detailed data are shown in Table S1, ESI.† | ||
2.2. Synthesis of [V(N-2,6-X2C6H3)(CHSiMe3){OC(CF3)3}(NHC)] [X = F (3), Cl (4)] and their use as catalysts for ROMP of NBE, tetracyclododecene (TCD).
It was demonstrated that the PMe3-supported fluorinated alkoxide, [V(N-2,6-Cl2C6H3)(CHSiMe3){OC(CF3)3}(PMe3)2] (Scheme 1), exhibited exclusive cis specificity in the ROMP of NBE and the high cis-specificity did not decrease even at 80 °C in the presence of PMe3. It was assumed that both the small arylimido ligand and the large fluorinated alkoxide ligands are responsible for the observed stereospecificity by favouring an enesyn addition of NBE to a syn-alkylidene. We thus have an interest in the question whether the corresponding alkylidene NHC complexes, [V(N-2,6-X2C6H3)(CHSiMe3){OC(CF3)3}(NHC)] [X = F (3), Cl (4)], would display the similar high cis specificity or not.These complexes (3 and 4) could be prepared by treating the corresponding dialkyl complexes, [V(N-2,6-X2C6H3)–(CH2SiMe3)2],6g with NHC [1,3-bis-(2,6-dimethylphenyl)-imidazol-2-ylidene (IXy)] in benzene at 50 °C for 8 h (through α-hydrogen abstraction, Scheme 2), whereas the similar reactions for syntheses of 1 and 2 from [V(N-2,6-Cl2C6H3)–(CH2SiMe3)2(OC6X5)] completed within 1 h in benzene at room temperature. The resultant complexes were characterized by NMR spectroscopy and elemental analysis. As shown in Fig. 3 (shown below), the structures of 3 and 4 were determined by X-ray crystallographic analysis.
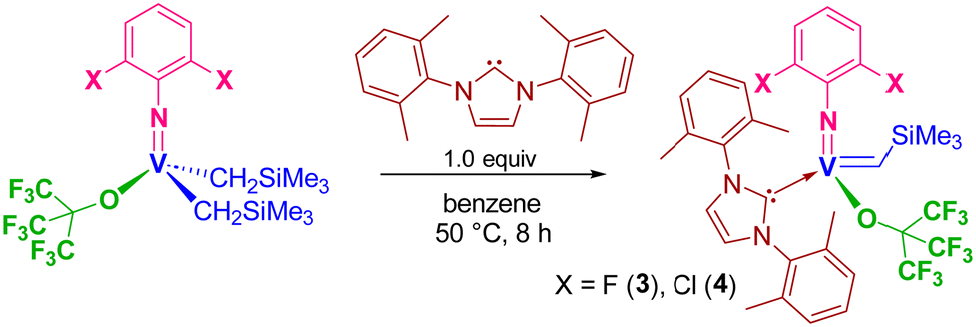 | ||
| Scheme 2 Synthesis of [V(N-2,6-X2C6H3)(CHSiMe3){OC(CF3)3}(NHC)] [X = F (3), Cl (4)]. | ||
 | ||
| Fig. 3 ORTEP drawings for [V(N-2,6-X2C6H3)(CHSiMe3)[OC(CF3)3](NHC)] [X = F (3, left), Cl (4, right)]. Hydrogen atoms were omitted for clarity and thermal ellipsoids are drawn at 30% probability level. Additional data including crystal data and collection parameters for structural analysis are shown in ESI.† | ||
The structural analyses revealed that these complexes fold a distorted tetrahedral geometry around vanadium [N–V–C(7), O–V–C(15), N(1)–V(1)–C(15), C(7)–V(1)–O(1) = 108.98(18)°, 111.05(13)°, 102.43(14)°, 111.76(18)° (complex 3); 114.2(3)°, 105.98(17)°, 103.95(17)°, 107.6(3)° (complex 4), respectively]. The V–N–C bond angles in the arylimido ligand were rather linear, only slightly bent [164.8(3)°, 175.1(4)° for complexes 3, 4, respectively] with a rather short V–N distance [1.673(3) Å, 1.652(4) Å for complexes 3, 4, respectively]. These values are comparable to those reported for [V(N-1-adamantyl)(CHSiMe3)–(OC6F5)(PMe3)2] [173.3(3)°, 1.660(3) Å, respectively]12 and [V(N-adamantyl)(CHSiMe3)(CH2SiMe3)(NHC-DIP)] [165.0(2)°, 1.637(2) Å, respectively; NHC-DIP = 1,3-bis-(2,6-diisopropylphenyl)-imidazol-2-ylidene].8b The V–C distances in the alkylidene [1.829(4), 1.784(7) Å, for complexes, 3, 4, respectively] and NHC [2.124(3), 2.142(4) Å, respectively] are close to that found in [V(N-adamantyl)(CHSiMe3)(CH2SiMe3)–(NHC-DIP)]8b [1.829(3), 2.172(2) Å, respectively]. These findings are also corresponded to the observation of broad resonances ascribed to the alkylidene carbon in the 13C NMR spectra (341.7, 340.3 ppm, respectively), clearly indicating the presence of vanadium-alkylidene bond, whereas the NHC ligand coordinates to vanadium as a neutral donor (Table 2).
| X = F (3) | X = Cl (4) | |
|---|---|---|
| a CCDC 2358163 (complex 3) and 2358164 (complex 4) contain the supplementary crystallographic data in detail. | ||
| Bond distances (Å) | ||
| V(1)–N(1) | 1.673(3) | 1.652(4) |
| V(1)–C(7) | 1.829(4) | 1.784(7) |
| V(1)–O(1) | 1.882(3) | 1.946(5) |
| V(1)–C(15) | 2.124(3) | 2.142(4) |
| Bond angles (°) | ||
| N(1)–V(1)–C(15) | 102.43(14) | 103.95(17) |
| N(1)–V(1)–O(1) | 119.34(15) | 117.4(2) |
| N(1)–V(1)–C(7) | 108.98(18) | 114.2(3) |
| C(7)–V(1)–C(15) | 101.44(17) | 106.8(2) |
| C(7)–V(1)–O(1) | 111.76(18) | 107.6(3)) |
| O(1)–V(1)–C(15) | 111.05(13) | 105.98(17) |
| C(1)–N(1)–V(1) | 164.8(3) | 175.1(4) |
The ROMPs of NBE by catalysts 3 and 4 were conducted in toluene, and the selected results are summarised in Table 3. It was revealed that the 2,6-dichlorophenyl complex (4) exhibited low catalytic activities even under the high initial NBE concentration conditions (run 25–28), whereas the resultant polymers possess olefinic double bond with high cis selectivity (92–93%); nevertheless, the observed cis selectivity in the olefinic double bonds was lower than that by catalyst 2. The activity improved at 50 °C (run 29), whereas the cis selectivity slightly decreased upon increase in the reaction temperature.
| Run | Cat. (μmol) | Temp./°C | Time | Yield/mg | TONb | TOFb/h−1 (s−1) | cis /% |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: NBE 200 mg (2.12 mmol), toluene 1.2 mL. b TON (turnovers) = NBE reacted (mol)/mol-V. TOF (turnover frequency) = TON/h. c cis percentage (%) estimated by 1H NMR spectra. d Benzene 4.8 mL in place of toluene 1.2 mL. e Yield > 99%. | |||||||
| 25d | 4 (10) | 25 | 0.5 h | Trace | |||
| 26 | 4 (10) | 25 | 3 h | 13.8 | 15 | 5 | 93 |
| 27 | 4 (10) | 25 | 16 h | 35 | 37 | 2 | 92 |
| 28 | 4 (10) | 25 | 21 h | 42.3 | 45 | 2 | 93 |
| 29 | 4 (10) | 50 | 1.0 h | 16.6 | 18 | 18 | 90 |
| 30 | 3 (5.0) | 25 | 1.0 min | 95 | 201 | 720000 (200) |
88 |
| 31 | 3 (5.0) | 25 | 1.5 min | 106 | 225 | 539000 (150) |
88 |
| 32 | 3 (10) | 50 | 0.20 min | 216e | 220 | 66000 (18.3) |
79 |
| 33 | 3 (5.0) | 50 | 0.25 min | 40 | 84 | 121000 (337) |
81 |
| 34 | 3 (10) | 80 | 0.10 min | 202e | 200 | 126000 (35.0) |
65 |
In contrast, the 2,6-difluorophenylimido complex (3) exhibited superior catalytic activity at 25 °C (TOF = 150–337 s−1), and the activity increased at 50 °C (run 33); the polymerisation run at 80 °C completed even after 6 s (run 34). However, the cis selectivity decreased upon increasing the polymerisation temperature. In all cases, the resultant polymers were hardly soluble in THF or chloroform for GPC analysis, suggesting that the resultant polymers possessed very high molecular weights.6f
Table 4 summarises the results in ROMPs of tetracyclododecene (TCD) by catalysts 3 and 4 in benzene at 25 °C. As observed in the ROMPs of NBE, catalyst 4 showed low catalytic activity, whereas the polymerisation with high catalyst loading resulted in poly(TCD) with a rather high trans content (trans 82%, runs 35, 36). In contrast, catalyst 3 showed much higher catalytic activity compared to 4, affording the ring opened polymers with substantial cis selectivity (61–66%). We assume that these results are probably due to rotational isomer (syn/anti), once formed syn isomer would convert to (thermodynamically stable) anti form before TCD insertion; these probably affect the cis specificity in these catalyses. As observed in the ROMPs of TCD by the other vanadium-alkylidene catalysts,6a,b as well as niobium–alkylidene catalysts,13 the resultant polymers were hardly soluble in THF (chloroform) for GPC analysis.
| Run | Cat. (μmol) | Time | Yield/mg | TONb | TOFb/h−1 | cis /% |
|---|---|---|---|---|---|---|
| a Reaction conditions: TCD 340 mg (2.12 mmol), benzene 1.2 mL. b TON (turnovers) = TCD reacted (mol)/mol-V. TOF (turnover frequency) = TON/h. c cis percentage (%) estimated by 1H NMR spectra. | ||||||
| 35 | 4 (5.0) | 24 h | 17.9 | 22.3 | 0.9 | 18 |
| 36 | 4 (10) | 24 h | 100.3 | 62.6 | 2.6 | 18 |
| 37 | 3 (5.0) | 4 min | 38.5 | 48.0 | 721 | 61 |
| 38 | 3 (10) | 3 min | 61.0 | 38.1 | 761 | 62 |
| 39 | 3 (10) | 4 min | 83.4 | 52.0 | 781 | 64 |
| 40 | 3 (20) | 3 min | 113.8 | 35.5 | 710 | 64 |
| 41 | 3 (20) | 3 min | 116.0 | 36.2 | 724 | 66 |
Nevertheless, selected poly(TCD)s were successfully hydrogenated by 4-MeC6H4SO2NHNH2 (excess) in toluene at 130 °C.9,10 The corresponding DSC thermograms showed that the resultant hydrogenated polymers are amorphous and possessed sole glass transition temperature at 168.6–169.6 °C (Fig. 4) in all cases, clearly suggesting that the resultant polymers possess syndiotactic stereobase.9b,10
 | ||
| Fig. 4 DSC thermograms for hydrogenated ring opened poly(TCD) prepared by catalysts 3 and 4. | ||
3. Conclusions
We have shown that perhalogenated phenoxide alkylidene complexes, V(N-2,6-Cl2C6H3)(CHSiMe3)(OC6X5)(NHC)6f [X = F (1), Cl (2)], exhibited remarkable catalytic activities [TOF = 133–193 s−1 (1), 90.9–126 s−1 (2)] for ring-opening metathesis polymerisation (ROMP) of norbornene (NBE), and the resultant ring opened polymers prepared at 25 °C possessed high cis selectivity (93–98%); the pentachloro-phenoxide catalyst (2) showed rather higher cis specificity (95–98%). Under diluted polymerisation conditions, linear relationships between the Mn values and the polymer yields were demonstrated (Fig. 1), strongly suggesting that these polymerisations proceeded in a living manner without significant catalyst decomposition. The catalytic activities by 1, 2 slightly increased at 50 °C, whereas the cis specificity decreased upon increasing the polymerisation temperature. Further increase in the activity was observed upon addition of 1.0 equiv. of PMe3 with decreasing the cis specificity. The resultant polymers possessed exclusive syndiotactic stereoregularity irrespective of the polymerisation temperature on the basis of 13C NMR spectra for the hydrogenated polymers (Fig. 2).Synthesis and structural analysis of the perfluorinated alkoxide complexes, [V(CHSiMe3)(N-2,6-X2C6H3){OC(CF3)3}(NHC)] [X = F (3), Cl (4)], have been studied, and these complexes possessed a distorted tetrahedral geometry around vanadium containing NHC ligand as a neutral donor. The 2,6-difluorophenylimido complex (3) showed comparable catalytic activities for ROMP of NBE conducted at 25 °C, whereas the 2,6-dichlorophenylimido complex (4) showed extremely the low catalytic activities. The resultant polymers possessed high cis selectivities (88–93%) which were lower than those conducted by 1, 2. The activity by 3 increased at 50 °C with decreasing the cis specificity. The resultant ring opened polymers prepared by ROMP of tetracyclododecene (TCD) by 4 possessed highly trans selectivity (82%) in the olefinic double bond, whereas polymers prepared by 3 possessed cis (61–66%). We assume that these results are probably due to rotational isomer (syn/anti), once formed syn isomer would convert to (thermodynamically stable) anti form before TCD insertion; these probably affect the cis specificity in these catalyses. The resultant poly(TCD) possessed syndiotactic stereoregularity confirmed by DSC thermograms for hydrogenated polymers.
As described above, the cis selectivity in the resultant poly(NBE) prepared by the fluorinated alkoxides (3,4) at 25 °C were comparable or rather low (cis 88–93%, Table 3) compared to those prepared by the perhalogenated phenoxides (cis 91–97%, Table 1) under the same conditions. The observed fats are clear contrast to those reported in the PMe3-supported catalysts.6a,b As discussed in the ROMPs of TCD by catalysts 3 and 4, isomerization of syn rotational isomer to the (thermally stable) anti form can be thus considered before coordination of cyclic olefins. These should be clarified as the future subject.
Through this research, we have demonstrated that the present vanadium–alkylidene catalysts show promising potential for ring opening metathesis polymerisation and the ligand modification plays an important role for control of cis/trans and the tacticity control. In particular, on the basis of results by the fluorinated alkoxide catalysts (3, 4), we are currently exploring more ligand modification for development of catalysts affording exclusive cis, syndiotactic polymers. Moreover, these catalysts also showed promising characteristics in ring closing metathesis reactions.14 We highly hope to introduce further results in the near future.
4. Experimental
4.1. General procedures
The experiments were conducted under a nitrogen atmosphere on Vacuum Atmospheres Drybox. Anhydrous grade of octane, dichloromethane, n-hexane, benzene, and toluene (Kanto Chemical Co., Inc.) were transferred into bottles containing a mixture of molecular sieves (3A 1/16, 4A 1/8, and 13 × 1/16) inside the drybox. These solvents were purified by flushing an alumina short column under nitrogen stream in the drybox prior to use. [V(N-2,6-Cl2C6H3)(CH2SiMe3)2{OC(CF3)3}],6a [V(N-2,6-Cl2C6H3)(CHSiMe3)(OC6X5)(NHC)]6f [X = F (1), Cl (2)] and NHC (IXy) ligand6f used were prepared following the previously reported procedures. Norbornene (NBE, Tokyo Chemical Industry Co., Ltd.) was distilled in the presence of sodium before use. Tetracyclododecene (TCD, Zeon corporation) were purified by passing through alumina short column, and then dried in the presence of molecular sieves. p-Toluenesulfonyl hydrazide (4-MeC6H4SO2NHNH2, Sigma-Aldrich Co. LLC.) for hydrogenation of polymers was used without further purification.All 1H, 13C, 19F, 31P and 51V NMR spectra were recorded on a Bruker AV500 spectrometer (500.13 MHz for 1H, 125.77 MHz for 13C, 470.54 MHz for 19F, 202.45 MHz for 31P and 131.55 MHz for 51V). All spectra were obtained in the solvent indicated at 25 °C unless otherwise noted. Chemical shifts are given in ppm and are referenced to SiMe4 (δ 0.00 ppm, 1H, 13C), CFCl3 (δ 0.00, 19F), and VOCl3 (δ 0.00, 51V). Coupling constants and half-width values, Δν1/2, are given in Hz. The ratio of cis/trans of polymers were calculated by the integration of 1H NMR spectra. 13C NMR spectra of hydrogenated poly(NBE)s were recorded on a Bruker Avance III 500 MHz spectrometer (500.13 MHz for 1H, 125.77 MHz for 13C) with CryoprobeTM DCH 500/3 at 50 °C (reference chloroform at 77.2 ppm for 13C).
Gel-permeation chromatography (GPC) was performed to estimate the molecular weights (Mw, Mn) and their distributions, HPLC grade THF (degassed prior to use) was used as eluent. GPC was performed at 40 °C on a Shimadzu SCL-10A using a RID-10A detector (Shimadzu Co. Ltd.) in THF (containing 0.03 wt% of 2,6-di-tert-butyl-p-cresol, flow rate 1.0 mL min−1). GPC columns (ShimPAC GPC-806, 804 and 802, 30 cm × 8.0 mm diameter, spherical porous gel made of styrene/divinylbenzene copolymer, ranging from <102 to 2 × 107 MW) were calibrated versus polystyrene standard samples.
4.2. Synthesis of [V(N-2,6-F2C6H3)(CH2SiMe3)2{OC(CF3)3}]6g
Into a n-hexane solution (30 mL) containing V(N-2,6-F2C6H3)(CH2SiMe3)3 (501 mg, 1.14 mmol) was added (CF3)3COH (538 mg, 2.28 mmol, 2.0 equiv.) at −30 °C. The solution was then gradually warmed to room temperature and was stirred for 12 h. The solution was passed through a Celite pad and washed with n-hexane. The combined filtrate and the wash were taken to dryness in vacuo resulted in dark-brown oil product. Yield: 635 mg (1.08 mmol, 94.9% yield). 1H NMR (500 MHz, C6D6, 25 °C): δ 6.40 (t, 2H, J = 8.00 Hz, Ar-H), 6.26 (t, 1H, J = 7.20 Hz, Ar-H), 3.03 (br, 4H, V–CH2SiMe3), 0.11 (s, 18H, SiMe3). 19F NMR (C6D6, 25 °C): δ −74.11 (s), −117.49 (s). 51V NMR (C6D6, 25 °C): δ 725.32 (Δν1/2 = 291 Hz).4.3. Synthesis of [V(CHSiMe3)(N-2,6-F2C6H3){OC(CF3)3}(NHC)] (3)
Into a benzene solution (10 mL) containing [V(N-2,6-F2C6H3)(CH2SiMe3)2{OC(CF3)3}] (635 mg, 1.08 mmol), NHC [NHC = 1,3-bis-(2,6-dimethylphenyl)-imidazol-2-ylidene, 298 mg, 1.08 mmol, 1.0 equiv.] was added at 50 °C, and the reaction mixture was further stirred for 8 h. The solution was then placed to a rotary evaporator in the drybox to remove volatiles. The benzene solution containing the resultant residue was passed through a Celite pad. The filter cake was washed twice with benzene, and volatiles in the combined filtrate and the wash were removed in vacuo. The resultant solid was then dissolved in a minimum amount of benzene, and the solution was added dropwise into a stirred cold n-hexane solution (pre-cooled at −30 °C) resulted in dark-brown solid. Yield: 261 mg (0.336 mmol, 31.1% yield). 1H NMR (500 MHz, C6D6, 25 °C): δ 16.24 (s, 1H, V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHSiMe3), 6.84 (s, 6H, Ar-H), 6.41 (t, 2H, J = 7.52 Hz, Ar-H), 6.27 (t, 1H, J = 7.00 Hz, Ar-H), 5.93 (s, 2H, NCH), 2.07 (s, 6H, Ar-CH3), 1.85 (s, 6H, Ar-CH3), 0.26 (s, 9H, SiMe3). 13C{1H} NMR (C6D6, 25 °C): δ 340.3 (br), 161.5, 159.5, 138.7, 135.6, 135.3, 134.7, 133.6, 130.9, 129.5, 128.7, 125.6, 123.2, 123.0, 122.3, 121.7 (t), 120.9, 118.6, 110.4 (dd), 84.5 (br), 17.8, 17.6, 1.5. 19F NMR (C6D6, 25 °C): δ −74.59 (s), −118.80 (s). 51V NMR (C6D6, 25 °C): δ 166.53 (Δν1/2 = 230 Hz). Anal. calcd. for C33H33F11N3OSiV (+0.08 × n-hexane): C, 51.39; H, 4.40; N, 5.37. Found: C, 51.27; H, 4.58; N, 5.39.
CHSiMe3), 6.84 (s, 6H, Ar-H), 6.41 (t, 2H, J = 7.52 Hz, Ar-H), 6.27 (t, 1H, J = 7.00 Hz, Ar-H), 5.93 (s, 2H, NCH), 2.07 (s, 6H, Ar-CH3), 1.85 (s, 6H, Ar-CH3), 0.26 (s, 9H, SiMe3). 13C{1H} NMR (C6D6, 25 °C): δ 340.3 (br), 161.5, 159.5, 138.7, 135.6, 135.3, 134.7, 133.6, 130.9, 129.5, 128.7, 125.6, 123.2, 123.0, 122.3, 121.7 (t), 120.9, 118.6, 110.4 (dd), 84.5 (br), 17.8, 17.6, 1.5. 19F NMR (C6D6, 25 °C): δ −74.59 (s), −118.80 (s). 51V NMR (C6D6, 25 °C): δ 166.53 (Δν1/2 = 230 Hz). Anal. calcd. for C33H33F11N3OSiV (+0.08 × n-hexane): C, 51.39; H, 4.40; N, 5.37. Found: C, 51.27; H, 4.58; N, 5.39.
4.4. Synthesis of [V(CHSiMe3)(N-2,6-Cl2C6H3){OC(CF3)3}(NHC)] (4)
Into a benzene solution (10 mL) containing [V(N-2,6-Cl2C6H3)(CH2SiMe3)2{OC(CF3)3}] (423 mg, 0.682 mmol), NHC [NHC = 1,3-bis-(2,6-dimethylphenyl)-imidazol-2-ylidene, 188 mg, 0.680 mmol, 1.0 equiv.] was added at 50 °C, and the solution was stirred for 8 h. The solution was then placed to a rotary evaporator in the drybox to remove volatiles. The benzene solution containing the resultant residue was passed through a Celite pad. The filter cake was washed twice with benzene, and volatiles in the combined filtrate and the wash were removed in vacuo. The resultant solid was then dissolved in a minimum amount of benzene, and the solution was added dropwise into a stirred cold n-hexane solution (pre-cooled at −30 °C) resulted in dark-brown solid. Yield 158 mg (0.195 mmol, 28.7% yield). 1H NMR (500 MHz, C6D6, 25 °C): δ 15.85 (s, 1H, VCHSiMe3), 6.93–6.79 (m, 8H, Ar-H), 6.25 (t, 1H, J = 7.40 Hz, Ar-H), 5.94 (s, 2H, NCH), 2.03 (s, 6H, Ar-CH3), 1.89 (s, 6H, Ar-CH3), 0.17 (s, 9H, SiMe3). 13C NMR (C6D6, 25 °C): δ 341.7 (br), 155.2 (br), 138.6, 135.5, 135.4, 134.3, 133.0, 131.4, 129.6, 129.0, 128.5, 128.3, 127.3, 125.7, 123.3, 122.9, 122.3, 121.0, 118.7, 84.4 (br), 18.1, 17.9, 1.5. 19F NMR (C6D6, 25 °C): δ −73.74 (s). 51V NMR (C6D6, 25 °C): δ 196.00 (Δν1/2 = 242 Hz). Anal. calcd. for C33H33Cl2F9N3OSiV (+0.12 × n-hexane): C, 49.46; H, 4.27; N, 5.13. Found: C, 49.33; H, 4.35; N, 5.35.
4.5. ROMP of NBE and TCD
Into a 15-mL vial, NBE (4.24, 2.12 or 1.06 mmol) or TCD (2.12 mmol) was dissolved in benzene (4.8 or 9.6 mL). A prescribed amount of catalyst dissolved in benzene was added to the above mixture to start the polymerisation for a prescribed time. The polymerisation was then quenched by addition of benzaldehyde in excess. The resultant mixture was stirred for an additional 30 min at room temperature for completion, then poured into 100 mL of methanol. The resultant solid was collected by filtration, washed with methanol, and then dried in vacuo.Poly(NBE): 1H NMR (CDCl3 at 25 °C): δ 5.35 (1H, trans), 5.21 (1H, cis) 2.80 (1H, cis), 2.44 (1H, trans), 1.89–1.80 (3H), 1.36 (2H), 1.04 (1H). 13C NMR (CDCl3 at 25 °C): δ 134.0, 133.2, 43.6, 42.8, 38.8, 38.6, 33.4, 33.1, 32.5, 32.3. Detailed assignments are shown in the ESI† (Fig. S2 and S3, ESI†).
Poly(TCD): 1H NMR (CDCl3 at 25 °C): δ 5.51 (2H), 2.97–2.70 (2H), 2.28–2.20 (2H), 2.02 (2H), 1.73–1.65 (1H), 1.43–1.28 (4H), 1.05 (2H), 0.91 (1H). 13C NMR (CDCl3 at 25 °C): δ 135.2, 131.2, 130.4, 53.0, 48.6, 46.2, 37.9, 36.0, 29.8.
4.6. Hydrogenation of poly(NBE) and poly(TCD).9
The resultant polymers (120 mg) were transferred to a Schlenk tube and dissolved in toluene (20 mL). 4-MeC6H4SO2NHNH2 (4 equiv.) was added. The Schlenk tube was then placed into an oil bath heated to 130 °C, and the solution mixture was stirred for 12 h. After the reaction was complete, the mixture was cooled to room temperature and poured into 100 mL of methanol. The resultant solid was collected by filtration, washed with methanol, and then dried in vacuo.Author contributions
KN designed the project (conceptualization, catalyst design, methodology, supervision) including funding acquisition and project administration. MMB contributed to the conceptualization and the catalyst design. SK and JS conducted synthesis of complexes 3 and 4, polymerisation, analysis of polymers in investigation and YK prepared complexes 1, 2. DS helped crystallographic analysis. KN wrote the original manuscript, and both MMB and KN revised the manuscript. All authors have discussed the results and approved the final version of the manuscript.Data availability
Data is contained within the article and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was partly supported by Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS, Grant 21H01942). MRB thanks the Deutsche Forschungsgemeinschaft DFG (project ID 358283783 – CRC 1333/2 2022). The authors express their thank to Dr Shigetaka Hayano (Zeon Co.) for careful 13C NMR spectral analysis of the hydrogenated poly(NBE) sample.Notes and references
- For selected example (book), see: (a) Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, Germany, 2003 Search PubMed; (b) Olefin Metathesis: Theory and Practice, ed. K. Grela, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2014 Search PubMed; (c) Handbook of Metathesis, ed. R. H. Grubbs and A. G. Wenzel, Wiley-VCH, Weinheim, Germany, 2nd edn, 2015, vol. 1 Search PubMed.
- For selected example (reviews in polymerisation), see: (a) M. R. Buchmeiser, Chem. Rev., 2000, 100, 1565–1604 CrossRef CAS PubMed; (b) Handbook of Metathesis, ed. R. H. Grubbs and A. G. Wenzel, Wiley-VCH, Weinheim, Germany, 2nd edn, 2015, vol. 3 Search PubMed; (c) K. Nomura and M. M. Abdellatif, Polymer, 2010, 51, 1861–1881 CrossRef CAS; (d) A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS; (e) H. Mutlu, L. M. de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404 RSC; (f) M. D. Schulz and K. B. Wagener, Macromol. Chem. Phys., 2014, 215, 1936 CrossRef CAS; (g) R. H. Grubbs and E. Khosravi, Handbook of Metathesis, Wiley-VCH, Weinheim, Germany, 2nd edn, 2015, vol. 3 CrossRef; (h) Y. Chen, M. M. Abdellatif and K. Nomura, Tetrahedron, 2018, 74, 619–692 CrossRef CAS.
- (a) R. R. Schrock, Acc. Chem. Res., 1990, 23, 158–165 CrossRef CAS; (b) R. R. Schrock, Chem. Rev., 2002, 102, 145–180 CrossRef CAS PubMed; (c) C. Pietraszuk, Olefin Metathesis: Theory and Practice, ed. K. Grela, John Wiley & Sons, Inc., Hoboken, New Jergy, USA, 2014, pp. 371–396 Search PubMed; (d) D. Mindiola, Acc. Chem. Res., 2006, 39, 813–821 CrossRef CAS; (e) R. R. Schrock, Chem. Rev., 2009, 109, 3211–3226 CrossRef CAS; (f) R. R. Schrock, Acc. Chem. Res., 2014, 47, 2457–2466 CrossRef CAS PubMed; (g) M. J. Benedikter, F. Ziegler, J. Groos, P. M. Hauser, R. Schowner and M. R. Buchmeiser, Coord. Chem. Rev., 2020, 415, 213315 CrossRef CAS; (h) K. Nomura, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O’hare, Elsevier B.V., 2022, vol. 5, pp. 587–650 Search PubMed; (i) R. R. Schrock, M. R. Buchmeiser, J. Groos and J. Benedikter, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O’hare, Elsevier B.V., 2022, vol. 5, pp. 671–773 Search PubMed.
- (a) K. Nomura and S. Zhang, Chem. Rev., 2011, 111, 2342–2362 CrossRef CAS; (b) K. Nomura and W. Zhang, Chem. Sci., 2010, 1, 161–173 RSC; (c) K. Nomura and X. Hou, Dalton Trans., 2017, 46, 12–24 RSC; (d) S. Zhang, W. Zhang and K. Nomura, Adv. Organomet. Chem., 2017, 68, 93–136 CrossRef; (e) K. Nomura and S. Mekcham, Adv. Organomet. Chem., 2023, 79, 1–39 CAS.
- (a) M. R. Buchmeiser, Chem. Eur. J., 2018, 24, 14295–14301 CrossRef CAS; (b) M. R. Buchmeiser, Macromol. Rapid Commun., 2019, 40, 1800492 CrossRef.
- (a) X. Hou and K. Nomura, J. Am. Chem. Soc., 2015, 137, 4662–4665 CrossRef CAS; (b) X. Hou and K. Nomura, J. Am. Chem. Soc., 2016, 138, 11840–11849 CrossRef CAS; (c) K. Nomura and X. Hou, Organometallics, 2017, 36, 4103–4106 CrossRef CAS; (d) S. Chaimongkolkunasin and K. Nomura, Organometallics, 2018, 37, 2064–2074 CAS; (e) K. Nomura and S. Chaimongkolkunasin, Catal. Sci. Technol., 2020, 10, 5840–5846 RSC; (f) Y. Kawamoto, I. Elser, M. R. Buchmeiser and K. Nomura, Organometallics, 2021, 40, 2017–2022 CrossRef CAS; (g) S. Mekcham and K. Nomura, J. Am. Chem. Soc., 2023, 145, 17001–17006 CrossRef CAS PubMed.
- (a) S. Sen, J. Unold, W. Frey and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2014, 53, 9384–9388 Search PubMed; (b) R. Schowner, W. Frey and M. R. Buchmeiser, J. Am. Chem. Soc., 2015, 137, 6188–6191 CAS; (c) K. Herz, J. Unold, J. Hänle, R. Schowner, S. Sen, W. Frey and M. R. Buchmeiser, Macromolecules, 2015, 48, 4768–4778 CrossRef CAS; (d) I. Elser, R. Schowner, W. Frey and M. R. Buchmeiser, Chem. Eur. J., 2017, 23, 6398–6405 CAS; (e) C. Lienert, W. Frey and M. R. Buchmeiser, Macromolecules, 2017, 50, 5701–5710 CAS; (f) I. Elser, B. R. Kordes, W. Frey, K. Herz, R. Schowner, L. Stçhr, H. J. Altmann and M. R. Buchmeiser, Chem. Eur. J., 2018, 24, 12652–12659 CrossRef CAS; (g) M. J. Benedikter, R. Schowner, I. Elser, P. Werner, K. Herz, L. Stöhr, D. A. Imbrich, G. M. Nagy, D. Wang and M. R. Buchmeiser, Macromolecules, 2019, 52, 4059–4066 CrossRef CAS; (h) K. Herz, M. Podewitz, L. Stöhr, D. Wang, W. Frey, K. R. Liedl, S. Sen and M. R. Buchmeiser, J. Am. Chem. Soc., 2019, 141, 8264–8276 CrossRef CAS; (i) R. Schowner, I. Elser, M. Benedikter, M. Momin, W. Frey, T. Schneck, L. Stçhr and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2020, 59, 951–958 CrossRef CAS; (j) M. Benedikter, J. Musso, M. K. Kesharwani, K. L. Sterz, I. Elser, F. Ziegler, F. Fischer, B. Plietker, W. Frey, J. Kästner, M. Winkler, J. van Slageren, M. Nowakowski, M. Bauer and M. R. Buchmeiser, ACS Catal., 2020, 10, 14810–14823 CrossRef CAS; (k) M. J. Benedikter, J. V. Musso, W. Frey, R. Schowner and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2021, 60, 1374–1382 CrossRef CAS PubMed.
- (a) C. D. Abernethy, G. M. Codd, M. D. Spicer and M. K. A. Taylor, J. Am. Chem. Soc., 2003, 125, 1128–1129 CrossRef CAS PubMed; (b) W. Zhang and K. Nomura, Organometallics, 2008, 27, 6400–6402 CrossRef CAS; (c) C. Lorber and L. Vendiera, Dalton Trans., 2009, 6972–6984 RSC; (d) S. Zhang, W.-C. Zhang, D.-D. Shang, Z.-Q. Zhang and Y.-X. Wu, Dalton Trans., 2015, 44, 15264–15270 RSC.
- For example, (a) V. C. Gibson and T. Okada, Macromolecules, 2000, 33, 655–656 CrossRef CAS; (b) B. Autenrieth and R. R. Schrock, Macromolecules, 2015, 48, 2493–2503 CrossRef CAS.
- We acknowledge Dr S. Hayano (Zeon Co.) for careful NMR analysis of our typical poly(NBE) sample (using cryoprobe). Estimation of tacticity in the hydrogenated poly(NBE): S. Hayano and Y. Nakama, Macromolecules, 2014, 47, 7797–7811 CrossRef CAS.
- A reviewer commented that PDI (Mw/Mn) values in the resultant polymers were large and increased over time course. The values were apparently large compared to those reported in the ROMP by using the PMe3-supported catalysts.6a,b It is clear that the present catalysts (1, 2) showed much (>10 times) higher catalytic activities and conversion in the ROMP by 1 already reached more than 40% even after 1 min with high Mn value (Mn = 394000, Table S1 (ESI†), initial molar ratio charged: NBE/V = 5300). Therefore, it seems likely that reason for the large PDI values would be due to the rather slow initiation compared to the very rapid propagation. As communicated previously, increase in the viscosity of the reaction mixture can also be highly considered additionally.
- K. Nomura, K. Suzuki, S. Katao and Y. Matsumoto, Organometallics, 2012, 31, 5114–5120 CAS.
- K. Chatchaipaiboon and K. Nomura, J. Jpn. Petrol. Inst., 2021, 64, 238–244 CrossRef CAS.
- M. Unoki and K. Nomura, the results were partly introduced in 13th International Vanadium Symposium, Lisbon, Portugal, November 2023.
Footnotes |
| † Electronic supplementary information (ESI) available: (i) Additional results for ring-opening metathesis polymerisation of norbornene, (ii) selected NMR spectra of the polymers, (iii) selected DSC thermograms of the polymers after hydrogenation, and (iv) additional adda including crystal data and collection parameters for structural analysis of [V(CHSiMe3)(N-2,6-X2C6H3){OC(CF3)3}(NHC)] (X = F, Cl)]. CCDC 2358163 and 2358164. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj02537g |
| ‡ SK and JS: equal contribution. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |