
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembled π-conjugated chromophores: preparation of one- and two-dimensional nanostructures and their use in photocatalysis
David
Cappelletti
 ,
Marianna
Barbieri
,
Alessandro
Aliprandi
,
Michele
Maggini
and
Luka
Đorđević
*
,
Marianna
Barbieri
,
Alessandro
Aliprandi
,
Michele
Maggini
and
Luka
Đorđević
*
Department of Chemical Sciences, University of Padova, via Marzolo 1, 35131, Padova, Italy. E-mail: luka.dordevic@unipd.it
First published on 4th April 2024
Abstract
Photocatalytic systems have attracted research interest as a clean approach to generate energy from abundant sunlight. In this context, developing efficient and robust photocatalytic structures is crucial. Recently, self-assembled organic chromophores have entered the stage as alternatives to both molecular systems and (in)organic semiconductors. Nanostructures made of self-assembled π-conjugated dyes offer, on the one hand, molecular customizability to tune their optoelectronic properties and activities and on the other hand, provide benefits from heterogeneous catalysis that include ease of separation, recyclability and improved photophysical properties. In this contribution, we present recent achievements in constructing supramolecular photocatalytic systems made of chromophores for applications in water splitting, H2O2 evolution, CO2 reduction, or environmental remediation. We discuss strategies that can be used to prepare ordered photocatalytic systems with an emphasis on the effect of packing between the dyes and the resulting photocatalytic activity. We further showcase supramolecular strategies that allow interfacing the organic nanostructures with co-catalysts, molecules, polymers, and (in)organic materials. The principles discussed here are the foundation for the utilization of these self-assembled materials in photocatalysis.
 Luka Đorđević | Luka obtained his Ph.D. from the University of Trieste, working with Prof. Bonifazi. During his Ph.D. he was a visiting scholar at the Broad Institute of MIT and Harvard University. After graduation, he performed postdoctoral research with Prof. Prato (University of Trieste) and Prof. Bonifazi (Cardiff University). He then joined the group of Prof. Stupp (Northwestern University) as a fellow at the Center for Bio-inspired Energy Science. Recently, Luka joined the Department of Chemical Sciences at the University of Padova. Here, thanks to the ERC starting grant, he is creating a team to investigate strategies to convert, store and use solar energy. |
Introduction
Due to increasing global energy demands, in addition to limited fossil fuels and environmental concerns, the conversion of solar light into energy or chemicals is gaining prominence. A constant source of inspiration for developing light-harvesting systems is natural photosynthesis, in which a preorganized assembly of chromophores performs conversion of light into chemical energy.1–8 While natural light-harvesting structures are usually organized via complex protein environments, some systems lack the support of a protein matrix. For example, in green sulphur photosynthetic bacteria, the so-called chlorosomes are based solely on pigments made of self-assembled chromophores.9 In these organisms, the energy absorbed by molecular antennas is ultimately used to transform substrates, such as carbon dioxide and hydrogen sulphide, into organic compounds, water and sulphur. While achieving such complex transformations in artificial systems is still far from our reach, the ability to prepare photoactive assemblies simply through self-assembly of chromophores has fascinated scientists and brought forward several exceptional systems in the last few years.This contribution provides an overview of the emerging field of self-assembled organic chromophores and their application in photocatalysis. Supramolecular polymers composed of chromophores are arrays formed by monomeric dye units interconnected through directional secondary interactions, such as metal–ligand coordination, π–π stacking, hydrogen bonding, or a combination of these. It is noteworthy that the first observation of supramolecular dye polymers dates back to the late 1930s, when Scheibe10 and Jelley11 reported studies on cyanine aggregates and their photophysical properties. Other seminal contributions date back to the late 1980s, when Aida and co-workers reported on an amphiphilic porphyrin that formed co-facial stacks in aqueous media.12 Since these pioneering works and with the advent of supramolecular polymers,13 the field has been constantly evolving.14–25 Supramolecular polymers are also finding applications in energy-related applications26 with recent literature reviewing the photocatalytic application of perylene diimides (PDIs)27 and other systems for H2 production and CO2 reduction.28
Here, we will focus on organic dyes that self-assemble through π–π stacking, as they can form robust and ordered aggregates in water. We organise our discussion by strategies that can be used to prepare ordered aggregates with photocatalytic applications. In these aggregates, the constituent monomeric chromophores can undergo electronic coupling, leading to emerging properties that enable solar energy conversion (Fig. 1). The spotlight of this work is on one- or two-dimensional nanostructures obtained through dye self-assembly in water, aqueous solutions containing salts, or solvent mixtures containing water. The ability to perform modern chemical reactions in water, paired with effective recycling strategies for catalysts, represents a cornerstone for sustainable and green chemistry. Additionally, water is a unique medium for self-assembly since biological systems excel at controlling supramolecular polymerization in aqueous media. In this context, synthetic self-assembled chromophores can combine the bioinspired aspect from natural systems with the synthetic accessibility and tunability of artificial molecules.
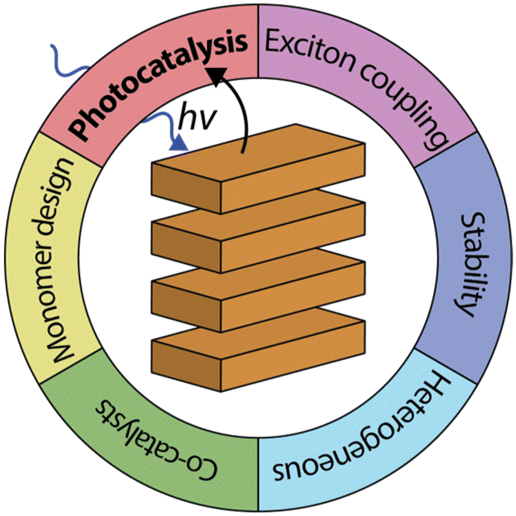 | ||
| Fig. 1 Organic dye aggregates used in photocatalysis have several advantages compared to their monomeric or semiconductor counterparts. | ||
Self-assembled chromophores in photocatalysis
When dyes self-assemble through π–π stacking (Fig. 2a), their photophysical properties can be drastically affected. In comparison to the monomer, aggregates can either exhibit blue-shifted (hypochromic shift) or red-shifted (bathochromic shift) absorption bands and are commonly called H- or J-aggregates, respectively. These changes can be explained through Kasha's exciton coupling theory,29 which is summarized in Fig. 2b. Taking a co-facially stacked dimer as an example, the transition dipole moments (TDM) of the monomers are oriented parallel to each other (slip angle of θ = 90°, H-aggregate, right side). In the other borderline case, the transition dipole moments are in line (θ = 0°, J-aggregates, left side). In addition, there are several deviations from the two ideal situations, but these are discussed elsewhere.30 While we exemplified the case of a dimer, the discussion on transition dipole moments and the transition energy also applies to large aggregates. Therefore, in aggregates the question of which chromophore is excited becomes irrelevant, as the excitation is rather delocalized and should be treated as a superposition of locally excited states.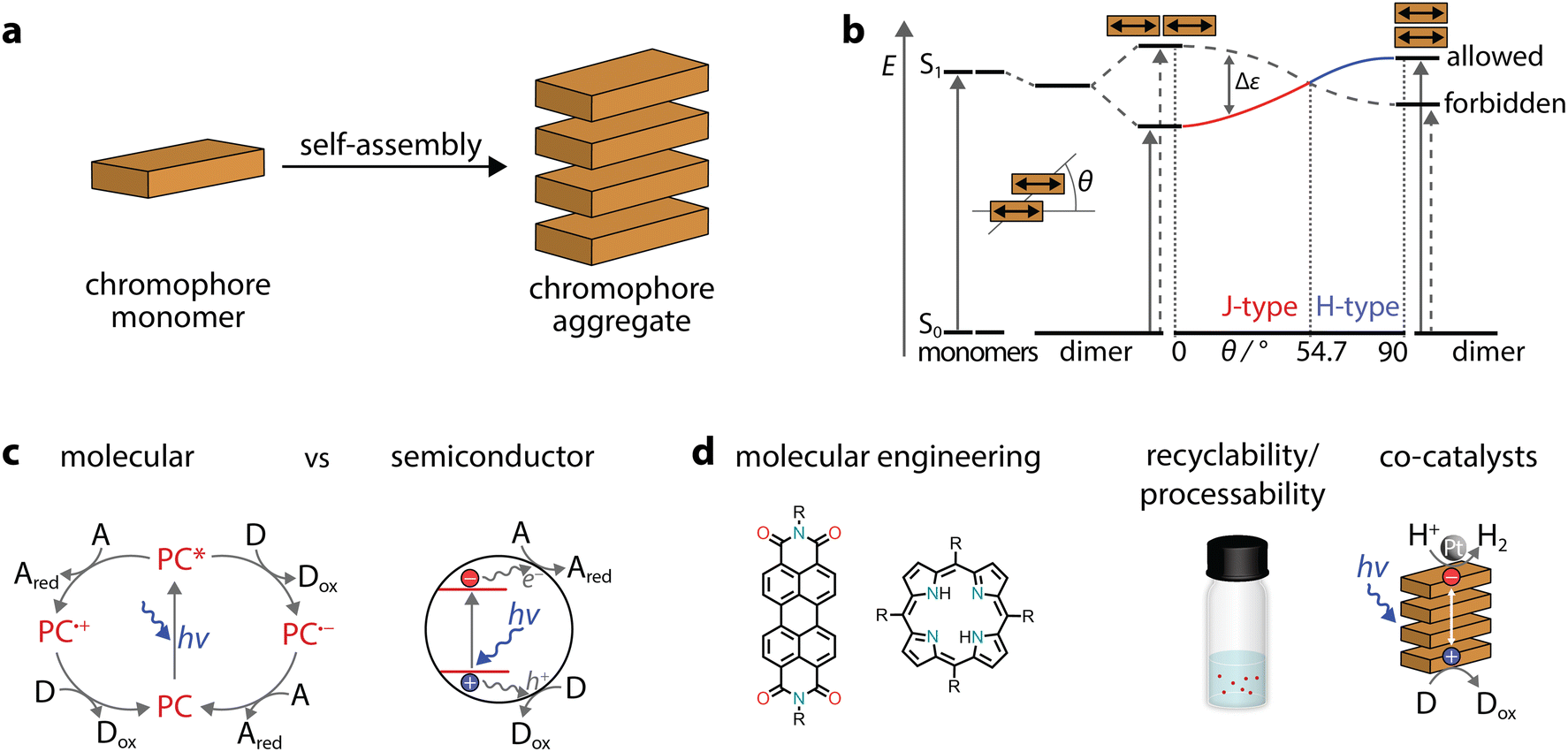 | ||
| Fig. 2 Overview of self-assembled chromophores and their use in photocatalysis. (a) Schematic representation of the self-assembly of a chromophore into an aggregate. (b) Schematic energy diagram for the exciton coupling of dyes with transition dipole moments and allowed transitions in J-type and H-type dimers. (c) Schematic representation of molecular versus semiconductor photocatalytic processes. (d) Self-assembled dye nanostructures combine advantages of homogeneous molecular photosensitizers (molecular engineering of monomers) and heterogeneous catalysts (stability, recyclability, and processability) for sacrificial photocatalysis. | ||
In molecular photoredox catalysis,31 the photocatalyst is promoted to the excited state, which can undergo two possible pathways (Fig. 2c). Through oxidative or reductive quenching mechanisms, radicals are formed and are more reactive than the neutral precursors. Unlike in molecular photocatalysts, in semiconductors, excitation through incident photons leads to the generation of electron–hole pairs (excitons), followed by their separation into free charges, which ultimately drive redox reactions (Fig. 2c).32 Depending on the distance between the electron–hole pairs, excitons can be either localized on the same monomer (Frenkel exciton) or diffused over several monomers (charge transfer excitons). Indeed, the formation of π–π stacks could allow enhanced separation and migration of the photogenerated carriers, minimizing recombination which is highly desirable in photocatalysis.25 For example, crystalline nanostructures, obtained through interchain π–π stacking of polymeric fluorenes,33,34 were found to exhibit long-range exciton transport that is beneficial for light-driven H2 evolution.35 Thus, highly ordered organic aggregates could have advantages in photocatalytic processes.
Using self-assembled dye polymers could offer additional benefits in photocatalysis (Fig. 2d). First, molecular engineering and customizability can be achieved through a multitude of organic chemistry procedures, with changes in the chemical structure then being translated to an aggregate. Second, further chemical modifications can increase surface wettability, addressing the low activity in the inherent apolar nature of various heterogeneous photocatalysts.36 Similarly, good dispersion can facilitate the easy processing of organic molecules. Third, the heterogeneous nature of extended aggregates can be used to recover and recycle the photocatalysts as well as to tune the interface for photocatalytic reactions.37 Fourth, supramolecular protocols for the formation of aggregates could allow relatively easy purification and eliminate batch-to-batch variations. Finally, organic “soft materials” benefit from properties such as relatively low toxicity, light weight, general affordability, and abundance on Earth.38
It should also be taken into consideration that the light absorption depends on the band energy gap (HOMO–LUMO for molecules and valence–conduction bands for semiconductors) and that the absolute bands positions determine the thermodynamic driving force for specific reactions. In the following sections we will also refer to co-catalysts and sacrificial agents. Indeed, in most cases it is necessary to add a dedicated co-catalyst (electrocatalyst) and a sacrificial agent that interact with photogenerated electrons and holes, making the light-absorbing unit essentially a photosensitiser. The addition of electrocatalysts enables certain photocatalytic reactions, such as H2 evolution, O2 production or CO2 reduction. In other cases, such as environmental remediation and H2O2 production, the addition of co-catalysts can be avoided since the direct interaction of the semiconductor with molecular O2 can occur. Finally, we also mention photocatalytic apparent quantum yields (AQYs), sometimes also called apparent quantum efficiencies, which are calculated as the ratio between the number of reacted electrons (e.g., 2× number of H2 molecules produced) and the number of incident photons of a certain wavelength, in the system. Since photocatalysis can be affected by a variety of parameters, including light sources and illumination conditions, we focus on the AQY at certain wavelengths rather than the product evolution rate.
Self-assembly of chromophores in photocatalytic nanostructures
Chromophores, made of π-conjugated systems, usually possess low solubility in water and their tendency is to self-assemble into robust aggregates due to a combination of hydrophobic and π–π stacking interactions. Large hydrophobic surfaces tend to aggregate in aqueous media both through entropic (hydration water being released in the bulk) and enthalpic (decrease of interfacial area with water) driving forces.20 Therefore, to prepare self-assembled nanostructures in aqueous media, the chromophore monomers (or discrete aggregates) need to be first solubilized (or dispersed) and then their aggregation can be triggered. This can be achieved through various protocols. For instance, supramolecular artificial antennas have been prepared using different procedures, including precipitation from good to bad solvents, ionic self-assembly, acid–base neutralization, and surfactant-assisted self-assembly (see Fig. 3). These protocols, that have been exploited to prepare other systems as well, will be presented and discussed in the following sections.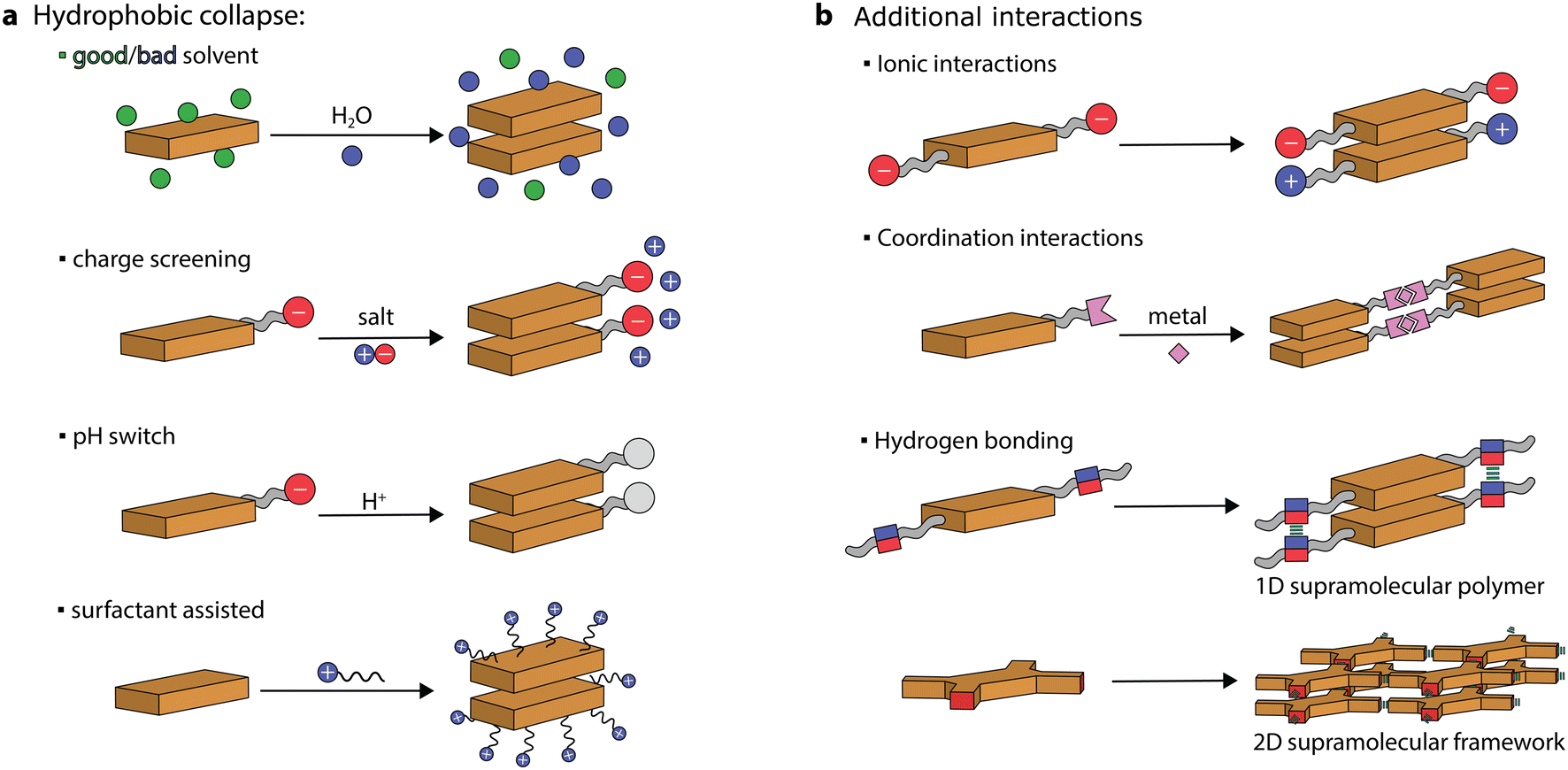 | ||
| Fig. 3 Chromophore aggregation triggered in aqueous media through various protocols. (a) Precipitation, charge screening, acid–base neutralization, and surfactant-assisted self-assembly. (b) Aggregation of dyes through hydrophobic collapse and π–π stacking aided through additional interactions, which include complementary charges, coordination interactions, and hydrogen bonding. | ||
Precipitation
The main approach is to dissolve the chromophore in a good solvent and then to induce its self-assembly by addition of a bad solvent. This is usually achieved by injecting an organic solution of the dye into water, which induces the hydrophobic collapse of monomers into supramolecular structures.39–41Organic molecules comprised of electron-rich donor and electron-deficient acceptor units can facilitate charge transfer in the excited state,42,43 ultimately improving exciton dissociation and enhancing visible light absorption. One of the most exploited self-assembled dye system in photocatalysis is based on perylene diimides (PDIs, perylene-3,4,9,10-tetracarboxylic diimides). PDIs are characterized by a strong conjugation between the electron-rich perylene core and the electron-poor imide groups, resulting in an exceptional acceptor–donor–acceptor scaffold. Unsubstituted PDIs can be initially dissolved in concentrated sulfuric acid, and then its hydrophobic collapse can be induced by the addition of water.44 Crystalline materials can be obtained due to the strong π–π stacking of H-aggregated molecules aided by lateral hydrogen bonding (–N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ). Strong π–π stacking also results in a high orbital overlap between PDI molecules, leading to a deeper valence band of the self-assembled organic semiconductor (+2.20 eV vs. NHE) compared to the LUMO of the monomeric PDI (+1.61 eV vs. NHE), which was exploited for water oxidation to O2, upon visible light irradiation and in the presence of a sacrificial acceptor (Ag+).44 It was suggested that light irradiation leads to photogenerated electrons and holes that are delocalized along the π–π stacking direction.
). Strong π–π stacking also results in a high orbital overlap between PDI molecules, leading to a deeper valence band of the self-assembled organic semiconductor (+2.20 eV vs. NHE) compared to the LUMO of the monomeric PDI (+1.61 eV vs. NHE), which was exploited for water oxidation to O2, upon visible light irradiation and in the presence of a sacrificial acceptor (Ag+).44 It was suggested that light irradiation leads to photogenerated electrons and holes that are delocalized along the π–π stacking direction.
Several other chromophores, consisting of donor–acceptor motifs, have been reported to self-assemble into nanostructures through precipitation protocols and possess photocatalytic activity, ranging from H2 evolution45 to the photooxidation of organic substrates46 and environmental remediation.47 Recently, organic donor–acceptor–donor molecules based on thiophene–benzothiadiazole–thiophene (TBT) were precipitated (THF/H2O) and used for photocatalytic H2 evolution (Fig. 4a).48,49 In the presence of the triethanolamine (TEOA) sacrificial donor, the rod-like aggregates photoproduced H2 with an AQY of approximately 2% (400–450 nm), which was attributed to a combination of broad visible light absorption, a small band gap, wettability, and long exciton lifetimes. Furthermore, it was observed that no additional co-catalyst was added, and the residual palladium from cross-coupling was found to impact the photocatalytic activity.
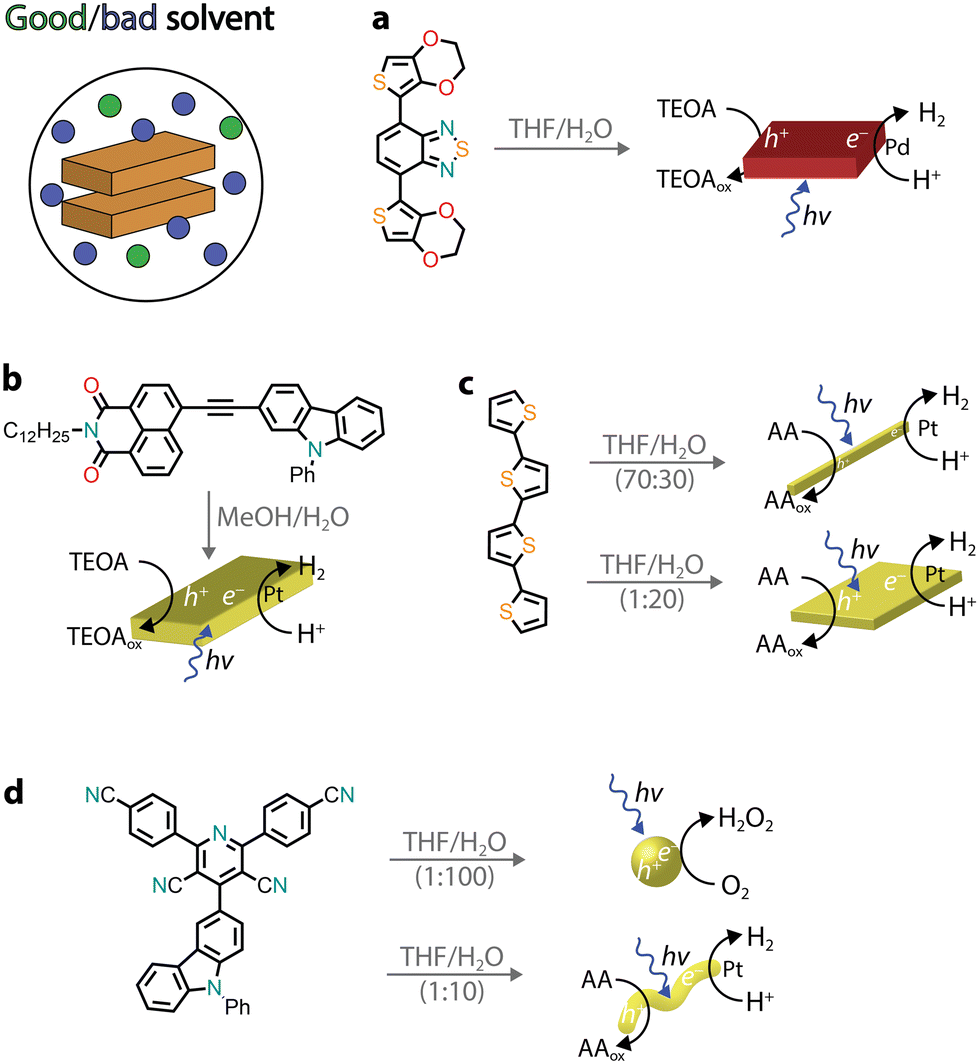 | ||
| Fig. 4 Representative examples of self-assembled nanostructures used in photocatalysis prepared by precipitation. Abbreviations: THF: tetrahydrofuran, TEOA: triethanolamine, and AA: ascorbic acid. | ||
The presence of donor–acceptor groups in monomer dyes can result in molecules with large dipole moments, facilitating the self-assembly process through dipole–dipole interactions. A naphthalene monoimide–phenyl carbazole monomer (NMI–Czl, Fig. 4b) was found to form nanoribbons upon precipitation, driven by alternate stacking of electron-rich moieties (carbazole, Czl) with electron-deficient groups (naphthalene monoimide, NMI).50 It was observed that long-lived charge-separated excitons were generated upon photoexcitation, which were responsible for the photocatalytic activity. Specifically, H2 evolution was observed in the presence of a platinum co-catalyst and TEOA as a sacrificial donor with an AQY of 1.3% at 400 nm.
By precipitating a chromophore from a good solvent to a bad solvent, the volume-to-volume ratio between these two solvents can play an important role in tuning the hydrophobicity and, consequently, the packing between monomers. It is perhaps not surprising that different packings, which usually translate into different morphologies, can lead to dissimilar photocatalytic activity.51–54 In one example, a quaterthiophene (QTH) molecule was observed to form crystalline nanorods in a THF/H2O mixture (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v), whereas a nanosheet morphology was noted when a THF solution was injected into H2O (Fig. 4c).54 It was proposed that the monomers arrange either in a head-to-tail fashion, leading to nanorods, or in a slipped stacking arrangement, resulting in a nanosheet morphology. In the latter, enhanced photoexcited charge transfer was observed, leading to higher H2 photocatalytic activity (in the presence of Pt co-catalysts and the ascorbic acid sacrificial donor). Notably, for quaterthiophene nanosheets, it was claimed that a solar-to-hydrogen efficiency of 18% could be reached when a Pt co-catalyst and a 4-methylbenzyl alcohol sacrificial donor were used. Another recent example is a donor–acceptor organic molecule that was shown to exhibit two different aggregate states, depending on the volume ratio of THF/H2O used for precipitation (Fig. 4d).55 Due to the difference in packing, the two morphologies also showed different photocatalytic activities with the nanospheres producing H2O2 and the nanofibers promoting H2 production. It was proposed that the excitons underwent charge separation facilitated by the nanofiber crystalline structure, while the amorphous nanospheres showed better electron transfer to O2 (critical in the photocatalytic H2O2 production).
:3 v/v), whereas a nanosheet morphology was noted when a THF solution was injected into H2O (Fig. 4c).54 It was proposed that the monomers arrange either in a head-to-tail fashion, leading to nanorods, or in a slipped stacking arrangement, resulting in a nanosheet morphology. In the latter, enhanced photoexcited charge transfer was observed, leading to higher H2 photocatalytic activity (in the presence of Pt co-catalysts and the ascorbic acid sacrificial donor). Notably, for quaterthiophene nanosheets, it was claimed that a solar-to-hydrogen efficiency of 18% could be reached when a Pt co-catalyst and a 4-methylbenzyl alcohol sacrificial donor were used. Another recent example is a donor–acceptor organic molecule that was shown to exhibit two different aggregate states, depending on the volume ratio of THF/H2O used for precipitation (Fig. 4d).55 Due to the difference in packing, the two morphologies also showed different photocatalytic activities with the nanospheres producing H2O2 and the nanofibers promoting H2 production. It was proposed that the excitons underwent charge separation facilitated by the nanofiber crystalline structure, while the amorphous nanospheres showed better electron transfer to O2 (critical in the photocatalytic H2O2 production).
Charge screening
Chromophores that carry charged headgroups can dissolve in aqueous solutions due to the electrostatic repulsion between the ionic groups on the monomers. However, upon the addition of salts, the electrostatic repulsion can be screened, which leads to the hydrophobic collapse of chromophores into ordered aggregates.The self-assembly of several perylene monoimides (PMIs), carrying a carboxylate alkyl chain at the imide position, has been thoroughly studied in the presence of salts. A decade ago, a pioneering study reported the formation of crystalline nanoribbons after the addition of salts to aqueous solutions of a PMI amphiphile (Fig. 5a).56 The addition of salt is responsible for screening the negatively charged headgroups of PMIs, causing hydrophobic collapse into ordered nanostructures. Another notable observation was that the formation of extended nanostructures, and their entanglement, led to the formation of hydrogels. It was also reported that hydrated nanostructures, in which the supramolecular photosensitizers and nickel DuBois catalysts were co-localized, led to better photocatalytic H2 evolution in the presence of sacrificial ascorbic acid compared to dried gels on solid substrates.
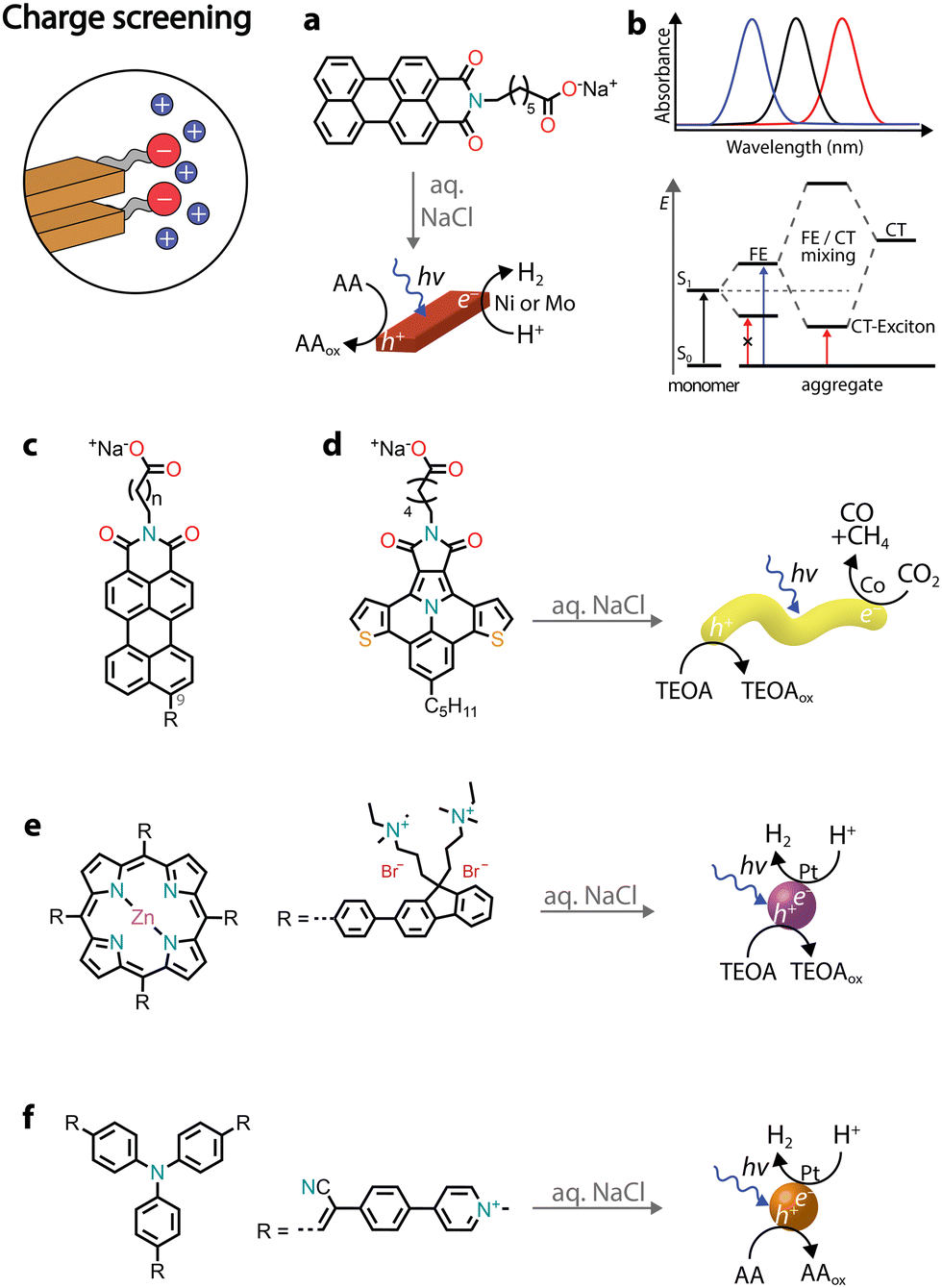 | ||
| Fig. 5 Illustrative examples in which charge screening was employed to self-assemble amphiphilic chromophores and used for the photoproduction of solar fuels. | ||
Recently, it was reported that enhancing the porous scaffold within the PMI hydrogel led to better photocatalytic properties.57 Thermal annealing (at 95 °C for 1 hour) of an aqueous PMI amphiphile solution containing salts led to the growth of large crystalline nanoribbons. The resulting crystalline polymers exhibited a more porous structure compared to control samples, leading to enhanced diffusion of [Mo3S13]2− catalysts and the ascorbic acid sacrificial donor, ultimately contributing to a better photocatalytic H2 evolution.
PMI crystalline assemblies have interesting photophysical properties.58 As mentioned above, chromophore assemblies can form H- and J-aggregates that display blue- and red-shifted absorption relative to the monomer absorbance maxima, respectively. In addition to the long-range coulombic coupling of the chromophore's transition dipole moments (TDMs), in organic crystals also short-range charge transfer (CT) coupling between π-orbitals can affect the formation of excitons. While TDM coupling is primarily influenced by the orientation of chromophores in space, CT coupling is very sensitive to sub-Å molecular displacements. When crystalline PMI assemblies are formed, the orbital orientation between pairs of molecules becomes identical across the crystal, resulting in pronounced CT coupling. The two modes of coupling can even mix, giving rise to the creation of a completely new electronic structure known as the CT exciton (Fig. 5b). Therefore, in PMI assemblies, in addition to the typical absorption blue-shifted peak of H-aggregates, a new red-shifted absorbance feature is observed due to the mixing of TDM and CT states.
Several chemical modifications were reported on PMI amphiphiles, both on the charged headgroup and the perylene core (Fig. 5c). For example, increasing the distance between the charged headgroup and the aromatic core resulted in aggregates with higher orbital overlap and more exciton splitting, leading to better photocatalytic H2 evolution.59 Additionally, different substituents were introduced at position 9 of the perylene core, influencing the dipole moments and energy levels of the chromophores.60,61 Electron-poor substituents (such as –CN) led to nanostructures with no photocatalytic activity, due to the decreased dipole–dipole interactions between monomers and the lowered conduction band below the catalyst's reduction potential. Electron-rich substituents (such as –NH2 or –OCH3) gave nanostructures with photocatalytic H2 activity, but lower when compared to the unsubstituted PMI amphiphiles. Increased dipole–dipole interactions were found to result in a different crystalline packing with lower exciton splitting.60,62 Introducing substituents with varying steric demands was found to influence crystal packing and, subsequently, the formation of CT excitons.63,64 Less sterically demanding substituents led to molecules that quickly crystallized in water, but with no CT exciton formation and a low H2 photocatalytic activity. Bulkier substituents needed addition of salt and thermal annealing to crystallize, but resulted in crystalline nanoribbons with red-shifted CT excitons that could efficiently photosensitize the [Mo3S13]2− catalyst for H2 evolution in the presence of ascorbic acid.
Due to the LUMO energy of PMIs, it is expected that the corresponding nanostructures could predominantly photosensitize proton reduction catalysts. By substituting the perylene core with more electron-rich ones, such as diareno-fused ullazines, supramolecular polymers capable of sensitizing CO2 reduction electrocatalysts could be obtained (Fig. 5d).65 Ullazine imides were found to drive the reduction of CO2 to CO and CH4 in the presence of dicobalt catalysts and sacrificial TEOA over longer periods of time when compared to homogeneous photosensitizers.
The chromore's photophysical properties, other than CT formation, could also be affected by the addition of salts, ultimately contributing to improved H2 evolution. In one example, an octacationic zinc porphyrin (Zn tetraphenylporphyrin fluorene derivative, ZnTPP-FN) was charge screened in the presence of sodium chloride, leading to the formation of aggregated microspheres (Fig. 5e).66 While no long-range order was observed, the aggregation resulted in structures with enhanced communication with the Pt catalysts and faster transfer of photogenerated holes to the TEOA donor. The charge screening by various halide salts was also found to influence the photocatalytic nanostructures (Fig. 5f).67 Specifically, addition of iodide salts was found to increase intersystem crossing, resulting in triplet states with longer lifetimes and achieving an AQY of almost 10% at 500 nm in the presence of Pt co-catalysts and sacrificial ascorbic acid.
pH switch
Similarly to charge screening, acid–base neutralization of charged chromophore amphiphiles also leads to their hydrophobic collapse into nanostructures. Most commonly, chromophores substituted with carboxylic acids are dissolved in water by the addition of a base (such as NaOH), which is followed by the addition of an acid to induce self-assembly through hydrophobic collapse.Several perylene diimide (PDI) derivatives have been well studied as model chromophore amphiphiles for the formation of photocatalytic nanostructures through acid–base neutralization (Fig. 6a).68,69 PDIs can be synthesized in high yields through condensation reactions between perylene dianhydride and substituted amines. A PDI substituted with propanoic acid, in particular, has been exploited for its photocatalytic properties in O2 evolution,701O2 generation,71 and even in environmental remediation.72,73 In one example, the relatively deep valence band (+1.52 V vs. NHE) of the PDI nanofibers was exploited for the water oxidation reaction (+1.23 V vs. NHE).70 In the presence of Ag+ sacrificial acceptors, it was observed that the O2 quantum yield could reach 0.5% at 600 nm. Also in this case, the catalytic activity could be attributed to the formation of π–π stacks that allow separation and migration of the photogenerated carriers. It is interesting to note that the photogenerated electron–hole separation and migration could be tuned by modifying the linker length between the PDI core and the carboxylic acid71 or by self-sorting with nanostructures made of other chromophore amphiphiles (Fig. 6b).74
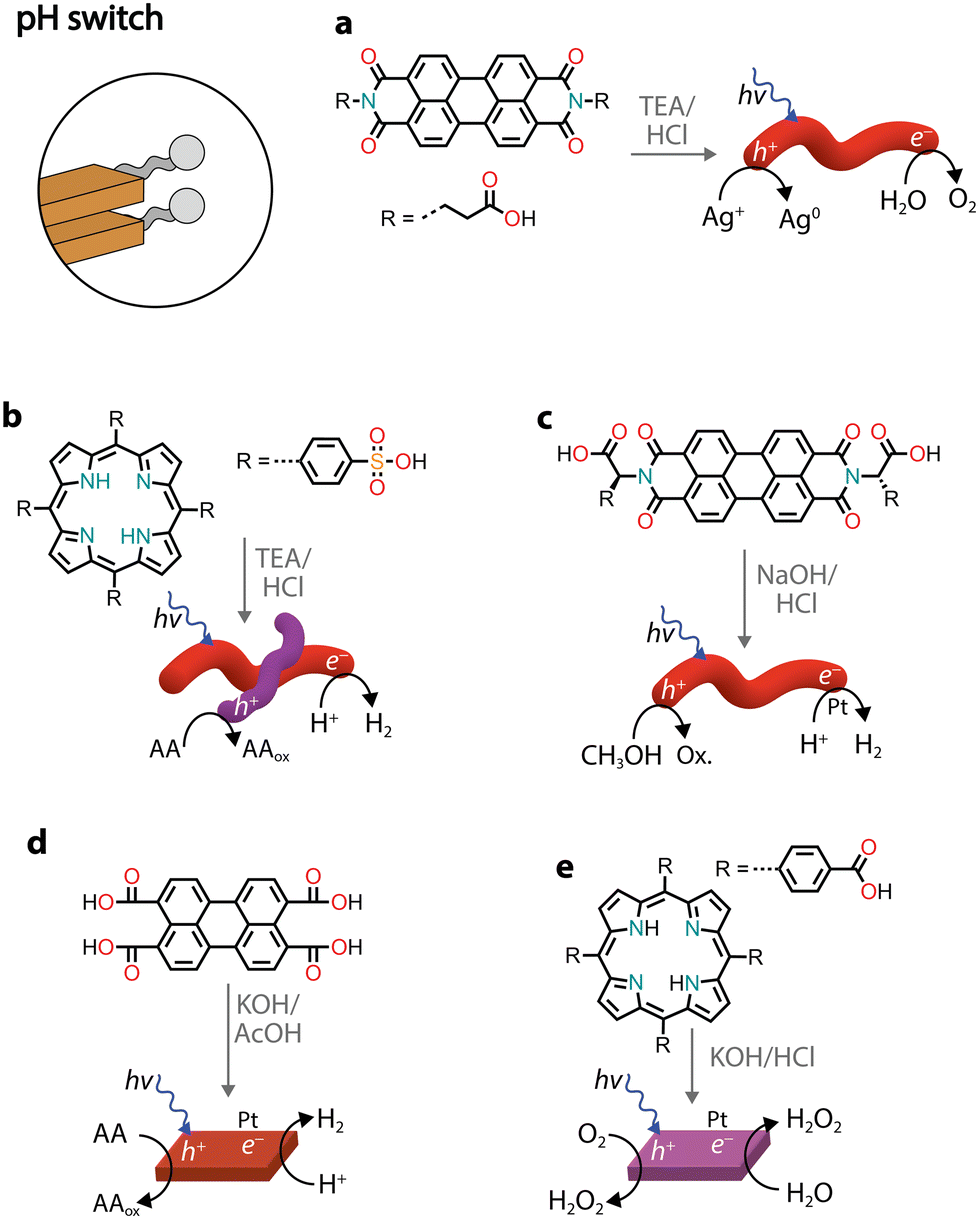 | ||
| Fig. 6 Representative examples of supramolecular nanostructures obtained through acid–base neutralization. Abbreviations: TEA: triethylamine and AcOH: acetic acid. | ||
Conjugates of natural amino acids and PDIs have been carefully studied for their self-assembly into nanofibers (Fig. 6c).75–77 The carboxylic acid group on the amino acid side groups allow the chromophores to be solubilized at high pH. The addition of a dilute acid then reduces solubility through protonation of the side groups, initiating further self-assembly into one-dimensional nanostructures. In addition to pH, the aggregation state can also be controlled by the addition of different amounts of methanol, which serves as a sacrificial electron donor in photocatalytic H2 evolution with Pt co-catalysts.77 It was also noted that the amino acid side chain also played an important role in the local packing within the aggregates, resulting either in strong or weak mixing of Frenkel and CT excitons.
Recently, perylene tetracarboxylic acid (PTCA) was first dissolved in water through hydrolysis of the anhydride precursor and, upon the addition of acetic acid, self-assembled nanosheets were formed (Fig. 6d).78 The nanostructures in the presence of Pt co-catalysts and sacrificial ascorbic acid showed H2 evolution up to AQY = 13.5% at 420 nm. The activity was attributed to a combination of exciton coupling between π–π stacked molecules and hydrophilic carboxylic acids on the surface that come into direct contact with protons used for H2 production.
Porphyrin nanosheets, prepared through acid–base neutralization, have been reported for H2 or O2 evolution79 and very recently for H2O2 photoproduction as well (Fig. 6e).80 In the latter case, it was proposed that H2O2 could be produced through both reductive (O2 to H2O2) and oxidative (H2O to H2O2) pathways. The involvement of both photogenerated electrons and holes allowed the system to achieve a quantum efficiency of 14.9% at 420 nm.80 While the reductive H2O2 production occurred as expected through O2 reduction to superoxide radicals, which subsequently react with protons to form H2O2, the oxidative process did not necessitate a sacrificial donor and managed to use the holes in water oxidation to H2O2. It was proposed that the carboxylic groups on the chromophores could be oxidized to peroxy acid groups, which in the presence of water could thermally decompose into H2O2. This study paves the way forward for the use of organic nanostructures in solar fuel production without the need for additional co-catalysts and sacrificial donors.
Surfactant-assisted self-assembly
Among various protocols, surfactant-assisted self-assembly has been particularly studied in the formation of porphyrin nanostructures.81 In particular, using surfactants allows the use of hydrophobic chromophores that have not been chemically modified. It permits good morphological tuning, stabilizes nanostructures in water, enables controlled growth of nanostructures, and is compatible with the protocols already discussed.A zinc tetra(4-pyridyl)porphyrin, first solubilized in an acidic solution and then injected into a basic solution, yields amorphous irregular particles.82 However, in the presence of surfactants, porphyrin nanocrystals can be obtained (Fig. 7a).82–84 Furthermore, it was observed that nanocrystal nucleation and growth are under kinetic control, which was used to obtain different morphologies. Similarly, a deprotonation–protonation reaction in a porphyrin carrying acidic groups can lead to the formation of nanocrystalline aggregates.85–88 For example, 5,10,15,20-tetrakis(4-(hydroxyl)phenyl)porphyrin (THPP) in the presence of a surfactant (cetyltrimethylammonium bromide, CTAB) yielded aggregates confined within surfactant micelles (Fig. 7b).86 The formation of crystalline nanostructures exhibited higher photocatalytic H2 evolution with a Pt co-catalyst and sacrificial ascorbic acid, especially when compared to the original porphyrin powders.
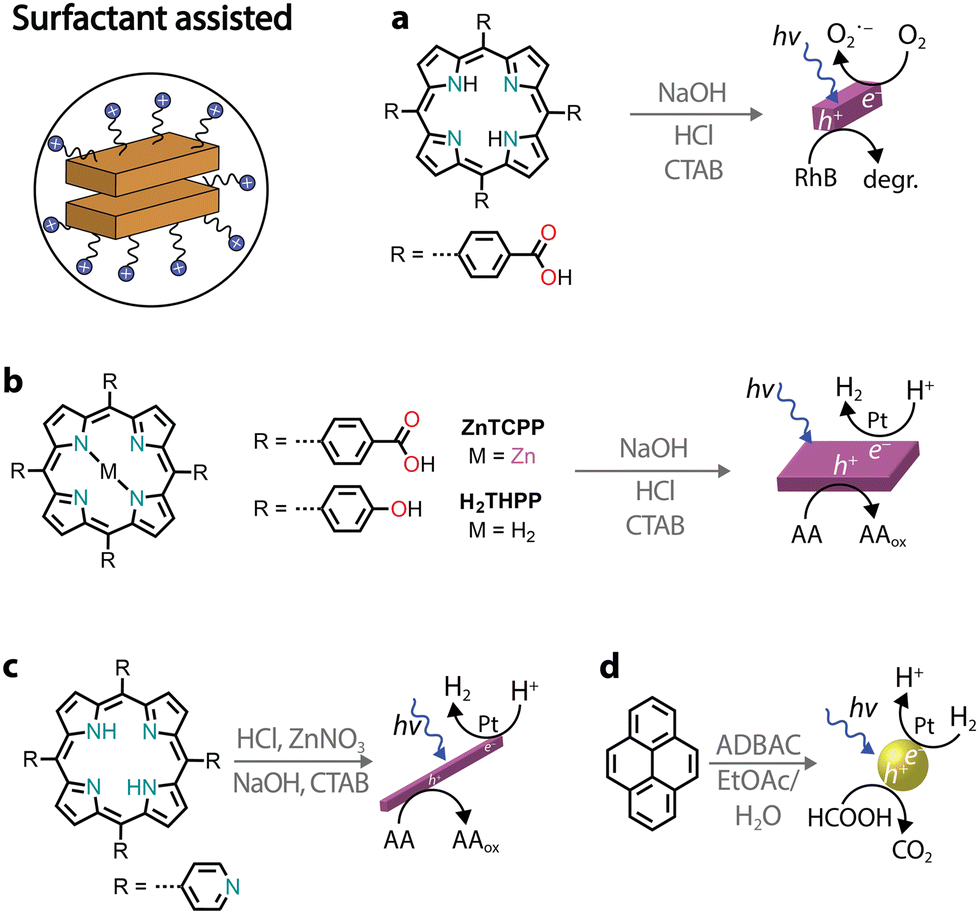 | ||
| Fig. 7 Illustrative examples of photocatalytic nanostructures obtained through surfactant-assisted self-assembly. | ||
As an alternative to the acid–base neutralization of substituted porphyrins, surfactants can also be used to aid precipitation89,90 or facilitate the formation of microemulsions.91 In the latter, oil-in-water emulsions can be heated to evaporate the organic solvent, triggering the self-assembly of nanostructures within the surfactant droplets (Fig. 7c).92,93 Recently, it was also shown that chromophores other than porphyrins can also be benefit from aggregation through the surfactant microemulsion procedure.94 For example, the photocatalytic H2 activity of pyrene aggregates can reach an AQY of 20% at 400 nm in the presence of a platinum co-catalyst and sacrificial formic acid (Fig. 7d).
Hydrogen bonding-aided self-assembly
While π–π interactions can drive the formation of chromophore stacks in aqueous solutions, by introducing lateral groups that are able to hydrogen bond,95 monomers can be guided to form supramolecular nanostructures with improved exciton coupling between the dyes.For instance, a tricationic zinc porphyrin was modified with aramid linkers, which guided the self-assembly of the chromophore amphiphiles into micelles (Fig. 8a).96,97 The quadruple hydrogen bonding between monomers led to photostable nanostructures, while the solubilized monomer was found to photobleach quickly.96 When a negatively charged cobalt porphyrin catalyst was added to interact with a positively charged photosensitizer, the hierarchical nanostructure created a micro-environment capable of driving CO2 reduction to H2, CO, and CH4 with an initial AQY of 15% (at 450 nm) in the presence of a triethylamine sacrificial donor.96 Similar to the aramid linker, chromophores with side chains capable of hydrogen bonding (such as peptide nucleic acids and peptides) were reported to lead to nanostructures with photosensitizing capabilities.98–100
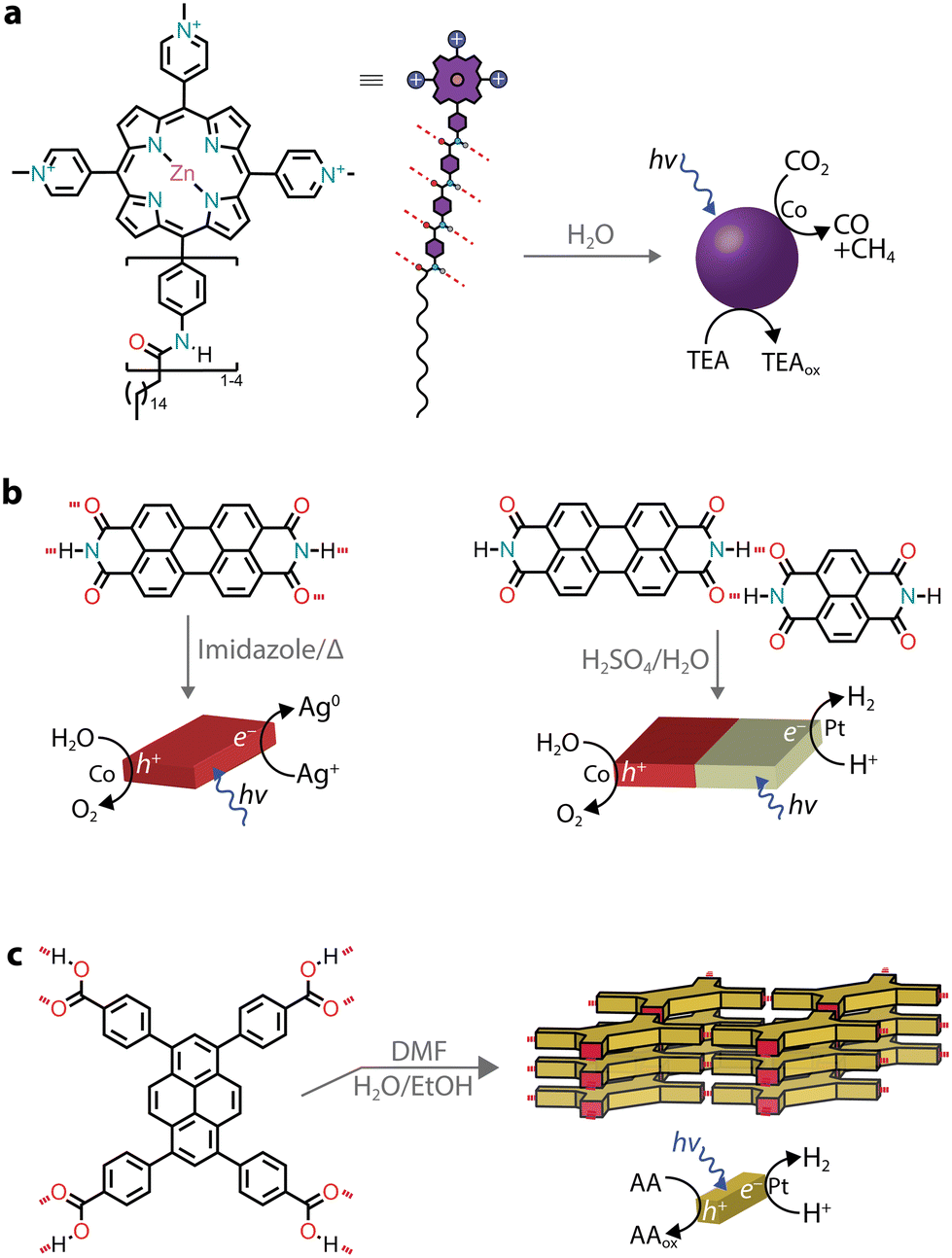 | ||
| Fig. 8 Hydrogen bonding can be used to aid the supramolecular organization of chromophores. | ||
Several PDI derivatives were reported to form lateral hydrogen bonding between the dye stacks, resulting in nanosheets or nanoribbon morphologies.44,68–70,101–103 While most of them were discussed in previous sections, since excitonic coupling occurs along the π–π stacking directions, it has recently been shown that lateral hydrogen bonds can be exploited for the formation of heterojunctions (Fig. 8b).104 The hydrogen bonding between two organic self-assembled nanostructures was reported to create a Z-scheme heterostructure for overall water splitting.104 The photogenerated traps on PDI nanocrystals could catalyse the production of O2 together with a cobalt co-catalyst, while the electrons on naphthalene diimides lead to the production of H2 aided by a platinum co-catalyst. The AQY for O2 production reached 5.2% (at 380 nm) in the presence of an Ag+ sacrificial acceptor.
An interesting case is when discrete tectons are pre-designed to self-assemble with intermolecular hydrogen bonds to obtain crystalline and porous materials, known as hydrogen-bonded organic frameworks (HOF, Fig. 8c). When organic chromophores are devised to carry multiple highly directional complementary H-bonding interactions, frameworks can be obtained. These frameworks take advantage of the π–π stacking between the layers for excitonic coupling and the porous nature of the scaffold.105–108 Recently, a HOF based on 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy) was reported to reach an AQY of 28.6% (at 420 nm) in sacrificial photocatalytic H2 evolution (ascorbic acid and Pt co-catalysts).109 This high activity was attributed to the fast transfer of photogenerated excitons to the adjacent hydrophilic micropores, which reduces the loss of excitons through recombination during their migration. Previously, it was also shown that the same crystalline framework had a 200-fold higher photocatalytic activity compared to the amorphous material.110 It was further reported that by changing the linker length between the pyrene core and the carboxylic acids, two main effects can be observed: (i) the pores are enlarged, aiding the diffusion of molecules within the frameworks111,112 and (ii) the additional π–π stacking between linkers can greatly enhance the chemical and thermal stability of the HOFs.111 In addition to H2 photoproduction, pyrene-based HOFs were also reported for photooxidation of sulfides to sulfoxides through singlet oxygen production.111,113
Interaction with catalysts, (macro)molecules and materials
For improved photocatalytic activities, it is crucial to interface the self-assembled chromophores with metals, catalysts, and materials. Alternatively, the self-assembly of dyes can be assisted, and even templated, by addition of other molecules or polymers.Ionic interactions between complementary charges are commonly used to drive interactions between charged photosensitizers and co-catalysts (Fig. 9a).114–116 It has been suggested that placing complementary positive or negative charges either on the nanostructures or the catalysts is not interchangeable and can lead to a difference of up to 5 times in the photocatalytic production of H2.117
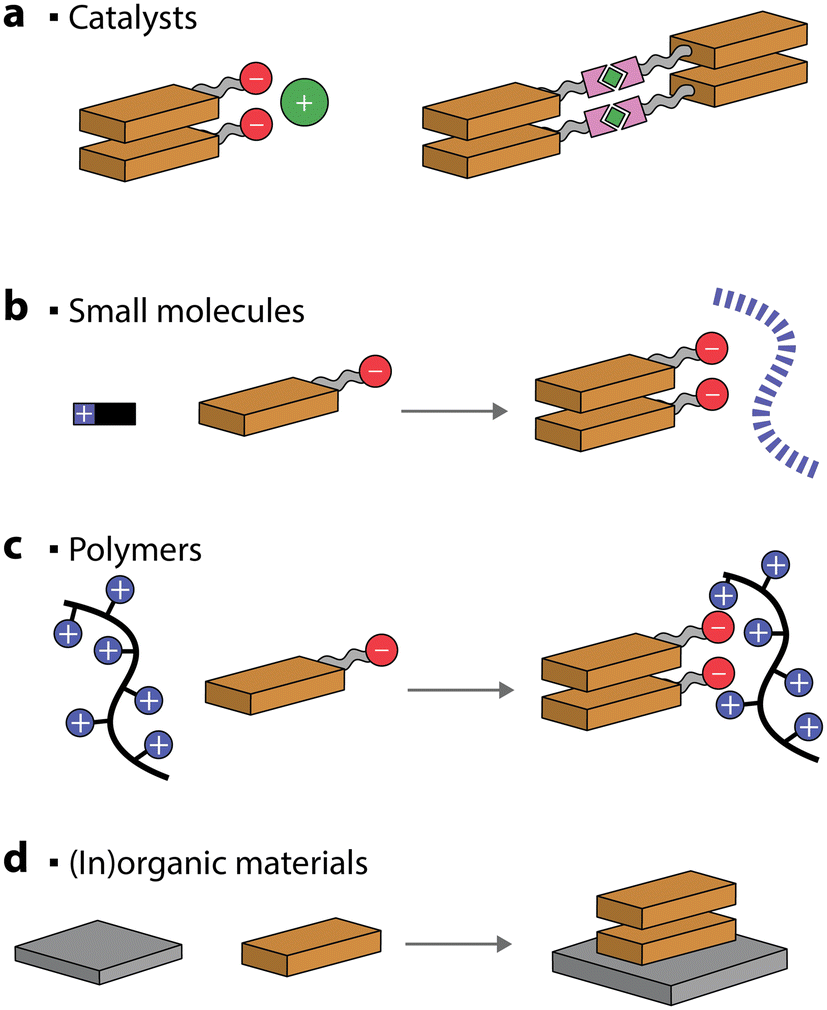 | ||
| Fig. 9 Schematic representation of self-assembled chromophores that can be co-assembled with co-catalysts, small molecules, polymers, or (in)organic materials. | ||
An interesting approach to creating self-assembled chromophore photocatalytic nanostructures is to take advantage of coordination bonds. Metal–ligand coordination can be exploited to decrease the solubility of the dye in the solvent system and thus drive the organized aggregation through π–π interactions.118,119 Even more stimulating is the case where the metals added also serve as catalysts for various chemical reactions (Fig. 9a).120,121 For example, a coordination polymer gel, made through ruthenium–polypyridyl complexes, was utilized to drive CO2 to CH4 photoreduction with an AQY of 7.7% (at 400 nm) in the presence of triethylamine and 1-benzyl-1,4-dihydronicotinamide sacrificial donor. Similarly, a PDI–zirconium self-assembled nanostructure showed good photocatalytic H2 ability, reaching 11.7% AQY (at 630 nm), in the presence of sacrificial ascorbic acid.122 Another strategy used to enhance the photocatalytic performance is anchoring co-catalysts to self-assembled nanostructures through metal–ligand interactions.122–125
Oppositely charged small molecules and chromophores can spontaneously self-assemble into hierarchical nanostructures in the presence of salts and acids.126–128 Small molecules, such as peptides, can typically form supramolecular polymers by themselves. However, when chromophores are added, a long-range order of the latter can be obtained (Fig. 9b). This can result in the excitons being delocalized over multiple porphyrin molecules, which can be utilized for photocatalytic H2 evolution in the presence of Pt catalysts and sacrificial ascorbic acid.126–128 Similarly, hierarchical self-assembled nanostructures were obtained through ionic interactions between Fmoc-dipeptides and tin porphyrins.129 The resulting nanostructures were found to photooxidize H2O to O2 in the presence of iridium oxide as a co-catalyst and persulfate as a sacrificial electron acceptor.
Charged covalent polymers were also shown to act as templates for inducing the self-assembly of chromophores (Fig. 9c).130 For example, PMI amphiphiles were shown to self-assemble into crystalline nanoribbons when loaded into positively charged covalent hydrogels.130 It was observed that the presence of a charged polymer had a similar effect on the chromophore self-assembly to charge screening with salts. Also in this case, the photocatalytic H2 evolution, in the presence of a thiomolybdate catalyst and sacrificial ascorbic acid, was observed. In another example, a positively charged porphyrin was self-assembled on anionic poly(styrene sulfonate) polymeric brushes.131 The polymer, acting as a nanoscale support for controlled porphyrin aggregation, was utilized for light-induced iodide oxidation.
Complementary charges were also employed to self-assemble positively- and negatively-charged chromophores, constructing photosensitizing nanostructures for H2 evolution (in the presence of Pt and TEOA),132,133 to form donor–acceptor nanostructures that promote charge separation134 and could potentially be used for nanostructures interaction with electron mediators in photocatalytic applications.135
Preparation of organic–inorganic composites is another common strategy to improve the photocatalytic activity, mainly due to the improved separation efficiency of photogenerated electron–hole pairs (Fig. 9d). One of the most common self-assembled organic–inorganic materials is PDI/TiO2.136–138 It was proposed that the two can interact through hydrogen bonding and it was noted that the hybrid could reach an AQY for H2 production of 70% (at 365 nm) in the presence of Pt as a co-catalyst and methanol as a sacrificial donor.139 TiO2 could also be deposited in situ on one-dimensional PDI nanofibers and the composite was utilized for H2 evolution in the presence of a Pt co-catalyst and an amine sacrificial donor.140,141 Heterojunctions of PDIs have also been reported with Ag2S quantum dots,142 SnO2 quantum dots,143 Pd quantum dots,144 indium tin oxide,145 Bi2WO6,146 and Ti3C2Tx MXenes,147 to name a few. Another possibility for preparing heterojunction photocatalysts is to self-assemble PDIs in the presence of organic materials, such as graphene148 and carbon nitride.149 Other commonly utilized self-assembled chromophores to form composited are porphyrins, which have been reported in conjunction with, for example, TiO2150–154 and graphene.155,156 The common observation for all the mentioned composited is that their photocatalytic activity was higher when compared to the individual components, demonstrating that this is a viable strategy for further improving the efficiency of self-assembled chromophores.
Energy landscapes of supramolecular photocatalysts
Depending on the strength and dynamics of non-covalent bonds, there can be a subtle interplay between kinetics and thermodynamics during aggregation. Therefore, a supramolecular aggregate might follow the kinetic pathway rather than the thermodynamic one. Experimental studies have shown that the outcome between kinetic and thermodynamic aggregates depends on the preparation methods, which span from temperature variation to solvent processing.157 In the context of supramolecular chromophore nanostructures, the kinetic and thermodynamic aggregates might possess different photocatalytic properties, especially if the excitonic coupling in the two states is different.This was the case for a PMI amphiphile with a propyl tail at position 9, which was observed to form two distinct supramolecular architectures with different photocatalytic activities (Fig. 10a).158 When the chromophore was dissolved in an aqueous salt solution, it formed crystalline ribbons. Upon thermal annealing, the ribbons were transformed into crystalline scroll-like nanostructures. Importantly, the transformation led to a three-fold improvement in photocatalytic H2 production with a thiomolybdate catalyst and ascorbic acid as a sacrificial donor. Only in the stable phase, the formation of CT excitons was observed, which were responsible for the enhancement of H2 evolution.
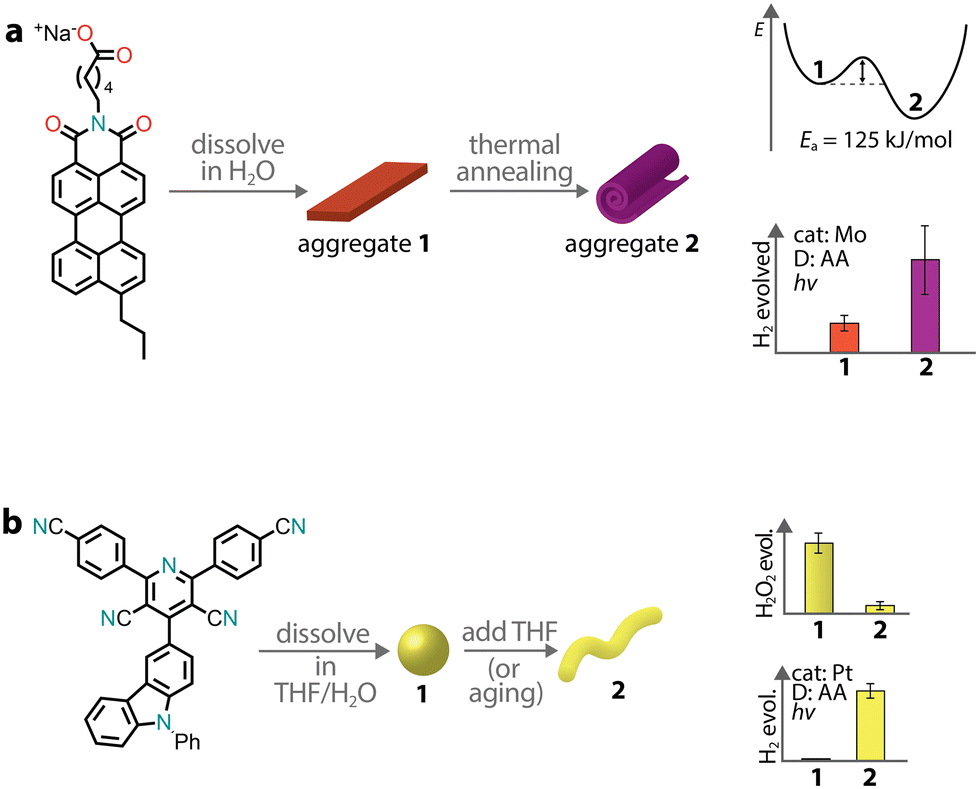 | ||
| Fig. 10 Aggregation leading to the formation of kinetic and thermodynamic products, which can have different photocatalytic activities. | ||
We already mentioned a donor–acceptor organic molecule that exhibited the formation of two different aggregate states in aqueous dispersion, depending on the THF/water ratio used in the precipitation procedure.55 It was also observed that amorphous nanospheres could transform into crystalline nanofibers upon aging, suggesting that the former are the kinetic product while the latter the thermodynamic one (Fig. 10b). This case is quite peculiar as one morphology (amorphous nanospheres) promoted H2O2 photoproduction, while the other (crystalline nanofibers) supported sacrificial H2 evolution.
In addition to aging and thermal annealing, there is also a need to develop alternative strategies to obtain thermodynamic aggregates. Since rapidly injecting a solution into a bad solvent could lead to the formation of kinetic aggregates, an in situ enzymatic hydrolysis method for self-assembled chromophores was proposed.159 Diketopyrrolopyrrole, functionalized with amino acid methyl esters, was found to produce supramolecular hydrogels after enzymatic hydrolysis. The hydrogels were used to produce 1O2, which was exploited for the oxidation of sulfides to sulfoxides.
Conclusions
In this contribution, we have discussed the use of self-assembled chromophores in the field of photocatalytic energy conversion, with the representative examples summarized in Table 1. Due to their large π-conjugated structures, dye–dye stacking is driven by π–π and hydrophobic interactions, enabling the formation of aggregates. A particularly interesting case of chromophore amphiphile self-assembly is the formation of hydrogels with their three-dimensional porous networks allowing co-localization and easy diffusion of the species involved in catalysis.| Compounda | Prep.b | Morphology | Gas evolution ratec/mmol g−1 h−1 | TONc/molprod molcat−1) | Light source | AQYd (nm)/% | Catalyste | Cond.f | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a For brevity, here it is reported the abbreviation of the compound used in the main text and, for the complete chemical structure, it is suggested either to check the figures or the references. b Solvents and reagents (or their mixtures) used for the preparation of the nanostructures. c Values as reported (or estimated by us from figures when not explicitly mentioned) from the references; TON stands for turnover number; n.r. stands for not reported. d Apparent quantum yield also referred to as the quantum efficiency and photonic efficiency in some of the references; n.r. stands for not reported. e Metals and type of co-catalyst reported for photocatalytic activity (synthesis stands for the residual content from synthesis; photodep. stands for photodeposition; NPs stands for nanoparticles). f Conditions used for photocatalytic testing which include sacrificial agents and solvents; TEOA is triethanolamine, AA is ascorbic acid, MBA is 4-methyl benzyl alcohol, and TEA is triethylamine. | |||||||||
| D–A molecule | THF/H2O | Nano/micro-rods | H2: 0.142 | n.r. | Monochromatic LED (λ ≥ 420 nm) | 2.0 (400–450 nm) | Pd (synthesis) | TEOA in H2O | 48 |
| NMI–Czl | MeOH/H2O | Nanoribbons | H2: 0.417 (3 h) | n.r. | Xe lamp (λ ≥ 400 nm) | 1.3 (400 nm) | Pt (photodep.) | TEOA in H2O | 50 |
| QTH | THF/H2O | Nanosheets | H2: 1.21 (12 h) | n.r. | Xe lamp (110 mW cm−2) | 3 (365 nm) | Pt (photodep.) | MBA in H2O | 54 |
| D–A molecule | THF/H2O | Nanospheres | H2O2: 3.20 (1 h) | n.r. | Solar simulator (AM1.5G) | n.r. | — | H2O (O2 atm) | 55 |
| THF/H2O | Nanofibers | H2: 31.85 (1 h) | n.r. | 4.43 (420 nm) | Pt (photodep.) | AA in H2O | |||
| PMI | NaCl aq. | Nanoribbons | H2: 0.63 (18 h) | 3.4 × 102 (18 h) | Halogen bulb (250 W cm−2) | n.r. | Ni DuBois (molecular) | AA in H2O | 56 |
| PMI | NaCl aq. | Nanoribbons | n.r. | 7.5 × 103 (110 h) | Halogen bulb (250 W cm−2) | 0.06 (505 nm) | [Mo3S13]2− (molecular) | AA in H2O | 57 |
| NaCl aq. (thermal annealing) | n.r. | 1.35 × 104 (110 h) | 0.13 (505 nm) | ||||||
| PMI | NaCl aq. (thermal annealing) | Nanoribbons | n.r. | H2: 1.5 × 103 (18 h) | Halogen bulb (250 W cm−2) | n.r. | [Mo3S13]2− (molecular) | AA in H2O | 63 |
| PMI | NaCl aq. (thermal annealing) | Nanoribbons (helical) | n.r. | H2: 1.1 × 104 (18 h) | Halogen bulb (250 W cm−2) | n.r. | [Mo3S13]2− (molecular) | AA in H2O | 158 |
| Ullazine imides | NaCl aq. (thermal annealing) | Nanofibers | n.r. | CO: 4.6 × 104 | 450 nm LED (200 mW cm−2) | n.r. | Dicobalt criptate (molecular) | TEOA in CH3CN/H2O (CO2 atm) | 65 |
| CH4: 1.5 × 104 | |||||||||
| H2: 26 (144 h) | |||||||||
| ZnTPP-FN | NaCl aq. | Microspheres | H2: 10.8 (6 h) | n.r. | Xe lamp (AM1.5G, 100 mW cm−2) | n.r. | Pt (photodep.) | TEOA in H2O | 66 |
| D–A molecule | NaI aq. | Nanoparticles | H2: 460 (ca. 2.5 h) | Ca. 9 × 103 (12 h) | Xe lamp (3.3 mW cm−2, λ ≥ 450 nm) | 9.8 (500 nm) | Pt (photodep.) | AA in H2O | 67 |
| PDI | H2SO4/H2O | Nanosheets | H2: 1.1 (ca. 1.5 h) | n.r. | Xe lamp (385 mW cm−2, λ ≥ 420 nm) | n.r. | Pt (photodep.) | AA in H2O | 68 |
| PDI | TEA/HCl and H2O | Nanofibers | O2: ca. 0.02 | n.r. | Xe lamp (48 mW cm−2, 600 nm) | 0.56 (600 nm) | — | AgNO3 in H2O | 70 |
| PDI/TPPS | TEA/HCl and H2O | Nanofibers | H2: 30.36 (3 h) | n.r. | Xe lamp (600 mW cm−2) | 3.81 (650 nm) | Pt (photodep.) | AA in H2O | 74 |
| PDI | NaOH/HCl and H2O | Nanofibers | H2: 0.003 (4 h) | 1.58 × 102 (307 h) | Xe lamp (100 mW cm−2) | 0.018 (365 nm) | Pt (NPs) | MeOH in H2O | 75 |
| PTCA | KOH/CH3COOH and H2O | Nanosheets | H2: 41.8 (2.5 h) | n.r. | Xe lamp (100 mW cm−2, λ ≥ 420 nm) | 13.5 (420 nm) | Pt (photodep.) | AA in H2O | 78 |
| TCPP | KOH/HCl and H2O | Nanosheets | H2: 0.041 (6 h) | n.r. | Xe lamp (152 mW cm−2) | n.r. | — | TEOA in H2O | 79 |
| O2: 0.036 (6 h) | AgNO3 in H2O | ||||||||
| TCPP | KOH/HCl and H2O | Nanosheets | H2O2: 1.25 (4 h) | n.r. | Xe lamp (λ ≥ 420 nm) | 14.9 (420 nm) | — | H2O (O2 atm) | 80 |
| ZnTPyP | HCl/NaOH and CTAB H2O | Nanocrystals | H2: 47.1 (5 h) | n.r. | Xe lamp (λ ≥ 420 nm) | n.r. | Pt (photodep.) | AA in H2O | 83 |
| THPP | NaOH/HCl and CTAB H2O | Nanorods | H2: 19.5 (5 h) | n.r. | Xe lamp (90 mW cm−2, λ ≥ 420 nm) | n.r. | Pt (photodep.) | AA in H2O | 86 |
| ZnTCPP/THPP | NaOH/HCl and CTAB H2O | Nanosheets | H2: 41.4 (9 h) | n.r. | Xe lamp (632 mW cm−2) | 2.07 (420 nm) | Pt (photodep.) | AA in H2O | 88 |
| InTPP | CHCl3/H2O and CTAB | Nanorods | H2: 845.4 (5 h) | n.r. | Xe lamp (λ ≥ 400 nm) | n.r. | Pt (photodep.) | AA in H2O | 93 |
| Pyrene | EtOAc/ADBAC and H2O | Nanoparticles | H2: ca. 0.16 (4 h) | n.r. | LED lamp (50 W, 400 nm) | 20.8 (400 nm) | Pt (photodep.) | Formic acid | 94 |
| Zn porphyrin aramid | H2O | Nanomicelles | CH4: 8.5, CO: 2.8, H2: 1.6 (30 d) | CH4: 6.6 × 103, CO: 2.2 × 103, H2: 1.2 × 103 (30 d) | Xe lamp (AM1.5G, λ ≥ 420 nm) | 15.1 (450 nm) | Co complex (molecular) | TEA in H2O (CO2 atm) | 96 |
| NDINH/PDINH | H2SO4/H2O–ethylene glycol | Nanowires | H2: 0.32 | n.r. | Xe lamp (783 mW cm−2) | 3.36 (Pt, TEOA, 380 nm) | — | H2O | 104 |
| O2: 0.15 (4 h) | 5.16 (Co, AgNO3, 380 nm) | ||||||||
| TBAPy | DMF/H2O–EtOH | Framework | H2: 358 (2.5 h) | n.r. | Xe lamp (500 mW cm−2) | 28.6 (420 nm) | Pt (photodep.) | AA in H2O | 109 |
| TBAPy | DMF/CHCl3–acetone | Framework | H2: 3.1 (20 h) | n.r. | Xe lamp (λ ≥ 420 nm) | 4.1 (420 nm) | Pt (photodep.) | AA in H2O | 110 |
The overlap of monomer molecular orbitals in the aggregate is beneficial for the formation of band-like electronic energy structures. Upon irradiation with visible light, the electrons in the valence band of the aggregates transition to the conduction band, generating electron–hole pairs. These pairs then undergo separation into free charges, ultimately driving redox reactions. Most commonly, these reactions lead to the production of H2, O2 evolution, and photodegradation reactions. However, examples of photooxidation of organic substrates, H2O2 production, and CO2 reduction have also been reported lately.
Despite the progress in this emerging field, several challenges lie ahead. While synthetic feasibility allows rational incorporation of functional groups, their effect on self-assembly, packing, and photocatalysis is not immediate and often is difficult to predict. Further efforts should also be directed toward the structural characterization of the aggregates. While the intrinsic nature of chromophores and their aggregates allows them to be studied by photophysical means (especially when coupled with theoretical calculations), the packing mode between the molecules is often not fully elucidated. In this context, having highly ordered assemblies can certainly be of help, but often this comes at the expense of the dynamic and reversible nature typical of supramolecular systems. For further advancements, it is also important to study the supramolecular polymerization processes, especially regarding the formation of kinetic and thermodynamic aggregates.
The photocatalytic activity of self-assembled chromophores also faces several challenges. Among these, stability against photodegradation, recyclability, photocatalytic mechanisms, and apparent quantum yields for a photocatalytic reaction are not always reported. A key question to be addressed in future research is whether self-assembled organic semiconductors can carry out the catalytic conversion by themselves or with the assistance of Earth-abundant, low-cost metal co-catalysts. While there are some indications that these systems can be integrated into electrodes, it is yet to be determined if photoelectrochemical systems can eliminate the need for sacrificial agents or enable new redox strategies. Expanding the portfolio of accessible chemical reactions, including organic photoredox transformations and photobiocatalysis, is another open question for the field. To further optimize the photocatalytic activity, attention should be placed on tuning the band-like structure of these materials, in which robotic experimentation could be of great help.160,161 The band structure could be tuned through chromophore co-assembling and alloying between crystalline aggregates,162,163 as well as attaining precise integration of the catalytic components (already demonstrated with covalent polymers).164 Improving the mechanical (and photophysical) properties of self-assembled chromophores, which could be achieved with hybrid supramolecular covalent materials,165–167 could allow the preparation of robust photocatalysts that might be even 3D printed.168 Finally, the dynamic nature of the aggregates could be used to obtain photocatalytic activity in an out-of-equilibrium state.169,170
Towards the full exploitation of self-assembled dyes for photocatalysis, it is desirable to address these open challenges. However, organic self-assembled materials have already provided a unique platform in photocatalysis. Promising research has shown the virtually infinite possibilities to tune these soft supramolecular photocatalysts for solar fuel generation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union (ERC, PhotoDark, 101077698). Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them.References
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- J. Otsuki, J. Mater. Chem. A, 2018, 6, 6710–6753 RSC.
- T. Keijer, T. Bouwens, J. Hessels and J. N. H. Reek, Chem. Sci., 2021, 12, 50–70 RSC.
- P. T. Smith, E. M. Nichols, Z. Cao and C. J. Chang, Acc. Chem. Res., 2020, 53, 575–587 CrossRef CAS PubMed.
- A. Bhattacharyya, S. De Sarkar and A. Das, ACS Catal., 2021, 11, 710–733 CrossRef CAS.
- C. Wang, M. P. O'Hagan, B. Willner and I. Willner, Chem. – Eur. J., 2022, 28, e202103595 CrossRef CAS PubMed.
- R. W. Jadhav, D. N. Nadimetla, V. K. Gawade, L. A. Jones and S. V. Bhosale, Chem. Rec., 2023, 23, e202200180 CrossRef CAS PubMed.
- M. Rudolf, S. V. Kirner and D. M. Guldi, Chem. Soc. Rev., 2016, 45, 612–630 RSC.
- J. T. Nielsen, N. V. Kulminskaya, M. Bjerring, J. M. Linnanto, M. Rätsep, M. Ø. Pedersen, P. H. Lambrev, M. Dorogi, G. Garab, K. Thomsen, C. Jegerschöld, N.-U. Frigaard, M. Lindahl and N. Chr. Nielsen, Nat. Commun., 2016, 7, 12454 CrossRef CAS PubMed.
- G. Scheibe, Angew. Chem., 1937, 50, 212–219 CrossRef CAS.
- E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS.
- T. Aida, A. Takemura, M. Fuse and S. Inoue, J. Chem. Soc., Chem. Commun., 1988, 391 RSC.
- C. Fouquey, J. Lehn and A. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS.
- V. L. Malinovskii, D. Wenger and R. Häner, Chem. Soc. Rev., 2010, 39, 410–422 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- N. Sakai and S. Matile, Beilstein J. Org. Chem., 2012, 8, 897–904 CrossRef CAS PubMed.
- J. D. Tovar, Acc. Chem. Res., 2013, 46, 1527–1537 CrossRef CAS PubMed.
- S. Sengupta and F. Würthner, Acc. Chem. Res., 2013, 46, 2498–2512 CrossRef CAS PubMed.
- A. Jain and S. J. George, Mater. Today, 2015, 18, 206–214 CrossRef CAS.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- S. Chen, R. Costil, F. K. Leung and B. L. Feringa, Angew. Chem., Int. Ed., 2021, 60, 11604–11627 CrossRef CAS PubMed.
- L. MacFarlane, C. Zhao, J. Cai, H. Qiu and I. Manners, Chem. Sci., 2021, 12, 4661–4682 RSC.
- M. Coste, E. Suárez-Picado and S. Ulrich, Chem. Sci., 2022, 13, 909–933 RSC.
- Y. Guo, Q. Zhou, B. Zhu, C. Y. Tang and Y. Zhu, EES Catal., 2023, 1, 333–352 RSC.
- O. Dumele, J. Chen, J. V. Passarelli and S. I. Stupp, Adv. Mater., 2020, 32, 1907247 CrossRef CAS PubMed.
- B. Yang, L. Lu, S. Liu, W. Cheng, H. Liu, C. Huang, X. Meng, R. D. Rodriguez and X. Jia, J. Mater. Chem. A, 2024, 12, 3807–3843 RSC.
- J. Tian, J. Yu, Q. Tang, J. Zhang, D. Ma, Y. Lei and Z.-T. Li, Mater. Futures, 2022, 1, 042104 CrossRef.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- T. Brixner, R. Hildner, J. Köhler, C. Lambert and F. Würthner, Adv. Energy Mater., 2017, 7, 1700236 CrossRef.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- Y. Zhang, H. Shaikh, A. J. Sneyd, J. Tian, J. Xiao, A. Blackburn, A. Rao, R. H. Friend and I. Manners, J. Am. Chem. Soc., 2021, 143, 7032–7041 CrossRef CAS PubMed.
- X.-H. Jin, M. B. Price, J. R. Finnegan, C. E. Boott, J. M. Richter, A. Rao, S. M. Menke, R. H. Friend, G. R. Whittell and I. Manners, Science, 2018, 360, 897–900 CrossRef CAS PubMed.
- Y. Zhang, J. Tian, H. Shaikh, H. K. MacKenzie, Y. He, C. Zhao, S. Lei, Y. Ren and I. Manners, J. Am. Chem. Soc., 2023, 145, 22539–22547 CrossRef CAS PubMed.
- Y. Bai, L. Wilbraham, H. Gao, R. Clowes, H. Yang, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 19958–19964 RSC.
- Z. Jiang, P. Wang, G. Liang, X. Wen, G. Huang, H. Song, B. Jiang, S. Jin, F. Xu, X. Ding, T. K. Kim, H. Chen, J. Yu, J. Ye and S. Wang, Adv. Funct. Mater., 2023, 33, 2303335 CrossRef CAS.
- V. S. Vyas, V. W. Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- G. Geng, Z. Wang, P. Chen, B. Guan, C. Yang and M. Liu, Phys. Chem. Chem. Phys., 2018, 20, 8488–8497 RSC.
- D. D. La, R. W. Jadha, N. M. Gosavi, E. R. Rene, T. A. Nguyen, B. Xuan-Thanh, D. D. Nguyen, W. J. Chung, S. W. Chang, X. H. Nguyen, L. D. Tran and S. V. Bhosale, J. Water Process Eng., 2021, 40, 101876 CrossRef.
- D. Zhang, L.-Y. Peng, K.-X. Teng, L.-Y. Niu, G. Cui and Q.-Z. Yang, ACS Mater. Lett., 2023, 5, 180–188 CrossRef CAS.
- L. Wang and W. Zhu, Adv. Sci., 2023, 2307227 Search PubMed.
- Y. Jiao, L. Đorđević, H. Mao, R. M. Young, T. Jaynes, H. Chen, Y. Qiu, K. Cai, L. Zhang, X.-Y. Chen, Y. Feng, M. R. Wasielewski, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 8000–8010 CrossRef CAS PubMed.
- D. Liu, J. Wang, X. Bai, R. Zong and Y. Zhu, Adv. Mater., 2016, 28, 7284–7290 CrossRef CAS PubMed.
- L. Liu, X. Chen, Y. Chai, W. Zhang, X. Liu, F. Zhao, Z. Wang, Y. Weng, B. Wu, H. Geng, Y. Zhu and C. Wang, Chem. Eng. J., 2022, 444, 136621 CrossRef CAS.
- S. Dadwal, H. Deol, M. Kumar and V. Bhalla, Sci. Rep., 2019, 9, 11142 CrossRef PubMed.
- G. Liu, S. You, Y. Zhang, H. Huang and H. Spanjers, J. Colloid Interface Sci., 2019, 553, 666–673 CrossRef CAS PubMed.
- X. Yuan, C. Wang, L. Vallan, A. T. Bui, G. Jonusauskas, N. D. McClenaghan, C. Grazon, S. Lacomme, C. Brochon, H. Remita, G. Hadziioannou and E. Cloutet, Adv. Funct. Mater., 2023, 33, 2211730 CrossRef CAS.
- X. Yuan, K. Yang, C. Grazon, C. Wang, L. Vallan, J. Isasa, P. M. Resende, F. Li, C. Brochon, H. Remita, G. Hadziioannou, E. Cloutet and J. Li, Angew. Chem., Int. Ed., 2024, 63, e202315333 CrossRef CAS PubMed.
- H. Lin, J. Wang, J. Zhao, Y. Zhuang, B. Liu, Y. Zhu, H. Jia, K. Wu, J. Shen, X. Fu, X. Zhang and J. Long, Angew. Chem., Int. Ed., 2022, 61, e202117645 CrossRef CAS PubMed.
- V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Chem. Commun., 2021, 57, 4055–4058 RSC.
- E. Orfanos, K. Ladomenou, P. Angaridis and A. G. Coutsolelos, Dalton Trans., 2022, 51, 8009–8014 RSC.
- Y. He, R. Li, A. Cao, X. Xu, W. Huang, M. Sun, J. Li, X. Chen and L. Kang, Cryst. Growth Des., 2022, 22, 2620–2627 CrossRef CAS.
- B. Kommula, P. Durairaj, S. Mishra, S. Kar, A. Sury, A. Kumar, A. K. De, S. Sarkar and S. Bhattacharyya, ACS Appl. Nano Mater., 2022, 5, 14746–14758 CrossRef CAS.
- H. Yang, C. Li, T. Liu, T. Fellowes, S. Y. Chong, L. Catalano, M. Bahri, W. Zhang, Y. Xu, L. Liu, W. Zhao, A. M. Gardner, R. Clowes, N. D. Browning, X. Li, A. J. Cowan and A. I. Cooper, Nat. Nanotechnol., 2023, 18, 307–315 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- A. J. Dannenhoffer, H. Sai, B. Harutyunyan, A. Narayanan, N. E. Powers-Riggs, A. N. Edelbrock, J. V. Passarelli, S. J. Weigand, M. R. Wasielewski, M. J. Bedzyk, L. C. Palmer and S. I. Stupp, Nano Lett., 2021, 21, 3745–3752 CrossRef CAS PubMed.
- N. J. Hestand, R. V. Kazantsev, A. S. Weingarten, L. C. Palmer, S. I. Stupp and F. C. Spano, J. Am. Chem. Soc., 2016, 138, 11762–11774 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, D. J. Fairfield, A. R. Koltonow and S. I. Stupp, J. Am. Chem. Soc., 2015, 137, 15241–15246 CrossRef CAS PubMed.
- A. S. Weingarten, A. J. Dannenhoffer, R. V. Kazantsev, H. Sai, D. Huang and S. I. Stupp, J. Am. Chem. Soc., 2018, 140, 4965–4968 CrossRef CAS PubMed.
- L. Đorđević, H. Sai, Y. Yang, N. A. Sather, L. C. Palmer and S. I. Stupp, Angew. Chem., Int. Ed., 2023, 62, e202214997 CrossRef PubMed.
- B. Harutyunyan, A. Dannenhoffer, S. Kewalramani, T. Aytun, D. J. Fairfield, S. I. Stupp and M. J. Bedzyk, J. Phys. Chem. C, 2017, 121, 1047–1054 CrossRef CAS.
- R. V. Kazantsev, A. J. Dannenhoffer, T. Aytun, B. Harutyunyan, D. J. Fairfield, M. J. Bedzyk and S. I. Stupp, Chem, 2018, 4, 1596–1608 CAS.
- H. Sai, G. C. Lau, A. J. Dannenhoffer, S. M. Chin, L. Đorđević and S. I. Stupp, Nano Lett., 2020, 20, 4234–4241 CrossRef CAS PubMed.
- O. Dumele, L. Đorđević, H. Sai, T. J. Cotey, M. H. Sangji, K. Sato, A. J. Dannenhoffer and S. I. Stupp, J. Am. Chem. Soc., 2022, 144, 3127–3136 CrossRef CAS PubMed.
- X. Yang, Z. Hu, Q. Yin, C. Shu, X. Jiang, J. Zhang, X. Wang, J. Jiang, F. Huang and Y. Cao, Adv. Funct. Mater., 2019, 29, 1808156 CrossRef.
- H. Lee, A. Abudulimu, J. C. Roldao, H. Nam, J. Gierschner, L. Lüer and S. Y. Park, ChemPhotoChem, 2023, 7, e202200177 CrossRef CAS.
- Y. Sheng, W. Li, Y. Zhu and L. Zhang, Appl. Catal., B, 2021, 298, 120585 CrossRef CAS.
- H. Xu, Z. Wang, S. Feng, X. Liu, X. Gong and J. Hua, Int. J. Hydrogen Energy, 2023, 48, 8071–8081 CrossRef CAS.
- J. Wang, W. Shi, D. Liu, Z. Zhang, Y. Zhu and D. Wang, Appl. Catal., B, 2017, 202, 289–297 CrossRef CAS.
- J. Wang, D. Liu, Y. Zhu, S. Zhou and S. Guan, Appl. Catal., B, 2018, 231, 251–261 CrossRef CAS.
- P. Chen, L. Blaney, G. Cagnetta, J. Huang, B. Wang, Y. Wang, S. Deng and G. Yu, Environ. Sci. Technol., 2019, 53, 1564–1575 CrossRef CAS PubMed.
- Q. Zhang, L. Jiang, J. Wang, Y. Zhu, Y. Pu and W. Dai, Appl. Catal., B, 2020, 277, 119122 CrossRef CAS.
- J. Yang, J. Jing, W. Li and Y. Zhu, Adv. Sci., 2022, 9, 2201134 CrossRef CAS PubMed.
- M. C. Nolan, J. J. Walsh, L. L. E. Mears, E. R. Draper, M. Wallace, M. Barrow, B. Dietrich, S. M. King, A. J. Cowan and D. J. Adams, J. Mater. Chem. A, 2017, 5, 7555–7563 RSC.
- E. R. Draper, L. Wilbraham, D. J. Adams, M. Wallace, R. Schweins and M. A. Zwijnenburg, Nanoscale, 2019, 11, 15917–15928 RSC.
- D. McDowall, B. J. Greeves, R. Clowes, K. McAulay, A. M. Fuentes-Caparrós, L. Thomson, N. Khunti, N. Cowieson, M. C. Nolan, M. Wallace, A. I. Cooper, E. R. Draper, A. J. Cowan and D. J. Adams, Adv. Energy Mater., 2020, 10, 2002469 CrossRef CAS.
- Y. Guo, Q. Zhou, J. Nan, W. Shi, F. Cui and Y. Zhu, Nat. Commun., 2022, 13, 2067 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, X. Chen, H. Zhang and J. Wang, Adv. Mater., 2019, 31, 1806626 CrossRef PubMed.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- C. Zhang, P. Chen, H. Dong, Y. Zhen, M. Liu and W. Hu, Adv. Mater., 2015, 27, 5379–5387 CrossRef CAS PubMed.
- Y. Zhong, J. Wang, R. Zhang, W. Wei, H. Wang, X. Lü, F. Bai, H. Wu, R. Haddad and H. Fan, Nano Lett., 2014, 14, 7175–7179 CrossRef CAS PubMed.
- J. Wang, Y. Zhong, L. Wang, N. Zhang, R. Cao, K. Bian, L. Alarid, R. E. Haddad, F. Bai and H. Fan, Nano Lett., 2016, 16, 6523–6528 CrossRef CAS PubMed.
- D. Wang, L. Niu, Z.-Y. Qiao, D.-B. Cheng, J. Wang, Y. Zhong, F. Bai, H. Wang and H. Fan, ACS Nano, 2018, 12, 3796–3803 CrossRef CAS PubMed.
- S. Mandal, S. K. Nayak, S. Mallampalli and A. Patra, ACS Appl. Mater. Interfaces, 2014, 6, 130–136 CrossRef CAS PubMed.
- N. Zhang, L. Wang, H. Wang, R. Cao, J. Wang, F. Bai and H. Fan, Nano Lett., 2018, 18, 560–566 CrossRef CAS PubMed.
- J. Jing, J. Yang, Z. Zhang and Y. Zhu, Adv. Energy Mater., 2021, 11, 2101392 CrossRef CAS.
- J. Jing, J. Yang, W. Li, Z. Wu and Y. Zhu, Adv. Mater., 2022, 34, 2106807 CrossRef CAS PubMed.
- X. Tian, C. Lin, Z. Zhong, X. Li, X. Xu, J. Liu, L. Kang, G. Chai and J. Yao, Cryst. Growth Des., 2019, 19, 3279–3287 CrossRef CAS.
- R. Cao, G. Wang, X. Ren, P.-C. Duan, L. Wang, Y. Li, X. Chen, R. Zhu, Y. Jia and F. Bai, Nano Lett., 2022, 22, 157–163 CrossRef CAS PubMed.
- P. Guo, P. Chen, W. Ma and M. Liu, J. Mater. Chem., 2012, 22, 20243 RSC.
- Y. Zhong, Z. Wang, R. Zhang, F. Bai, H. Wu, R. Haddad and H. Fan, ACS Nano, 2014, 8, 827–833 CrossRef CAS PubMed.
- Y. Liu, L. Wang, H. Feng, X. Ren, J. Ji, F. Bai and H. Fan, Nano Lett., 2019, 19, 2614–2619 CrossRef CAS PubMed.
- T. He, Y. Zhang, H. Zhang, J. Zhao, H. Shi, H. Yang and P. Yang, ChemSusChem, 2023, 16, e202300500 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Chem. Commun., 2016, 52, 6860–6872 RSC.
- J. Yu, L. Huang, Q. Tang, S.-B. Yu, Q.-Y. Qi, J. Zhang, D. Ma, Y. Lei, J. Su, Y. Song, J.-C. Eloi, R. L. Harniman, U. Borucu, L. Zhang, M. Zhu, F. Tian, L. Du, D. L. Phillips, I. Manners, R. Ye and J. Tian, Nat. Catal., 2023, 6, 464–475 CrossRef CAS.
- J. Ortony, J. Tian, D.-Y. Kim, W. Lindemann, T. Christoff-Tempesta, A. Lew and Y. Cho, USPTO, 20200298194A1, 2020 Search PubMed.
- E. Nikoloudakis, K. Karikis, J. Han, C. Kokotidou, A. Charisiadis, F. Folias, A. M. Douvas, A. Mitraki, G. Charalambidis, X. Yan and A. G. Coutsolelos, Nanoscale, 2019, 11, 3557–3566 RSC.
- M. Xu, K. Kong, H. Ding, Y. Chu, S. Zhang, F. Yu, H. Ye, Y. Hu and J. Hua, J. Mater. Chem. C, 2020, 8, 930–934 RSC.
- S. Adorinni, G. Goti, L. Rizzo, F. Grassi, S. Kralj, F. Matroodi, M. Natali, R. De Zorzi, S. Marchesan and L. Dell'Amico, Chem. Commun., 2023, 59, 7619–7622 RSC.
- K. Kong, S. Zhang, Y. Chu, Y. Hu, F. Yu, H. Ye, H. Ding and J. Hua, Chem. Commun., 2019, 55, 8090–8093 RSC.
- H. Li, C. Wang, X. Bai, X. Wang, B. Sun, D. Li, L. Zhao, R. Zong and D. Hao, Mater. Chem. Front., 2020, 4, 2673–2687 RSC.
- Y. Sheng, W. Li, L. Xu and Y. Zhu, Adv. Mater., 2022, 34, 2102354 CrossRef CAS PubMed.
- X. Xu, L. Meng, J. Zhang, S. Yang, C. Sun, H. Li, J. Li and Y. Zhu, Angew. Chem., Int. Ed., 2023, e202308597 Search PubMed.
- A. Zhang, D. Si, H. Huang, L. Xie, Z. Fang, T. Liu and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202203955 CrossRef CAS PubMed.
- L. Yang, J. Yuan, G. Wang, Q. Cao, C. Zhang, M. Li, J. Shao, Y. Xu, H. Li and J. Lu, Adv. Funct. Mater., 2023, 33, 2300954 CrossRef CAS.
- M. Hu, C. Wu, S. Feng and J. Hua, Molecules, 2023, 28, 6850 CrossRef CAS PubMed.
- H. Zhao, Z. Zhou, X. Feng, C. Liu, H. Wu, W. Zhou and H. Wang, Nano Res., 2023, 16, 8809–8816 CrossRef CAS.
- Q. Zhou, Y. Guo and Y. Zhu, Nat. Catal., 2023, 6, 574–584 CrossRef CAS.
- C. M. Aitchison, C. M. Kane, D. P. McMahon, P. R. Spackman, A. Pulido, X. Wang, L. Wilbraham, L. Chen, R. Clowes, M. A. Zwijnenburg, R. S. Sprick, M. A. Little, G. M. Day and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7158–7170 RSC.
- K. Ma, P. Li, J. H. Xin, Y. Chen, Z. Chen, S. Goswami, X. Liu, S. Kato, H. Chen, X. Zhang, J. Bai, M. C. Wasson, R. R. Maldonado, R. Q. Snurr and O. K. Farha, Cell Rep. Phys. Sci., 2020, 1, 100024 CrossRef.
- L. Tong, Y. Lin, X. Kou, Y. Shen, Y. Shen, S. Huang, F. Zhu, G. Chen and G. Ouyang, Angew. Chem., Int. Ed., 2023, 62, e202218661 CrossRef CAS PubMed.
- J. Li, Q. Yin, S.-Y. Gao, Y. Feng, S. Ye, H.-F. Li and R. Cao, ACS Sustainable Chem. Eng., 2023, 11, 4389–4397 CrossRef CAS.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11, 146–153 CrossRef CAS PubMed.
- F. Arcudi, L. Đorđević, B. Nagasing, S. I. Stupp and E. A. Weiss, J. Am. Chem. Soc., 2021, 143, 18131–18138 CrossRef CAS PubMed.
- F. Arcudi, L. Đorđević, N. Schweitzer, S. I. Stupp and E. A. Weiss, Nat. Chem., 2022, 14, 1007–1012 CrossRef CAS PubMed.
- H. Lin, Z. Ma, J. Zhao, Y. Liu, J. Chen, J. Wang, K. Wu, H. Jia, X. Zhang, X. Cao, X. Wang, X. Fu and J. Long, Angew. Chem., Int. Ed., 2021, 60, 1235–1243 CrossRef CAS PubMed.
- L. Zeng, T. Liu, C. He, D. Shi, F. Zhang and C. Duan, J. Am. Chem. Soc., 2016, 138, 3958–3961 CrossRef CAS PubMed.
- P. Verma, A. Singh, F. A. Rahimi, P. Sarkar, S. Nath, S. K. Pati and T. K. Maji, Nat. Commun., 2021, 12, 7313 CrossRef CAS PubMed.
- Z. Zhong, R. Li, W. Lin, X. Xu, X. Tian, X. Li, X. Chen and L. Kang, Appl. Catal., B, 2020, 260, 118135 CrossRef CAS.
- P. Verma, D. Samanta, P. Sutar, A. Kundu, J. Dasgupta and T. K. Maji, ACS Appl. Mater. Interfaces, 2023, 15, 25173–25183 CrossRef CAS PubMed.
- H. Ding, Z. Wang, K. Kong, S. Feng, L. Xu, H. Ye, W. Wu, X. Gong and J. Hua, J. Mater. Chem. A, 2021, 9, 7675–7683 RSC.
- Z. Wang, L. E. Lybarger, W. Wang, C. J. Medforth, J. E. Miller and J. A. Shelnutt, Nanotechnology, 2008, 19, 395604 CrossRef PubMed.
- V. Kunz, V. Stepanenko and F. Würthner, Chem. Commun., 2015, 51, 290–293 RSC.
- E. Nikoloudakis, M. Pigiaki, M. N. Polychronaki, A. Margaritopoulou, G. Charalambidis, E. Serpetzoglou, A. Mitraki, P. A. Loukakos and A. G. Coutsolelos, ACS Sustainable Chem. Eng., 2021, 9, 7781–7791 CrossRef CAS.
- K. Liu, R. Xing, C. Chen, G. Shen, L. Yan, Q. Zou, G. Ma, H. Möhwald and X. Yan, Angew. Chem., Int. Ed., 2015, 54, 500–505 CrossRef CAS PubMed.
- K. Liu, R. Xing, Y. Li, Q. Zou, H. Möhwald and X. Yan, Angew. Chem., Int. Ed., 2016, 55, 12503–12507 CrossRef CAS PubMed.
- Y. Xiu, X. Zhang, Y. Feng, R. Wei, S. Wang, Y. Xia, M. Cao and S. Wang, Nanoscale, 2020, 12, 15201–15208 RSC.
- J. H. Kim, D. H. Nam, Y. W. Lee, Y. S. Nam and C. B. Park, Small, 2014, 10, 1272–1277 CrossRef CAS.
- Q. Tang, Y. Han, L. Chen, Q. Qi, J. Yu, S. Yu, B. Yang, H. Wang, J. Zhang, S. Xie, F. Tian, Z. Xie, H. Jiang, Y. Ke, G. Yang, Z. Li and J. Tian, Angew. Chem., Int. Ed., 2024, e202315599 CAS.
- S. Frühbeißer and F. Gröhn, J. Am. Chem. Soc., 2012, 134, 14267–14270 CrossRef PubMed.
- Z. Wang, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 16720–16721 CrossRef CAS PubMed.
- Y. Tian, K. E. Martin, J. Y.-T. Shelnutt, L. Evans, T. Busani, J. E. Miller, C. J. Medforth and J. A. Shelnutt, Chem. Commun., 2011, 47, 6069 RSC.
- J. Yang, J. Jing and Y. Zhu, Adv. Mater., 2021, 33, 2101026 CrossRef CAS PubMed.
- D.-L. Jiang, C.-K. Choi, K. Honda, W.-S. Li, T. Yuzawa and T. Aida, J. Am. Chem. Soc., 2004, 126, 12084–12089 CrossRef CAS PubMed.
- W. Wei, D. Liu, Z. Wei and Y. Zhu, ACS Catal., 2017, 7, 652–663 CrossRef CAS.
- Y. Wang, Z. Zhao, R. Sun, J. Bian, Z. Zhang and L. Jing, Nanoscale, 2022, 14, 8041–8049 RSC.
- Y. Liang, W. Gui, Z. Yang, K. Cheng, X. Zhou, C. Yang, J. Xu and W. Zhou, RSC Adv., 2023, 13, 11938–11947 RSC.
- X. Li, X. Lv, Q. Zhang, B. Huang, P. Wang, X. Qin, X. Zhang and Y. Dai, J. Colloid Interface Sci., 2018, 525, 136–142 CrossRef CAS PubMed.
- S. Chen, D. L. Jacobs, J. Xu, Y. Li, C. Wang and L. Zang, RSC Adv., 2014, 4, 48486–48491 RSC.
- S. Chen, Y. Li and C. Wang, RSC Adv., 2015, 5, 15880–15885 RSC.
- J. Yang, H. Miao, W. Li, H. Li and Y. Zhu, J. Mater. Chem. A, 2019, 7, 6482–6490 RSC.
- D. Liu, Y. Li, X. Zhang, L. Yang and X. Luo, ChemCatChem, 2022, 14, e202200087 CrossRef CAS.
- W. Wei, Z. Wei, D. Liu and Y. Zhu, Appl. Catal., B, 2018, 230, 49–57 CrossRef CAS.
- J. T. Kirner, J. J. Stracke, B. A. Gregg and R. G. Finke, ACS Appl. Mater. Interfaces, 2014, 6, 13367–13377 CrossRef CAS PubMed.
- K. Zhang, J. Wang, W. Jiang, W. Yao, H. Yang and Y. Zhu, Appl. Catal., B, 2018, 232, 175–181 CrossRef CAS.
- S. H. Goudar, S. Bhoi, S. K. Sahoo, K. V. Rao and N. Kurra, Small, 2024, 2309905 CrossRef PubMed.
- J. Yang, H. Miao, Y. Wei, W. Li and Y. Zhu, Appl. Catal., B, 2019, 240, 225–233 CrossRef CAS.
- Q. Gao, J. Xu, Z. Wang and Y. Zhu, Appl. Catal., B, 2020, 271, 118933 CrossRef CAS.
- T. Hasobe, H. Sakai, K. Mase, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 4441–4449 CrossRef CAS.
- P. Guo, P. Chen and M. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 5336–5345 CrossRef CAS PubMed.
- D. D. La, T. A. Nguyen, X. S. Nguyen, T. N. Truong, H. P. Nguyen T., H. D. Ninh, H. T. Vo, S. V. Bhosale, S. W. Chang, E. R. Rene, T. H. Nguyen, S. M. Lee, L. D. Tran and D. D. Nguyen, J. Environ. Chem. Eng., 2021, 9, 106034 CrossRef CAS.
- V. Nikolaou, G. Charalambidis, K. Ladomenou, E. Nikoloudakis, C. Drivas, I. Vamvasakis, S. Panagiotakis, G. Landrou, E. Agapaki, C. Stangel, C. Henkel, J. Joseph, G. Armatas, M. Vasilopoulou, S. Kennou, D. M. Guldi and A. G. Coutsolelos, ChemSusChem, 2021, 14, 961–970 CrossRef CAS PubMed.
- H. Zhu, W. Zhu, S.-Z. Kang, L. Qin, T. Zhang and X. Li, Appl. Surf. Sci., 2024, 644, 158783 CrossRef CAS.
- D. D. La, A. Rananaware, M. Salimimarand and S. V. Bhosale, ChemistrySelect, 2016, 1, 4430–4434 CrossRef CAS.
- N. El-Shafai, M. E. El-Khouly, M. El-Kemary, M. S. Ramadan and M. S. Masoud, Photochem. Photobiol. Sci., 2019, 18, 2071–2079 CrossRef CAS PubMed.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2019, 4, 38–53 CrossRef.
- R. V. Kazantsev, A. J. Dannenhoffer, A. S. Weingarten, B. T. Phelan, B. Harutyunyan, T. Aytun, A. Narayanan, D. J. Fairfield, J. Boekhoven, H. Sai, A. Senesi, P. I. O'Dogherty, L. C. Palmer, M. J. Bedzyk, M. R. Wasielewski and S. I. Stupp, J. Am. Chem. Soc., 2017, 139, 6120–6127 CrossRef CAS PubMed.
- S. Biswas, M. Kumar, A. M. Levine, I. Jimenez, R. V. Ulijn and A. B. Braunschweig, Chem. Sci., 2020, 11, 4239–4245 RSC.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- W. Zhang, M. Yu, T. Liu, M. Cong, X. Liu, H. Yang, Y. Bai, Q. Zhu, S. Zhang, H. Gu, X. Wu, Z. Zhang, Y. Wu, H. Tian, X. Li, W.-H. Zhu and A. I. Cooper, Nat. Synth., 2024 DOI:10.1038/s44160-024-00494-9.
- B. Adelizzi, N. J. Van Zee, L. N. J. De Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110–6121 CrossRef CAS PubMed.
- A. Dannenhoffer, H. Sai, E. P. Bruckner, L. Đorđević, A. Narayanan, Y. Yang, X. Ma, L. C. Palmer and S. I. Stupp, Chem, 2023, 9, 170–180 CAS.
- J. Tian, Y. Zhang, L. Du, Y. He, X.-H. Jin, S. Pearce, J.-C. Eloi, R. L. Harniman, D. Alibhai, R. Ye, D. L. Phillips and I. Manners, Nat. Chem., 2020, 12, 1150–1156 CrossRef CAS PubMed.
- J. Seo, C. Kantha, J. F. Joung, S. Park, R. Jelinek and J. Kim, Small, 2019, 15, 1901342 CrossRef PubMed.
- A. Ashcraft, K. Liu, A. Mukhopadhyay, V. Paulino, C. Liu, B. Bernard, D. Husainy, T. Phan and J. Olivier, Angew. Chem., Int. Ed., 2020, 59, 7487–7493 CrossRef CAS PubMed.
- E. P. Bruckner, T. Curk, L. Đorđević, Z. Wang, Y. Yang, R. Qiu, A. J. Dannenhoffer, H. Sai, J. Kupferberg, L. C. Palmer, E. Luijten and S. I. Stupp, ACS Nano, 2022, 16, 8993–9003 CrossRef CAS PubMed.
- N. A. Sather, H. Sai, I. R. Sasselli, K. Sato, W. Ji, C. V. Synatschke, R. T. Zambrotta, J. F. Edelbrock, R. R. Kohlmeyer, J. O. Hardin, J. D. Berrigan, M. F. Durstock, P. Mirau and S. I. Stupp, Small, 2021, 17, 2005743 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. Sato and T. M. Hermans, Nat. Commun., 2017, 8, 15899 CrossRef CAS PubMed.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |