
Synthesis and catalysis of a Co13 disk surrounded by vanadium–oxygen crown†
Yuji
Kikukawa
 *a,
Isshin
Yoshida
a,
Ryoji
Mitsuhashi
b,
Masaru
Yamane
a and
Yoshihito
Hayashi
*a
*a,
Isshin
Yoshida
a,
Ryoji
Mitsuhashi
b,
Masaru
Yamane
a and
Yoshihito
Hayashi
*a
aDepartment of Chemistry, Graduate School of Natural Science and Technology, Kanazawa University, Kakuma, Kanazawa 920-1192, Japan. E-mail: kikukawa@se.kanazawa-u.ac.jp
bInstitute of Liberal Arts and Science, Kanazawa University, Kakuma, Kanazawa 920-1192, Japan
First published on 6th August 2024
Abstract
By the reaction of the binuclear cobalt-containing polyoxovanadate [Co2(H2O)2V10O30]6− with methanol, a tridecanuclear cobalt containing polyoxovanadate, [Co13(OH)6(OCH3)6V24O72]10− (Co13), was synthesized. Single crystals suitable for X-ray crystallographic analysis were obtained as trimethylphenylammonium salts. The tridecanuclear cobalt core exhibited a planar disk structure and 24 vertex-sharing VO4 units surrounded the edge of the disk. This is the largest VO4-based polyoxovanadate as far as we know. Compound Co13 was stable in the solution state. Electrochemical analysis showed the possibility of Co13 to act as an oxidation catalyst for alcohols. Compound Co13 acted as a catalyst for oxidation of several alcohols. After the reaction, the Co13 catalyst was retrieved by the addition of an excess amount of ethyl acetate and filtration. The structure of the catalyst was maintained and reused. The oxidation of not only aromatic but also aliphatic alcohols proceeded with Co13. Compound Co13 showed catalytic performance for the oxidation of secondary alcohols even in the presence of alkenic and primary alcoholic functions.
Introduction
Vanadium–oxygen species exist as multinuclear species, called polyoxovanadates, in the aqueous solution, except for under strong acidic and strong basic conditions.1 Octahedrally coordinated VO6-based decavanadates and tetrahedrally coordinated VO4-based oligomers are the dominant species in acidic and basic solutions, respectively. The VO4-based structures are bridged by the vertex-sharing oxygen atoms to form circular structures. The circular polyoxovanadates act as inorganic ligands with nucleophilicity to stabilize cationic species at the centre of the circles.2 Protons and various metal cores stabilized by circular polyoxovanadates, such as [HV4O12]3−, [PdV6O18]4−, [Cu2V8O24]4−, [Ni4V10O30(OH)2(H2O)6]4− (Ni4), [Mn2(V5O15)2]6−, [Co2(H2O)2V10O30]6− (Co2, Fig. 1), [(H2O)M(V4O12)2]5− (M = Y, Ho), [LnV9O27]6− (Ln = Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy), and [LnV10O30]7− (Ln = La, Er, Tm, Yb, Lu), were reported.2–8 Although the obtained structures showed versatility, the synthetic procedures are almost the same. Tetravanadate [V4O12]4− is dissolved in an organic solvent and the addition of metal cations gives the metal-containing polyoxovanadates. Tetravanadate easily changes the number of VO4 units and the conformations of the rings.9 In comparison with the general organic ligands, circular polyoxovanadate ligands show high flexibility as if the ligands adjust their own structures to the central core structures. Lanthanide-surrounding polyoxovanadates vary the conformations depending on the ionic radii.6,7 In the case of manganese and cobalt, the number of manganese and cobalt cations increased by the reaction of the additional metal cations to form [M3(H2O)OAcV10O30]5− (M = Co (Co3), Mn, OAc = acetate) and [Mn6O6V8O26(OAc)2]6−.10,11 | ||
| Fig. 1 Anion structures of (a) Co2 and (b) Co13. The green, orange, red, and black spheres represent cobalt, vanadium, oxygen, and carbon atoms, respectively. Orange tetrahedra represent VO4 units. | ||
Layered cobalt oxides and hydroxides have attracted attention for application in sustainable and renewable energy generation and storage, superconductors, and catalysts.12–15 The separation of the monolayers produced a hydroxide ion conductor and electrochemical catalysts, especially for water splitting.16 To prepare discrete monolayers, peeling reagents and/or protecting ligands for the flat surface of monolayers are important.17,18 In these cases, the monolayers show size distribution. With the appropriate ligands surrounding the edge of the monolayer, single species were obtained. Up to now, planar multinuclear cobalt–oxygen complexes have been reported.19 Planar heptanuclear cobalt hydroxide stabilized with functionalized 6-methoxyphenoxides showed the unique electron transfer, and it was used as an oxidation catalyst.19,20 With isobutyrate and N-butyldiethanolamine, a decanuclear cobalt disk was synthesized. The disk contained both bridging hydroxide and alkoxide ligands.21 By the reaction of cobalt sources with tris(hydroxymethyl)alkane, hepta- and tridecanuclear cobalt disks were prepared.22,23 Triazacyclononane (tacn) also acted as a terminal ligand. Terminal mononuclear cobalt–tacn complexes were prepared in the first step. Then, the coordination of 6 monomers to the heptanuclear cobalt core afforded the tridecanuclear cobalt disk under basic media. The terminal ligands prevented further hydrolysis.24 Not only organic ligands but also rigid polyoxometalates can stabilize the planar cobalt cores, such as [Co4(H2O)2(XW9O34)2]10− (X = P, V) and [Co4(OH)2(H2O)6(H2SiW10O36)2]6−.25–28 Compound [Co4(H2O)2(XW9O34)2]10− acted as an efficient homogeneous water oxidation catalyst. In this work, synthesis of a multinuclear cobalt disk surrounded by VO4-based polyoxovanadate and its catalytic properties for alcohol oxidation were investigated.
Results and discussion
Synthesis of tridecanuclear cobalt containing polyoxovanadate
The solid of the previously reported binuclear cobalt containing polyoxovanadate Co2 showed water-induced thermochromism.5 The original colour of Co2 was light green and, by exposure to water vapor, the solid colour changed to brown and, by heating to evaporate the adsorbed water, the light green colour was retrieved.5 This result inspired us to investigate the colour change by addition of a substrate to the Co2 solution. Methanol or 2-propanol was added to the acetonitrile solution of Co2. In the case of methanol, the solution colour changed from green to brown. Addition of 2-propanol caused no colour change. From the mixed solution of acetonitrile and methanol, tiny amounts of brown crystals were obtained. The IR spectrum of the crystalline sample was different from that of Co2, indicating the structure conversion (Fig. S1, ESI†). Although the quality of the crystals was too low to complete the X-ray crystallographic analysis, the anion structure was confirmed. It contained a tridecanuclear cobalt disk surrounded by 24 VO4 units (Co13, Fig. 1). To improve the yield, triethylamine was added to the synthetic solution to assist the proton removal of methanol. The product Co13 was selectively precipitated from the synthetic solution. The low yield of Co13 was due to the unidentified by-products in the solution. The yield clearly increased with increasing the amount of added triethylamine, while an excess amount of triethylamine caused impurities. After dissolution of 200 mg of Co2 in 0.8 mL of the mixed solvent of acetonitrile and methanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), addition of 2, 3, 4, and 5 equiv. of triethylamine with respect to Co2 and allowing the solution to stand for 3 days at room temperature yielded 5.5, 9.4, 13, and 14 mg of the product, respectively. Under the optimized conditions, the yield reached 24%. From the elemental analysis and thermogravimetric analysis, the formula of the product was (Et4N)5(HEt3N)5[Co13(OH)6(OCH3)6V24O72]·10H2O. From the electrospray ionization mass spectrometry (ESI-MS) spectrum, the corresponding ion pairs due to Co13 were observed (Fig. S2, ESI†). The distribution of the ion pairs indicates that the bridging methoxide ligands and/or hydroxide ligands are exchangeable.
:1, v/v), addition of 2, 3, 4, and 5 equiv. of triethylamine with respect to Co2 and allowing the solution to stand for 3 days at room temperature yielded 5.5, 9.4, 13, and 14 mg of the product, respectively. Under the optimized conditions, the yield reached 24%. From the elemental analysis and thermogravimetric analysis, the formula of the product was (Et4N)5(HEt3N)5[Co13(OH)6(OCH3)6V24O72]·10H2O. From the electrospray ionization mass spectrometry (ESI-MS) spectrum, the corresponding ion pairs due to Co13 were observed (Fig. S2, ESI†). The distribution of the ion pairs indicates that the bridging methoxide ligands and/or hydroxide ligands are exchangeable.
Single crystals suitable for X-ray crystallographic analysis were obtained by the cation exchange to trimethylphenylammonium. The IR spectra in the fingerprint region of polyoxovanadates were the same between ethylammonium salts and trimethylphenylammonium salts, showing that the anion structure was maintained during the cation exchange (Fig. S1, ESI†). The BVS values of cobalt and vanadium atoms were 2.00–2.10 and 4.96–5.17, showing cobalt and vanadium atoms were divalent and pentavalent, respectively.29 Oxygen atoms surrounding the centred cobalt atom were hydroxide ligands. They bridged the centred cobalt atom and 6 cobalt atoms. The 6 cobalt atoms were bridged by methoxide ligands. The methyl groups were located vertically to the tridecanuclear cobalt disk, and the directions were opposite to the next methoxide ligands. Methoxide ligands also bonded to the outer 6 cobalt atoms. The arrangement of the 13 cobalt atoms was similar to that of [Co13(OH)24(tacn)6]8+.24 The tridecanuclear cobalt disk was surrounded by the [V24O72]24− crown. The crown was composed of 24 vertex-sharing VO4 units. The 48 membered V24O24 ring could be drawn with a single stroke. The top view of the ring represented a gear shape (Fig. 2). The side view of the ring contained three waves. The numbers of waves of the surrounding polyoxovanadate ligands in Co2 and Co3 were 5 and 4, respectively.10 With increasing number of cobalt atoms in the crown, the number of waves of the vanadium–oxygen ring decreased (Fig. 2). The conformation relationship between the central disk and surrounding polyoxovanadate crowns in Co13 was different from that in previously reported planar core-containing polyoxovanadate Ni4.4 The polyoxovanadate crown of Co13 was located in the same plane as the tridecanuclear cobalt disk, while the crown of Ni4 was perpendicular to the tetranuclear nickel plane.
 | ||
| Fig. 2 Top and side views and schematic representations of (a) the V10O10 ring in Co2 and (b) the V24O24 ring in Co13. The orange and red spheres represent vanadium and oxygen atoms, respectively. | ||
The direct current magnetic measurement of the trimethylphenylammonium salts of Co13 was carried out using the crystalline sample. The χMT value of 48.3 cm3 K mol−1 was much larger than the value calculated from the spin-only equation for 13 high-spin CoII ions (24.4 cm3 K mol−1) due to the significant spin–orbit coupling of the octahedral CoII ion (Fig. S3, ESI†). The χMT value gradually decreased to a minimum at 34 K and slightly increased to reach a value of 40.1 cm3 K mol−1 at 19 K. Upon further cooling, the χMT abruptly decreased down to 24.6 cm3 K mol−1. The slight increase at 34 K can be attributed to the ferromagnetic exchange coupling between CoII centers. This feature competed with the sharp decrease owing to the zero-field splitting. The alternating current susceptibility measurement was performed to observe slow magnetic relaxation behavior. However, no frequency dependence was observed for the out-of-phase signal  , indicating that Co13 is not a single-molecule magnet (Fig. S4, ESI†).
, indicating that Co13 is not a single-molecule magnet (Fig. S4, ESI†).
Oxidation properties
The electrochemical properties of Co13 were investigated. In the range of 0–2.0 V vs. ferrocene/ferrocenium (Fc/Fc+), a weak and broad oxidation peak around 1.5 V was observed. By the addition of methanol, an increase of the catalytic current over 1.0 V was observed. In addition, an irreversible oxidation peak at 1.5 V was clearly observed (Fig. 3). The IR spectra show that the Co13 structure is maintained after treatment with an excess amount of methanol in the solution state (Fig. S5, ESI†). Without Co13 or with a typical metavanadate, [V4O12]4−, the current increase was not observed. With Co(NO3)2, the catalytic current increase was observed over 1.4 V without a peak top (Fig. S6, ESI†). With Co2, addition of methanol caused the decomposition of the structure. The second cycle of Co13 solution with methanol showed a weak current probably due to the adsorption of the inactive species on the surface of the electrode (Fig. S7, ESI†). In the case of water addition to the Co13 solution, the peak current over 1.0 V increased (Fig. S8, ESI†). Addition of non-oxidative tert-butyl alcohol did not show the current increase (Fig. S8, ESI†). After the treatment of 5000 equiv. of water or 10000 equiv. of tert-butyl alcohol, the Co13 structure was maintained (Fig. S5, ESI†).
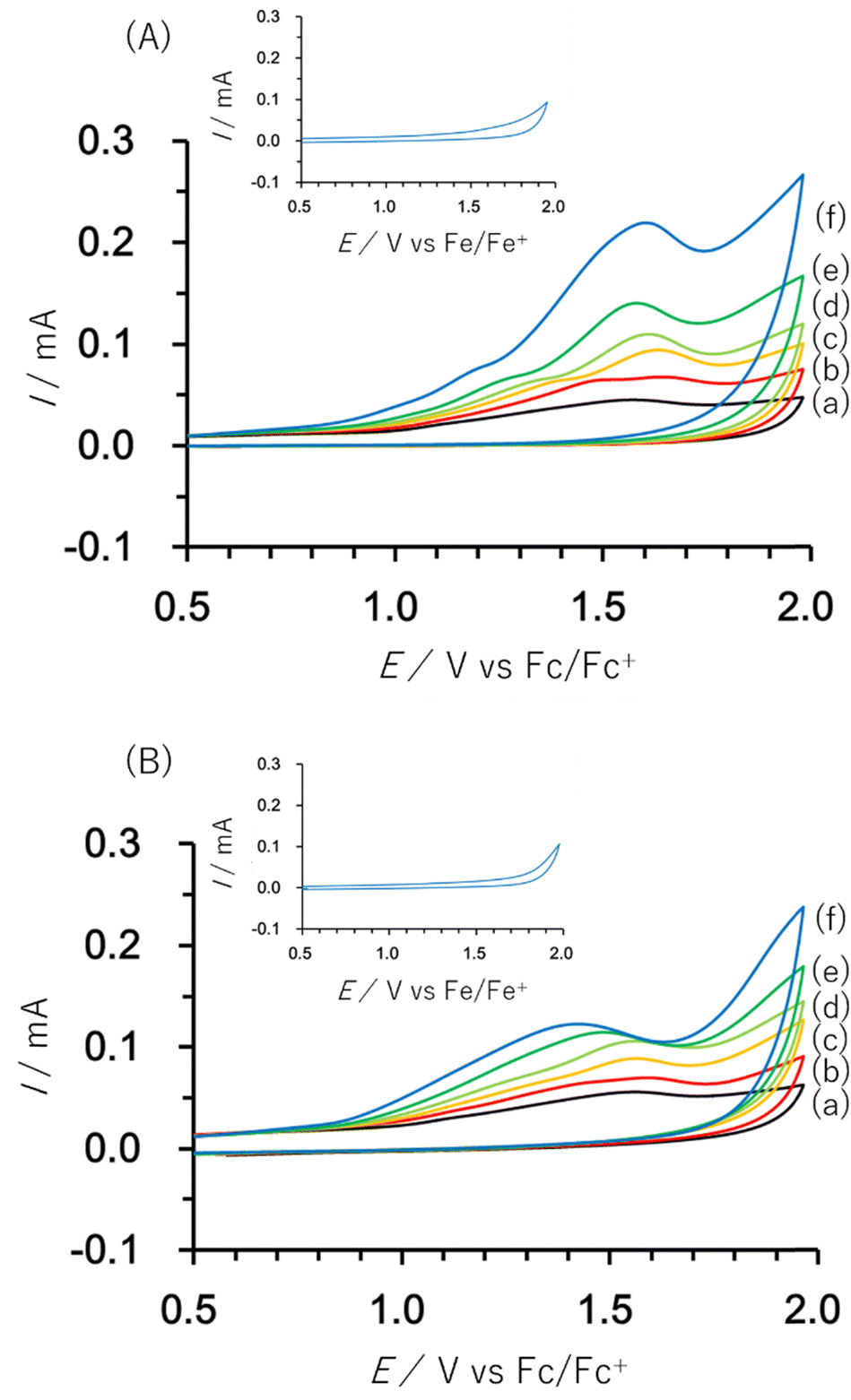 | ||
| Fig. 3 First cycle of cyclic voltammograms of Co13 in the presence of (a) 0 equiv., (b) 100 equiv., (c) 300 equiv., (d) 500 equiv., (e) 1000 equiv., and (f) 3000 equiv. of (A) methanol and (B) 1-butanol. The insets show voltammograms under the condition (f) without Co13. | ||
By the addition of 1-butanol to the Co13 solution, the peak top potential shifted to 1.4 V, indicating the ligand exchange (Fig. 3). The IR spectra also supported the ligand exchange (Fig. S9, ESI†). After the reaction with 1-butanol, 2-butanol, and benzyl alcohol, the peak at 860 cm−1 of the original Co13 shifted to 843, 847, and 853 cm−1, respectively. In the case of bulky tert-butyl alcohol, no peak shift was observed (Fig. S5, ESI†). After the constant potential electrolysis at 1.5 V in the presence of 1-butanol with Co13, a trace amount of butanal was detected by the GC–MS measurement.
Next, chemical oxidation of alcohols was investigated (Table 1). tert-Butyl hydroperoxide (TBHP) was used as an oxidant. The IR spectrum of the solid collected by filtration after the addition of ethyl acetate into the Co13 solution in the presence of 500 equiv. of TBHP was identical to that of the original samples of Co13, showing the stability of Co13 towards TBHP (Fig. S10, ESI†). The reaction was carried out in the mixed solvent of propylene carbonate and acetonitrile (1:1, v/v) to dissolve the catalysts. With Co13 as a catalyst, oxidation of diphenyl methanol proceeded smoothly to afford the corresponding ketone in 99% yield in 24 h (entry 1).
| Entry | Substrate | Product | Catalyst (μmol) | Time/h | Conversion/% | Yield/% |
|---|---|---|---|---|---|---|
| a Reaction conditions: substrate (1 mmol), catalysts (2–26 μmol), 5.5 M of TBHP in decan (1 mmol), acetonitrile (1 mL), propylene carbonate (1 mL), internal standard (0.2 mmol), 32 °C, 800 rpm with a Teflon-coated magnetic stirrer bar. Conversion and yields were determined by GC. b The primary alcohol oxidation product was not detected. c The alcohol was selectively oxidized without the formation of epoxide. | ||||||
| 1 |

|

|
Co13 (2) | 24 | 99 | 99 |
| 2 | Co(NO3)2 (26) | 24 | 45 | 45 | ||
| 3 | Co2 (13) | 24 | 91 | 88 | ||
| 4 | {Et4N}4[V4O12] (12) | 24 | 93 | 93 | ||
| 5 | Co(NO3)2 (26) {Et4N}4[V4O12] (12) | 24 | 78 | 78 | ||
| 6 | — | 24 | 8 | 7 | ||
| 7 |

|

|
Co13 (2) | 36 | 96 | 95 |
| 8 | R = p-Me | R = p-Me | Co13 (2) | 36 | 95 | 92 |
| 9 | R = p-Cl | R = p-Cl | Co13 (2) | 36 | 99 | 99 |
| 10 | R = m-NO2 | R = m-NO2 | Co13 (2) | 36 | 98 | 98 |
| 11 |

|

|
Co13 (2) | 35 | 94 | 94 |
| 12 |

|

|
Co13 (2) | 2 | 33 | 32 |
| 13 | {Et4N}4[V4O12] (12) | 2 | 24 | 15 | ||
| 14 |

|

|
Co13 (2) | 24 | 54 | 54 |
| 15 |

|

|
Co13 (2) | 21 | 46 | 46 |
| 16b |

|

|
Co13 (2) | 10 | 44 | 29 |
| 17c |

|

|
Co13 (2) | 10 | 71 | 47 |
After the reaction, the catalyst was collected by filtration after the addition of an excess amount of ethyl acetate. The IR spectrum of the retrieved catalyst was the same as those of the original samples (Fig. S11, ESI†). The ESI-MS spectrum of the retrieved catalyst showed the related peak sets of Co13 (Fig. S12, ESI†). The retrieved catalyst was reused for alcohol oxidation. The oxidation of diphenyl methanol with the retrieved catalyst gave a 93% yield (Fig. S13, ESI†). The reactivity of Co(NO3)2 was lower than that of Co13 (entry 2). Although the reactivities of Co2 and [V4O12]4− were similar to that of Co13, their structures were decomposed during the reaction (entries 3 and 4; Fig. S14 and S15, ESI†). The second run with retrieved catalysts of Co2 and [V4O12]4− gave lower yields (Fig. S13, ESI†). The simple mixture of Co(NO3)2 and [V4O12]4− gave a medium yield of the product (entry 5). Without catalysts, the reaction hardly proceeded (entry 6). The oxidation of 1-phenyl ethanol and its derivatives smoothly proceeded to give the corresponding acetophenone derivatives (entries 7–10). The derivatives with both electron donating and electron accepting groups were efficiently oxidized. The oxidation of the radical-clock substrate exclusively produced cyclopropyl phenyl ketone without ring-opening products, indicating that free-radical intermediates were not involved in the present alcohol oxidation (entry 11).30 The oxidation of benzyl alcohol with Co13 gave a higher yield of benzaldehyde than that with [V4O12]4− in 2 h, although the yield was low due to substrate inhibition (entries 12 and 13, Fig. S9, ESI†). The oxidation reactions of cyclic and linear aliphatic alcohols proceeded to afford the corresponding aliphatic ketones with medium yields with stopping the reactions probably due to the inhibition of substrates by the coordination to active centres (entries 14–17). The alcohol oxidation with Co13 was secondary selective. The oxidation of 1,3-butanediol selectively took place at the secondary position (entry 16). 2-Cyclohexene-1-ol possesses two oxidative functions. In the presence of Co13, alcohol oxidation selectively proceeded without the formation of epoxy products (entry 17). The oxidative product observed in the GC chart was only 2-cyclohexene-1-one.
After the treatment of Co13 with TBHP, the IR peak at 860 cm−1 was slightly shifted, indicating the coordination of TBHP (Fig. S10, ESI†). The isolated samples after the treatment with TBHP were utilized for the oxidation of diphenyl methanol. Without an additional oxidant, ca. 6 equiv. of benzophenone was obtained, showing the coordination of TBHP to Co13 was included in the reaction paths. In the previous work, it was reported that a tetrahedrally coordinated vanadium centre has the potential to activate TBHP.31 Based on the results reported herein and previous reports, we propose the possible reaction paths. The oxidant TBHP was activated by the coordination to vanadium and/or cobalt centres. The approach of the substrates to the activated TBHP gives the products.
Experimental
Materials
All reagents were obtained from commercial suppliers and used without further purification unless otherwise noted. {Et4N}4[V4O12] and {Et4N}6[Co2(H2O)2V10O30] (Co2) were synthesized following the literature method.5,32 7-Oxabicyclo[4.1.0]heptan-2-ol (2,3-epoxycyclohexanol) was obtained with the previously reported system with {n-Bu4N}5[V18O46(NO3)]5−.33Synthesis of the tridecanuclear cobalt-containing polyoxovanadate [Co13(OH)6(OCH3)6V24O72]10− (Co13)
{Et4N}6[Co2(H2O)2V10O30] 200 mg (0.10 mmol) was dissolved in 0.8 mL of the mixed solvent of acetonitrile and methanol (3:1, v/v). After the addition of 58 μL triethylamine (0.40 mmol), the solution was stirred for 1 h. Then the solution was filtered to remove the insoluble materials. The solution was kept standing for 2 days at 35 °C. Co13 (21 mg) was obtained as a brown powder (24% yield based on cobalt). Elemental analysis calcd for {Et4N}6{Et3HN}4[Co13(OH)6(OCH3)6V24O72]·12H2O: C 19.37%, H 4.84%, N 2.90%; found: C 19.44%, H 4.61%, N 2.82%. Ethylammonium salts of Co13 were used for the electrochemical analysis and catalytic reactions. The crystalline samples suitable for the single crystal X-ray analysis were obtained by cation exchange. Co13 and 100 equiv. of trimethylphenylammonium bromide with respect to Co13 were dissolved in DMSO at 60 °C. Vapor diffusion of ethyl acetate to the Co13 solution at 35 °C gave red crystals. Elemental analysis calcd for {Me3PhN}10[Co13(OH)6(OCH3)6V24O72]·4Me2SO·10H2O: C 23.64%, H 3.97%, N 2.65%, S 2.43%; found: C 23.37%, H 3.99%, N 2.60%, S 2.45%. CCDC 2364056 contains the supplementary crystallographic data for Co13 (Table S1, ESI†).
Conclusions
Methanol-induced structure conversion from binuclear to tridecanuclear cobalt-containing polyoxovanadates under basic conditions was discovered by monitoring the colour change of the solution with additives. Among cobalt-containing VO4-based polyoxovanadate crowns, the numbers of waves in the V–O rings decrease with increasing number of central cobalt atoms. Compound Co13 acted as an oxidation catalyst for the alcohol. The ligand exchanges might assist the oxidation reactions. The size and conformation of polyoxovanadate crowns were controllable to adjust the central metal cores. Conversion of the central metal core structures induces structure dependent properties. Polyoxovanadate crowns are some of the candidates to support such phenomena.Author contributions
Y. K.: conceptualization, funding acquisition; R. M.: characterization; I. Y.: spectroscopic analysis and catalytic reactions; M. Y.: synthesis and electrochemistry; Y. K. and Y. H.: supervision. All authors have contributed to writing of the manuscript.Data availability
Crystallographic data for Co13 have been deposited at CCDC under 2364056 and can be obtained from https://www.ccdc.cam.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr H. Aihara for the preliminary experiments. This research was partly supported by the JSPS KAKENHI Grant No. JP23H04621 and JP23K04776, JSPS Core-to-Core program, and the JST FOREST Program Grant No. JPMJFR2023720466.Notes and references
-
C. F. Baes and R. E. Mesmer, The hydrolysis of cations, John Wiley, New York, 1976 Search PubMed
.
- Y. Hayashi, Coord. Chem. Rev., 2011, 255, 2270 CrossRef CAS
- J. Fuchs, S. Mahjour and J. Pickardt, Angew. Chem., Int. Ed. Engl., 1976, 15, 374 CrossRef
- T. Kurata, A. Uehara, Y. Hayashi and K. Isobe, Inorg. Chem., 2005, 44, 2524 CrossRef CAS PubMed
- S. Inami, M. Nishio, Y. Hayashi, K. Isobe, H. Kameda and T. Shimoda, Eur. J. Inorg. Chem., 2009, 5253 CrossRef CAS
- M. Nishio, S. Inami, M. Katayama, K. Ozutsumi and Y. Hayashi, Inorg. Chem., 2012, 51, 784 CrossRef CAS PubMed
- M. Nishio, S. Inami and Y. Hayashi, Eur. J. Inorg. Chem., 2013, 1876 CrossRef CAS
- T. Maruyama, H. Kawabata, Y. Kikukawa and Y. Hayashi, Eur. J. Inorg. Chem., 2019, 529 CrossRef CAS
- Y. Kikukawa, H. Kawabata and Y. Hayashi, RSC Adv., 2021, 11, 31688 RSC
- T. Maruyama, Y. Kikukawa, H. Sakiyama, M. Katayama, Y. Inada and Y. Hayashi, RSC. Adv, 2017, 7, 37666 RSC
- T. Maruyama, A. Namekata, H. Sakiyama, Y. Kikukawa and Y. Hayashi, New J. Chem., 2019, 43, 17703 RSC
- Y. Zhang and H. Ohta, NPG Asia Mater., 2023, 15, 67 CrossRef
- Y. Lyu, X. Wu, K. Wang, Z. Feng, T. Cheng, Y. Liu, M. Wang, R. Chen, L. Xu, J. Zhou, Y. Lu and B. Guo, Adv. Energy Mater., 2021, 11, 2000982 CrossRef CAS
- J. V. Badding, Nat. Mater., 2003, 2, 208 CrossRef CAS PubMed
- G. Chen, H. Wan, W. Ma, N. Zhang, Y. Cao, X. Liu, J. Wang and R. Ma, Adv. Energy Mater., 2020, 10, 1902535 CrossRef CAS
- G. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef PubMed
- R. Ma, Z. Liu, L. Li, N. Iyi and T. Sasaki, J. Mater. Chem., 2006, 16, 3809 RSC
- N. Tarutani, S. Kimura, T. Sakata, K. Suzuki, K. Katagiri and K. Inumaru, ACS Mater. Lett., 2022, 4, 1430 CrossRef CAS
- A. M. Ulman and D. F. Nocera, J. Am. Chem. Soc., 2013, 135, 15053 CrossRef PubMed
- T. S. Mahapatra, D. Basak, S. Chand, J. Lengyel, M. Shatruk, V. Bertolasi and D. Ray, Dalton Trans., 2016, 45, 13576 RSC
- S. Schmitz, T. Secker, M. Batool, J. van Leusen, M. A. Nadee and P. Kögerler, Inorg. Chim. Acta, 2018, 482, 522 CrossRef CAS
- K.-D. Leng, S.-K. Xing, R. Herchel, J.-L. Liu and M.-L. Tong, Inorg. Chem., 2014, 53, 5458 CrossRef PubMed
- Y. Peng, C.-B. Tian, Y.-H. Lan, N. Magnani, Q.-P. Li, H.-B. Zhang, A. K. Powell and S.-W. Du, Eur. J. Inorg. Chem., 2013, 5534 CrossRef CAS
- Sugiarto, Y. Imai and Y. Hayashi, Inorg. Chem., 2023, 62, 1845 CrossRef CAS PubMed
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickes, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268 CrossRef CAS PubMed
- Y. Kikukawa, K. Suzuki, K. Yamaguchi and N. Mizuno, Inorg. Chem., 2013, 52, 8644 CrossRef CAS PubMed
- Y. Kuriyama, Y. Kikukawa, K. Suzuki, K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2016, 22, 3962 CrossRef CAS PubMed
- D. Altermatt and I. D. Brown, Acta Crystallogr., 1985, B41, 244 Search PubMed
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317 CrossRef CAS
- Y. Kikukawa, Y. Sakamoto, H. Hirasawa, Y. Kurimoto, H. Iwai and Y. Hayashi, Catal. Sci. Technol., 2022, 12, 2438 RSC
- H. Nakano, T. Ozeki and A. Yagasaki, Acta Crystallogr., 2002, C58, m464 CAS
- I. Yoshida, Y. Kikukawa, R. Mitsuhashi and Y. Hayashi, Nanoscale, 2024, 16, 10584 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2364056. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj02900c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |